 Zhenjun Li
Zhenjun Li Xu-ming Wang2†
Xu-ming Wang2† Zhi Guo Liu
Zhi Guo Liu- 1State Key Laboratory for Infectious Disease Prevention and Control, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
- 2Hainan Provincial People’s Hospital, Haikou, China
- 3Sanya People’s Hospital, Sanya, China
- 4Ulanqab Centre for Endemic Disease Prevention and Control, Jining, China
- 5Inner Mongolia Autonomous Region Center for Comprehensive Disease Control and Prevention, Huhhot, China
Brucellosis has been reported in several regions of Hainan Province, but the extent of the disease has not been fully elucidated. Conventional biotyping methods, multiple locus variable number tandem repeats analysis (MLVA), and single-nucleotide polymorphisms (SNPs) from draft genome sequencing were employed to characterize the strains. There were four biovars (Brucella melitensis bv. 1, 2, and 3 and Brucella suis bv. 3) detected, which showed that the biovar diversity of Brucella in Hainan is higher than in other areas of China. Both B. melitensis bv. 3 and B. suis bv. 3 were dominant species and showed epidemiology patterns that were compatible with both southern and northern China. Eight of MLVA-11 genotypes were known (31, 111, 116, 120, 136, 291, 297, and 345), and the remaining seven were novel (HN11-1 to HN11-7); these data showed that Brucella strains in this study had multiple geographic origins and exhibited characteristics of origin and evolution of co-existing imported and Hainan specific lineage. A total of 41 strains were found, belonging to 37 unique genotypes that each represented a single strain, which suggests that these strains were not directly related epidemiologically and indicates that the epidemic characteristics of human brucellosis in Hainan was dominated by sporadic strains. The high HGDI values were observed in MLVA-8, MLVA-11, and MLVA-16 among two species, suggesting considerable genetic diversity among these species. MST is characterized based on MLVA-16 that was found both throughout China and on a global level and showed that strains of this study had significant genetic differences with strains from many parts of the globe and seemingly represent a unique genetic lineage. Whole-genome SNP analysis showed that four B. melitensis were closely related to strains from China’s northern provinces, and the source of infection was partly of human brucellosis in this province that may have been from these regions. The B. suis were closely related to strains from the United States, and further investigation of the transportation of animals, such as pigs, is needed to elucidate the origins of these strains.
Introduction
Globally, brucellosis is a zoonosis affecting a wide range of domestic and wild animals and humans (Marvi et al., 2018). Due to its complex nature, brucellosis remains a serious threat to public health and livestock in developing countries (Khan and Zahoor, 2018). The genus Brucella has traditionally been divided into six species based on host preference, B. abortus, cattle; B. melitensis, sheep and goats; B. suis, pigs; B. canis, dogs; B. ovis, sheep; and B. neotomae, wood desert rats (De Figueiredo et al., 2015). Recent isolates from humans (B. inopinata), aquatic mammals (B. pinnipedialis and B. ceti), and a common vole (B. microti) have been recognized as new Brucella species (De Figueiredo et al., 2015). At present, human brucellosis cases had been reported in almost all China provinces since 2010 (Lai et al., 2017). In Hainan Province, which is located at the southernmost part of China, there were no historically reported brucellosis cases, and only one human brucellosis case was reported in Danzhou in 1985, brucellosis cases significantly increased in 2017 between humans (Wang et al., 2019), but the available information on the extent of the disease and relatedness among strains is unknown. Characterizing the circulating strains is a critical sector in understanding brucellosis in the epidemic area (Ledwaba et al., 2019). Accurate discrimination relatedness among strains with the MLVA assay is necessary to determine the source, origin, and geographical spread of infection (Whatmore et al., 2016). Moreover, the characterization of the genome of Brucella species with WGS provides excellent genetic resolution and resolve intraspecies relationships for closely related species (Wattam et al., 2014; Abou Zaki et al., 2017). Presently, whole-genome data from many species of Brucella spp. strains have been published and are available in NCBI (Delvecchio et al., 2002; Pisarenko et al., 2018). The purpose of this study is to analyze these Brucella strains using bio-typing and to arrange the molecular scheme including MLVA and WGS to elucidate prevalence characteristics and relatedness among strains for epidemiological purposes, and to provide useful information for the control and prevention of brucellosis in Hainan Province.
Materials and Methods
Ethics Statement
This research was conducted according to the principles of the Declaration of Helsinki. The study is a retrospective investigation of historical strain collections using molecular typing methods, and the study protocol was approved by the Ethics Committees of the National Institute for Communicable Disease Control and Prevention. Informed consent was obtained from all of the patients prior to diagnosis and all patient data were anonymized. Brucella spp. were isolated from patients’ blood samples and were used for diagnosis of disease and following the confirmation of their consent.
Bacterial Strains
A total of 41 strains were examined in this study. These strains were collected from nine counties in Hainan Province from 2009 to 2019. A total of 41 strains were recovered from 41 patients: 39 of them were collected from the blood of patients, and the other two strains were recovered from cerebrospinal fluid and vertebral puncture fluid. The isolates and biovar of the strains were identified using standard procedures (Alton et al., 1975). All isolates were identified as Brucella species on the basis of morphology and conventional identification methods according to standard biotyping procedures, including the need for CO2 for growth, H2S production, sensitivity to thionin (10 and 20 μg/ml), basic fuchsin (20 μg/ml), and agglutination with mono-specific antiserum for A and M antigens and phage lysis test [Tbilisi (Tb); Berkeley (Bk2); Weybridge (Wb)] (Al Dahouk et al., 2003; OIE, 2018). Reagents and pages were obtained from the China Institute of Veterinary Drug Control, which is China’s National Reference Laboratory for Animal Brucellosis. B. melitensis 16M (BM), B. abortus 544 (BA), and B. suis 1330 (BS) reference strains were used as experimental controls. The DNA of the strains was extracted with a Nucleic Acid Automatic Extraction System (LLXBIO China, Ltd., China) using a single loop of fresh Brucella cells that were grown for 48 h according to the manufacturer’s instructions.
Brucella MLVA-16 Genotyping Scheme
MLVA-16 was performed as previously described (Liu et al., 2017). The 16 primer pairs were divided into three groups: Panel 1 (MLVA-8: eight loci including bruce06, bruce08, bruce11, bruce12, bruce42, bruce43, bruce45, and bruce55), panel 2A (three loci including bruce18, bruce19, and bruce21), and panel 2B (five loci including bruce04, bruce07, bruce09, bruce16, and bruce30); MLVA-11 (panels 1 and 2A), and MLVA-16 (panels 1, 2A, and 2B). PCR amplifications were performed in 20-μl reaction volumes. A total of 5 μl of PCR products for the 16 loci were denatured and resolved by capillary electrophoresis on an ABI Prism 3130 automated fluorescent capillary DNA sequencer (Applied Biosystems). Fragments were sized after comparison to a ROX (carboxy-X-rhodamine)-labeled molecular ladder (MapMaker 1000; Bioventures, Inc., Murfreesboro, TN, United States) and Gene Mapper software version 4.0 (Applied Biosystems). The fragment sizes were subsequently converted to repeat unit numbers using a published allele numbering system (Scholz and Vergnaud, 2013).
Analysis of the MLVA Data
BioNumerics version 7.6 software (Applied Maths, Belgium) was used to analyze the MLVA-16 assay data. Both the categorical coefficient and the unweighted pair group methods were applied to the clustering analysis. MLVA-11 was used to investigate the geographical origins between our isolates (De Massis et al., 2015; Liu et al., 2019) (Supplementary Table S1) and 528 isolates from the MLVA bank including the non-available group (2), the Africa group (4), the America group (75), the West Mediterranean group (77), the China group (97), and the East Mediterranean group (273) (Supplementary Table S2). Minimum-spanning trees were constructed using the goeBURST algorithm with PHYOVIZ 2.0 (Nascimento et al., 2016). The MLVA-16 approach was applied to the genetically related investigation between strains from those reported in China (n = 367) (Supplementary Table S3) and at the global level (n = 2687; 2124 in B. melitensis and 563 in B. suis) (Supplementary Tables S4_1, S4_2). The genetic diversity of Brucella strains in this study was calculated based on the Hunter–Gaston Diversity Index (HGDI) according to a previously published method (Hunter, 1990).
Single-Nucleotide Polymorphism (SNP) Analyses Based on the Draft Genome Sequence
A total of 21 Brucella strains (15 in B. melitensis and six in B. suis) were selected for whole-genome draft sequencing, since these strains were isolated from recent years and represented the predominant species in this region. Genomic DNA of these strains were extracted with the SDS method (Lim et al., 2016). The harvested DNA was detected using agarose gel electrophoresis and quantified by Qubit® 2.0 Fluorometer (Thermo Scientific). Sequencing libraries were generated using NEBNext® UltraTM DNA Library Prep Kit for Illumina (NEB, United States) in accordance with the manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. The draft genome of 21 Brucella strains was sequenced using Illumina NovaSeq PE150 at the Beijing Novogene Bioinformatics Technology, Co., Ltd. Illumina PCR adapter reads and low-quality reads from the paired end were filtered for quality control using readfq (version 10). All good-quality paired reads were assembled using the SOAP de novo (Li et al., 2008, 2010),1 SPAdes (Bankevich et al., 2012),2 and ABySS (Simpson et al., 2009)3 into a number of scaffolds. Filtered reads were then subjected to gap-closing. Draft genomic alignment between the sample genome and reference genome [B. melitensis 16M (Assembly ID: GCA_000007125.1) and B. suis 1330 (Assembly ID: GCA_000007505.1)] were performed using the MUMmer (Kurtz et al., 2004) and LASTZ (Chiaromonte et al., 2002) tools. Whole-genome SNPs were found using the results of genomic alignment among samples by the MUMmer and LASTZ, as mentioned above. In addition, 132 B. melitensis and 31 B. suis genomes were retrieved from GenBank and used for comparison and preliminary phylogenetic analyses. The phylogenetic tree was constructed using the TreeBeST (Vilella et al., 2009) based on Maximum-Likelihood Phylogenies (PHYML) with 1000 bootstrap replicates. Subsequently, the B. melitensis strains present discernible genetic differences in the SNP phylogenetic tree were removed; then, a simple phylogenetic tree was reconstructed based on 34 B. melitensis strains (Supplementary Table S5).
