Abstract
Thermosynechococcus is a genus of thermophilic unicellular cyanobacteria that are dominant in microbial mats at about 50–65°C in alkaline hot springs of eastern Asia. We used PacBio SMRT Sequencing to sequence the complete genome of a novel strain of thermophilic cyanobacterium, Thermosynechococcus sp. CL-1, isolated from the Chin-Lun hot spring (pH 9.3, 62°C) in Taiwan. Genome-scale phylogenetic analysis and average nucleotide identity (ANI) results suggested that CL-1 is a new species in the genus Thermosynechococcus. Comparative genome analysis revealed divergent genome structures of Thermosynechococcus strains. In addition, the distinct genetic differences between CL-1 and the other Thermosynechococcus strains are related to photosynthesis, transporters, signal transduction, the chaperone/usher system, nitric oxide protection, antibiotic resistance, prokaryotic immunity systems, and other physiological processes. This study suggests that Thermosynechococcus strains have actively acquired many putative horizontally transferred genes from other bacteria that enabled them to adapt to different ecological niches and stressful conditions in hot springs.
Introduction
Thermophilic cyanobacteria grow photosynthetically under high-temperature and very stressful environments in hot springs (Ward et al., 2012). The underlying mechanisms of how thermophilic cyanobacteria adapt to different stressful conditions of hot springs are still not fully understood. In addition, the enzymes of hot-spring cyanobacteria are highly stable and can catalyze enzymatic reactions under high-temperature conditions (Patel et al., 2019). Therefore, hot-spring cyanobacteria and their bioproducts may have high value for biotechnology and industrial applications (Patel et al., 2019).
Genomic studies of hot-spring cyanobacteria have been conducted in several countries such as the United States and Japan (Nakamura et al., 2002; Bhaya et al., 2007; Stolyar et al., 2014; Olsen et al., 2015). Molecular analysis of the microbial mat community in Octopus Spring of Yellowstone National Park revealed three unrelated Synechococcus phylogenetic lineages (>10% 16S rRNA sequence variation), A/B, C1, and C9 (Papke et al., 2003). The dominant Synechococcus in the Yellowstone hot springs were the Synechococcus A/B genotypes. Representative strains among A/B genotypes with sequenced genome information are JA-3-3-Ab and JA-2-3Ba (Bhaya et al., 2007). A recent comparative genomic study of four Synechococcus strains of Mushroom Spring, Yellowstone National Park, within the A lineage revealed distinct differences in gene content and alleles between high-light- and low-light-adapted strains (Olsen et al., 2015). This study suggested that strains of closely related putative ecotypes have developed different genomic adaptations that enable them to inhabit distinct ecological niches in microbial mats of Yellowstone hot springs.
Thermosynechococcus were dominant in microbial mats at about 50–65°C in alkaline hot springs of eastern Asia and also found at low abundance in some hot springs of North American (Papke et al., 2003; Liao et al., 2006; Everroad et al., 2012; Tang et al., 2018). Representative strains with sequenced genome information are Thermosynechococcus elongatus BP-1 (BP-1), Thermosynechococcus vulcanus NIES-2134 (T. vulcanus) and Thermosynechococcus sp. NK55 (NK55) isolated from hot springs in Japan (Nakamura et al., 2002; Stolyar et al., 2014); T. elongatus PKUAC-SCTE542 (SCTE542) isolated from a hot spring in western Sichuan in China (Liang et al., 2019); and Synechococcus lividus PCC6715 isolated from a hot spring in Yellowstone National Park in the United States (Dyer and Gafford, 1961). Thermosynechococcus strains have been widely used for photosynthesis and other scientific research (Liang et al., 2019; Patel et al., 2019). In addition, previous studies demonstrated that several Thermosynechococcus strains (BP-1, T. vulcanus and SCTE542) could perform natural transformation of foreign DNA via homologous recombination (Iwai et al., 2004; Liang et al., 2019).
Thermosynechococcus sp. CL-1 (CL-1) was isolated in the Chin-Lun hot spring (pH 9.3, 62°C) in eastern Taiwan (Hsueh et al., 2007a; Figure 1A). The 16S rRNA gene of CL-1 was very similar (three to six mismatches) to that of representative Thermosynechococcus strains (BP1, T. vulcanus, NK55 and SCTE542). CL-1 has been studied in terms of CO2 elimination in the packed tower with potassium hydroxide to enhance mass transfer of CO2 (about fivefold) and also to regenerate alkaline solution by photosynthesis (Hsueh et al., 2007b). CL-1 showed high performance of carbon bio-fixation and also carbohydrate production (the highest content is 61%) under N-limiting conditions with sufficient dissolved inorganic carbon (Hsueh et al., 2009). In terms of types of bioreactors, CL-1 was applied to a continuous column cultivation system with 1.7 g/L/d of the highest cell mass productivity obtained (Su et al., 2012). The growth of CL-1 seems to be limited by illumination because its cell mass productivity can reach about 2.8 g/L/d under a 1.5-cm light path flat-plate photobioreactor and optimal biomass concentration (about 3 g/L) (Su et al., 2013). After modifying the composition ratios of medium, the cell mass productivity, CO2 fixation rate, and carbohydrate productivity can be up to 3.3, 5.3, and 1.8 g/L/d, respectively (Su et al., 2017). CL-1 seems to be a good candidate for CO2 elimination and simultaneous production of bioenergy precursor (such as carbohydrates) (Su et al., 2017).
FIGURE 1
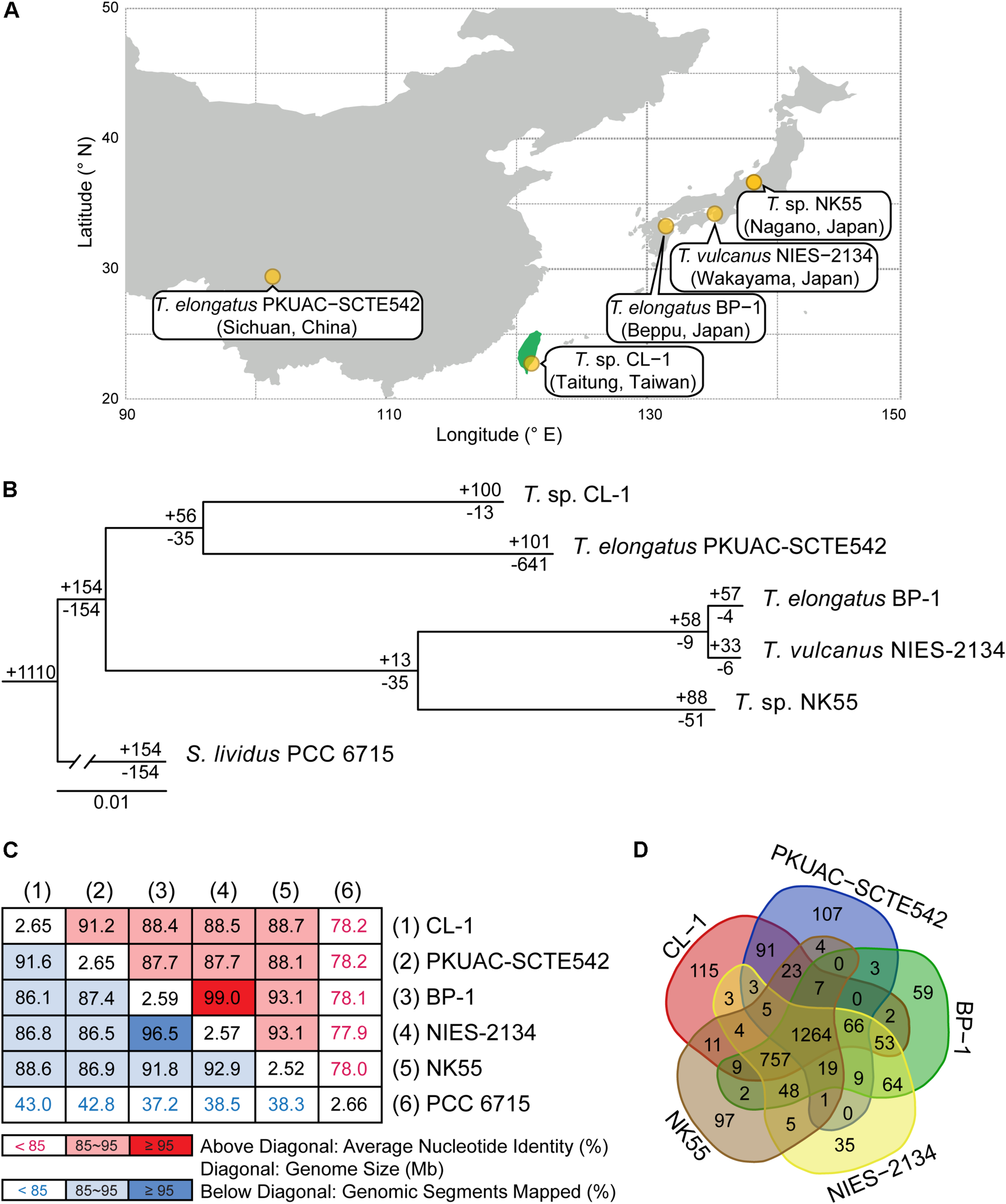
Comparisons between CL-1 and other representative Thermosynechococcus strains. (A) Geographic locations of isolation. (B) Maximum-likelihood phylogeny and phylogenetic distribution of homologous gene clusters. The molecular phylogeny was inferred by concatenated protein alignment of 1,085 single-copy genes shared by all genomes (281,438 aligned sites). All branches received 100% bootstrap support based on 1,000 replicates. Synechococcus lividus PCC 6715 was included as an outgroup to root the tree. The numbers above a branch and preceded by a “+” sign indicate the number of homologous genes uniquely present in all daughter lineages. The numbers below a branch and preceded by a “−” sign indicate the number of homologous genes uniquely absent in all daughter lineages. For example, the strain CL-1 has 100 homologous gene clusters that are not found in any of the other genomes and 13 gene clusters are shared by all genomes except CL-1. (C) Pairwise genome sequence similarity scores. The numbers along the diagonal indicate the genome sizes (Mb). The numbers above diagonal indicate the average nucleotide identity (ANI) values (%). The numbers below the diagonal indicate the proportion of segments that could be mapped between the two genomes for ANI calculation. (D) Number of shared and unique homologous gene clusters.
