 Liliana R. Teixeira1
Liliana R. Teixeira1 Cristina M. Cordas2
Cristina M. Cordas2 Marta P. Fonseca1Norma E. C. Duke3†
Marta P. Fonseca1Norma E. C. Duke3† Phani Raj Pokkuluri3*†
Phani Raj Pokkuluri3*† Carlos A. Salgueiro1*
Carlos A. Salgueiro1*- 1UCIBIO-Requimte, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade NOVA de Lisboa, Caparica, Portugal
- 2LAQV, REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade NOVA de Lisboa, Caparica, Portugal
- 3Biosciences Division, Argonne National Laboratory, Lemont, IL, United States
The monoheme outer membrane cytochrome F (OmcF) from Geobacter sulfurreducens plays an important role in Fe(III) reduction and electric current production. The electrochemical characterization of this cytochrome has shown that its redox potential is modulated by the solution pH (redox-Bohr effect) endowing the protein with the necessary properties to couple electron and proton transfer in the physiological range. The analysis of the OmcF structures in the reduced and oxidized states showed that with the exception of the side chain of histidine 47 (His47), all other residues with protonatable side chains are distant from the heme iron and, therefore, are unlikely to affect the redox potential of the protein. The protonatable site at the imidazole ring of His47 is in the close proximity to the heme and, therefore, this residue was suggested as the redox-Bohr center. In the present work, we tested this hypothesis by replacing the His47 with non-protonatable residues (isoleucine – OmcFH47I and phenylalanine – OmcFH47F). The structure of the mutant OmcFH47I was determined by X-ray crystallography to 1.13 Å resolution and showed only minimal changes at the site of the mutation. Both mutants were 15N-labeled and their overall folding was confirmed to be the same as the wild-type by NMR spectroscopy. The pH dependence of the redox potential of the mutants was measured by cyclic voltammetry. Compared to the wild-type protein, the magnitude of the redox-Bohr effect in the mutants was smaller, but not fully abolished, confirming the role of His47 on the pH modulation of OmcF’s redox potential. However, the pH effect on the heme substituents’ NMR chemical shifts suggested that the heme propionate P13 also contributes to the overall redox-Bohr effect in OmcF. In physiological terms, the contribution of two independent acid–base centers to the observed redox-Bohr effect confers OmcF a higher versatility to environmental changes by coupling electron/proton transfer within a wider pH range.
Introduction
c-type cytochromes are key elements for the extracellular electron transfer processes in Geobacter species (Methé et al., 2003; Morgado et al., 2012). The genome of Geobacter sulfurreducens encodes for 128 c-type cytochromes out of which 31 are predicted to be located in the outer membrane (Methé et al., 2003). The outer membrane cytochrome F (OmcF) is one of these proteins. Genetic and proteomics studies have suggested that OmcF is an important protein for extracellular electron transfer in the respiratory pathways responsible for Fe(III) reduction and current production by G. sulfurreducens (Kim et al., 2005, 2008). However, the results obtained suggested that OmcF is not directly involved in these respiratory pathways being necessary for the transcription of the appropriate genes encoding for proteins directly involved in Fe(III) reduction (OmcB and OmcC) or electricity production in microbial fuel cells (OmcE and OmcS) (Kim et al., 2005, 2008).
OmcF is a 104 amino acids protein with a predicted small lipid anchor at the N-terminus formed by the first 19 residues, a soluble domain consisting of residues 20–104 and a low-spin c-type heme group with axial His-Met coordination (Pokkuluri et al., 2009). The reduced and oxidized structures of the soluble part of OmcF have been determined by NMR and X-ray crystallography (Pokkuluri et al., 2009; Dantas et al., 2017). The structure of OmcF showed high similarity to those of cytochromes c6 from photosynthetic algae and cyanobacteria, particularly from Scenedesmus obliquus (Schnackenberg et al., 1999) and Monoraphidium braunii (Banci et al., 1998). Although the amino acid sequence of OmcF shows a higher homology with the cytochrome c6 of S. obliquus, the geometry of the heme axial methionine is more similar to that of cytochrome c6 from M. braunii (Pokkuluri et al., 2009).
The oxidized and reduced structures of OmcF are globally similar (Dantas et al., 2017). However, local redox-linked conformational changes were identified, in particular for the polypeptide segments Ala53-Ile62, Asn74-Gly78, Glu84-Ala90, and the C-terminus region (residues Val100-Pro104). In addition, the analysis of the pH-dependence of the backbone and side chain NH NMR signals also identified important pH-linked conformational changes, particularly in the vicinity of the heme group (Dantas et al., 2017). The most affected NH signals near the heme were those from residues His47, Glu49, Leu52, and Gly76.
The redox potential of OmcF was determined by visible potentiometric redox titrations and cyclic voltammetry at pH 7 and 8 using the normal hydrogen electrode (NHE) as reference (Pokkuluri et al., 2009; Teixeira et al., 2018). The values obtained by the two techniques were similar: + 180 (pH 7) and + 140 mV (pH 8) by cyclic voltammetry and + 180 (pH 7) and + 127 (pH 8) by potentiometric redox titrations. The values obtained indicate that the redox potential of OmcF is significantly modulated in this pH range (pH 7–8). The observed pH modulation of the formal redox potential (known as the redox-Bohr effect) indicates that the protein is able to thermodynamically couple electron and proton transfer. A complete electrochemical study covering a wider pH range, between 3.3 and 9.0, was also carried out and showed that the redox potential only varies considerably in the physiological pH range for G. sulfurreducens cellular growth (between pH 6 and 8) (Teixeira et al., 2018). This study also permitted the determination of the pKa values for the redox-Bohr center in the reduced (pKred = 7.6) and oxidized (pKox = 6.7) states. However, the molecular determinant(s) responsible for the pH modulation of the OmcF redox potential have not yet been identified.
The identification of the redox-Bohr center is important to elucidate the functional mechanism of OmcF and to contribute to the understanding of the extracellular electron transfer processes in G. sulfurreducens. The protonatable site of the imidazole ring of histidine 47 (His47) has been hypothesized as the redox-Bohr center based: (i) on the typical pKa value for a histidine side chain and (ii) on its spatial location near the heme in the structure of OmcF (Figure 1; Dantas et al., 2017). To test this hypothesis, in the present work, we used site-directed mutagenesis to replace the His47 by isoleucine (OmcFH47I) and phenylalanine (OmcFH47F) residues. In both cases, the side chain of the substituting residues is not protonatable and, therefore, will permit the evaluation of the contribution of His47 to the observed redox-Bohr effect. Non-labeled (hereafter referred as natural abundance) and 15N-labeled mutants were produced. Their overall folding was confirmed to be the same as the wild-type cytochrome by NMR and their electrochemical characterization was carried out in the physiological pH range. In addition, the crystal structure of the OmcFH47I mutant was determined.
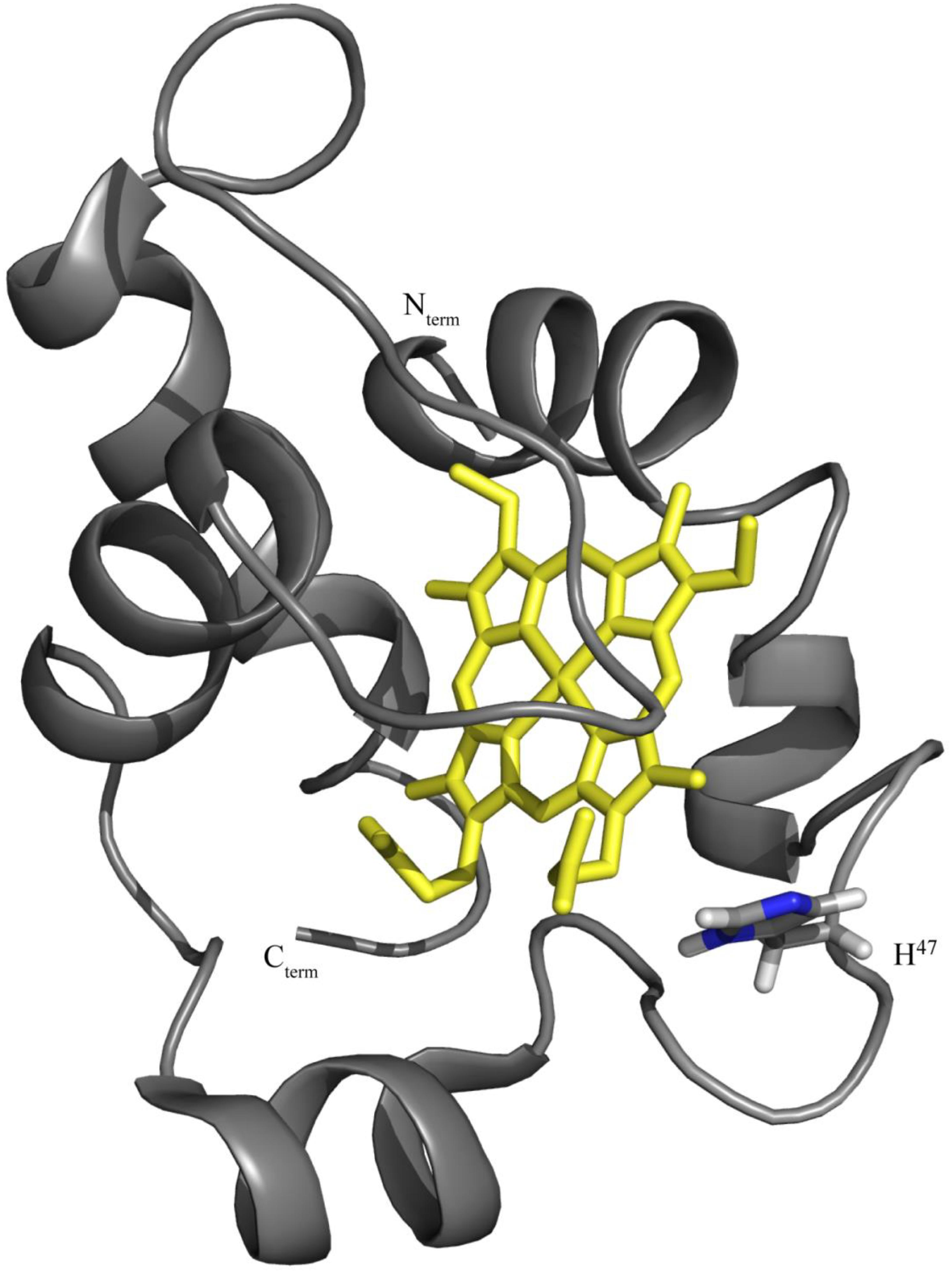
Figure 1. OmcF’s lowest energy solution structure [PDB code 5MCS (Dantas et al., 2017)] highlighting the location of residue His47. The OmcF polypeptide chain and the heme group are colored gray and yellow, respectively. The N- and C-termini, as well as side chain and Hα proton of His47 are labeled. Figure was generated with PyMOL (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC).
