 Dev Paudel1
Dev Paudel1 Fengxia Liu1
Fengxia Liu1 Liping Wang1Matthew Crook2Stephanie Maya1
Liping Wang1Matthew Crook2Stephanie Maya1 Ze Peng1
Ze Peng1 Karen Kelley3Jean-Michel Ané4
Karen Kelley3Jean-Michel Ané4 Jianping Wang1,5*
Jianping Wang1,5*- 1Agronomy Department, University of Florida, Gainesville, FL, United States
- 2Department of Microbiology, Weber State University, Ogden, UT, United States
- 3Interdisciplinary Center for Biotechnology Research, University of Florida, Gainesville, FL, United States
- 4Departments of Bacteriology and Agronomy, University of Wisconsin–Madison, Madison, WI, United States
- 5Plant Molecular and Cellular Biology Program, Genetic Institute, University of Florida, Gainesville, FL, United States
In many legumes, the colonization of roots by rhizobia is via “root hair entry” and its molecular mechanisms have been extensively studied. However, the nodulation of peanuts (Arachis hypogaea L.) by Bradyrhizobium strains requires an intercellular colonization process called “crack entry,” which is understudied. To understand the intercellular crack entry process, it is critical to develop the tools and resources related to the rhizobium in addition to focus on investigating the mechanisms of the plant host. In this study, we isolated a Bradyrhizobium sp. strain, Lb8 from peanut root nodules and sequenced it using PacBio long reads. The complete genome sequence was a circular chromosome of 8,718,147 base-pair (bp) with an average GC content of 63.14%. No plasmid sequence was detected in the sequenced DNA sample. A total of 8,433 potential protein-encoding genes, one rRNA cluster, and 51 tRNA genes were annotated. Fifty-eight percent of the predicted genes showed similarity to genes of known functions and were classified into 27 subsystems representing various biological processes. The genome shared 92% of the gene families with B. diazoefficens USDA 110T. A presumptive symbiosis island of 778 Kb was detected, which included two clusters of nif and nod genes. A total of 711 putative protein-encoding genes were in this region, among which 455 genes have potential functions related to symbiotic nitrogen fixation and DNA transmission. Of 21 genes annotated as transposase, 16 were located in the symbiosis island. Lb8 possessed both Type III and Type IV protein secretion systems, and our work elucidated the association of flagellar Type III secretion systems in bradyrhizobia. These observations suggested that complex rearrangement, such as horizontal transfer and insertion of different DNA elements, might be responsible for the plasticity of the Bradyrhizobium genome.
Introduction
Rhizobia are soil-borne bacteria that colonize plant tissues inter- or intra-cellularly and trigger the development of new organs called nodules, which generally happens on legume roots. Inside of these nodules, the rhizobia differentiate into bacteroids and convert N2 gas into ammonium (Oldroyd and Downie, 2008; Kuever et al., 2015; Ormeño-Orrillo et al., 2015; Peix et al., 2015; Mus et al., 2016). This legume–rhizobium symbiosis is fundamental to sustainable agriculture as this relationship can alleviate the need to provide synthetic nitrogen fertilizers in agricultural systems (Howieson and Dilworth, 2016). In most legume–rhizobium associations, flavonoid molecules in the rhizosphere, released by plant roots, serve as signals to activate bacterial transcriptional regulator protein, NodD. The nodD gene controls the expression of nod common and specificity genes (Dénarié et al., 1996). The products of nod genes are involved in the synthesis and secretion of lipo-chitooligosaccharides (LCOs), called Nod factors, which are the primary determinant of specificity between rhizobia and their legume hosts (Downie and Walker, 1999; Oldroyd, 2013). Nod factor recognition at the root surface leads to the initiation of a nodule primordium in the root cortex. However, in some associations, Nod factors are not required, and the rhizobia use other strategies, such as the use of effectors (Masson-Boivin and Sachs, 2018). Nevertheless, in all associations, the rhizobia go through the epidermis and the cortex to gain access to developing nodule primordia, where the bacteria are released into cortical cells (Oldroyd et al., 2011). Rhizobial infection in the majority of legume species, such as soybean (Glycine max), Medicago truncatula, and Lotus japonicus, follows a “root hair entry” pathway, which involves penetrating via intracellular infection threads (Bonaldi et al., 2011; Oldroyd et al., 2011). However, intracellular infection threads are not found in the roots of legumes such as peanut (Arachis hypogaea L.) and Aeschynomene, where infection occurs intercellularly via “crack entry.” In peanut, the rhizobia enter the root, where root hairs emerge and occupy the space between the root hair wall and adjoining epidermal and cortical cells. They colonize the intercellular spaces and invade sub-epidermal cortical cells via an endocytosis-like process (Okubo et al., 2012a). The invaded cells divide and are incorporated in the nodule tissue. Adjacent cells separate at the middle lamellae, and this space is filled with bacteria, thus distributing the bacteria intercellularly (Chandler, 1978). This intercellular infection is considered an evolutionarily ancient invasion mechanism (Groth et al., 2010; Madsen et al., 2010), which might be a key to facilitate transforming non-legumes for biological nitrogen fixation via symbiosis (Charpentier and Oldroyd, 2010).
Rhizobia are known to occur in eight bacterial families: Rhizobiaceae, Brucellaceae, Phyllobacteriaceae, Xanthobacteraceae, Bradyrhizobiaceae, Hyphomicrobiaceae, Methylobacteriaceae, and Burkholderiaceae (de Lajudie et al., 2019). Among these families, Bradyrhizobiaceae contains the genus Bradyrhizobium, a slow-growing (doubling time >8 h), non-acid-producing root nodule bacterium of leguminous plants (Jordan, 1982). Bradyrhizobia are described as gram-negative, aerobic, non-spore forming, short rod-shaped (0.5–0.9 μm by 1.2–3.0 μm), and mobile by one polar or subpolar flagellum (Jordan, 1982). Currently, a total of 41 species have been assigned to the genus Bradyrhizobium (Parte, 2014). Bradyrhizobium diazoefficiens, recently reclassified from B. japonicum (Delamuta et al., 2013) forms symbiotic, nitrogen-fixing relationships with soybean plants and has been used in agriculture for decades (Stacey, 1995; Kaneko et al., 2002). Considering its essential role in nitrogen fixation, the complete genome of B. diazoefficiens USDA 110T, has been sequenced (Kaneko et al., 2002). Furthermore, according to the National Center for Biotechnology Information Database, 37 Bradyrhizobium strains isolated from different species, including soybean, Aeschynomene, and wheat have been sequenced. In general, clustering of nodulation and nitrogen fixation genes in plasmids or symbiotic islands is common in rhizobial genomes that colonize roots by “root hair entry” (Giraud et al., 2007). Some rhizobia of the genus Bradyrhizobium efficiently nodulate peanuts, the second most important legume crop in the world after soybeans (Urtz and Elkan, 1996). Even though the peanut–rhizobium symbiosis is essential and supplies 55% of nitrogen needs for optimal peanut growth (Hardarson, 1993), there is a shortage of genomic information on peanut rhizobia.
Unraveling the genomes of peanut rhizobia is particularly critical because the infection mechanism that takes place during peanut nodulation is proposed to be an ancestral state (Sprent, 2007). This extracellular mode of colonization could be a more plausible target to engineer entry of rhizobia into non-legumes than the more advanced root-hair entry (Charpentier and Oldroyd, 2010). While molecular mechanisms in intracellular root-hair entry species like M. truncatula and L. japonicus have been under extensive investigation, many questions regarding the symbiotic pathway for evolutionarily ancient extracellular pathway remain unaddressed. To further study the extracellular infection path, it is critical to not just investigate the host mechanisms in controlling infection, but also to develop the tools and resources related to rhizobium. Therefore, the objective of this study was to increase the knowledge on native bradyrhizobia that nodulate peanut by studying the genome of strain Lb8 and its phylogenetic characterization. We utilized long reads from single-molecule real-time (SMRT) sequencing (Pacific Biosciences) to assemble the complete genome of Bradyrhizobium sp. Lb8. Here, we provide an analysis of the first complete genome sequence of an isolated strain of Bradyrhizobium that efficiently fixes nitrogen in peanuts.
Results
Isolation of the Rhizobia Strains From Peanut Nodules
Out of 370 colony PCRs, 359 colonies showed PCR products, without a difference in amplicon size. Five PCR products that were randomly selected for sequencing revealed that the five isolates belonged to the genus Bradyrhizobium, with two unique 16S rRNA sequences. Strains L1, L3, and L4 had identical 16S rRNA sequences, while strains Lb8 and Ls1 had identical 16S rRNA sequences.
Characterization of the Isolated Strains
These five Bradyrhizobium strains showed three different growth curves (Supplementary Figure 1). Lb8 grew the fastest in the first 8 days followed by L1, L3, and L4. The growth rate of L1, L3, and L4 continued after day 8 and surpassed Lb8 on day 9. Nodules were seen on the roots of all peanut plants inoculated with the five strains separately, at 13 days after inoculation (DAI) (Supplementary Figure 2A). Nodules were harvested at 22 DAI and immediately cut in half. The inside of the nodules was dark pink (Supplementary Figure 2B), indicating that all the five rhizobial strains isolated from peanut were able to infect peanuts to form functional nodules.
The number of nodules was significantly higher (p ≤ 0.05, LSD) on seedlings infected by strain Lb8, as compared to the remaining isolates (Figure 1A). No significant difference in the diameters of nodules induced by the other four isolates was observed (p > 0.05, LSD). The efficiency of nitrogen fixation was significantly higher (p ≤ 0.05, LSD) for nodules induced by strain Lb8 than those induced by L1, L3, L4, and Ls1 (Figure 1B). The average length of gram-negative rod-shaped form cells (Supplementary Figure 3A) was 1.43 ± 0.44 μm long (Supplementary Figure 3B), 1.45 ± 0.6 and 1.21 ± 0.26 μm, for Lb8, Ls1, and L1, respectively.
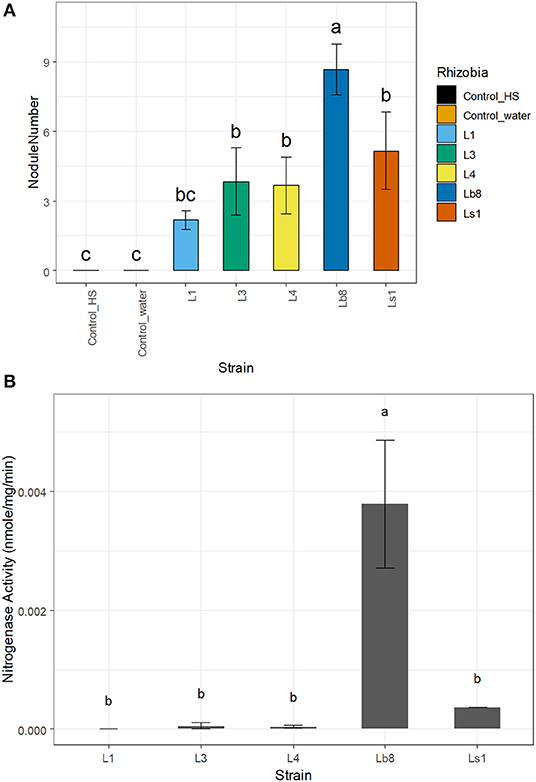
Figure 1. Bar plots showing the number of nodules per plant (A) and nitrogenase activity (B) of the different strains. Same letters on top of each bar plot are not significantly different (LSD, p > 0.05).
