 Ana G. Abril1
Ana G. Abril1 Mónica Carrera2*
Mónica Carrera2* Karola Böhme3
Karola Böhme3 Jorge Barros-Velázquez4
Jorge Barros-Velázquez4 Benito Cañas5
Benito Cañas5 Jose L. R. Rama1
Jose L. R. Rama1 Tomás G. Villa1
Tomás G. Villa1 Pilar Calo-Mata4*
Pilar Calo-Mata4*- 1Department of Microbiology and Parasitology, Faculty of Pharmacy, University of Santiago de Compostela, Santiago de Compostela, Spain
- 2Department of Food Technology, Spanish National Research Council, Marine Research Institute, Vigo, Spain
- 3Agroalimentary Technological Center of Lugo, Lugo, Spain
- 4Department of Analytical Chemistry, Nutrition and Food Science, School of Veterinary Sciences, University of Santiago de Compostela, Lugo, Spain
- 5Department of Analytical Chemistry, Complutense University of Madrid, Madrid, Spain
The present work focuses on LC-ESI-MS/MS (liquid chromatography-electrospray ionization-tandem mass spectrometry) analysis of phage-origin tryptic digestion peptides from mastitis-causing Streptococcus spp. isolated from milk. A total of 2,546 non-redundant peptides belonging to 1,890 proteins were identified and analyzed. Among them, 65 phage-origin peptides were determined as specific Streptococcus spp. peptides. These peptides belong to proteins such as phage repressors, phage endopeptidases, structural phage proteins, and uncharacterized phage proteins. Studies involving bacteriophage phylogeny and the relationship between phages encoding the peptides determined and the bacteria they infect were also performed. The results show how specific peptides are present in closely related phages, and a link exists between bacteriophage phylogeny and the Streptococcus spp. they infect. Moreover, the phage peptide M∗ATNLGQAYVQIM∗PSAK is unique and specific for Streptococcus agalactiae. These results revealed that diagnostic peptides, among others, could be useful for the identification and characterization of mastitis-causing Streptococcus spp., particularly peptides that belong to specific functional proteins, such as phage-origin proteins, because of their specificity to bacterial hosts.
Introduction
Streptococcus spp. are among the main mastitis pathogens present in dairy products (Forsman et al., 1997; Böhme et al., 2012). The genus Streptococcus includes numerous mastitis-causing species that are responsible for high economic losses as well as human health issues (Lopez-Sanchez et al., 2012; Richards et al., 2014). The major species involved in both clinical and subclinical mastitis are Streptococcus agalactiae, Streptococcus canis, Streptococcus dysgalactiae and Streptococcus uberis (Lundberg et al., 2014; Richards et al., 2014). Additionally, Streptococcus gallolyticus (Dumke et al., 2015) and Streptococcus parauberis (Park et al., 2013) have been reported as minor mastitis agents.
It is well known that Streptococcus spp. may carry temperate bacteriophages in their genomes (Brüssow and Desiere, 2001; Romero et al., 2004; Fortier and Sekulovic, 2013). These phages are usually integrated into bacterial chromosomes as prophages, wherein they may provide new and beneficial properties to the host, or in contrast, they may disrupt genes, thus affecting their expression (Fortier and Sekulovic, 2013). Phage genome excisions and integrations are mediated by phage-encoded DNA recombinases (Menouni et al., 2015), which can act at specific phage attachment sites in the bacterial genome that are identical to those present in the phage genome. Some phages can integrate randomly within the bacterial genome; for example, phage Mu (as long as a particular gene is not expressed). It is evident that the interaction of streptococcal species with bacteriophages may greatly alter the variability in bacterial populations (Feiner et al., 2015).
Only 3% of phage genomes in the NCBI nucleotide database represent active phages against Streptococcus spp. (Harhala et al., 2018). Some phages have been reported and described in S. uberis (Hill and Brandy, 1989), S. agalactiae (Domelier et al., 2009; Bai et al., 2013), and S. dysgalactiae (Davies et al., 2007) by means of different methods, such as molecular characterization and complete genome sequencing. There are some well-known Siphoviridae phages of Streptococcus spp., including the species Sfi21, Sfi11, and Sfi19, which are mainly found in Streptococcus thermophilus (Brüssow and Desiere, 2001; Canchaya et al., 2004). Additionally, the genome sequence of EJ-1, a Myoviridae phage of Streptococcus pneumoniae, has been reported (Romero et al., 2004).
The interaction between bacteria and phages, including pathogenic and industrially applicable bacteria, is a rapid coadaptation that allows highly dynamic coevolution (Koskella and Brockhurst, 2014). It is known that closely related phages normally occupy the same genome insertion site in different bacteria. A specific site can be occupied by a phage, empty in another strain or replaced by an altogether different phage (Canchaya et al., 2004).
Classic culture-based methods used for the detection of pathogenic bacteria (Ivnitski et al., 1999; Chakravorty et al., 2007) and their phages (Gantzer et al., 1998; Ackermann, 2007) are time-consuming and laborious procedures. Therefore, new rapid molecular microbial diagnostic methods based on genomics and proteomics tools have been developed to achieve faster and more efficient detection, identification and strain biotyping of bacterial species (Lasch et al., 2009; Böhme et al., 2011, 2012; Branquinho et al., 2014; Quintela-Baluja et al., 2014). As bacteriophages are highly specific for their host pathogens, phage typing is a classic technique used for bacterial detection and characterization (Craigie and Yen, 1938). Phage receptor-binding proteins (RBPs), as well as the study of their nucleic acids, the use of antibodies, and phage-display peptides (PDPs), have been studied as biosensors for pathogen detection (Rees and Voorhees, 2005; Lavigne et al., 2009; Singh et al., 2013; Chanishvili, 2015; Richter et al., 2018).
Proteomics techniques using matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) and liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) instruments have been used for the analysis and detection of pathogenic bacterial strains based on specific diagnostic peptides (Calo-Mata et al., 2016; Pfrunder et al., 2016). In addition, LC-ESI-MS/MS techniques have been employed for the identification and detection of bacterial bacteriophages such as the bacteriophage lambda (Serafim et al., 2017). However, no study has been published for Streptococcus spp. phage detection and identification by LC-ESI-MS/MS so far without previously phage purification for the analysis. With this method, putative temperate phages in addition to virulent phages present in the analyzed strains were identified.
In this work, we aimed to study for the first time the proteomics of specific peptides of streptococcal species for the identification of both phage and bacterial strains by LC-ESI-MS/MS.
Materials and Methods
In this study, tryptic digestion peptides from the mastitis-causing strain Streptococcus spp. isolated from milk were analyzed by LC-ESI-MS/MS. A total of 100 μg of protein extraction was digested with trypsin, cleaned on a C18 microSpinTM, and analyzed by LC-MS/MS. Proteomic data were processed by SEQUEST (Proteome Discoverer 1.4 package, Thermo Fisher Scientific) against bacteria in the UniProt/TrEMBL database. The overall time of culture and protein extraction and purification was 3 days (depending on the strain, the time of growth may have been 1 or 2 days, another day for protein extraction and an additional day for protein purification). Mass spectrometry analysis and peptide detection took 2–3 h, according to the methodology employed in the present work.
Bacteria
A total of 14 Streptococcus spp. strains (Table 1) were used in this study. Five reference strains were obtained from different culture collections (ST1, ST2, ST3, and ST14 from the Spanish culture collection and ST4 and ST5 from the German culture collection). Additionally, 9 strains were isolated from the milk of mastitis cows at the LHICA (Laboratorio de Higiene, Inspección y Control de Alimentos) (ST6, ST7, ST8, ST9, ST10, and ST11) laboratory, Faculty of Veterinary Sciences, University of Santiago de Compostela, Spain. These strains were genetically identified as Streptococcus spp. based on the VITEK 2 system and 16S rRNA gene sequencing (Alnakip et al., 2014; Quintela-Baluja et al., 2014). The strains were reactivated in brain heart infusion (BHI, Oxoid Ltd., Hampshire, United Kingdom) and incubated at 31°C for 24 h. Bacterial cultures were then grown on plate count agar (PCA, Oxoid) at 31°C for 24 h.
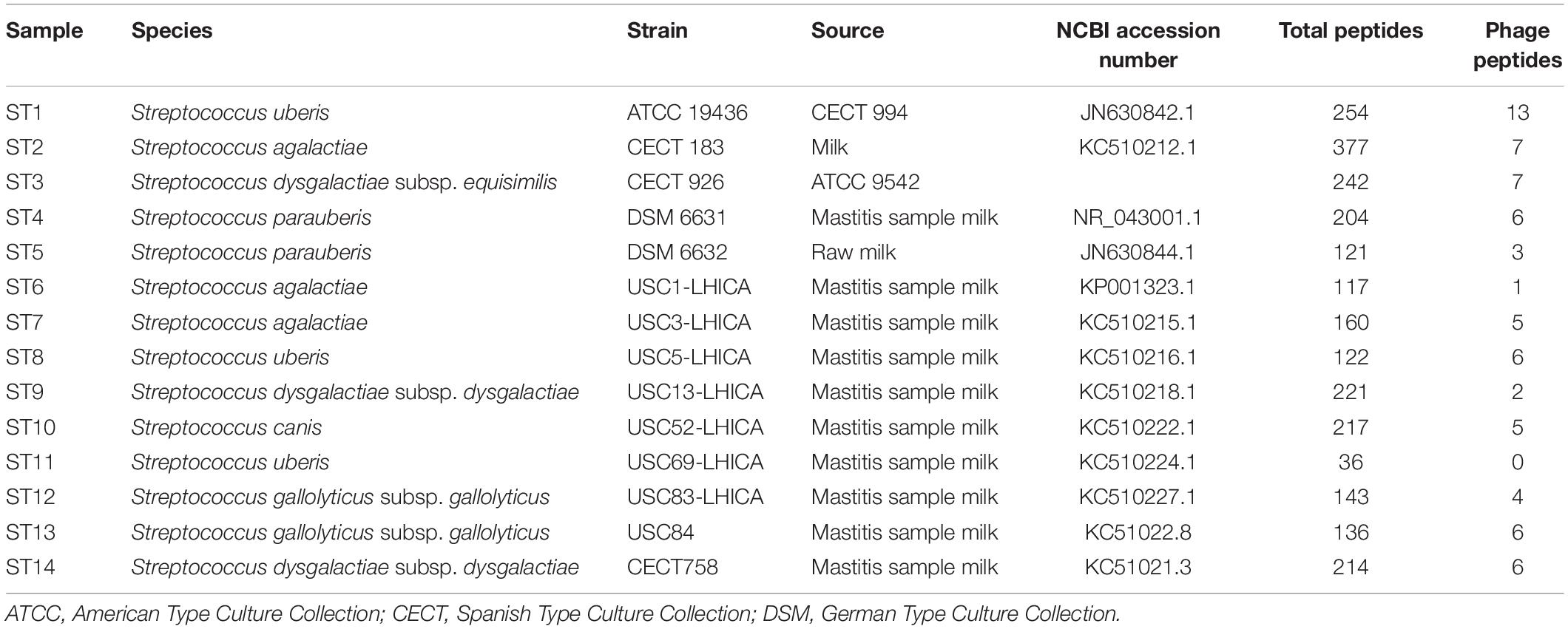
Table 1. Streptococcus spp. strains used in this study; total peptides were identified by LC-ESI-MS/MS and number of phage peptides was analyzed by BLASTp.