Materials and Methods
Site-Directed Mutagenesis
The residue His47 was replaced by isoleucine (OmcFH47I) and phenylalanine (OmcFH47F) using the NZYMutagenesis kit (NZYTech) and the pCK32 vector (encoding for the soluble part of OmcF formed by residues 20–104) as template (Pokkuluri et al., 2009). The necessary oligonucleotides were designed by the QuikChange Primer Design program (Agilent Technologies). For the OmcFH47I and OmcFH47F mutants, the following pairs of primers were used: 5′-agggtcttttccgggatgacggtgttgcctcc-3′/5′-ggaggcaacaccgtcatcccggaaaagaccct-3′ (OmcFH47I) and 5′-agggtc ttttccgggaagacggtgttgcctcc-3′/5′-ggaggcaacaccgtcttcccggaaaagacc ct-3′ (OmcFH47F). All primers were synthesized by Invitrogen and the mutations were confirmed by DNA sequencing (STAB VIDA).
Expression and Purification of OmcF and Mutants
Natural abundance and 15N-labeled proteins were expressed and purified as previously described (Fernandes et al., 2008; Pokkuluri et al., 2009). A summary of the procedure is presented here. Escherichia coli cells BL21 (DE3) containing the plasmids pEC86 (encoding for the expression of c-type cytochrome maturation proteins) (Arslan et al., 1998) and pCK32 were grown in 2xYT medium to an OD600 nm value between 1.5 and 1.8. From this point forward, distinct protocols were used for production of natural abundance and labeled proteins. In the first case, the expression was induced with 20 μM isopropyl β-D-thiolgalactopyranoside (IPTG), and the cultures were incubated overnight at 30°C. In the second case, to produce 15N-labeled proteins, cells were collected by centrifugation and transferred to M9 minimal medium supplemented with 15NH4Cl and α-aminolevulinic acid. Protein expression was then induced with 100 μM IPTG and the cultures were incubated overnight at 30°C.
In both cases, cells were first harvested by centrifugation (6400 ×g for 20 min) and lyzed using a buffer containing 100 mM Tris-HCl (pH 8), 0.5 mM EDTA, 20% sucrose, and 0.5 mg/mL of lysozyme. The periplasmic fraction was then recovered by centrifugation (14,700 ×g for 20 min) followed by an ultracentrifugation step (225,000 × for 1 h). This fraction was dialyzed against 20 mM sodium acetate (pH 5). After the dialysis step, the protein was loaded onto cation-exchange columns (2 × 5 mL Econo-Pac High S cartidges, Bio-Rad) pre-equilibrated with the same buffer and eluted with a linear sodium chloride gradient (0–200 mM). The fractions containing the targeted protein were then loaded onto a XK 16/70 SuperdexTM 75 prep grade column (GE Healthcare Life Sciences) pre-equilibrated with the same buffer. The protein’s purity was accessed by dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) stained with BlueSafe (NZYTech) and its concentration determined by visible spectroscopy using an absorption molar coefficient of 23.8 × 103 M–1cm–1 (Lukat et al., 2008).
NMR Studies
NMR spectra were acquired on a Bruker Avance 600 MHz spectrometer with a triple-resonance cryoprobe at 25°C. To assist the assignment of the backbone and side chain NH signals in each mutant, 15N-labeled samples were prepared in 45 mM sodium phosphate (pH 7) with 100 mM final ionic strength in 92% H2O/8% 2H2O. Natural abundance samples of the mutants were prepared in the same buffer to assist the assignment of the heme substituent signals. OmcF wild-type samples were also prepared in the same buffer (pH 6.1 and 9.4) in 2H2O (99.9%) to study the pH dependence of the heme substituents’ signals.
Reduction of the proteins was achieved by adding an equimolar solution of sodium dithionite, after degassing the samples with a continuous flow of argon. The full reduction of the samples was confirmed by 1D 1H NMR. 2D 1H,15N-HSQC spectra were acquired for 15N-labeled samples, whereas 2D 1H, 1H-TOCSY (60 ms) and 2D 1H, 1H-NOESY (80 ms) were acquired for natural abundance samples.
The water signal was used to calibrate the 1H chemical shifts. 15N chemical shifts were calibrated using indirect referencing (Wishart et al., 1995). The data were processed using TOPSPIN (Bruker Biospin, Karlsruhe, Germany) and analyzed with Sparky (TD Goddard and DG Kneller, Sparky 3, University of California, San Francisco, CA, United States).
Electrochemical Studies
The cyclic voltammetry measurements were performed inside a Faraday cage using a three electrodes’ configuration in a single compartment electrochemical cell, as described previously (Teixeira et al., 2018). Briefly, a neomycin sulfate (3 μL of a 2 mM stock) and protein solutions (5 μL of a 255 μM stock) were placed on the working electrode, followed by evaporation at room temperature (solvent casting technique). The thin layer procedure was implemented by the entrapment of the protein using a cellulose membrane (spectra/Pro) with a 6 to 8 kDa cut-off. The electrodes were immersed into a buffer solution of 32 mM sodium phosphate (pH 6, 7, and 8) with 100 mM final ionic strength, previously degassed with a continuous flow of argon. All assays were carried out at room temperature and at least three independent replicates were performed for each assay at scan rates between 2.5 and 100 mVs–1. From the midpoint redox potential obtained for each scan rate using the second cycle [using E0’ = (Epa + Epc)/2], the average and standard deviation values were calculated. Controls were performed applying the same methodology but in the absence of protein. The measured redox potential values were corrected for the NHE (Sawyer et al., 1995; Liu et al., 1997; Lindgren et al., 2000).
Crystallization, Data Collection, and Structure Determination
Crystallization trials for both mutants were carried out by hanging drop vapor diffusion method at room temperature using protein sample concentrations of 20 mg/mL. Attempts to grow crystals using the same conditions as the wild-type OmcF (1.2 M trisodium citrate dihydrate, 0.1 M Tris pH 8.5) were unsuccessful. However, very thin needle clusters in case of OmcFH47I were obtained using the conditions reported for OmcF consisting of an N-terminus Strep-tag II (Lukat et al., 2008). The drops consisted of 1 μL of protein plus 1 μL precipitating reagent [1.8 M ammonium sulfate, 0.1 M HEPES pH 7.5, and 2%(v/v) PEG400] equilibrated over 0.5 mL of reagent solution in the well. The crystals were optimized for data collection by microseeding technique. A thin needle cluster grown from the above-mentioned condition was placed on a glass slide in a 10 μL drop of reservoir solution. Viewing under an optical microscope, this crystal cluster was crushed with a razor blade. The droplet containing the pieces of the crushed crystals was collected from the glass slide. This solution is named seed I. The glass slide was then washed with another fresh 10 μL drop of well solution and collected (named seed II solution). New crystallization trials were conducted using the same conditions as above, except hanging drops now consisted of 1 μL of protein, 1 μL well reagent, and 0.2 μL seed solution. Best crystals for OmcFH47I were obtained from a drop supplemented with seed II solution. Microseeding also yielded crystals as needle clusters. A piece cleaved from a cluster was used for data collection. In case of OmcFH47F mutant, seeding the crystallization drops with OmcFH47I seed solution was unsuccessful.
