 Ying Zhang
Ying Zhang Guangzhu Yang
Guangzhu Yang Meiling Fang1,2
Meiling Fang1,2 Ke-Qin Zhang
Ke-Qin Zhang Jianping Xu
Jianping Xu- 1State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, and Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan University, Kunming, China
- 2School of Life Sciences, Yunnan University, Kunming, China
- 3Department of Biology, McMaster University, Hamilton, ON, Canada
Predatory fungi in Orbiliaceae (Ascomycota) have evolved a diversity of trapping devices that enable them to trap and kill nematodes, other small animals, and protozoans. These trapping devices include adhesive hyphae, adhesive knobs, adhesive networks, constricting rings, and non-constricting rings. Their diversity and practical importance have attracted significant attention from biologists, making them excellent model organisms for studying adaptative evolution and as biological control agents against parasitic nematodes. The putative origins and evolutionary relationships among these carnivorous fungi have been investigated using nuclear protein-encoding genes, but their patterns of mitogenome relationships and divergences remain unknown. Here we analyze and compare the mitogenomes of 12 fungal strains belonging to eight species, including six species representing all four types of nematode trapping devices and two from related but non-predatory fungi. All 12 analyzed mitogenomes were of circular DNA molecules, with lengths ranging from 146,101 bp to 280,699 bp. Gene synteny analysis revealed several gene rearrangements and intron transfers among the mitogenomes. In addition, the number of protein coding genes (PCGs), GC content, AT skew, and GC skew varied among these mitogenomes. The increased number and total size of introns were the main contributors to the length differences among the mitogenomes. Phylogenetic analyses of the protein-coding genes indicated that mitochondrial and nuclear genomes evolved at different rates, and signals of positive selection were found in several genes involved in energy metabolism. Our study provides novel insights into the evolution of nematode-trapping fungi and shall facilitate further investigations of this ecologically and agriculturally important group of fungi.
Introduction
Nematode-trapping fungi (NTF) are a taxonomically heterogeneous group of asexual ascomycetes that can form special structures (traps) to capture free-living nematodes in soil (Barron, 1977). Members of the Orbiliaceae family represent the largest group of NTF, which include at least 96 species belonging to genera Arthrobotrys (53 species, producing adhesive three-dimensional networks), Dactylellina (28 species, producing adhesive knobs, non-constricting rings, and adhesive column), and Drechslerella (14 species, producing constricting rings) (Zhang and Hyde, 2014). These NTF species were originally defined based primarily on conidial characteristics such as size, septation, and type of conidiogenous cells (Subramanian, 1963). Recent research showed that trapping devices are also phylogenetically informative for classifying NTF species (Rubner, 1996; Scholler et al., 1999; Li et al., 2005; Yang and Liu, 2006; Yang et al., 2007).
Because of their unique life style and potential as biocontrol agents against parasitic nematodes, NTF have been studied over several decades. Those studies cover a range of topics, including their substrate preference, the origin and development of trapping devices, and the molecular mechanisms of pathogenesis against nematodes. Previous studies based on multiple nuclear gene phylogeny indicated that the trapping mechanisms within the Orbiliales have evolved along two major lineages, one leading to species with constricting rings and the other to species with adhesive traps, including three-dimensional networks, adhesive knobs, and adhesive branches (Yang et al., 2007). Furthermore, molecular clock calibration based on two fossil records estimated that the two major lineages diverged from each other ∼246 million years ago (Mya) (Yang et al., 2012).
So far, the genome sequences of several NTF have been obtained, assembled, and annotated, including strains from Arthrobotrys conoides, Arthrobotrys oligospora, Dactylellina appendiculata, Dactylellina drechsleri, Dactylellina haptotyla, Drechslerella stenobrocha, and Dactylellina cionopaga. Such a rich dataset has made it possible for evolutionary studies at the genomic level (Zhang W. et al., 2016). Genome sequence comparisons have shown that NTF tend to have expanded gene families coding chitin-degrading enzymes and proteases, possess a well-developed cellulose-degrading metabolism, but have relatively few plant pathogenesis-related genes. Together, these features are consistent with their saprotrophic ancestry (Liu et al., 2014). However, nothing is known about the evolutionary patterns and divergences of NTF from the mitochondrial genomic perspective.
Mitochondria are energy-generating organelles that play a critical role in numerous cellular functions, including ATP production, cellular homeostasis, and apoptosis (Wallace, 2005). Due to their distinct genome structure, inheritance pattern, and rates of evolution, mitochondrial genomes (mitogenomes) have been frequently used in evolutionary biology and systematic studies (Pantou et al., 2006; Kouvelis et al., 2008; Basse, 2010). However, among the estimated 2.2–3.8 million fungal species (Hawksworth and Lücking, 2017), only 375 records of fungal mitogenomes are published by the end of 20191. These mitogenomes show a large variation in genome size, ranging from 12,055 bp in Rozella allomycis to 19,431 bp in the fission yeast Schizosaccharomyces pombe (Chen and Butow, 2005) and 235,849 bp in the common filamentous plant pathogen Rhizoctonia solani (Losada et al., 2014). Despite the large genome size variation, the fungal mitogenomes usually contain 14 genes that encode oxidative phosphorylation system proteins, the large (rnl) and small (rns) ribosomal RNA subunits, and a fairly constant set of tRNA genes (Gray et al., 1999; Lavín et al., 2008). The major contributor to the large genome size variations among the fungal mitogenomes are the varying number of introns, as well as the genes within those introns such as the endonuclease genes containing either the GIY-YIG or the LAGLIDADG motifs (Wu et al., 2009; Mardanov et al., 2014; Sandor et al., 2018). So far, most of these mitogenomes have been reported separately and there have been very few comparative analyses of mitogenomes of a specific group of fungi, including NTF.
In this study, we analyzed the complete mitochondrial genomes of six NTF species representing all four types of nematode trapping devices and two other closely related species incapable of forming any nematode trapping devices. To provide a better understanding of the mitogenomic diversity among NTF, for two species A. oligospora and Drechslerella brochopaga, we also analyzed the mitogenomes from four and two natural isolates, respectively. The 12 mitogenomes were annotated, and their gene contents, structures, and gene orders were compared to assess variation and conservation among NTF strains and species. All the mitogenomes we analyzed are largely syntenic, but the size and the intron content differ greatly. The Ka/Ks ratios obtained clearly suggested that though purifying selection was the dominant force driving the evolution of most protein-coding genes, signals of positive selection were found for several genes across NTF species. Our comparative analyses of mitogenomes of NTF provide an important foundation for future studies on NTF population genetics, taxonomy, mechanisms of trap formation, and biocontrol application.
Materials and Methods
Sampling, DNA Extraction, and Genome Sequencing
The published mitochondrial genomes of six NTF species representing all four types of nematode trapping devices were analyzed and compared with each other as well as with two closely related species but are incapable of producing any nematode trapping devices (Jiang et al., 2018; Zhou et al., 2018; Deng and Yu, 2019a, b; Fang et al., 2019; Li and Yu, 2019; Wang S. et al., 2019; Zhang and Yu, 2019). In addition to the nine published mitogenomes, we generated the mitogenome sequences of three more natural strains of A. oligospora and included them for both inter- and intra-species comparisons among NTFs (Table 1). All the strains and species were identified based on their morphological features and their sequences at the internal transcribed spacer (ITS) regions of the ribosomal RNA gene cluster (Zhang Y. et al., 2016). To obtain their mitogenome sequences, their total genomic DNA was extracted from the mycelia collected from single-spore cultures growing on cellophane membrane on PDA according to the method described by Zhang et al. (2011). Sequencing libraries and the whole genome sequencing were performed with an Illumina HiSeq 2500 Platform by Novegene Co., Ltd. (Beijing, China).
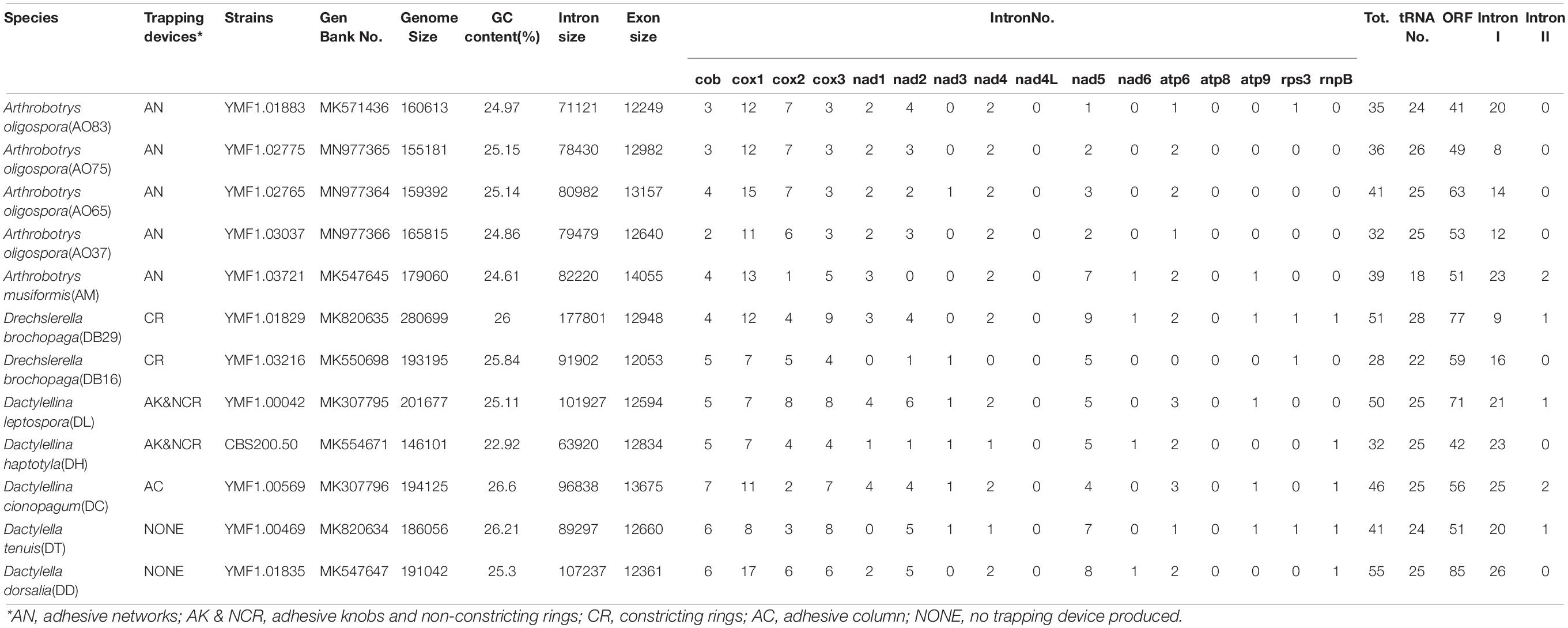
Table 1. Mitogenome features in six nematode-trapping fungal species and two related non-predatory species.