Owing to the relatively fast-growing nature, and its ability to induce higher number of nodules with nitrogenase activity, the Lb8 isolate was selected for whole genome sequencing. Based on a 16S rRNA gene sequence analysis, Lb8 was most related to Bradyrhizobium kavangenese isolated from the nodules of cowpea (Grönemeyer et al., 2015), with a sequence identity of 99%. It was clustered within a group of non-photosynthetic nod gene-containing bradyrhizobia, which included B. diazoefficiens, and was separated from those of photosynthetic nod gene-lacking strains, such as Bradyrhizobium sp. ORS278 (host: A. sensitive in Africa) and Bradyrhizobium sp. BTAi1 (host: A. indica in North America) (Giraud et al., 2007; Figure 2). No nodule was observed on soybean cultivars inoculated by Lb8 by 20 DAI indicating this strain may be specific to peanut.
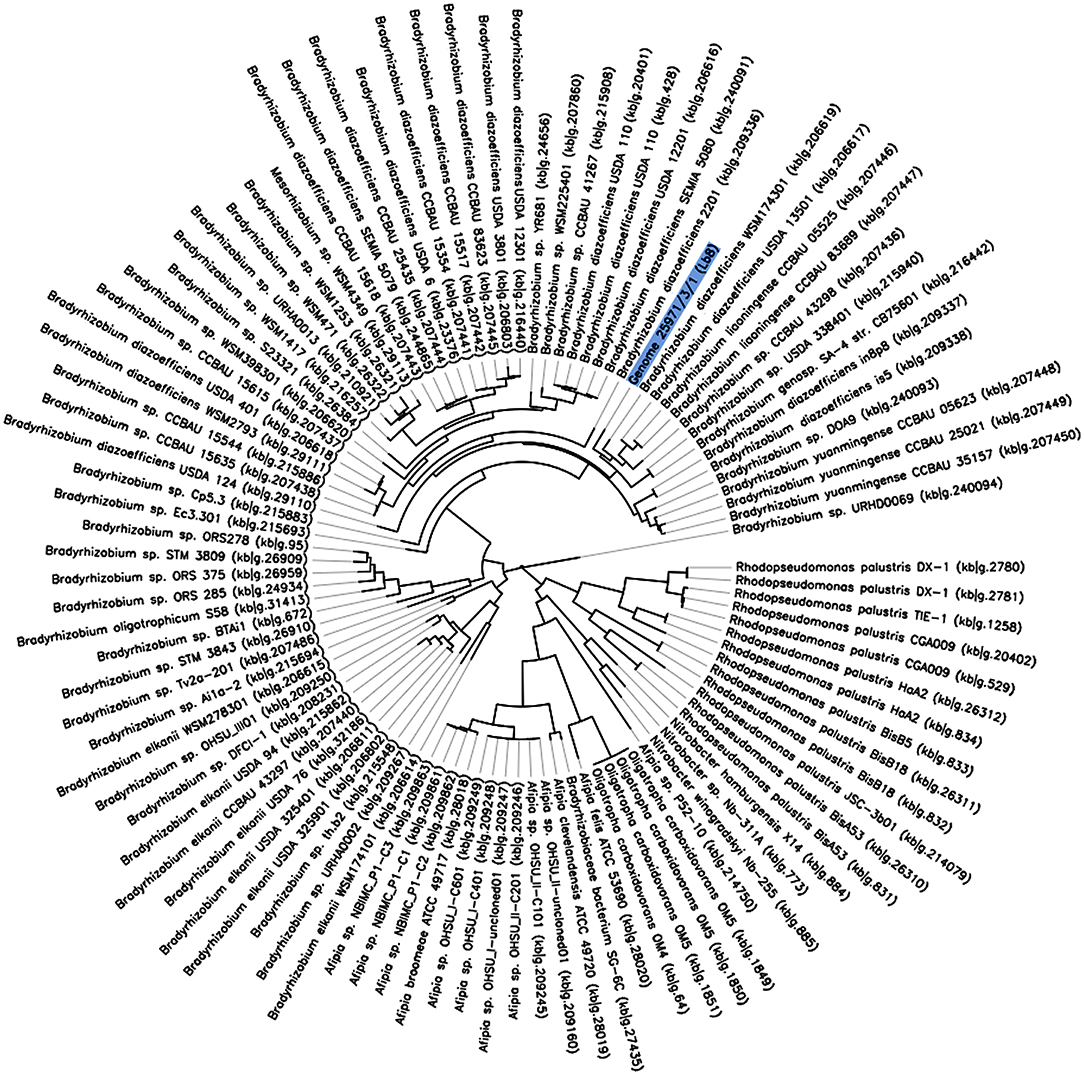
Figure 2. The phylogeny of Bradyrhizobium sp. Lb8 in comparison to a set of closely related genomes selected from all public KBase genomes. The phylogenetic tree was constructed using FastTree. The genome of Bradyrhizobium sp. Lb8 is highlighted in blue.
Genome Sequence of Lb8 Strain
A total of 377,698 raw sequences, with approximately 2.36 Gb size, were obtained by PacBio sequencing. The mean read length was 6,253 bp, and the most extended read was 39,742 bp. The final genome assembly consisted of one circular chromosome of 8,718,147 bp with 63.14% GC content. The GC content was unevenly distributed in several locations. A shift of GC skew in two regions of the genome at coordinates 17 Kb and 4.7 Mb (Figure 3, the innermost circle) were assigned as a putative oriC location and terminus location (Supplementary Figure 4), respectively. No plasmid sequence was detected in the PacBio sequences obtained. The Lb8 genome was mostly collinear with B. diazoefficiens USDA 110T except at the site of the symbiosis island (SI) (Supplementary Figure 5).
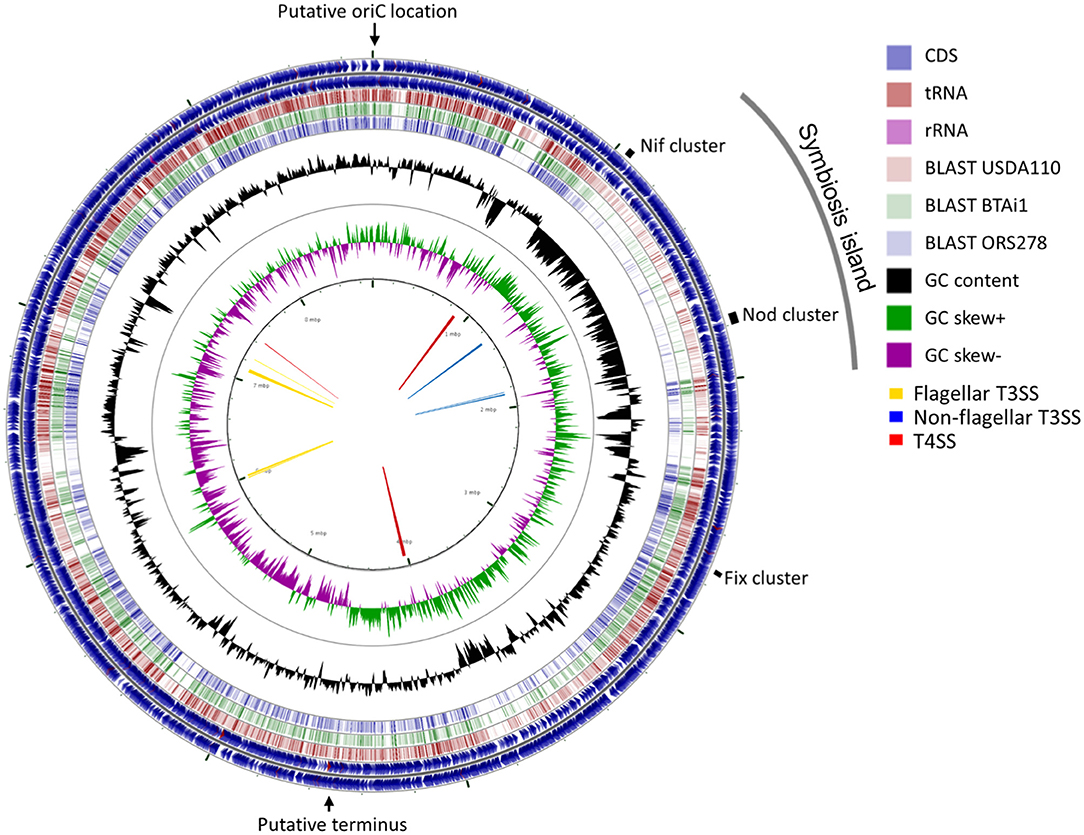
Figure 3. A circular representation of the chromosome of Bradyrhizobium sp. Lb8. The outermost and the second circle show the positions of the putative protein-encoding genes in clockwise and counter-clockwise directions, respectively. The third, fourth, and fifth circles from the outside represent BLASTn comparisons with Bradyrhizobium strains USDA 110T, BTAi1, and ORS278, respectively (E < 10−10). The innermost circle shows GC skew (higher than average GC content in green, lower than average in purple) and the second innermost circle shows the GC content of the entire sequence. Markings inside the innermost circle represent genome positions in Mb. Arrows in the center represent clusters of protein secretion systems. The positions of the putative replication origin, putative replication terminus, symbiosis island, and nitrogen fixation genes are shown outside of the outermost circle.
Genome Annotation
A total of 8,433 protein-encoding genes (PEG)s were predicted, and 4,915 genes with clear known functions were classified into 27 categories, each with different biological roles, according to classic RAST (Table 1; Aziz et al., 2008). The average gene density was one gene per 1,027 bp. A total of 54 RNAs (non-PEG) were detected in the genome, including an rRNA gene cluster containing the 5S, 23S, and 16S rRNA genes (at coordinates 7,959,101–7,964,425 of the genome), and 51 tRNA genes scattered throughout the genome, for transferring the 20 standard amino acids.

Table 1. Number of identified protein encoding genes of known functions in Bradyrhizobium sp. Lb8 that are present in different subsystems according to the RAST server subsystem classification (Aziz et al., 2008).
A total of 84 PEGs were categorized into nitrogen metabolism (Table 1). Out of these, 11 were involved in cyanate hydrolysis, 20 were involved in nitrogen fixation, one in nitrosative stress, 18 in nitrate and nitrite ammonification, 16 in ammonium assimilation, and one was an amidase with predicted urea and nitrile hydratase functions. Seventeen PEGs were involved in denitrification. A region of 1,015 Kb (1,170,906–2,186,497) had consistently lower GC content (59.22%) compared to the average GC content (63.14%) of the whole genome. This region hosted two clusters of nitrogen fixation related genes and was termed a “symbiosis island” (SI). A met-tRNA gene was identified at one end of the region (2,185,706–2,185,781). Of the 711 putative PEGs assigned to the SI, 455 (64%) showed sequence similarity to genes of known functions and 18 PEGs were related to nitrogen metabolism.