The X-ray diffraction data were collected at the 22ID beam line of the SER-CAT, Advanced Photon Source (Argonne, IL, United States). X-ray diffraction data were collected to a high resolution of 1.13 Å. Data reduction and scaling was achieved with the program HKL 2000 (Otwinowski and Minor, 1997). Structure solution was achieved by molecular replacement using the wild-type OmcF coordinates [PDB code 3CU4 (Pokkuluri et al., 2009)], after removing the side chain atoms beyond Cβ of His47 using the Phaser-MR (McCoy et al., 2007) routine within Phenix package (Adams et al., 2010). Auto-build routine within Phenix was used for model building. The program Coot (Emsley and Cowtan, 2004) was used to inspect the electron density maps and make the necessary adjustments in the model and solvent molecules. Refinement was carried out by the program Phenix. The final model included residues 23–104, one heme-c, 91 water molecules, and a sulfate ion. The data collection and refinement statistics are presented in Table 1.
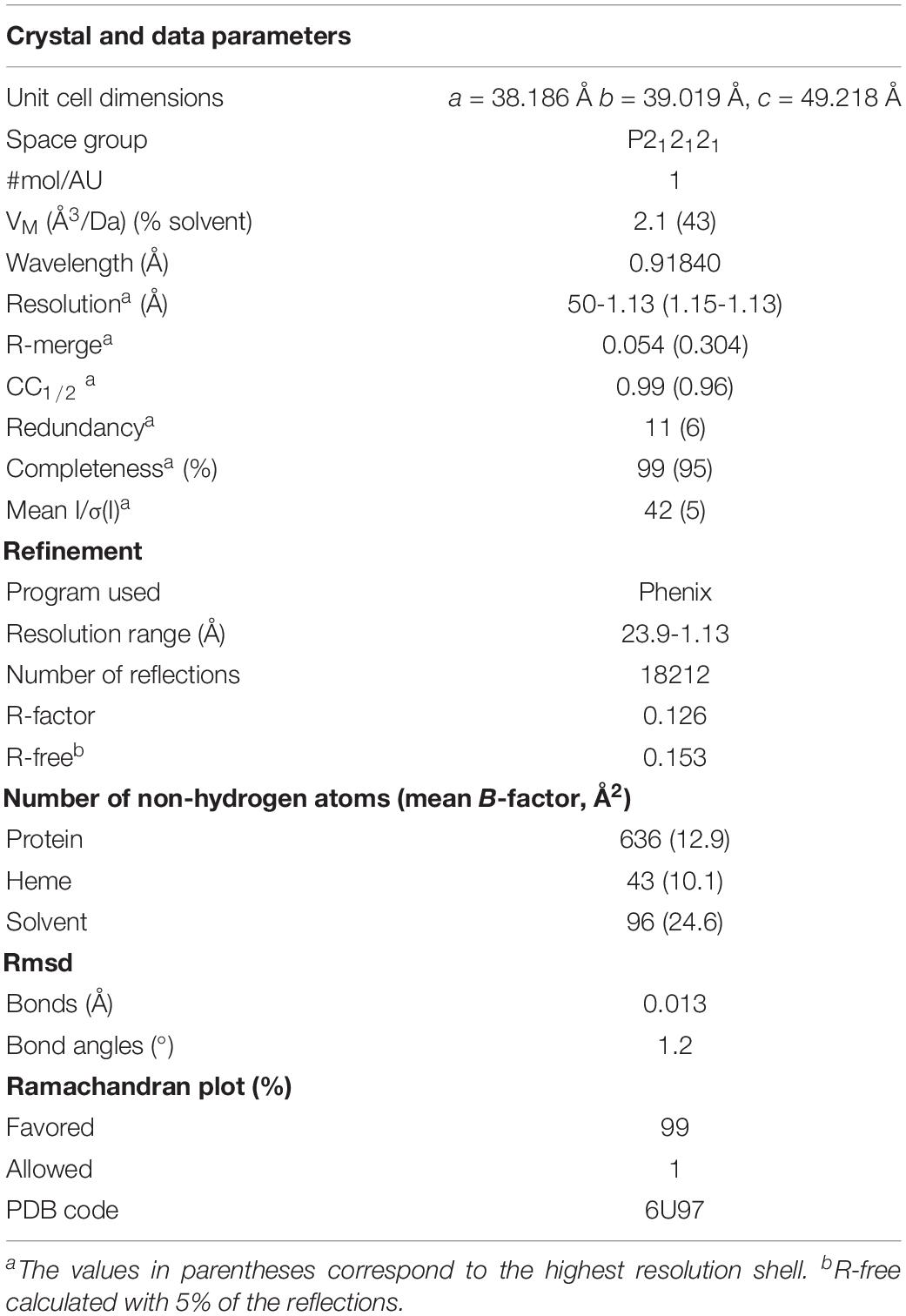
Table 1. Crystallographic parameters and refinement statistics for OmcFH47I mutant.
Results and Discussion
Impact of the Mutations on the Global Folding of the Protein
The protein yields obtained for each mutant (OmcFH47I and OmcFH47F) were similar to the ones obtained for the wild-type protein, approximately 10 and 3 mg of protein per liter of cell culture for natural abundance and 15N-labeled proteins, respectively. The UV–visible spectra of the mutants showed similar features compared to the wild-type protein (Supplementary Table S1).
The NMR chemical shifts of the nuclei of the molecule are very sensitive to changes in their chemical environment and, consequently, can be explored to fingerprint the overall structure of the proteins in solution. Thus, before undertaking the electrochemical characterization of the mutants, the impact of each mutation on the protein conformation was evaluated by 2D 1H,15N-HSQC NMR experiments. The backbone and side chain NH signals of each mutant were assigned (Figure 2) using the same methodology described for the wild-type protein (Dantas et al., 2015). The comparison of the spectra obtained for the wild-type and the mutants showed a similar dispersion of signals, indicating that the overall folding is maintained (Figure 2). The analysis of the 1H and 15N combined chemical shift variation showed that the residues located in the polypeptide segment Val46-Lys50 and Gly78 were the most affected (Figure 3A). The polypeptide segment Val46-Lys50 includes the mutated residue (Ile47 and Phe47 for OmcFH47I and OmcFH47, respectively) and a proline residue (Pro48). This residue does not possess NH due to its cyclic nature and for this reason no chemical shift variation was observed. On the other hand, Gly78 is closely located to Val46, whose side chain, together with the imidazole ring of His47, forms a pocket around the heme methyl 121CH3 (see inset in Figure 3A). Thus, the replacement of His47 residue introduced only small rearrangements at the vicinity of the mutation site, without affecting the global folding of the protein. Both mutants showed a similar variation of their chemical shift suggesting that the local rearrangements are comparable. Following the analysis of the impact of the mutated residues in the backbone and side chain NH signals, their impact was further evaluated regarding the heme 1H NMR signals. The heme signals were assigned using 2D 1H, 1H-TOCSY and 2D 1H, 1H-NOESY NMR spectra, as described for the wild-type protein (Dantas et al., 2015). The assignment of these signals and their comparison with those obtained for the wild-type protein are indicated in Supplementary Table S2 and Figure 3B, respectively. In both mutants, the most affected signal is the heme methyl 121CH3, which is the closest to the mutated residue (cf. insets in Figures 3A,B). The other affected heme signals, although to a smaller extent, correspond to propionate protons (13CH2 groups), the meso proton 10H and the thioether proton 81H, which are allocated to the heme face containing the methyl 121CH3 (see inset in Figure 3B). Therefore, the heme proton signals showing the largest chemical shift variations are in the vicinity of most affected residues, namely, the polypeptide segment Val46-Lys50 and Gly78 (cf. insets in Figures 3A,B), which further confirmed that the small conformational changes caused by the amino acid replacements are restricted to the neighboring regions of the mutated residue.