Protein Extraction
Protein extraction was carried out as described previously (Böhme et al., 2010a, b; Carrera et al., 2017). Briefly, a loop full of each bacterial culture was harvested, and the biomass was mixed with 100 μL of a solution containing 50% acetonitrile (50% ACN) (Merck, Darmstadt, Germany) and 1% aqueous trifluoroacetic acid (1% TFA) (Acros Organics, NJ, United States). After vortexing and centrifugation, the supernatant was treated with a solution of lysis buffer [60 mM Tris-HCl pH 7.5, 1% lauryl maltoside, 5 mM phenylmethanesulfonyl fluoride (5 mM PMSF) and 1% dithiothreitol (1% DTT)]. The supernatants were transferred to a new tube and then quantified using the bicinchoninic acid method (Sigma Chemical Co., United States). All analyses were performed in triplicate.
Peptide Sample Preparation
Protein extracts were subjected to in-solution tryptic digestion (Carrera et al., 2013). A total of 100 μg of protein extract was dried under vacuum in a SpeedVac (CentriVap, Labconco Co., United States) and resuspended for denaturation in 25 μL of 8 M urea in 25 mM ammonium bicarbonate (pH 8.0). After 5 min of sonication, DTT was added to a final concentration of 10 mM, and the solution was incubated at 37°C for 1 h. Alkylation was performed with the addition of iodoacetamide to a final concentration of 50 mM, and the solution was incubated for 1 h at room temperature in darkness. Then, the sample was diluted fourfold with 25 mM ammonium bicarbonate, pH 8.0, to decrease the urea concentration. Finally, proteins were digested with trypsin (1:100 protease-to-protein ratio) (Promega, WI, United States) at 37°C overnight.
Shotgun LC-ESI-MS/MS Analysis
Peptide digests were acidified with formic acid (FA), cleaned on a C18 MicroSpinTM column (The Nest Group, Southborough, MA, United States) and analyzed by LC-ESI-MS/MS using a Proxeon EASY-nLC II Nanoflow system (Thermo Fisher Scientific, San Jose, CA, United States) coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) (Calo-Mata et al., 2016; Carrera et al., 2017). The peptide separation (2 μg) was performed on a reverse-phase (RP) column (EASY-Spray column, 50 cm × 75 μm ID, PepMap C18, 2 μm particles, 100 Å pore size, Thermo Fisher Scientific) with a 10 mm precolumn (Accucore XL C18, Thermo Fisher Scientific) using a linear 120 min gradient from 5 to 35% solvent B (solvent A, 98% water, 2% ACN, 0.1% FA; solvent B, 98% ACN, 2% water, 0.1% FA) at a flow rate of 300 nL/min. For ionization, a spray voltage of 1.95 kV and a capillary temperature of 230°C were used. Peptides were analyzed in positive mode from 400 to 1,600 amu (1 μscan), followed by 10 data-dependent collision-induced dissociation (CID) MS/MS scans (1 μscan) using an isolation width of 3 amu and a normalized collision energy of 35%. Fragmented masses were set in dynamic exclusion for 30 s after the second fragmentation event, and unassigned charged ions were excluded from MS/MS analysis.
LC-ESI-MS/MS Mass Spectrometry Data Processing
The results from the LC-ESI-MS/MS spectra were searched against the Streptococcus UniProt/TrEMBL database (13,528 reviewed and 1,290,635 unreviewed protein sequence entries) using SEQUEST-HT (Proteome Discoverer 2.1, Thermo Fisher Scientific). The following constraints were used for the searches: semitryptic cleavage with up to two missed cleavage sites and tolerance windows of 1.2 Da for precursor ions and 0.6 Da for MS/MS fragment ions. The variable modifications that were allowed were as follows: (M∗) methionine oxidation (+15.99 Da), (C∗) carbamidomethylation of Cys (+57.02 Da) and acetylation of the N-terminus of the protein (+42.0106 Da). The database search results were subjected to statistical analysis with the Percolator algorithm (Käll et al., 2007). To validate the peptide assignments, the false discovery rate (FDR) was kept below 1%.
Selection of Potential Peptide Biomarkers
To study specificity, we used the BLASTp algorithm on each peptide that was identified by LC-ESI-MS/MS to determine homologies and exclusiveness with protein sequences registered in the NCBI database (Altschul et al., 1990). To search for BLASTp parameters, we included and excluded Streptococcaceae taxa with the aim of identifying which peptides belong to Streptococcus spp. bacteriophages.
Phage Genome Comparison and Relatedness
Genomes of all studied Streptococcus spp. phages were downloaded from the GenBank database and analyzed and compared with the server VICTOR (Virus Classification and Tree Building Online Resource1); the intergenomic distances were calculated, and a phylogenomic tree was constructed (Meier-Kolthoff and Göker, 2017).
Holin gene sequences of the studied bacteriophages from the GenBank database were compared. Sequences were aligned using Clustal_X software (Thompson et al., 1997), and distances were calculated according to Kimura’s two-parameter model (Kimura, 1980). The phylogenetic trees were inferred using maximum likelihood (ML) models (Saitou and Nei, 1987; Rogers and Wofford, 1998). MEGA 7.09 software (Kumar et al., 2016) was used for the analyses.
Results
Streptococcus spp. Proteome Repository
Protein mixtures from each of the 14 different Streptococcus spp. strains were digested with trypsin and analyzed by LC-ESI-MS/MS.
A total of 2,546 non-redundant peptides corresponding to 1,890 different annotated proteins were identified for all Streptococcus spp. strains. Among them, 69 different peptides were identified as phage peptides. These phage peptides were selected, and the BLASTp algorithm was used to determine the homology and exclusiveness of these peptides with protein sequences registered in the NCBI database (Altschul et al., 1990). For the BLASTp parameters, the term Streptococcaceae was included and excluded with the aim of finding peptides belonging to Streptococcus spp. bacteriophages. Supplementary Table S1 in Supplementary Data 1 summarizes the list of 65 possible specific streptococcal bacteriophage peptides, bacterial peptides with putative phage origin, and bacteria and phages with 100% homology with respect to the protein NCBI database. All streptococcal phage peptides with 100% homology were found to belong mainly to Siphoviridae (Sfi21 and Sfi11 genera) and to Podoviridae (unclassified genera). These Siphoviridae and Podoviridae family genomes are usually organized into functional modules such as lysogeny, DNA replication, packaging, morphogenesis and lysis (Lucchini et al., 1999b; Xia and Wolz, 2014).
In some cases, peptides assigned to phage proteins did not correspond to any described phages in the NCBI database; for example, peptide LIDAELTKAGVRDAEIFGK from strain ST1 (Supplementary Table S1 in Supplementary Data 1). Furthermore, four phage peptides were identified only in streptococcal strains analyzed in this study but were not described in bacteria in the literature; for example peptide VKEKIAAIVNDEELLAK from strain ST1. Strikingly, 64 peptides found in this study were described for the first time in Streptococcus spp. analyzed; for peptide ITTGIISAARIGAEAITADKLK from strain ST1 (Supplementary Table S1 in Supplementary Data 1). This means that only one identified peptide was previously described from the species analyzed. Furthermore, the peptide M∗ATNLGQAYVQIM∗PSAK is highly specific to S. agalactiae in the NCBI database and S. agalactiae ST2 analyzed in this study (Supplementary Table S1 in Supplementary Data 1). Figure 1 shows the MS/MS spectrum for the M∗ATNLGQAYVQIM∗PSAK species-specific peptide biomarker from S. agalactiae.
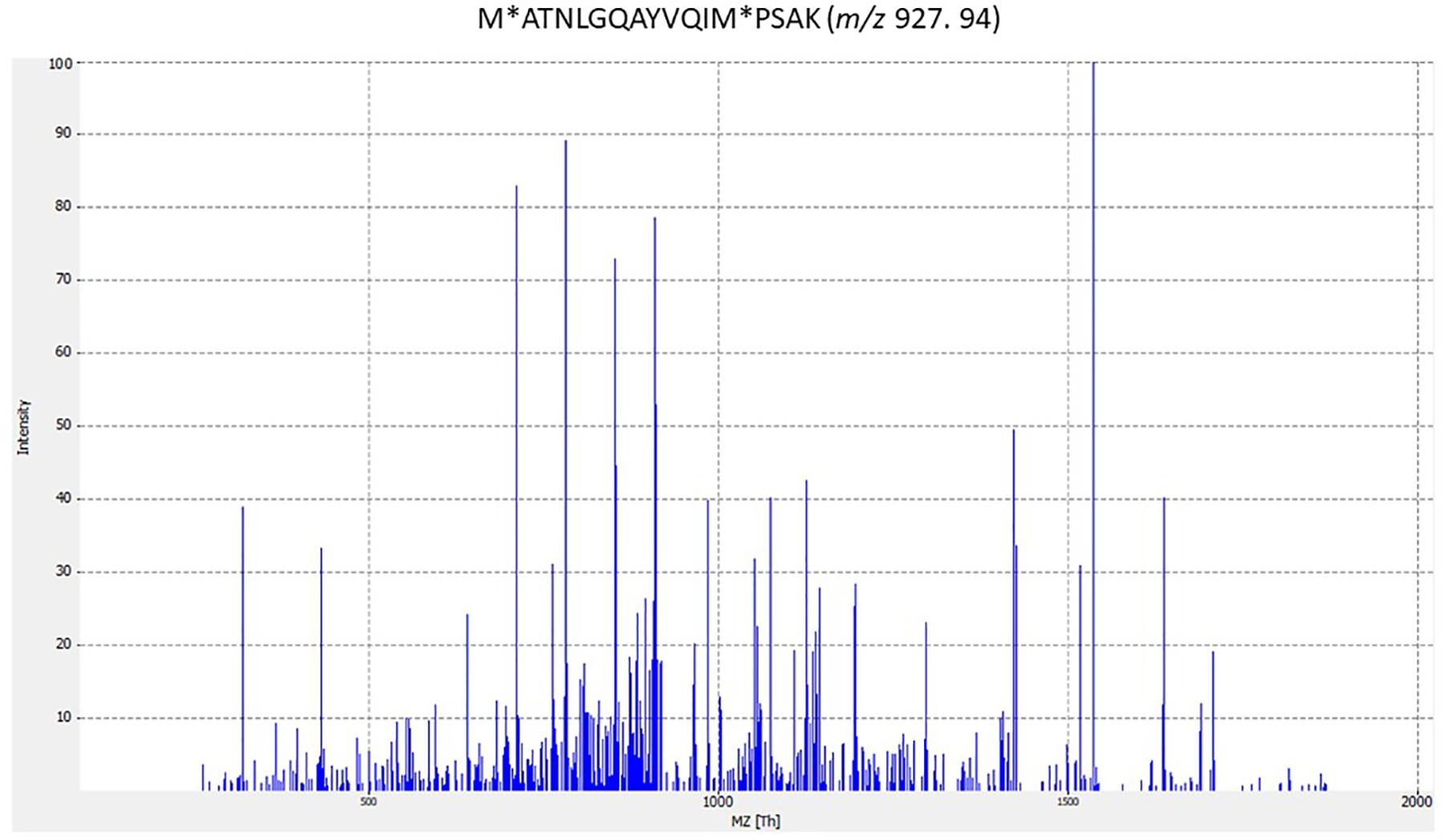
Figure 1. MS/MS spectrum for M∗ATNLGQAYVQIM∗PSAK species-specific peptide biomarker from Streptococcus agalactiae. The corresponding peptides were tested for specificity to S. agalactiae using the BLASTp algorithm. (M∗ methionine oxidation).