Results
Identification and Distribution of Brucella Strains
All of the 41 Brucella strains were identified as B. melitensis bv. 1 (n = 5), B. melitensis bv. 2 (n = 1), B. melitensis bv. 3 (n = 23), and B. suis bv. 3 (n = 12) using classical biotyping methods (Table 1). From 2009 to 2019, a total of 41 Brucella strains were isolated. The number of strains isolated included 4 strains in 2009, 2 strains in 2010, 4 strains in 2012, 11 strains in 2013, 5 strains in 2014, 1 strain in 2015, 2 strains in 2016, 6 strains in 2017, 4 strains in 2018, and 2 strains in 2019. The 41 Brucella strains were observed in nine counties in Hainan Province, including Ledong (n = 1), Lingshui (n = 1), Wanning (n = 2), Chengmai (n = 2), Dingan (n = 3), Sanya (n = 3), LinGao (n = 7), Haikou (n = 8), and Dongfang (n = 13), and the original locations associated with the other strains are unknown (Figure 1).
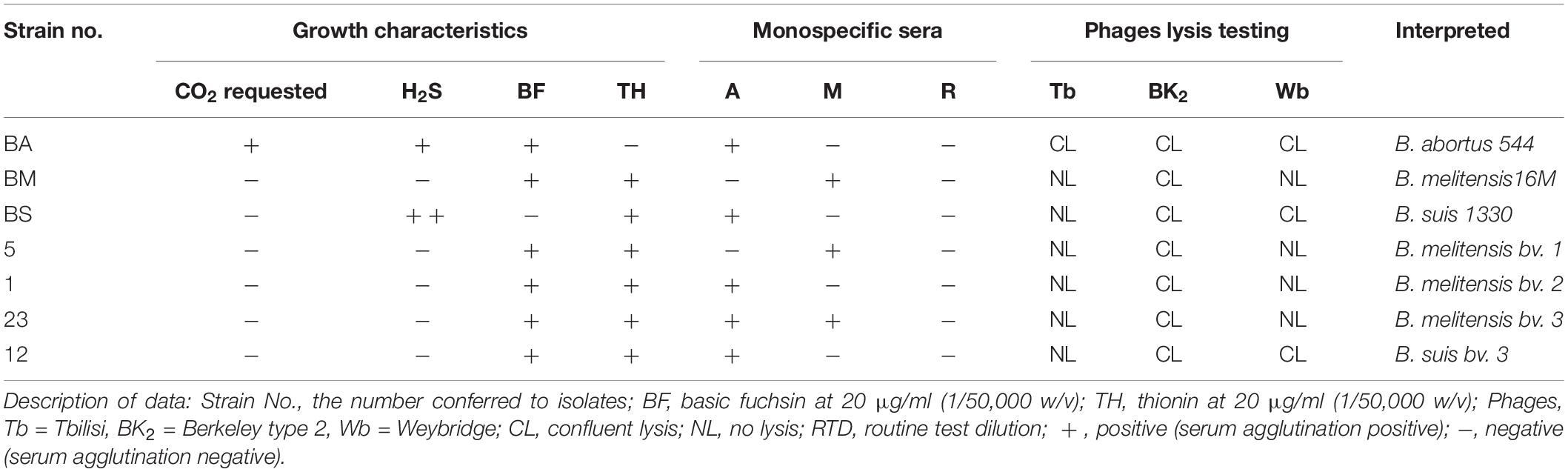
Table 1. Biotyping characteristics of Brucella species isolates in Hainan Province, China.
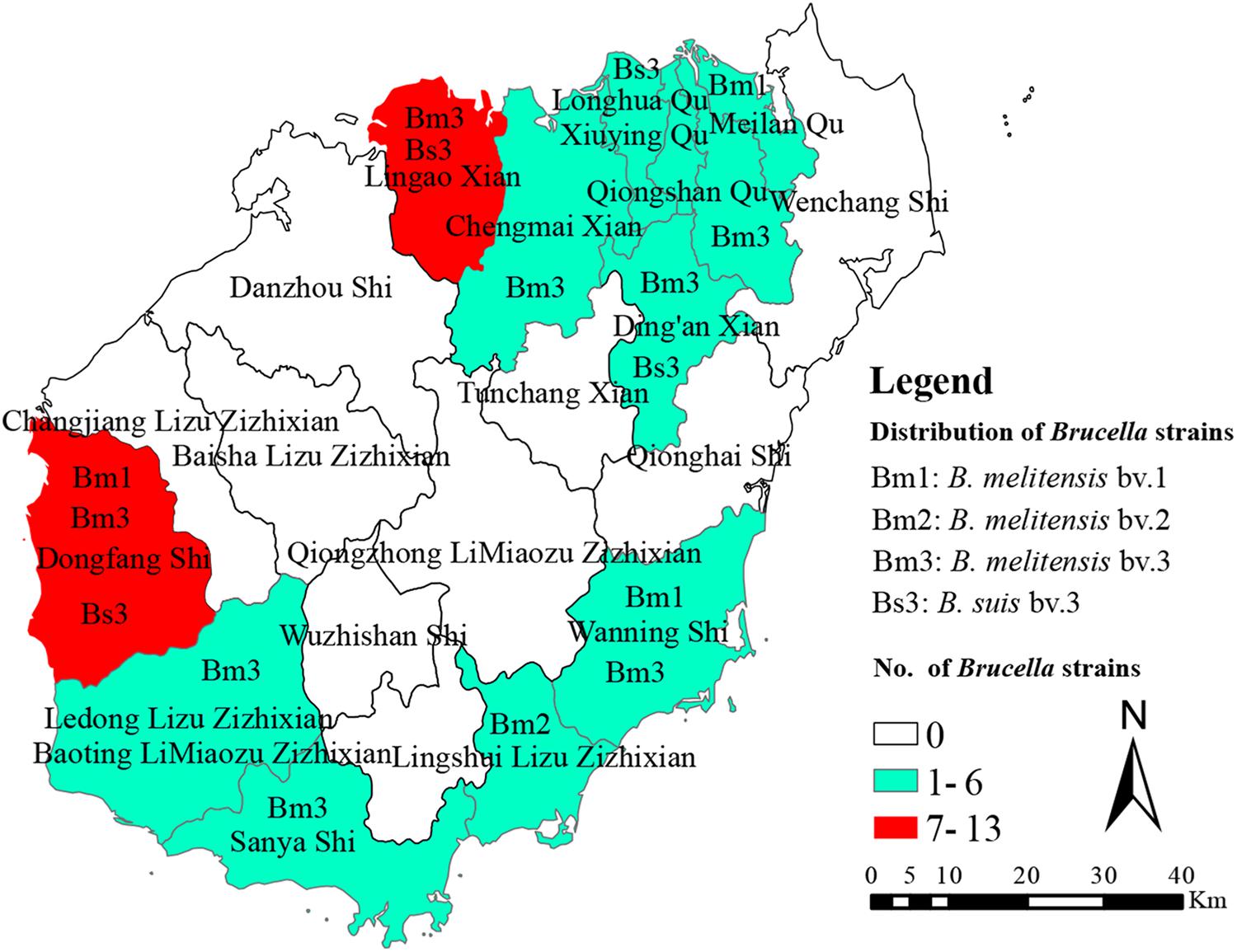
Figure 1. The geographic distribution of Brucella samples in Hainan, China. The map of Hainan province in this study was download from open source map (https://commons.wikimedia.org/wiki/Atlas_of_the_world).
MLVA-16 Genotyping Results
Based on the complete MLVA-16 scheme, 41 strains were divided into two groups (I and II). Twenty-nine B. melitensis strains clustered in group I, and all B. suis strains clustered in group II (Figure 2). A total of 29 B. melitensis isolates clustered into 27 distinct MLVA-16 genotypes (GT1-27), and 25 of them were represented by singular independent strains. The other two genotypes (GT12 and GT26) were shared by two isolates obtained from the same family (Figure 2). Following analysis of the data pertaining to MLVA-8, 29 B. melitensis strains formed nine MLVA-8 genotypes, including six known genotypes [83 (n = 1), 114 (n = 1), 47 (n = 2), 63 (n = 4), 58 (n = 9), and 42 (n = 9)] and three novel genotypes: HN8-1 (2-3-4-11-2-2-3-2; n = 1), HN8-2 (2-2-4-10-2-2-3-2, n = 1), HN8-5 (3-4-2-12-2-2-3-2, n = 1). The phylogeographic relationships of the 29 Hainan Brucella isolates were assessed with MLVA-11, and 11 different genotypes were identified (Figure 3). Seven of these genotypes were previously described (111, 116, 120, 136, 291, 297, and 345) and the remaining four novel (HN11-1 to HN11-3 and HN11-7) genotypes represent one to two strains with single locus variants of these genotypes. Genotypes 120 and 116 were the predominant genotypes representing 55% (16/29) of the total isolates; the two genotypes were distributed in six areas of Hainan Province. Minimum spanning tree analysis revealed that the MLVA-11 genotypes of B. melitensis strains were clustered into the “East Mediterranean” and “America” groups and strains from the former were dominant (Figure 3). In B. melitensis strains, the HGDI value of seven loci was >0.5, and the HGDI value of Panel 1, MLVA-11, and MLVA-16 was 0.8054, 0.8424, and 0.9951, respectively (Table 2).