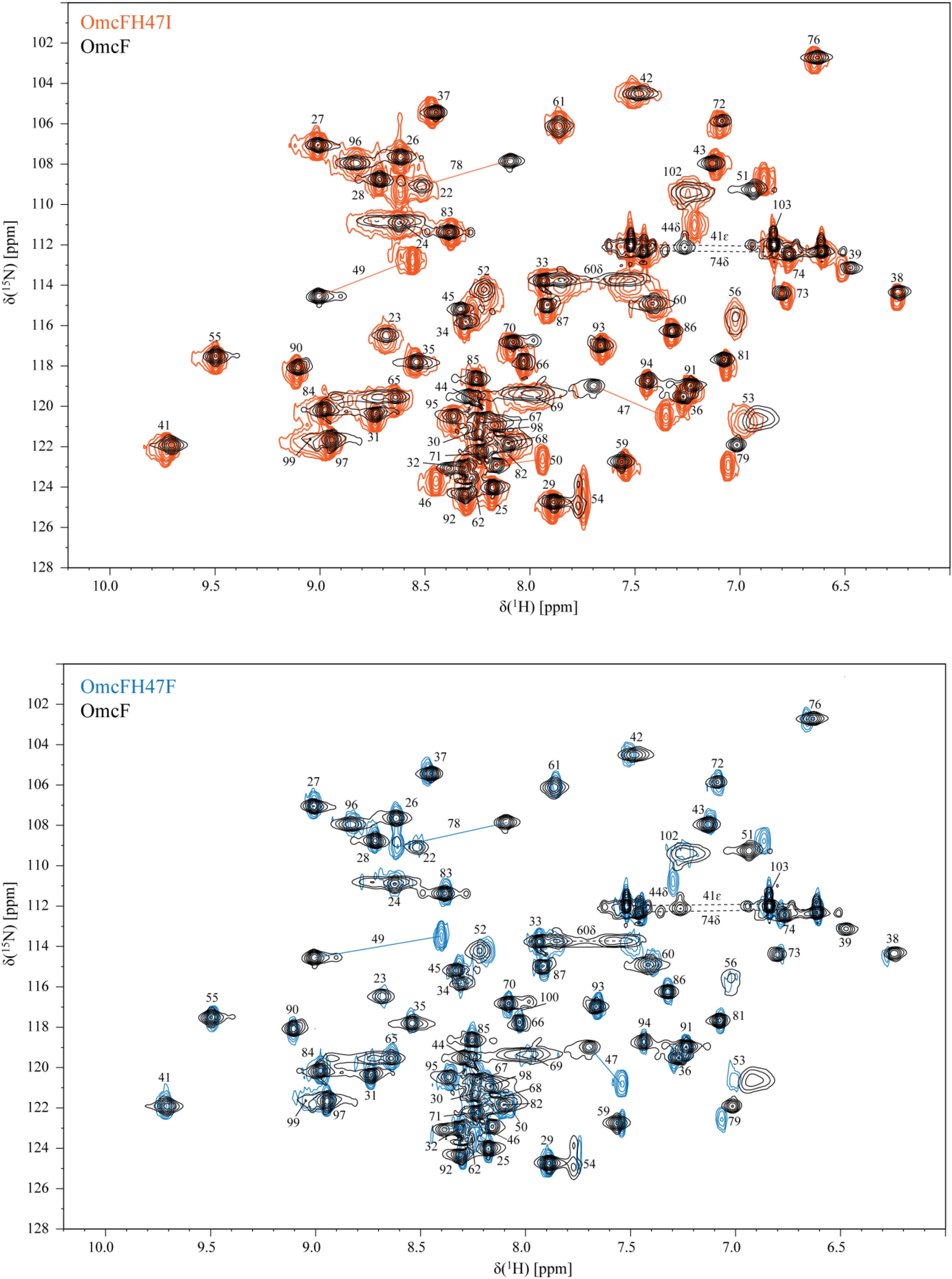
Figure 2. 2D 1H,15N-HSQC NMR spectra of OmcF (black contours), OmcFH47I (orange contours), and OmcFH47F (blue contours) in the reduced state (25°C, pH 7). The most affected signals in the mutants, compared to the wild-type spectrum, are connected by a straight line. Dashed lines correspond to side chains of amino acids.
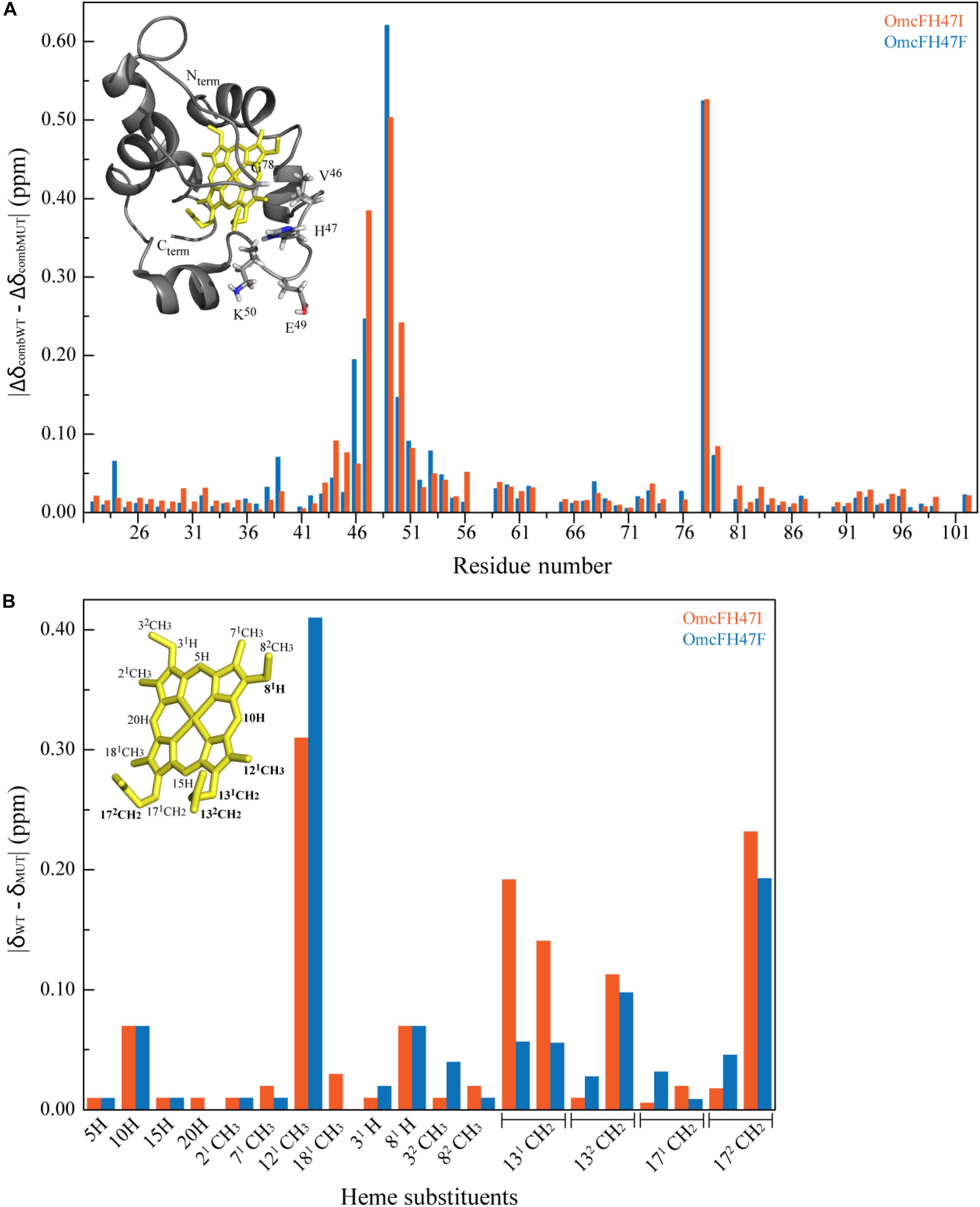
Figure 3. Effects on the polypeptide and heme substituent’s NMR signals caused by the replacement of His47 by isoleucine or phenylalanine. (A) Comparison between the combined 1H and 15N chemical shifts observed in the 2D 1H,15N HSQC NMR spectra of OmcF mutants (ΔδcombMUT) and those of OmcF (ΔδcombWT). The differences were calculated using the equation Δδcomb = [(ΔδH)2 + wi(ΔδN)2]1/2, where ΔδH is the difference between 1H shifts, ΔδN the difference between 15N shifts, and wi = ∣γ15N∣/∣γ1H∣ a weighting factor that accounts for the differences in nuclei sensitivity (Schumann et al., 2007). The color code of the NMR spectra indicated in Figure 2 was used for each mutant: OmcFH47I (orange bars) and OmcFH47F (blue bars). The most affected residues are shown in the solution structure of cytochrome OmcF in the inset. (B) Comparison of the heme 1H chemical shifts observed in the 2D 1H NOESY NMR spectra of OmcF mutants (δMUT) and those of wild-type OmcF (δWT). The heme substituents are numbered according to the IUPAC-IUB nomenclature (Moss, 1988) and the most affected heme substituent signals are highlighted in bold in the heme diagram. In both panels the structural data correspond to OmcF’s lowest energy solution structure [PDB code 5MCS (Dantas et al., 2017)] generated with PyMOL (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC).
Effect of the Mutations on the Redox-Bohr Center Properties
The redox potential values of cytochromes can be modulated by the solution pH which would be functionally relevant within a physiological pH range. This modulation is designated redox-Bohr effect, in analogy with the Bohr effect in the hemoglobin (Perutz, 1989). From a simple electrostatic view of the heme iron charge, the reduced form is expected to be stabilized by the protonation of the redox-Bohr center while its deprotonation stabilizes the oxidized form.
Previous studies have shown that the redox potential of OmcF is strongly modulated in the pH range 6–8, i.e., at the physiological range for the G. sulfurreducens growth (Pokkuluri et al., 2009; Teixeira et al., 2018). Thus, in the present work, after confirming that only local conformational changes were observed in the region of the mutated residues, the redox potential values of the OmcF mutants were measured by cyclic voltammetry in the same pH range. For proper comparison, measurements for the wild-type protein were carried out in the same experimental conditions as the mutants in the present study and are reported in Table 2. The cyclic voltammograms were recorded in the potential window between 0 and 0.35 V, with scan rates between 2.5 and 100 mVs–1. At all pH values, well-defined redox pairs were visible and, by comparison with the controls, were assigned to the OmcF heme group. The data obtained for the OmcF mutants at pH 7 are illustrated in Figure 4 (cyclic voltammograms obtained for OmcF at pH 7 and corresponding control are shown in Supplementary Figure S1). The signals show a quasi-reversible electrochemical behavior between the scan rates 2.5–20 mVs–1, just like the wild-type cytochrome (Teixeira et al., 2018). The quasi-reversible electrochemical behavior was verified through the usual criteria for the thin layer regime, namely: the ratio of the cathodic (Ipc) and anodic (Ipa) peak current intensities are approximately 1; the Ipc and Ipa are linearly proportional to the scan rate, and the separation between the cathodic (Epc) and anodic (Epa) peak potential (ΔEp) increases with the applied scan rate. The estimated thin layer thickness was in average 2.5 μm (Brett and Brett, 1993; Bard and Faulkner, 2001; Correia dos Santos et al., 2003; Santos et al., 2015). The formal potential values of OmcFH47I and OmcFH47F mutants were determined by the average of the anodic and cathodic peak potentials (at the point of maximum current intensity) and are indicated in Table 2. As it is possible to observe, the formal potential values for both mutants were lower than the value found for the wild-type cytochrome. This indicates that the oxidized form in the mutants is more stabilized as expected by the replacement of the positively charged side chain of His47 in the wild-type by an uncharged side chain in both mutants.