Here we sequenced and analyzed the genome of CL-1 isolated from the Chin-Lun hot spring in Taiwan. The genome of CL-1 was compared to published genomes of four other Thermosynechococcus strains and S. lividus in the C1 genotype to reveal genetic diversity and adaption mechanisms of hot-spring cyanobacteria.
Materials and Methods
Sample Source and Genome Sequencing
The strain CL-1 was isolated from the Chin-Lun hot spring (pH 9.3, 62°C) in Taitung, Taiwan as described (Hsueh et al., 2007a). Axenic culture of the strain was performed at 45°C on a BG-11 agar plate or in BG-11 liquid medium supplemented with 20 mM TES (pH 8.0) under continuous white LED light (20 μmol photons m–2 s–1).
Total genomic DNA was extracted and purified by using the DNeasy Plant Maxi Kit (QIAGEN, Germany). Quality and quantity of purified genomic DNA were assessed by using the NanoDrop 2000 spectrophotometer (ThermoFisher, United States) and 1% agarose gel electrophoresis. Whole-genome shotgun sequencing involved using the PacBio Sequel platform (Pacific Biosciences, United States). One gel-plus (20 kb) library and one SMRT cell was used. The de novo genome assembly involved using the Hierarchical Genome Assembly Process (HGAP) assembler v4 (Chin et al., 2013). Gene prediction and annotation involved using the NCBI prokaryotic genome annotation pipeline (Tatusova et al., 2016). We also used the RAST annotation system to minimize poor calls (Aziz et al., 2008). All bioinformatics tools were used with the default settings unless stated otherwise.
Genome Analysis
The procedures for genome analysis were based on those described in our previous work (Chung et al., 2013; Lo et al., 2013; Tsai et al., 2018). Briefly, the genome map was prepared by using Circos v0.69-6 (Krzywinski et al., 2009). To identify genes that may have originated from horizontal gene transfer (HGT), we performed BLASTP searches (Camacho et al., 2009) against the NCBI non-redundant protein database (Benson et al., 2018). Only hits with high-scoring pairs accounting for at least 90% of the query length and overall amino acid sequence similarity of at least 40% were retained to ensure that the hits represent likely homologs rather than non-homologous genes sharing only conserved domains. Genes with the best hit that lacked a taxonomic assignment at the genus level or was derived from metagenomics surveys were manually examined for more reliable inference. A gene is classified as putatively acquired if more than half of the top-five hits were from other genera. Among the putatively acquired genes, those with non-Thermosynechococcus sequences as the best hits are classified as being recently acquired.
For comparative analysis within the genus, a list of closely related strains with genome sequences available was compiled from the NCBI genome database (Benson et al., 2018) and a literature search (Table 1). The homologous gene clusters among these genomes were identified by using OrthoMCL (Li et al., 2003). The KEGG database (Kanehisa et al., 2010) was used for examining annotation and gene function. For pairwise genome alignments, the NUCleotide MUMmer (NUCmer) program of the MUMmer package v3.23 (Kurtz et al., 2004) was used with the setting “–maxmatch –mincluster 200.” The average nucleotide identity (ANI) and percentage of genome segments mapped for each genome pair were calculated by using FastANI (Jain et al., 2018). For phylogenetic analysis, MUSCLE v3.8.31 (Edgar, 2004) was used to generate multiple sequence alignments and PhyML v3.3 (Guindon and Gascuel, 2003) for maximum-likelihood inference. The packages ggplot2 v3.2.0 (Wickham, 2009) and gggenes v0.4.0 (Wilkins and Kurtz, 2019) were used to visualize gene locations and syntenies. The distributions of genes by functional category were plotted by using the “barplot” function of R (R Core Team, 2019).
TABLE 1
| Strain | T. sp. | T. elongatus | T. elongatus | T. vulcanus | T. sp. | S. lividus |
| CL-1 | PKUAC-SCTE542 | BP-1 | NIES-2134 | NK55 | PCC 6715 | |
| Origin | Taitung, | Sichuan, | Beppu, | Wakayama, | Nagano, | Yellowstone |
| Taiwan | China | Japan | Japan | Japan | National Park, | |
| United States | ||||||
| GenBank accession | CP040671 | CP032152 | NC_004113 | NZ_AP018202 | NC_023033 | NZ_CP018092 |
| Genome size (bp) | 2,647,823 | 2,648,728 | 2,593,857 | 2,571,271 | 2,520,064 | 2,659,739 |
| G + C content (%) | 53.5 | 53.3 | 53.9 | 53.9 | 53.8 | 53.5 |
| Coding density (%) | 88.9 | 48.9 | 89.5 | 88.7 | 85.9 | 76.6 |
| Number of rRNA genes | 3 | 3 | 3 | 3 | 3 | 3 |
| Number of tRNA genes | 41 | 41 | 42 | 40 | 41 | 41 |
| Number of protein-coding genes | 2,465 | 1,625 | 2,476 | 2,413 | 2,287 | 2,227 |
| Number of pseudogenes | 84 | 944 | 0 | 70 | 112 | 321 |
Genome statistics of representative thermophilic cyanobacteria.
All strains in the genus Thermosynechococcus with available genome sequences are included; Synechococcus lividus PCC 6715 was included as an outgroup. All strains were isolated from hot springs; the locations are listed.
Chemical Analysis
The water sample was taken from the Chin-Lun hot spring located in the east of Taiwan. Temperature, pH, and conductivity were in situ measured by probes. Other items, heavy metals, major ions, and non-purgeable organic carbon (NPOC) underwent laboratory analyses after standard pretreatments. The pretreatments and analyses were performed according to Taiwan National Institute of Environmental Analysis (NIEA) methods1. The analysis of As was performed by hydride generation/atomic absorption spectrometry. Mercury (Hg) was measured by cold-vapor atomic absorption spectrometry. Other heavy metals (Cr, Cd, Cu, and Zn) were measured by inductively coupled plasma-atomic emission spectrometry. Major ions (Na+, , K+, Mg2+, Ca2+, Cl–, , , /, ) were measured by ion chromatography. NPOC was measured by the combustion oxidation/non-dispersive infrared absorption method.
Results and Discussion
Complete Genome Sequence of Thermosynechococcus sp. CL-1
Whole-genome shotgun sequencing of Thermosynechococcus sp. CL-1 generated 47,666 filtered reads (average length = 7,536 bp, N50 length = 8,319 bp, total length = 359,209,730 bp). These reads provided ∼138-fold coverage of the genome, and the de novo assembly produced a single 2,647,823-bp circular chromosome (Figure 2). No plasmid was found. The annotation included 41 tRNA genes, one complete set of 16S-23S-5S rRNA genes, and 2,465 protein-coding genes (Table 1).
FIGURE 2
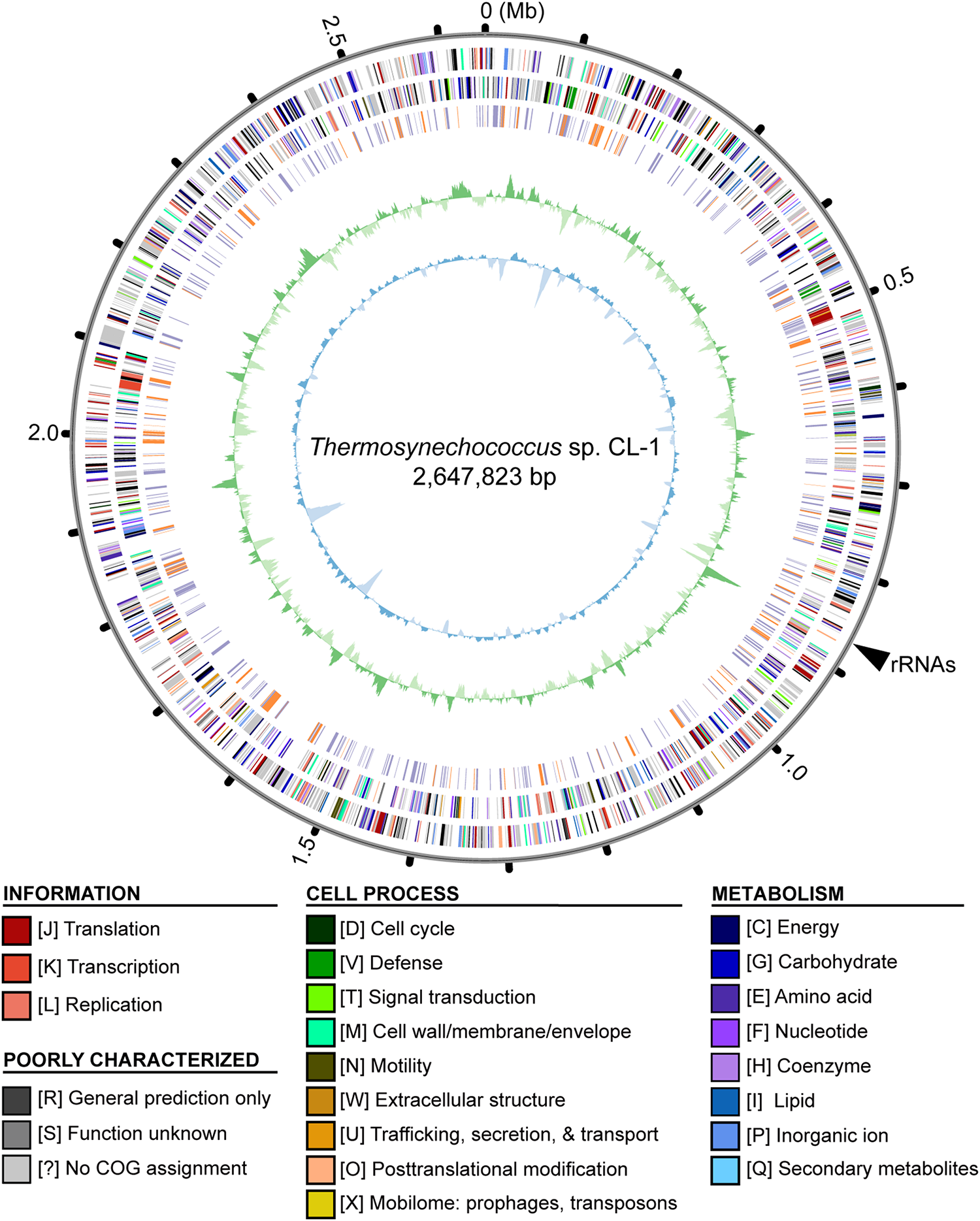
Genome map of Thermosynechococcus sp. CL-1. Rings from outside in: (1) Scale marks in Mb; (2, 3), protein-coding genes on the forward and reverse strand, respectively (color-coded by functional categories); (4) locations of putatively acquired genes (orange: recent; purple: other); (5) GC skew (positive: dark green; negative: dark green); (6) GC content (above average: dark blue; below average: light blue). The position of rRNA genes (at ∼0.87 Mb) is marked by a black triangle outside of the rings. Note that the regions with low GC content (e.g., at ∼0.14, 1.62, and 1.83 Mb) often correspond to putatively acquired genes in the “recent” category. These putatively acquired DNA segments may came from donor genomes with lower GC content, and have not been ameliorated yet due to their recent acquisition.