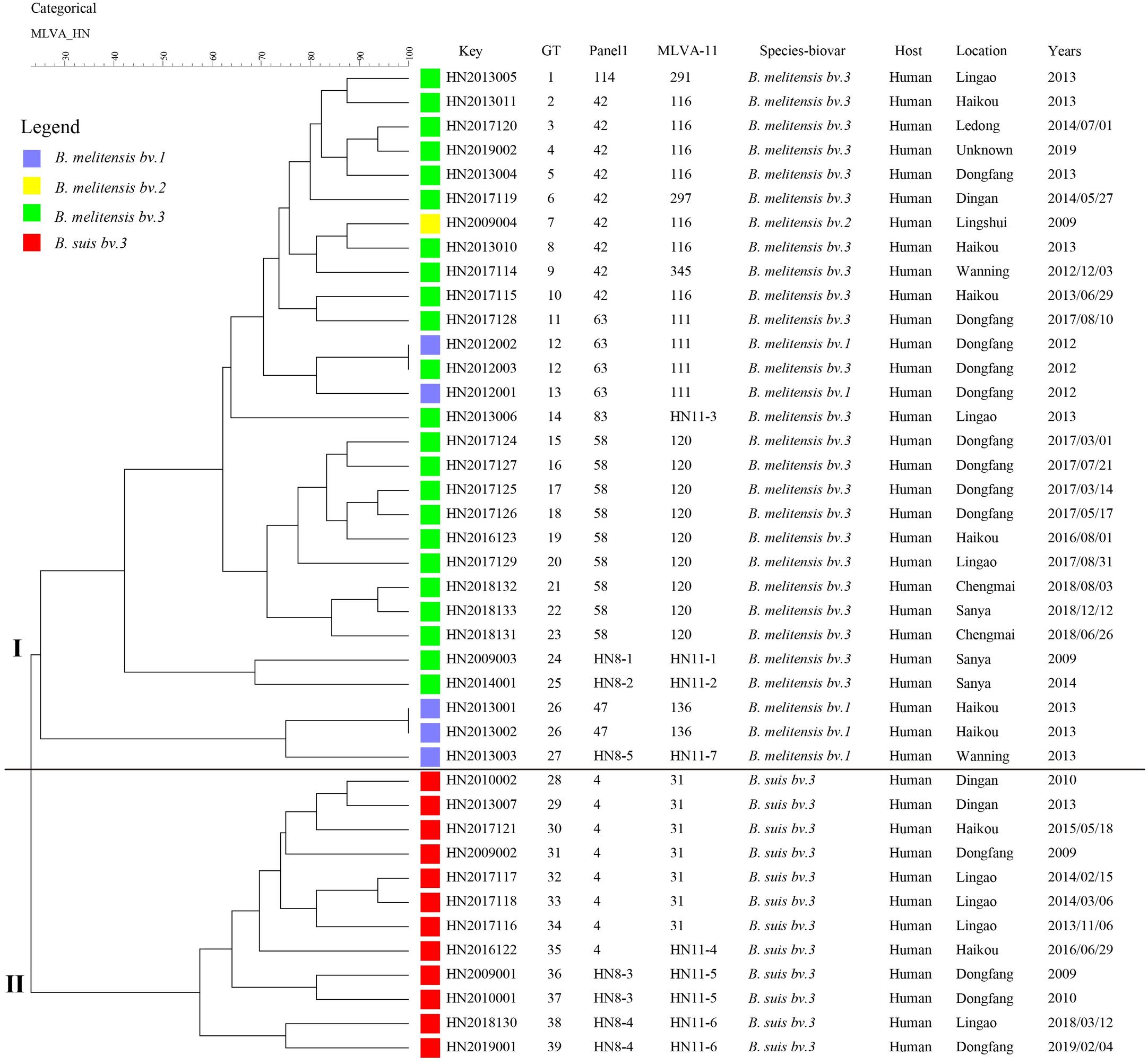
Figure 2. A dendrogram based on the MLVA-16 genotyping assay (UPGMA method) showing the relationships between the 41 Brucella isolates. The columns show the identification numbers (Key), MLVA-16 genotypes (GT), panel 1 genotypes and MLVA-11 (panels 1 and 2A) genotypes, species biovar, host, location, and the year the strains were isolated.
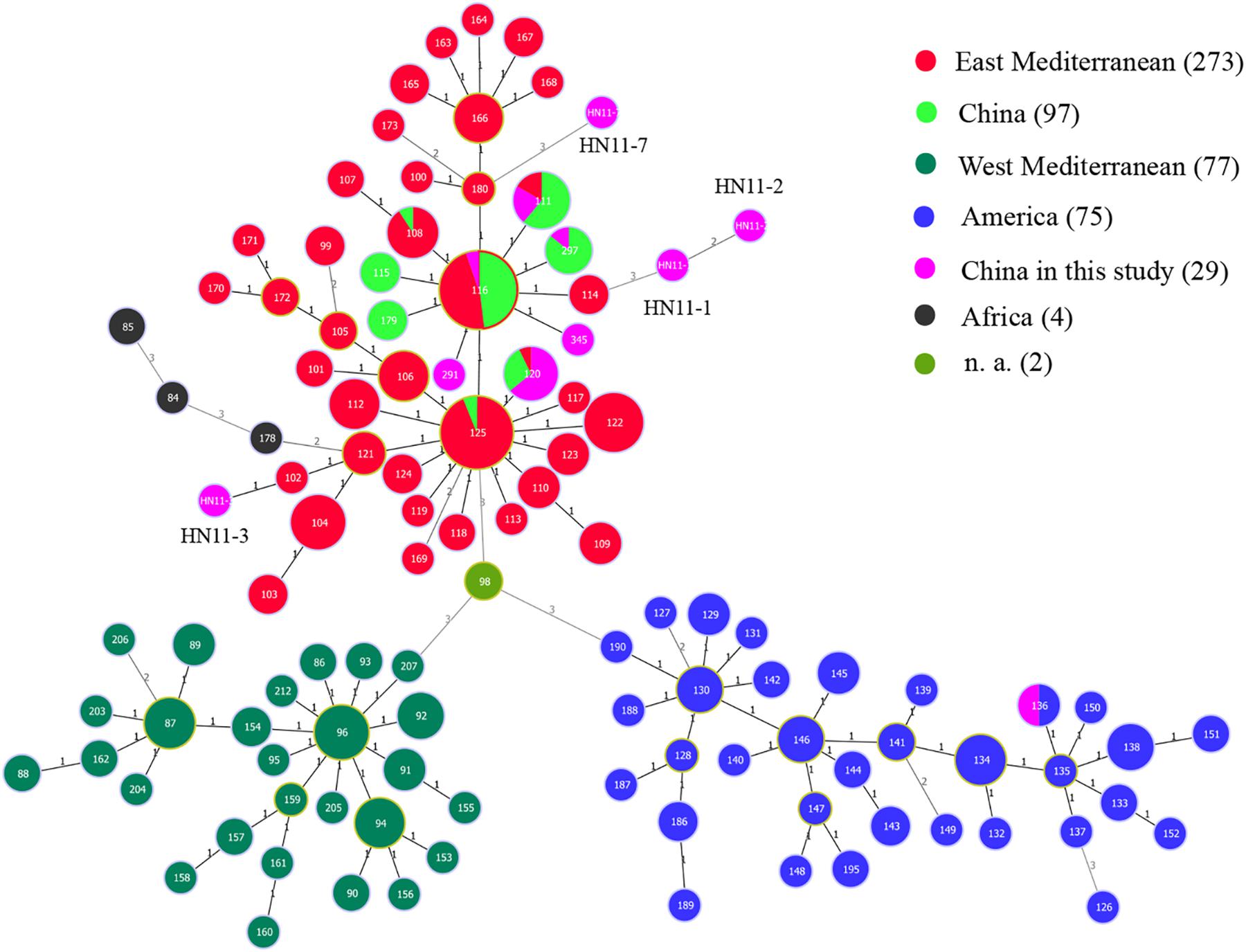
Figure 3. A MST for B. melitensis using the MLVA-11 data with the East Mediterranean group (red), the America group (blue), the Africa group (black), and the West Mediterranean group (blackish green) and compared to the China genotypes (green) and China isolates from our study (pink). n. a., not available (light green).
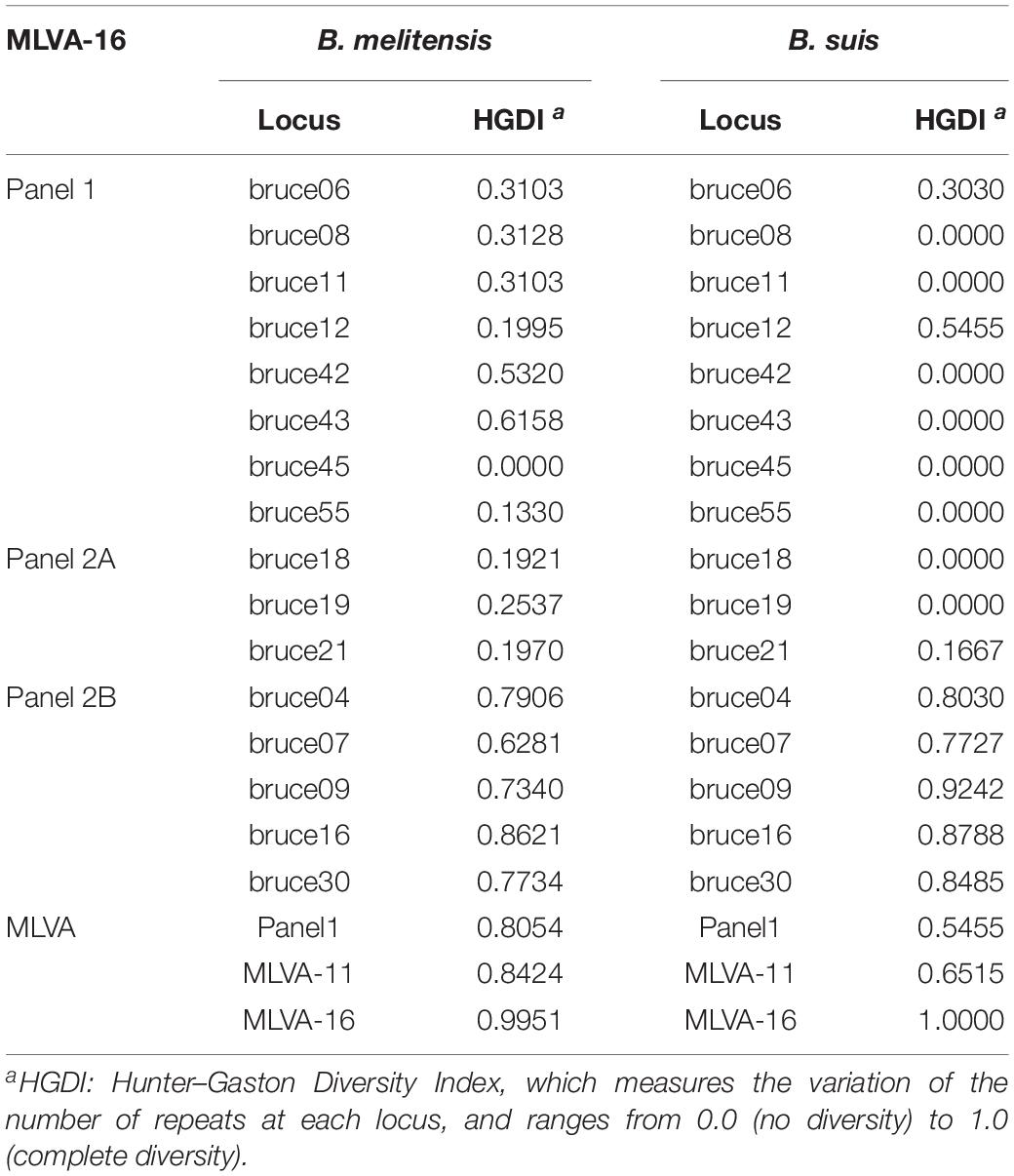
Table 2. HGDI values of 29 B. melitensis isolates and 12 B. suis isolates.
