 Shuang Wang1
Shuang Wang1 Lei Sun1
Lei Sun1 Ning Ling2Chen Zhu2Fengqin Chi1Weiqun Li1Xiaoyu Hao1Wu Zhang3Jingyang Bian4Lei Chen1Dan Wei5*
Ning Ling2Chen Zhu2Fengqin Chi1Weiqun Li1Xiaoyu Hao1Wu Zhang3Jingyang Bian4Lei Chen1Dan Wei5*- 1Key Lab of Soil Environment and Plant Nutrition of Heilongjiang Province, Heilongjiang Fertilizer Engineering Research Center, Institute of Soil Fertilizer and Environment Resources, Heilongjiang Academy of Agricultural Sciences, Harbin, China
- 2Jiangsu Provincial Key Lab for Organic Solid Waste Utilization, Nanjing Agricultural University, Nanjing, China
- 3Heihe Branch of Heilongjiang Academy of Agricultural Sciences, Heihe, China
- 4Daqing Branch of Heilongjiang Academy of Agricultural Sciences, Daqing, China
- 5Institute of Plant Nutrition and Resources, Beijing Academy of Agriculture and Forestry Sciences, Beijing, China
Songnen Plain is originally one of the three major glasslands in China and has now become one of the three most concentrated distribution areas of sodic-saline soil worldwide. The soil is continuously degraded by natural and anthropogenic processes, which has a negative impact on agricultural production. The investigation of microbial diversity in this degraded ecosystem is fundamental for comprehending biological and ecological processes and harnessing the potential of microbial resources. The Illumina MiSeq sequencing method was practiced to investigate the bacterial diversity and composition in saline-alkali soil. The results from this study show that the change in pH under alkaline conditions was not the major contributor in shaping bacterial community in Songnen Plain. The electrical conductivity (EC) content of soil was the most important driving force for bacterial composition (20.83%), and the second most influencing factor was Na+ content (14.17%). Bacterial communities were clearly separated in accordance with the EC. The dominant bacterial groups were Planctomycetes, Proteobacteria, and Bacteroidetes among the different salinity soil. As the salt concentration increased, the indicators changed from Planctomycetes and Bacteroidetes to Proteobacteria and Firmicutes. Our results suggest that Proteobacteria and Firmicutes were the main indicator species reflecting changes of the main microbial groups and the EC as a key factor drives the composition of the bacterial community under alkaline conditions in saline-alkali soil of Songnen Plain.
Introduction
Salinity and/or sodicity is one of the main problems causing soil degradation, which is a grievous environmental problem with negative impacts on agricultural sustainable development. All over the world, over 800 million ha of land is estimated to be affected by salinity, which includes saline and alkaline soil (Yadav et al., 2011). It is reported that ~20% of the agricultural land worldwide is salt affected, and there is an increase in saline land of 1–1.5 × 106 ha worldwide every year (Sumner, 2000; Munns and Tester, 2008). If this continues, about half of cultivable land will be taken out of production by the middle of the 21st century (Mahajan and Tuteja, 2005). As one of the three most concentrated alkaline soil distribution areas worldwide, the Songnen Plain is located in the northeast of China (Wang et al., 2009). In this region, several environmental problems such as soil salinization, alkalinization, and desertification occur due to soil parent material, hydrological conditions, and overgrazing (Zhou et al., 2011). The annual rainfall exceeds the potential evaporation and poor management such as overgrazing and irrational utilization are common in the area (Gao and Liu, 2010; Liu et al., 2011a); thus, the affected alkali-saline area has been increasing in size by 20,000 ha per year (Zhang et al., 2015). Not only about two-thirds of the land in this area is salinized, but also increased by 1.5–2% annually (Ma et al., 2015). Under increase aridity and soil alkalization, large areas of croplands have been abandoned (Shang et al., 2003) and large proportions of grasslands have degraded seriously to unprecedented levels (Li et al., 2014).
Soil microorganisms participate in multiple aspects in the adjustment of ecological processes, for instance, degradation of organic matter, nutrient element transformations, enzyme production, and maintenance of soil quality (Sleator et al., 2008). Given that microorganisms are rapidly affected by the changes of their environment (Jiang et al., 2012), some biological characteristics (such as microbial diversity, composition, and structure) of soil are often considered to be sensitive and early indicators of dynamic environmental changes and soil ecological stress status (Li et al., 2011; Liu and Kang, 2014). For instance, soil microbial diversity was shaped by land-use changes, such as urbanization, agriculture, deforestation, and desertification. Many studies have shown that current environmental factors, for example, nutritional status, metalloid contamination, soil pH, the plant secretion (Hansel et al., 2008; Rousk et al., 2010; Xiong et al., 2010), and geographic distance (Xiong et al., 2012), influence structure and composition of microbial community.
Among the environmental factors, the pH was proposed to be a driver force for bacterial horizontal distribution in the soil (Shen et al., 2013; Liu et al., 2014). As soil salinization and alkalinization frequently co-occur, meta-analysis has been conducted merely on microbial diversity and composition (including bacteria and archaea) in saline soil habitats (Lozupone and Knight, 2007; Ma and Gong, 2013). Salinity is a dominant factor in determining bacterial community composition (Lozupone and Knight, 2007), however, relatively little is known about how the bacterial community composition response to the salinity gradients. Especially, the microbial structure under different salinities at similar pH has not been investigated.
Several recent studies on the composition of soil microbial diversity in saline soils revealed that soil salinization had negative effects not merely on soil biochemical properties, but also on the structure of microbial communities (Foti et al., 2007; Yuan et al., 2007; Hidri et al., 2013; Wang et al., 2014; Zhang et al., 2015). Regarding the soil microbial community in saline soils of Songnen Plain, only some representative new species of halophilic and halotolerant bacteria and archaea have been reported by pure culture methods (Wu et al., 2008; Wang et al., 2010; Liu et al., 2011b; He et al., 2014; Pan et al., 2016). These microorganisms have gradually formed adaptations, including unique structures and physiological functions, such as the accumulation of osmotic adjustment-related substances (Yan and Marschner, 2012). However, physical and chemical properties in saline-alkaline environments and microbial composition in Songnen Plain have not been sufficiently explored.
Systematic analysis of microbial diversity and composition in saline-alkaline soil of Songnen Plain is essential for gaining insight into the biological and ecological processes, saline adaption mechanisms, and digging the potential microbial resources from such environments. We collected 29 soil samples with different salinities across Songnen Plain and performed high throughput sequencing (Illumina MiSeq sequencing) to investigate the microbial diversity and composition in this under-studied system and to identify the key factors controlling the distribution of bacterial communities. The aim was to clarify the direct effects of environmental factors in shaping bacterial communities under geographic scale.
Materials and Methods
Site Description
The study area locates in the Songnen Plain (42°30′–51°20′N, 121°40′–128°30′E), Northeast China and belongs to a transitional zone between semi-humid and semi-arid regions, and is typically influenced by continental monsoon climate with mean annual temperature of 4.7°C (Shang et al., 2003; Yang et al., 2010). The average annual evaporation in this region is four times greater than the annual precipitation. The groundwater average mineral content is 2–5 g/L, with a maximum of 10 g/L, and the major anions present are CO32− and HCO3− (Zhang, 2014). The saline-sodic soils are characterized by a high pH (up to 10) and a large exchangeable Na percentage (Chi and Wang, 2010). The dominant zonal soils include meadow carbonate chernozem, deep chernozem, sodium carbonate-salinized soil, and dark chestnut soil. These soils are mainly distributed in the west part of Jilin and Heilongjiang provinces (Chi et al., 2011), including Zhenglai, Da’an, Changling, Qianguo, and Tongyu prefectures in Jilin province, and Zhaoyuan, Zhaozhou, Dumeng, Daqing, and Anda prefectures in Heilongjiang province (Wang et al., 2009). There are no naturally growing tree species because of salinization, only some salt/sodium-tolerant grass species such as Leymus chinensis, Puccinellia tenuiflora, and Suaeda corniculata are able to grow in the study area.