Similar to other cyanobacteria, such as the closely related T. elongatus BP-1 (Nakamura et al., 2002) or the more distant Anabaena sp. PCC 7120 (Kaneko et al., 2001), the chromosome organization did not exhibit strong patterns of GC-skew (Figure 2). One high positive peak at ∼0.87 Mb corresponded to the location of the rRNA gene cluster. Regarding the GC content, several low GC regions were found (e.g., at ∼0.14, 1.62, and 1.83 Mb). These regions all correspond to DNA segments that may have been acquired recently. These putatively acquired segments may have originated from donors with lower GC content and have not been ameliorated yet due to their recent acquisition.
Comparison With Other Cyanobacteria
Four other strains with complete genome sequences are available for the genus Thermosynechococcus (Table 1). We selected these genomes as well as the outgroup S. lividus for comparative analysis. Examination of gene content revealed 1,110 homologous gene clusters shared by all these genomes (Supplementary Table S1); 1,085 were present as single-copy genes in all strains. A concatenated alignment of these genes showed 281,438 aligned amino acid sites and produced a maximum-likelihood phylogeny with 100% bootstrap support for all branches (Figure 1B). From the genome-scale phylogeny, CL-1 from Taiwan is most closely related to SCTE542 from China, whereas the three other Thermosynechococcus strains from Japan form a sister clade. Intriguingly, two strains of T. elongatus (i.e., BP-1 and SCTE542) do not form a monophyletic clade, which indicates a conflict between phylogeny and taxonomy. Results from genome-wide ANI (Figure 1C) and the number of shared genes (Figure 1D) are similar to the patterns observed from molecular phylogeny. Pairwise genome alignments (Figure 3) indicated very low conservation in chromosomal organization among these cyanobacteria. BP-1 and T. vulcanus showed extensive proliferation of mobilome-related elements (e.g., insertion sequences, integrase, and transposases) (Figure 4), but the locations of these mobile genetic elements do not fully explain the disruption of synteny (Figure 3).
FIGURE 3
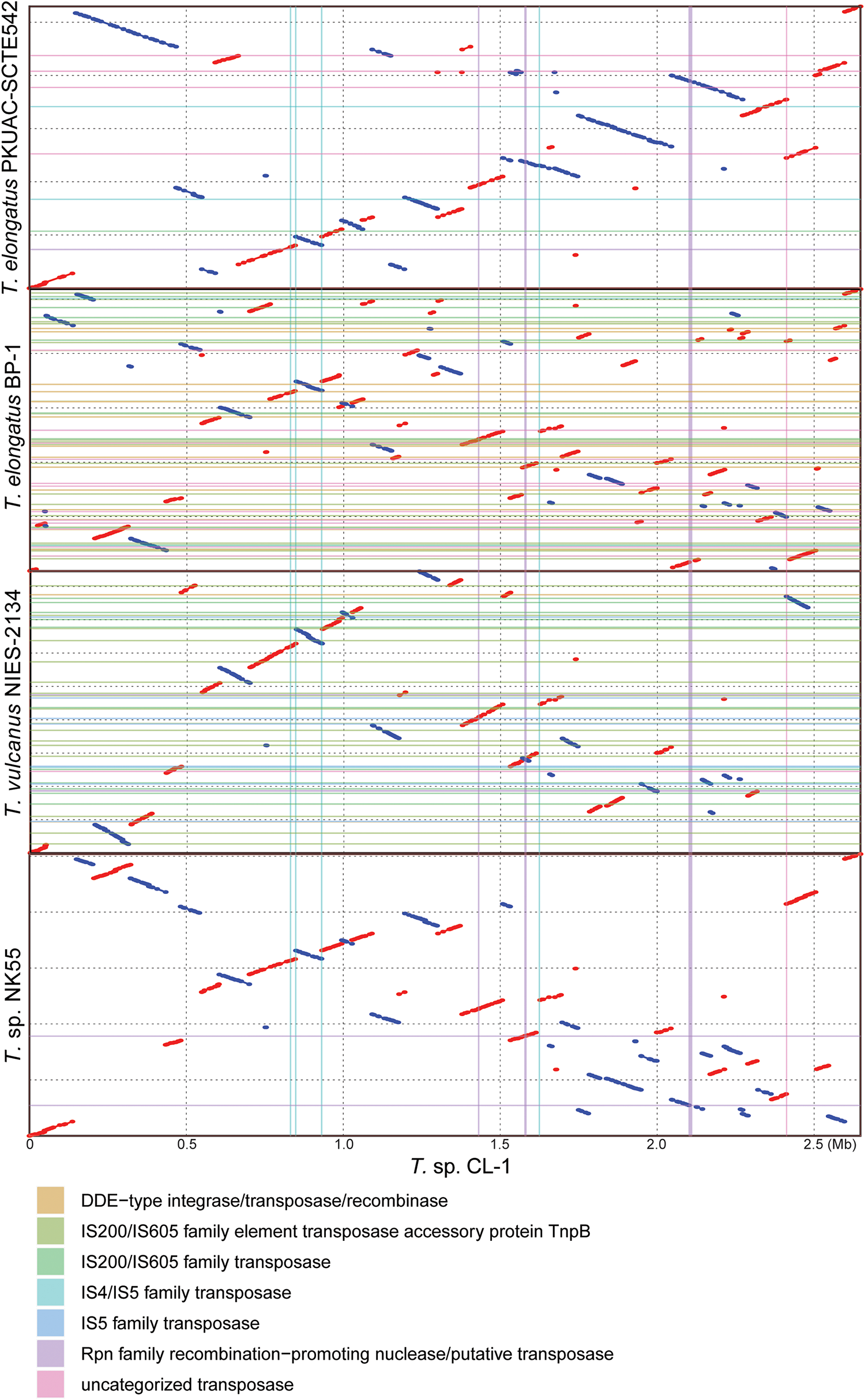
Pairwise genome alignments. The CL-1 genome is used as the reference for pairwise alignment with each of the other available Thermosynechococcus genomes. Red dots indicate matches in the same orientation, blue dots indicate matches in the opposite orientations. Mobilome-related segments (e.g., prophages, transposons, etc.) are represented by color lines (vertically for those found in the CL-1 genome and horizontally for those found in the other genomes) for visualization of correspondence between mobilome and chromosomal synteny breakpoints.