The Lb8 genome possessed both Type III secretion systems (T3SS) and Type IV secretion systems (T4SS). Three clusters of non-flagellar T3SS (with 12, 5, and 4 genes, respectively), four clusters of flagellar T3SS (with 7, 7, 27, and 5 genes, respectively), and three clusters of P-type T4SS (with 16, 13, and 4 genes, respectively), were identified in the genome (Figure 3). T6SS secretion system was not identified in the Lb8 genome. T3SS secreted inner membrane proteins homologous to flagellar export components such as yscQRSTUJL, spaRS, hrcRTUJV, excRT, epaR1, excRU, ssaUV, escRJV, and pscJ which were located on the SI (peg.1219, peg.1224–peg.1226, peg.1230–peg.1232, peg.1234–peg.1235, and peg.1744–peg.1748). Similarly, homologs of T3SS, host injection protein (yopB), secretion bridge (yscJ), secretion cytoplasmic protein (yscL), and inner membrane channel protein (lcrD, hrcV, escV, and ssaV) were also included in the SI (peg.1757, peg.1759, peg.1762, and peg.1766). Homologs of T4SS (virD4B11, tadAZC, cpaFEC, and rcpA) were located outside the SI (peg.844, peg.1001, peg.1003–peg.1004, peg.1006, peg.3484–peg.3485, peg.3492, peg.3752, peg.31, and peg.1004), except one homolog, rcpA/cpaE, which was identified in the SI (peg.1236). The two paralogous virD4 proteins (peg.844 and peg.3752) were 67% identical to each other and homologous to conjugal transfer protein traG in B. diazoefficiens. All the 17 Bradyrhizobium sp. with complete genome sequence available had clusters of flagellar T3SS while non-flagellar T3SS was present in 11 genomes, T4SS in 13 genomes, and T6SS was present in 8 genomes (Table 2).
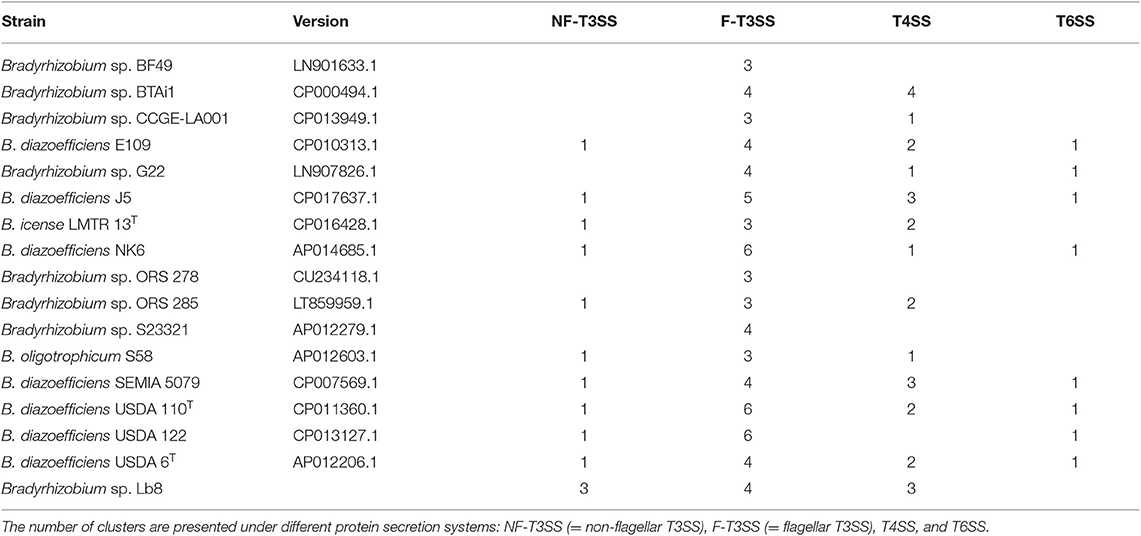
Table 2. Number of clusters of different protein secretion systems in Bradyrhizobium strains predicted by T346 Hunter (Martínez-García et al., 2015).
Lb8 possesses nodulation genes including nodA, nodC, nodD, nodI, nodJ, nodN, nodS, nodT, nodU, nodZ, nolA, nolY, and nolO. Most of these genes were located in the SI except nodN and nodZ. Lb8 also possessed fixA, fixB, fixC, fixJ, fixK, fixL fixR, and fixX genes. A cluster of fixJ and fixL was located outside of the SI (coordinates of 2,967,208–2,969,821), while a paralogous fixJ (two-component nitrogen fixation transcriptional regulator) was identified inside the SI (coordinates 2,083,915–2,084,499). These genes shared 52% sequence identity with 99% query sequence coverage. A fixC gene (probable electron transfer flavoprotein-quinone oxidoreductase) was also identified in the genomic island (coordinates 1,264,204–1,265,511). A cluster of hydrogenase uptake (hup) genes hupF-hypC-hoxLO-hyaE-hoxQV-hupK-hypABFCDE was located on the SI (coordinates 1,214,329–1,227,503). These hup genes reduce energy loss associated with symbiotic nitrogen fixation and contribute to increased efficiency of nitrogen-fixation (Albrecht et al., 1978; Evans et al., 1987). Genes related to flagella formation (fliJRQEPMLXIDKFGN, flhAFB, flgCBFGAHJLKED, flbT, and flaF), including flagellar motor rotation genes (motAB) were found in the Lb8 genome. Lb8 had periplasmic nitrate reductase component genes, napE and napD (coordinates 7,255,262–7,255,773) and norEQD (nitric oxide reductase activation protein) formed a cluster (coordinates 3,452016–3,457,609). A total of 21 genes were annotated as putative transposases and eight of them were insertion sequences (ISs), though none of them were classified in the subsystem related to transposable element by RAST due to the limited curation of the RAST system (Table 1, Aziz et al., 2008). Homologs of trbBCDEJLFGI, which are essential for conjugative transfer of Ti-plasmid in A. tumefaciens (Li and Everhart, 1998), were found in two clusters (4,041,083–4,050,188 and 885,151–894,557). The first cluster included one T4SS cluster. A cluster of trbIGFL was also identified at another site of the genome (coordinates 7,430,109–7,434,552) showing gene duplications.
Phylogenetic Analysis
A phylogenetic tree based on 49 highly conserved Clusters of Orthologous Groups (COG) placed Bradyrhizobium strain Lb8 with non-photosynthetic nod gene-containing bradyrhizobia (Figure 2) closely related to B. diazoefficiens WSM174301, which was isolated from soybean root nodules. B. diazoefficiens USDA 110T from soybean root nodules was clustered in a separate node. A further phylogenetic analysis, based on concatenated nod genes, suggested that strain Lb8 was closely related to strains USDA 110T, SEMIA 5079, and USDA 6T (Supplementary Figure 6).
Comparative Genomics
The analysis of average nucleotide identity (ANI) between Bradyrhizobium sp. Lb8 and 17 other Bradyrhizobium strains with complete genome sequences showed that Bradyrhizobium sp. Lb8 represented a novel species (ANI < 95%, Supplementary Table 2). The closest fully assembled genome was Bradyrhizobium sp. CCGE-LA001 (ANI = 88.85%).
A reciprocal BLAST comparison was conducted between Lb8, and three other nodulating Bradyrhizobium strains with complete genome sequence availability, namely, USDA 110T, ORS278, and BTAi1. Overall, 5,712 (68.6%), 4,534 (54.4%), and 4,409 (52.9%) genes showed the same best reciprocal hit between Lb8 and USDA 110T, BTAi1, and ORS278, respectively. To further evaluate the relationship between Lb8 and the three other strains, OrthoMCL was used to cluster the orthologous groups (gene families) among the four strains. In total, 7,571 orthologous groups were clustered from 25,050 protein sequences. These four strains shared 3,784 gene families. Among the grouped gene families, Lb8 shared 5,539 (92.41%) gene families with USDA 110T, followed by 84,365 (72.82 %) gene families with BTAi1, and 4,212 (70.27%) gene families with ORS278. There were 93 (1.55%) gene families corresponding to 298 protein sequences unique to Lb8 (Figure 4).
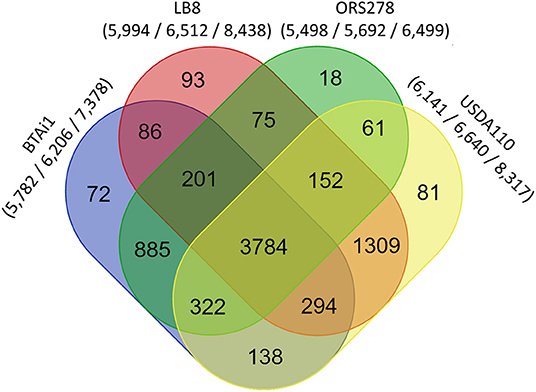
Figure 4. Comparative genomics analysis of Bradyrhizobium strains Lb8, USDA 110T, BTAi1, and ORS278. An oval represents each genome, and the number of shared and unique genes are shown by overlapping and non-overlapping parts of the ovals (OrthoMCL 2.0.7, E < 10−5). The numbers under species names indicate the number of gene families, the gene number in the families, and the total number of genes (protein sequences) input into OrthoMCL.
Out of the 152 genes related to nitrogen metabolism, 75 genes were present in all four strains (Supplementary Table 1) with 42 genes (56%) highly conserved across these four genomes (Supplementary Table 1, Supplementary Figure 7). Out of these 42 genes, 15 were related to nitrogen fixation, five were related to cyanate hydrolysis, one was amidase, five were related to nitrate and nitrite ammonification, five were related to ammonium assimilation, two were in the denitrifying reductase gene cluster, and the remaining nine genes were related to denitrification. In the Lb8 strain, genes related to nitrogen metabolism such as cynR (cyanate hydrolysis), nifT, and ginE (ammonia assimilation) were unique in comparison to other three Bradyrhizobium strains (ORS278, BTAi1, and USDA 110T).
Homologs of nodQ, nodM, nodI, nodD1, nodD2, nodN, nodV, and nodW were present in all the four genomes compared. nodY gene did not have homologs in Lb8, ORS278, and BTAi1 but was present in USDA 110T. nodS and nodU were present in both Lb8 and USDA 110T but were absent in ORS278 and BTAi1. nod genes present in Lb8 were collinear with those in USDA 110T (Figure 5A). Three copies of the nodU genes were present in the Lb8 genome. Interestingly, gene homologs of nodN and nodT were found in all four genomes analyzed. nodT was inverted in ORS278. Similarly, nif and fix genes were collinear between Lb8 and USDA 110T (Figure 5B). nif and fix genes were also collinear between BTAi1 and ORS278 genomes. However, nif and fix genes were inverted in the BTAi1 genome, and rearrangement of genes was seen on the BTAi1 genome as compared to the USDA 110T or Lb8 genome.
