 Karin Bischof†Doris Schiffer†Sarah Trunk†
Karin Bischof†Doris Schiffer†Sarah Trunk† Thomas Höfler
Thomas Höfler Anja HopferGerald Rechberger
Anja HopferGerald Rechberger Günther Koraimann*
Günther Koraimann*- Institute of Molecular Biosciences, University of Graz, Graz, Austria
In conjugative elements such as integrating conjugative elements (ICEs) or conjugative plasmids (CPs) transcription of DNA transfer genes is a prerequisite for cells to become transfer competent, i.e., capable of delivering plasmid DNA via bacterial conjugation into new host bacteria. In the large family of F-like plasmids belonging to the MobF12A group, transcription of DNA transfer genes is tightly controlled and dependent on the activation of a single promoter, designated PY. Plasmid encoded TraJ and chromosomally encoded ArcA proteins are known activators, whereas the nucleoid associated protein heat-stable nucleoid structuring (H-NS) silences the PY promoter. To better understand the role of these proteins in PY promoter activation, we performed in vitro DNA binding studies using purified H-NS, ArcA, and TraJR1 (TraJ encoded by the conjugative resistance plasmid R1). All proteins could bind to R1PY DNA with high affinities; however, only ArcA was found to be highly sequence specific. DNase I footprinting studies revealed three H-NS binding sites, confirmed the binding site for ArcA, and suggested that TraJ contacts a dyad symmetry DNA sequence located between −51 and −38 in the R1PY promoter region. Moreover, TraJR1 and ArcA supplied together changed the H-NS specific protection pattern suggesting that these proteins are able to replace H-NS from R1PY regions proximal to the transcription start site. Our findings were corroborated by PY-lacZ reporter fusions with a series of site specific R1PY promoter mutations. Sequential changes of some critical DNA bases in the TraJ binding site (jbs) from plasmid R1 to plasmid F led to a remarkable specificity switch: The PY promoter became activatable by F encoded TraJ whereas TraJR1 lost its activation function. The R1PY mutagenesis approach also confirmed the requirement for the host-encoded response-regulator ArcA and indicated that the sequence context, especially in the −35 region is critical for PY regulation and function.
Introduction
Horizontal gene transfer by conjugation is ubiquitous among microorganisms belonging to the kingdoms of bacteria and archaea. If a transfer competent cell harboring a mobile genetic element contacts a recipient in a suitable environment, subsequent transfer of genetic information can decisively expand the metabolic, resistance, or virulence capabilities of the recipient bacterium. In canonical conjugation systems, single-stranded DNA is transported unidirectionally by means of a cell envelope-spanning multi-protein transport complex [termed type IV secretion system (T4SS)]. The process of DNA transfer through the T4SS is ATP dependent and coupled to DNA replication. Mechanistic and structural aspects are detailed in excellent recent reviews (Wong et al., 2012; Christie, 2016; Zechner et al., 2017; Waksman, 2019). The transported DNA is usually guided by at least one protein and derived from an autonomously replicating conjugative plasmid (CP) or from an integrating conjugative element (ICE). Importantly, additional cargo genes integrated into the mobile DNA element can rapidly spread throughout bacterial populations.
Within the Enterobacteriaceae, F-like plasmids are highly prevalent and have been found to be frequently associated with a variety of cargo genes such as antibiotic, biocide, and metal resistance genes. In addition, colicin and microcin genes as well as virulence and enterotoxin genes can be present, providing advantageous traits to their hosts (Koraimann, 2018). Due to the presence of a common backbone with DNA transfer, replication, toxin-antitoxin, and partitioning genes, this group of CPs represent highly successful mobile genetic elements that spread and persist in many enterobacterial species and importantly also in pathogenic strains of Escherichia coli (for a recent review, see Koraimann, 2018). Based on a classification scheme for CPs, F-like plasmids with the classical F plasmid or the antibiotic resistance plasmid R1 belong to the MOBF12A group. They harbor conjugation genes that are phylogenetically related and share a similar arrangement (Fernandez-Lopez et al., 2016; Koraimann, 2018; Figure 1A). Nine subgroups have been defined within the MOBF12A based on the amino acid sequence variability of the DNA transfer gene activator TraJ (Koraimann, 2018).