Assembly and Annotation of Mitochondrial Genomes
The sequencing, assembly, and annotation for nine of the 12 mitogenomes analyzed in this study have been described previously (Jiang et al., 2018; Zhou et al., 2018; Deng and Yu, 2019a, b; Fang et al., 2019; Li and Yu, 2019; Wang S. et al., 2019; Zhang and Yu, 2019). To assemble and annotate the three remaining mitogenomes of A. oligospora strains, their mitogenome sequence reads were first identified for homology with the published A. oligospora mitogenome (GenBank accession number MK571436) using the NCBI BLAST algorithm. For each strain, all mitogenome reads were extracted from the whole genome sequencing data according to our previously described methods (Deng and Yu, 2019a). We then used the SPAdes 3.9.0 software with a kmer size of 17 to de novo assemble the three A. oligospora strains’ mitogenomes with the obtained clean reads (Bankevich et al., 2012). The gaps were filled by separate PCR and sequencing with primers on regions flanking the gaps, resulting in one circular mitochondrial genome for each strain. The mitogenomes were annotated using both MFannot2 and GLIMMER3. The tRNAs were identified and annotated using tRNAscan-SE (Brooks and Lowe, 2005). All ORFs were searched and identified by ORFFinder4. Syntenic blocks in all 12 mitogenomes were identified using MAUVE 2.3.1 based on whole mitogenome sequence alignments (Darling et al., 2004). For each strain, we determined the relative synonymous codon usage (RSCU) in 14 mitochondrial protein coding genes (PCGs) using CodonW1.4.25 with the fungal mitochondrial genetic code 4. The following formulas were used to assess mitogenome strand asymmetry: AT skew = [A − T]/[A + T]; GC skew = [G − C]/[G + C].
The non-synonymous (Ka) and synonymous substitution rates (Ks) for all 14 mitochondrial PCGs of the NTF mitogenomes were calculated using KaKs_Calculator2.0 (Wang et al., 2010). The calculated Ka/Ks ratios were then analyzed to infer the potential selection pressure on each gene pair. Generally, a Ka/Ks ratio greater than, equal to, or less than 1 indicates positive (diversifying) selection, neutral evolution, or purifying (negative) selection, respectively.
Transition transversion biases were estimated for both the mitogenomic PCGs and the three single-copy nuclear protein-coding genes: the second largest subunit of RNA polymerase II gene (rpb2), the translation elongation factor 1-α gene (tef1-α), and the β tubulin gene (β-tub). These estimates were obtained by employing Maximum Composite Likelihood (MCL) statistical method with the Tamura-Nei model and gaps/missing data were excluded. In addition, we also obtained the Maximum Likelihood estimation of transition transversion biases by employing Kimura 2-parameter substitution model, where evolution rates were set as Gamma distributed with Invariant sites (G + I) and the number of discrete gamma categories was 5. The transition transversion bias estimates by MCL and ML were carried out in MEGA6 (Tamura and Nei, 1993; Tamura et al., 2013).
Phylogenetic Analysis
To determine the evolutionary relationships of our selected NTF and other species from the phylum Ascomycota, amino acid sequences at each of the 14 PCGs (atp6, 8–9, cob, cox1–3, nad1–6, and nad4L) were individually analyzed for all selected taxa. The 14 gene trees were compared among themselves for their tree topology congruences. As there was no evidence of phylogenetic incongruence, the concatenated amino acid sequences of these 14 PCGs were used to construct a more robust phylogeny among the taxa. Here we included 94 species from other representative phyla and classes, including Pezizomycotina, Saccharomycotina, Taphrinomycotina, Zygomycota, Blastocladiomycota, and Basidiomycota. Our 14 individual gene trees showed no evidence of significant incongruence. Thus, the multiple protein sequences were concatenated and aligned using MUSCLE in software MEGA6.0 (Edgar, 2004; Tamura et al., 2013). MrBayes v3.2.6 (Huelsenbeck and Ronquist, 2001) was used to construct the phylogenetic tree using the Bayesian inference (BI) method based on the combined gene set. In this phylogenetic analysis, the first 25% trees were discarded as burn-in, and the remaining trees were used to calculate Bayesian posterior probabilities (BPP) in a 50% majority-rule consensus tree.
Results
Genome Size and Intron Distribution
The mitogenomes of the 12 fungal strains investigated here were all composed of circular DNA molecules with genome sizes ranging from 146,101 bp to 280,699 bp (Table 1). The two species incapable of forming trapping devices have mitogenomes sized 186,056 bp and 191,042 bp, respectively, well within the range of the mitogenomic sizes of those with nematode trapping ability. However, despite their large mitogenome size difference (almost two-fold difference between the largest and the smallest mitogenomes), the concatenated lengths of all exons were very similar among the strains, from DB16’s 12,053 bp to AM’s 14,055 bp, a difference of about 2 kb. Indeed, strain DB29 has the largest mitogenome at 280,699 bp but an intermediate concatenated exon size of 12,948 bp. We note that the concatenated exons excluded those in the open reading frames (ORFs) of introns. Indeed, some of the introns contain ORFs coding for potential endonucleases (group I) or reverse-transcriptases (group II). Most of the mitochondrial introns we annotated in these 12 strains belong to group I, only 1 or 2 introns distributed in strains AM21, DB29, DL42, DT, and DC are group II introns.
Among the 14 protein coding genes (cox1, cox2, cox3, cob, atp6, atp8, atp9, nad1, nad2, nad3, nad4, nad4L, nad5, nad6), 12 contained at least one intron in one of the 12 strains (Table 1). The gene containing the most introns was cox1. However, two genes, nad4L and atp8, have no intron in any of the 12 strains. The variations in intron numbers were found both among species as well as among strains within species, in this case A. oligospora and Dr. brochopaga. Overall, though there are differences in how introns are distributed among species, there is no clear pattern for association between the total number of introns and specific type of trapping devices (Figure 1). Our results are consistent with the dynamic nature of intron distribution in fungal mitogenomes.
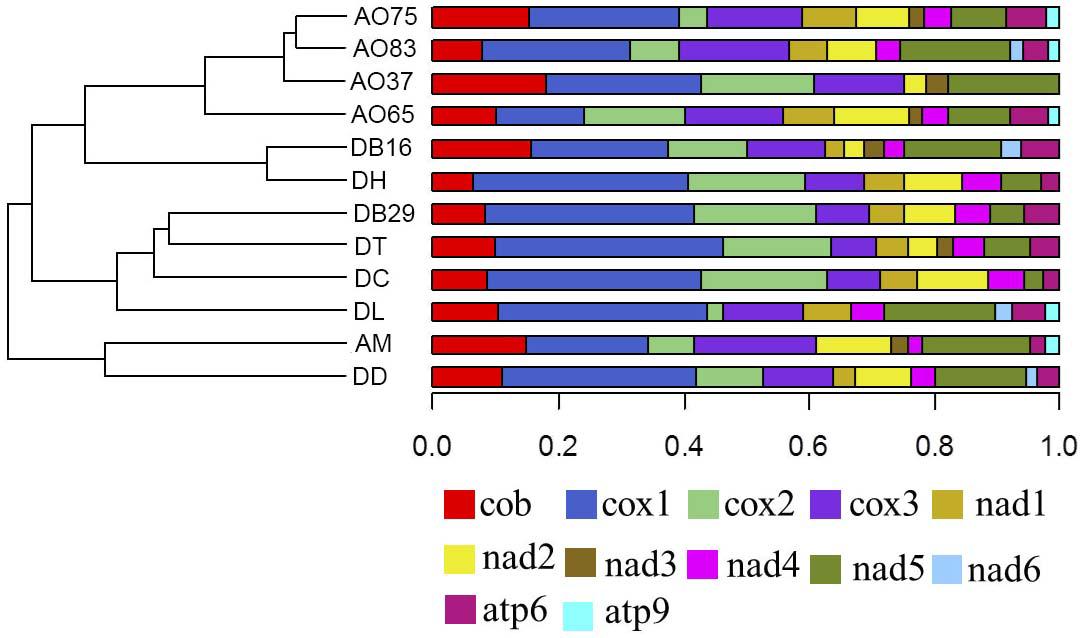
Figure 1. Dendrograms showing the relationships among strains based on their relative intron numbers in each of the 14 mitochondrial core protein coding genes. The “plotTree.barplot” function from the phytools R package (Revell, 2011) was used to estimate the similarities between samples. A distance matrix was first calculated using the intron numbers distributed in the 14 protein coding genes within each strain.