Site Selection and Soil Sampling
A total of 29 soil samples (with a mean depth of 0–15 cm) with GPS located site information from 12 counties (cities) across Jilin and Heilongjiang provinces were randomly collected in September 2013 (Figure 1). At each site, the analyzed soil samples mixture was from three soil samples collected in the vertices of 1 m side equilateral triangle. Then, the soil sample mixture from each site was divided into two subsamples: one was air dried, 2 mm sieved for soil physical and chemical analysis, and the other subsample was stored at −80°C for subsequent high-throughput sequencing.
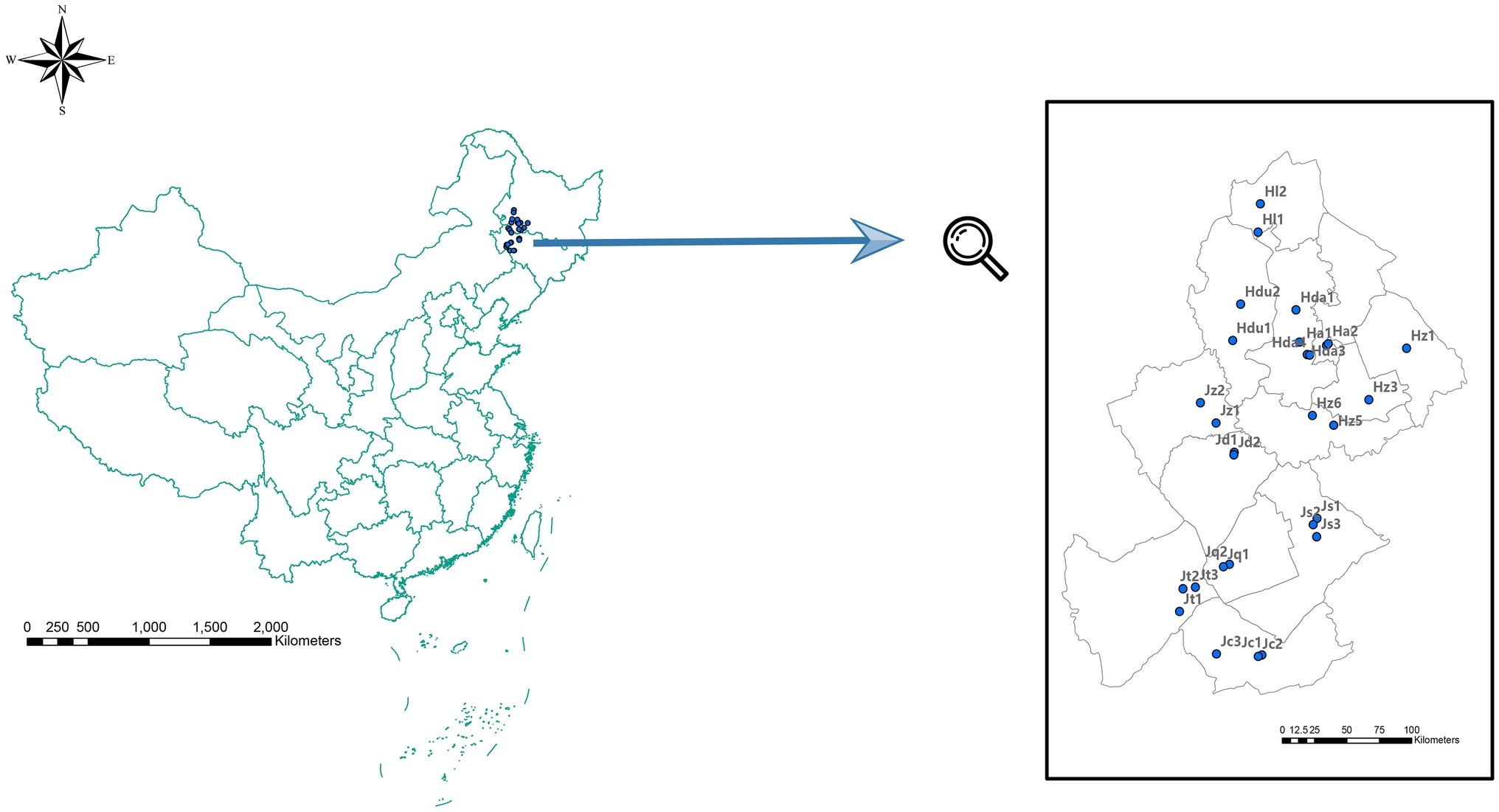
Figure 1. Sampling positions in Jilin Province [including Zhenglai (Jz), Da’an (Jd), Qianguo (Jq), Changling (Jc), Songyuan (Js), and Tongyu (Jt) prefectures], and in Heilongjiang Province [including Dumeng (Hdu), Lindian (Hl), Daqing (Hda), Anda (Ha) Zhaodong (Hz1), and Zhaozhou (Hz3) prefectures].
Soil Physical and Chemical Analysis
Soil organic carbon (SOC) was measured on a Total Organic Carbon (TOC) Analyzer (Multi N/C2100, Analytik-Jena, Germany). The electrical conductivity of the saturated paste extraction (EC) was measured using a conductivity meter (DDS-307A, REX, Shanghai), and pH was determined with air-dried soil (soil:water, 1:5) by pH meter (PHS-3C, REX, Shanghai). Soil exchangeable sodium(ammonium acetate exchange method) was determined by an atomic absorption spectrophotometer (TAS-990, Persee, Shanghai). Potentiometric titration was used to determine CO32−, HCO3−, Cl−, and SO42− with air-dried soil (soil:water, 1:5). The cations Ca2+ and Mg2+ were detected using Ethylenediaminetetraacetic acid (EDTA) titration and Na+ and K+ were measured using an atomic absorption spectrophotometer (TAS-990, Persee, Shanghai). Total content of dissolved salt (TDS) was determined by the drying-weighing method. The soil:water 1:5 tilt rate was placed in an oven at 105°C until constant weight was reached.
Soil DNA Extraction
The total DNA was extracted from 0.25 g soil samples using the Power Soil DNA Isolation Kit (Mo Bio Laboratories Inc., Carlsbad, CA, USA), followed by electrophoresis using a 1% agarose gel. The quality and quantity of DNA extracts (final volume, 100 μl) were examined using a Nano Drop spectrophotometer (Nano Drop Technologies, Wilmington, DE, USA).
Illumina Sequencing for Communities of Bacteria
The primer pair 515F (5′-GTGCCAGCMGCCGCGG-3′)/907R (5′-CCGTCAATTCMTTTRAGTTT-3′) was used to amplify the V4-V5 region of bacterial 16S rRNA genes (Yao et al., 2017). A 6-bp unique barcode unique to each sample was added into the reverse primers. Amplicon sequencing was performed using the Illumina MiSeq platform at Majorbio Inc. (Shanghai, China).
The sequencing data analysis was performed according to previous study (Zhu et al., 2016). Briefly, raw high-throughput sequencing data was processed using QIIME toolkit and the UPARSE pipeline (Caporaso et al., 2010; Edgar, 2013). After filtering DNA sequences using quality files, the remaining sequences were trimmed to remove barcodes and forward primers. The low quality (quality score < 20, length < 300 bp) sequences were excluded. The sequencing data were pre-treated to remove the chimeras from the datasets. After optimizing the sequences, the UPARSE pipeline was used to make an operational taxonomic unit (OTU) table. The identity threshold to bin the sequences into OTUs was 97%, and the most abundant sequence from each OTU was selected as the representative sequence for that OTU. The assignment of taxonomic data to microbial representative sequences was based on the Ribosomal Data Project (RDP) database. All sequencing data were deposited in the NCBI Sequence Read Archive (SRA) database (accession number: SRP172399).