Phage Peptides Identified From the Analyzed S. uberis Strains
ST1, ST8, and ST11 were the strains analyzed for the species S. uberis. For strains ST1 and ST8, thirteen and five peptides were identified, respectively. The S. uberis tryptic peptides from strains ST1 and ST8 were found to possibly belong to twelve and twenty-five different described phages, respectively, based on a similarity search. The different peptides from strains ST1 and ST8 could belong to the same bacteriophage, P4761. The peptides identified in each strain and the bacteriophages in which the peptides were present based on similarity are shown in Supplementary Table S1 in Supplementary Data 1.
The majority of the determined ST1 peptides belonged to structural proteins such as head and tail proteins, a phage minor structural protein from the GP20 family, a Gp58-like protein and a tape measure protein. Gp58 is a minor structural protein previously described in Lactobacillus delbrueckii subsp. lactis bacteriophage LL-H (Mikkonen and Alatossava, 1994). The tape measure protein (TMP) determines the tail length and facilitates DNA entry into the cell during infection, and it has been reported that some TMPs carry lysozyme-like and peptidase domains (Rodriguez-Rubio et al., 2012; Mahony et al., 2016). A peptide from the RecT protein was also identified from an ST1 strain. RecT is a RecA-dependent, RecA-independent DNA single-strand annealing protein (SSAP) that belongs to the RecT/Redβ superfamily and is involved in DNA recombination pathways. All the mentioned SSAPs seem to be of bacteriophage origin; RecT proteins, on the other hand, have been described in several bacteria, while Redβ has only been found in lambdoid phages (Iyer et al., 2002; Menouni et al., 2015). Additionally, the phage endopeptidase and matrix-binding protein EbhB was determined from S. uberis strain ST1. It was reported that EbhB is an adhesin consisting of several domains encoding the largest protein in Staphylococcus aureus and is involved in agglutination, attachment to host extracellular matrix, biofilm accumulation, and escape from phagocytosis (Walker et al., 2013). For strain S. uberis ST8, the determined peptides belonged to a Gp10 family protein, a Gp348 protein, a phage integrase and two uncharacterized phage proteins. One of these uncharacterized phage peptides (Supplementary Table S1 in Supplementary Data 1) presents the same sequence as peptides from twenty-three different phages described in the NCBI database. As an identified protein in strain ST8, Gp10 is a putative tail component found in Caudovirales, and Gp348 is definitively the main capsid protein E (GpE); upon assembly, GpE acquires icosahedral symmetry, thus stabilizing the DNA in the head (Dokland and Murialdo, 1993).
Determined Phage Peptides From the Analyzed S. parauberis Strains
ST4 and ST5 were the strains analyzed for the species S. parauberis. For strain ST4, four peptides were determined, and three peptides were determined for the ST5 strain. The peptides from S. parauberis strains ST4 and ST5 were found to belong to sixteen and five different described phages, respectively, as determined with the NCBI database. In summary, the determined peptides based on similarity for each strain and the bacteriophages in which these peptides are present are shown in Supplementary Table S1 in Supplementary Data 1.
Among the ST4-determined peptides, a phage peptide from a tail protein, an uncharacterized phage peptide and a peptide from a DUF1372 domain of unknown function were identified. It should be noted that there was a large number of uncharacterized protein sequences in databases and that more than 20% of all protein domains are annotated as “domains of unknown function” (DUFs) (Bateman et al., 2010; Goodacre et al., 2014). Several uncharacterized phage proteins and DUFs from Streptococcus spp. bacteriophages were identified from the analyzed strains (Bateman et al., 2010; Goodacre et al., 2014). The DUF1372 domain-containing proteins included several sequences belonging to Streptococcus spp. bacteriophage as well as other related proteins in Streptococcus spp. In addition, for strain ST4, a peptide from the GDSL-like family of lipases/acylhydrolases identical to some Streptococcus spp. phages was identified; GDSL-like family proteins are hydrolases that exhibit a variety of properties, including known broad substrate specificity and regiospecificity (Akoh et al., 2004).
For S. parauberis strain ST5, the determined peptides included a phage tail fiber PblB peptide (surface protein). In this sense, the surface proteins PblB and PblA, which encode streptococcal phages, are incorporated into phage particles and are involved in tail assembly, suggesting that both proteins are of bacterial prophage origin. These proteins were reported in Streptococcus mitis and increase platelet-binding ability (Bensing et al., 2001; Mitchell et al., 2007). Furthermore, PblA was also found in Streptococcus oralis, Streptococcus pyogenes and S. pneumoniae. However, both proteins, PblB and PblA, are uncommon since no known protein has strong similarity to these bacterial adhesins. For strain ST5, an uncharacterized protein peptide and peptides of the Cro/CI family of phage repressors were also identified. As integrases and regulator proteins, CI and Cro are encoded in the lysogeny module. If the CI repressor predominates, the phage will remain in the lysogenic state, but if Cro outnumbers CI, the phage will enter the lytic cycle. The most extended regulator in the prokaryotic world is the xenobiotic XRE, which contains a helix-turn-helix (HTH) conformation, is involved in DNA binding and shows similarity to the Cro repressor of phage λ (Durante-Rodríguez et al., 2016). In the NCBI database, peptides of the CI/Cro repressor types are usually named XRE-family proteins for bacteria.
Phage Peptides Identified From the Analyzed S. dysgalactiae Strains
Streptococcus dysgalactiae strains ST3, ST9 and ST14 were analyzed in this study. For strains ST3 and ST9, seven and two peptides were identified, respectively. For ST14, five peptides were determined, and one of them could tentatively belong to two previously described phages. The different peptides from S. dysgalactiae strains ST3 and ST9 were found to belong to six and one different described phage, respectively, according to the NCBI database. In summary, the determined peptides from each strain and the bacteriophages in which the peptides are present, based on similarity, are shown in Supplementary Table S1 in Supplementary Data 1.
Four peptides identified from the ST3 strain belong to proteins of the structural module, including a major tail protein, a phage capsid protein, a head-tail connector and a phage assembly chaperone protein. This last protein is required in the morphogenesis of long-tailed phages (Pell et al., 2013). A phage integrase and a phage repressor of the Cro/CI type from the lysogeny module were also identified from the ST3 strain. Furthermore, a phage-like HicB family antitoxin was identified from this strain. This protein comes from the toxin–antitoxin loci that encode toxin and antitoxin proteins (the antitoxin has a DNA-binding domain and autoregulates the operon). This operon exhibits a complex phylogeny due to HGT (Horizontal gene transfer), and proteins of this family are often detected in phages, plasmids and bacterial genomes (Jørgensen et al., 2009).
The peptides of strain ST9 were identified as a prophage pi2 protein peptide from the morphogenesis module and an uncharacterized protein peptide. For ST14, a phage integrase family peptide, a phage tail tape measure peptide, a repressor of the Cro/CI type peptide and one uncharacterized protein peptide were determined by LC-ESI-MS/MS (Supplementary Table S1 in Supplementary Data 1).
Phage Peptides Identified From the Analyzed S. agalactiae Strains
The strains analyzed for S. agalactiae were ST2, ST6, and ST7. For strain ST2, seven peptides were identified, and for ST6 and ST7, one and five peptides were determined, respectively (Supplementary Data 1). Additionally, S. agalactiae tryptic peptides from strains ST2 and ST7 were found to belong to seven and one different described phage, respectively. For S. agalactiae ST6, a phage peptide was determined, but it was not associated with a described characterized phage in the NCBI database.
Some of the identified peptides from ST2 belong to proteins of the morphogenesis module, including a phage tail protein, phage envelope protein and a minor phage structural protein. The identified peptide from the pblA tail fiber protein from strain ST2 is very specific for the species S. agalactiae (M∗ATNLGQAYVQIM∗PSAK) (Figure 1), in contrast to the analyzed peptide sequences that are present in streptococcal species (Supplementary Data 1). A phage replication protein peptide, phage-related chromosomal island protein (PrCI) peptide, and an uncharacterized protein peptide were also identified for strain ST2. PrCIs are characterized by a specific set of phage-related functions that allow transductional recombination events and carry genes involved in biofilm formation, superantigen synthesis, antibiotic resistance and phage resistance and encoding ATP-binding cassette (ABC) transporters. Several PrCIs have been identified in S. aureus, S. pyogenes, Enterococcus. faecalis and Lactococcus lactis (Novick et al., 2012). In addition, phages can carry CRISPR (clustered regularly interspaced short palindromic repeats) arrays that, with associated Cas proteins, target a chromosomal island of the bacterial host. S. agalactiae contains two CRISPR/Cas systems in its genome. One of them is present in several strains, and the other is ubiquitous. The S. agalactiae 2-A CRISPR1/Cas system preferentially targets ICE (integrative conjugative elements) and bacteriophages normally widespread among S. agalactiae. Likewise, S. pyogenes and S. thermophilus spacers match described sequences in viruses (Lopez-Sanchez et al., 2012; Koskella and Brockhurst, 2014).
For strain ST6, a peptide from an uncharacterized protein was identified. Identified peptides from strain ST7 belong to a major capsid protein, a tape measure protein and a phage integrase. Additionally, a DnaD and phage-associated domain-containing protein peptide and a tail fiber pblB peptide were identified in S. agalactiae ST7. This tail fiber pblB peptide was also identified in S. parauberis ST5, and this was the first time that this protein was identified in S. agalactiae and S. parauberis species. The aforementioned DnaD and phage-associated domain-containing proteins are DNA replication proteins that are involved in the primosomal step of DNA replication and DNA remodeling; these proteins can also bind to DNA and form large nucleoprotein complexes. DnaD-like motifs have been found in proteins of unknown function both in bacterial and phage proteins. Phage 7201 of S. thermophilus has a DnaD-like domain that is linked to a RepA-like domain thus far associated with the initiation of DNA replication, and as such, it is likely that such a domain is in fact an essential common structural module in the primosomal complex (Carneiro et al., 2006; Huang Y. H. et al., 2016).