Gene Arrangement Analysis
All the mitogenomes from the eight fungal species encode an essential set of conserved genes including three cytochrome c oxidase subunits (cox1, cox2, cox3), apocytochrome b (cob), three ATP synthase subunits (atp6, atp8, atp9), seven subunits of NADH dehydrogenase (nad1, nad2, nad3, nad4, nad4L, nad5, nad6), the small and large ribosomal RNA subunits (rns, rnl). The mitogenome-encoded ribosomal small subunit protein 3 (rps3) and the ORF coding for a putative rnpB were also found among our strains. The number of tRNA genes in the mitochondrial genomes of the NTF species ranges from 18 to 28. All the genes are located on the same DNA strand. The gene order of the 14 major PCGs was highly conserved across the mitogenomes of the six NTF species, with the exception of switched locations of atp9 and nad1 genes in DB16 (Figure 2). However, compared to the six NTF species, the two non-predatory fungal species DD and DT had a large rearranged mitogenome region, with atp8 being from the upstream of the nad4 gene changed to the downstream of cox1 gene. Based on the gene orders, the 12 mitogenomes could be divided into three types as shown in Figure 2. The first type contains the four strains of A. oligospora and the representative of each of the following species AM, DL, DH, DC, as well as DB29. The second type is represented by DB16, and the third type by DD and DT. The high gene synteny among these mitogenomes is also reflected by the Mauve alignment (Figure 3). All 12 mitogenomes could be divided into four homologous regions. The red, light green and blue regions are all similar in their sizes, except for the large light green region in DD that is also inverted compared to all other strains. In contrast, the sizes of the largest syntenic regions (in dark green) differed substantially among these species according to their mitochondrial genome sizes (Figure 3).
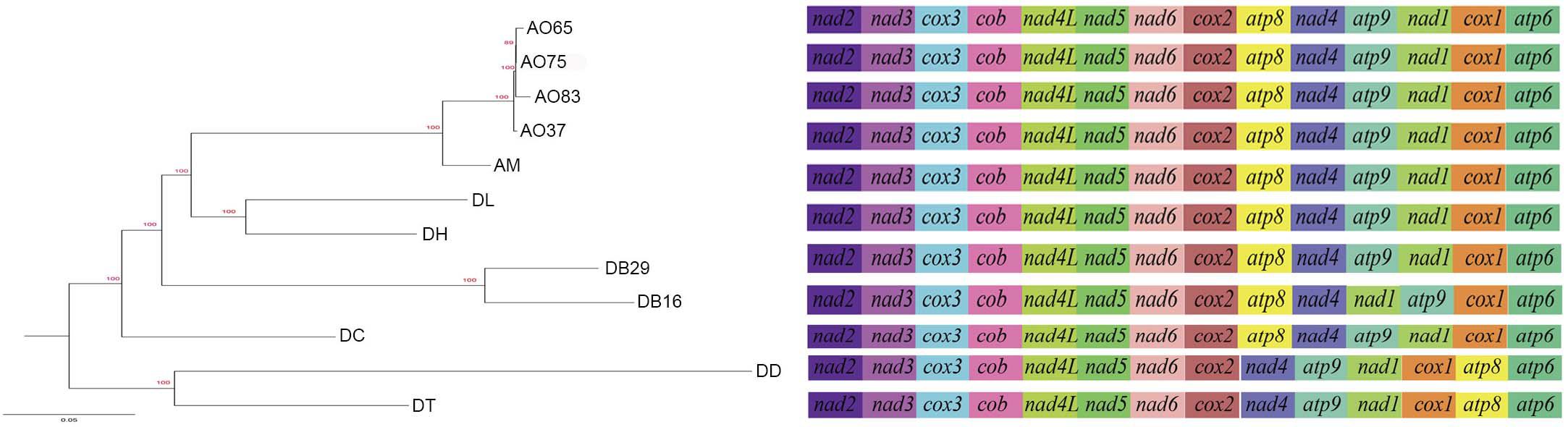
Figure 2. Phylogeny of nematode trapping fungi and their close relatives based on their concatenated nucleotide sequences of the 14 mitochondrial core protein coding genes. The gene order in each mitogenome is indicated.

Figure 3. Co-linearity analysis of 12 fungal mitogenomes analyzed in this study, generated with Mauve 2.3.1.
AT and GC Skews
The lengths and GC contents of the 15 core PCGs varied among the 12 mitogenomes (Figure 4). The low GC content of all 12 strains (ranging from 22.92% to 26.6%) is similar to other fungal mitogenomes in Pezizomycotina (Lin et al., 2015). Here, the mitogenome from DC contained the highest GC content (Supplementary Table S1). Our analyses identified that the AT skews of most genes in most of our samples were negative. The only exceptions were the rps3 gene in most strains (Supplementary Table S2), the atp9 gene in three samples (DC, AM, and DD), and the nad4L gene in five samples (all AO samples and AM). Conversely, GC skew was positive in most PCGs except at atp8 gene in all strains, and the cob, cox2-3, and nad3 genes for some of the strains. Our results suggested that the nucleotide compositions varied among NTF species and PCGs. Interestingly, in contrast to the skews calculated for other species of NTF, all adhesive network- producing strains have more Ts than As in their protein coding genes, indicating that their mitogenomes were likely subjected to different selection pressures than other NTF species and the two non-predatory species.
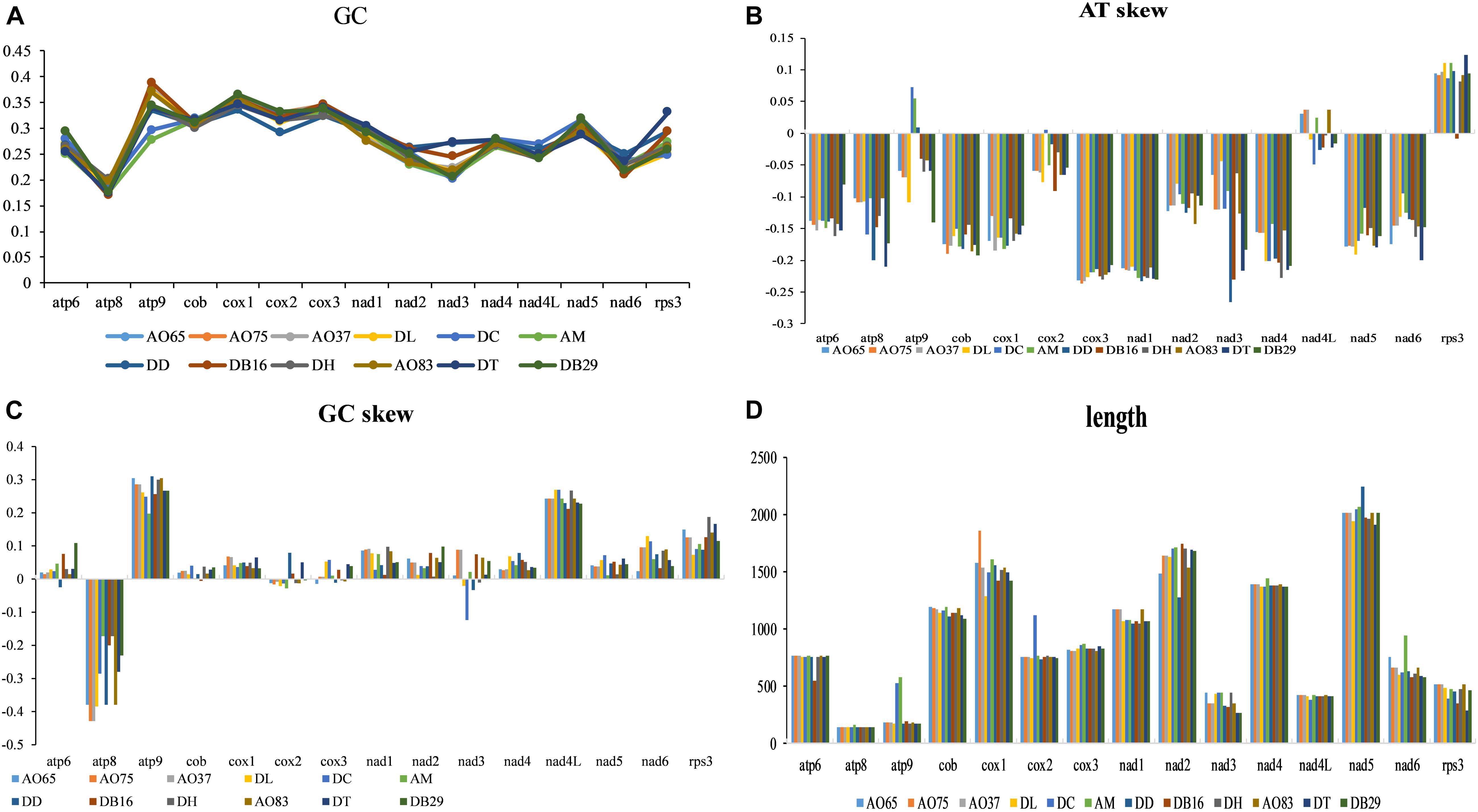
Figure 4. Variation in the length and base composition of each of the 15 protein-coding genes among 12 fungal mitochondrial genomes. (A) GC content across the 15 genes; (B) AT skew; (C) GC skew; (D) gene length variation.