Statistical Analysis
Analysis of variance was performed for each measured soil variable, and variance was compared between groups by a Fisher’s least significant difference test (α = 0.05). The richness index (Chao1 index), α-diversity (Shannon diversity) index, and rarefaction curves of soil samples were calculated by QIIME with normalized data. Non-metric multidimensional scaling (NMDS), and one-way PERMANOVA were used to analyze the β-diversity of bacteria in different treatments according to Bray-Curtis distance and Canonicla Correlation Analysis (CCA) was used to examined the relationships between the environmental factors and bacterial community structure with the R (2.15.3) packages ape and vegan. Aggregated boosted tree (ABT) analysis (De'ath, 2007), was performed using the gbmplus package (with 500 trees used for the boosting, 0.02-fold shrinkage rate and three-way interactions) to determine the relative influence of environmental variables on bacterial community composition (NMDS axis 1).
Network analyses were used to dissect the interrelationship and interaction between bacterial species along the different electrical conductivity (EC) gradients. All pair wise Pearson correlation coefficients were calculated by Mothur (version 1.29.2) for analysis of the networks. The correlation coefficient of Pearson correlation was further filtered with the cut-off as an absolute value of 0.6–0.93. After applying multiple hypothesis correction by the BH method (Benjamini and Hochberg, 1995), edges with adjusted values of p below 0.05 were kept and were further used to improve the veracity of the networks. Interactive networks were visualized by Gephi with a Fruchterman-Reingold layout (Bastian et al., 2009). The average clustering coefficient, average path length, and modularity of the network were calculated (Newman, 2006). We referred to the active species as the species that most strongly interacts with the other species within the networks (Magurran and Henderson, 2003; Montoya et al., 2006; Barberán et al., 2012). According to the network analysis, we selected the first 10 hubs under different salinity treatments. Times of iteration were 10,000. The indicator status of OTUs from each of the salinity treatments was assessed, and indicator of a treatment was conducted under the significance threshold of 0.05.
Results
Physical and Chemical Properties for All Collected Samples
To explore the soil factors that affect bacterial community composition, we first survey the physical and chemical properties in over 29 soil samples in different salinities collected from Jilin and Heilongjiang provinces (Figure 1). In all the studied sites, soil samples showed differences in pH. One sample from Ha2 had a pH of 8.84, whereas the pH of others was ranged from 9.89 to 10.66. However, the remaining physical and chemical properties, including various ion concentrations (Cl−, Na+, SO42−, CO32−, HCO3−, Ca2+, Mg2+, and Na+), SOC, EC, and SAR, varied more substantially than the pH. In particular, the lowest concentration of SO42− was 0.480 g/kg from Ha1, and the highest was 21.6 g/kg in Hda3, a difference reached 48 times. The same conspicuous change was observed in EC, the highest EC (Hda4: 10.867 ms/cm) was 26.3 times higher than the lowest value (Jc1: 0.413 ms/cm) (Supplementary Table S1).
Bacterial Community Analysis
The widespread change of the physical and chemical properties leads us to ask whether these properties would be associated with bacterial community in the salinity soils. The soil bacterial communities under different salinization levels were compared by sequencing of the bacterial 16S rRNA amplifications. Therefore, we sought to identify environmental factors that contribute to the ecological variation of bacterial community by analyzing characteristics of different soil samples (Supplementary Table S1), and creating ABT models to evaluate the relative impact of environmental factors on the bacterial composition NMDS axis 1. The NMDS result showed that the bacterial community structure was significantly separated in three EC levels (PERMANOVA, F = 0.2248, p < 0.001) (Figure 2). Soil EC was the most important driving force for microbial composition (20.83%, Figure 3), and the second most influencing factor was Na+ (14.17%) followed by Cl−. Given the key role of salinity in shaping the bacterial community, we tentatively divided all samples into three groups depending on the EC level as low (L)-, medium (M)- and high (H)-level treatment. The L, M, and H treatments represent EC of 0–2, 2–4, and >4 ms/cm, respectively. However, there was no significant diffidence in the α-diversity, including Chao1 index and Shannon index, among the three different salinity levels (Figure 4).
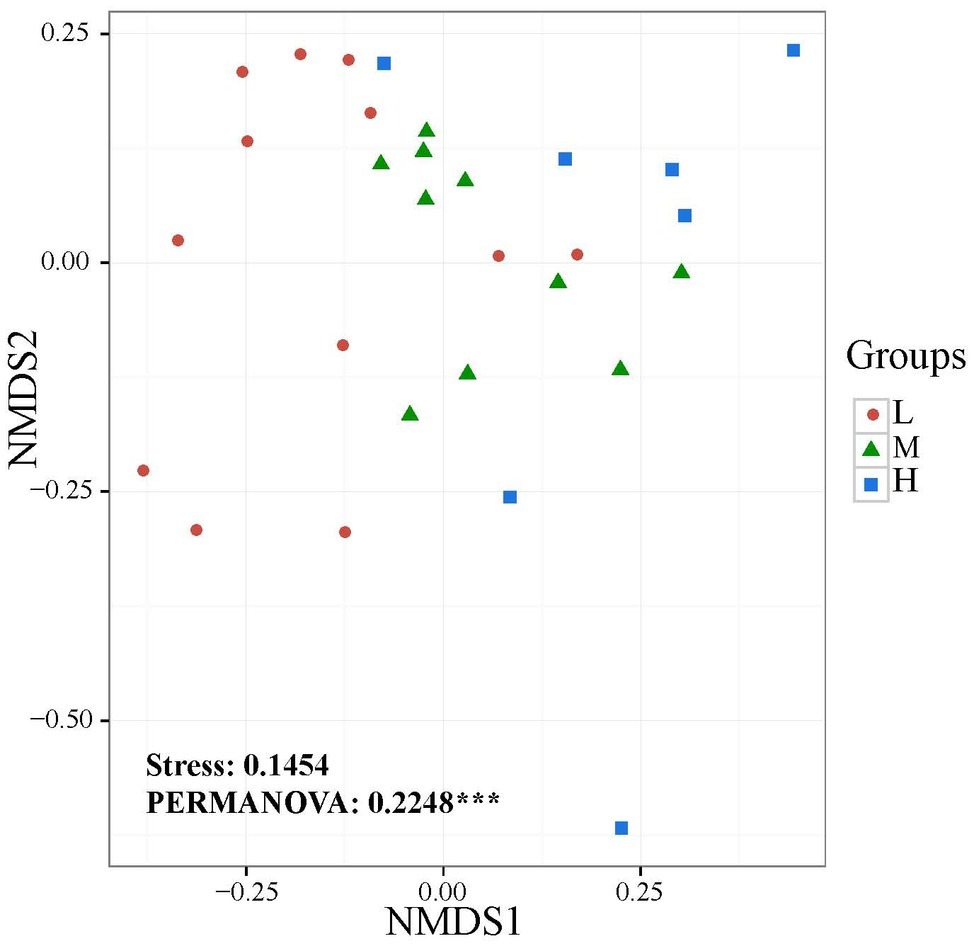
Figure 2. Non-metric multidimensional scaling (NMDS) ordination plot of soil bacteria community structure based on the number of OTUs detected by sequencing.