Phage Peptides Identified From the Analyzed S. canis Strain
Five peptides were determined from the analyzed S. canis strain ST10, including phage repressors of the Cro/CI type, a Gp58-like protein, a major tail protein, a phage integrase and an uncharacterized protein peptide. The peptides from S. canis strain ST10 were found to belong to three different described phages (Supplementary Table S1 in Supplementary Data 1). The function of these proteins was described above.
Phage Peptides Identified From the Analyzed S. gallolyticus Strains
The S. gallolyticus strains analyzed were ST12 and ST13. Four peptides were identified from strain ST12, and six peptides were identified from strain ST13. Two ST12 peptides shared the same peptide sequence with two described phages in the NCBI database (Supplementary Table S1 in Supplementary Data 1), and the same applied for strain ST13.
Among the ST12 peptides, a phage endopeptidase peptide and an uncharacterized protein phage from phage phiARI0131-1 were determined. Additionally, a cytosine methyltransferase peptide was identified in strain ST12. Cytosine methyltransferase is a type II DNA methyltransferase (MTase) C5-methylcytosine (Class III) currently found in the prokaryotic world and associated with a restriction endonuclease that forms a restriction modification (R/M) system, thus protecting bacterial cells from foreign DNA. These methyltransferases have also been found in phage genomes, and they exhibit the ability to confer protection from REases (cognate restriction endonuclease) of their bacterial host(s) (Murphy et al., 2013). Furthermore, for S. gallolyticus ST12, a PBSX family phage terminase peptide was identified, and it is involved in DNA packaging and has also been implicated in endonuclease, ATPase, and double-stranded DNA binding activities (Gual et al., 2000). A PBSX family peptide, a phage endopeptidase peptide, a head tail connector peptide, a phage tape measure peptide and two uncharacterized protein peptides were identified in strain ST13.
Additionally, different strains from different species presented peptide sequences that belong to the same described phages (Supplementary Table S1 in Supplementary Data 1).
Streptococcus spp. Phage Genome Comparison and Their Relatedness
A phylogenomic tree of the complete genome of Streptococcus spp. phages from the NCBI database (accession numbers can be found in Supplementary Table S2 in Supplementary Data 1) with 100% similarity to those found in this study was built (Figure 2). Many phages found in this study were classified in the order Caudovirales but not in families, and some were unclassified. Genomes of well-known phages of different families including Siphoviridae, Myoviridae, and Podoviridae, such as phage lambda, T4 and T7, respectively, were added for comparison purposes. The analysis of genomes showed five well-defined clusters. To the best of our knowledge, this is the first time that phages from mastitis-causing streptococci were grouped in a phylogenetic tree. Two principal branches separated clusters A, B, C, and D from cluster E. Clusters A and B were located in the same secondary branch, and the same occurred with clusters C and D. Cluster A was formed by Streptococcus/Siphoviridae viruses, most of which were classified as Sfi21 dt1 viruses. Cluster B was formed by unclassified Streptococcus phages, and 2 Streptococcus phages were classified as Siphoviridae. Cluster C was formed by unclassified Streptococcus phages, and cluster D, which is related to cluster C, was formed by phage lambda, T4 and T7. Finally, cluster E was formed by Streptococcus/Podoviridae phages and unclassified Streptococcus phages.
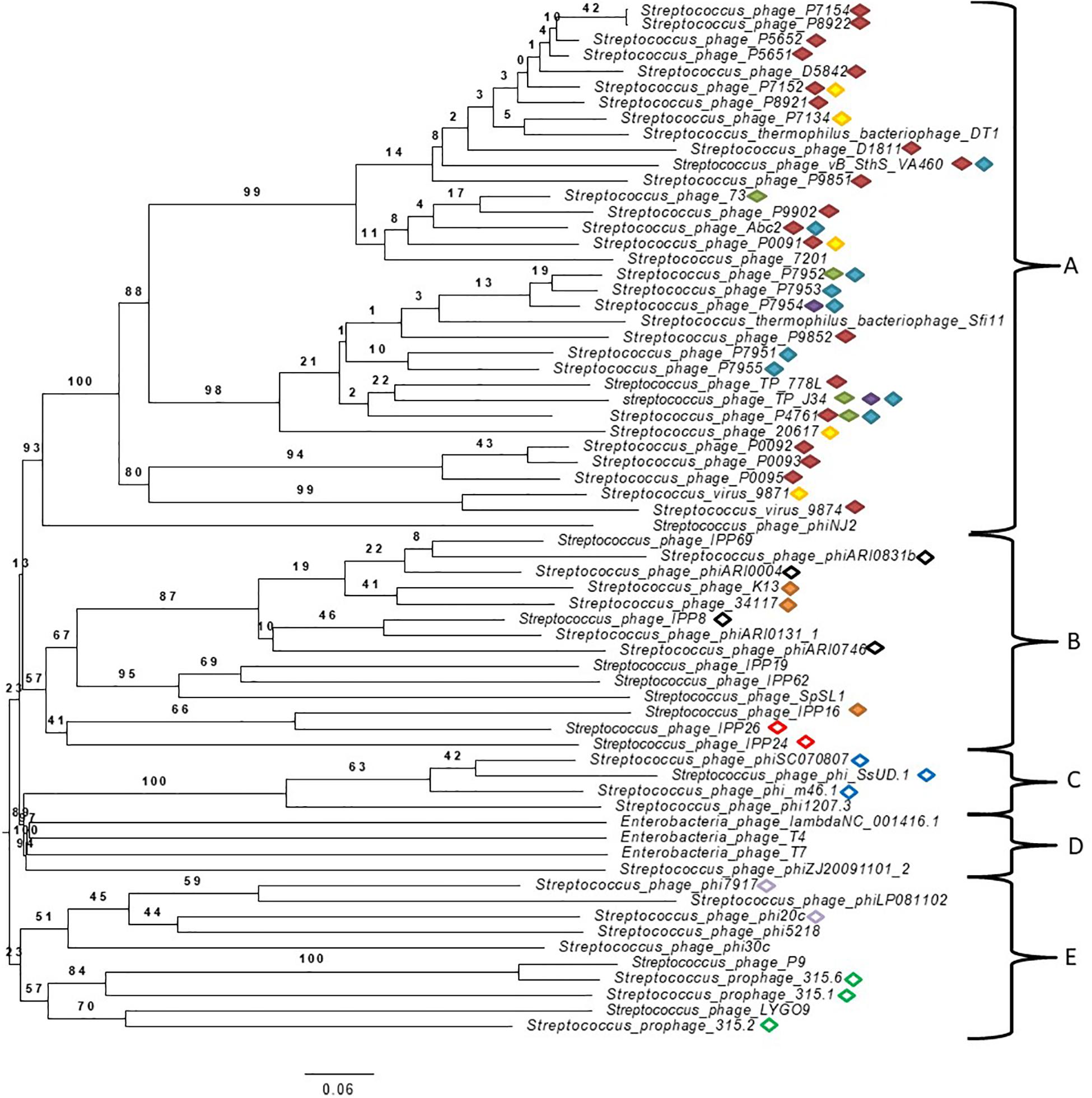
Figure 2. Phylogenetic tree generated by VICTOR using the complete genomic sequences of Streptococcus virus phiAbc2 (NC_013645.1), Streptococcus phage 315.1 (NC_004584.1), Streptococcus phage 34117 (FR671407.1), Streptococcus phage 73 (KT717083.1), Streptococcus phage D5842 (MH000602.1), Streptococcus phage IPP26 (KY065467.1), Streptococcus phage IPP8 (KY065450.1), Streptococcus phage K13 (NC_024357.1), Streptococcus phage P0091 (KY705251.1), Streptococcus phage P0092 (KY705252.1), Streptococcus phage P0093 (KY705253.1), Streptococcus phage P0095 (KY705255.1), Streptococcus phage P4761 (KY705258.1), Streptococcus phage P5651 (KY705260.1), Streptococcus phage P5652 (KY705261.1), Streptococcus phage P7134 (KY705264.1), Streptococcus phage P7152 (KY705266.1), Streptococcus phage P7154 (KY705267.1), Streptococcus phage P7951 (KY705277.1), Streptococcus phage P7952 (KY705278.1), Streptococcus phage P7953 (KY705279.1), Streptococcus phage P7954 (KY705280.1), Streptococcus phage P7955 (KY705281.1), Streptococcus phage P8921 (KY705282.1), Streptococcus phage P8922 (KY705283.1), Streptococcus phage P9851 (KY705284.1), Streptococcus phage P9852 (KY705285.1), Streptococcus phage P9902 (KY705289.1), Streptococcus phage phi7917 (KC348601.1), Streptococcus phage phiARI0004 (NC_031920.1), Streptococcus phage phiARI0746 (NC_031907.1), Streptococcus phage phi-m46.1 (FM864213.1), Streptococcus phage phiSC070807 (KT336321.1), Streptococcus phage phiZJ20091101-2 (KX077893.1), Streptococcus phage TP-778L (NC_022776.1), Streptococcus phage TP-J34 (HE861935.1), Streptococcus phage TP-J34 (HE861935.1), Streptococcus virus 7201 (NC_002185.1), Streptococcus virus 9871 (NC_031069.1), Streptococcus virus 9874 (NC_031023.1), Streptococcus virus DT1 (AF085222.2), Streptococcus phage 20617 (NC_023503.1), Streptococcus phage 315.2 (NC_004585.1), Streptococcus phage 315.6 (NC_004589.1), Streptococcus phage D1811 (MH000604.1), Streptococcus phage IPP16 (KY065457.1), Streptococcus phage IPP19 (Y065460.1), Streptococcus phage IPP24 (KY065465.1), Streptococcus phage IPP62 (KY065498.1), Streptococcus phage IPP69 (KY065505.1), Streptococcus phage LYGO9 (JX409894.1), Streptococcus phage P9 (NC_009819.1), Streptococcus phage phi1207.3 (AY657002.1), Streptococcus phage phi20c (KC348598.1), Streptococcus phage phi30c (KC348599.1), Streptococcus phage phi5218 (KC348600.1), Streptococcus phage phiARI0131-1 (NC_031901.1), Streptococcus phage phiARI0831b (KT337369.1), Streptococcus phage phiLP081102 (KX077890.1), Streptococcus phage phiNJ2 (NC_019418.1), Streptococcus phage phi-SsUD.1 (FN997652.1), Streptococcus phage SpSL1 (NC_027396.1), Streptococcus phage vB_SthS_VA460 (MG708275.1), Streptococcus virus Sfi11 (NC_002214.1) and phage Lambda (NC_001416.1), phage T4 (NC_000866.4) and phage T7 (NC_001604.1) as well-known bacteriophages of different studied families. Specific cluster peptides are represented by different color forms:  ; red filled diamond YPNSFTAYIMDVKGC*K (cluster A specific),
; red filled diamond YPNSFTAYIMDVKGC*K (cluster A specific),  ; green filled diamond VKEKIAAIVNDEELLAK (cluster A specific),
; green filled diamond VKEKIAAIVNDEELLAK (cluster A specific),  ; purple filled diamond ISGITLSDTNVVAGNLISSSEHFVQM*LSNIK (cluster A specific),
; purple filled diamond ISGITLSDTNVVAGNLISSSEHFVQM*LSNIK (cluster A specific),  ; blue filled diamond FQLALLNTTTTFAQYQCSAYIDFEGQR (cluster A specific),
; blue filled diamond FQLALLNTTTTFAQYQCSAYIDFEGQR (cluster A specific),  ; yellow filled diamond DNVLIETVKM*QGEQAM*QLVK (cluster A specific),
; yellow filled diamond DNVLIETVKM*QGEQAM*QLVK (cluster A specific),  ; orange filled diamond TTTELTTITHTVGDIQKEMVRM*NDR (cluster B specific),
; orange filled diamond TTTELTTITHTVGDIQKEMVRM*NDR (cluster B specific),  ; black outlined diamond M*LANGITLSYGESKETYTKLVGLK (cluster B specific),
; black outlined diamond M*LANGITLSYGESKETYTKLVGLK (cluster B specific),  ; red outlined diamond LSDKPKSYTEILNNIINSQK (cluster B specific),
; red outlined diamond LSDKPKSYTEILNNIINSQK (cluster B specific),  ; blue outlined diamond ITTGIISAARIGAEAITADKLK (cluster C specific),
; blue outlined diamond ITTGIISAARIGAEAITADKLK (cluster C specific),  ; gray outlined diamond HAQGDVGWNINSVTQRIVPANINAESEIWSK (cluster E specific),
; gray outlined diamond HAQGDVGWNINSVTQRIVPANINAESEIWSK (cluster E specific),  ; green outlined diamond TLITMTEQIKNLTDDVK (cluster E specific). (M*methionine oxidation; C* carbamidomethylation of Cys).