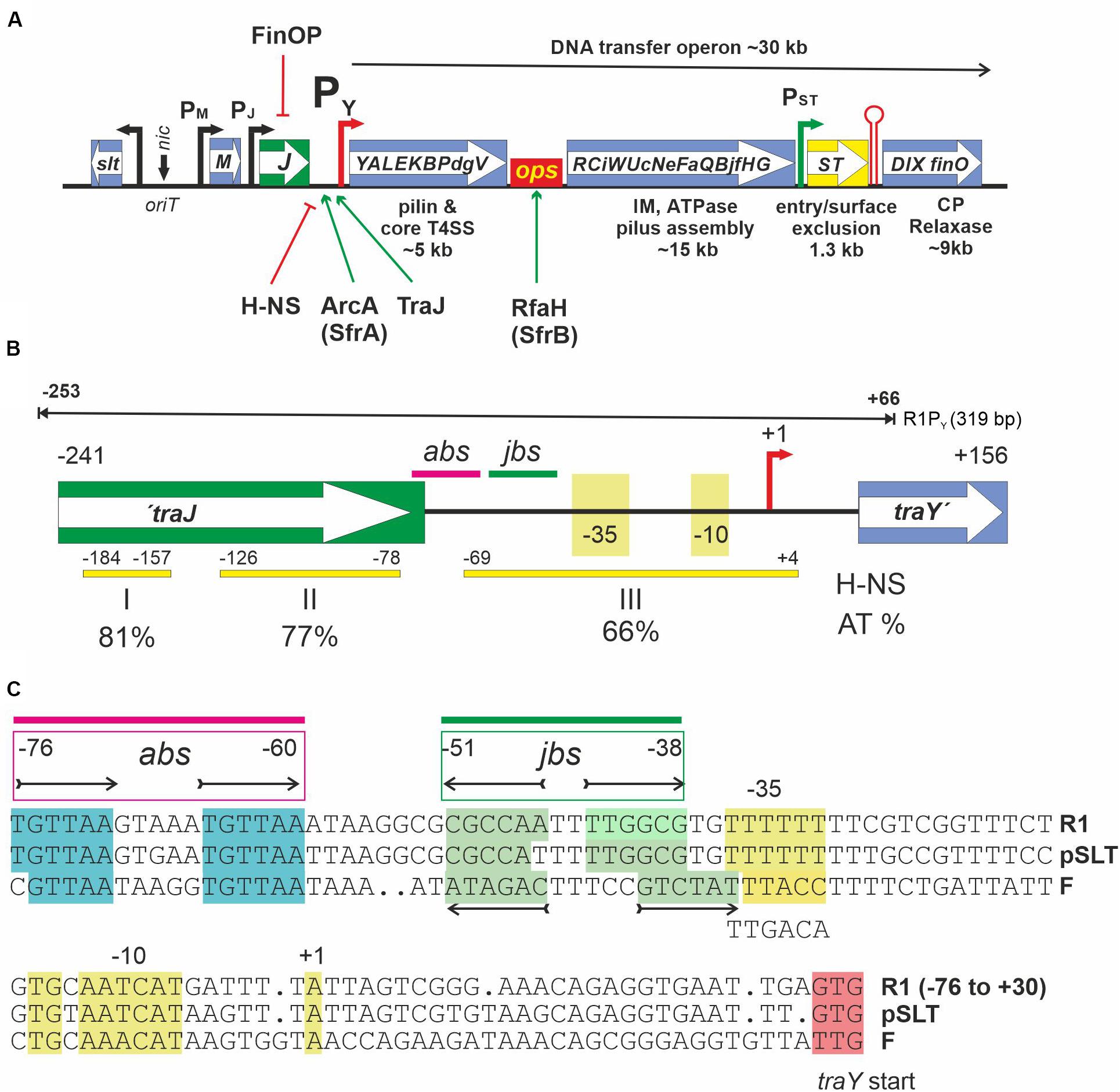
Figure 1. (A) Schematic representation of the DNA transfer region of F-like MOBF12A plasmids. DNA transfer (tra) genes are indicated by capital letters inside the boxes. oriT is the origin of DNA transfer, representing a DNA region where several DNA binding proteins such as TraM, TraY, and TraI (the relaxase-helicase) bind. nic denotes the nick site which is recognized by the relaxase for the initiation of ssDNA transfer into recipients via a T4SS. Arrows indicate known promoters. PY is the only known promoter for transcription of the approximately 35 DNA transfer operon genes. It is regulated by the indicated host and plasmid encoded proteins. H-NS acts as a silencing factor whereas both ArcA (a.k.a. SfrA, host encoded) and TraJ (plasmid encoded) are needed for activation. Transcription of the complete operon additionally requires the host encoded anti-termination protein RfaH and its recognition site ops (operon polarity suppressor). Entry/surface exclusion genes traS and traT are constitutively transcribed in plasmid carrying cells. The transcriptional terminator (red hairpin) is functional only for transcripts initiating at promotor PST. For a more in-depth discussion of DNA transfer genes and their regulation in MOBF12A plasmids, the reader is referred to a recent review (Koraimann, 2018). (B) A schematic overview of the PY promoter region of the MOBF12A prototype plasmid R1 from −241 to +156 relative to the transcription start site (+1, red arrow) of the tra operon is shown. This fragment is present in the promoter test plasmid pRSYZ4. Yellow bars with numbers indicating their location represent H-NS binding regions (I,II,III) as determined in this work. AT percentage of these regions is shown below. abs, binding site for ArcA; jbs, binding site for TraJ. These elements are also present on the 319 bp DNA fragment—termed R1PY—used for EMSA and DNase I footprinting experiments (black line above). (C) Sequence representation of the PY promoter of MOBF12A plasmids R1 (KY749247), pSLT (AE006471), and F (AP001918). To access a complete plasmid sequence in the NCBI nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore), please use the accession number provided in parentheses. The non-template DNA strand is shown. abs and jbs sites as determined in this work are given above the sequences for the plasmid R1 PY promoter. The tandem repeat for ArcA binding [consensus: TGTTAA(N5)TGTTAA] is highlighted in cyan and the jbs inverted repeat sequences are highlighted in green. −10, extended −10 (TG), −35 as well as the first transcribed nucleotide (+1) are highlighted in yellow.