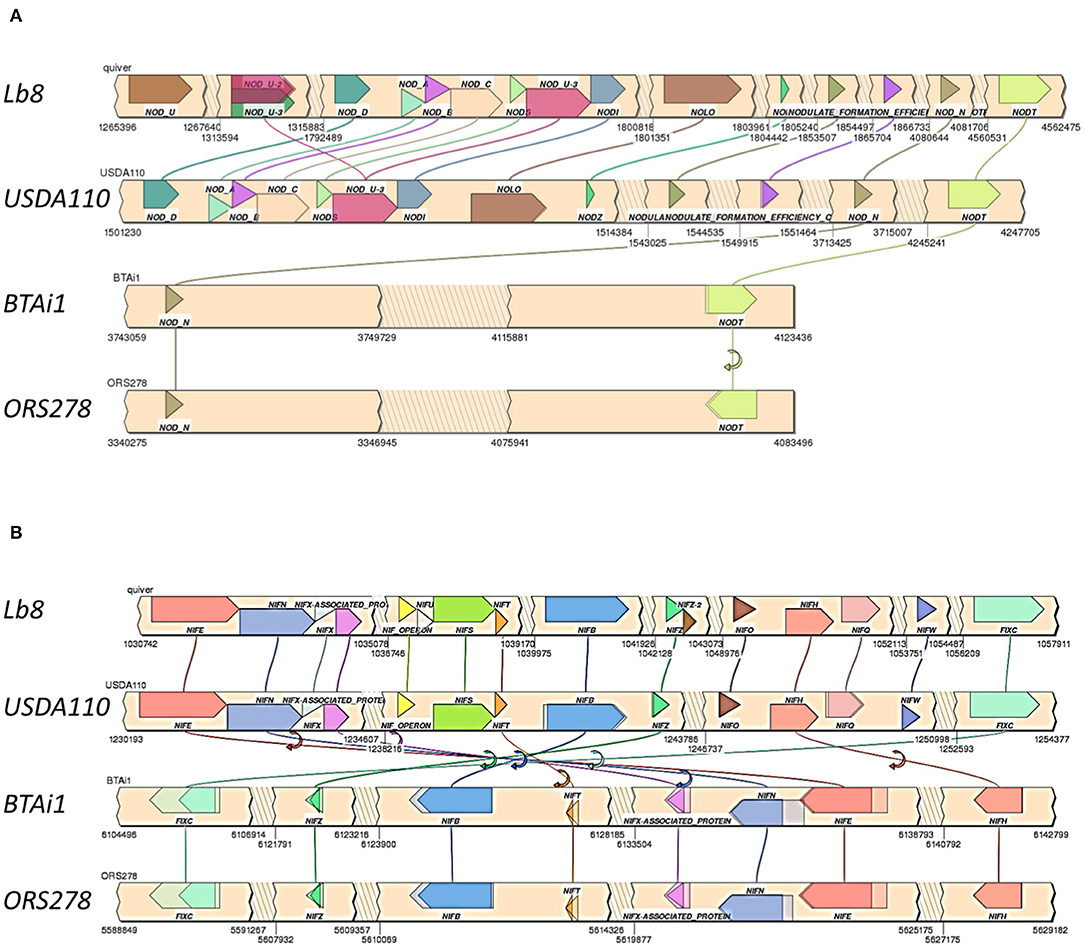
Figure 5. Synteny of clustered nod (A) and nif /fix (B) genes in Lb8, USDA 110T, BTAi1, and ORS278 genomes. Genes are color coded with labels listed above or below the genes. Gene direction is indicated as pointed ends. Flip arrows indicate a change in direction between genomes. Jagged edges indicate skipped sections.
Discussion
Nitrogen is an essential and often growth-limiting nutrient for plants (Gresshoff and Ferguson, 2017). The association of nitrogen-fixing rhizobia with legumes is a sustainable and environmentally friendly alternative to synthetic nitrogen fertilizers (Gresshoff and Ferguson, 2017). Many Bradyrhizobium sp. can nodulate peanuts, where a high degree of correlation exists between nitrogenase activity and shoot weight, nodule number, nodule mass, and total nitrogen (Wynne et al., 1983; Nigam et al., 1985; Li et al., 2015, 2019). Approximately 90% of the world peanut crop is produced in developing countries where the pod yield is around four times less than that in developed countries (Boogerd and Van Rossum, 1997). Therefore, the selection of appropriate rhizobial strains could lead to an improvement in nodule number, shoot weight, and plant color (Wynne et al., 1983). This will eventually increase the amount of nitrogen available for the plants, subsequently improving pod yield. Moreover, nodulation in peanuts occurs via a “crack entry” mechanism (Howieson and Dilworth, 2016), which is believed to be evolutionarily more ancient than infection via root hairs (Sprent, 2007). Therefore, unraveling the mysteries of this ancestral form of colonization would increase our knowledge in identifying suitable symbiosis clusters that potentially could be transferred to other non-legume crops.
A high level of diversity and heterogeneity exist in peanut symbionts. Several fast-growing endophytic isolates of Gammaproteobacteria that were unable to elicit root nodules were recovered from peanut nodules in Argentina (Ibáñez et al., 2009), as well as some slow-growing Bradyrhizobium isolates that have been characterized (Muñoz et al., 2011). However, complete genomes of root-nodulating bacteria that live in symbiosis with peanuts are not available. On the other hand, B. diazoefficens USDA 110T that fixes nitrogen in economically important crops such as soybean, has been completely sequenced (Kaneko et al., 2002). Other Bradyrhizobium strains with complete genome sequences include Bradyrhizobium sp. BTAi1 and Bradyrhizobium sp. ORS 278, which form nodules on Aeschynomene (Giraud et al., 2007). Complete sequences of Bradyrhizobium sp. CCGE-LA001, which was obtained from a wild Phaseolus sp., and non-nodulating isolate, Bradyrhizobium sp. S23321, which was isolated from paddy fields, are also available (Okubo et al., 2012b; Rogel and Moral, 2016). Limited knowledge on the interaction of peanut with rhizobia restricts our ability to fully utilize the nitrogen-fixing capacity of rhizobia in crack entry species. Therefore, this study was initiated to sequence the whole genome of an efficiently growing Bradyrhizobium strain capable of inducing peanut root nodules and to characterize the strain.
Sequence Features of the Lb8 Genome
An isolate, Lb8, was selected for whole-genome sequencing because of its relatively fast growth rate and high nitrogenase activity compared to the other isolates from peanut nodules. Lb8 had identical 16S rRNA sequence to Ls1, but they had different growth curves and nitrogenase activity. Despite having identical 16S rRNA, bacteria could show variable biosynthetic properties. For example, multiple strains in Brevundimonas alba with identical 16S rRNA sequence had different growth rate and cell yield (Jaspers and Overmann, 2014). Similarly, various species in Streptomyces that had identical 16S rRNA sequence showed different phenotypic and morphologic properties (Antony-Babu et al., 2017). In our research, the differences in the growth curve of these isolates could be explained by variations in other parts of the genome. Phylogeny results from 16S rRNA and housekeeping genes suggested that the Lb8 genome resembled more closely the non-photosynthetic nod gene-containing bradyrhizobia than the photosynthetic nod gene-lacking bradyrhizobia. This was corroborated with RAST annotations on whole genome sequencing data, where we could not find any genes related to photosynthesis (light-harvesting complexes, photosynthesis, or electron transport and photophosphorylation). Although symbiotic genes do not offer taxonomic information because they are located in easily interchangeable elements (Chahboune et al., 2011), a phylogenetic tree based on nod genes showed that Lb8 belongs to the same symbiovar as strains SEMIA 5079, USDA 110T, and USDA 6T. ANI values of Lb8 compared with other sequenced Bradyrhizobium genomes indicated that Bradyrhizobium sp. Lb8 was a novel species that fixes nitrogen in peanut nodules.
We didn't find any plasmid sequences in the PacBio sequences of the DNA sample, which was prepared from the ChargeSwitch gDNA mini bacteria kit to target the genomic DNA of the bacteria. In addition, bradyrhizobia typically have no plasmid, which is a standard feature of bradyrhizobia (MacLean et al., 2007). Plasmids are more common in fast-growing rhizobia such as Sinorhizobium fredii (Scholla and Elkan, 2009). The symbiotic island in Lb8 was integrated in its genome, which is a typical feature of non-photosynthetic Bradyrhizobium strains, such as USDA 110T. Even though the Lb8 genome shared 92% of the gene families with USDA 110T, it showed several key differences. The genome of Bradyrhizobium sp. Lb8 was ~400 kb smaller than that of USDA 110T and it possessed a much larger symbiotic island than USDA 110T. Unlike USDA 110T, Lb8 did not possess homologs of nodY gene and the T6SS was not present in Lb8. Presence of genes like cynR, nifT, and ginE in Lb8 make it unique. Lb8 lacks nitrous oxide reductase genes like nosDXFRY that are present in USDA 110T. Lb8 had only one cluster of nif genes whereas in USDA 110T they were present in two clusters.
Symbiosis Island
Acquisition of the SI converts a saprophyte into a symbiont. Therefore, the island is likely to contain other genes that contribute to successful plant-microbe interactions, in addition to genes required for nodule formation and symbiotic nitrogen fixation (Sullivan and Ronson, 1998). SIs were identified in M. loti ICMP3153 and M. japonicum MAFF303099 as DNA segments of 500 to 611 Kb with low GC content and flanked by a complete and partial phe-tRNA gene (Sullivan and Ronson, 1998; Hubber et al., 2004). A split SI of 410 Kb plus 6 Kb was identified in a 680 Kb DNA region of low GC content in USDA 110T (Kaneko et al., 2002). Sequence analysis of the Lb8 genome also revealed the presence of a 1,015 Kb genomic island with relatively low GC content, including a complete 777 Kb symbiotic DNA region. Genomic islands are usually integrated at the tRNA loci, with an integrase gene at the 5′ junction (Hacker and Kaper, 2000; Zhang and Zhang, 2004). In M. loti, a partially duplicated phe-tRNA gene was identified while a partially duplicated val-tRNA gene was identified in USDA 110T. The presence of a partially duplicated met-tRNA gene and a putative integrase gene near the end of this island in Lb8 reinforces the presence of an SI in the genome. The association of SIs with a phage-related integrase implies that SIs may have originated from an ancient integration of bacteriophage. Most genomic islands have mobile genes like transposase and integrase (Hacker and Kaper, 2000). It was remarkable that 16 out of the 21 transposases detected (76%) and six out of 16 integrase genes (38%) were located in the presumptive SI in Lb8 with low GC content. Therefore, we hypothesize that an ancient SI was transferred by horizontal gene transfer, which is well-known to contribute to microbial evolution (Hacker and Kaper, 2000).
Nitrogen Metabolism-Related Genes
Genes related to different steps of the symbiotic nitrogen fixation process were identified in the Lb8 genome. Specifically, cynR (cyanate hydrolysis), nifT, and ginE (ammonium assimilation) were unique in comparison to the other Bradyrhizobium strains such as ORS278, BTAi1, and USDA 110T. Although the exact function of nifT has not been identified, it has been suggested to have a limited role in nitrogenase maturation in Klebsiella pneumoniae (Simon et al., 1996).