Among 12 B. suis strains, 12 single MLVA-16 genotypes (GT28-39) were observed (Figure 2). These strains were sorted into three MLVA-8 genotypes, 4 (n = 8), HN8-3 (2-3-4-12-3-1-5-2, n = 2), and HN8-4 (1-3-4-13-3-1-5-2, n = 2), and formed four MLVA-11 genotypes, one genotype was previously described (31) and the remaining three were novel (HN11-4 to HN11-6). MLVA-11 genotypes of B. suis belong to the genotype 31 clone group (Figure 4). In B. suis strains, all Panel 2B loci were > 0.77, 0.3030 in bruce06, 0.5455 in bruce12, and 0.1667 in bruce21. The other eight loci showed no diversity: the HGDI values of Panel 1, MLVA-11, and MLVA-16 were 0.5455, 0.6515, and 1.0000, respectively (Table 2). A dendrogram of the 41 Brucella strains shows the strain identification features, the MLVA-8 genotype, panel 1 + panel 2A genotype, their geographical origins, and the year of isolation (Figure 2).
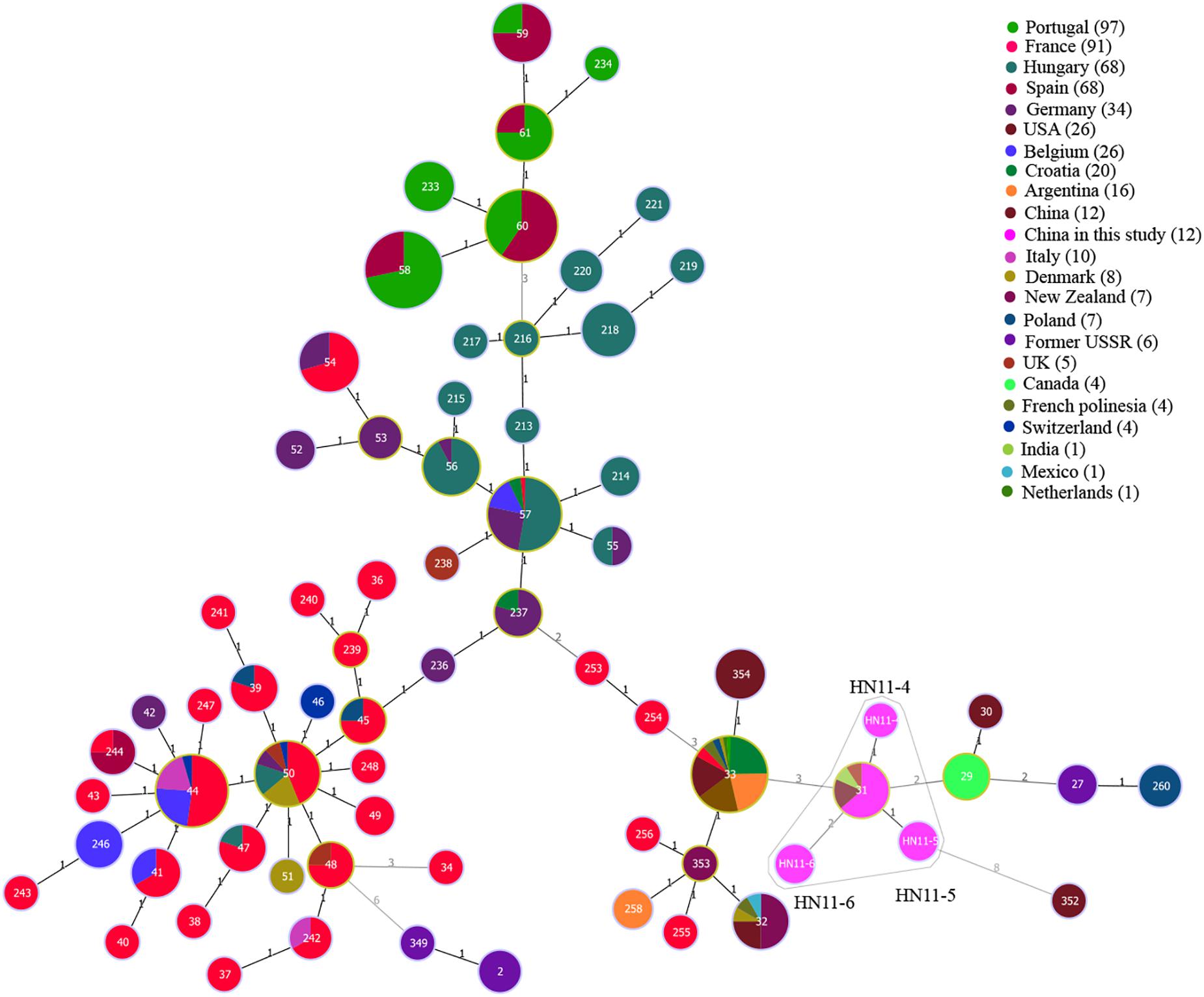
Figure 4. A MST for B. suis using the MLVA-11 data with the Portugal isolates (light green), France isolates (red), Hungary isolates (dark green), Spain isolates (Danish red), Germany isolates (dark purple), United States isolates (brown red), Belgium isolates (blue), Croatia isolates (emerald-green), Argentina isolates (orange), Italy isolates (dark pink), Denmark isolates (canary yellow), New Zealand isolates (dark Brown), Poland isolates (blue-gray), former USSR isolates (light purple), United Kingdom isolates (light red), Canada isolates (green), French Polynesia isolates (soil orange), Switzerland isolates (dark blue), India isolates (Yellow-green), Mexico isolates (lake blue), Netherlands isolates (green onion), China isolates (brown), and China isolates from our study (pink).
Relevance of Genotyping for Clinical Cases and Trace-Back Analysis of Brucella
GT4 represents single strains obtained from a patient’s blood and his household registry in Anhui Province. There was no contact history with the infected source, and he worked in Guangdong Province. This strain had completely identical MLVA-16 genotypes with strains 2011Jiang#013 and 2011Jiang#017 [MLVAbank (Brucella_4_3)] from Guangdong province. GT22 contained a single strain that was isolated from a sheep farm in Saya, and this strain belonged to MLVA-11 genotype 120, which was the dominant genotype in Hainan Province. There was a similar MLVA-16 genotype with strain 2011Jiang#059 [MLVAbank (Brucella_4_3)] from Yunnan Province, China. Two genotypes (GT32 and GT33) each represent single B. suis strains and were obtained from two patients in the same family. These people raised pigs, and two strains showed a single locus difference in highly variable bruce16 only.
Epidemiological Relationships Between Brucella Strains
Multiple locus VNTR was used to determine the epidemiological links between 41 Brucella strains from Hainan Province and 367 Brucella strains (including B. melitensis and B. suis) from other provinces of China. Two shared genotypes were observed among the B. melitensis strains from this study and two different provinces, that is, Xinjiang and Guangdong (Figure 5). There is no shared MLVA-16 genotype among B. suis strains. Domestic isolates from Hainan Province were compared to foreign strains using an MLVA-16 assay. There were only two identical MLVA-16 genotypes from B. melitensis strains among those observed in this study and the strains from the United States and Argentina had discernible genetic difference from strains from Italy and France (Figure 6). However, B. suis in this study formed terminal subclades with strains from the United States and other parts of China, and it had significant genetic differences from strains from France, Italy, Spain, Germany, and Portugal (Figure 7).