Codon Usage Analysis
Codon usage analysis indicated that the most frequently used codons were UUU (for Phenylalanine; Phe), UUA (for Leucine; Leu), AUU (for Isoleucine; Ile), AUG (for Methionine; Met), GUU (for Valine; Val), and AGA (for Serine; Ser), CCU (for Proline; Pro), ACU (for Threonine; Thr), GCU (for Alanine; Ala), UAU (for Tyrosine; Tyr), CAU (for Histidine; His), GGU (for Arginine; Arg), AAU (for Asparagine; Asp), UGU (for Cysteine; Cys), CAA (for Glutamine; Gln), GAA (for Glutamic acid; Glu), GGA (for Glycine; Gly), AAA (for Lysine; Lys) (Supplementary Table S3). Our analyses showed that the codon usage patterns were highly similar among the 12 mitogenomes, with the most commonly used codon for each amino acid all being the same across all 12 strains (Figure 5). The bias in high A + T ratios in these mitogenomes have likely contributed to the preference of codons with high A + T content.
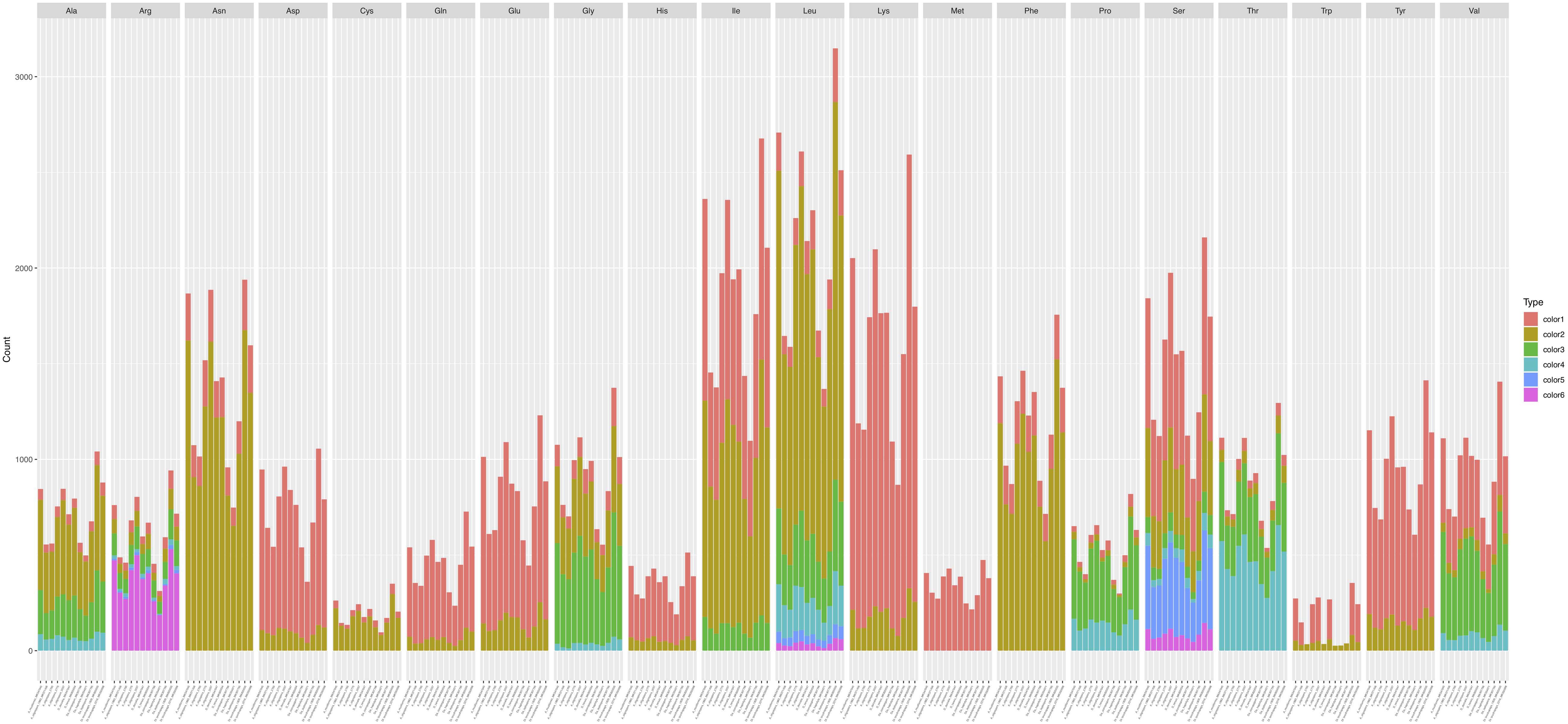
Figure 5. Codon usage in the 12 fungal mitochondrial genomes. Frequency of codon usage is plotted on the y-axis.
Evolutionary Rates of PCGs and Nuclear Genes
The mean nucleotide frequencies of the three analyzed nuclear genes are A = 24.66%, T/U = 24.70%, C = 26.38%, and G = 24.27%, and those of mitogenomic PCGs are 29.31, 41.12, 14.03, and 15.54%, respectively. The rates of transitional and transversional substitutions among mitochondrial and nuclear genes vary, with the rates of transitional substitutions from T to C and from A to G at much higher frequencies in nuclear genes than in mitochondrial genes. Similarly, much higher rates of transversional substitutions from A to C, T to G, C to G and G to C were found in nuclear genes than mitochondrial genes. In contrast, rates for remaining types of transitional and transversional substitutions were found much higher in mitogenomes than in nuclear genomes of the same group of species (Table 2).
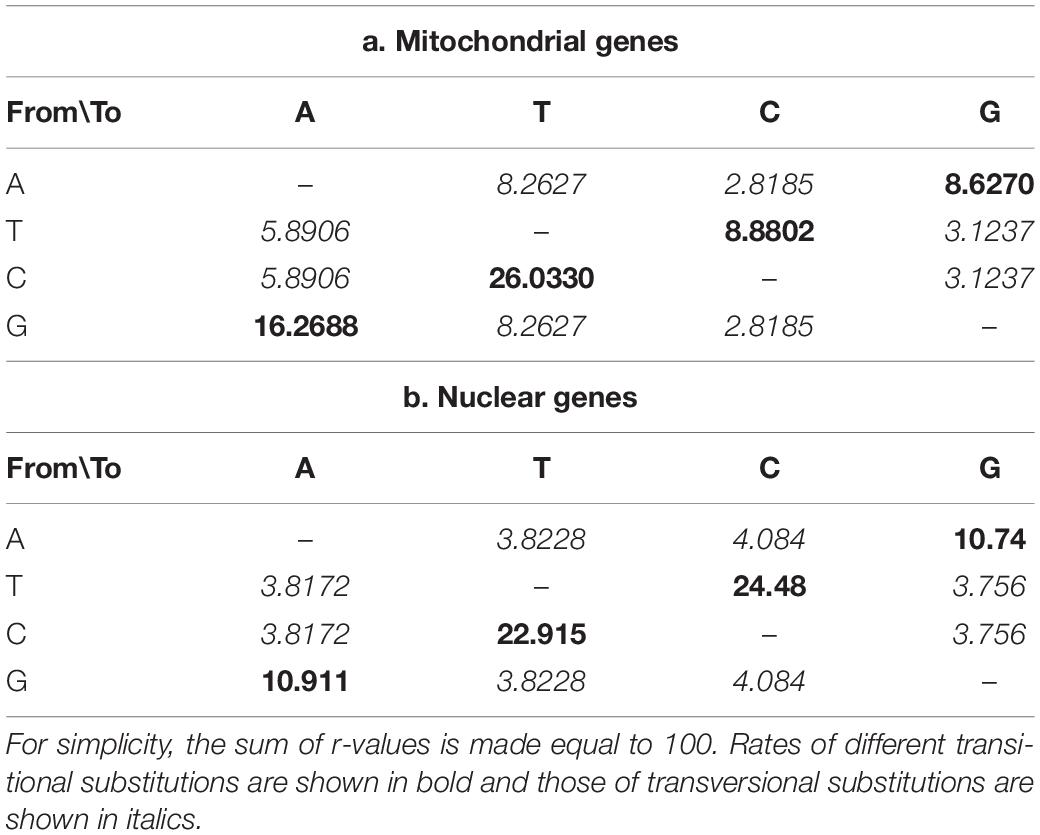
Table 2. Probability of substitution (r) from one base (row) to another base (column).
Among the 15 mitochondrial PCGs, seven (rps3, cox1-2, nad2-3, and nad5-6) had a higher non-synonymous substitution rate (Ka) than synonymous substitution rate (Ks) between several species pairs. Our results indicate that besides purifying selection, NTF species were subject to positive selection at specific gene loci (Figure 6).