In most rhizobia, nod genes are essential for Nod factor biosynthesis and transportation. Bacterial mutants defective in Nod factor production can no longer associate with host legumes, and legume mutants that are defective in the perception of Nod factors can no longer host the bacteria (Oldroyd, 2013), with few exceptions. In peanuts, nodC gene was essential to induce cortical cell division leading to formation of nodule primordium. The absence of the gene, however, did not affect the bacteria's ability to colonize peanut roots (Ibáñez and Fabra, 2011). nodD is essential for the expression of the other nod genes (Stacey et al., 1995). NodD1 appears to be the only functional nodD gene in B. diazoefficiens, and its activation is dependent on the presence of isoflavones (Stacey, 1995). NodV and nodW may also be able to activate nod gene expression (Stacey, 1995). In the Lb8 genome, nod genes were present in two clusters. Cluster I contained most of the nod genes (nodU1U2DABCSU3I-nolO-nodZ-nfeC). NodT and nodN were found in a separate cluster in the symbiosis island. The presence of nodABC genes, which are required for the synthesis of the lipo-chitooligosaccharidic backbone, suggests that Lb8 nodulates peanuts in a Nod factor-dependent manner, but it is also possible that these genes have been retained to colonize alternative legume hosts. Photosynthetic Bradyrhizobium strains, such as BTAi1 and ORS278, without the nodABC genes required for Nod factor biosynthesis can still colonize their host legumes (Giraud et al., 2007), indicating the possibility of an alternative pathway to initiate symbiosis (Masson-Boivin and Sachs, 2018).
In addition to the nod genes, nif and fix are essential genes involved in nitrogen fixation among Bradyrhizobium species and other rhizobia. nif genes found in bradyrhizobia have similar function and structure with the related genes found in Klebsiella pneumoniae, a free-living diazotroph (Fischer and Hennecke, 1987; Hennecke, 1990). nifW and nifZ genes are involved in MoFe protein maturation, and their requirement varies significantly among diazotrophs (Li et al., 2016). nifBENH are involved in the synthesis of FeMo-co from iron, sulfur, molybdenum, and homocitrate under reducing conditions (Rubio and Ludden, 2008). nifQ is an iron–sulfur protein with a redox-responsive [Fe–S] cluster and is also a molybdoprotein that donates molybdenum for FeMo-co synthesis in the simultaneous presence of nifH and nifEN (Rubio and Ludden, 2008). nifS directs the assembly of nifU, and with nifB sequentially provides the iron–sulfur core while nifX binds a variety of structurally related factors (Rubio and Ludden, 2008). Fix genes are essential for symbiotic nitrogen fixation. In B. diazoefficiens strain USDA 110T, the nif and fix genes were found in at least two different clusters on the chromosome. Cluster I included most of the nif genes. Cluster II contained three fix genes (Hennecke, 1990). However, in Lb8, nif genes were present in a single cluster, including nifWQHOZBTSUXNE. Presence of this gene cluster shows an active role of Lb8 in nitrogen fixation. One fix gene (fixC) was also detected in the cluster. The genome also contained fixJ, which controlled symbiotically important genes other than those whose expression was dependent on the NifA protein (Anthamatten and Hennecke, 1991). Nif A protein activates nif genes in B. diazoefficiens strain USDA 110T and is sensitive to oxygen (Fischer and Hennecke, 1987). The presence of the hydrogenase gene cluster increases the efficiency of nitrogen fixation because Ni-Mo hydrogenase mediates uptake of dihydrogen via nitrogenase (Kaneko et al., 2002). Plants with nodules containing hydrogenase systems were shown to fix more nitrogen and produced higher dry weights than plants nodulated with strains lacking hydrogen uptake genes (Albrecht et al., 1978). The presence of hup genes in Lb8 might promote higher efficiency of nitrogen fixation.
Lb8 possesses a nirV gene, which is required for nitrite reduction and is found in bacteria with copper-containing nitrite reductases (Jain and Shapleigh, 2001). A copper-containing nitrite reductase was observed immediately upstream of the nirV gene in Lb8. Thus, Lb8 should have the capacity to reduce nitrite. Putative products of periplasmic nitrate reductase genes are likely to be involved in denitrification from nitrate to N2 (Black et al., 2012). A homolog of a tfdB-like gene (2,4-dichlorophenol 6-monooxygenase), which is implicated in the degradation of 2,4-D (2,4-dichlorophenol; Vallaeys et al., 1999), was also found in Lb8.
Protein Secretion Systems
Protein secretion systems play a crucial role in the interaction of bacteria and their eukaryotic hosts (Eichinger et al., 2016). T3SSs are involved in host colonization, virulence, and symbiotic interactions and they can be horizontally transferred (Poggio et al., 2007; Cole et al., 2017; Levy et al., 2018). T4SSs are involved in bacterial conjugation, DNA uptake and release, and effector translocation (Fronzes et al., 2009). Lb8 possesses both T3SS and T4SS genes. Interestingly, the genes for the non-flagellar T3SS were located on the symbiosis island, while the genes for the flagellar T3SS and the T4SS were located outside the symbiosis island. This feature is similar to USDA 110T (Kaneko et al., 2002), except that USDA 110T also possesses a single T6SS cluster outside the symbiosis island (Banerjee et al., 2019). USDA 110T has been classified as “Root Nodular II” based on its presence of T3SS, T4SS, and T6SS secretome (Banerjee et al., 2019). On the other hand, the absence of T6SS and the presence of T3SS and T4SS makes Lb8 closer to the “Root nodular I” type genomes. Flagellar T3SSs drive cell mobility while non-flagellar T3SSs deliver bacterial effectors into host cells via channels that cross the bacterial envelope and the host cell membrane to establish trans-kingdom interactions (Abby and Rocha, 2012; Deng et al., 2017). Rhizobial T3 effectors suppress plant defense responses against rhizobia and promote symbiosis-related processes (Staehelin and Krishnan, 2015). Type IV effectors secreted by the T4SS in M. japonicum MAFF303099 promoted nodule formation on its host, Lotus corniculatus (Hubber et al., 2004; Martínez-Hidalgo et al., 2016). The T4SS and T3SS may be involved in fine-tuning of host-specific nodulation in peanuts. In M. japonicum MAFF303099, genes for both T3SS and T4SS were located inside the symbiosis island (Sullivan and Ronson, 1998; Martínez-Hidalgo et al., 2016). Presence of the flagellar T3SS in all 17 Bradyrhizobium strains analyzed in this study indicates that the flagellar T3SS might be an integral component of symbiosis.
In summary, current study revealed a whole genome analysis of Lb8, a native Bradyrhizobium strain isolated from peanut nodules. The availability of this genome will facilitate the understanding of how rhizobia work and interact with peanut. To ultimately clarify the unique mechanisms of crack entry, it will be necessary to characterize the infection path of rhizobia into peanut roots. While previous studies have shown that Nod factors might not be needed for colonization of peanut roots, Nod factors are known to be involved in the initiation of cortical cell division (Ibáñez and Fabra, 2011). The nod genes identified in this study could also shed light on subsequent studies on the requirement for Nod factors in peanut–rhizobium interactions. Crack entry is believed to be evolutionarily ancient and is the simplest entry method, so it holds high promise to be used as a donor in extending the nitrogen fixation symbiosis to important non-legume crops. We want to emphasize that Bradyrhizobium sp. Lb8 could be a model microorganism for nodulating soil bradyrhizobia for crack entry species, providing a valuable tool for experiments in genetics, physiology, and ecology of such species. Additional investigations into the crack entry mechanism will not only impact biotechnological applications and enhance our comprehension of plant–microbe interactions but may also be vital to illustrate the evolution of symbiosis.
Materials and Methods
Strain Isolation
Two cultivated peanut lines, Tifrunner and UF 487A were grown in the field of the Plant Science Research and Education Unit of University of Florida at Citra, FL, which was inoculated with a commercial peanut inoculum. Nodules from 30-day-old plants were harvested from these two peanut lines. A total of 20 individual nodules were obtained from 10 plants of two peanut lines, Tifrunner and UF 487A. The procedure used for rhizobia isolation followed the method described previously (Taurian et al., 2002, 2006) with some modifications. In brief, the detached nodules from roots were surface sterilized by immersing in 95% ethanol for 30 s and in 0.1% HgCl2 for 4 min, followed by washing six times with sterile distilled water. The surface-sterilized nodules were individually crushed in a drop of sterile water. The suspension of the crushed nodules after centrifugation was streaked on 20 yeast extract mannitol (YEM) agar plates with one nodule suspension per plate and incubated at 28°C for 7 days (Vincent, 1970). Strains were streaked to single colonies three times to ensure that it was a pure culture. Colonies of different sizes and colors were representatively picked for colony PCR to amplify 16s rRNA gene.
Colony PCR and Sequencing
For colony PCR, a pair of primers designed from 16S rRNA gene were synthesized by Invitrogen: F27: 5′-AGAGTTTGATCATGGCTCAG-3′, and R1541: 5′-AAGGAGGTGATCCAGCCGCA-3′ (Weisburg et al., 1991). A total volume of 20 μL reaction mixture was composed of the single colony of bacteria, 2 μL of 10× buffer [0.1 mol/L Tris-HCl (pH = 9.0), 0.5 mol/L KCl, 7.5 mmol/L MgCl2, 0.1% Triton X-100], 5 μmol of each primer, 2.5 mmol/L each of deoxynucleotide triphosphates (dNTP), and 1 unit of Taq DNA polymerase (Promega). Amplification was carried out on the program for the initial denaturing step with 94°C for 5 min, followed by 35 cycles for 30 s at 94°C, 45 s at 55°C, 1 min at 72°C, with a final extension at 72°C for 10 min. The amplicons were separated on a 1.5% agarose gel for 1 h at 150 V. Selected representative PCR products were purified by using a QIAgen PCR purification kit (Cat. No. 28104) according to the manufacturer's protocol and then sequenced by the Sanger method at the Interdisciplinary Center of Biotechnology Research at the University of Florida. The trimmed clean sequences were BLASTed against the NCBI GenBank nucleotide database to identify the best hit sequences with known annotation. The taxonomy-confirmed colonies were picked and stored in 20% glycerol solution at −80°C.
Gram Staining, Morphological Observation, and Growth Curve Characterization
Gram staining was done following the standard protocol (Christopher and Bruno, 2003). The slides were observed under an Olympus BX41 microscope (Olympus America Inc., Melville, NY, USA). Scanning electron microscopy observation was performed according to published protocol (Mazia et al., 1975). Briefly, cultures were deposited onto poly-L-lysine treated 0.2 μm Millipore filters and fixed by immersion into 4% (v/v) paraformaldehyde, 2.5% (v/v) glutaraldehyde in 0.1 M cacodylate buffer, pH 7.3 (Electron Microscopy Sciences). Filters containing cells were rinsed twice in 0.1 M cacodylate buffer, water washed thrice, and dehydrated through a gradient of ethanol (25, 50, 75, 95, 100%). Dehydrated cells were critical point dried (Autosamdri-915, Tousimis) and mounted with a carbon adhesive tab and graphite paste onto a 12 mm aluminum SEM stub (Ted Pella, Inc.). The samples were further rendered conductive by sputter coating with Au/Pd and argon gas (DeskV, Denton Vacuum) and imaged on a Hitachi SU5000 Schottky Field-Emission Variable Pressure SEM (Hitachi High Technologies, America). The cell size was measured on 20–30 randomly selected individual cells.