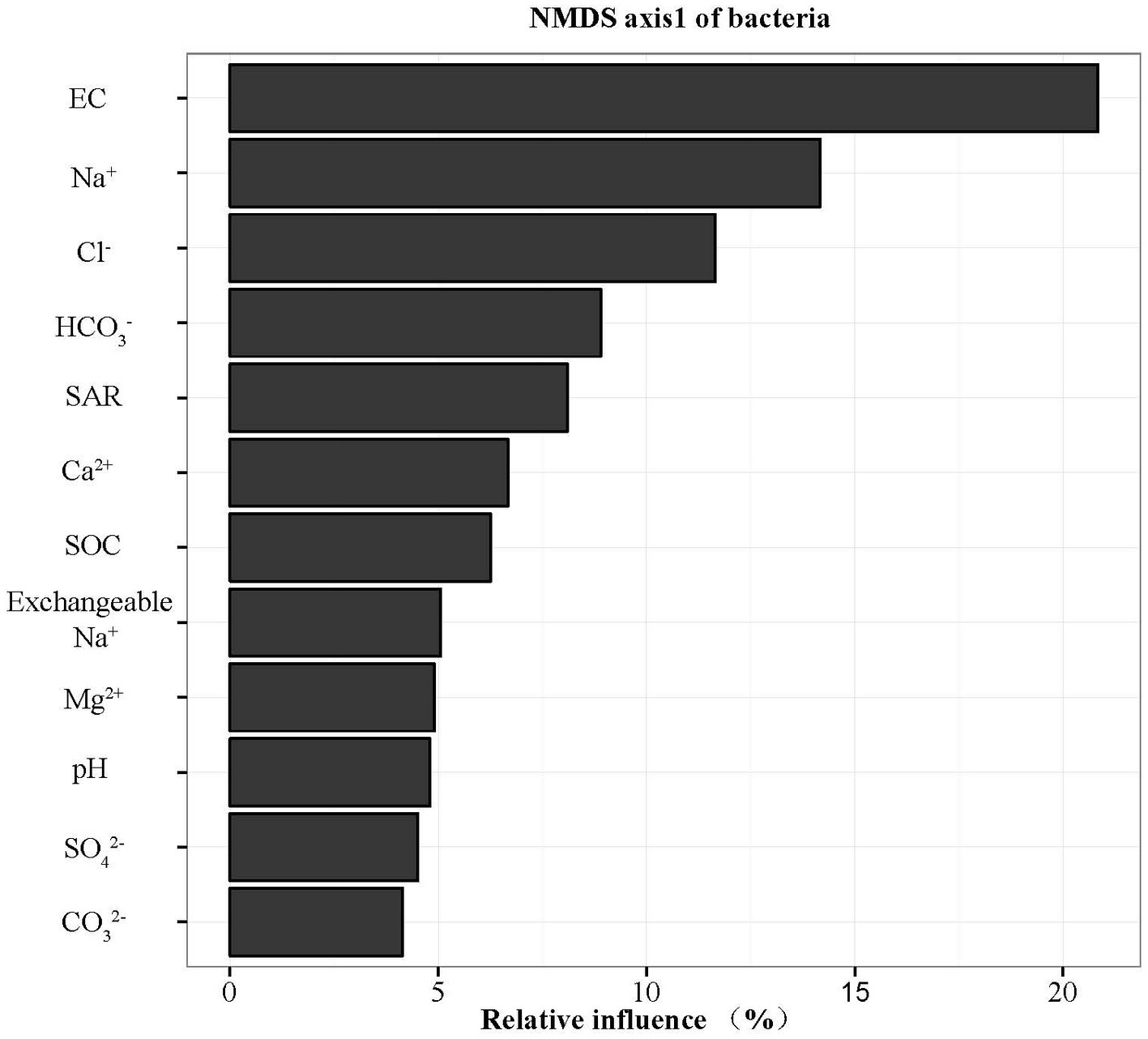
Figure 3. Relative variable importance plot (%) of environmental driving factors for composition of bacteria by ABT models.
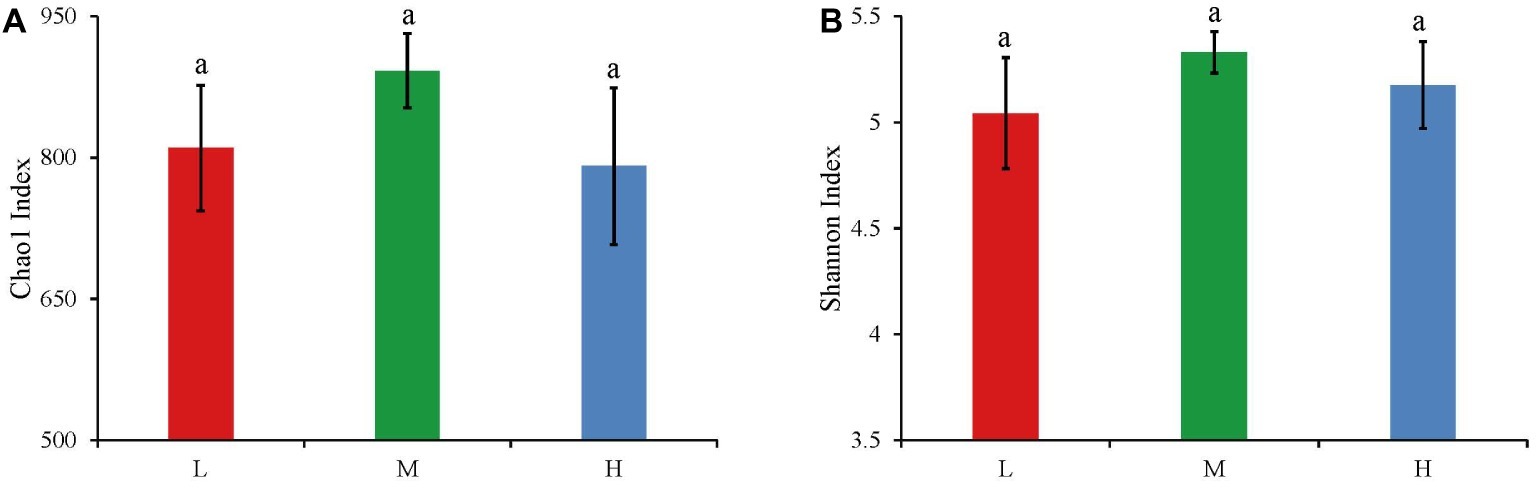
Figure 4. The Chao1 index (A) and Shannon index (B) of bacteria in three salinity rates. The values are the means of three replicates, error bars represent standard errors (L: n = 12; M: n = 10; H: n = 7), and different letters above the bars indicate significance differences at the p < 0.05 level. The L, M, and H treatments represent EC of 0–2, 2–4, and >4 ms/cm, respectively.
The Co-occurrence of Bacteria in Three-Salinity Gradient Treatments
Bacterial species usually work corporately to regulate soil properties. Based on the abovementioned statement about dividing the samples into three groups, we divided all samples into three salinity treatments (L, M, and H treatments) and the three treatment groups were used for network analysis. The network parameters of the three salinity levels were showed significant differences (Figure 5). In the L treatment, the network had 1,208 nodes and 29,183 edges, and the modularity was 0.72, with 10 modules. For the M treatment, the network presented 982 nodes and 12,819 edges, and the modularity was 0.82, with 9 modules. Finally, 878 nodes and 10,871 edges were found in the H treatment, for which the modularity was 0.74, with 7 modules. Nodes, edges, modularity, and modules all declined with increasing salinity, suggesting that a higher concentration of salt led to reducing connectivity of the bacterial network (Figure 5).
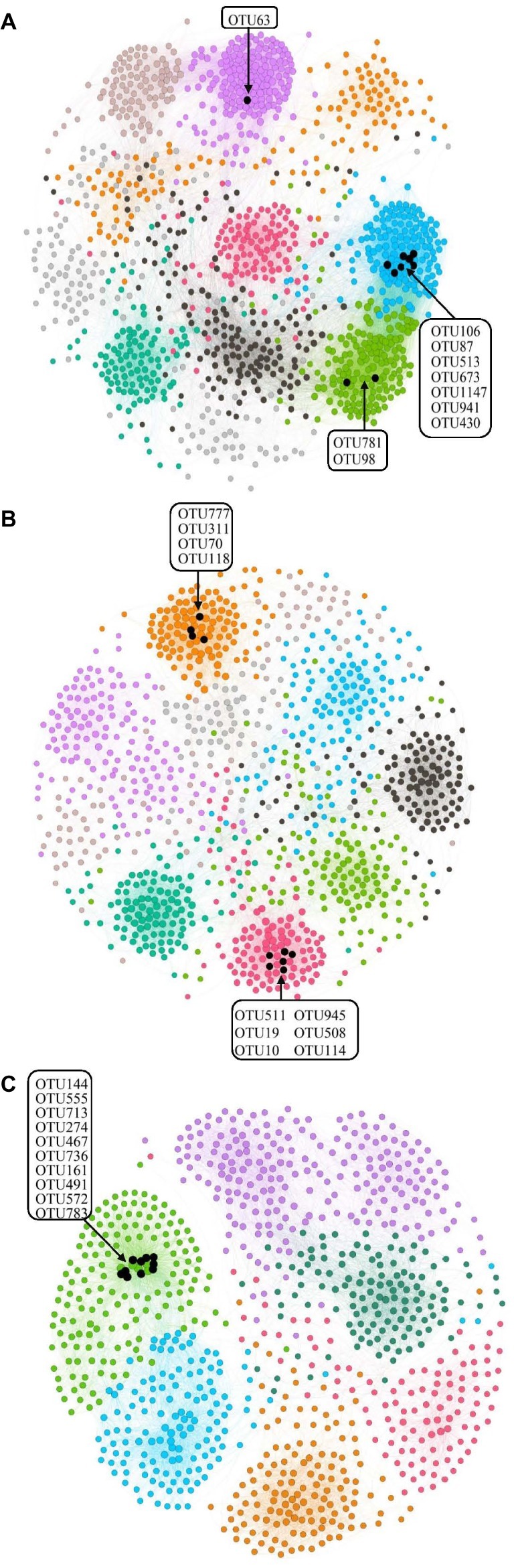
Figure 5. Network analysis of the three different salinity rates treatments based on Pearson correlations. The different color edges belong to different modules. (A) Low-salinity treatment (EC: 0–2) with modularity resolution of 0.72 and 10 modules; (B) middle-salinity treatment (EC: 2–4) with modularity resolution of 0.82 and 9 modules; and (C) high-salinity treatment (EC: >4) with modularity resolution of 0.74 and 7 modules. The black nodes indicate the OTUs belonging to active hubs in the network.