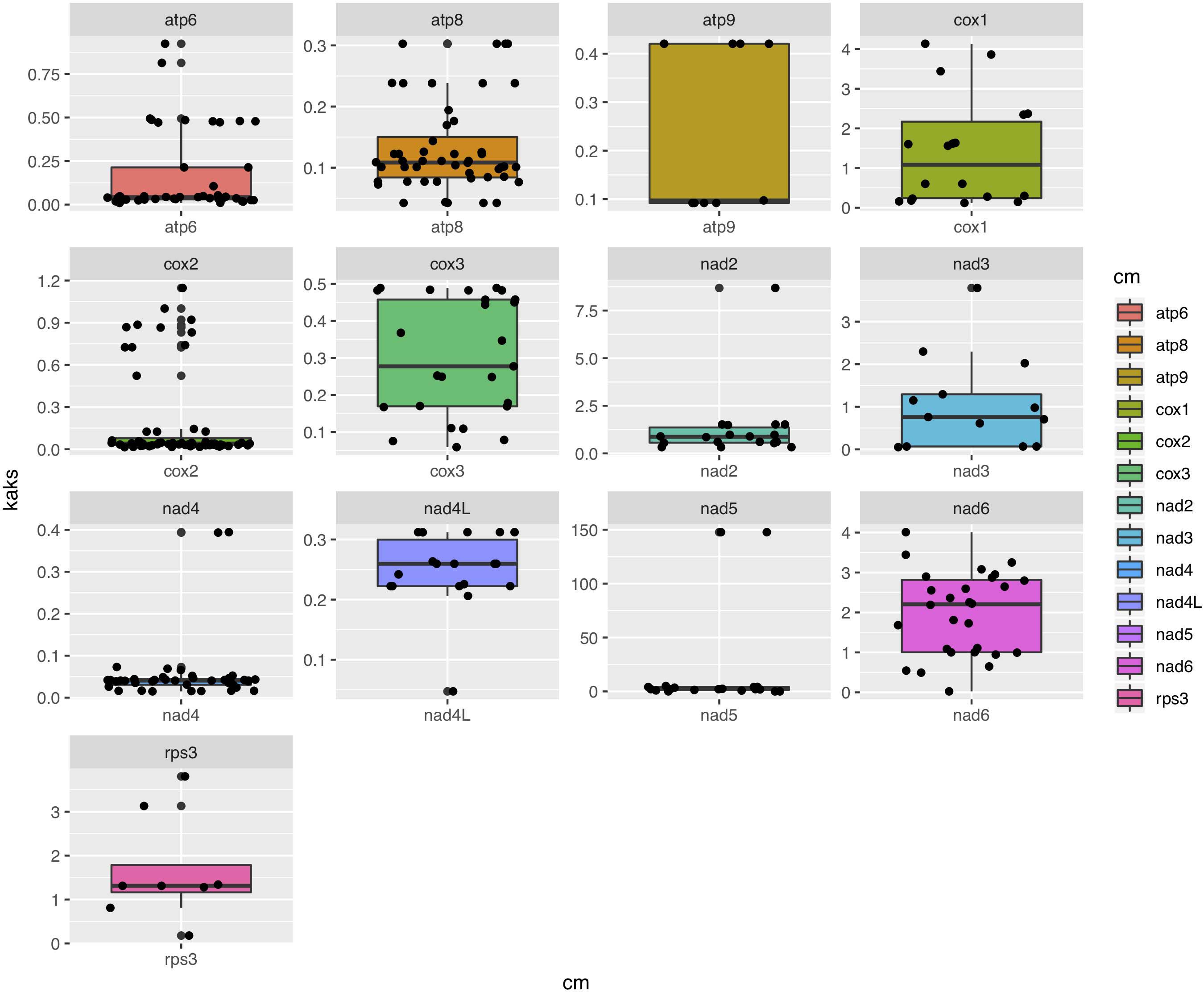
Figure 6. Box-plot comparisons of the Ka/Ks ratios estimated in the various mitochondrial genes. The estimates were based on pairwise alignments of samples.
Phylogenetic Analysis
Our single gene based phylogenetic analyses revealed no evidence for statistically significant incongruences among the 14 protein-coding gene trees. Thus, here we focus on analyzing the concatenated mitochondrial protein sequences. Our results revealed many well-support backbone nodes in Ascomycota, similar to those revealed by nuclear genes (Schoch et al., 2009; Figure 7). For example, high statistical supports were found for subclasses Saccharomycotina and Pezizomycotina. Interestingly, Saccharomycotina was found to be more closely related to Taphrinomycotina than to Pezizomycotina. Data presented here suggested that the common ancestor of Orbiliomycetes and Pezizomycetes likely represented an early diverging lineage of the Pezizomycotina (James et al., 2006), with the remaining four classes sampled here forming a well-supported clade. Moreover, in both the mitochondrial and nuclear phylogenies, these NTFs were found to be within a well-supported monophyletic group (Li et al., 2005; Yang et al., 2007). Our results suggest that non-predatory species with similar morphological characters (except the traps) to these NTFs (such as our DT and DD), may have differentiated from other Orbiliomycetes at an early stage during evolution. In our phylogeny based on concatenated mitochondrial protein sequences, the species producing adhesive column (DC) was found to be the first one diverging from the non-predatory species DT and DD. Therefore, DC likely represents one of the early branching members of NTFs. Other predators producing constricting rings and adhesive knobs and networks seemed to form a well-support clade that emerged later.
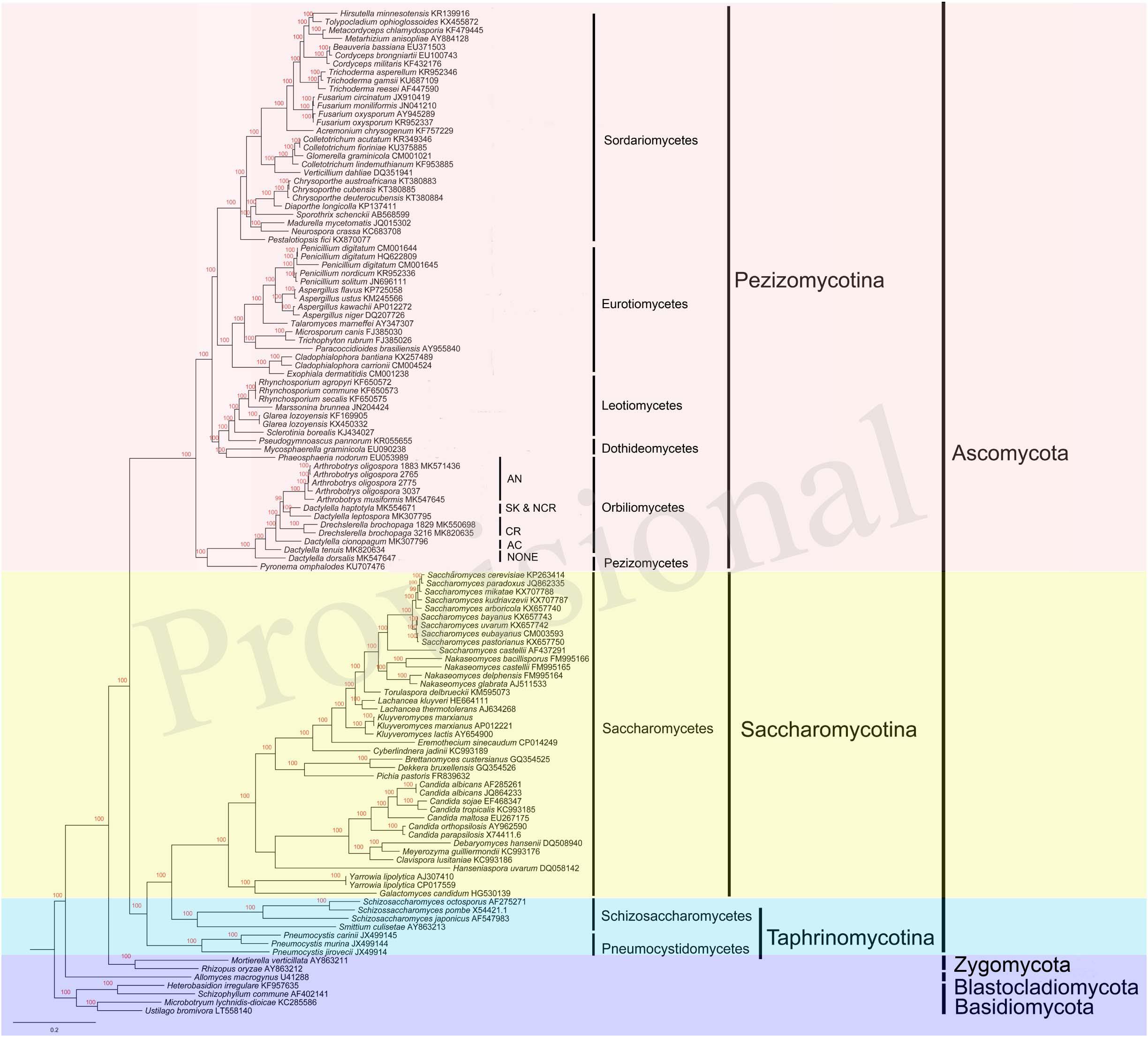
Figure 7. Bayesian tree of 104 species from the five main classes of Pezizomycotina, as well as representative species from other fungal phyla. The tree was constructed based on concatenated amino acid sequences of 14 conserved protein coding genes in fungi atp6, 8–9, cob, cox1–3, nad1–6, and nad4L. The resulting Bayesian posterior probabilities (BPP) ≥ 70% are shown above internal branches. GenBank accession numbers of all sequences are given. AN, adhesive networks; AK & NCR, Adhesive knobs and non-constricting rings; CR, constricting rings; AC, adhesive column; NONE, no trapping device produced.
Discussion
In this study, we obtained the mitogenome sequences of three strains of A. oligospora. These three mitogenome sequences were analyzed together with nine previously published mitochondrial genomes. Together, these 12 mitogenomes represent six species of NTF and two related species of non-predatory fungi. Our analyses indicated that these 12 fungal mitogenomes are among the largest and most variable in length of the known eukaryotic mitogenomes (Paquin et al., 1997; Salavirta et al., 2014). The size of published fungal mitogenomes varies from 12.06 kb to 235.85 kb, which represents a 19.6-fold difference in size. Likewise, great mitogenome size variations (146–280 kb) were revealed among NTF species, largely due to differences in the number and size of introns (28–55 introns per species).