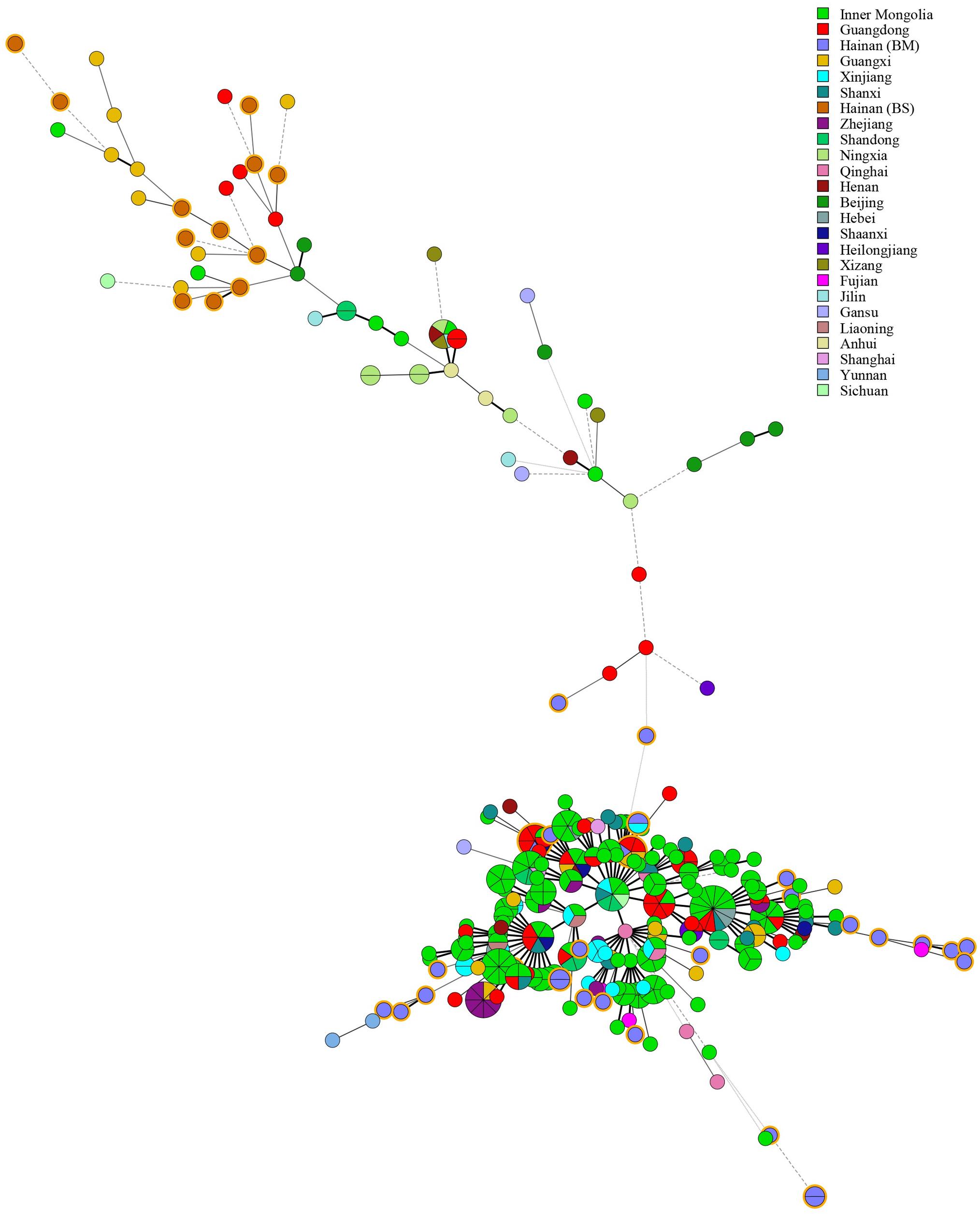
Figure 5. Minimum Spanning Tree characteristics based on Brucella strains throughout China. The tree was constructed using MLVA-16 data from 367 profiles of Brucella available in the MLVA international database. The nodes including isolates from this study are highlighted in yellow.
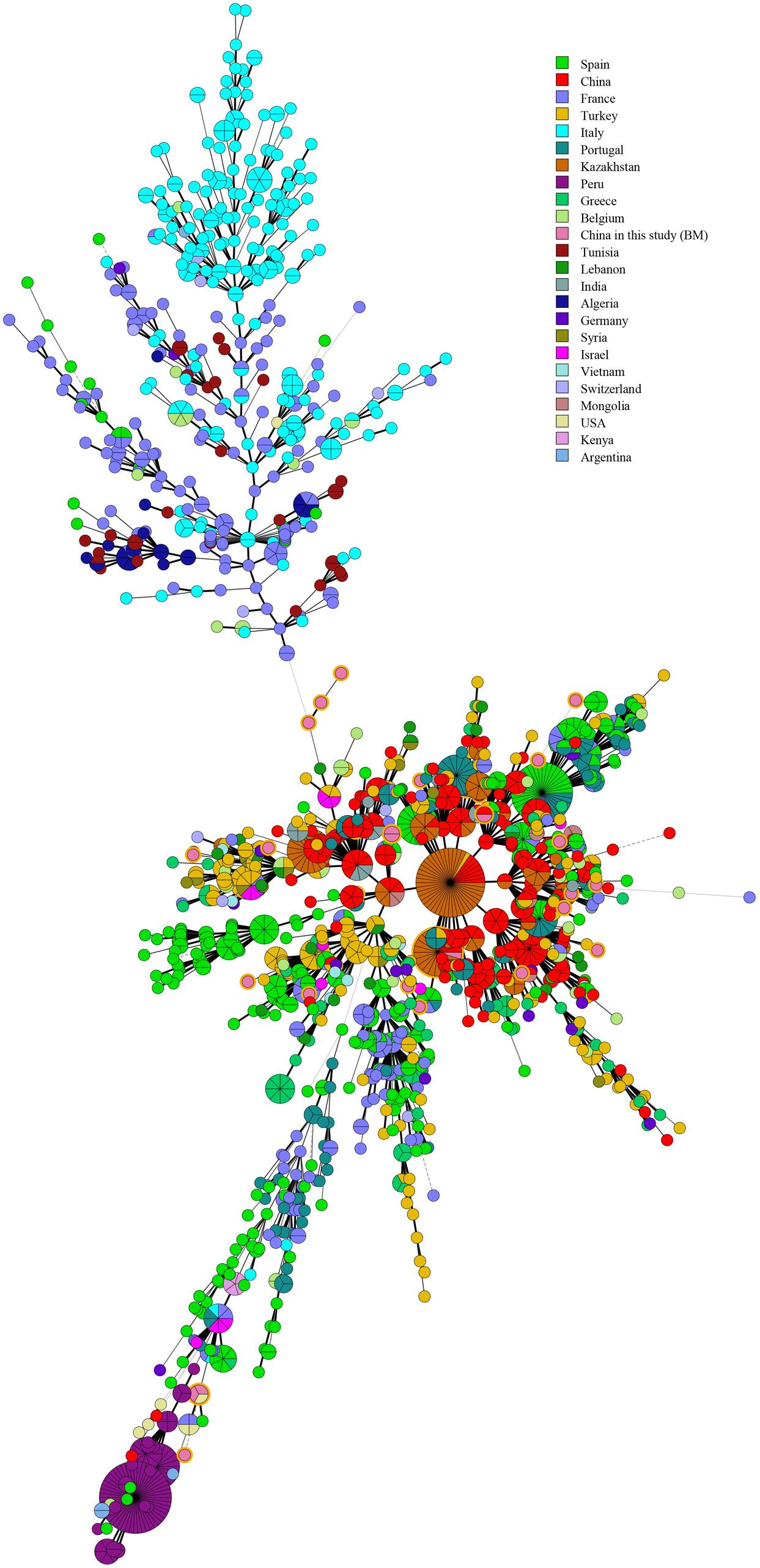
Figure 6. Minimum Spanning Tree characteristics based on global B. melitensis strains.
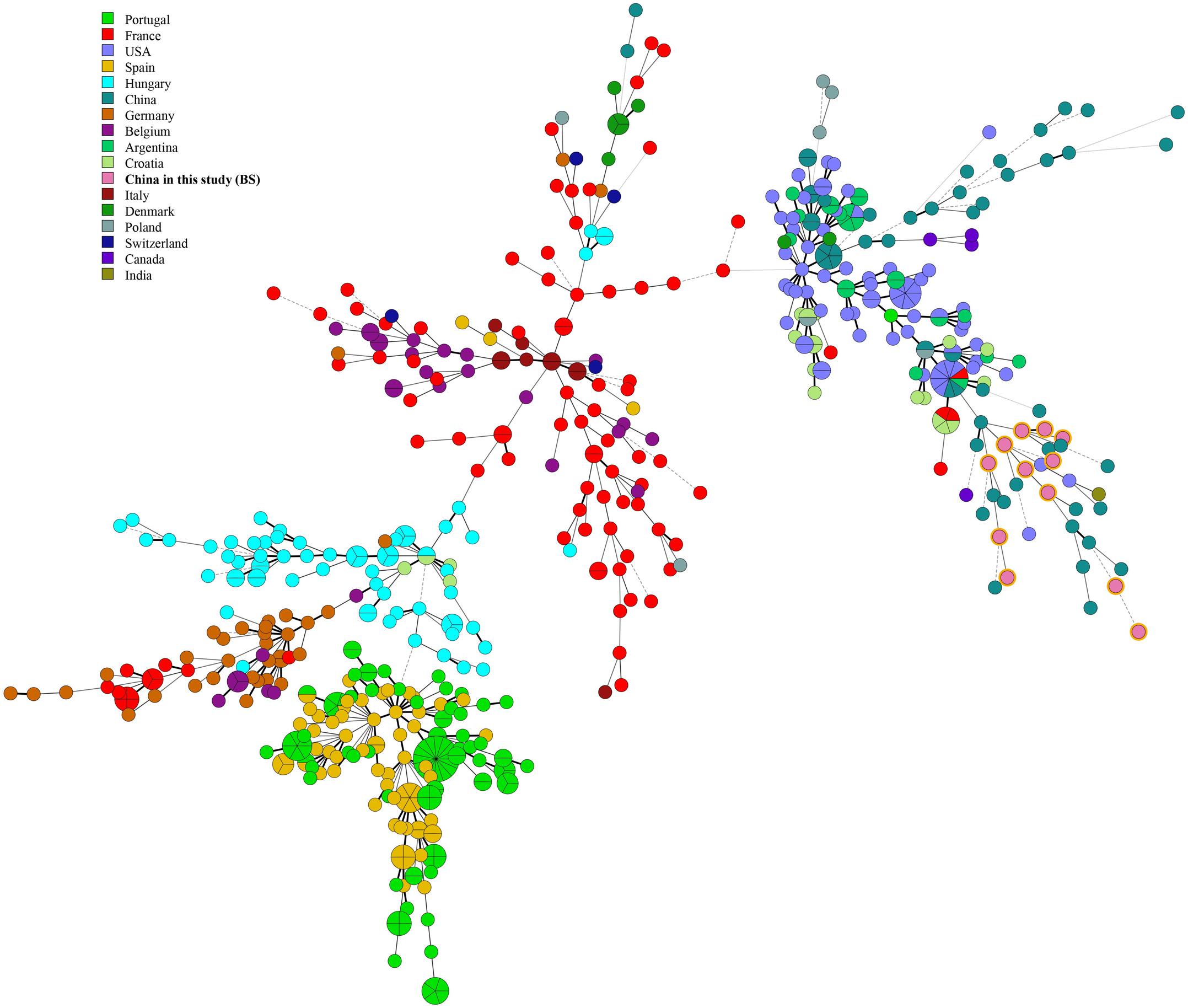
Figure 7. Minimum Spanning Tree characteristics based on global B. suis strains.