To evaluate the growth rate of each isolated bacterial strain, we streaked a line on YEM medium in stock solution and cultured at 28°C until a colony was seen after about 7 days. Single colonies were picked with tips and kept in a 50 ml tube with 30 ml YEM medium. Each strain had at least three replicates. The cultures were diluted to 2x, 4x, and 10x on day two, three, and four, respectively. The tubes were shaken for 28 days, and the OD600 value of each strain solution was measured at the same time every day to evaluate the growth rate of strains.
Nodule Formation Test
To test whether the isolated strains can infect peanut and form functional nodules, each isolated rhizobial strain was inoculated into 200 ml YEM broth in a shaking incubator at 28°C. When the OD600 value reached 0.05–0.1, the liquid cultures were spun down at 4,000 × g for 15 min. Bacterial cells were then washed with sterile water and re-suspended to OD600 = 0.05–0.1, with ~106 cells per ml as inoculum.
Seeds of the cultivated peanut genotype Tifrunner and soybean cultivars, Bennings and Boggs, were sterilized in 0.1% HgCl2 solution for 7 min, and then washed for three times using ddH2O for 5 min each time. Sterilized seeds were immersed in a 500 ml glass flask for 2 days and then transferred to germination box with germination paper soaked with ddH2O and incubated at 25°C. After 4–5 days, germinated seeds were transferred to a growth bag (Ziploc plastic bag with filter paper) with 40 ml of 25% Hoagland's solution without nitrogen and grown in a growth chamber with 12 h light/12 h dark cycles and at 25°C. The growth bags were supported upright on a board by clippers. When the root length was ~6 cm, plants were selected to be inoculated by each rhizobial isolate separately with three biological replicates. For rhizobial inoculation, a 1-ml bacterial suspension (OD600 = 0.05–0.1) was applied to the surface of the root in the growth bag for inoculation. Water and Hoagland's solution were used as controls. Nodules were counted and then harvested for weight and size measurement and dissection and color observation at 2 weeks after inoculation. The diameter of mature nodules was evaluated by measuring the total diameters of 5 mature nodules on each plant using a Digital Caliper. All analysis and plots were done in software R, v 3.4.4 (http://www.R-project.org/) unless noted otherwise.
Nitrogenase Activity Measurement
Seeds of cultivated peanut genotype Tifrunner were inoculated with different strains to test nitrogenase activities of these strains during symbiosis by acetylene reduction assay (ARA) (Hardy et al., 1973). The inoculation procedure was the same as described above. At 12 days post-inoculation, primary roots with nodules inoculated by different strains were cut and put into three 140 ml bottles with a rubber stopper. Each bottle had 2–3 roots with nodules. From each bottle, 10 ml air was removed and replaced with the same amount of acetylene (10 ml) by syringe injection. The bottles were then sealed and incubated at 28°C for 4 h. After the reaction, 1 ml of air from each bottle was taken out by syringe, and the amount of ethylene formed was measured by using a Gas Chromatographer (5890 Series). After ethylene measurement, nodules were cut from roots in each bottle to get fresh weight. Nitrogenase activity (nmol/mg/min) was calculated using the following formulae:
Sequencing and Data Analysis
Genomic DNA was extracted from a 50 ml liquid culture of a single colony using ChargeSwitch gDNA mini bacteria kit (Life Technologies) for whole-genome sequencing using the PacBio RS II system (Pacific Biosciences). An approximately 20 Kb insert PacBio library was constructed and sequenced in three SMRT cells. The PacBio reads were de novo assembled using the HGAP (Chin et al., 2013) protocol, which generated an initial assembly of 24 contigs with a total length of 11,944,080 bp. Plasmid Finder (v1.3) (Carattoli et al., 2014) was used to check for the presence of any plasmids. BLASR (Chaisson and Tesler, 2012) was used to map reads on contigs. The average depth per contig ranged from 43× to 69×, except for unitig_0, which had 403× coverage. Reads that mapped to unitig_0 (a contaminant contig which was closely related to Microbacterium sp. CGR1) were removed. Remaining reads were then extracted and used for assembly using the HGAP version 1 protocol under SMRT Analysis 2.1.1 and polished using Quiver (Pacific Biosciences). The contig was circularized using Circlator/1.5.1 (Hunt et al., 2015) and reverse complemented with EMBOSS/6.5.7 to get the final assembly. Annotation of the genome was done according to the RAST protocol (Aziz et al., 2008). NCBI-BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to further identify any additional symbiosis related genes in Lb8 with default settings. A putative oriC was identified by using Ori-Finder (Gao and Zhang, 2008). rRNA genes were predicted using the RNAmmer 1.2 (http://www.cbs.dtu.dk/services/RNAmmer/) web server. Protein secretion systems were identified using T346 Hunter (Martínez-García et al., 2015).
Genome Comparison and Ortholog Analysis
A circular genome map showing GC skew and GC content was created using CGview server with default parameters (Grant and Stothard, 2017). Putative orthologous genes were identified using bidirectional BLASTn comparisons with an E-value cutoff of 10−20. OrthoMCL software (Fischer et al., 2011) was used to identify orthologous gene groups between the rhizobial genomes being compared. Protein sequences of Lb8 were downloaded from the results of the RAST server, and the protein sequences of other genomes were downloaded from the NCBI website (BTAi1: NC_009485.1, USDA 110T: NZ_CP011360.1, and ORS278: CU234118.1). OrthoMCL analysis on these protein sequences was done by following “Basic Protocol 2” from (Fischer et al., 2011), with default parameters. An E-value of “1e−5” was used for all-versus-all BLASTp comparisons. Orthologous relationships were depicted in a Venn diagram.
Average Nucleotide Identity (ANI) between Lb8 and 17 Bradyrhizobium strains with complete genome sequences (Genbank ID: LT859959.1, CP013949.1, CP013127.1, CP016428.1, LN907826.1, LN901633.1, CP017637.1, CP011360.1, AP014685.1, CP010313.1, CP007569.1, AP012603.1, CU234118.1, CP000494.1, BA000040.2, AP012279.1, AP012206.1) was calculated using ANI calculator (Rodriguez and Konstantinidis, 2016).
Phylogeny of Nodulation and Nitrogen Fixation Related Genes
Phylogenetic analysis was conducted using KBase (Arkin et al., 2016) with the “Insert Genome Into Species Tree” app that used 49 highly conserved Clusters of Orthologous Groups (COG) families to find matching corresponding sets of sequences for a specific genome. Sequences from the selected genomes were then inserted into the reference alignments, and closest neighbors were extracted and concatenated. After that, a tree was rendered using the FastTree 2 algorithm (Price et al., 2010). Phylogenetic analysis of nod genes, including nodU1U2DABCSU3I-nolO-nodZ-nfeC-nodNT in selected species, was constructed using Geneious v.10.2.3 (Drummond et al., 2011). A multiple alignment for each gene was done using ClustalW (Cost matrix–BLOSUM; Cap open cost−10; Gap extend cost−0.1). The alignments were concatenated, and the concatenated alignment was used to construct a phylogenetic tree (Alignment type–Global alignment with free end gaps; Cost matrix–BLOSUM62; Genetic Distance Model–Jukes-Cantor; Tree build Method–Neighbor-Joining; Outgroup–No Outgroup; Bootstrap−1000).
Data Availability Statement
The whole genome sequence of Bradyrhizobium sp. Lb8 is available from RAST server with genome ID 6666666.283533.
Author Contributions
JW conceived the research and designed the experiments. DP, FL, LW, MC, SM, ZP, and KK performed the experiments and analyzed the data. DP prepared the manuscript draft. JW, MC, and J-MA critically revised the manuscript. All authors approved the final version of the manuscript.
Funding
This study was funded by the Florida Peanut Producers Association, the University of Florida IFAS Early Career Scientists Award, and USDA National Institute of Food and Agriculture, Hatch Project 1011664. Publication of this article was funded in part by the University of Florida Open Access Publishing Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We highly appreciate Dr. Zenglu Li at University of Georgia and Dr. Brian Diers at University of Illinois at Urbana Champaign for providing the soybean seeds. We want to thank Hanh Nguyen for providing 16S primers.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00093/full#supplementary-material
Supplementary Figure 1. A growth curve of five peanut nodule isolates grown at 28°C in YEM medium.
Supplementary Figure 2. (A) Nodules formed on Tifrunner roots 13 days after inoculation by five different strains. (B) Root nodules showing active nodulation after 22 days of inoculation with the different strains.
Supplementary Figure 3. (A) A scanning electron micrograph of the three strains L1, Lb8, and Ls1. (B) Bar plots showing the length of the different strains (n = 44). Same letters on top of each bar plot are not significantly different (HSD, p > 0.05).
Supplementary Figure 4. A GC-skew plot showing the putative locations of the oriC and terminus in the Bradyrhizobium sp. Lb8 genome.
Supplementary Figure 5. A dot plot showing the similarity between Bradyrhizobium sp. Lb8 and B. diazoefficiens USDA 110T.
Supplementary Figure 6. Neighbor-joining tree based on concatenated sequences of nod genes present in Bradyrhizobium sp. Lb8 (nodU2DABCSU3I-effC-nodZNT) and closely related species within the genus Bradyrhizobium. The significance of each branch is indicated by a bootstrap value calculated for 1,000 subsets. Bar, 3 substitutions per 1,000 nucleotides.
Supplementary Figure 7. The number of nitrogen metabolism-related genes that are present in the different Bradyrhizobium genomes.
Supplementary Table 1. Nitrogen metabolism related genes in four Bradyrhizobium genomes. The numbers in the list refer to locus numbers on the RAST server. Numbers in column names after the species name refer to accession number (In Excel sheet).
Supplementary Table 2. ANI (%) values for comparison of Bradyrhizobium sp. Lb8 with 17 complete genomes of Bradyrhizobium (In Excel sheet).