; green outlined diamond TLITMTEQIKNLTDDVK (cluster E specific). (M*methionine oxidation; C* carbamidomethylation of Cys).
Peptide biomarkers were found in related Streptococcus phages (Table 2) that were located closely in the phylogenomic tree (Figure 2). The peptides YPNSFTAYIM∗DVKGC∗K and VKEKIAAIVNDEELLAK were found in 23 and 4 Streptococcus phages of cluster A, respectively. The peptide ISGITLSDTNVVAGNLISSSEHFVQM∗LSNIK was also found in 2 closely related phages (Streptococcus phage P7954 and Streptococcus phage TP-J34) of cluster A. The peptides FQLALLNTTTTFAQYQC∗SAYIDFEGQR and DNVLIETVKM∗QGEQAM∗QLVK were found in 9 and 5 phages of cluster A, respectively. The peptide TTTELTTITHTVGDIQKEM∗VRM∗NDR was found in 3 Streptococcus phages (Streptococcus phage IPP16, Streptococcus phage K13, and Streptococcus phage 34117) of cluster B. The peptide M∗LANGITLSYGESKETYTKLVGLK was found in 4 phages of the firs branch of cluster B, and the peptide LSDKPKSYTEILNNIINSQK was found in 2 phages (Streptococcus phage IPP24 and Streptococcus phage IPP26) of the second branch of cluster B. The peptide ITTGIISAARIGAEAITADKLK was found in 3 (Streptococcus phage phi-SsUD.1, Streptococcus phage phi-m46.1, and Streptococcus phage phiSC070807) of 4 Streptococcus phages of cluster C. The peptide HAQGDVGWNINSVTQRIVPANINAESEIWSK was found in 2 phages (Streptococcus phage phi20c and Streptococcus phage phi7917) of the first branch of cluster E, and the peptide TLITM∗TEQIKNLTDDVK was found in 3 phages (Streptococcus phage 315.2 and Streptococcus phage 315.6, Streptococcus phage 315.1) of the second branch.

Table 2. Phage biomarker peptides that belong to bacteriophages, and phylogenomic tree clusters are located.
In addition, a correlation that related all peptides found in bacteriophages with 100% similarity and with bacterial species for each cluster was inferred. The results showed that the clustered phages were related to specific species of Streptococcus. Cluster A phage peptides were found to be related mainly to S. thermophilus. All cluster B phage peptides were found to be related mainly to S. pneumoniae. Cluster C phage peptides were found to be mainly related to S. uberis and Streptococcus suis. Cluster E phage peptides were found in different Streptococcus species. However, the bacterial species S. uberis and S. parauberis were often found to be related in all clusters of the phylogenomic tree. These results support previously reported data on studied phages and where they were isolated from Brueggemann et al. (2017), McDonnell et al. (2017).
To further analyze phage clustering, we selected the holin gene to construct a phylogenetic relationship tree and compared the phylogenomic relationship among phages with 100% similarity to the peptides. The holin protein is essential for host lysis and constitutes one of the most diverse functional proteins in phage genomes analyzed previously (Wang et al., 2000; Goerke et al., 2009). The comparison of the holin gene sequences of the studied bacteriophages (Figure 3) showed that the phylogenomic and phylogenetic clusters were similar (Figure 3). Cluster A of the phylogenomic tree was well defined in the holin tree. Previous analyses revealed two holin-like sequences, class I and class II, immediately upstream of the endolysin gene (Wang et al., 2000). This was observed for some of the bacteriophage holins that appeared in different clusters of the phylogenetic tree.
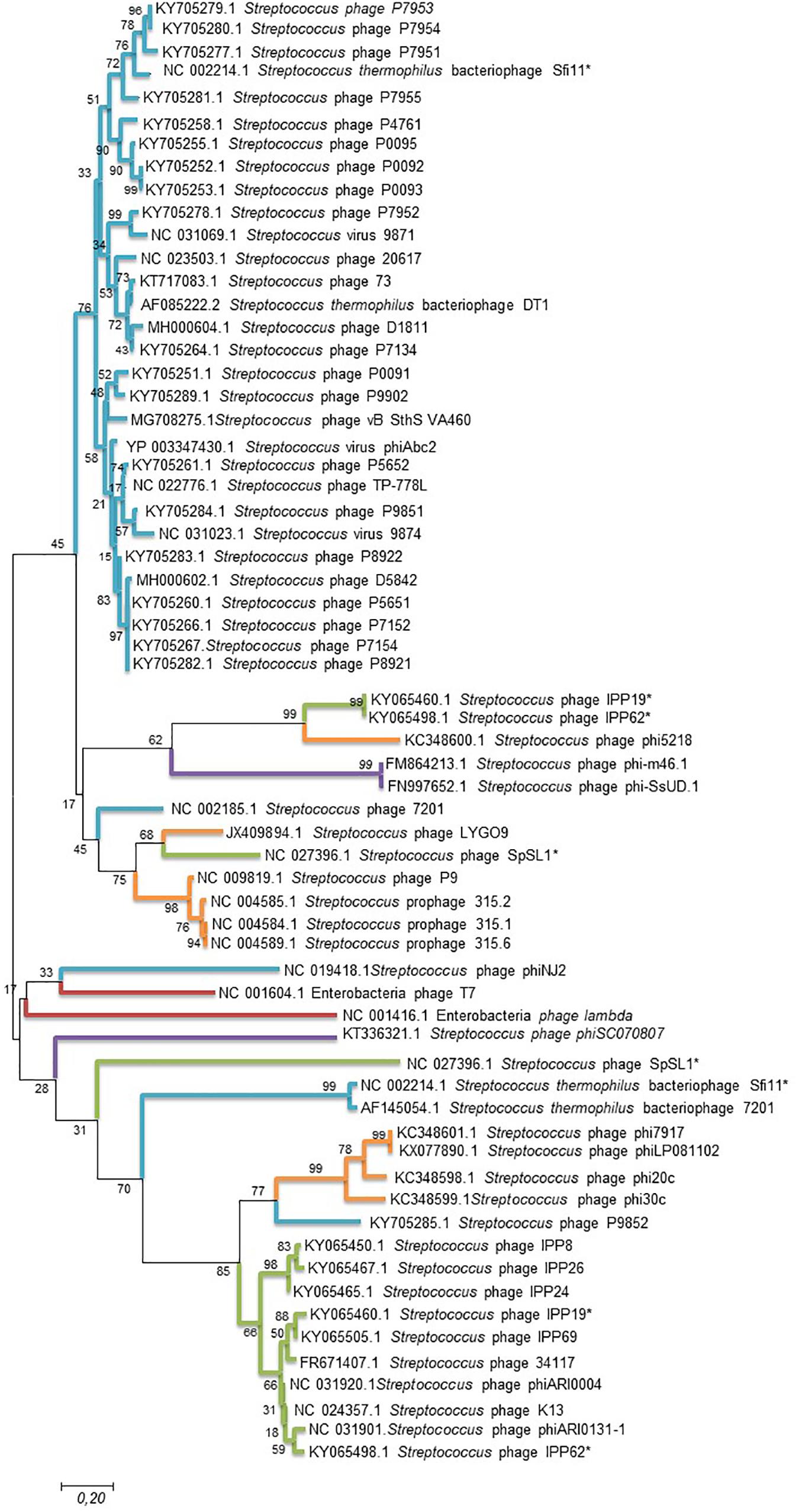
Figure 3. Maximum likelihood phylogenetic tree based on sequences of the holin gene of Streptococcus phages. Streptococcus phages of the different clusters displayed in the phylogenetic tree generated by VICTOR (Figure 2) are represented by different color branches; blue; cluster A, green; cluster B, purple; cluster C, red; cluster D and, orange; cluster E. The significance of each branch is indicated by a bootstrap value calculated as a percentage for 1,000 subsets. Bar, 20 nt substitutions per 100 nt. *two holin-like sequences.
Discussion
Proteomics techniques using matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) and liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) instruments have been used for the analysis and detection of pathogenic bacterial pathogen strains based on specific diagnostic peptides (Lasch et al., 2009; Böhme et al., 2011, 2012; Branquinho et al., 2014; Quintela-Baluja et al., 2014; Calo-Mata et al., 2016; Pfrunder et al., 2016; Carrera et al., 2017). In addition, LC-ESI-MS/MS techniques have been employed for the identification and detection of bacterial bacteriophages such as the bacteriophage lambda (Serafim et al., 2017).
LC-MS/MS-based methods offer several advantages compared with other approaches because one can directly identify bacteriophage peptides without the necessity of inferring conclusions based on other approaches such as genomics tools. Additionally, in this study, we implemented a precise method that may be useful for further research and analysis, without the requirement of growing bacteria; instead, the samples can be collected directly from food. The total time estimated to complete the analysis since the beginning of the experiments is 3 days, and the direct analysis from foodstuffs should take less than 1 day.