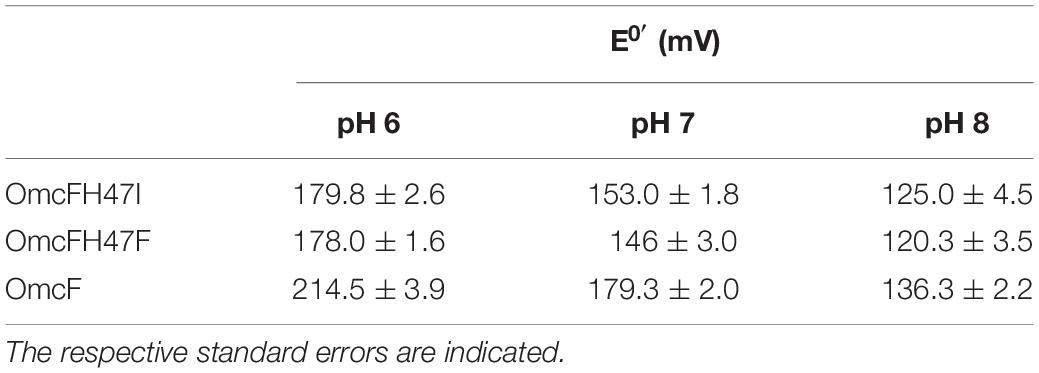
Table 2. Formal potential values (versus NHE) for OmcF and His47 mutants determined by cyclic voltammetry at pH 6, 7, and 8.
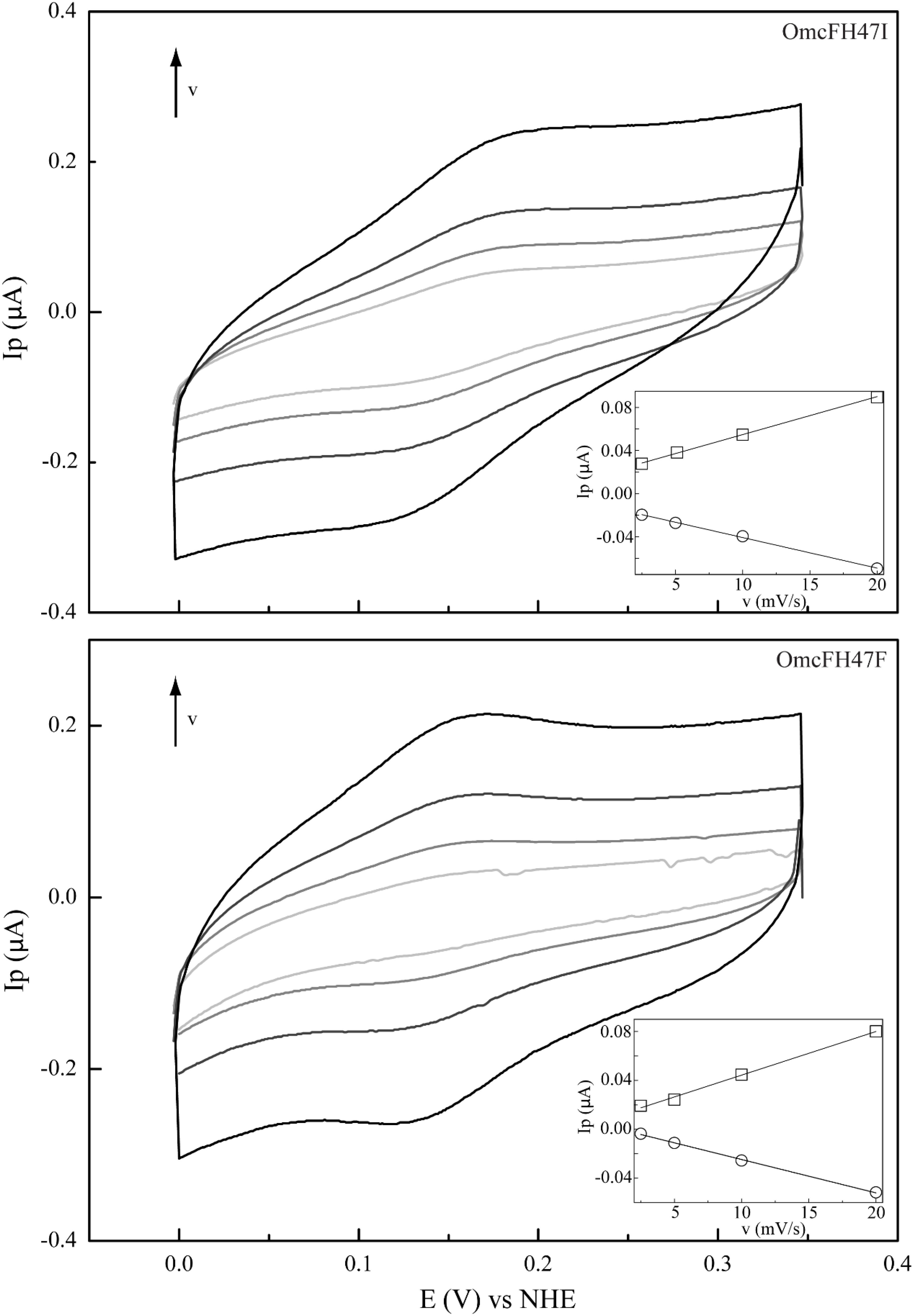
Figure 4. Cyclic voltammograms of mutants OmcFH47I (top) and OmcFH47F (bottom) at scan rates (ν) from 2.5 to 20 mV s–1 (pH 7). Inset: peak current as a function of the scan rate. Anodic and cathodic peak currents are represented by squares and circles, respectively.
As observed for the wild-type cytochrome, the redox potential values of the mutants were also pH-dependent. However, the difference between the redox potential values of the mutants and those of the wild-type decreases with the increase in pH, which indicates that the replacements have a smaller impact on the redox properties of the heme group at higher pH. Overall, the data obtained suggested that the redox-Bohr center is protonated at pH 6 and deprotonated at pH 8. This is also in agreement with the pKa values previously determined for the redox-Bohr center (pKox = 6.7; pKred = 7.6) (Teixeira et al., 2018) and explains the higher impact of the substitutions at pH 6. The additional positive charge in the redox-Bohr center at pH 6 stabilizes the reduced state (higher redox potential values) whereas the loss of the positive charge at higher pH facilitates the oxidation of the heme and stabilizes the oxidized state (lower redox potential values). This is also reflected in the differences observed for the redox potential values of the mutants compared to the wild-type cytochrome at pH 6 (34.7 and 36.5 mV for OmcFH47I and OmcFH47F, respectively) and 8 (11.3 and 16.0 mV for OmcFH47I and OmcFH47F, respectively). In fact, in the wild-type at pH 6, the side chain of His47 is protonated and positively charged, whereas at pH 8, the side chain of His47 is deprotonated and uncharged. In the mutants, the replacements do not introduce any charge variation in the vicinity of the heme group at both pH values.
From the analysis of Table 2, it is also clear that the pH dependence of the redox potential values is smaller in the mutants compared to the wild-type cytochrome but confirms unequivocally the involvement of His47 in the pH modulation of the redox potential of OmcF. However, the fact that the redox-Bohr effect was not completely abolished by the replacement of His47 by non-protonatable residues, suggested that additional acid group(s) might be involved in the global redox-Bohr effect.
Structural Probe of the Redox-Bohr Center in OmcF
The analysis of the OmcF structures showed that, in addition to the side chain of His47, other protonatable groups include the N- and C-termini, six arginine, two aspartic acid, five glutamic acid, one histidine, two lysine, two tyrosine residues, and the heme propionate groups at positions 13 (P13) and 17 (P17). With the exception of heme propionate groups, all other protonatable groups are distant from the heme iron, and therefore are unlikely to affect the redox potential of OmcF. Since the heme propionates are the best additional candidates for the redox-Bohr effect, we evaluated the pH dependence of the heme substituent signals of OmcF in the reduced state. The assignment of the entire set of the heme substituents in the oxidized state is more complex compared to the reduced state due to the paramagnetic effect of the heme unpaired electron (Salgueiro et al., 1997). This often impairs the full assignment of the heme signals in the oxidized state, which is in fact the case for OmcF. Because the assignment of the heme substituent signals of OmcF was fully obtained for the reduced state at pH 7 (Dantas et al., 2015), this state was selected to evaluate the pH dependence of the heme substituent NMR signals. Considering the pKa value of the redox-Bohr center in the reduced state (pKred = 7.6) (Teixeira et al., 2018), the pH values of 6.1 and 9.4 were selected to ensure a proper comparison of the heme substituent chemical shifts when the redox-Bohr center is protonated and deprotonated, respectively.