References
Abby, S. S., and Rocha, E. P. C. (2012). The non-flagellar Type III secretion system evolved from the bacterial flagellum and diversified into host-cell adapted systems. PLoS Genet. 8:e1002983. doi: 10.1371/journal.pgen.1002983
Albrecht, S., Maier, R., Hanus, J., Russell, S., Emerich, D., and Evans, H. (1978). Hydrogenase in Rhizobium japonicum increases nitrogen. Science 203, 1255–1257. doi: 10.1126/science.203.4386.1255
Anthamatten, D., and Hennecke, H. (1991). The regulatory status of the fixL- and fixJ-like genes in Bradyrhizobium japonicum may be different from that in Rhizobium meliloti. MGG Mol. Gen. Genet. 225, 38–48. doi: 10.1007/bf00282640
Antony-Babu, S., Stien, D., Eparvier, V., Parrot, D., Tomasi, S., and Suzuki, M. T. (2017). Multiple Streptomyces species with distinct secondary metabolomes have identical 16S rRNA gene sequences. Sci. Rep. 7, 1–8. doi: 10.1038/s41598-017-11363-1
Arkin, A. P., Stevens, R. L., Cottingham, R. W., Maslov, S., Henry, C. S., Dehal, P., et al. (2016). The DOE Systems Biology Knowledgebase (KBase). bioRxiv 96354. doi: 10.1371/journal.pone.0124501
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Banerjee, G., Basak, S., Roy, T., and Chattopadhyay, P. (2019). Intrinsic role of bacterial secretion systems in phylogenetic niche conservation of Bradyrhizobium spp. FEMS Microbiol Ecol. 95, 1–11. doi: 10.1093/femsec/fiz165
Black, M., Moolhuijzen, P., Chapman, B., Barrero, R., Howieson, J., Hungria, M., et al. (2012). The genetics of symbiotic nitrogen fixation: comparative genomics of 14 rhizobia strains by resolution of protein clusters. Genes 3, 138–166. doi: 10.3390/genes3010138
Bonaldi, K., Gargani, D., Prin, Y., Fardoux, J., Gully, D., Nouwen, N., et al. (2011). Nodulation of Aeschynomene afraspera and A. indica by photosynthetic Bradyrhizobium sp. Strain ORS285 : the nod-dependent versus the nod-independent symbiotic interaction. Mol. Plant-Microbe Interact. 24, 1359–1371. doi: 10.1094/MPMI-04-11-0093
Boogerd, F. C., and Van Rossum, D. (1997). Nodulation of groundnut by Bradyrhizobium: a simple infection process by crack entry. FEMS Microbiol. Rev. 21, 5–27. doi: 10.1111/j.1574-6976.1997.tb00342.x
Carattoli, A., Zankari, E., Garcia-Fernandez, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Chahboune, R., Carro, L., Peix, A., and Barrijal, S. (2011). Bradyrhizobium cytisi sp. nov., isolated from effective nodules of Cytisus villosus. Int. J. Syst. Evol. Microbiol. 61, 2922–2927. doi: 10.1099/ijs.0.027649-0
Chaisson, M. J., and Tesler, G. (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13:238. doi: 10.1186/1471-2105-13-238
Chandler, M. R. (1978). Some observations on infection of Arachis hypogaea L. by Rhizobium. J. Exp. Bot. 29, 749–755. doi: 10.1093/jxb/29.3.749
Charpentier, M., and Oldroyd, G. (2010). How close are we to nitrogen-fixing cereals? Curr. Opin. Plant Biol. 13, 556–564. doi: 10.1016/j.pbi.2010.08.003
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Christopher, K., and Bruno, E. (2003). “Identification of bacterial species,” in Tested Studies for Laboratory Teaching. Proceedings of the 24th Workshop/Conference of the Association for Biology Laboratory Education, ed M. O'Donnell (Louisiana), 103–130.
Cole, B. J., Feltcher, M. E., Waters, R. J., Wetmore, K. M., Mucyn, T. S., Ryan, E. M., et al. (2017). Genome-wide identification of bacterial plant colonization genes. PLoS Biol. 15:e2002860. doi: 10.1371/journal.pbio.2002860
de Lajudie, P. M., Andrews, M., Ardley, J., Eardly, B., Jumas-Bilak, E., Kuzmanović, N., et al. (2019). Minimal standards for the description of new genera and species of rhizobia and agrobacteria. Int. J. Syst. Evol. Microbiol. 69, 1852–1863. doi: 10.1099/ijsem.0.003426
Delamuta, J. R. M., Ribeiro, R. A., Ormeño-Orrillo, E., Melo, I. S., Martínez-Romero, E., and Hungria, M. (2013). Polyphasic evidence supporting the reclassification of Bradyrhizobium japonicum group Ia strains as Bradyrhizobium diazoefficiens sp. nov. Int. J. Syst. Evol. Microbiol. 63, 3342–3351. doi: 10.1099/ijs.0.049130-0
Dénarié, J., Debellé, F., and Promé, J.-C. (1996). Rhizobium lipo-chitooligosaccharide nodulation factors: signaling molecules mediating recognition and morphogenesis. Annu. Rev. Biochem. 65, 503–535. doi: 10.1146/annurev.bi.65.070196.002443
Deng, W., Marshall, N. C., Rowland, J. L., McCoy, J. M., Worrall, L. J., Santos, A. S., et al. (2017). Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microbiol. 15, 323–337. doi: 10.1038/nrmicro.2017.20
Downie, J. A., and Walker, S. A. (1999). Plant responses to nodulation factors. Curr. Opin. Plant Biol. 2, 483–489. doi: 10.1016/s1369-5266(99)00018-7
Drummond, A. J., Ashton, B., Buxton, S., Cheung, M., Cooper, A., Duran, C., et al. (2011). Geneious, Version 10.2.3. Auckland: Geneious.
Eichinger, V., Nussbaumer, T., Platzer, A., Arnold, R., and Rattei, T. (2016). EffectiveDB - updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 44, 669–674. doi: 10.1093/nar/gkv1269
Evans, H. J., Harker, A. R., Papen, H., Sterling, A., Hanus, F. J., Zuber, M., et al. (1987). Physiology, biochemistry, and genetics of the uptake hydrogenase in rhizobia. Annu. Rev. Microbiol. 41, 335–361. doi: 10.1146/annurev.mi.41.100187.002003
Fischer, H. M., and Hennecke, H. (1987). Direct response of Bradyrhizobium japonicum nifA-mediated nif gene regulation to cellular oxygen status. MGG Mol. Gen. Genet. 209, 621–626. doi: 10.1007/bf00331174
Fischer, S., Brunk, B. P., Chen, F., Gao, X., Harb, O. S., Iodice, J. B., et al. (2011). Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinformatics. 12, 1–19. doi: 10.1002/0471250953.bi0612s35
Fronzes, R., Christie, P. J., and Waksman, G. (2009). The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 7, 703–714. doi: 10.1038/nrmicro2218
Gao, F., and Zhang, C.-T. (2008). Ori-Finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinformatics 9:79. doi: 10.1186/1471-2105-9-79
Giraud, E., Moulin, L., Vallenet, D., Barbe, V., Cytryn, E., Avarre, J.-C., et al. (2007). Legumes symbioses: absence of Nod genes in photosynthetic Bradyrhizobia. Science 316, 1307–1312. doi: 10.1126/science.1139548
Grant, J. R., and Stothard, P. (2017). The CGView Server : a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, 181–184. doi: 10.1093/nar/gkn179
Gresshoff, P. M., and Ferguson, B. J. (2017). Molecular signals in nodulation control. Int. J. Mol. Sci. 18, 10–12. doi: 10.3390/ijms18010125
Grönemeyer, J. L., Hurek, T., and Reinhold-Hurek, B. (2015). Bradyrhizobium kavangense sp. Nov., a symbiotic nitrogen-fixing bacterium from root nodules of traditional namibian pulses. Int. J. Syst. Evol. Microbiol. 65, 4886–4894. doi: 10.1099/ijsem.0.000666
Groth, M., Takeda, N., Perry, J., Uchida, H., Draxl, S., Brachmann, A., et al. (2010). NENA, a Lotus japonicus homolog of Sec13, is required for rhizodermal infection by arbuscular mycorrhiza fungi and rhizobia but dispensable for cortical endosymbiotic development. Plant Cell 22, 2509–2526. doi: 10.1105/tpc.109.069807
Hacker, J., and Kaper, J. B. (2000). Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54, 641–679. doi: 10.1146/annurev.micro.54.1.641
Hardarson, G. (1993). Methods for enhancing symbiotic nitrogen fixation. Plant Soil 152, 1–17. doi: 10.1007/BF00016329
Hardy, R. W. F., Burns, R. C., and Holsten, R. D. (1973). Applications of the acetylene-ethylene assay for measurement of nitrogen fixation. Soil Biol. Biochem. 5, 47–81. doi: 10.1016/0038-0717(73)90093-X
Hennecke, H. (1990). Nitrogen fixation genes involved in the Bradyrhizobium japonicum-soybean symbiosis. FEBS Lett. 268, 422–426. doi: 10.1016/0014-5793(90)81297-2
Howieson, J. G., and Dilworth, M. J., (eds.). (2016). Working With Rhizobia Australian Centre for International Agricultural Research. Australian Centre for International Agricultural Research.
Hubber, A., Vergunst, A. C., Sullivan, J. T., Hooykaas, P. J. J., and Ronson, C. W. (2004). Symbiotic phenotypes and translocated effector proteins of the Mesorhizobium loti strain R7A VirB/D4 type IV secretion system. Mol. Microbiol. 54, 561–574. doi: 10.1111/j.1365-2958.2004.04292.x
Hunt, M., Silva, N., De Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S.R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16:294. doi: 10.1186/s13059-015-0849-0
Ibáñez, F., Angelini, J., Taurian, T., Tonelli, M. L., and Fabra, A. (2009). Endophytic occupation of peanut root nodules by opportunistic Gammaproteobacteria. Syst. Appl. Microbiol. 32, 49–55. doi: 10.1016/j.syapm.2008.10.001
Ibáñez, F., and Fabra, A. (2011). Rhizobial Nod factors are required for cortical cell division in the nodule morphogenetic programme of the Aeschynomeneae legume Arachis. Plant Biol. 13, 794–800. doi: 10.1111/j.1438-8677.2010.00439.x
Jain, R., and Shapleigh, J. P. (2001). Characterization of nirV and a gene encoding a novel pseudoazurin in Rhodobacter sphaeroides 2.4.3. Microbiology 147, 2505–2515. doi: 10.1099/00221287-147-9-2505
Jaspers, E., and Overmann, J. (2014). Ecological significance of microdiversity: identical 16S rRNA gene sequences can be found in bacteria with highly divergent genomes and ecophysiologies. Appl. Environ. Microbiol. 70, 4831–4839. doi: 10.1128/AEM.70.8.4831-4839.2004
Jordan, D. C. (1982). Transfer of Rhizobium japonicum Buchananm 1980 to Bradyrhizobium japonicum gen. nov., a genus of slow growing root nodule bateria. Int. J. Syst. Bacteriol. 32, 378–380. doi: 10.1099/00207713-32-1-136
Kaneko, T., Nakamura, Y., Sato, S., Minamisawa, K., Uchiumi, T., Sasamoto, S., et al. (2002). Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 9, 189–197. doi: 10.1093/dnares/9.6.189
Kuever, J., Rainey, F. A., and Widdel, F. (2015). Bergey's Manual of Systematics of Archaea and Bacteria. Hoboken, NJ: John Wiley & Sons, Ltd.
Levy, A., Gonzalez, I. S., Mittelviefhaus, M., Clingenpeel, S., Paredes, S. H., Miao, J., et al. (2018). Genomic features of bacterial adaptation to plants. Nat. Genet. 50, 138–150. doi: 10.1038/s41588-017-0012-9
Li, P., and Everhart, D. M. (1998). Genetic and sequence analysis of the pTiC58 trb locus, encoding a mating-pair formation system related to members of the Type IV secretion family. J. Bacteriol. 180, 6164–6172.