We defined the 10 nodes with largest number of edges as network hubs, which were active mediators in the bacterial community network (Supplementary Table S2). Taken as a whole, Planctomycetes, Proteobacteria, and Bacteroidetes were the three dominant phyla among the different salinity rate treatments, including 63.3% network hubs at the phylum level (Figure 6). Half of the network hubs in the L treatment belonged to Planctomycetes and Bacteroidetes, such as OTU513, OTU106, OTU63, OTU781, and OTU1147. The hubs of M treatment had similar phylogenetic classifications to the L treatment, with half assigned to Planctomycetes and Bacteroidetes. However, at the highest salinity, more than half hubs were classified as Proteobacteria, for instance, OTU713, OTU274, OTU572, OTU555, and OTU783. However, only one indicator (OTU144) belonged to the classification of phyla at low salt concentrations, while a second indicator (OTU777) belonged to the classification of phyla in moderate salt concentrations, which were identified as Planctomycetes and Bacteroidetes, respectively. As the salt concentration increased, the indicators changed from Planctomycetes and Bacteroidetes to Proteobacteria and Firmicutes. Supplementary Figure S2 shows that the Rhodospirillaceae, belonging to a family of Proteobacteria, had a strong correlation with the EC (R2 = 0.3365, p < 0.001). At the higher taxonomic level, the genus of Marinicella, assigned to Proteobacteria, also had a positive correlation with EC. The results revealed that multiple microbes belonging to Proteobacteria are well adapted to the high-salinity environment.
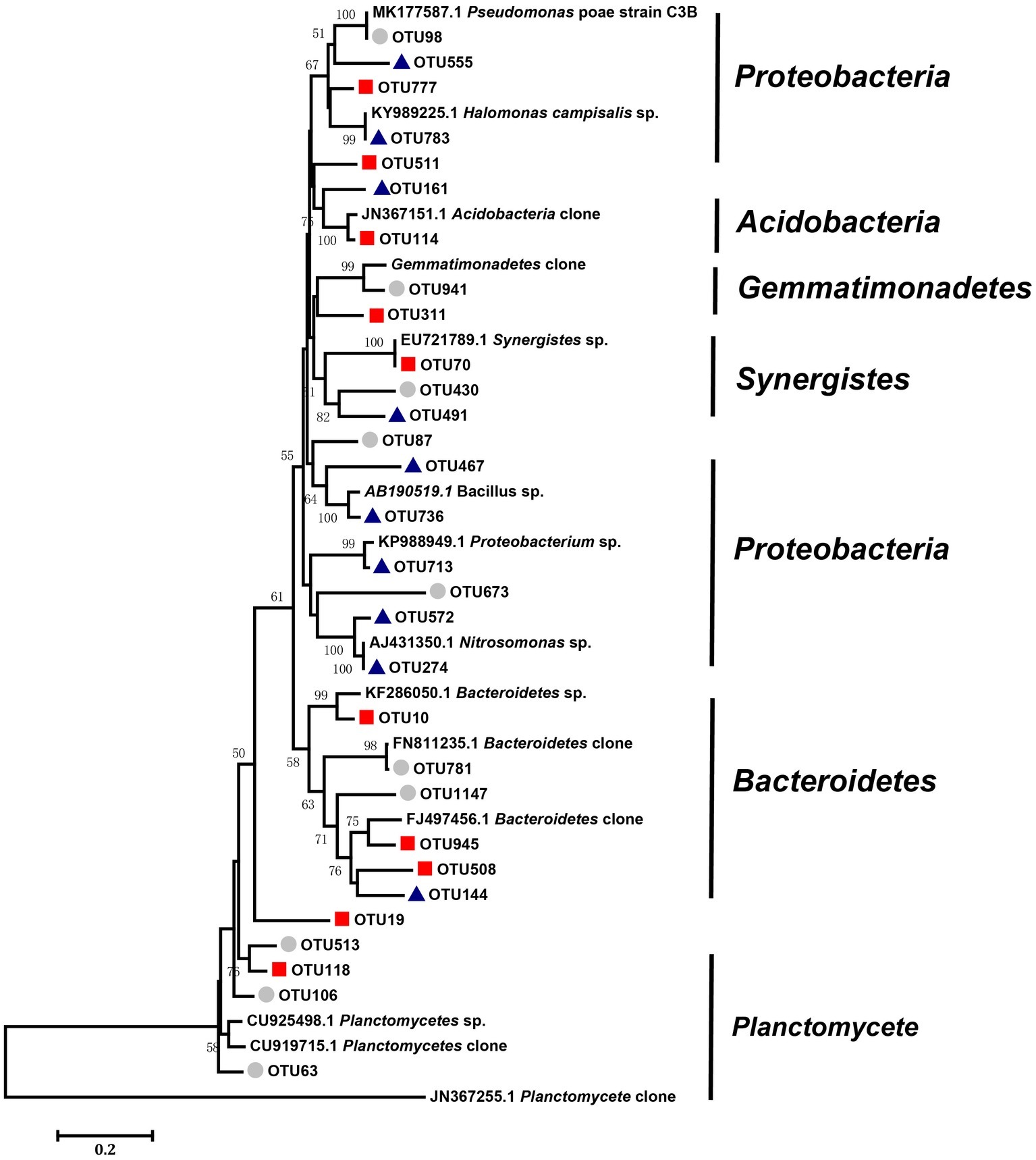
Figure 6. Neighbor-joining analysis of key OTUs forms the networks of three different salinity treatments (low, middle, and high). Bootstrap values higher than 40% are indicated at branch nodes. The scale bar represents 2% nucleic acid sequence divergence. The key OTUs as circles represent OTUs in the low-salinity treatment (EC: 0–2); the key OTUs as squares represent OTUs in the middle-salinity treatment (EC: 2–4); and the key OTUs as triangles represent OTUs in the high-salinity treatment (EC:>4).
Discussion
Driving Force for Microbial Community Composition in Saline Soils
We collected a total of 29 soil samples from Jilin and Heilongjiang provinces to investigate the distribution characteristics of microbes and reveal the general rule of microbial community composition under different salinities. Saline soils are characterized by high salt concentrations as well as an uneven temporal and spatial water distribution. The high salt concentrations shape the special environment patterns for microbes, causing the microbes in saline soils to vary from those found in the non-saline environment (Canfora et al., 2014). Firstly, we have analyzed the physical and chemical properties among the samples with different salinities. Although pH is a potential important determinant of salinity, we found that pH does not vary substantially among these samples, while the concentration of CO32− varies greatly, from 0.01 to 2.82 g/kg. Besides, the concentration of other ions, SOC, EC, and SAR are all different to some extent in the 29 soil samples, encouraging us to investigate whether the variation of physical and chemical properties was associated with salinity.