Interestingly, the NTFs and their closely related two non-predatory fungal species are among the species with large mitogenomes, with strain DB29 having the largest mitogenome of all known fungi sequenced to date. Furthermore, two strains of the same species, DB29 and DB16, showed a large difference in their genome sizes, 280,699 and 193,195 bp, respectively. Based on mitogenome sequence comparisons, almost all the size differences among the 12 mitogenomes could be attributed to mitochondrial introns. However, it’s currently not known why certain strains and species accumulate more introns than other strains and species. Here, the two strains of Dr. brochopaga with different mitogenome sizes came from different sources: DB29 was isolated from the ascospore germination culture of its teleomorphic form Orbilia orientalis (Raitv.) Baral (Yu et al., 2006), whereas DB16 was directly obtained from a single conidium in a soil sample (Guo et al., 2009). In the lab, strain DB29 is capable of sexual reproduction while strain DB16 is incapable of sexual reproduction. It’s tempting to speculate that the ability of strain DB29 to reproduce sexually through mating may have contributed to the spread of introns to its mitogenome and consequently to its large mitogenome size.
Similarly, the mitogenome size differences among strains of A. oligospora were primarily due to the size and distribution of introns. In A. oligospora, strains belong to one of two mating types. The mating type genes regulate mating (hyphal fusion) and meiosis. Recent research has shown increasing evidence for mitochondrial DNA recombination under both laboratory and field conditions, though it is widely believed that the inheritance of mitochondrial DNA in fungi is uniparental and non-recombining (Wilson and Xu, 2012; Breton and Stewart, 2015; Xu and Wang, 2015; Wang et al., 2017). Indeed, mutation of a specific gene involved in sex-determination caused the rapid spread of multiple introns in the human pathogenic yeast Cryptococcus neoformans (Yan et al., 2018). In the case of NTF and their close relatives, further studies are needed to investigate the mobility of introns in genetic crosses. In addition, mitochondrial- and nuclear-associated plasmids and double-stranded RNA (dsRNA) elements are common in fungi and they can spread during hyphal fusion, potentially contributing to the diversity and size expansion in the mitogenomes observed in this study.
Because all mitogenomes are believed to have originated from a common ancestor, the mitochondrial gene order can also reflect the evolutionary relationships among organisms (Zheng et al., 2018). A comparative alignment of 12 mitogenomes with the Mauve program clearly showed a largely syntenic relationship among all samples we investigated, but the sizes and relative positions of homologous fragments showed slight variation among some of the species. Here, the two non-predatory species had a distinct location for atp8 that’s different from that in other species. Closely related plants often contain mitochondrial genomes with different gene orders, which have been attributed to homologous recombination within individual mitochondrial genomes mediated by their high proportions of repeat sequences (Maréchal and Brisson, 2010). However, despite their potential importance, the mitochondrial gene order variations among fungi have rarely been evaluated (Aguileta et al., 2014). It is possible that the DD and DT gene arrangement represents the ancestral condition and those of the NTF are an evolutionarily derived condition. As indicated in the phylogeny, DD is located on the basal branch in the Orbiliomycetes clade, this change in gene order could be caused by an intra-molecular recombination within the mitochondrial genome of the common ancestor of the analyzed NTF.
The GC content of mitochondrial genomes varies among organisms, and can be affected by mutation bias, selection, and biases of reconstitution-related DNA repair (Chen et al., 2014). Interestingly, in almost all fungal mitogenomes sequenced so far, codon usage is biased strongly toward codons ending in A or T (Li et al., 2018a, b). Indeed, over 94% of the optimal codons in the 10 NTF mitogenomes end in A or T (Supplementary Table S2). This was likely due to the high AT content found in fungal mitogenomes, where the rates of transitional and transversional substitution from C, G to T, A are much higher than those of the reverse direction and those of nuclear genes.
As one of the most important organelles, mitochondrion generates the universal energy currency ATP and plays an important role in almost all biological activities. Even though mitochondria were originated from a common ancestral alpha- proteobacterium, they may have evolved together with different nuclear genomes and adapted to different environments (Yan et al., 2007; Einer et al., 2017; Zhang B. et al., 2017). Previous studies have shown that different groups of eukaryotes seem to have different mitogenome mutation rates, and the rates of mutation in fungal mitochondrial genomes are generally considered intermediate between those in animals (highest mutation rate) and plants (lowest mutation rate) (Aguileta et al., 2014). However, there are significant differences among fungal groups in their relative nuclear-mitochondrial genome mutation rates (Sandor et al., 2018). Regardless, mitochondrial genes have been used for evolutionary studies in a variety of fungi, such as the endophytic fungus Phialocephala scopiformis (Robicheau et al., 2016), the entomopathogens Hirsutella vermicola (Zhang Y.-J. et al., 2017), Pochonia chlamydosporia (Lin et al., 2015), and H. thompsonii (Wang et al., 2018). For example, a mitogenomic phylogeny showed that all of the invertebrate-pathogenic fungi cluster together to form a monophyletic group in Hypocreales, which is noticeably distinguished from a cluster comprising of plant fungal pathogens (Lin et al., 2015). Based on mitochondrial gene sequences, our NTFs were also found to be in a well-supported monophyletic group different from the two non-predatory species, consistent with these invertebrate-pathogenic fungi sharing a common ancestor.
At present, there are several hypotheses on the origin and evolution of nematode-trapping lifestyles in the Ascomycota, and these hypotheses differ in the order of emergence of individual trap types, including what the ancestral trap type might be (Rubner, 1996; Ahren et al., 1998; Li et al., 2005; Yang et al., 2007). Based on the phylogenetic relationship revealed in our study, predatory fungi evolved from non-predatory ancestors and we propose that adhesive column was likely the first type to emerge. This was then followed by the emergence of constricting rings (CR). Our results are consistent with the hypothesis of Yang et al. (2007) that the trapping mechanisms within the Orbiliales have evolved along two major lineages, one leading to species with constricting rings and the other to species with adhesive traps, including three-dimensional networks, knobs, and branches. Indeed, physiological and microscopic observations indicate that CR and various adhesive devices have distinctly different materials. For example, microscopically within trap cells, CR contains unique, oblong-shaped, electron-dense inclusions, which are absent in the ring cells after nematodes are captured, whereas cells of adhesive devices exhibit multiple globose electron-dense bodies that persist (Tzean and Estey, 1981; Nordbring-Hertz, 1988). Our analyses demonstrated that, though with similar morphology, non-constricting rings (NCR) were phylogenetically distant from the CR, which is consistent with the phylogeny reconstructed based on multiple nuclear gene sequences. Although the rates of transitional and transversional substitutions between mitochondrial and nuclear genes varied among our study organisms, the framework established by the mitogenome sequences showed that the origins and divergence patterns of various predatory fungi in Ascomycota were similar to those inferred based on nuclear gene sequences. Overall, our results indicate that mitochondrial genes can be useful molecular markers for NTF taxonomy, systematics, population genetics, and evolutionary studies.
Due to their functional constraints, mitochondrially encoded protein sequences typically evolve slower than predicted by the high nucleotide substitution rate in the mitochondrial genome (Giorgi et al., 1999). Thus, we expected to observe prevalent evidence for purifying selection on non-synonymous mutations during mitochondrial evolution (Meiklejohn et al., 2007). However, our analyses showed a mixed pattern. Interestingly, a recent analysis of diverse animal taxa showed that 26% of non-synonymous mitochondrial substitutions may have been fixed by adaptive evolution (James et al., 2015). Furthermore, frequent non-synonymous polymorphisms in both atp6 and VAR1 genes were observed in yeast, consistent with relaxed purifying selection on either protein-coding genes or the intergenic regions (Jung et al., 2012), resulting in fast evolution of yeast mitochondrial genomes. Similar to most yeasts, NTF can grow as saprophytes in soils. However, different from yeasts, NTF can enter the parasitic stage by developing specific traps. During the switch between different lifestyles, multiple fungal signal transduction pathways are activated by its nematode prey to further regulate downstream genes associated with diverse cellular processes such as energy metabolism, biosynthesis of the cell wall and adhesive proteins, cell division, glycerol accumulation, and peroxisome biogenesis (Yang et al., 2011; Meerupati et al., 2013). Therefore, genes involved in energy metabolism (e.g., cytochrome c oxidase, ATP synthase and NADH dehydrogenase subunits) might be targets of natural selection and adaptation to meet the huge change in energy demand, facilitating genetic changes in these genes.
Other than these energy metabolism-related genes with high Ka/Ks ratios, our analyses also showed a high Ka/Ks ratio in the rps3 gene among the 12 mitogenomes. Interestingly, the mushroom species Hypsizygus marmoreus also has a high Ka/Ks ratio (1.4) in rps3 gene, consistent with positive selection of this gene and its functional importance (Wang G. et al., 2019). Similarly, the rps3 gene in the entomopathogenic fungus P. chlamydosporia has also likely experienced positive selection, leading to a unique evolutionary pattern in Hypocreales (Lin et al., 2015). The RPS3 protein plays a critical role in ribosome biogenesis and DNA repair in eukaryotes (Kim et al., 2013). This is the only mitochondrial ribosomal protein encoded by mitogenomes in NTFs. It’s currently unknown why this protein underwent diversifying selection among several distinct groups of fungi. This and other issues such as the relevance of our findings on NTF mitogenomes to other functionally important groups of fungi (e.g., those parasitizing insects, plants, and other animals) remain to be investigated.
Data Availability Statement
The datasets generated in this study can be found in GenBank (Accession number MK571436).
Author Contributions
JX, ZY, K-QZ, and YZ conceived and designed the study. YZ and JX wrote the manuscript. GY, CD, and MF conducted the experiments. YZ, GY, CD and MF analyzed the data. YZ and JX revised the manuscript. All authors read and approved the final manuscript.