Phage peptides such as M∗ATNLGQAYVQIM∗PSAK could be good biomarkers because they are unique and specific for a particular bacterial species, in this case for S. agalactiae. Additionally, some determined phage-origin peptides in this study could be useful as specific biomarker peptides for the distinct detection of pathogenic bacteria and/or their phages in a sample. As bacteriophages are signal amplifiers because of their life cycle, bacterial peptide and phage peptide detection is significant for the identification of pathogens.
As presented in this manuscript, LC-ESI-MS/MS analysis is a good strategy for the rapid characterization of phage-origin proteins present in Streptococcus spp. In addition, the peptides in this study may be very useful as peptide biomarkers for the identification and characterization of mastitis-causing Streptococcus spp.
Additionally, the results show that a specific peptide biomarker is present in near-related phages, and bacteriophages that present the same peptides as specific Streptococcus spp. are located very close in the phylogenomic tree. This suggests that a link exists between phage phylogeny and bacteriophages that could infect the same bacterial species. Due to bacteriophage activity against host species, there is a high possibility that phages will be effective against related host species within the genus Streptococcus. These data support the use of bacteriophages in what is known as “phage therapy” to control pathogenic bacterial populations.
Moreover, this study shows how phylogenomic trees based on genome analysis provide useful information and corroborates previous investigations. These previous studies suggested that this type of analysis as well as phylogenetic studies is an appropriate tool for studying and reconstructing the viral history of phages, and this tool may be considered a significant complement to virus classification based on properties such as morphology, genome composition (DNA/RNA), orientation (+/− sense), segmentation, gene sets and replication strategies (Simmonds, 2015). On the other hand, phage genomic content is not currently well known (Argov et al., 2017), making phage analysis more difficult. The first priority then must be guided by the acquisition of large amounts of data for phage-infecting bacteria (Brüssow and Desiere, 2001).
Finally, it is known that bacteria-phage interactions are mutually beneficial, and their coevolution maintains phenotypic and genetic diversity by contributing dramatically to lateral gene transfer (Koskella and Brockhurst, 2014). As the majority of bacteria contain bacteriophage DNA in their genomes, it is expected that additional and novel interactions will be observed in the future, thus contributing to a better understanding of bacteriophage biology (Argov et al., 2017).
Data Availability Statement
The datasets generated for this study can be found in the Uniprot and GenBank: complete genomic sequences of Streptococcus virus phiAbc2 (NC_013645.1), Streptococcus phage 315.1 (NC_004584.1), Streptococcus phage 34117 (FR671407.1), Streptococcus phage 73 (KT717083.1), Streptococcus phage D5842 (MH000602.1), Streptococcus phage IPP26 (KY065467.1), Streptococcus phage IPP8 (KY065450.1), Streptococcus phage K13 (NC_024357.1), Streptococcus phage P0091 (KY705251.1), Streptococcus phage P0092 (KY705252.1), Streptococcus phage P0093 (KY705253.1), Streptococcus phage P0095 (KY705255.1), Streptococcus phage P4761 (KY705258.1), Streptococcus phage P5651 (KY705260.1), Streptococcus phage P5652 (KY705261.1), Streptococcus phage P7134 (KY705264.1), Streptococcus phage P7152 (KY705266.1), Streptococcus phage P7154 (KY705267.1), Streptococcus phage P7951 (KY705277.1), Streptococcus phage P7952 (KY705278.1), Streptococcus phage P7953 (KY705279.1), Streptococcus phage P7954 (KY705280.1), Streptococcus phage P7955 (KY705281.1), Streptococcus phage P8921 (KY705282.1), Streptococcus phage P8922 (KY705283.1), Streptococcus phage P9851 (KY705284.1), Streptococcus phage P9852 (KY705285.1), Streptococcus phage P9902 (KY705289.1), Streptococcus phage phi7917 (KC348601.1), Streptococcus phage phiARI0004 (NC_031920.1), Streptococcus phage phiARI0746 (NC_031907.1), Streptococcus phage phi-m46.1 (FM864213.1), Streptococcus phage phiSC070807 (KT336321.1), Streptococcus phage phiZJ20091101-2 (KX077893.1), Streptococcus phage TP-778L (NC_022776.1), Streptococcus phage TP-J34 (HE861935.1), Streptococcus phage TP-J34 (HE861935.1), Streptococcus virus 7201 (NC_002185.1), Streptococcus virus 9871 (NC_031069.1), Streptococcus virus 9874 (NC_031023.1), Streptococcus virus DT1 (AF085222.2), Streptococcus phage 20617 (NC_023503.1), Streptococcus phage 315.2 (NC_004585.1), Streptococcus phage 315.6 (NC_004589.1), Streptococcus phage D1811 (MH000604.1), Streptococcus phage IPP16 (KY065457.1), Streptococcus phage IPP19 (Y065460.1), Streptococcus phage IPP24 (KY065465.1), Streptococcus phage IPP62 (KY065498.1), Streptococcus phage IPP69 (KY065505.1), Streptococcus phage LYGO9 (JX409894.1), Streptococcus phage P9 (NC_009819.1), Streptococcus phage phi1207.3 (AY657002.1), Streptococcus phage phi20c (KC348598.1), Streptococcus phage phi30c (KC348599.1), Streptococcus phage phi5218 (KC348600.1), Streptococcus phage phiARI0131-1 (NC_031901.1), Streptococcus phage phiARI0831b (KT337369.1), Streptococcus phage phiLP081102 (KX077890.1), Streptococcus phage phiNJ2 (NC_019418.1), Streptococcus phage phi-SsUD.1 (FN997652.1), Streptococcus phage SpSL1 (NC_027396.1), Streptococcus phage vB_SthS_VA460 (MG708275.1), Streptococcus virus Sfi11 (NC_002214.1), and phage Lambda (NC_001416.1), phage T4 (NC_000866.4), phage T7 (NC_001604.1).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work has received financial support from the Xunta de Galicia and the European Union (European Social Fund – ESF), by the Spanish Ministry of Economy and Competitivity Project AGL 2.013-48.244-R and by the European Regional Development Fund (ERDF) (2007–2013). This work was also supported by the GAIN-Xunta de Galicia Project (IN607D 2017/01). MC is supported by the Ramón y Cajal contract (Ministry of Science and Innovation of Spain).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01241/full#supplementary-material
Footnotes
References
Achigar, R., Magadán, A. H., Tremblay, D. M., Pianzzola, M. J., and Moineau, S. (2017). Phage-host interactions in Streptococcus thermophilus: genome analysis of phages isolated in Uruguay and ectopic spacer acquisition in CRISPR array. Sci. Rep. 7:43438.
Ackermann, H. (2007). 5500 Phages examined in the electron microscope. Arch. Virol. 152, 227–243. doi: 10.1007/s00705-006-0849-1
Akoh, C. C., Leem, G. C., Liaw, Y. C., Huang, T. H., and Shaw, J. F. (2004). GDSL family of serine esterases/lipases. Prog. Lipid Res. 43, 534–552. doi: 10.1016/j.plipres.2004.09.002
Ali, Y., Koberg, S., Heßner, S., Sun, X., Rabe, B., Back, A., et al. (2014). Temperate Streptococcus thermophilus phages expressing superinfection exclusion proteins of the Ltp type. Front. Microbiol. 5:98. doi: 10.3389/fmicb.2014.00098
Alnakip, M. E., Quintela-Baluja, M., Böhme, K., Fernández-No, I. C., Amer, I. H., Elsayed, M. S., et al. (2014). “Comparative identification of streptococci of dairy origin by VITEK-2 system and 16S rRNA gene sequencing,” in Proceedings of the First Inter. Conf of Impact of Environmental Hazards on Food Safety (Zagazig: Zagazig University), 30–58.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Argov, T., Azulay, G., Pasechnek, A., Stadnyuk, O., Ran-Sapir, S., Borovok, I., et al. (2017). Temperate bacteriophages as regulators of host behavior. Curr. Opin. Microbiol. 38, 81–87. doi: 10.1016/j.mib.2017.05.002
Arioli, S., Eraclio, G., Della Scala, G., Neri, E., Colombo, S., Scaloni, A., et al. (2018). Role of temperate bacteriophage Φ20617 on Streptococcus thermophilus DSM 20617T autolysis and biology. Front. Microbiol. 9:2719. doi: 10.3389/fmicb.2018.02719
Bai, Q., Yang, Y., and Lu, C. (2016). Isolation and characterization of siphovirusphages infecting bovine Streptococcus agalactiae. Acta Microbiol. Sin. 56, 317–326.
Bai, Q., Zhang, W., Yang, Y., and Lu, C. (2013). Characterization and genome sequencing of a novel bacteriophage infecting Streptococcus agalactiae with high similarity to a phage from Streptococcus pyogenes. Arch. Virol. 158, 1733–1741. doi: 10.1007/s00705-013-1667-x
Bateman, A., Coggill, P., and Finn, R. D. (2010). DUFs: families in search of function. Acta Crystallogr. Sec. F Sect. F Struct. Biol. Cryst. Commun. 66, 1148–1152. doi: 10.1107/S1744309110001685
Bensing, B. A., Siboo, I. R., and Sullam, P. M. (2001). Proteins PblA and PblB of Streptococcus mitis, which promote binding to human platelets, are encoded within a lysogenic bacteriophage. Infect. Immun. 69, 6186–6192. doi: 10.1128/IAI.69.10.6186-6192.2001
Beres, S. B., Sylva, G. L., Barbian, K. D., Lei, B., Hoff, J. S., Mammarella, N. D., et al. (2002). Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. PNAS 99, 10078–10083. doi: 10.1073/pnas.152298499
Böhme, K., Fernández-No, I. C., Barros-Velázquez, J., Gallardo, J. M., Calo-Mata, P., and Cañas, B. (2010a). Species differentiation of seafood spoilage and pathogenic gram-negative bacteria by MALDI-TOF mass fingerprinting. J. Proteome Res. 9, 3169–3183. doi: 10.1021/pr100047q
Böhme, K., Fernández-No, I. C., Barros-Velázquez, J., Gallardo, J. M., Cañas, B., and Calo-Mata, P. (2010b). Comparative analysis of protein extraction methods for the identification of seafood-borne pathogenic and spoilage bacteria by MALDI-TOF mass spectrometry. Anal. Methods. 2, 1941–1947. doi: 10.1039/c0ay00457j
Böhme, K., Fernández-No, I. C., Barros-Velázquez, J., Gallardo, J. M., Cañas, B., and Calo-Mata, P. (2011). Rapid species identification of seafood spoilage and pathogenic Gram-positive bacteria by MALDI-TOF mass fingerprinting. Electrophoresis 32, 2951–2965. doi: 10.1002/elps.201100217
Böhme, K., Morandi, S., Cremonesi, P., Fernández-No, I. C., Barros-Velázquez, J., Castiglioni, B., et al. (2012). Characterization of Staphylococcus aureus strains isolated from Italian dairy products by MALDI-TOF mass fingerprinting. Electrophoresis 33, 2355–2364. doi: 10.1002/elps.201100480
Branquinho, R., Sousa, C., Lopes, J., Pintado, M. E., Peixe, L. V., and Osorio, H. (2014). Differentiation of Bacillus pumilus and Bacillus safensis using MALDI-TOF-MS. PLoS One 9:e0110127. doi: 10.1371/journal.pone.0110127
Brenciani, A., Bacciaglia, A., Vignaroli, C., Pugnaloni, A., Varaldo, P. E., and Giovanetti, E. (2010). Phim46.1, the main Streptococcus pyogenes element carrying mef(A) and tet(O) genes. Antimicrob. Agents Chemother. 54, 221–229. doi: 10.1128/aac.00499-09
Brueggemann, A. B., Harrold, C. L., Rezaei Javan, R., van Tonder, A. J., McDonnell, A. J., and Edwards, B. A. (2017). Pneumococcal prophages are diverse, but not without structure or history. Sci. Rep. 7, 1–13.