Phylogenetic Analysis Based on WGS-SNP
In the present study, the whole-genome draft sequences of 21 isolated strains, including 15 B. melitensis strains and 6 B. suis strains, were found. This was done to determine the genetic and evolutionary characteristics of this population. Briefly, the contigs from 21 strains ranged in length (>500 bp) from 19 to 25, with an average of 23. The average N50 Length (bp) (274,284–462,157) and N90 Length (bp) (102,688–117,359) were 329,690 (bp) and 116,049 (bp); the average GC content of 21 strains was 57.24% (57.18%–57.29%); the range of number of genes in these strains was 3210–3325, the average number of genes was 3276. B. melitensis 16M and B. suis 1330 served as reference genomes for sequent datasets. The SNP numbers of 34 B. melitensis strains and 37 B. suis strains analyzed were 98,262 and 66,166. The whole-genome SNP analysis showed that 15 B. melitensis strains were grouped into two clusters (I and II) (Figure 8), 4 of which in cluster I were closely related with strains from Inner Mongolia, while the remaining 11 strains were divided into cluster II. However, the analyzed B. suis strains were sorted into four groups (I–IV) (Figure 9), strains from this study fell into groups II and IV, and three B. suis from group II were closely related with strains from the United States, while the other three strains in group IV showed an independent branch.
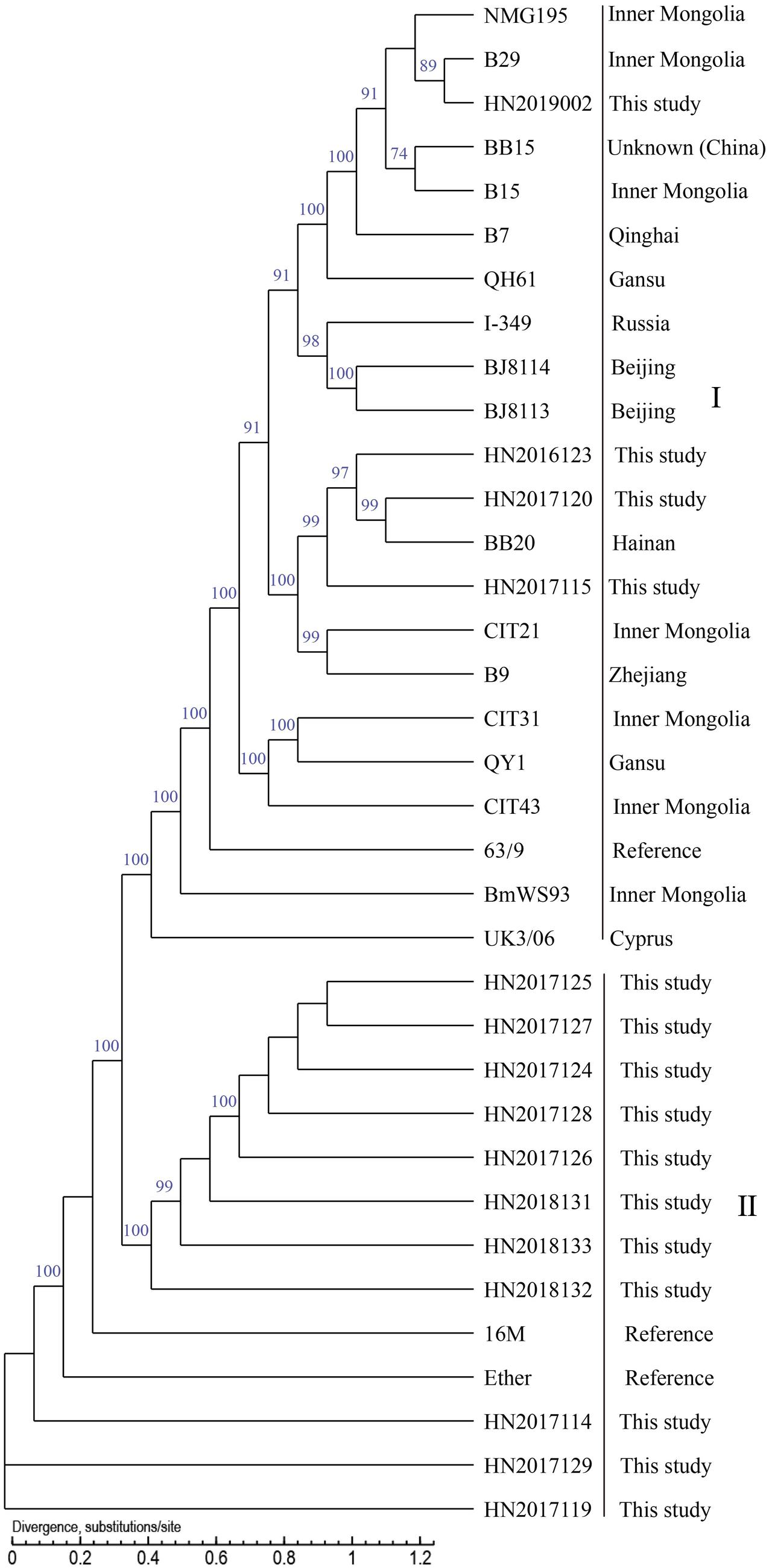
Figure 8. Phylogenetic trees of Brucella melitensis using SNPs from whole genomes.
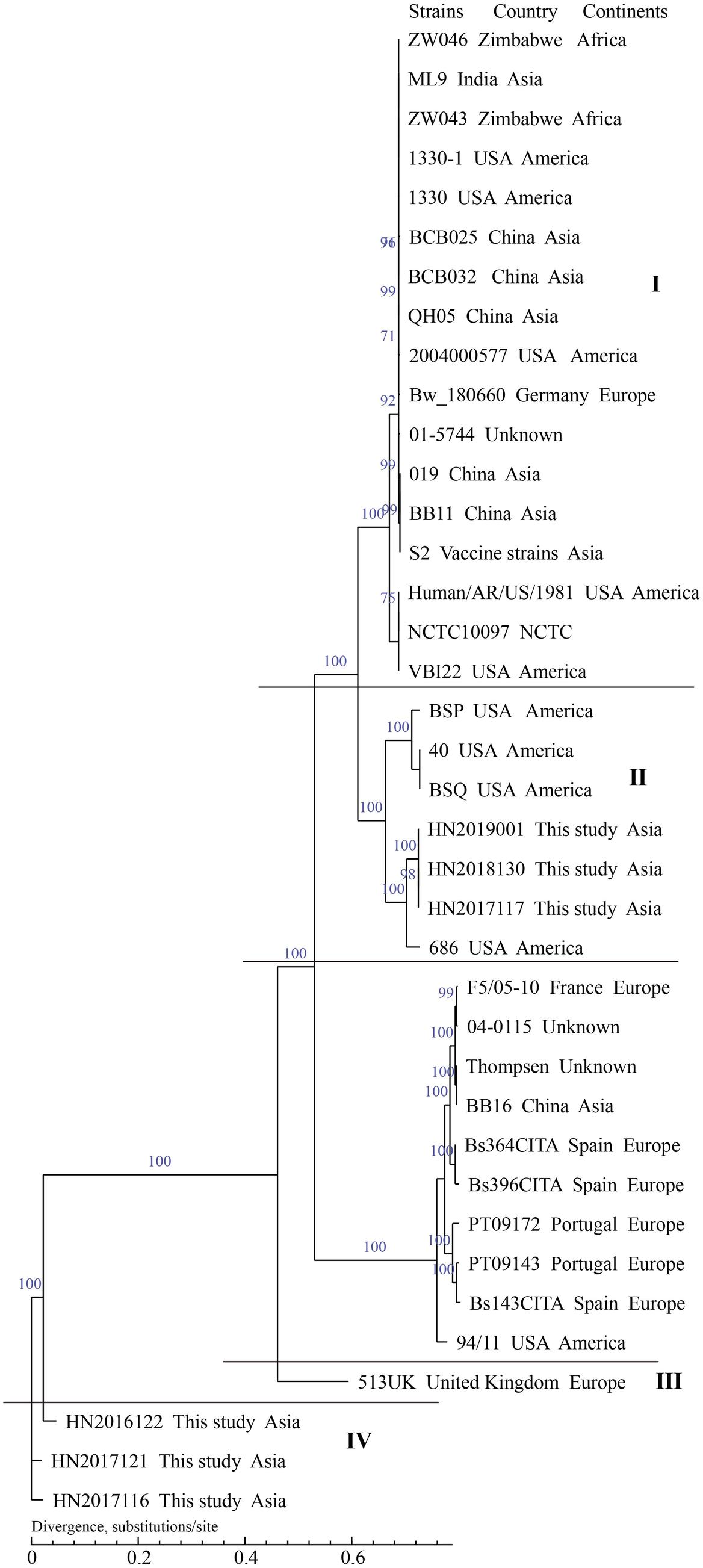
Figure 9. Phylogenetic trees of Brucella suis using SNPs from whole genomes. The dendrograms were generated using maximum likelihood with 1000 bootstrap replicates.