The ability to transport DNA from donors into recipient cells requires the expression of DNA transfer genes. A variety of regulatory mechanisms ensure that only a few cells in a genetically homogeneous population undergo this developmental process and become transfer competent. Thereby, at the population level, negative consequences of transfer gene expression are reduced without compromising horizontal gene transfer to novel hosts (Koraimann and Wagner, 2014; Stingl and Koraimann, 2017). In F-like plasmids DNA transfer genes are controlled through a network of regulatory elements ensuring that under optimal conditions only a few cells in a population—approximately 0.1 to 1%—proceed to transfer competence (Frost and Koraimann, 2010; Koraimann and Wagner, 2014). The key plasmid encoded activator that has to escape negative control is TraJ, a plasmid encoded protein. As schematically depicted in Figure 1A, TraJ and chromosomally encoded ArcA proteins are absolutely required to overcome heat-stable nucleoid structuring (H-NS) protein silencing and to activate transcription of DNA transfer genes from the main transfer operon promoter PY (Will and Frost, 2006; Wagner et al., 2013; Lu et al., 2018).
Here we characterize DNA binding characteristics of these proteins and focus on defining the binding sites in the PY promoter region of plasmid R1 (TraJR1), a conjugative resistance plasmid discovered 1963 in London in a clinical Salmonella enterica serovar Paratyphi B isolate (Datta and Kontomichalou, 1965). Originally named R7268, it was subsequently renamed R1 (Meynell and Datta, 1966). Only recently the complete sequence of this plasmid has been determined (Cox and Schildbach, 2017, accession: KY749247). In order to characterize DNA binding, purified H-NS, ArcA, and TraJR1 proteins were used in both electrophoretic mobility shift assays (EMSA) and DNase I footprinting experiments. EMSA revealed DNA binding by H-NS and TraJ with low sequence specificity, whereas ArcA binding to R1PY was highly sequence specific. In DNase I footprinting experiments, the TraJR1 binding site (jbs) in the PY promoter region was found to span the region from −51 to −38, immediately adjacent to the ArcA binding site (abs), located at −76 to −60 (see Figures 1B,C). Furthermore, TraJ and ArcA together were found to be capable of displacing the silencing factor H-NS from promoter proximal sites of the PY promoter region in vitro. In order to corroborate these findings, we mutated the proposed crucial sequence elements of the PY promoter and tested the mutants in a well-established reporter system. This approach confirmed the binding site for TraJR1 in the region −51 to −38 and revealed that specificity for binding by TraJ solely resides in this locus. Second, it confirmed the essential contribution of ArcA in PY promoter activation and finally demonstrated that the non-canonical −35 element is important for control and regulation of PY.