Funding
This research is jointly supported by the National Natural Science Foundation of China (31760010 and 31770026) to YZ and ZY, the Top Young Talents Program of the Ten Thousand Talents Plan in Yunnan Province to YZ, and the “Double First Class” Research Project in Yunnan University (C176280107) to JX.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00617/full#supplementary-material
TABLE S1 | Dataset of AT skew, GC skew, GC content and length of the 15 protein coding genes in the 12 analyzed mitogenomes.
TABLE S2 | Locations of the rps3 gene in each of the 12 analyzed mitogenomes.
TABLE S3 | Dataset of the relative synonymous codon usage (RSCU) in the 14 mitochondrial protein-coding genes.
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?opt=organelle&taxid4751&sort=Genome
- ^ https://megasun.bch.umontreal.ca/cgi-bin/dev_mfa/mfannotInterface.pl
- ^ https://www.ncbi.nlm.nih.gov/genomes/MICROBES/glimmer_3.cgi
- ^ https://www.ncbi.nlm.nih.gov/orffinder/
- ^ http://codonw.sourceforge.net/index.html
References
Aguileta, G., de Vienne, D., Ross, O., Hood, M., Giraud, T., Petit, E., et al. (2014). High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 6, 451–465. doi: 10.1093/gbe/evu028
Ahren, D., Ursing, B. M., and Tunlid, A. (1998). Phylogeny of nematode-trapping fungi based on 18S rDNA sequences. FEMS Microbiol. Lett. 158, 179–184. doi: 10.1016/S0378-1097(97)00519-3
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A., Dvorkin, M., Kulikov, A., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barron G. L. (ed.) (1977). The Nematode-Destroying Fungi. [Topics in Mycology No. 1.]. Guelph: Canadian Biological Publications.
Basse, C. W. (2010). Mitochondrial inheritance in fungi. Curr. Opin. Microbiol. 13, 712–719. doi: 10.1016/j.mib.2010.09.003
Breton, S., and Stewart, D. (2015). Atypical mitochondrial inheritance patterns in eukaryotes. Genome 58, 423–431. doi: 10.1139/gen-2015-0090
Brooks, A., and Lowe, T. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Chen, H., Sun, S.-C., Norenburg, J., and Sundberg, P. (2014). Mutation and selection cause codon usage and bias in mitochondrial genomes of ribbon worms (Nemertea). PLoS One 9:e85631. doi: 10.1371/journal.pone.0085631
Chen, X. J., and Butow, R. A. (2005). The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 6, 815–825. doi: 10.1038/nrg1708
Darling, A., Mau, B., Blattner, F., and Perna, N. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Deng, C., and Yu, Z. (2019a). The complete mitochondrial genomes of Dactylellina leptospora (Orbiliales, Orbiliaceae). Mitochondrial DNA B 4, 1615–1616. doi: 10.1080/23802359.2019.1604098
Deng, C., and Yu, Z. (2019b). The complete mitochondrial genomes of the nematode-trapping fungus Dactylellina cionopaga. Mitochondrial DNA B 4, 866–867. doi: 10.1080/23802359.2019.1573115
Edgar, R. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Einer, C., Hohenester, S., Wimmer, R., Wottke, L., Artmann, R., Schulz, S., et al. (2017). Mitochondrial adaptation in steatotic mice. Mitochondrion 40, 1–12. doi: 10.1016/j.mito.2017.08.015
Fang, M., Wang, S., Xu, J., Jiang, L., Zhou, D., Zhang, K.-Q., et al. (2019). Characterization of the complete mitochondrial genome of Drechslerella brochopaga, a fungal species trapping nematodes with constricting rings. Mitochondrial DNA B 4, 858–859. doi: 10.1080/23802359.2019.1572466
Giorgi, C., Gissi, C., Pesole, G., and Reyes, A. (1999). Evolutionary genomics in Metazoa: the mitochondrial DNA as a model system. Gene 238, 195–209. doi: 10.1016/s0378-1119(99)00270-x
Gray, M., Burger, G., and Lang, B. (1999). Mitochondrial evolution. Science 283, 1476–1481. doi: 10.1126/science.283.5407.1476
Guo, J.-W., Yu, Z. F., Li, C., and Zhang, K.-Q. (2009). The evaluation of sexual reproduction capacity of orbiliaceous anamorphs. Mycosystema 28, 692–697.
Hawksworth, D. L., and Lücking, R. (2017). Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 5:FUNK-0052-2016. doi: 10.1128/microbiolspec.FUNK-0052-2016
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
James, J., Piganeau, G., and Eyre-Walker, A. (2015). The rate of adaptive evolution in animal mitochondria. Mol. Ecol. 25, 67–78. doi: 10.1111/mec.13475
James, T., Kauff, F., Schoch, C., Matheny, P., Valerie, H., Cox, C., et al. (2006). Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443, 818–822. doi: 10.1038/nature05110
Jiang, L., Zhang, Y., Xu, J., Zhang, K.-Q., and Zhang, Y. (2018). The complete mitochondrial genomes of the nematode-trapping fungus Arthrobotrys oligospora. Mitochondrial DNA B 3, 968–969. doi: 10.1080/23802359.2018.1507651
Jung, P., Friedrich, A., Reisser, C., Hou, J., and Schacherer, J. (2012). Mitochondrial genome evolution in a single protoploid yeast species. G3 2, 1103–1111. doi: 10.1534/g3.112.003152
Kim, Y., Kim, H., and Kim, J. (2013). Cytoplasmic ribosomal protein S3 (rpS3) plays a pivotal role in mitochondrial DNA damage surveillance. BBA Bioenergetics 1833, 2943–2952. doi: 10.1016/j.bbamcr.2013.07.015
Kouvelis, V. N., Sialakouma, A., and Typas, M. A. (2008). Mitochondrial gene sequences alone or combined with ITS region sequences provide firm molecular criteria for the classification of Lecanicillium species. Mycol. Res. 112, 829–844. doi: 10.1016/j.mycres.2008.01.016
Lavín, J. L., Oguiza, J. A., Ramírez, L., and Pisabarro, A. G. (2008). Comparative genomics of the oxidative phosphorylation system in fungi. Fungal Genet. Biol. 45, 1248–1256. doi: 10.1016/j.fgb.2008.06.005
Li, Q., Liao, M., Yang, M., Xiong, C., Jin, X., Chen, Z., et al. (2018a). Characterization of the mitochondrial genomes of three species in the ectomycorrhizal genus Cantharellus and phylogeny of agaricomycetes. Int. J Biol. Macromol. 118, 756–769. doi: 10.1016/j.ijbiomac.2018.06.129
Li, Q., Wang, Q., Cheng, C., Jin, X., Chen, Z., Xiong, C., et al. (2018b). Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. Int. J Biol. Macromol. 119, 792–802. doi: 10.1016/j.ijbiomac.2018.07.197
Li, W.-J., and Yu, Z.-F. (2019). The complete mitochondrial genomes of Dactylella tenuis, a fungus phylogenetically close to nematode-trapping fungus. Mitochondrial DNA B 4, 2704–2705. doi: 10.1080/23802359.2019.1644229
Li, Y., Hyde, K. D., Jeewon, R., Cai, L., Vijaykrishna, D., and Zhang, K. (2005). Phylogenetics and evolution of nematode-trapping fungi (Orbiliales) estimated from nuclear and protein coding genes. Mycologia 97, 1034–1046. doi: 10.3852/mycologia.97.5.1034
Lin, R., Liu, C., Shen, B., Bai, M., Ling, J., Chen, G., et al. (2015). Analysis of the complete mitochondrial genome of Pochonia chlamydosporia suggests a close relationship to the invertebrate-pathogenic fungi in Hypocreales. BMC Microbiol. 15:5. doi: 10.1186/s12866-015-0341-8
Liu, K., Zhang, W., Lai, Y., Xiang, M., Wang, X., Zhang, X., et al. (2014). Drechslerella stenobrocha genome illustrates the mechanism of constricting rings and the origin of nematode predation in fungi. BMC Genomics 15:114. doi: 10.1186/1471-2164-15-114
Losada, L., Pakala, S. B., Fedorova, N. D., Joardar, V., Shabalina, S. A., Hostetler, J., et al. (2014). Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol. Lett. 352, 165–173. doi: 10.1111/1574-6968.12387
Mardanov, A. V., Beletsky, A. V., Kadnikov, V. V., Ignatov, A. N., and Ravin, N. V. (2014). The 203 kbp mitochondrial genome of the phytopathogenic fungus sclerotinia borealis reveals multiple invasions of introns and genomic duplications. PLoS One 9:e107536. doi: 10.1371/journal.pone.0107536
Maréchal, A., and Brisson, N. (2010). Recombination and the maintenance of plant organelle genome stability. New Phytol. 186, 299–317. doi: 10.1111/j.1469-8137.2010.03195.x
Meerupati, T., Andersson, K.-M., Friman, E., Kumar, D., Tunlid, A., and Ahrén, D. (2013). Genomic mechanisms accounting for the adaptation to parasitism in nematode-trapping fungi. PLoS Genet. 9:e1003909. doi: 10.1371/journal.pgen.1003909
Meiklejohn, C., Montooth, K., and Rand, D. (2007). Positive and negative selection on the mitochondrial genome. Trends genet. 23, 259–263. doi: 10.1016/j.tig.2007.03.008
Nordbring-Hertz, B. (1988). Nematophagous fungi: strategies for nematode exploitation and for survival. Microbiol. Sci. 5, 108–116.