Discussion
Brucellosis remains one of the major risks of public health in China, and human brucellosis has been reported in all 32 mainland provinces (regions) (Chen et al., 2014). Hainan province is located in the southernmost region of the nation and human brucellosis was rare in this region before 2009. The characterization of the geographic distribution and species of circulating strains is a critical process in understanding the epidemiology of brucellosis in the epidemic area (Kang et al., 2017). At least four species/biovars were detected in nine different areas, which suggests that Brucella strains were widely distributed in this province and exhibited high biovar diversity, which may have contributed to the spread and expansion of Brucella strains in this region. B. melitensis bv. 3 (n = 23) and B. suis bv. 3 (n = 12) were dominant species, and these results were different from studies from other major areas of China (Ma et al., 2016; Sun et al., 2016). Previous reports show that B. melitensis were the most common in the northwest and northeast China, but B. suis were often observed in the southern regions (Liu et al., 2018; Piao et al., 2018). This result indicates that brucellosis in Hainan has the unique epidemiology patterns of compatible southern and northern China. Comparative genomic analysis of the representative strains is needed to reveal the origin and phylogenetic profile. Moreover, the detection of brucellosis in animals is essential for the prevention of the disease. We consider that efficient preventive measures should be established to control the disease.
Pertaining to MLVA-8 data, 12 different MLVA-8 genotypes were identified, and 7 of them were previously described and widely distributed in other provinces in China. The remaining five genotypes were novel genotypes, which included three in B. melitensis and two in B. suis. Seven of 12 MLVA-8 genotypes were exclusively found in the Hainan Province, and these results suggest that the epidemiology of brucellosis in this region may be distinctive. Eleven MLVA-11 genotypes were found in strains from 29 B. melitensis isolates, suggesting that strains from this region have multiple geographic origins, but the East Mediterranean group was dominant. MLVA-11 genotype 136 was common in France, the United States, and Spain and belongs in the “America” group (Le Fleche et al., 2006). MLVA-11 genotypes 291, 297, and 345 were distributed in Guangdong, Shandong, Inner Mongolia, and other norther regions; MLVA-11 genotype 120 was predominantly distributed in Yunnan, Guangxi, Fujian, and Turkey. The strains representing this genotype may be imported from the vicinity of Yunnan, Guangxi, and Fujian provinces, which is consistent with the current epidemic features of human brucellosis. In addition, the HGDI value of seven loci in B. melitensis strains was over 0.5, and the HGDI value of Panel 1, MLVA-11, and MLVA-16 were all higher than those of strains from other regions within China (Jiang et al., 2011).
Four MLVA-11 genotypes were observed in 12 B. suis strains, of which the MLVA-11 genotype 31 was the dominant genotype. It was also common in the United States, United Kingdom, and India, indicating that some B. suis strains in this area had a common geographic origin with strains from these nations (Ferreira et al., 2017). Moreover, a total of seven new MLVA-11 genotypes were found in two species, four in B. melitensis strains and three in B. suis strains, and these genotypes may be unique in Hainan province. These data indicate that the Brucella strains in this study had multiple geographic origins, and this population exhibited characteristics of origin and evolution of co-existing imported and Hainan specific lineage. The diversity index of MLVA-16 in B. suis strains was 1.0000, which suggests that there was considerable genetic diversity among the B. suis population.
Based on the MLVA-16 scheme, 90% (25 + 12/41) strains formed unique genotypes and represented as single independent strains, which suggests that these strains had no related epidemiology and the epidemic characteristics of brucellosis in this province were predominated by sporadic occurrences (Liu et al., 2017). There were two shared genotypes (GT12 and GT26) that each consisted of two B. melitensis strains from the same family, and this finding confirmed the occurrence of the brucellosis outbreak events of the family. These two families were all engaged in breeding sheep and had regular contact with sheep every day, which is in agreement with information that was previously reported. Cluster outbreaks of a family of human brucellosis is common in southern China, and contact with infected sheep or its products is one of the main reasons for infection brucellosis (Kong, 2018). HN2019002 (GT4) was obtained from the blood of a patient in Hainan Provincial People’s Hospital and a household was registered to this patient in Anhui Province. During his work at Guangdong Province, the strain had a completely identical MLVA-16 genotype with strains from Guangdong Province, and we considered that this patient was infected with Brucella in Guangdong. HN2018133 (GT22) was isolated from the veterinary sample of a sheep farm in Sanya and showed almost identical genotypes with strains from Yunnan Province, China, and infected sheep imported from Yunnan Province was the source of infection for this case.
Among B. suis strains, both HN2017117 (GT32) and HN2017118 (GT33) strains were obtained from two patients in the same family, and there was only a single locus difference in the bruce16 locus, which suggests that there was a common source of infection among the two cases, but a mutation event occurs in highly variable loci (Kilic et al., 2011).
Based on an epidemiology investigation throughout China, two B. melitensis strains from this region had MLVA-16 genotypes identical to those of strains from Xinjiang and Guangdong Provinces, so these regions are the most likely sources of infection for human brucellosis in Hainan Province (Tian et al., 2017), but no identical MLVA-16 genotypes were found in B. suis species. Subsequent comparison of the relatedness of strains at a global level was performed, and two MLVA-16 genotypes from B. melitensis strains were observed between strains in this report and the United States and Argentina; B. suis strains formed terminal subclades with strains from the United States and other provinces of China, indicating that strains from Hai province had visible genetic differences compared to strains from many parts of the globe.
Whole-genome SNP analysis showed that 15 B. melitensis strains were grouped into two clusters (I and II), which indicated that strains from each group had great genetic differences. The four strains in cluster I were closely related with strains from Inner Mongolia, which suggests that strains from Inner Mongolia were potential sources of infection for human brucellosis in Hainan Province. This result coincides with the conclusion from the MLVA analysis, where a previous study reported that strains from Inner Mongolia had a complete and identical MLVA-16 genotype with strains from many provinces in China (Liu et al., 2017); the other 11 strains were divided into cluster II, which occupied the basal node of the phylogenetic tree. We suggest that a comparison of the genome of B. melitensis strains from many different regions is warranted.
However, the analyzed B. suis strains were sorted into four groups (I–IV): B. suis from this study fell into two groups (II and IV) and three B. suis from group II were closely related to strains from the United States. Further investigation of the movement of animals, especially pigs, should be performed to identify the origins of these strains; the other strains in group IV showed an independent branch and are located in the basal node of the phylogenetic tree and had a previous historical occurrence. We observed that strains in this region exhibited unique characteristics of origin and evolution. Further efforts, examination of more strains, WGS, and collection of epidemiological data from the south are needed to accurately outline the pattern of transmission of brucellosis in Hainan, China.
Moreover, our study has some limitations. First, the data were collected from passive diagnosis that might have been influenced by laboratory tests or the physician’s understanding of the disease. Second, due to variability in the number of strains collected among different counties and for different years in this study, further research with additional strains is essential. Third, our research had no strains from animals in this region, and a genetic comparison in strains from a different host is lacking. Therefore, a whole-province survey on the human and animal infections with Brucella should be initiated.
Conclusion
In the present study, molecular analysis of human Brucella strains from Hainan Province was performed. Our research showed that there was considerable diversity of biovar and genotypes among Brucella strains from Hainan Province. The epidemic characteristics of brucellosis in this region were predominated by sporadic traits, but also confirmed the occurrence of a brucellosis family outbreak. B. melitensis may have been imported from northern China. B. suis strains showed unique characteristics of origin and evolution, and they are of a lineage native to Hainan Province. Our work not only contributes to better understanding of the epidemiology of human brucellosis in Hainan Province, China, but it also provides considerable information that could be used to formulate control strategies for this disease.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committees of the National Institute for Communicable Disease Control and Prevention. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
ZGL performed strain identification, MLVA genotyping and cluster analysis, and drafted the manuscript. XW and MW conducted epidemiological investigations and data analysis. XZ and HC prepared the DNA samples. ZJL and ZGL participated in the design of the study and critically reviewed the manuscript. ZJL and DL participated in the design of the study and managed the project. All authors have read and approved the final version of the manuscript.
Funding
This study was supported by the National Key R&D Program of China (Grant Nos. 2019YFC1200705, 2018ZX10734401, and 2018ZX10734404), the Natural Science Fund of the Inner Mongolia Autonomous Region (No. 2018MS08004), and the Natural Science Fund of the Hainan Province (No. 817319). The funding agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00452/full#supplementary-material
Abbreviations
HGDI, Hunter–Gaston discrimination index; MLVA, multiple locus variable-number tandem repeat analysis; MST, minimum spanning tree; PCR, polymerase chain reaction; SNPs, single-nucleotide polymorphisms; VNTR, variable-number tandem repeat analysis; WGS, whole-genome sequencing.
Footnotes
- ^ https://github.com/aquaskyline/SOAPdenovo2
- ^ http://cab.spbu.ru/software/spades/
- ^ http://www.bcgsc.ca/platform/bioinfo/software/abyss
References
Abou Zaki, N., Salloum, T., Osman, M., Rafei, R., Hamze, M., and Tokajian, S. (2017). Typing and comparative genome analysis of Brucella melitensis isolated from Lebanon. FEMS Microbiol. Lett. 364:fnx199. doi: 10.1093/femsle/fnx199
Al Dahouk, S., Tomaso, H., Nockler, K., Neubauer, H., and Frangoulidis, D. (2003). Laboratory-based diagnosis of brucellosis–a review of the literature. Part I: techniques for direct detection and identification of Brucella spp. Clin. Lab. 49, 487–505.