Brüssow, H., and Desiere, F. (2001). Comparative phage genomics and the evolution of Siphoviridae: insights from dairy phages. Mol. Microbiol. 39, 213–222. doi: 10.1046/j.1365-2958.2001.02228.x
Calo-Mata, P., Carrera, M., Böhme, K., Caamaño-Antelo, S., Gallardo, J. M., Barros-Velázquez, J., et al. (2016). Novel peptide biomarker discovery for detection and identification of bacterial pathogens by LC-ESI-MS MS. J. Anal. Bioanal. Tech. 7, 1–9. doi: 10.4172/2155-9872.1000296
Canchaya, C., Fournous, G., and Brüssow, H. (2004). The impact of prophages on bacterial chromosomes. Mol. Microbiol. 53, 9–18. doi: 10.1111/j.1365-2958.2004.04113.x
Carneiro, M. J. V. M., Zhang, W., Ioannou, C., Scott, D. J., Allen, S., Roberts, C. J., et al. (2006). The DNA-remodelling activity of DnaD is the sum of oligomerization and DNA-binding activities on separate domains. Mol. Microbiol. 60, 917–924. doi: 10.1111/j.1365-2958.2006.05152.x
Carrera, M., Böhme, K., Gallardo, J. M., Barros-Velázquez, J., Cañas, B., and Calo-Mata, P. (2017). Characterization of foodborne strains of Staphylococcus aureus by shotgun proteomics: functional networks, virulence factors and species-specific peptide biomarkers. Front. Microbiol. 8:2458. doi: 10.3389/fmicb.2017.02458
Carrera, M., Cañas, B., and Gallardo, J. M. (2013). The sarcoplasmic fish proteome: pathways, metabolic networks and potential bioactive peptides for nutritional inferences. J. Proteomics 78, 211–220. doi: 10.1016/j.jprot.2012.11.016
Chakravorty, S., Helb, D., Burday, M., Connell, N., and Alland, D. (2007). A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 69, 330–339. doi: 10.1016/j.mimet.2007.02.005
Chanishvili, N. (2015). “Bacteriophage-based products and techniques for identification of biological pathogens,” in Nanotechnology to Aid Chemical and Biological Defense, ed. T. A. Camesano (Dordrecht: Springer), 17–33. doi: 10.1007/978-94-017-7218-1_2
Craigie, J., and Yen, C. H. (1938). The demonstration of types of B. typhosus by means of preparations of Type II Vi phage. Can. J. Public Health 29, 484–496.
Croucher, N. J., Hanage, W. P., Harris, S. R., McGee, L., van der Linden, M., de Lencastre, H., et al. (2014). Variable recombination dynamics during the emergence, transmission and ‘disarming’ of a multidrug-resistant pneumococcal clone. BMC Biol. 12:49. doi: 10.1186/1741-7007-12-49
Croucher, N. J., Harris, S. R., Fraser, C., Quail, M. A., Burton, J., van der Linden, M., et al. (2011). Rapid pneumococcal evolution in response to clinical interventions. Science 331, 430–434. doi: 10.1126/science.1198545
Croucher, N. J., Mostowy, R., Wymant, C., Turner, P., Bentley, S. D., and Fraser, C. (2016). Horizontal DNA transfer mechanisms of bacteria as weapons of intragenomic conflict. PLoS Biol. 14:e1002394. doi: 10.1371/journal.pbio.1002394
da Silva Duarte, V., Giaretta, S., Treu, L., Campanaro, S., Vidigal, P. M. P., Tarrah, A., et al. (2018). Draft genome sequences of three virulent Streptococcus thermophilus bacteriophages isolated from the dairy environment in the Veneto region of Italy. Genome Announc. 6:e0045-18.
Davies, M. R., Mcmillan, D. J., Domselaar, G. H., Van Jones, M. K., and Sriprakash, K. S. (2007). Phage 3396 from a Streptococcus dysgalactiae subsp. equisimilis pathovar may have its origins in Streptococcus pyogenes. J. Bacteriol. 189, 2646–2652. doi: 10.1128/JB.01590-06
Dokland, T., and Murialdo, H. (1993). Structural transitions during maturation of lambda capsids. J. Mol. Biol. 233, 682–694. doi: 10.1006/jmbi.1993.1545
Domelier, A., Mee-marquet, N., Van Der Sizaret, P., Mereghetti, L., and Quentin, R. (2009). Molecular characterization and lytic activities of Streptococcus agalactiae bacteriophages and determination of lysogenic-strain features. J. Bacteriol. 191, 4776–4785. doi: 10.1128/JB.00426-09
Dumke, J., Hinse, D., Vollmer, T., Schulz, J., and Knabbe, C. (2015). Potential transmission pathways of Streptococcus gallolyticus subsp. gallolyticus. PLoS One 10:e0126507. doi: 10.1371/journal.pone.0126507
Duplessis, M., and Moineau, S. (2001). Identification of a genetic determinant responsible for host specificity in Streptococcus thermophilus bacteriophages. Mol. Microbiol. 41, 325–336. doi: 10.1046/j.1365-2958.2001.02521.x
Durante-Rodríguez, G., Mancheño, J. M., Díaz, E., and Carmona, M. (2016). Refactoring the λ phage lytic/lysogenic decision with a synthetic regulator. Microbiol. Open 5, 575–581. doi: 10.1002/mbo3.352
Feiner, R., Argov, T., Rabinovich, L., Sigal, N., Borovok, I., and Herskovits, A. A. (2015). A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol 13, 641–650. doi: 10.1038/nrmicro3527
Forsman, P., Tilsala-Timisjärvi, A., and Alatossava, T. (1997). Identification of staphylococcal and streptococcal causes of bovine mastitis using 16S-23S rRNA spacer regions. Microbiology 143, 3491–3500. doi: 10.1099/00221287-143-11-3491
Fortier, L. C., and Sekulovic, O. (2013). Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365. doi: 10.4161/viru.24498
Gantzer, C., Maul, A., Audic, J. M., and Pharmacie, D. (1998). Detection of infectious enteroviruses, enterovirus genomes, somatic coliphages and Bacteroides fragilis phages in treated Wastewater. Appl. Environ. Microbiol. 64, 4307–4312. doi: 10.1128/aem.64.11.4307-4312.1998
Goerke, C., Pantucek, R., Holtfreter, S., Schulte, B., Zink, M., Grumann, D., et al. (2009). Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. bacterial. 191, 3462–3468. doi: 10.1128/jb.01804-08
Goodacre, N. F., Gerloff, D. L., and Uetz, P. (2014). Protein domains of unknown function are essential in Bacteria. mBio 5:e0744-13. doi: 10.1128/mB.00744-13.Editor
Gual, A., Camacho, A. G., and Alonso, J. C. (2000). Functional analysis of terminase large subunit, G2P, of Bacillus subtilis bacteriophage SPP1. J. Biol. Chem. 275, 35311–35319. doi: 10.1074/jbc.M004309200
Guglielmotti, D. M., Deveau, H., Binetti, A. G., Reinheimer, J. A., Moineau, S., and Quiberoni, A. (2009). Genome analysis of two virulent Streptococcus thermophilus phages isolated in Argentina. Int. J. Food Microbiol. 136, 101–109. doi: 10.1016/j.ijfoodmicro.2009.09.005
Harhala, M., Barylski, J., Humi, K., Lecion, D., Wojciechowicz, J., Lahutta, K., et al. (2018). Two novel temperate bacteriophages infecting Streptococcus pyogenes: their genomes, morphology and stability. PLoS One 13:e0205995. doi: 10.1371/journal.pone.0205995
Hill, A. W., and Brandy, C. A. (1989). A note on the isolation and propagation of lytic phages from Streptococcus uberis and their potential for strain typing. J. Appl. Bacteriol. 67, 425–431. doi: 10.1111/j.1365-2672.1989.tb02513.x
Huang, J., Ma, J., Shang, K., Hu, X., Liang, Y., Li, D., et al. (2016). Evolution and diversity of the antimicrobial resistance associated mobilome in Streptococcus suis: a probable mobile genetic elements reservoir for other streptococci. Front. Cell Infect. Microbiol. 6:118. doi: 10.3389/fcimb.2016.00118
Huang, Y. H., Lien, Y., Huang, C. C., and Huang, C. Y. (2016). Characterization of Staphylococcus aureus primosomal DnaD protein: highly conserved C-terminal region is crucial for ssDNA and PriA helicase binding but not for DnaA protein-binding and self-tetramerization. PLoS One 11:e0157593. doi: 10.1371/journal.pone.0157593
Hynes, A. P., Rousseau, G. M., Agudelo, D., Goulet, A., Amigues, B., Loehr, J., et al. (2018). Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat. Commun. 9:2919. doi: 10.1038/s41467-018-05092-w
Iannelli, F., Santagati, M., Santoro, F., Oggioni, M. R., Stefani, S., and Pozzi, G. (2014). Nucleotide sequence of conjugative prophage Φ1207. 3 (formerly Tn1207. 3) carrying the mef (A)/msr (D) genes for efflux resistance to macrolides in Streptococcus pyogenes. Front. Microbiol. 5:687. doi: 10.3389/fmicb.2014.00687
Ivnitski, D., Abdel-hamid, I., Atanasov, P., and Wilkins, E. (1999). Biosensors for detection of pathogenic bacteria. Biosens. Bioelectron. 14, 599–624. doi: 10.1016/s0956-5663(99)00039-1
Iyer, L., Koonin, E. V., and Aravind, L. (2002). Classification and evolutionary history of the single-strand annealing proteins, RecT, Redβ, ERF and RAD52. BMC Genomics 3:8. doi: 10.1186/1471-2164-3-8
Jørgensen, M. G., Pandey, D. P., Jaskolska, M., and Gerdes, K. (2009). HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 191, 1191–1199. doi: 10.1128/JB.01013-08
Käll, L., Canterbury, J. D., Weston, J., Noble, W. S., and MacCoss, M. J. (2007). Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925. doi: 10.1038/nmeth1113
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/bf01731581
Koskella, B., and Brockhurst, M. A. (2014). Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931. doi: 10.1111/1574-6976.12072
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7. 0 for bigger datasets brief communication. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lasch, P., Beyer, W., Nattermann, H., Stämmler, M., Siegbrecht, E., Grunow, R., et al. (2009). Identification of Bacillus anthracis by using matrix-assisted laser desorption ionization-time of flight mass spectrometry and artificial neural networks. J. Appl. Environ. Microbiol. 75, 7229–7242. doi: 10.1128/AEM.00857-09