The assignment of the heme substituent signals at pH 6.1 and 9.4 was carried out using the same strategy as previously described at pH 7 (Dantas et al., 2015) and are listed in Supplementary Table S3. The variation of the heme substituent’s chemical shifts at pH 6.1 and 9.4 is indicated in Figure 5 and shows that the heme propionate groups and methyl 121CH3 are the most affected ones. However, the pH dependence of the heme propionates is quite distinct. Indeed, while the four protons of P17 were clearly affected by the pH, only one proton from P13 heme was affected. Such behavior, together with the fact that the heme methyl 181CH3 is essentially pH independent, suggests that P13 is the best additional candidate for contribution to the redox-Bohr effect. In summary, electrochemical studies were carried out on OmcFH47I and OmcFH47F mutants and the pH dependence of the heme substituent NMR signals indicated that the two main contributors for the redox-Bohr effect in OmcF are the side chain of His47 and the heme propionate group P13. The deprotonation/protonation of the histidine side chain explains the large chemical shift variation on heme methyl 121CH3 that extends also to heme protons 10H and 81H, while the deprotonation/protonation of P13 is responsible for the significant variations observed for the chemical shift of the P17 protons (Figure 5).
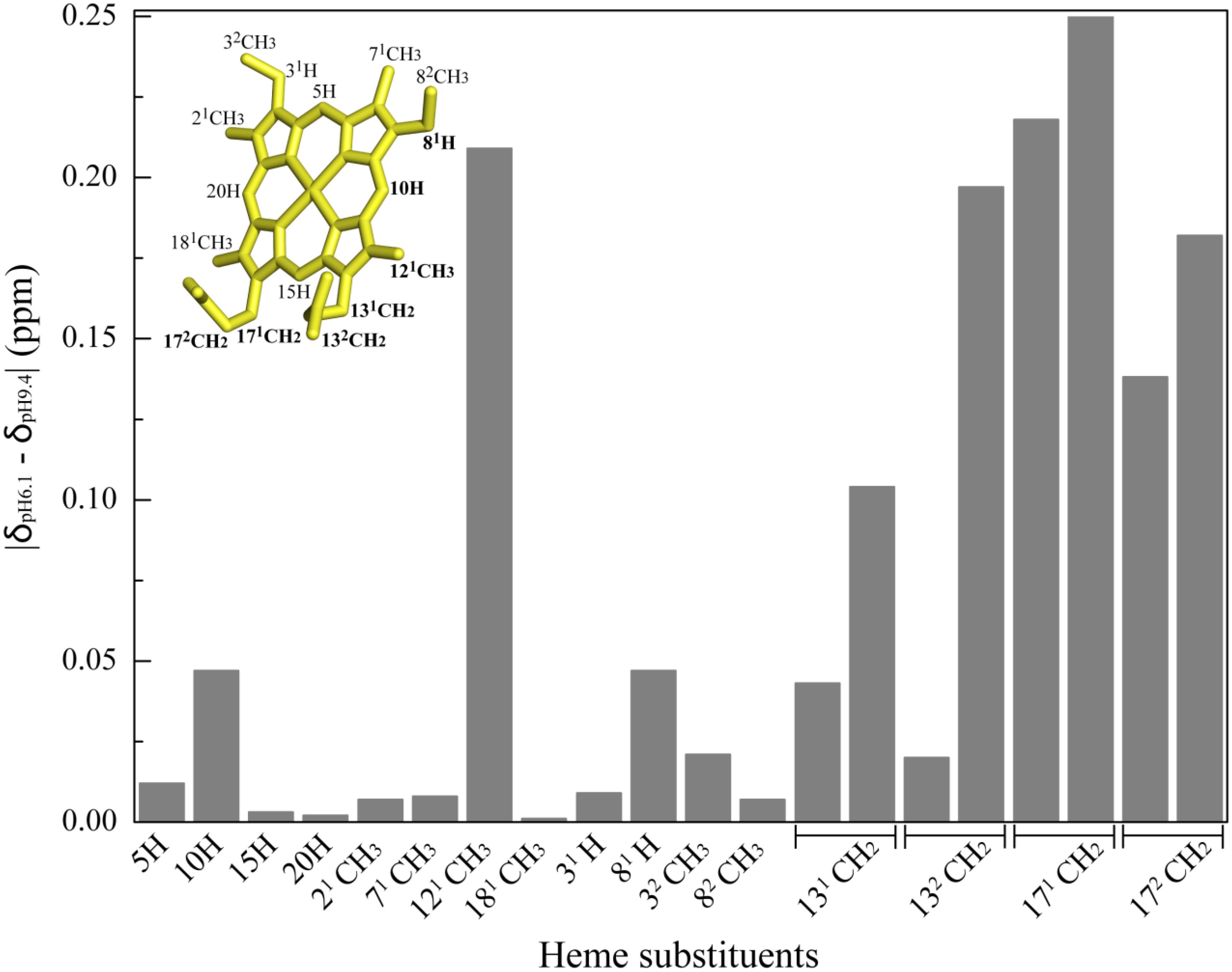
Figure 5. Variation of OmcF’s heme substituent’s proton chemical shifts at pH 6.1 (δpH6.1) and 9.4 (δpH9.4). The heme substituents are numbered according to the IUPAC-IUB nomenclature (Moss, 1988) and the most affected heme substituent signals are highlighted in bold in the heme diagram. The heme group in the inset was taken from OmcF’s lowest energy solution structure [PDB code 5MCS (Dantas et al., 2017)] and was generated with PyMOL (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC).
Crystal Structure of OmcFH47I
The OmcFH47I mutant crystallized in the same space group as the wild-type OmcF but with a slight difference in the unit cell dimensions resulting in a 1% decrease in unit cell volume of the mutant crystals. The overall structure of the OmcFH47I mutant in the oxidized state is very close to that of the wild-type with an overall root-mean-square deviation (rmsd) of 0.4 Å for all Cα atoms (residues 23–104). A Cα carbon trace of the overlap of the OmcFH47I mutant structure on the wild-type OmcF is shown in Supplementary Figure S2. The electron density for the side chain of Ile47 clearly showed two conformations, refined at occupancies of 0.7 and 0.3. The two conformations of the isoleucine side chain differed primarily in the location of the CD1 methyl group. Deviations higher than the overall rmsd (ranging from 0.6 to 0.8 Å) were observed near the mutation site from residues 47–50, and from residues 76 to 78, which are in line with the analysis of the 1H and 15N combined NMR chemical shift variation (Figure 3A). Deviations ranging from 1.0 to 1.3 Å were also seen in residues 84–86, which are distant from the heme, caused by different interactions across the crystal interface.
The replacement of the polar histidine residue by non-polar isoleucine did not cause any significant changes in the interactions of the heme with the protein. The isoleucine side chain(s) forms van der Waals contact(s) with the heme atoms. The propionate D (P13), which is closest to the mutation site, forms water mediated hydrogen bonds with Tyr71 and the other propionate in both wild-type and mutant structures. It also hydrogen bonds with other water molecules. Although this propionate can hydrogen bond with the side chain of His47 in the wild-type OmcF structure, this was not observed. On the other hand, the propionate A (P17) forms a salt bridge with Lys50 (NZ) with one of its carboxyl oxygen atoms and hydrogen bonds with Asn60 (ND2) with the other oxygen atom. These interactions involving the P17 carboxyl oxygen atoms are observed in both native and OmcFH47I mutant structures. The interactions formed by the heme propionates are shown in Figure 6.