Alton, G. G., Jones, L. M., and Pietz, D. E. (1975). Laboratory techniques in brucellosis. Monogr. Ser. World Health Organ. 1975, 1–163.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Chen, S., Zhang, H., Liu, X., Wang, W., Hou, S., Li, T., et al. (2014). Increasing threat of brucellosis to low-risk persons in urban settings. China Emerg. Infect. Dis. 20, 126–130. doi: 10.3201/eid2001.130324
Chiaromonte, F., Yap, V. B., and Miller, W. (2002). Scoring pairwise genomic sequence alignments. Pac. Symp. Biocomput. 2002, 115–126.
De Figueiredo, P., Ficht, T. A., Rice-Ficht, A., Rossetti, C. A., and Adams, L. G. (2015). Pathogenesis and immunobiology of brucellosis: review of Brucella-host interactions. Am. J. Pathol. 185, 1505–1517. doi: 10.1016/j.ajpath.2015.03.003
De Massis, F., Ancora, M., Atzeni, M., Rolesu, S., Bandino, E., Danzetta, M. L., et al. (2015). MLVA as an epidemiological tool to trace back Brucella melitensis biovar 1 Re-emergence in Italy. Transbound Emerg. Dis. 62, 463–469. doi: 10.1111/tbed.12397
Delvecchio, V. G., Kapatral, V., Redkar, R. J., Patra, G., Mujer, C., Los, T., et al. (2002). The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc. Natl. Acad. Sci. U.S.A. 99, 443–448.
Ferreira, A. C., Correa De Sa, M. I., Dias, R., and Tenreiro, R. (2017). MLVA-16 typing of Brucella suis biovar 2 strains circulating in Europe. Vet. Microbiol. 210, 77–82. doi: 10.1016/j.vetmic.2017.09.001
Hunter, P. R. (1990). Reproducibility and indices of discriminatory power of microbial typing methods. J. Clin. Microbiol. 28, 1903–1905. doi: 10.1128/jcm.28.9.1903-1905.1990
Jiang, H., Fan, M., Chen, J., Mi, J., Yu, R., Zhao, H., et al. (2011). MLVA genotyping of Chinese human Brucella melitensis biovar 1, 2 and 3 isolates. BMC Microbiol. 11:256. doi: 10.1186/1471-2180-11-256
Kang, S. I., Her, M., Erdenebaataar, J., Vanaabaatar, B., Cho, H., Sung, S. R., et al. (2017). Molecular epidemiological investigation of Brucella melitensis circulating in Mongolia by MLVA16. Comp. Immunol. Microbiol. Infect. Dis. 50, 16–22. doi: 10.1016/j.cimid.2016.11.003
Khan, M. Z., and Zahoor, M. (2018). An overview of brucellosis in cattle and humans, and its serological and molecular diagnosis in control strategies. Trop. Med. Infect. Dis. 3:65. doi: 10.3390/tropicalmed3020065
Kilic, S., Ivanov, I. N., Durmaz, R., Bayraktar, M. R., Ayaslioglu, E., Uyanik, M. H., et al. (2011). Multiple-locus variable-number tandem-repeat analysis genotyping of human Brucella isolates from Turkey. J. Clin. Microbiol. 49, 3276–3283. doi: 10.1128/JCM.02538-10
Kong, W. (2018). Brucellosis infection increasing in Southern China. Eur. J. Intern. Med. 51, e16–e18. doi: 10.1016/j.ejim.2018.03.004
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12.
Lai, S., Zhou, H., Xiong, W., Gilbert, M., Huang, Z., Yu, J., et al. (2017). Changing epidemiology of human brucellosis, China, 1955-2014. Emerg. Infect. Dis. 23, 184–194. doi: 10.3201/eid2302.151710
Le Fleche, P., Jacques, I., Grayon, M., Al Dahouk, S., Bouchon, P., Denoeud, F., et al. (2006). Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol. 6:9. doi: 10.1186/1471-2180-6-9
Ledwaba, M. B., Gomo, C., Lekota, K. E., Le Fleche, P., Hassim, A., Vergnaud, G., et al. (2019). Molecular characterization of Brucella species from zimbabwe. PLoS Negl. Trop. Dis. 13:e0007311. doi: 10.1371/journal.pntd.0007311
Li, R., Li, Y., Kristiansen, K., and Wang, J. (2008). SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714. doi: 10.1093/bioinformatics/btn025
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., et al. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Lim, H. J., Lee, E. H., Yoon, Y., Chua, B., and Son, A. (2016). Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J. Appl. Microbiol. 120, 379–387. doi: 10.1111/jam.13011
Liu, Z. G., Di, D. D., Wang, M., Liu, R. H., Zhao, H. Y., Piao, D. R., et al. (2017). MLVA genotyping characteristics of human Brucella melitensis isolated from Ulanqab of inner Mongolia, China. Front. Microbiol. 8:6. doi: 10.3389/fmicb.2017.00006
Liu, Z. G., Wang, H., Wang, M., and Li, Z. J. (2019). Investigation of the molecular epizootiological characteristics and tracking of the geographical origins of Brucella canis strains in China. Transbound Emerg. Dis. 67, 834–843. doi: 10.1111/tbed.13404
Liu, Z. G., Wang, L. J., Piao, D. R., Wang, M., Liu, R. H., Zhao, H. Y., et al. (2018). Molecular investigation of the transmission pattern of Brucella suis 3 From Inner Mongolia, China. Front. Vet. Sci. 5:271. doi: 10.3389/fvets.2018.00271
Ma, J. Y., Wang, H., Zhang, X. F., Xu, L. Q., Hu, G. Y., Jiang, H., et al. (2016). MLVA and MLST typing of Brucella from Qinghai, China. Infect. Dis. Poverty 5:26. doi: 10.1186/s40249-016-0123-z
Marvi, A., Asadi-Aliabadi, M., Darabi, M., Abedi, G., Siamian, H., and Rostami-Maskopaee, F. (2018). Trend analysis and affecting components of human brucellosis incidence during 2006 to 2016. Med. Arch. 72, 17–21. doi: 10.5455/medarh.2018.72.17-21
Nascimento, M., Sousa, A., Ramirez, M., Francisco, A. P., Carrico, J. A., and Vaz, C. (2016). PHYLOViZ 2.0: providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 33, 128–129. doi: 10.1093/bioinformatics/btw582
Piao, D. R., Liu, X., Di, D. D., Xiao, P., Zhao, Z. Z., Xu, L. Q., et al. (2018). Genetic polymorphisms identify in species/biovars of Brucella isolated in China between 1953 and 2013 by MLST. BMC Microbiol. 18:7. doi: 10.1186/s12866-018-1149-0
Pisarenko, S. V., Kovalev, D. A., Volynkina, A. S., Ponomarenko, D. G., Rusanova, D. V., Zharinova, N. V., et al. (2018). Global evolution and phylogeography of Brucella melitensis strains. BMC Genom. 19:353. doi: 10.1186/s12864-018-4762-2
Scholz, H. C., and Vergnaud, G. (2013). Molecular characterisation of Brucella species. Rev. Sci. Tech. 32, 149–162. doi: 10.20506/rst.32.1.2189
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Sun, M. J., Di, D. D., Li, Y., Zhang, Z. C., Yan, H., Tian, L. L., et al. (2016). Genotyping of Brucella melitensis and Brucella abortus strains currently circulating in Xinjiang, China. Infect. Genet. Evol. 44, 522–529. doi: 10.1016/j.meegid.2016.07.025
Tian, G. Z., Cui, B. Y., Piao, D. R., Zhao, H. Y., Li, L. Y., Liu, X., et al. (2017). Multi-locus variable-number tandem repeat analysis of Chinese Brucella strains isolated from 1953 to 2013. Infect. Dis. Poverty 6:89. doi: 10.1186/s40249-017-0296-0
Vilella, A. J., Severin, J., Ureta-Vidal, A., Heng, L., Durbin, R., and Birney, E. (2009). EnsemblCompara Genetrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19, 327–335. doi: 10.1101/gr.073585.107
Wang, X. M., Huang, M. H., and Cui, B. Y. (2019). Analysis on brucellosis epidemiological characteristics in hainan province. Zhong. Liu Xing Bing Xue Za Zhi. 40, 350–353.
Wattam, A. R., Foster, J. T., Mane, S. P., Beckstrom-Sternberg, S. M., Beckstrom-Sternberg, J. M., Dickerman, A. W., et al. (2014). Comparative phylogenomics and evolution of the Brucellae reveal a path to virulence. J. Bacteriol. 196, 920–930. doi: 10.1128/JB.01091-13
Whatmore, A. M., Koylass, M. S., Muchowski, J., Edwards-Smallbone, J., Gopaul, K. K., and Perrett, L. L. (2016). Extended multilocus sequence analysis to describe the global population structure of the genus Brucella: phylogeography and relationship to biovars. Front. Microbiol. 7:2049. doi: 10.3389/fmicb.2016.02049
Keywords: Brucella melitensis, Brucella suis, multiple loci variable number tandem repeats analysis, single-nucleotide polymorphism, Hainan
Citation: Li Z, Wang X, Zhu X, Wang M, Cheng H, Li D and Liu ZG (2020) Molecular Characteristics of Brucella Isolates Collected From Humans in Hainan Province, China. Front. Microbiol. 11:452. doi: 10.3389/fmicb.2020.00452
Received: 13 November 2019; Accepted: 02 March 2020;
Published: 27 March 2020.
Edited by:
Narjol González-Escalona, United States Food and Drug Administration, United StatesReviewed by:
David O’Callaghan, Université de Montpellier, FranceLuca Freddi, Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail (ANSES), France
Ana Cristina Ferreira, National Institute of Agricultural and Veterinary Research (INIAV), Portugal
Copyright © 2020 Li, Wang, Zhu, Wang, Cheng, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi Guo Liu, bGl1emhpZ3VvQGljZGMuY24=; d2xjYmx6Z0AxMjYuY29t
†These authors have contributed equally to this work