Identifying environmental factors contributing to the variation of microbial communities is a central aim in ecology. Several studies have shown that environmental factors, including soil pH (Rousk et al., 2010) and trophic status (Hansel et al., 2008; Xiong et al., 2010) affect the microbial community. As pH is the most important driver in bacterial community change (Shen et al., 2013; Liu et al., 2014), the explanation of pH for community variation only accounted for 4.80% in this study. ABT models were used to analyze the influence of the environmental factors on the bacterial community composition. EC content of soil was the most important driving force for microbial community composition (Figure 3) rather than soil pH. In CCA analysis, the EC value was the highest factor explaining the bacterial community by Monte Carlo permutation (Supplementary Table S3). This result showed gradient distribution of salinity along our sampling sites. Thus, EC could be the major factor controlling the differences. It has been reported recently that both low and high EC will affect bacterial growth in high-salinity soils (Rath et al., 2019), the influence of EC might be different among bacterial species, supporting our finding that EC is a causal factor for bacterial community in our salinity soil samples.
A previous study has shown that bacterial composition was associated with pH changes in both acidic soils and alkaline lake sediment (Xiong et al., 2012). While the current study shows that pH is unlikely to be the principal contributor to the considerable microbial community changes according to the ABT model. It should be noted that effect of pH in shaping bacterial communities is affected by local features. It is well known that salinization consists of salt accumulation by one or more natural processes including high salt content of the parent material or in groundwater, and human interventions such as inappropriate irrigation practices or inappropriate use of fertilization regimes. However, no matter which way, Na+ is a type of salt, which is the main component. For alkaliphilic bacterial taxa, the Na+ could serve as important proton replacement to cope with the high external pH (Canfora et al., 2014). As the relative abundance of these microbes are associated with the carbon mineralization rate in the soil (Fierer et al., 2007), we speculate the changes of dominant taxa that occur across the salinity gradient probably play a role in regulating ecosystem functions.
Changes in Diversity and Structure of Microbial Community in Saline Soils
In general, soil microbial diversity changes distinctly in response to environmental variations (Liu and Kang, 2014). However, in this study, the α-diversity did not change remarkably, whereas there was significant variation in β-diversity under the different salinity. Moreover, some studies have reported that α-diversity and β-diversity did not change simultaneously β-diversity transforms prior to α-diversity (Van Diepen et al., 2011; Xiong et al., 2012; Zhu et al., 2016). Furthermore, some researchers have suggested that the change of community structure (β-diversity) was typically correlated with shifts in functional behavior (Van Diepen et al., 2011; Fierer et al., 2013; Griffiths and Philippot, 2013). We speculated that the function responds sensitively and rapidly in the environment, which shows through β-diversity rather than α-diversity. In summary, these results suggest that the microbial community functions and structure respond initially to the environment variations, and then it takes a longer time for microbial α-diversity to change in the saline-alkali soils.
It has been a great challenge to dissect the association between microbial community structure and soil ecosystem. In this study, we used co-occurrence network analysis based on correlation to thoroughly dissect the microbial associations under salinity (Barberán et al., 2012; Faust and Raes, 2012; Friedman and Alm, 2012). Network analyses were conducted to reveal positive and negative interactions among different OTUs. A microbial community in the low-salinity soil was found to have a network with higher connectivity, suggesting more operational community with a greater number of functionally interrelated members (10 major modules) compared to the microbial community network in the high-salinity soil (7 major modules) (Figure 5). Given that the highly connected microbes within a module that co-occur might share similar ecological characteristics within communities (Chaffron et al., 2010; Williams et al., 2014; Yao et al., 2014), our results suggest that the high-salinity soil could harbor less ecologically similar functional groups.
Another important benefit of network analysis in microbial ecology studies is the ability to identify central organisms in maintaining soil ecosystems, according to the network theory, the “hub” species, as hotspots of connections in the microbial network, are the most important mediators for the complicated interactions among different species constituting the soil ecosystem (Montoya et al., 2006). According to the method described by the previous study (Zhu et al., 2018), 10 nodes with the most edges were defined as network hubs. Planctomycetes, Proteobacteria, and Bacteroidetes were the predominant phyla occupying 63.3% of the network hubs among the different salinity treatments (Figure 6). Ma and Gong (2013) reported that six phyla (Proteobacteria, Actinobacteria, Firmicutes, Acidobacteria, and Bacteroidetes) contained 90% of the bacterial sequences in saline soils; here, we detected five out of the six major phyla, with the exception for Acidobacteria. Proteobacteria, one common bacterial taxa in saline soil identified by a previous study (Valenzuela-Encinas et al., 2009), was the common denominator in our experimental sites, being especially dominant in highly saline soils, with 50% frequency in the indicators. In addition, Proteobacteria was reported as “salinity related” in a previous study (Yang et al., 2018), our results confirmed this finding, as Rhodospirillaceae and Marinicella, assigned to Proteobacteria, had good correlation with salt concentration. In addition, Firmicutes can also be considered special indicators specifically for the high salinity rate soil, which was absent in various hyper saline environments in previous studies (Demergasso et al., 2004; Ramette, 2007). Bacillus stands out among the prevailing genera assigned to Firmicutes, as it has shown to be an important resource for exploring halophilic enzymes and metabolic pathways for pollutant remediation in saline soil (Liszka et al., 2012). Species within Proteobacteria and Firmicutes may be good indicators, reflecting changes of the main microbial groups in saline-alkali soil. Although other studies have reported Gemmatimonadetes and Bacteroidetes to be an important participant in biogeochemical transformations in soils under salinity (Fierer et al., 2012; Ma and Gong, 2013), they were not detected in the high salinity soils of the current study. Different regions form different ecological environments, resulting in various microbial compositions. Therefore, our result suggests the requirement of future study on a wide range of spatial scales.
Our examination on the variability of the microbial community in saline soils successfully revealed microbial community subdivision across micro-environmental gradients. We expect future studies using metagenome sequencing data could identify similar patterns of bacterial composition variation at finer taxonomic resolution.
Conclusion
Taken together, this study presents an attempt to explore bacterial composition in saline-alkali soils across Jilin and Heilongjiang provinces. We show that bacterial β-diversity and community structure correlate with the salt gradient. We demonstrate that EC, instead of pH predicts bacterial community structure in saline-alkaline soils. Microbes belonging to the phyla Proteobacteria and Firmicutes were predominant and may be good groups of indicator species, reflecting changes of the main microbial groups in saline-alkali soil. Our results revealed local geochemical features as driving force of bacterial composition in the soil, whereas the EC as a key dominant factor in regulating microbial composition at a regional spatial scale. Correlating population of microbes with environmental parameters could facilitate reconstructing the formation of bacterial communities under specific environmental conditions like salinity. In this study, we provide a framework for future research to deeply analyze microbial composition in extreme environments.
Data Availability Statement
The datasets generated for this study can be found in the NCBI Sequence Read Archive (SRA) database/SRP172399.
Author Contributions
DW and FC contributed to the conception of the study. SW and LS contributed significantly to analysis and manuscript preparation. WL, XH, WZ, and JB performed the collection of soil samples and data analyses. NL revised the manuscript. LC played an important role in interpreting the results. CZ helped perform the analysis with constructive discussions.
Funding
This work was supported by the National Key Research and Development Program of China (2017YFD0200604), the National Natural Science Foundation of China (41771284), the Major Projects of Research and Development Program of Application Technology of Heilongjiang Province of China (GA19B105), the Heilongjiang Natural Science Foundation (ZD2016008), the Special projects for the central government to guide local scientific and technological development (ZY18A04).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02902/full#supplementary-material
References
Barberán, A., Bates, S. T., Casamayor, E. O., and Fierer, N. (2012). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351. doi: 10.1038/ismej.2011.119
Bastian, M., Heymann, S., and Jacomy, M. (2009). Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media, 361–362. Available at: https://www.aaai.org/ocs/index.php/ICWSM/09/paper/view/154/1009 (Accessed October 23, 2018).