Lavigne, R., Ceyssens, P., and Robben, J. (2009). “Phage proteomics: applications of mass spectrometry,” in Bacteriophages, eds M. R. Clokie and A. M. Kropinski (Totowa, NJ: Humana Press), 239–251. doi: 10.1007/978-1-60327-565-1_14
Lopez-Sanchez, M. J., Sauvage, E., Da Cunha, V., Clermon, D., Ratsima Hariniaina, E., Gonzalez-Zorn, B., et al. (2012). The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol. Microbiol. 85, 1057–1071. doi: 10.1111/j.1365-2958.2012.08172.x
Lucchini, S., Desiere, F., and Brüssow, H. (1999a). Comparative genomics of Streptococcus thermophilus phage species supports a modular evolution theory. J. Virol. 73, 8647–8656. doi: 10.1128/jvi.73.10.8647-8656.1999
Lucchini, S., Desiere, F., and Brüssow, H. (1999b). Similarly organized lysogeny modules in temperate Siphoviridae from low GC content gram-positive bacteria. Virology 263, 427–435. doi: 10.1006/viro.1999.9959
Lundberg, Å, Nyman, A., Unnerstad, H. E., and Waller, K. P. (2014). Prevalence of bacterial genotypes and outcome of bovine clinical mastitis due to Streptococcus dysgalactiae and Streptococcus uberis. Acta Vet. Scand. 56, 1–11. doi: 10.1186/s13028-014-0080-0
Mahony, J., Alqarni, M., Stockdale, S., Spinelli, S., Feyereisen, M., Cambillau, C., et al. (2016). Functional and structural dissection of the tape measure protein of lactococcal phage TP901-1. Sci. Rep. 6, 1–10. doi: 10.1038/srep36667
McDonnell, B., Mahony, J., Hanemaaijer, L., Neve, H., Noben, J. P., Kouwen, T., et al. (2016). Identification and analysis of a novel group of bacteriophages infecting the lactic acid bacterium Streptococcus thermophilus. J. Appl. Environ. Microbiol. 82, 5153–5165. doi: 10.1128/aem.00835-16
McDonnell, B., Mahony, J., Hanemaaijer, L., Neve, H., Noben, J. P., Lugli, G. A., et al. (2017). Global survey and genome exploration of bacteriophages infecting the lactic acid bacterium Streptococcus thermophilus. Front. Microbiol. 8:1754. doi: 10.3389/fmicb.2017.01754
Meier-Kolthoff, J. P., and Göker, M. (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33, 3396–3404. doi: 10.1093/bioinformatics/btx440
Menouni, R., Hutinet, G., Petit, M. A., and Ansaldi, M. (2015). Bacterial genome remodeling through bacteriophage recombination. FEMS Microbiol. Lett. 362, 1–10. doi: 10.1093/femsle/fnu022
Mikkonen, M., and Alatossava, T. (1994). Characterization of the genome region encoding structural proteins of Lactobacillus delbrueckii subsp. lactis bacteriophage LL-H. Gene 151, 53–59. doi: 10.1016/0378-1119(94)90632-7
Mitchell, J., Siboo, I. R., Takamatsu, D., Chambers, H. F., and Sullam, P. M. (2007). Mechanism of cell surface expression of the Streptococcus mitis platelet binding proteins PblA and PblB. Mol. Microbiol. 64, 844–857. doi: 10.1111/j.1365-2958.2007.05703.x
Murphy, J., Mahony, J., Ainsworth, S., Nauta, A., and van Sinderen, D. (2013). Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. J. Appl. Environ. Microbiol. 79, 7547–7555. doi: 10.1128/AEM.02229-13
Neve, H., Zenz, K. I., Desiere, F., Koch, A., Heller, K. J., and BruÈssow, H. (1998). Comparison of the lysogeny modules from the temperate Streptococcus thermophilus bacteriophages TP-J34 and Sfi21: implications for the modular theory of phage evolution. Virology 241, 61–72. doi: 10.1006/viro.1997.8960
Palmieri, C., Princivalli, M. S., Brenciani, A., Varaldo, P. E., and Facinelli, B. (2011). Different genetic elements carrying the tet (W) gene in two human clinical isolates of Streptococcus suis. Antimicrob. Agents Chemother. 55, 631–636. doi: 10.1128/aac.00965-10
Park, M. A., Kwon, G., Hwang, Y., Jung, H., Kim, D., Park, J., et al. (2013). Genome sequence of Streptococcus parauberis strain KCTC11980, isolated from diseased Paralichthys olivaceus. Genome Announc. 1, 5–6. doi: 10.1128/genomeA.00780-713
Pell, L. G., Cumby, N., Clark, T. E., Tuite, A., Battaile, K. P., Edwards, A. M., et al. (2013). A conserved spiral structure for highly diverged phage tail assembly chaperones. J. Mol. Biol. 425, 2436–2449. doi: 10.1016/j.jmb.2013.03.035
Pfrunder, S., Grossmann, J., Hunziker, P., Brunisholz, R., Gekenidis, M. T., and Drissner, D. (2016). Bacillus cereus Group-Type strain-specific diagnostic peptides. J. Proteome Res. 15, 3098–3107. doi: 10.1021/acs.jproteome.6b00216
Quintela-Baluja, M., Böhme, K., Fernández-No, I. C., Alnakip, M. E., Caamano, S., Barros-Velázques, J., et al. (2014). “MALDI-TOF mass spectrometry, a rapid and reliable method for the identification of bacterial species in food-microbiology laboratories,” in Novel Food Preservation and Microbial Assessment Techniques, ed. I. S. Boziaris (Boca Raton, FL: CRC Press), 353–385. doi: 10.1201/b16758-16
Rees, J. C., and Voorhees, K. J. (2005). Simultaneous detection of two bacterial pathogens using bacteriophage amplification coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass. Spectrom. 19, 2757–2761. doi: 10.1002/rcm.2107
Novick, R. P., Christie, G. E., and Penadés, J. R. (2012). The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 8, 541–551. doi: 10.1038/nrmicro2393
Richards, V. P., Palmer, S. R., Bitar, P. D. P., Qin, X., Weinstock, G. M., Highlander, S. K., et al. (2014). Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Biol. Evol. 6, 741–753. doi: 10.1093/gbe/evu048
Richter, Ł, Janczuk-richter, M., Niedzió, J., Paczesny, J., and Ho, R. (2018). Recent advances in bacteriophage- based methods for bacteria detection. Drug Discov. Today 23, 448–455. doi: 10.1016/j.drudis.2017.11.007
Rodriguez-Rubio, L., Gutierrez, D., Martinez, B., Rodriguez, A., Gotz, F., and Garcia, P. (2012). The tape measure protein of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA35 has an active muramidase domain. J. Appl. Environ. Microbiol. 78, 6369–6371. doi: 10.1128/AEM.01236-12
Rogers, J. S., and Wofford, D. L. (1998). A fast method for approximating maximum likelihoods of phylogenetic trees from nucleotide sequences. Syst. Biol. 47, 77–89. doi: 10.1080/106351598261049
Romero, P., López, R., and García, E. (2004). Genomic organization and molecular analysis of the inducible prophage EJ-1, a mosaic myovirus from an atypical pneumococcus. Virology 322, 239–252. doi: 10.1016/j.virol.2004.01.029
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Serafim, V., Pantoja, L., Ring, C., Shah, H., and Shah, A. J. (2017). Rapid Identification of E. coli bacteriophages using mass spectrometry. J. Proteomics Enzymol. 6, 1–5. doi: 10.4172/2470-1289.1000130
Simmonds, P. (2015). Methods for virus classification and the challenge of incorporating metagenomic sequence data. J. Gen. Virol. 96, 1193–1206. doi: 10.1099/jgv.0.000016
Singh, A., Poshtiban, S., and Evoy, S. (2013). Recent advances in bacteriophage based biosensors for food-borne pathogen detection. Sensors 13, 1763–1786. doi: 10.3390/s130201763
Stanley, E., Walsh, L., van der Zwet, A., Fitzgerald, G. F., and van Sinderen, D. (2000). Identification of four loci isolated from two Streptococcus thermophilus phage genomes responsible for mediating bacteriophage resistance. FEMS Microbiol. Lett. 182, 271–277. doi: 10.1111/j.1574-6968.2000.tb08907.x
Tang, F., Bossers, A., Harders, F., Lu, C., and Smith, H. (2013). Comparative genomic analysis of twelve Streptococcus suis (pro) phages. Genomics 101, 336–344. doi: 10.1016/j.ygeno.2013.04.005
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL _ X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Tiwari, R., Artiushin, S., and Timoney, J. F. (2006). P9, a temperate bacteriophage of Streptococcus equi. Int. Congr. Ser. 1289, 165–168. doi: 10.1016/j.ics.2005.11.086
Walker, J. N., Crosby, H. A., Spaulding, A. R., Salgado-Pabón, W., Malone, C. L., Rosenthal, C. B., et al. (2013). The Staphylococcus aureus ArlRS two-component system is a novel regulator of agglutination and pathogenesis. PLoS Pathog. 9:e1003819. doi: 10.1371/journal.ppat.1003819
Wang, I.-N., Smith, D. L., and Young, R. (2000). Holins: the protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 54, 799–825. doi: 10.1146/annurev.micro.54.1.799
Keywords: pathogen detection, LC-ESI-MS/MS, proteomics, mass spectrometry, phage peptide biomarker
Citation: Abril AG, Carrera M, Böhme K, Barros-Velázquez J, Cañas B, Rama JLR, Villa TG and Calo-Mata P (2020) Characterization of Bacteriophage Peptides of Pathogenic Streptococcus by LC-ESI-MS/MS: Bacteriophage Phylogenomics and Their Relationship to Their Host. Front. Microbiol. 11:1241. doi: 10.3389/fmicb.2020.01241
Received: 24 February 2020; Accepted: 14 May 2020;
Published: 09 June 2020.
Edited by:
David Rodriguez-Lazaro, University of Burgos, SpainReviewed by:
Ana Griselda Binetti, CONICET Instituto de Lactología Industrial (INLAIN), ArgentinaPeter Pristas, Pavol Jozef Šafárik University in Košice, Slovakia
Copyright © 2020 Abril, Carrera, Böhme, Barros-Velázquez, Cañas, Rama, Villa and Calo-Mata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mónica Carrera, bWNhcnJlcmFAaWltLmNzaWMuZXM=; Pilar Calo-Mata, cC5jYWxvLm1hdGFAdXNjLmVz