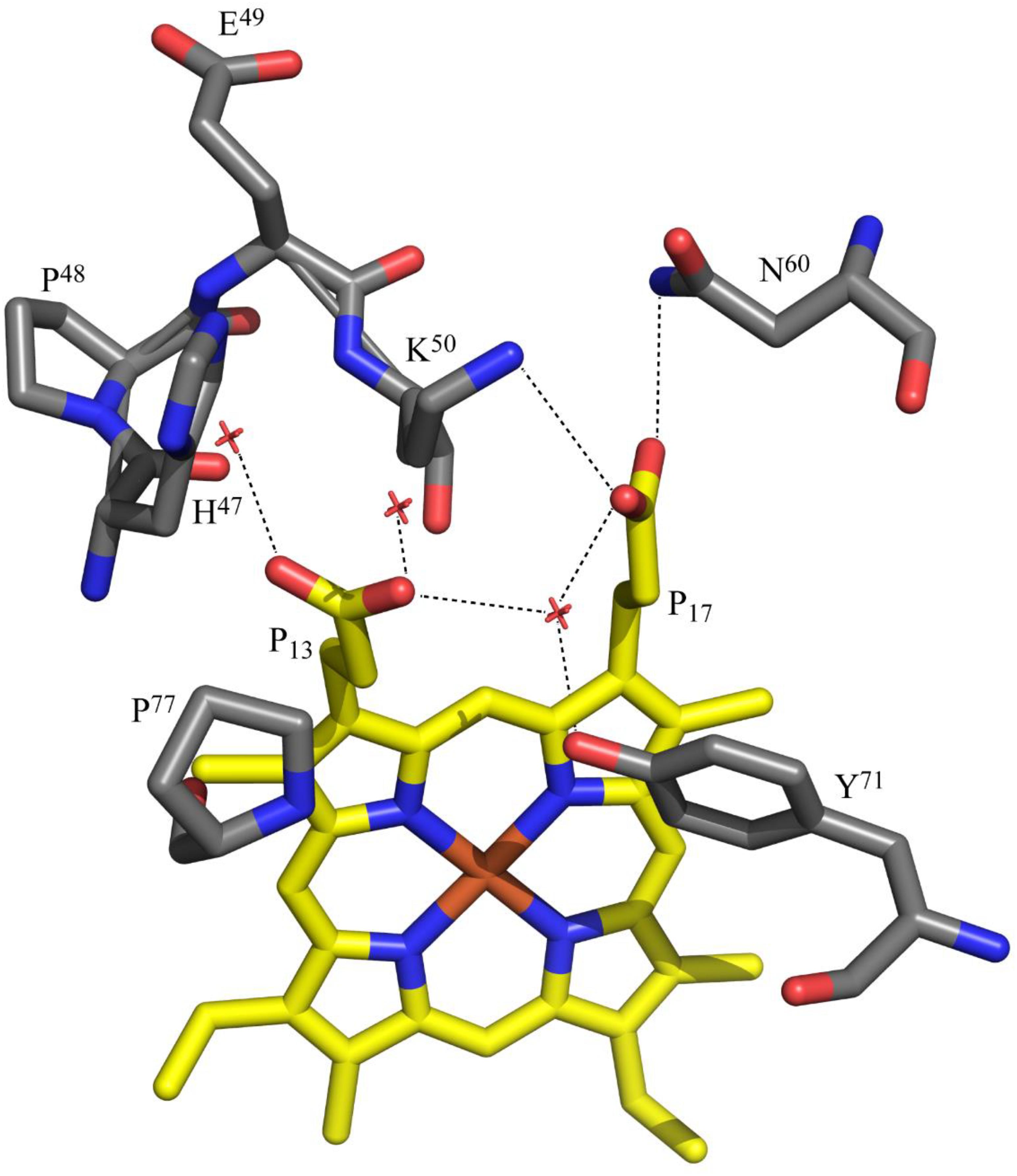
Figure 6. Polar interactions involving the heme propionates of OmcF. The propionates are labeled according to IUPAC nomenclature. The structural data corresponds to OmcF native structure [PDB code 3CU4 (Pokkuluri et al., 2009)] and was generated with PyMOL (The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC).
Heme Propionate P13 and His47 Are the Redox-Bohr Centers in OmcF
The observed hydrogen bond network established by the heme propionate groups further supports the conclusion that P13 contributes to the global redox-Bohr effect in OmcF. In fact, it is common that the heme propionic acids ionize with pKa values in the region 5–6 (Moore et al., 1980), and have been suggested as the groups responsible for the redox-Bohr effect in several monoheme cytochromes (Moore et al., 1980; Leitch et al., 1984; Costa et al., 1992; Santos et al., 2015). In the oxidized crystal structures of wild-type OmcF [PDB code, 3CU4 (Pokkuluri et al., 2009)] and in the OmcFH47I mutant (present work), one of the carboxyl atoms of the heme propionate P17 forms a salt bridge with NZ of Lys50 and the other carboxyl oxygen atom hydrogen bonds with ND2 of Asn60. Thus, this propionate is poised to be a hydrogen bond acceptor with both its carboxyl oxygen atoms, which stabilizes its deprotonated form. The stabilization offered by salt bridge with Lys50 and another hydrogen bond with Asn60 promotes to keep the pKa of this propionate group (P17) lower than the other propionate group (P13). The propionate P13 on the other hand does not form any interactions with the protein in the crystal structures. It is somewhat more exposed to solvent and only interacts with water molecules located in the crystal. This propionate is close to the side chain of His47 in the wild-type OmcF structure but does not form a hydrogen bond with it. In the solution structure of OmcF [PDB code, 5MCS (Dantas et al., 2017)], the carboxyl group of propionate P13 is positioned between the side chain of His47 and the main chain oxygen atom of Pro77. In majority of the conformations, the propionate P13 forms two hydrogen bonds via its two carboxyl oxygen atoms, one with ND1 atom of His47 and another with the main chain oxygen atom of Pro77. The latter interaction will be unfavorable if this propionate is not in the protonated (neutral) form. The interaction between propionate P13 and the main chain oxygen of Pro77 is not seen in the crystal structure due to the intermolecular interactions with a neighboring molecule in the crystal. Alternatively, the difference could be related to redox conformational changes between the two structures, namely, crystal (oxidized) and solution (reduced). Therefore, we propose that the propionate P13 has a pKa in the physiological range and, together with His47, the two acid–base groups contribute to extend the range of the overall redox-Bohr effect in OmcF. Upon removal of His47 in the OmcFH47 mutants, the magnitude of the redox-Bohr effect observed is approximately 30% less (54.8 and 57.7 mV for OmcFH47I and OmcFH47F, respectively, compared to 78.2 mV in OmcF – see Table 2).
Conclusion
The cytochrome OmcF from the bacterium G. sulfurreducens showed an important pH modulation of the heme reduction potential (redox-Bohr effect) in physiological range for G. sulfurreducens cellular growth. The spatial localization of His47 and the properties of its side chain suggested it as a good candidate for the redox-Bohr center in OmcF. This hypothesis was addressed in the present work by replacing the His47 with the non-protonatable residues isoleucine and phenylalanine. The global folding of the mutants was assessed by NMR spectroscopy and the comparison of the polypeptide and heme NMR signals showed that both the mutants were properly folded and that only local conformational changes were observed in the vicinity of the mutated residue regions. The crystal structure of OmcFH47I mutant determined at a high resolution also showed that the mutation did not affect the structure. Electrochemical cyclic voltammetry studies carried out for both the mutants, within the physiological pH range, showed that the redox potential values and the redox-Bohr effect were smaller compared to the wild-type cytochrome. This unequivocally confirms the role of His47 in the pH modulation of the OmcF heme redox potential and electron/proton transfer mechanisms. However, the redox-Bohr effect was not fully abolished in the mutants (approximately 30% less in the mutants) suggesting the existence of another redox-Bohr center in OmcF, which was attributed to the heme propionate P13. Therefore, these two acid–base groups with pKa in the physiological range (heme propionate P13 and His47) contribute to the overall observed redox-Bohr effect. Given the cellular location of OmcF at the outer membrane, the existence of two independent acid-base centers that contribute to redox-Bohr effect may permit the protein to be functionally active in a wider pH range, in response to environment changes. This study sheds light on how the cytochromes can extend the modulation of the redox potential within the physiological pH range not only through the interactions of their heme propionate groups with the neighboring protein atoms but also by utilizing strategic placement of additional protonatable residues near the heme. Delineation of such an intricate heme–protein interaction network is vital to a clearer understanding of the extracellular electron transfer processes mediated by these bacteria and the central role played by the multitude of cytochromes encoded within their genomes.
Data Availability Statement
The datasets generated for this study can be found in the PDB code 6U97.
Author Contributions
CS and PP conceived, designed, and supervised the project. LT and MF acquired and analyzed the NMR data. LT and CC acquired and analyzed the electrochemical data. ND and PP acquired and analyzed the X-ray data. LT, CC, PP, and CS wrote the manuscript.
Funding
This work was supported by the Fundação para a Ciência e a Tecnologia (FCT-MCTES) through Radiation Biology and Biophysics Doctoral Training Program (RaBBiT, PD/00193/2012), UID/FIS/00068/2019 (CEFITEC); PTDC/BBB-BQB/3554/2014 (to CS), PTDC/BIA-BQM/31981/2017 (to CS); and scholarship grant PD/BD/114445/2016 (to LT). This work was also supported by the Applied Molecular Biosciences Unit-UCIBIO by national funds from FCT/MCTES (UID/Multi/04378/2019). The NMR spectrometers at FCT NOVA are part of Rede Nacional de RMN (PTNMR), supported by FCT-MCTES (ROTEIRO/0031/2013 – PINFRA/22161/2016) co-funded by FEDER through COMPETE 2020, POCI, and PORL and FCT through PIDDAC. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02941/full#supplementary-material
References
Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. doi: 10.1107/S0907444909052925
Arslan, E., Schulz, H., Zufferey, R., Kunzler, P., and Thony-Meyer, L. (1998). Overproduction of the Bradyrhizobium japonicum c-type cytochrome subunits of the cbb3 oxidase in Escherichia coli. Biochem. Biophys. Res. Commun. 251, 744–747. doi: 10.1006/bbrc.1998.9549
Banci, L., Bertini, I., De la Rosa, M. A., Koulougliotis, D., Navarro, J. A., and Walter, O. (1998). Solution structure of oxidized cytochrome c6 from the green alga Monoraphidium braunii. Biochemistry 37, 4831–4843. doi: 10.1021/bi972765y
Bard, A. J., and Faulkner, L. R. (2001). Electrochemical Methods: Fundamentals and Applications. Hoboken, NJ: Wiley.