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 57, 289–300.
Canfora, L., Bacci, G., Pinzari, F., Papa, G. L., Dazzi, C., and Benedetti, A. (2014). Salinity and bacterial diversity: to what extent does the concentration of salt affect the bacterial community in a saline soil? PLoS One 9:e106662. doi: 10.1371/journal.pone.0106662
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335. doi: 10.1038/nmeth.f.303
Chaffron, S., Rehrauer, H., Pernthaler, J., and von Mering, C. (2010). A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 20, 947–959. doi: 10.1101/gr.104521.109
Chi, C., and Wang, Z. (2010). Characterizing salt-affected soils of Songnen plain using saturated paste and 1:5 soil-to-water extraction methods. Arid Land Res. Manag. 24, 1–11. doi: 10.1080/15324980903439362
Chi, C., Zhao, C., Sun, X., and Wang, Z. (2011). Estimating exchangeable sodium percentage from sodium adsorption ratio of salt-affected soil in the Songnen plain of Northeast China. Pedosphere 21, 271–276. doi: 10.1016/S1002-0160(11)60127-6
De'ath, G. (2007). Boosted trees for ecological modeling and prediction. Ecology 88, 243–251. doi: 10.1890/0012-9658(2007)88[243:BTFEMA]2.0.CO;2
Demergasso, C., Casamayor, E. O., Chong, G., Galleguillos, P., Escudero, L., and Pedrós-Alió, C. (2004). Distribution of prokaryotic genetic diversity in athalassohaline lakes of the Atacama Desert, Northern Chile. FEMS Microbiol. Ecol. 48, 57–69. doi: 10.1016/j.femsec.2003.12.013
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial ampliconreads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Faust, K., and Raes, J. (2012). Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550. doi: 10.1038/nrmicro2832
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Fierer, N., Ladau, J., Clemente, J. C., Leff, J. W., Owens, S. M., Pollard, K. S., et al. (2013). Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 342, 621–623. doi: 10.1126/science.1243768
Fierer, N., Leff, J. W., Adams, B. J., Nielsen, U. N., Bates, S. T., Lauber, C. L., et al. (2012). Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. PNAS 52, 21390–21395. doi: 10.1073/pnas.1215210110
Foti, M. J., Sorokin, D. Y., Lomans, B., Mussman, M., Zacharova, E. E., Pimenov, N. V., et al. (2007). Diversity, activity, and abundance of sulfate-reducing bacteria in saline and hypersaline soda lakes. Appl. Environ. Microbiol. 73, 2093–2100. doi: 10.1128/AEM.02622-06
Friedman, J., and Alm, E. J. (2012). Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8:e1002687. doi: 10.1371/journal.pcbi.1002687
Gao, J., and Liu, Y. (2010). Determination of land degradation causes in Tongyucounty, Northeast China via land cover change detection. Int. J. Appl. Earth Obs. Geoinf. 12, 9–16. doi: 10.1016/j.jag.2009.08.003
Griffiths, B. S., and Philippot, L. (2013). Insights into the resistance and resilience of the soil microbial community. FEMS Microbiol. Rev. 37, 112–129. doi: 10.1111/j.1574-6976.2012.00343.x
Hansel, C. M., Fendorf, S., Jardine, P. M., and Francis, C. A. (2008). Changes in bacterial and archaeal community structure and functional diversity along a geochemically variable soil profile. Appl. Environ. Microbiol. 74, 1620–1633. doi: 10.1128/AEM.01787-07
He, H., Liu, C., Zhao, J., Li, W., Pan, T., Yang, L., et al. (2014). Streptomyces zhaozhouensis sp. nov., an actinomycete isolated from candelabra aloe (Aloe arborescens mill). Int. J. Syst. Evol. Microbiol. 64, 1096–1101. doi: 10.1099/ijs.0.056317-0
Hidri, D. E., Guesmi, A., Najjari, A., Cherif, H., Ettoumi, B., Hamdi, C., et al. (2013). Cultivation-dependent assessment, diversity, and ecology of haloalkaliphilic bacteria in arid saline systems of southern Tunisia. BioMed Res. Int. 2013, 1–15. doi: 10.1155/2013/648141
Jiang, Y., Cheng, C., Xu, Z., and Liu, Y. (2012). Effects of single and mixed species forest ecosystems on diversity and function of soil microbial community in subtropical China. J. Soils Sediments 12, 228–240. doi: 10.1007/s11368-011-0442-4
Li, X., Liu, B., Cui, J., Liu, D., Ding, S., Gilna, B., et al. (2011). No evidence of persistent effects of continuously planted transgenic insect-resistant cotton on soil microorganisms. Plant Soil 339, 247–257. doi: 10.1007/s11104-010-0572-2
Li, Q., Yu, P., Chen, X., Li, G., Zhou, D., and Zheng, W. (2014). Facilitative and inhibitory effect of litter on seedling emergence and early growth of six herbaceous species in an early successional old field ecosystem. Sci. World J. 2014, 1–11. doi: 10.1155/2014/101860
Liszka, M., Clark, M., Schneider, E., and Clark, D. S. (2012). Nature versus nurture: developing enzymes that function under extreme conditions. Ann. Rev. Chem. Biomol. Eng. 3, 77–102. doi: 10.1146/annurev-chembioeng-061010-114239
Liu, S., and Kang, Y. (2014). Changes of soil microbial characteristics in saline-sodic soils under drip irrigation. J. Siol. Sci. Plant Nut. 14, 139–150.
Liu, S., Kang, Y., Wan, S., Wang, Z., Liang, Z., and Sun, X. (2011a). Water and salt regulation and its effects on Leymuschinensis growth under drip irrigation in saline-sodic soils of the Songnen plain. Agric. Water Manage. 98, 1469–1476. doi: 10.1016/j.agwat.2011.04.016
Liu, Y., Song, X., Jiang, J., Liu, Y., Xu, C., Li, H., et al. (2011b). Hydrocarboniphaga daqingensis sp. nov., isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 61, 408–411. doi: 10.1099/ijs.0.019380-0
Liu, J., Sui, Y., Yu, Z., Shi, Y., Chu, H., Jin, J., et al. (2014). High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of Northeast China. Soil Biol. Biochem. 70, 113–122. doi: 10.1016/j.soilbio.2013.12.014
Lozupone, C. A., and Knight, R. (2007). Global patterns in bacterial diversity. PNAS 104, 11436–11440. doi: 10.1073/pnas.0611525104
Ma, B., and Gong, J. (2013). A meta-analysis of the publicly available bacterial and archaeal sequence diversity in saline soils. World J. Microb. Biot. 29, 2325–2334. doi: 10.1007/s11274-013-1399-9
Ma, H., Yang, H., Liang, Z., and Ooi, M. K. J. (2015). Effects of 10-year management regimes on the soil seed bank in saline-alkaline grassland. PLoS One 10, 1–17. doi: 10.1371/journal.pone.0122319
Magurran, A. E., and Henderson, P. A. (2003). Expanding the excess of rare species in natural species abundance distributions. Nature 422, 714–716. doi: 10.1038/nature01547
Mahajan, S., and Tuteja, N. (2005). Cold, salinity and drought stresses: an overview. Arch. Biochem. Biophys. 444, 139–158. doi: 10.1016/j.abb.2005.10.018
Montoya, J. M., Pimm, S. L., and Sole, R. V. (2006). Ecological network and their fragility. Nature 442, 259–264. doi: 10.1038/nature04927
Munns, R., and Tester, M. (2008). Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 59, 651–681. doi: 10.1146/annurev.arplant.59.032607.092911
Newman, M. E. J. (2006). Modularity and community structure in networks. PNAS 103, 8577–8582. doi: 10.1073/pnas.0601602103
Pan, T., He, H., Li, C., Zhao, J., Zhang, Y., Li, J., et al. (2016). Streptomyces daqingensis sp. nov., isolated from saline-alkaline soil. Int. J. Syst. Evol. Microbiol. 66, 1358–1363. doi: 10.1099/ijsem.0.000887
Ramette, A. (2007). Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 62, 142–116. doi: 10.1111/j.1574-6941.2007.00375.x
Rath, K. M., Fierer, N., Murphy, D. V., and Rousk, J. (2019). Linking bacterial community composition to soil salinity along environmental gradients. ISME J. 13, 836–846. doi: 10.1038/s41396-018-0313-8
Rousk, J., Bååth, E., Brookes, P. C., Lauber, C. L., Lozupone, C., Caporaso, J. G., et al. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4, 1340–1351. doi: 10.1038/ismej.2010.58
Shang, Z., Gao, Q., and Dong, M. (2003). Impacts of grazing on the alkalinized-salinized meadow steppe ecosystem in the Songnen plain, China - a simulation study. Plant Soil 249, 237–251. doi: 10.1023/A:1022848329303
Shen, C., Xiong, J., Zhang, H., Feng, Y., Lin, X., Li, X., et al. (2013). Soil pH drives the spatial distribution of bacterial communities along elevation on Changbai Mountain. Soil Biol. Biochem. 57, 204–211. doi: 10.1016/j.soilbio.2012.07.013
Sleator, R. D., Shortall, C., and Hill, C. (2008). Metagenomics. Lett. Appl. Microbiol. 47, 361–366. doi: 10.1111/j.1472-765X.2008.02444.x
Valenzuela-Encinas, C., Neria-González, I., Alcántara-Hernández, R. J., Estrada-Alvarado, I., Zavala-Díaz de la Serna, F. J., Dendooven, L., et al. (2009). Changes in the bacterial populations of the highly alkaline saline soil of the former Lake Texcoco (Mexico) following flooding. Extremophiles 13, 609–621. doi: 10.1007/s00792-009-0244-4
Van Diepen, L. T. A., Lilleskov, E. A., and Pregitzer, K. S. (2011). Simulated nitrogen deposition affects community structure of arbuscular mycorrhizal fungi in northern hardwood forests. Mol. Ecol. 20, 799–811. doi: 10.1111/j.1365-294X.2010.04969.x
Wang, L., Seki, K., Miyazaki, T., and Ishihama, Y. (2009). The causes of soil alkalinization in the Songnen plain of Northeast China. Paddy Water Environ. 7, 259–270.