FIGURE 4
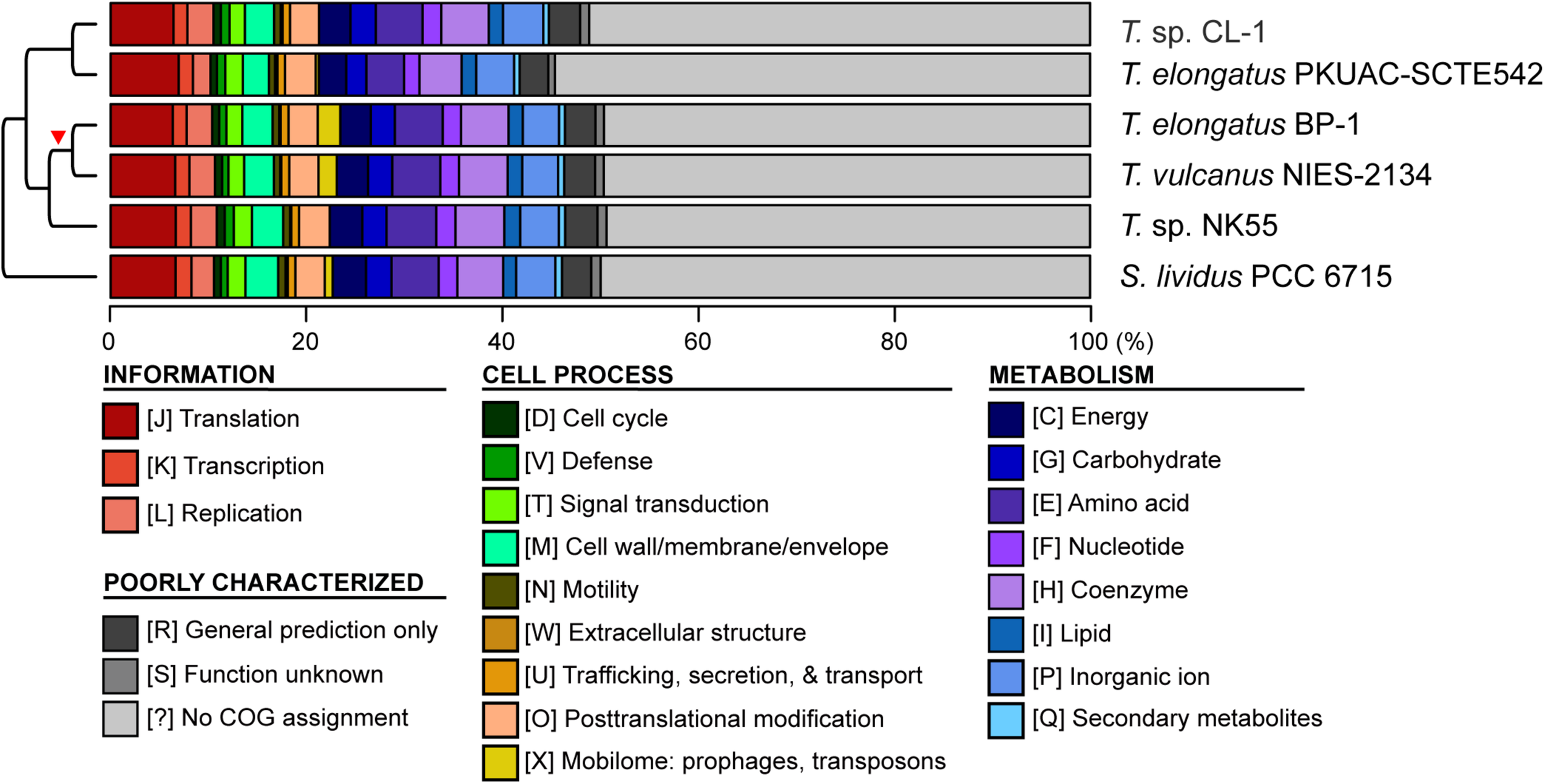
Functional classification of protein-coding genes. The functional categories were defined according to the COG database. The cladogram on the left side is based on Figure 1A. The red triangle indicates the putative origin of mobilome gene expansion.
Although CL-1 is most closely related to SCTE542, these two strains still show considerable divergence in genomes. The ANI value was only 91.2%, below the suggested cutoff of 95% for within-species comparison (Jain et al., 2018). Additionally, SCTE542 appeared to have undergone extensive genome degradation, such that it contains 944 annotated pseudogenes (Table 1) and lacks 641 genes shared by all other genomes compared (Figure 1D). Among the Japanese strains, T. elongatus BP-1 and T. vulcanus NIES-2134 are very similar in their genomes. These two strains share 96.5% of their genomic segments, and these segments have 99.0% ANI value (Figure 1C). Moreover, these two strains share ∼97% annotated genes (Figure 1D) and are unique compared with other Thermosynechococcus strains in their mobilome proliferation (Figure 4).
Taken together, these results suggest that some revisions of the Thermosynechococcus taxonomy may be necessary based on the 95% ANI cutoff (Jain et al., 2018). First, CL-1 likely represents a novel species within this genus. Second, SCTE542 is sufficiently divergent from BP-1 (i.e., ANI = 87.7%) to be considered as another novel species, rather than T. elongatus. Third, NIES-2134 has a highly similar genome and a close phylogenetic relationship to BP-1 (i.e., ANI = 99.0%) and thus could be re-classified as a strain of T. elongatus.
Horizontal Gene Transfer
The CL-1 genome is notable in having a high number of putatively acquired genes. Based on the high-throughput BLASTP-based screening, 458 of the 2,465 (19%) annotated protein-coding genes may have been acquired (Supplementary Table S2). Among these, 137 have the best hit from a putative donor that does not belong to the genus Thermosynechococcus, which suggests that these are recent acquisitions. For these 137 recently acquired genes, 42% of the putative donors are from the order Synechococcales, 29% are from other more divergent Cyanobacteria, and 17% are from the phylum Proteobacteria (Figure 5). In terms of functions, 72% of these do not have specific category assignment based on COG. For those with functional assignment, inorganic ion transport and metabolism (i.e., category “P”) is the most abundant category, followed by amino acid transport and metabolism (i.e., category “E”) (Figure 5).
FIGURE 5
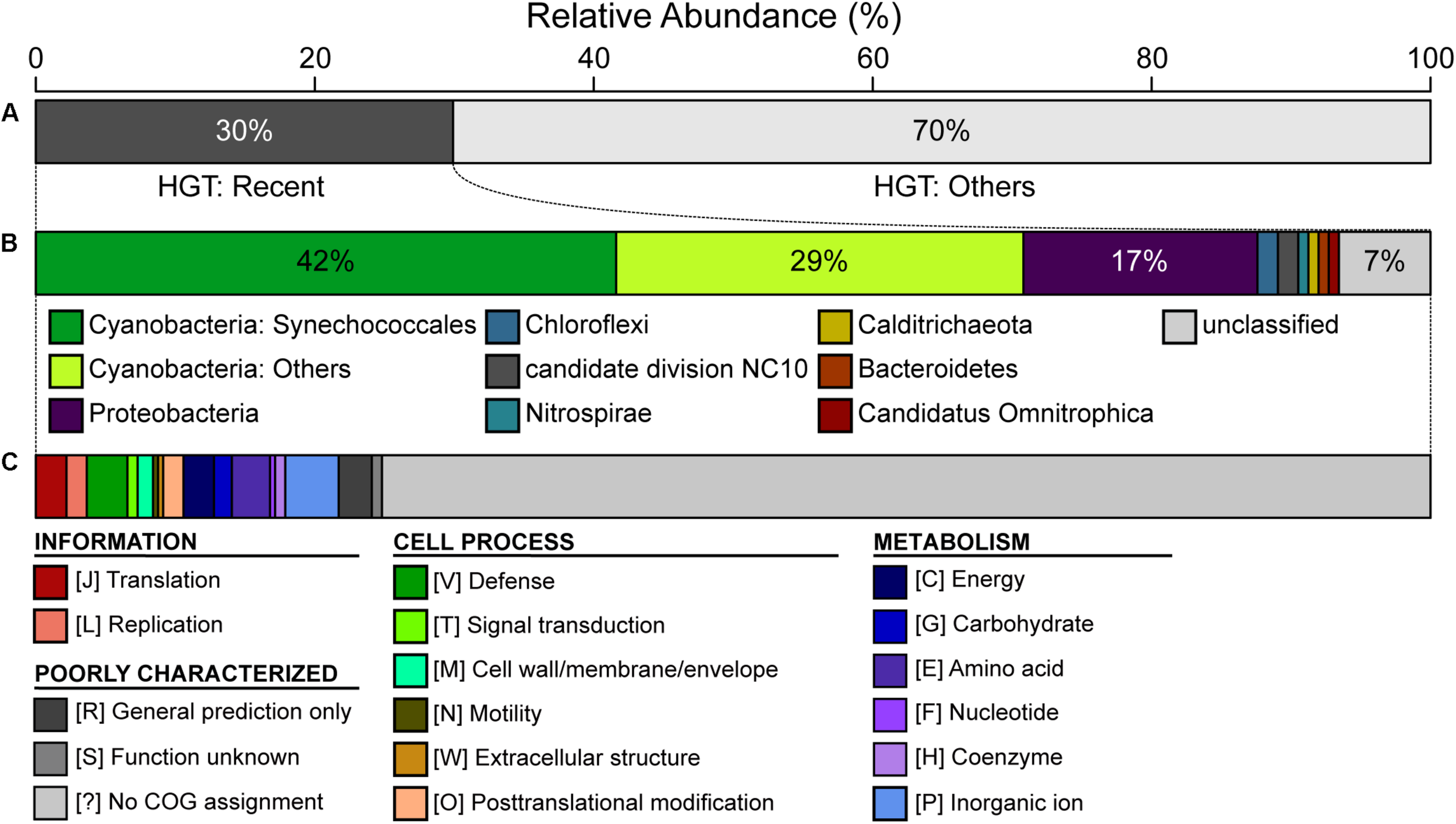
Summary of putative horizontal gene transfer (HGT). Among the 2,465 annotated protein-coding genes in the CL-1 genome, 458 were classified as putatively acquired by using a high-throughput BLASTP-based screening. (A) Classification based on the best hit; those with the best hit from outside of the genus Thermosynechococcus (137/458 = 30%) were classified as being recent acquisitions. (B) Taxonomic assignment of putative donors for recent HGT events. (C) Functional classification of recently acquired genes based on the COG database.
Because this high-throughput BLASTP-based approach may not be reliable due to biases in the taxon sampling of available genomes in the current database, we manually inspected these results to examine the hits and synteny information. Notable examples are visualized (Figure 6 and Supplementary Figure S1) and discussed in more detail below.