Brett, C. M. A., and Brett, A. M. O. (1993). Electrochemistry: Principles, Methods, and Applications. Oxford: Oxford University Press.
Correia dos Santos, M., Paes de Sousa, P., Simões Gonçalves, M., Krippahl, L., Moura, J., Lojou, E., et al. (2003). Electrochemical studies on small electron transfer proteins using membrane electrodes. J. Electroanal. Chem. 541, 153–162. doi: 10.1016/S0022-0728(02)01427-1424
Costa, H. S., Santos, H., Turner, D. L., and Xavier, A. V. (1992). Involvement of a labile axial histidine in coupling electron and proton transfer in Methylophilus methylotrophus cytochrome c. Eur. J. Biochem. 208, 427–433. doi: 10.1111/j.1432-1033.1992.tb17204.x
Dantas, J. M., Silva, M. A., Pantoja-Uceda, D., Turner, D. L., Bruix, M., and Salgueiro, C. A. (2017). Solution structure and dynamics of the outer membrane cytochrome OmcF from Geobacter sulfurreducens. Biochim. Biophys. Acta Bioenerg. 1858, 733–741. doi: 10.1016/j.bbabio.2017.03.007
Dantas, J. M., Silva e Sousa, M., Salgueiro, C. A., and Bruix, M. (2015). Backbone, side chain and heme resonance assignments of cytochrome OmcF from Geobacter sulfurreducens. Biomol. NMR Assign. 9, 365–368. doi: 10.1007/s12104-015-9611-9615
Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132. doi: 10.1107/S0907444904019158
Fernandes, A. P., Couto, I., Morgado, L., Londer, Y. Y., and Salgueiro, C. A. (2008). Isotopic labeling of c-type multiheme cytochromes overexpressed in E. coli. Protein Expr. Purif. 59, 182–188. doi: 10.1016/j.pep.2008.02.001
Kim, B.-C., Leang, C., Ding, Y.-H. R., Glaven, R. H., Coppi, M. V., and Lovley, D. R. (2005). OmcF, a putative c-type monoheme outer membrane cytochrome required for the expression of other outer membrane cytochromes in Geobacter sulfurreducens. J. Bacteriol. 187, 4505–4513. doi: 10.1128/JB.187.13.4505-4513.2005
Kim, B.-C., Postier, B. L., DiDonato, R. J., Chaudhuri, S. K., Nevin, K. P., and Lovley, D. R. (2008). Insights into genes involved in electricity generation in Geobacter sulfurreducens via whole genome microarray analysis of the OmcF-deficient mutant. Bioelectrochemistry 73, 70–75. doi: 10.1016/j.bioelechem.2008.04.023
Leitch, F. A., Moore, G. R., and Pettigrew, G. W. (1984). Structural basis for the variation of pH-dependent redox potentials of Pseudomonas cytochromes c-551. Biochemistry 23, 1831–1838. doi: 10.1021/bi00303a039
Lindgren, A., Larsson, T., Ruzgas, T., and Gorton, L. (2000). Direct electron transfer between the heme of cellobiose dehydrogenase and thiol modified gold electrodes. J. Electroanal. Chem. 494, 105–113. doi: 10.1016/S0022-0728(00)00326-320
Liu, Y., Seefeldt, L. C., and Parker, V. D. (1997). Entropies of redox reactions between proteins and mediators: the temperature dependence of reversible electrode potentials in aqueous buffers. Anal. Biochem. 250, 196–202. doi: 10.1006/ABIO.1997.2222
Lukat, P., Hoffmann, M., and Einsle, O. (2008). Crystal packing of the c6-type cytochrome OmcF from Geobacter sulfurreducens is mediated by an N-terminal Strep-tag II. Acta Crystallogr. Sect. D Biol. Crystallogr. 64, 919–926. doi: 10.1107/S0907444908021306
McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. doi: 10.1107/S0021889807021206
Methé, B. A., Nelson, K. E., Eisen, J. A., Paulsen, I. T., Nelson, W., Heidelberg, J. F., et al. (2003). Genome of Geobacter sulfurreducens: metal reduction in subsurface environments. Science 302, 1967–1969. doi: 10.1126/science.1088727
Moore, G. R., Pettigrew, G. W., Pitt, R. C., and Williams, R. J. P. (1980). pH dependence of the redox potential of Pseudomonas aeruginosa cytochrome c-551. Biochim. Biophys. Acta Bioenerg. 590, 261–271. doi: 10.1016/0005-2728(80)90030-90034
Morgado, L., Fernandes, A. P., Dantas, J. M., Silva, M. A., and Salgueiro, C. A. (2012). On the road to improve the bioremediation and electricity-harvesting skills of Geobacter sulfurreducens: functional and structural characterization of multihaem cytochromes. Biochem. Soc. Trans. 40, 1295–1301. doi: 10.1042/BST20120099
Moss, G. P. (1988). Nomenclature of tetrapyrroles (Recommendations 1986). Eur. J. Biochem. 178, 277–328. doi: 10.1111/j.1432-1033.1988.tb14453.x
Otwinowski, Z., and Minor, W. (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. doi: 10.1016/S0076-6879(97)76066-X
Perutz, M. F. (1989). Mechanisms of cooperativity and allosteric regulation in proteins. Q. Rev. Biophys. 22, 139–237. doi: 10.1017/S0033583500003826
Pokkuluri, P. R., Londer, Y. Y., Wood, S. J., Duke, N. E. C., Morgado, L., Salgueiro, C. A., et al. (2009). Outer membrane cytochrome c, OmcF, from Geobacter sulfurreducens: high structural similarity to an algal cytochrome c6. Proteins Struct. Funct. Bioinforma. 74, 266–270. doi: 10.1002/prot.22260
Salgueiro, C. A., Turner, D. L., and Xavier, A. V. (1997). Use of paramagnetic NMR probes for structural analysis in cytochrome c3 from Desulfovibrio vulgaris. Eur. J. Biochem. 244, 721–734. doi: 10.1111/j.1432-1033.1997.00721.x
Santos, T. C., de Oliveira, A. R., Dantas, J. M., Salgueiro, C. A., and Cordas, C. M. (2015). Thermodynamic and kinetic characterization of PccH, a key protein in microbial electrosynthesis processes in Geobacter sulfurreducens. Biochim. Biophys. Acta Bioenerg. 1847, 1113–1118. doi: 10.1016/J.BBABIO.2015.06.005
Sawyer, D. T., Sobkowiak, A., Roberts, J. L., and Sawyer, D. T. (1995). Electrochemistry for Chemists. Hoboken, NJ: Wiley.
Schnackenberg, J., Than, M. E., Mann, K., Wiegand, G., Huber, R., and Reuter, W. (1999). Amino acid sequence, crystallization and structure determination of reduced and oxidized cytochrome c6 from the green alga Scenedesmus obliquus. J. Mol. Biol. 290, 1019–1030. doi: 10.1006/jmbi.1999.2944
Schumann, F. H., Riepl, H., Maurer, T., Gronwald, W., Neidig, K.-P., and Kalbitzer, H. R. (2007). Combined chemical shift changes and amino acid specific chemical shift mapping of protein–protein interactions. J. Biomol. NMR 39, 275–289. doi: 10.1007/s10858-007-9197-z
Teixeira, L. R., Dantas, J. M., Salgueiro, C. A., and Cordas, C. M. (2018). Thermodynamic and kinetic properties of the outer membrane cytochrome OmcF, a key protein for extracellular electron transfer in Geobacter sulfurreducens. Biochim. Biophys. Acta Bioenerg. 1859, 1132–1137. doi: 10.1016/j.bbabio.2018.07.007
Keywords: site-directed mutagenesis, electron transfer proteins, c-type cytochrome, redox-Bohr effect, cyclic voltammetry, nuclear magnetic resonance, X-ray crystallography
Citation: Teixeira LR, Cordas CM, Fonseca MP, Duke NEC, Pokkuluri PR and Salgueiro CA (2020) Modulation of the Redox Potential and Electron/Proton Transfer Mechanisms in the Outer Membrane Cytochrome OmcF From Geobacter sulfurreducens. Front. Microbiol. 10:2941. doi: 10.3389/fmicb.2019.02941
Received: 19 September 2019; Accepted: 06 December 2019;
Published: 14 January 2020.
Edited by:
Amelia-Elena Rotaru, University of Southern Denmark, DenmarkReviewed by:
Pier-Luc Tremblay, Wuhan University of Technology, ChinaFalk Harnisch, Helmholtz Centre for Environmental Research (UFZ), Germany
Copyright © 2020 Teixeira, Cordas, Fonseca, Duke, Pokkuluri and Salgueiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Phani Raj Pokkuluri, cHBva2t1bHVyaUBnbWFpbC5jb20=; Carlos A. Salgueiro, Y3NhbGd1ZWlyb0BmY3QudW5sLnB0
†Present address: Norma E. C. Duke, SER-CAT and the Department of Biochemistry and Molecular Biology, University of Georgia, Athens, GA, United States Phani Raj Pokkuluri, X-ray Science Division, Argonne National Laboratory, Lemont, IL, United States