Pantou, M. P., Kouvelis, V. N., and Typas, M. A. (2006). The complete mitochondrial genome of the vascular wilt fungus Verticillium dahliae: a novel gene order for Verticillium and a diagnostic tool for species identification. Curr. Genet. 50, 125–136. doi: 10.1007/s00294-006-0079-9
Paquin, B., Laforest, M., Forget, L., Roewer, I., Wang, Z., Longcore, J., et al. (1997). The fungal mitochondrial genome project: evolution of fungal mitochondrial genomes and their gene expression. Curr. Genet. 31, 380–395. doi: 10.1007/s002940050220
Revell, L. (2011). Phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Robicheau, B., Young, A., Labutti, K., Grigoriev, I., and Walker, A. (2016). The complete mitochondrial genome of the conifer needle endophyte, Phialocephala scopiformis DAOMC 229536 confirms evolutionary division within the fungal Phialocephala fortinii s.l. – Acephala appalanata species complex. Fungal Biol. 121, 212–221. doi: 10.1016/j.funbio.2016.11.007
Rubner, A. (1996). Revision of predacious hyphomycetes in the Dactylella-Monacrosporium complex. Stud. Mycol. 39, 1–134.
Salavirta, H., Oksanen, I., Kuuskeri, J., Mäkelä, M., Laine, P., Paulin, L., et al. (2014). Mitochondrial genome of Phlebia radiata is the second largest (156 kbp) among fungi and features signs of genome flexibility and recent recombination events. PLoS One 9:e97141. doi: 10.1371/journal.pone.0097141
Sandor, S., Zhang, Y., and Xu, J. (2018). Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 102, 9433–9448. doi: 10.1007/s00253-018-9350-5
Schoch, C., Sung, G.-H., Lopez-Giraldez, F., Townsend, J., Miadlikowska, J., Valerie, H., et al. (2009). The ascomycota tree of life: a phylum-wide phylogeny clarifies the origin and evolution of fundamental reproductive and ecological traits. Syst. Biol. 58, 224–239. doi: 10.1093/sysbio/syp020
Scholler, M., Hagedorn, G., and Rubner, A. (1999). A reevaluation of predatory orbiliaceous fungi. II. A new generic concept. Sydowia 51, 89–113.
Subramanian, C. V. (1963). Dactylella, Monacrosporium and Dactyllina. J. Ind. Bot. Soc. 42, 291–300.
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substituions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526. doi: 10.1093/oxfordjournals.molbev.a040023
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tzean, S. S., and Estey, R. H. (1981). Species of Phytophthora and Pythium as Nematode-destroying Fungi. J. Nematol. 13, 160–163.
Wallace, D. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Ann. Rev. Genet. 39, 359–407. doi: 10.1146/annurev.genet.39.110304.095751
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., and Yu, J. (2010). KaKs_calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 8, 77–80. doi: 10.1016/s1672-0229(10)60008-3
Wang, G., Lin, J., Shi, Y., Chang, X., Wang, Y., Guo, L., et al. (2019). Mitochondrial genome in Hypsizygus marmoreus and its evolution in Dikarya. BMC Genomics 20:765. doi: 10.1186/s12864-019-6133-z
Wang, S., Fang, M., Xu, J., Jiang, L., Zhou, D., Zhang, K.-Q., et al. (2019). Complete mitochondrial genome and phylogenetic analysis of Orbilia dorsalia, a species producing mature sexual structures on culture. Mitochondrial DNA B 4, 573–574. doi: 10.1080/23802359.2018.1558121
Wang, L., Zhang, S., Li, J.-H., and Zhang, Y.-J. (2018). Mitochondrial genome, comparative analysis and evolutionary insights into the entomopathogenic fungus Hirsutella thompsonii: mitogenome and intron evolution in H. thompsonii. Environmen. Microbiol. 20, 3393–3405. doi: 10.1111/1462-2920.14379
Wang, P., Sha, T., Zhang, Y., Cao, Y., Mi, F., Liu, C., et al. (2017). Frequent heteroplasmy and recombination in the mitochondrial genomes of the basidiomycete mushroom Thelephora ganbajun. Sci. Rep. 7:1626. doi: 10.1038/s41598-017-01823-z
Wilson, A., and Xu, J. (2012). Mitochondrial inheritance: diverse patterns and mechanisms with an emphasis on fungi. Mycology 3, 158–166. doi: 10.1080/21501203.2012.684361
Wu, Y., Yang, J., Yang, F., Liu, T., Leng, W., Chu, Y., et al. (2009). Recent dermatophyte divergence revealed by comparative and phylogenetic analysis of mitochondrial genomes. BMC Genomics 10:238. doi: 10.1186/1471-2164-10-238
Xu, J., and Wang, P. (2015). Mitochondrial inheritance in basidiomycete fungi. Fungal Biol. Rev. 29, 209–219. doi: 10.1016/j.fbr.2015.02.001
Yan, Z., Li, Z., Yan, L., Yu, Y.-T., Yi, C., Chen, J., et al. (2018). Deletion of the sex-determining gene SXI1α enhances the spread of mitochondrial introns in Cryptococcus neoformans. Mobile DNA 9:24. doi: 10.1186/s13100-018-0129-0
Yan, Z., Sun, S., Shahid, M., and Xu, J. (2007). Environment factors can influence mitochondrial inheritance in the fungus Cryptococcus neoformans. Fungal Genet. Biol. 44, 315–322. doi: 10.1016/j.fgb.2006.10.002
Yang, E., Xu, L., Yang, Y., Zhang, X., Xiang, M., Wang, C., et al. (2012). Origin and evolution of carnivorism in the Ascomycota (fungi). Proc. Natl. Acad. Sci. U.S.A. 109, 10960–10965. doi: 10.1073/pnas.1120915109
Yang, J., Wang, L., Ji, X., Feng, Y., Li, X., Zou, C.-G., et al. (2011). Genomic and proteomic analyses of the fungus Arthrobotrys oligospora provide insights into nematode-trap formation. PLoS Pathog. 7:e1002179. doi: 10.1371/journal.ppat.1002179
Yang, Y., and Liu, X. (2006). New generic approach to the taxonomy of predatory anamorphic Orbiliaceae (Ascomycotina). Mycotaxon 97, 153–161.
Yang, Y., Yang, E. C., An, Z. Q., and Liu, X. Z. (2007). Evolution of nematode-trapping cells of predatory fungi of the Orbiliaceae based on evidence from rRNA-encoding DNA and multiprotein sequences. Proc. Natl. Acad. Sci. U.S.A. 104, 8379–8384. doi: 10.1073/pnas.0702770104
Yu, Z. F., Zhang, Y., Qiao, M., Baral, H. O., Weber, E., and Zhang, K. Q. (2006). Drechslerella brochopaga, the anamorph of Orbilia (Hyalinia) orientalis. Mycotaxon 96, 163–168.
Zhang, B., Zhang, Y.-H., Wang, X., Zhang, H., and Lin, Q. (2017). The mitochondrial genome of a sea anemone Bolocera sp. exhibits novel genetic structures potentially involved in adaptation to the deep-sea environment. Ecol. Evol. 7, 4951–4962. doi: 10.1002/ece3.3067
Zhang, Y.-J., Zhang, H.-Y., Liu, X., and Zhang, S. (2017). Mitochondrial genome of the nematode endoparasitic fungus Hirsutella vermicola reveals a high level of synteny in the family Ophiocordycipitaceae. Appl. Microb. Biotechnol. 101, 3295–3304. doi: 10.1007/s00253-017-8257-x
Zhang, W., Chen, X., Liu, X., and Xiang, M. (2016). Genome studies on nematophagous and entomogenous fungi in China. J. Fungi 2:9. doi: 10.3390/jof2010009
Zhang, Y., Zhang, Y. R., Dong, J. Y., He, X. X., Qiao, M., Baral, H. O., et al. (2016). Orbilia tianmushanensis sp nov., a new member of the O. luteorubella group with an unusual asexual morph. J. Microb. 54, 9–13. doi: 10.1007/s12275-016-5369-4
Zhang, Y., Yu, Z.-F., Xu, J., and Zhang, K.-Q. (2011). Divergence and dispersal of the nematode-trapping fungus Arthrobotrys oligospora from China. Environ. Microb. Rep. 3, 763–773. doi: 10.1111/j.1758-2229.2011.00297.x
Zhang, Y.-Q., and Yu, Z. (2019). The complete mitochondrial genomes of the nematode-trapping fungus Arthrobotrys musiformis. Mitochondrial DNA B 4, 979–980. doi: 10.1080/23802359.2019.1581106
Zheng, B.-Y., Cao, L.-J., Tang, P., van Achterberg, C., Hoffmann, A., Chen, H., et al. (2018). Gene arrangement and sequence of mitochondrial genomes yield insights into the phylogeny and evolution of bees and sphecid wasps (Hymenoptera: Apoidea). Mol. Phylogenet. Evol. 124, 1–9. doi: 10.1016/j.ympev.2018.02.028
Keywords: entomopathogenic fungi, anamorphic Orbiliaceae, mitochondrial phylogeny, adaptive evolution, rps3
Citation: Zhang Y, Yang G, Fang M, Deng C, Zhang K-Q, Yu Z and Xu J (2020) Comparative Analyses of Mitochondrial Genomes Provide Evolutionary Insights Into Nematode-Trapping Fungi. Front. Microbiol. 11:617. doi: 10.3389/fmicb.2020.00617
Received: 25 January 2020; Accepted: 19 March 2020;
Published: 15 April 2020.
Edited by:
Tomasz Kulik, University of Warmia and Mazury in Olsztyn, PolandReviewed by:
Yongjie Zhang, Shanxi University, ChinaCloe Pogoda, University of Colorado Boulder, United States
Copyright © 2020 Zhang, Yang, Fang, Deng, Zhang, Yu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zefen Yu, emZ5dXFtQGhvdG1haWwuY29t; Jianping Xu, anB4dUBtY21hc3Rlci5jYQ==; anB4dW1jbWFzdGVyQGdtYWlsLmNvbQ==