FIGURE 6
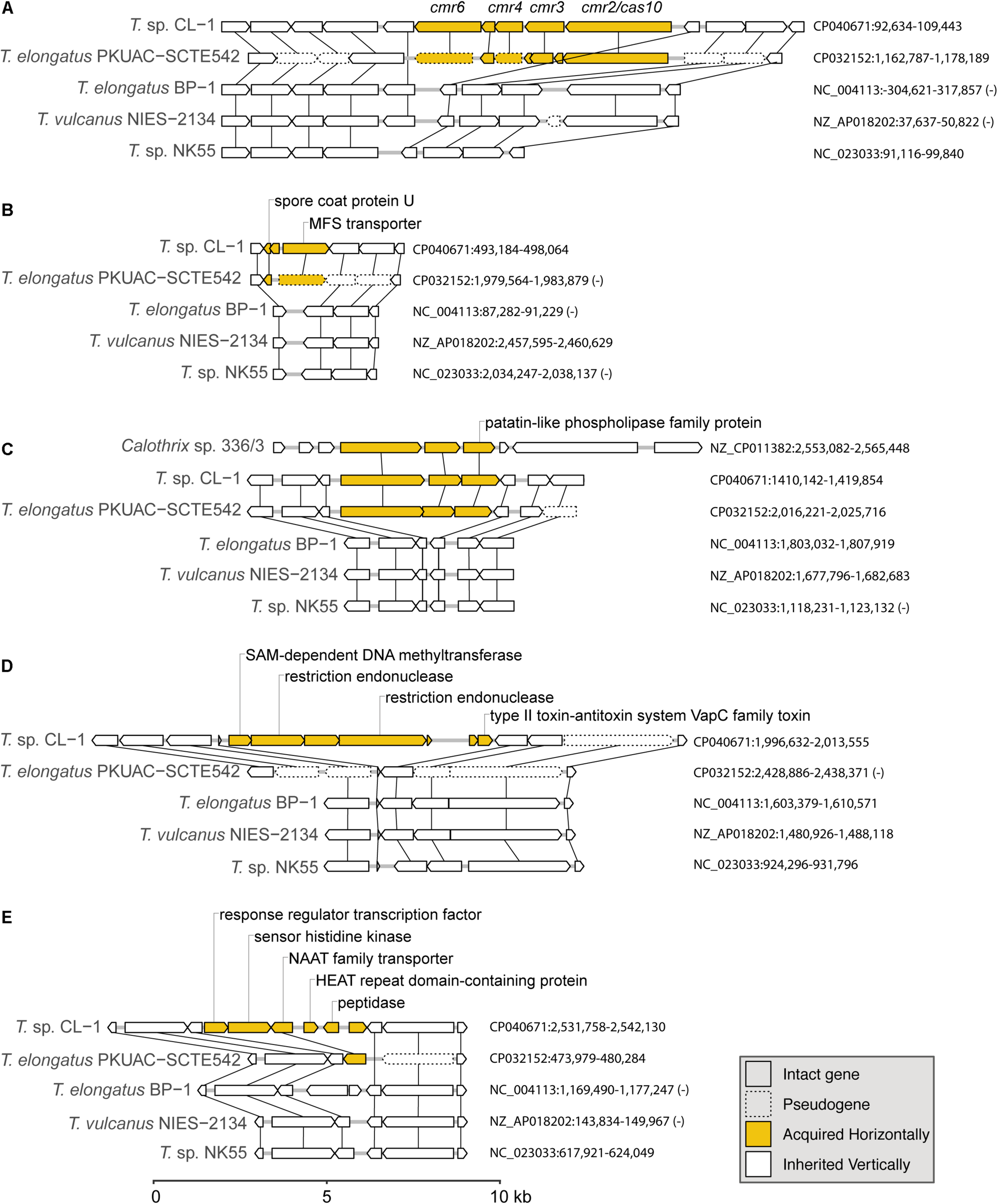
Examples of gene islands acquired by the CL-1 strain through horizontal gene transfer: (A) type III-B CRISPR-Cas system; (B) spore coat protein U and MFS transporter; (C) patatin-like phospholipase family protein; (D) multiple restriction endonucleases and type II toxin–antitoxin system; (E) NAAT family transporter. The sequence accession numbers and exact locations of these regions are labeled on the right. Genes in these regions are drawn to scale, and pseudogenes are drawn with dashed lines. Homologous genes across different genomes are linked by vertical lines. Putative acquired genes are in yellow. Locus tags of putatively acquired genes in the CL-1 genome: (A) FFX45_00470-FFX45_00490, (B) FFX45_02450-FFX45_02460, (C) FFX45_07120-FFX45_07130, (D) FFX45_09855-FFX45_09885, and (E) FFX45_12430-FFX45_12455.
The first example of gene acquisition is the type III-B CRISPR-Cas system found in CL-1 and SCTE542 genomes (Figure 6A; Makarova et al., 2011). The type I system in CL-1 and SCTE542 may have been inherited vertically because this system is also found in NK55. In contrast, the type III-B system may have been acquired horizontally in the common ancestor of CL-1 and SCTE542 (Figure 6A). Although the absence of these genes in the other three Thermosynechococcus genomes may be explained by one loss event, BLAST searches using the CL-1 homolog revealed that the best hits were from other families (i.e., not Synechococcaceae, which contains the genera Thermosynechococcus/Synechococcus) in the order Synechococcales. On the basis of this pattern, the absence of these genes in the most recent common ancestor (MRCA) of Thermosynechococcus, followed by horizontal gene acquisition in the MRCA of CL-1 and SCTE542 appears to be a reasonable hypothesis. However, this putatively acquired type III-B system lacks the CRISPR repeats and several other genes typically associated with the system (e.g., cmr1 and cmr5). Thus, this partial type III-B system probably is not functional.
Two other examples of gene acquisition at the same phylogenetic branch involve one gene island containing a spore coat protein U and a major facilitator superfamily (MFS) transporter (Figure 6B) and another one containing a patatin-like phospholipase family protein (Figure 6C). In the latter case, the same gene island was found in Calothrix sp. 336/3, a hydrogen-producing cyanobacterium isolated from a lake in Finland (Isojärvi et al., 2015). However, the Calothrix phospholipase homolog has a low level of sequence similarity (i.e., 48% identity and 64% similarity in protein sequences), which indicates that either Calothrix is a distant relative of the putative donor, or the gene acquisition in CL-1 occurred in the distant past.
Furthermore, we found one region of putative acquisition specific to CL-1. This region contains multiple restriction endonucleases and a set of type II toxin-antitoxin systems (i.e., vapBC) (Figure 6D). Although vapC was also found in SCTE542 (locus tag: D3A95_07350) and NK55 (locus tag: NK55_RS09295), these genes are located in different regions of the chromosome and may not share the same evolutionary origin. Finally, an island with six putatively acquired genes was found in CL-1 (Figure 6E). One of these genes encoding for a NAAT family transporter was also present in SCTE542, so we do not know whether this region was acquired in the common ancestor of these two strains (followed by degradation in SCTE542) or whether CL-1 experienced multiple acquisition events in this region.
Finally, a glycosyltransferase gene in the CL-1 genome (Locus tag: FFX45_09045) may have been acquired from a Proteobacteria donor (Supplementary Figure S1). Although distant homologs of this gene could be found in other Thermosynechococcus genomes, those homologs form a strongly supported monophyletic clade that is quite distant from the CL-1 homolog. Instead, the CL-1 homolog is more closely related to those found in Proteobacteria (e.g., Halomonas and Altererythrobacter).
Photosynthesis
The major structural components of photosynthesis genes are well conserved among Thermosynechococcus genomes. For example, two copies of cytochrome c-550 genes (psbV1and psbV2) (FFX45_09625, FFX45_09620) were tandemly arranged in genomes of all Thermosynechococcus and S. lividus strains. In addition, three copies of psbA genes (psbA1, psbA2, and psbA3) (FFX45_08385, FFX45_08390, FFX45_10905) encode the reaction center D1 protein of photosystem II. psbA1 and psbA2 were tandemly arranged in genomes of all Thermosynechococcus strains except SCTE542. In the SCTE542 genome, psbA1 and psbA2 are separated by a transposase gene. Only two copies of psbA genes (psbA1 and psbA3 homologs) were detected in the S. lividus genome. However, we found some significant variations in regulatory components of photosynthesis-related genes. For example, sbtA, the high-affinity sodium-dependent bicarbonate transport family permease gene, is present in only the genomes of CL-1 (FFX45_07280), SCTE542, and S. lividus but not in the three Japanese strains. In contrast, the high-affinity ABC-type bicarbonate transport system (encoded by cmpABCD operon) is present in the genomes of CL-1 (FFX45_03210-03225) and the three Japanese strains but not SCTE542 or S. lividus. Thus, only CL-1 has both types of high-affinity bicarbonate transporters and may have higher capacity for bicarbonate uptake under different growth environments. In addition, CL-1, SCTE542 and S. lividus have a distinct flavodoxin gene (fldA) (FFX45_02905), which is adjacent to a Crp/Fnr family transcriptional regulator gene (FFX45_02900) and may form an operon together. Flavodoxins are electron transfer proteins that may substitute the function of ferredoxin in the photosynthetic electron transport chain under iron-deficient conditions. Furthermore, iron stress-inducible proteins (IsiAs) are giant chlorophyll–protein complexes induced by iron deficiency in cyanobacteria (Bibby et al., 2001; Boekema et al., 2001). We found longer predicted N-terminal amino acid sequences of IsiA gene products in the three Japanese strains (with 15 extra amino acid residues) and CL-1, SCTE542 (pseudogene), and S. lividus (with 20 extra amino acid residues) than those of mesophilic cyanobacteria such as Synechocystis sp. PCC6803 and Synechococcus sp. PCC7942 (Figure 7). The structural differences among these IsiA proteins are consistent with the genome-scale phylogeny analysis that CL-1 from Taiwan is most closely related to SCTE542 from China, whereas the three other Thermosynechococcus strains from Japan form a sister clade. The physiological significance of structural differences among these IsiA proteins requires further study.