Wang, S., Sun, L., Wei, D., Zhou, B., Zhang, J., Gu, X., et al. (2014). Bacillus daqingensis sp. nov., a halophilic, alkaliphilic bacterium isolated from saline-sodic soil in Daqing, China. J. Microbiol. 52, 548–553. doi: 10.1007/s12275-014-3376-x
Wang, S., Yang, Q., Liu, Z., Sun, L., Wei, D., Zhang, J. Z., et al. (2010). Haloterrigena daqingensis sp. nov., an extremely haloalkaliphilic archaeon isolated from a saline-alkaline soil. Int. J. Syst. Evol. Microbiol. 60, 2267–2271. doi: 10.1099/ijs.0.013995-0
Williams, R. J., Howe, A., and Hofmockel, K. S. (2014). Demonstrating microbial cooccurrence pattern analyses within and between ecosystems. Front. Microbiol. 5:358. doi: 10.3389/fmicb.2014.00358
Wu, G., Wu, X. Q., Wang, Y. N., Chi, C. Q., Tang, Y. Q., Kida, K., et al. (2008). Halomonas daqingensis sp. nov., a moderately halophilic bacterium isolated from an oilfield soil. Int. J. Syst. Evol. Microbiol. 58, 2859–2865. doi: 10.1099/ijs.0.65746-0
Xiong, J., Liu, Y., Lin, X., Zhang, H., Zeng, J., Hou, J., et al. (2012). Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan plateau. Environ. Microbiol. 14, 2457–2466. doi: 10.1111/j.1462-2920.2012.02799.x
Xiong, J., Wu, L., Tu, S., Van Nostrand, J. D., He, Z., Zhou, J., et al. (2010). Microbial communities and functional genes associated with soil arsenic contamination and rhizosphere of the arsenic hyper-accumulating plant Pterisvittata L. Appl. Environ. Microbiol. 76, 7277–7284. doi: 10.1128/AEM.00500-10
Yadav, S., Irfan, M., Ahmad, A., and Hayat, S. (2011). Causes of salinity and plant manifestations to salt stress: a review. J. Environ. Biol. 32, 667–685.
Yan, N., and Marschner, P. (2012). Response of microbial activity and biomass to increasing salinity depends on the final salinity, not the original salinity. Soil Biol. Biochem. 53, 50–55. doi: 10.1016/j.soilbio.2012.04.028
Yang, J., Zhan, C., Li, Y., Zhou, D., Yu, Y., and Yu, J. (2018). Effect of salinity on soil respiration in relation to dissolved organic carbon and microbial characteristics of a wetland in the Liaohe River estuary, Northeast China. Sci. Total Environ. 642, 946–953. doi: 10.1016/j.scitotenv.2018.06.121
Yang, J., Zhang, S., Li, Y., Bu, K., Zhang, Y., Chang, L., et al. (2010). Dynamics of saline-alkali land and its ecological regionalization in western Songnen plain, China. Chin. Geogra. Sci. 20, 159–166. doi: 10.1007/s11769-010-0159-0
Yao, Q., Liu, J., Yu, Z., Li, Y., Jin, J., Liu, X., et al. (2017). Changes of bacterial community compositions after three years of biochar application in a black soil of Northeast China. Appl. Soil Ecol. 113, 11–21. doi: 10.1016/j.apsoil.2017.01.007
Yao, M., Rui, J., Li, J., Dai, Y., Bai, Y., Heděnec, P., et al. (2014). Rate-specific responses of prokaryotic diversity and structure to nitrogen deposition in the Leymuschinensis steppe. Soil Biol. Biochem. 79, 81–90. doi: 10.1016/j.soilbio.2014.09.009
Yuan, B. C., Li, Z. Z., Liu, H., Gao, M., and Zhang, Y. Y. (2007). Microbial biomass and activity in salt affected soils under arid conditions. Appl. Soil Ecol. 35, 19–328. doi: 10.1016/j.apsoil.2006.07.004
Zhang, J. (2014). Coastal saline soil rehabilitation and utilization based on forestry approaches in China. Berlin: Springer-Verlag Press.
Zhang, Y., Cao, C., Guo, L., Wu, Q., and Cui, Z. (2015). Soil properties, bacterial community composition, and metabolic diversity responses to soil salinization of a semiarid grassland in Northeast China. J. Soil Water Conserv. 70, 110–120. doi: 10.2489/jswc.70.2.110
Zhou, D., Li, Q., Song, Y., and Wang, X. (2011). Salinizaion-alkalization of Leymuschinensis glassland in Songnen plain of Northease China. Chin. J. Appl. Ecol. 22, 1423–1430.
Zhu, C., Ling, N., Guo, J., Wang, M., Guo, S., and Shen, Q. (2016). Impacts of fertilization regimes on arbuscular mycorrhizal fungal (AMF) community composition were correlated with organic matter composition in maize rhizospheresoil. Front. Microbiol. 7:1840. doi: 10.3389/fmicb.2016.01840
Keywords: bacterial community, driving force, electrical conductivity, saline-alkali soil, Songnen Plain
Citation: Wang S, Sun L, Ling N, Zhu C, Chi F, Li W, Hao X, Zhang W, Bian J, Chen L and Wei D (2020) Exploring Soil Factors Determining Composition and Structure of the Bacterial Communities in Saline-Alkali Soils of Songnen Plain. Front. Microbiol. 10:2902. doi: 10.3389/fmicb.2019.02902
Edited by:
Wen-Jun Li, Sun Yat-Sen University, ChinaReviewed by:
Junjie Liu, Northeast Institute of Geography and Agroecology (CAS), ChinaYu Shi, Institute of Soil Science (CAS), China
Osama Abdalla Abdelshafy Mohamad, Arish University, Egypt
Copyright © 2020 Wang, Sun, Ling, Zhu, Chi, Li, Hao, Zhang, Bian, Chen and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Wei, d2QyMDg3QDE2My5jb20=