Li, X. X., Liu, Q., Liu, X. M., Shi, H. W., and Chen, S. F. (2016). Using synthetic biology to increase nitrogenase activity. Microb. Cell Fact. 15:43. doi: 10.1186/s12934-016-0442-6
Li, Y. H., Wang, R., Sui, X. H., Wang, E. T., Zhang, X. X., Tian, C. F., et al. (2019). Bradyrhizobium nanningense sp. nov., Bradyrhizobium guangzhouense sp. nov. and Bradyrhizobium zhanjiangense sp. nov., isolated from effective nodules of peanut in Southeast China. Syst. Appl. Microbiol. 42:126002. doi: 10.1016/j.syapm.2019.126002
Li, Y. H., Wang, R., Zhang, X. X., Young, J. P. W., Wang, E. T., Sui, X. H., et al. (2015). Bradyrhizobium guangdongense sp. nov. and Bradyrhizobium guangxiense sp. nov., isolated from effective nodules of peanut. Int. J. Syst. Evol. Microbiol. 65, 4655–4661. doi: 10.1099/ijsem.0.000629
MacLean, A. M., Finan, T. M., and Sadowsky, M. J. (2007). Genomes of the symbiotic nitrogen-fixing bacteria of legumes. Plant Physiol. 144, 615–622. doi: 10.1104/pp.107.101634
Madsen, L. H., Jurkiewicz, A., Sullivan, J. T., Heckmann, A. B., Bek, A. S., Ronson, C. W., et al. (2010). The molecular network governing nodule organogenesis and infection in the model legume Lotus japonicus. Nat. Commun. 1:10. doi: 10.1038/ncomms1009
Martínez-García, P. M., Ramos, C., and Rodríguez-Palenzuela, P. (2015). T346Hunter: a novel web-based tool for the prediction of type III, type IV and type VI secretion systems in bacterial genomes. PLoS ONE 10:e0119317. doi: 10.1371/journal.pone.0119317
Martínez-Hidalgo, P., Ramírez-Bahena, M. H., Flores-Félix, J. D., Igual, J. M., Sanjuán, J., León-Barrios, M., et al. (2016). Reclassification of strains MAFF 303099 T and R7A into Mesorhizobium japonicum sp. nov. Int. J. Syst. Evol. Microbiol. 66, 4936–4941. doi: 10.1099/ijsem.0.001448
Masson-Boivin, C., and Sachs, J. L. (2018). Symbiotic nitrogen fixation by rhizobia — the roots of a success story. Curr. Opin. Plant Biol. 44, 7–15. doi: 10.1016/j.pbi.2017.12.001
Mazia, D., Schatten, G., and Sale, W. (1975). Adhesion of cells to surfaces coated with polylysine. J. Cell Biol. 66, 198–200. doi: 10.1083/jcb.66.1.198
Muñoz, V., Ibañez, F., Tonelli, M. L., Valetti, L., Anzuay, M. S., and Fabra, A. (2011). Phenotypic and phylogenetic characterization of native peanut Bradyrhizobium isolates obtained from Cordoba, Argentina. Syst. Appl. Microbiol. 34, 446–452. doi: 10.1016/j.syapm.2011.04.007
Mus, F., Crook, M. B., Garcia, K., Costas, A. G., Geddes, B. A., Kouri, E. D., et al. (2016). Symbiotic nitrogen fixation and the challenges to its extension to nonlegumes. Appl. Environ. Microbiol. 82, 3698–3710. doi: 10.1128/AEM.01055-16
Nigam, S. N., Dwivedi, S. L., Nambiar, P. T. C., Gibbons, R. W., and Dart, P. J. (1985). Combining ability analysis of N2-fixation and related traits in peanut. Peanut Sci. 12, 55–57. doi: 10.3146/pnut.12.2.0001
Okubo, T., Fukushima, S., and Minamisawa, K. (2012a). Evolution of Bradyrhizobium-Aeschynomene mutualism: living testimony of the ancient world or highly evolved state? Plant Cell Physiol. 53, 2000–2007. doi: 10.1093/pcp/pcs150
Okubo, T., Tsukui, T., Maita, H., Okamoto, S., Oshima, K., Fujisawa, T., et al. (2012b). Complete genome sequence of Bradyrhizobium sp. S23321: insights into symbiosis evolution in soil oligotrophs. Microbes Environ. 27, 306–315. doi: 10.1264/jsme2.me11321
Oldroyd, G. E. D. (2013). Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 11, 252–263. doi: 10.1038/nrmicro2990
Oldroyd, G. E. D., and Downie, J. A. (2008). Coordinating nodule morphogenesis with rhizobial infection in legumes. Annu. Rev. Plant Biol. 59, 519–546. doi: 10.1146/annurev.arplant.59.032607.092839
Oldroyd, G. E. D., Murray, J. D., Poole, P. S., and Downie, J. A. (2011). The rules of engagement in the legume-rhizobial symbiosis. Annu. Rev. Genet. 45, 119–144. doi: 10.1146/annurev-genet-110410-132549
Ormeño-Orrillo, E., Servín-Garcidueñas, L. E., Rogel, M. A., González, V., Peralta, H., Mora, J., et al. (2015). Taxonomy of rhizobia and agrobacteria from the Rhizobiaceae family in light of genomics. Syst. Appl. Microbiol. 38, 287–291. doi: 10.1016/j.syapm.2014.12.002
Parte, A. C. (2014). LPSN - list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 42, 613–616. doi: 10.1093/nar/gkt1111
Peix, A., Ramírez-bahena, M. H., Velázquez, E., Eulogio, J., Peix, A., Ram, M. H., et al. (2015). Bacterial associations with legumes. CRC Crit. Rev. Plant Sci. 34, 17–42. doi: 10.1080/07352689.2014.897899
Poggio, S., Abreu-Goodger, C., Fabela, S., Osorio, A., Dreyfus, G., Vinuesa, P., et al. (2007). A complete set of flagellar genes acquired by horizontal transfer coexists with the endogenous flagellar system in Rhodobacter sphaeroides. J. Bacteriol. 189, 3208–3216. doi: 10.1128/JB.01681-06
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Rodriguez, -R. L. M, and Konstantinidis, K. T. (2016). The enveomics collection : a toolbox for specialized analyses of microbial genomes and metagenomes. Peer J Prepr. 4:e1900v1. doi: 10.7287/peerj.preprints.1900
Rogel, M. A., and Moral, A. Z. (2016). Complete genome sequence of Bradyrhizobium sp. strain CCGE-LA001, isolated from field nodules of the enigmatic wild bean Phaseolus microcarpus. Genome 4:e00126-16. doi: 10.1128/genomeA.00126-16
Rubio, L. M., and Ludden, P. W. (2008). Biosynthesis of the iron-molybdenum cofactor of nitrogenase. Annu. Rev. Microbiol. 62:93–111. doi: 10.1146/annurev.micro.62.081307.162737
Scholla, M. H., and Elkan, G. H. (2009). Rhizobium fredii sp. nov., a fast-growing species that effectively nodulates soybeans. Int. J. Syst. Bacteriol. 34, 484–486. doi: 10.1099/00207713-34-4-484
Simon, H. M., Homer, M. J., and Roberts, G. P. (1996). Perturbation of nifT expression in Klebsiella pneumoniae has limited effect on nitrogen fixation. J. Bacteriol. 178, 2975–2977. doi: 10.1128/jb.178.10.2975-2977.1996
Sprent, J. I. (2007). Evolving ideas of legume evolution and diversity : a taxonomic perspective on the occurrence of nodulation. New Phytol. 174, 11–25. doi: 10.1111/j.1469-8137.2007.02015.x
Stacey, G. (1995). Bradyrhizobium japonicum nodulation genetics. FEMS Microbiol. Lett. 127, 1–9. doi: 10.1111/j.1574-6968.1995.tb07441.x
Stacey, G., Sanjuan, J., Luka, S., Dockendorff, T., and Carlson, R. W. (1995). Signal exchange in the Bradyrhizobium-soybean symbiosis. Soil Biol. Biochem. 27, 473–483. doi: 10.1016/0038-0717(95)98622-U
Staehelin, C., and Krishnan, H. B. (2015). Nodulation outer proteins: double-edged swords of symbiotic rhizobia. Biochem. J. 470, 263–274. doi: 10.1042/BJ20150518
Sullivan, J. T., and Ronson, C. W. (1998). Evolution of rhizobia by acquisition of a 500-kb symbiosis island that integrates into a phe-tRNA gene. Proc. Natl. Acad. Sci. U.S.A. 95, 5145–5149. doi: 10.1073/pnas.95.9.5145
Taurian, T., Fabra, A., and Aguilar, O. M. (2002). Isolation of acid tolerant peanut (Arachis hypogaea L.) rhizobia from soils of central Argentina. Nitrogen Fixat. Mol. Crop Product. 282:491. doi: 10.1007/0-306-47615-0_276
Taurian, T., Ibañez, F., Fabra, A., and Aguilar, O. M. (2006). Genetic diversity of rhizobia nodulating Arachis hypogaea L. in central Argentinean soils. Plant Soil 282, 41–52. doi: 10.1007/s11104-005-5314-5
Urtz, B. E., and Elkan, G. H. (1996). Genetic diversity among Bradyrhizobium isolates that effectively nodulate peanut (Arachis hypogaea). Can. J. Microbiol. 42, 1121–1130. doi: 10.1139/m96-144
Vallaeys, T., Courde, L., Gowan, C. M., Wright, A. D., and Fulthorpe, R. R. (1999). Phylogenetic analyses indicate independent recruitment of diverse gene cassettes during assemblage of the 2, 4-D catabolic pathway. FEMS Microbiol. Ecol. 28, 373–382. doi: 10.1111/j.1574-6941.1999.tb00591.x
Vincent, J. M. (1970). A Manual for the Practical Study of Root-Nodule Bacteria International Biological Programme. Oxford: Blackwell Scientific.
Weisburg, W., Barns, S., Pelletier, D., and Lane, D. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703. doi: 10.1128/jb.173.2.697-703.1991
Wynne, J. C., Elkan, G. H., Isleib, T. G., and Schneeweis, T. J. (1983). Effect of host plant, Rhizobium strain and host x strain interaction on symbiotic variability in peanut. Peanut Sci. 10, 110–114. doi: 10.3146/i0095-3679-10-2-18
Keywords: nodulation, peanut, Bradyrhizobium, genome, crack-entry
Citation: Paudel D, Liu F, Wang L, Crook M, Maya S, Peng Z, Kelley K, Ané J-M and Wang J (2020) Isolation, Characterization, and Complete Genome Sequence of a Bradyrhizobium Strain Lb8 From Nodules of Peanut Utilizing Crack Entry Infection. Front. Microbiol. 11:93. doi: 10.3389/fmicb.2020.00093
Received: 05 August 2019; Accepted: 16 January 2020;
Published: 07 February 2020.
Edited by:
Paula García-Fraile, Academy of Sciences of the Czech Republic (ASCR), CzechiaReviewed by:
Entao Wang, National Polytechnic Institute, MexicoMichael Göttfert, Dresden University of Technology, Germany
Copyright © 2020 Paudel, Liu, Wang, Crook, Maya, Peng, Kelley, Ané and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianping Wang, d2FuZ2pwQHVmbC5lZHU=