FIGURE 7
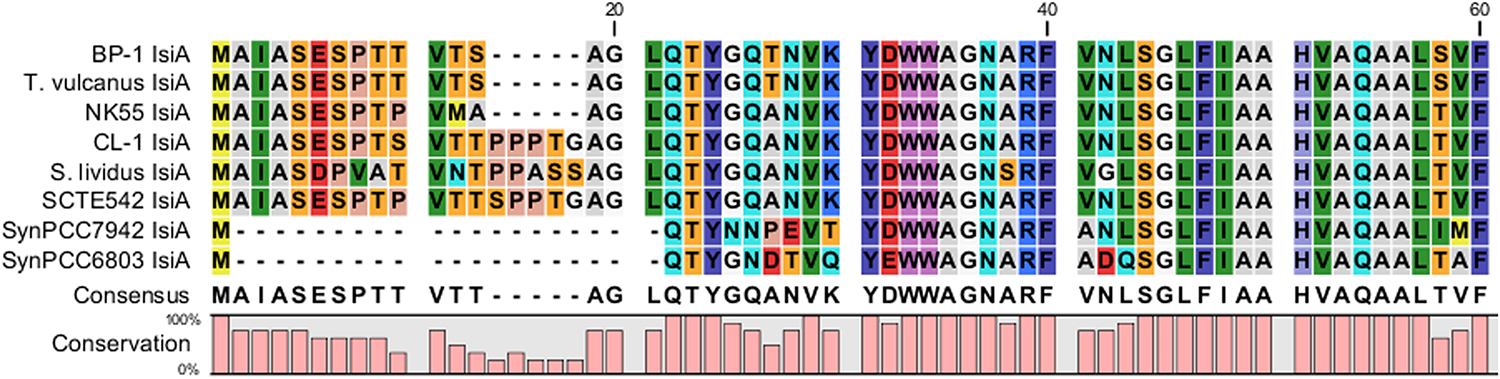
Comparative analysis of N-terminus amino acid sequences of iron stress-inducible (IsiA) proteins among five Thermosynechococcus strains, S. lividus, and two mesophilic cyanobacteria (i.e., Synechococcus sp. PCC7942 and Synechocystis sp. PCC6803) by using CLC Main Workbench (Qiagen Bioinformatics).
Metabolism
The core metabolism genes are highly conserved among Thermosynechococcus genomes and are similar to those found in other cyanobacteria (Beck et al., 2012). In addition, all the Thermosynechococcus genomes lack nitrogenases and hydrogenases (Stolyar et al., 2014). Furthermore, all the Thermosynechococcus genomes have a complete gene set (nrtABCD) of the nitrate transport system, nitrate reductase, and nitrite reductase for the assimilation of nitrate. Of note, CL-1 appeared to lose a complete gene set (urtABCDE) of the urea ABC transport system and several important components of urease genes (ureA, ureD, and ureG), which are well conserved in the genomes of the other Thermosynechococcus strains. The CL-1 strain is likely unable to import and utilize urea as a nitrogen source. In addition, the CL-1 genome has a distinct gene operon for nitric oxide protection, which is absent in genomes of the other Thermosynechococcus strains. The gene cluster contains dnrN or norA [encoding an iron-sulfur cluster repair di-iron protein (FFX45_09820) putatively involved in the repair of nitrosative damage to iron-sulfur clusters] and norB [encoding nitric-oxide reductase large subunit (FFX45_09815)]. This gene operon may participate in nitrogen metabolism and in defense against nitric oxide toxicity.
Transporters
CL-1, SCTE542, and S. lividus genomes have two distinct sets of gene clusters for the phosphate-specific transport-system pstSCAB operon (FFX45_00090-FFX45_00105, FFX45_04225-FFX45_04235). One shares high sequence similarity with the other Thermosynechococcus strains, but the other has a different origin. The multiple transporters for phosphate may have distinct affinity and/or activity under different phosphate concentrations. In addition, the CL-1 genome has two different types of Mg2+ transporter genes: mgtE (FFX45_01105) and corA (FFX45_03065). The SCTE542 genome has mgtE, and NK55 has corA. Moreover, CL-1, SCTE542, and S. lividus genomes have one gene operon that encodes a copper-translocating P-type ATPase (CopA) (FFX45_10400) and a Cu+ chaperone (CopZ) (FFX45_10405). The water analysis showed a small amount of arsenic (∼0.0183 mg/L) and nickel ions (0.212 mg/L) in the hot-spring water (see Supplementary Table S3). CL-1 and S. lividus genomes have one copy of an ArsB/NhaD family transporter (arsB) (FFX45_09985), but the other strains do not. Moreover, CL-1, NK55, and S. lividus genomes have one distinct gene operon that encodes the organoarsenical efflux MFS transporter ArsJ (FFX45_03530) and an associated glyceraldehyde-3-phosphate dehydrogenase (FFX45_03525). The ArsJ gene in the CL-1 genome exhibited 58% amino acid sequence identity to the ArsJ gene from Pseudomonas aeruginosa that conferred arsenate As(V) resistance (Chen et al., 2016). In addition, the CL-1 genome has one distinct MFS transporter gene (FFX45_02460; Figure 6B) that exhibited a significant degree of similarity to the nickel resistance gene (nreB) from Synechocystis sp. PCC6803 (54% amino acid sequence identity). Overall, CL-1 appeared to have several distinct transporter genes and various heavy metal efflux systems to cope with different stress conditions in hot-spring environments.
Signal Transduction
For the two-component signal transduction system, 15 and 26 potential genes for His kinases and response regulators, respectively, were identified in the CL-1 genome. Most are conserved in all Thermosynechococcus genomes. For example, the CL-1 genome contains two sets of motility-related (PixJ and PilJ homolog-associated) two-component signaling systems [including signal transduction His kinase/response regulators CheA (FFX45_00335 and FFX45_12785), methyl-accepting chemotaxis proteins (FFX45_00340 and 12790), CheW proteins (FFX45_00345 and FFX45_12795), and response regulator (CheY) proteins (FFX45_00350, FFX45_12800, and FFX45_12805)] that are conserved among all Thermosynechococcus strains except T. vulcanus. PixJ homolog-associated (blue-light-responsive) two-component signaling systems are truncated in the T. vulcanus genome. Of note, NK55 and S. lividus genomes have two additional sets of chemosensory two-component signaling systems, and BP1 and SCTE542 have one additional set. In addition, GGDEF/EAL domain proteins function as diguanylate cyclases/phosphodiesterases that synthesize/degrade cyclic di-GMP and participate in a cyclic-di-GMP signaling pathway that may regulate biofilm formation, motility, virulence, and cell cycle (Agostoni et al., 2013). Recent studies identified several cyanobacterichromes with GGDEF/EAL domains that mediated blue-light-induced cell aggregation in BP1 and T. vulcanus (Enomoto et al., 2015). The CL-1 and S. lividus genomes have 13 GGDEF/EAL domain protein genes, and the other Thermosynechococcus genomes only have 9–10 genes (Table 2). The hot-spring cyanobacteria JA-3-3-Ab and JA-2-3Ba (in the A/B lineage) have only four GGDEF or EAL domain proteins (see Table 2 and Agostoni et al., 2013). Thus, Thermosynechococcus and S. lividus strains (in the C1 linage) seem to have more complex cyclic-di-GMP signaling pathways than JA-3-3-Ab and JA-2-3Ba (in the A/B linage).
TABLE 2
| Strain | GGDEF | EAL | GGDEF + EAL | Total |
| T. sp. CL-1 | 6 | 1 | 6 | 13 |
| T. elongatus PKUAC-SCTE542 | 4 | 0 | 6 | 10 |
| T. elongatus BP1 | 4 | 0 | 5 | 9 |
| T. vulcanus NIES-2134 | 5 | 0 | 5 | 10 |
| T. sp. NK55 | 5 | 0 | 5 | 10 |
| S. lividus PCC 6715 | 5 | 0 | 8 | 13 |
| S. sp. JA-3-3Ab | 3 | 1 | 0 | 4 |
| S. sp. JA-2-3Ba | 4 | 0 | 0 | 4 |
Number of GGDEF and EAL domain-containing proteins in five Thermosynechococcus strains and representative Synechococcus strains from Yellowstone hot springs.
Chaperone–Usher System
CL-1, SCTE542, and S. lividus genomes have a unique gene cluster for a chaperone/usher system that is absent in the three Japanese strains. This gene cluster contains a molecular chaperone (fimC) (FFX45_06910), a fimbrial biogenesis outer membrane usher protein (fimD) (FFX45_06905), and three spore coat protein U domain-containing proteins (FFX45_06900, FFX45_06915, and FFX45_06920) (Geibel and Waksman, 2014). The chaperone/usher system is mainly present in proteobacteria but is also found in a few cyanobacteria. The function of the chaperone/usher system is to assemble proteinaceous filaments on the cell surface. These filaments could form fimbrial (pili) or non-fimbrial surface structures (e.g., a capsule or spore coat) (Leng et al., 2011).
Bipartite Aminoglycoside Nucleotidyltransferases
The CL-1 and SCTE542 genomes have three and one copies of putative bipartite aminoglycoside nucleotidyltransferase gene operons (FFX45_05120, FFX45_05125, FFX45_07775, FFX45_07780, FFX45_11395, and FFX45_11400 for CL-1; AXY68115.1 and AXY68116.1 for SCTE542) that may confer kanamycin resistance (Lehmann et al., 2003). Most kanamycin nucleotidyltransferases (KNTases) are a homodimer with each subunit composed of two domains (one for substrate biding, the other for nucleotide binding). However, the bipartite aminoglycoside nucleotidyltransferase gene operons of CL-1 and SCTE542 contain two different genes (one encodes a substrate binding domain and the other nucleotide binding domains of KNTases). CL-1 can grow in medium containing kanamycin (5 μg/mL). In contrast, BP-1 does not have the KNTase gene and was susceptible to kanamycin.
Prokaryotic Immunity Systems
The CL-1 and SCTE542 genomes encode both type I (FFX45_00470-00490) and type III-B CRISPR-Cas systems (FFX45_02320-02350) that confer resistance to foreign genetic elements (Makarova et al., 2011). The type I CRISPR-Cas system is also found in NK55. In addition, the CL-1 genome has type III restriction modification system (FFX45_09855, FFX45_09860, FFX45_09870, and FFX45_08355) in defense against foreign DNA molecules. BP1 and T. vulcanus have type I and type III restriction modification systems; the SCTE542 genome has type II restriction modification systems; and NK55 and S. lividus genomes both have type I to type III restriction modification systems. Furthermore, Cl-1 and NK55 genomes contain a distinct vapBC operon (FFX45_09880 and FFX45_09885) of the type II toxin–antitoxin system (Figure 6D). VapC is a toxin that induces RNA cleavage and is inhibited by the co-expression of the antitoxin VapB.
Other Distinct Features in the CL-1 Genome
One distinct feature in the CL-1 genome is the presence of 14 copies of RPN family genes that encode recombination-promoting nuclease/putative transposases (Kingston et al., 2017). Most are located in two tandem gene arrays (nine and four copies, respectively) (FFX45_10335-10375 and FFX45_07895-07910) except for one gene (FFX45_07205). However, there are only eight, seven, three and two copies of Rpn family gene(s) in T. vulcanus, BP1, SCTE542, and NK55 genomes, respectively. In addition, RPN family genes are absent in genomes of S. lividus, JA-3-3-Ab and JA-2-3Ba from springs of Yellowstone National Park. Furthermore, CL-1 and SCTE542 genomes share several distinct genes for a PQQ-dependent sugar dehydrogenase (FFX45_00065), a linear amide C-N hydrolase (FFX45_04715), an ADP-ribosylglycohydrolase family protein (FFX45_09525) and a gene cluster for pseudaminic acid biosynthesis (FFX45_00685-00695, FFX45_00730, and FFX45_00735). The physiological significance of these distinct genes in the CL-1 genome may require further study.
Conclusion
This work reported the comparative genomic analysis of a novel thermophilic cyanobacterium, Thermosynechococcus sp. CL-1, together with four other Thermosynechococcus strains and the outgroup S. lividus in the C1 genotype. Although the sequences of the 16S rRNA gene among these Thermosynechococcus strains are highly similar, the genome structures of these Thermosynechococcus strains exhibit extensive rearrangements. The genome-scale phylogenetic analysis and genome-wide ANI results both suggest that CL-1 is most closely related to SCTE542, and both are sufficiently divergent from other Thermosynechococcus lineages to be considered new species within this genus. In addition, we identified distinct genetic differences between CL-1 and the other Thermosynechococcus strains. Our results suggest that Thermosynechococcus strains actively acquired many functional genes via horizontal transfer to cope with the various types of stresses in alkaline hot springs.
Statements
Data availability statement
The genome sequence reported in this work was deposited in GenBank (accession no. CP040671).
Author contributions
H-TH provided the biological materials. Y-IC, LC, and Y-FC performed the experiments. Y-IC, LC, Y-FC, C-HK, H-TH, and H-AC analyzed the data. Y-IC, LC, H-TH, C-HK, and H-AC wrote the manuscript. C-HK and H-AC acquired the funding and supervised the project.
Funding
This funding for this work was provided by the Academia Sinica to C-HK and H-AC, and the Ministry of Science and Technology in Taiwan (MOST 108-2311-B-001-024) to H-AC. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgments
We thank the High-Throughput Sequencing Core in the Biodiversity Research Center at Academia Sinica (Taipei, Taiwan) for the PacBio sequencing service and assistance in genome assembly. We also thank Dr. Wen-Dar Lin in the Bioinformatics Core Facility of the Institute of Plant and Microbial Biology, Academia Sinica, for assistance in comparative genome analysis and Ms. Shu-Jen Chou in the Genomic Technology Core Lab of the Institute of Plant and Microbial Biology for assistance in genomic DNA library preparations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00082/full#supplementary-material
FIGURE S1Maximum-likelihood phylogeny of a putatively acquired glycosyltransferase gene (Locus tag: FFX45_09045). Bootstrap values above 70% are labeled. The CL-1 homolog is distantly related to other Thermosynechococcus homologs, while more closely related to homologs from Proteobacteria (e.g., Halomonas and Altererythrobacter).
TABLE S1Homologous gene clusters among representative thermophilic cyanobacteria genomes. Each row in the table lists one protein-coding gene; the cluster id (as assigned by OrthoMCL), and other information (e.g., genomic location and annotation) are included. Genes sharing the same cluster id are considered as homologs. Genes without any identifiable homolog were assigned to their own clusters with unique ids (i.e., singletons).
TABLE S2Top five BLASTP hits in the NCBI non-redundant protein database of all annotated protein-coding genes in the CL-1 genome. The putatively acquired genes are highlighted (red: recent; blue: other).
TABLE S3Chemical analysis of the water sample in the Chin-Lun hot spring.
Footnotes
References
1
Agostoni M. Koestler B. J. Waters C. M. Williams B. L. Montgomery B. L. (2013). Occurrence of cyclic di-GMP-modulating output domains in cyanobacteria: an illuminating perspective.mBio4:e0451-13. 10.1128/mBio.0045113
2
Aziz R. K. Bartels D. Best A. A. DeJongh M. Disz T. Edwards R. A. et al (2008). The RAST server: rapid annotations using subsystems technology.BMC Genom.9:75. 10.1186/1471-2164-9-75
3
Beck C. Knoop H. Axmann I. M. Steuer R. (2012). The diversity of cyanobacterial metabolism: genome analysis of multiple phototrophic microorganisms.BMC Genom.13:56. 10.1186/1471-2164-13-56
4
Benson D. A. Cavanaugh M. Clark K. Karsch-Mizrachi I. Ostell J. Pruitt K. D. et al (2018). GenBank.Nucleic Acids Res.46D41–D47. 10.1093/nar/gkx1094
5
Bhaya D. Grossman A. R. Steunou A. S. Khuri N. Cohan F. M. Hamamura N. et al (2007). Population level functional diversity in a microbial community revealed by comparative genomic and metagenomics analyses.ISME J.1703–713. 10.1038/ismej.2007.46
6
Bibby T. S. Nield J. Barber J. (2001). Iron deficiency induces the formation of an antenna ring around trimeric photosystem I in cyanobacteria.Nature412743–745. 10.1038/35089098
7
Boekema E. J. Hifney A. Yakushevska A. E. Piotrowski M. Keegstra W. Berry S. et al (2001). A giant chlorophyll-protein complex induced by iron deficiency in cyanobacteria.Nature412745–748. 10.1038/35089104
8
Camacho C. Coulouris G. Avagyan V. Ma N. Papadopoulos J. Bealer K. et al (2009). BLAST+: architecture and applications.BMC Bioinform.10:421. 10.1186/1471-2105-10-421
9
Chen J. Yoshinaga M. Garbinski L. D. Rosen B. P. (2016). Synergistic interaction of glyceraldehydes-3-phosphate dehydrogenase and ArsJ, a novel organoarsenical efflux permease, confers arsenate resistance.Mol. Microbiol.100945–953. 10.1111/mmi.13371
10
Chin C.-S. Alexander D. H. Marks P. Klammer A. A. Drake J. Heiner C. et al (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data.Nat. Meth.10563–569. 10.1038/nmeth.2474
11
Chung W.-C. Chen L.-L. Lo W.-S. Lin C.-P. Kuo C.-H. (2013). Comparative analysis of the peanut witches’-broom phytoplasma genome reveals horizontal transfer of potential mobile units and effectors.PLoS One8:e62770. 10.1371/journal.pone.0062770
12
Dyer D. L. Gafford R. D. (1961). Some characteristics of a thermophilic blue-green alga.Science134616–617. 10.1126/science.134.3479.616
13
Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput.Nucleic Acids Res.321792–1797. 10.1093/nar/gkh340
14
Enomoto G. Win N.-N. Narikawa R. Ikeuchi M. (2015). Three cyanobacteriochromes work together to form a light color-sensitive input system for c-di-GMP signaling of cell aggregation.Proc. Natl. Acad. Sci. U.S.A.1128082–8087. 10.1073/pnas.1504228112
15
Everroad R. C. Otaki H. Matsuura K. Haruta S. (2012). Diversification of bacterial community composition along a temperature gradient at a thermal spring.Microb. Environ.27374–381. 10.1264/jsme2.ME11350
16
Geibel S. Waksman G. (2014). The molecular dissection of the chaperone-usher pathway.Biochim. Biophys. Acta18431559–1567. 10.1016/j.bbamcr.2013.09.023
17
Guindon S. Gascuel O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood.Syst. Biol.52696–704. 10.1080/10635150390235520
18
Hsueh H. T. Chu H. Chang C. C. (2007a). Identification and characteristics of a cyanobacterium isolated from a hot spring with dissolved inorganic carbon.Environ. Sci. Technol.411909–1914. 10.1021/es0620639
19
Hsueh H. T. Chu H. Yu S. T. (2007b). A batch study on the bio-fixation of carbon dioxide in the absorbed solution from a chemical wet scrubber by hot spring and marine algae.Chemosphere66878–886. 10.1016/j.chemosphere.2006.06.022
20
Hsueh H. T. Li W. J. Chen H. H. Chu H. (2009). Carbon bio-fixation by photosynthesis of Thermosynechococcus sp CL-1 and Nannochloropsis oculta.J. Photochem. Photobiol. B9533–39. 10.1016/j.jphotobiol.2008.11.010
21
Isojärvi J. Shunmugam S. Sivonen K. Allahverdiyeva Y. Aro E.-M. Battchikova N. (2015). Draft genome sequence of Calothrix strain 336/3, a novel H2-producing cyanobacterium isolated from a Finnish lake.Genom. Announc.3:e001474-14. 10.1128/genomeA.01474-14
22
Iwai M. Katoh H. Katayama M. Ikeuchi M. (2004). Improved genetic transformation of the Thermophilic Cyanobacterium, Thermosynechococcus elongatus BP-1.Plant Cell Physiol.45171–175. 10.1093/pcp/pch015
23
Jain C. Rodriguez-R L. M. Phillippy A. M. Konstantinidis K. T. Aluru S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries.Nat. Commun.9:5114. 10.1038/s41467-018-07641-9
24
Kanehisa M. Goto S. Furumichi M. Tanabe M. Hirakawa M. (2010). KEGG for representation and analysis of molecular networks involving diseases and drugs.Nucleic Acids Res.38D355–D360. 10.1093/nar/gkp896
25
Kaneko T. Nakamura Y. Wolk C. P. Kuritz T. Sasamoto S. Watanabe A. et al (2001). Complete genomic sequence of the filamentous nitrogen-fixing cyanobacterium Anabaena sp. strain PCC 7120.DNA Res.8205–213. 10.1093/dnares/8.5.205
26
Kingston A. W. Ponkratz C. Raleigh E. A. (2017). Rpn (YhgA-Like) proteins of Escherichia coli K-12 and their contribution to RecA independent horizontal transfer.J. Bacteriol.199:e0787-16. 10.1128/JB.00787-16
27
Krzywinski M. Schein J. Birol I. Connors J. Gascoyne R. Horsman D. et al (2009). Circos: an information aesthetic for comparative genomics.Genome Res.191639–1645. 10.1101/gr.092759.109
28
Kurtz S. Phillippy A. Delcher A. L. Smoot M. Shumway M. Antonescu C. et al (2004). Versatile and open software for comparing large genomes.Genome Biol.5:R12. 10.1186/gb-2004-5-2-r12
29
Lehmann C. Lim K. Chalamasetty V. R. Krajewski W. Melamud E. Galkin A. et al (2003). The HI0073/HI0074 protein pair from Haemophilus influenzae is a member of a new nucleotidyltransferase family: structure, sequence analyses, and solution studies.Proteins50249–260. 10.1002/prot.10260
30
Leng X. Zhu W. Jin J. Mao X. (2011). Evidence that a chaperone–usher-like pathway of Myxococcus xanthus functions in spore coat formation.Microbiology1571886–1896. 10.1099/mic.0.047134-0
31
Li L. Stoeckert C. J. Roos D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes.Genom. Res.132178–2189. 10.1101/gr.1224503
32
Liang Y. Tang L. J. Luo Y. Kaczmarek M. B. Li X. Daroch M. (2019). Thermosynechococcus as a thermophilic photosynthetic microbial cell factory for CO2 utilization.Bioresour. Technol.278255–265. 10.1016/j.biortech.2019.01.089
33
Liao C.-C. Liu S.-L. Wang W.-L. (2006). Effects of temperature and pH on growth and photosynthesis of the thermophilic cyanobacterium Synechococcus lividus as measured by pulse-amplitude modulated fluorometry.Phycol. Res.54260–268. 10.1111/j.1440-1835.2006.00432.x
34
Lo W.-S. Chen L.-L. Chung W.-C. Gasparich G. E. Kuo C.-H. (2013). Comparative genome analysis of Spiroplasma melliferum IPMB4A, a honeybee-associated bacterium.BMC Genomics14:22. 10.1186/1471-2164-14-22
35
Makarova K. S. Haft D. H. Barrangou R. Brouns S. J. J. Charpentier E. Horvath P. et al (2011). Evolution and classification of the CRISPR–Cas systems.Nat. Rev. Microbiol.9467–477. 10.1038/nrmicro2577
36
Nakamura Y. Kaneko T. Sato S. Ikeuchi M. Katoh H. Sasamoto S. et al (2002). Complete genome structure of the Thermophilic cyanobacterium Thermosynechococcus elongatus BP-1.DNA Res.9123–130. 10.1093/dnares/9.4.123
37
Olsen M. T. Nowack S. Wood J. M. Becraft E. D. LaButti K. Lipzen A. et al (2015). The molecular dimension of microbial species: 3. Comparative genomics of Synechococcus strains with different light responses and in situ diel transcription patterns of associated putative ecotypes in the mushroom spring microbial mat.Front. Microbiol.6:604. 10.3389/fmicb.2015.00604
38
Papke R. T. Ramsing N. B. Bateson M. M. Ward D. M. (2003). Geographical isolation in hot spring cyanobacteria.Environ. Microbiol.5650–659. 10.1046/j.1462-2920.2003.00460.x
39
Patel A. Matsakas L. Rova U. Christakopoulos P. (2019). A perspective on biotechnological applications of Thermophilic microalgae and cyanobacteria.Bioresour. Technol.278424–434. 10.1016/j.biortech.2019.01.063
40
R Core Team (2019). R: A language and Environment for Statistical Computing.Vienna: R Foundation for Statistical Computing.
41
Stolyar S. Liu Z. Thiel V. Tomsho L. P. Pinel N. Nelson W. C. et al (2014). Genome sequence of the Thermophilic cyanobacterium Thermosynechococcus sp. strain NK55.Genome Announc.2:e001060-13. 10.1128/genomeA.01060-13
42
Su C. M. Hsueh H. T. Chen H. H. Chu H. (2012). Effects of dissolved inorganic carbon and nutrient levels on carbon fixation and properties of Thermosynechococcus sp. in a continuous system.Chemosphere88706–711. 10.1016/j.chemosphere.2012.04.011
43
Su C. M. Hsueh H. T. Li T. Y. Huang L. C. Chu Y. L. Tseng C. M. et al (2013). Effects of light availability on the biomass production, CO2 fixation, and bioethanol production potential of Thermosynechococcus CL-1.Bioresour. Technol.145162–165. 10.1016/j.biortech.2013.02.092
44
Su C. M. Hsueh H. T. Tseng C. M. Ray D. T. Shen Y. H. Chu H. (2017). Effects of nutrient availability on the biomass production and CO2 fixation in a flat plate photobioreactor.Aerosol. Air Qual. Res.171887–1897. 10.4209/aaqr.2016.09.0386
45
Tang J. Jiang D. Luo Y. Liang Y. Li L. Shah M. R. et al (2018). Potential new genera of cyanobacterial strains isolated from thermal springs of western Sichuan, China.Algal Res.3114–20. 10.1016/j.algal.2018.01.008
46
Tatusova T. DiCuccio M. Badretdin A. Chetvernin V. Nawrocki E. P. Zaslavsky L. et al (2016). NCBI prokaryotic genome annotation pipeline.Nucleic Acids Res.446614–6624. 10.1093/nar/gkw569
47
Tsai Y.-M. Chang A. Kuo C.-H. (2018). Horizontal gene acquisitions contributed to genome expansion in insect-symbiotic Spiroplasma clarkii.Genome Biol. Evol.101526–1532. 10.1093/gbe/evy113
48
Ward D. M. Castenholz R. W. Miller S. R. (2012). “Cyanobacteria in geothermal habitats,” in Ecology of Cyanobacteria, 2nd Edn, ed.WhittonB. A., (Heidleberg: Springer), 39–63. 10.1007/978-94-007-3855-3
49
Wickham H. (2009). ggplot2.New York, NY: Springer, 10.1007/978-0-387-98141-3
50
Wilkins D. Kurtz Z. (2019). Gggenes: Draw Gene Arrow Maps in “ggplot2”. Available at: https://CRAN.R-project.org/package=gggenes(accessed August 1, 2019).
Summary
Keywords
cyanobacterium, Thermosynechococcus , genome, comparative genomics, thermophilic cyanobacterium, adaption
Citation
Cheng Y-I, Chou L, Chiu Y-F, Hsueh H-T, Kuo C-H and Chu H-A (2020) Comparative Genomic Analysis of a Novel Strain of Taiwan Hot-Spring Cyanobacterium Thermosynechococcus sp. CL-1. Front. Microbiol. 11:82. doi: 10.3389/fmicb.2020.00082
Received
17 September 2019
Accepted
15 January 2020
Published
31 January 2020
Volume
11 - 2020
Edited by
Ludmila Chistoserdova, University of Washington, United States
Reviewed by
Sergey N. Gavrilov, Federal Research Centre Biotechnology (RAS), Russia; Eric Daniel Becraft, University of North Alabama, United States
Updates
Copyright
© 2020 Cheng, Chou, Chiu, Hsueh, Kuo and Chu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chih-Horng Kuo, chk@gate.sinica.edu.twHsiu-An Chu, chuha@gate.sinica.edu.tw
†These authors have contributed equally to this work
This article was submitted to Evolutionary and Genomic Microbiology, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.