Materials and Methods
Media, Growth Conditions, Bacterial Strains, Plasmids, and Oligonucleotides
LB medium (L–1: 10 g tryptone, 5 g yeast extract, 5 g NaCl) or 2xTY medium (per liter: 16 g tryptone, 10 g yeast extract, and 5 g NaCl) was used. For β-galactosidase assays, cells were grown in M9 minimal salt medium (Miller, 1972); L–1: 3 g KH2PO4, 12.8 g Na2HPO4 × 7 H2O, 1 g NH4Cl, 0,5 g NaCl, 3 g casaminoacids; 0.1 mg/mL thiamin; 0.2% glucose; 1 mM MgSO4. If appropriate, antibiotics such as 40 μg mL–1 kanamycin, 20 μg mL–1 chloramphenicol, or 50 μg mL–1 ampicillin were added. Unless otherwise indicated, cultures were grown aerated in a shaker-incubator at 180 r/min and 37°C. Cell densities (OD600) were measured in a Hitachi U5100 spectrophotometer. E. coli strains and plasmids are listed in Supplementary Table S1; oligonucleotides obtained from eurofins Genomics (Ebersberg, Germany) used in this study are listed in Supplementary Table S2.
DNA Manipulations, Cloning, Sequencing, and Sequence Analyses
DNA manipulations were done using standard techniques (Sambrook et al., 1989) or according to the manufacturers’ recommendations. PCR reactions for cloning purposes were performed using Phusion High-Fidelity DNA Polymerase (New England BioLabs). Construction of expression plasmids for TraJ (pSD1002), TraJI187T (pSD1002I187T), and H-NS (pSThns) are detailed in the Supplementary Methods section. ArcA was overproduced from plasmid pETarcA-1 (Strohmaier et al., 1998). Mutations in the R1PY-lacZ promoter test plasmid pRSYZ4 (Strohmaier et al., 1998; Wagner et al., 2013) were introduced according to a modified QuikChange protocol (Liu and Naismith, 2008; Laible and Boonrod, 2009). TraJ expressing plasmids in the R1PY promoter activation studies were the same as used previously (Wagner et al., 2013) except pJSlt2 containing the traJ gene from the Salmonella enterica plasmid pSLT. All resulting plasmids created in this study were verified by DNA sequencing using the Eurofins Genomics (Ebersberg, Germany) sequencing service. In silico construction of plasmids and sequence analysis was performed using SnapGene software.
Protein Expression and Purification
ArcA protein containing an N-terminal His6 tag was purified using the method described and activated by phosphorylation before use in EMSA and DNase I footprintig assays (Strohmaier et al., 1998). TraJ and His6-H-NS were produced and purified as described in the Supplementary Material.
DNA Fragments for EMSA and DNase I Footprinting Experiments
All DNA fragments were amplified by PCR using 5′-Cy5-labeled primers as indicated. Primers and their sequences are listed in Supplementary Table S2. R1PY DNA (319 bp) from −253 to +66 relative to the transcription start site of the tra operon containing the 3′ end of the traJ gene, the previously established ArcA-P binding site (Strohmaier et al., 1998), the predicted PY promoter (Strohmaier et al., 1998), and the 5′ end of the traY gene (Figure 1B) was amplified by PCR from E. coli harboring plasmid R1 using oligonucleotides PYTransfw, and PYTransrev1. For footprinting studies, only one of the two primers was 5′-Cy5-labeled. GZ DNA (261 bp) is a fragment generated from plasmid pGZ119EH DNA using oligonucleotides pTG-NF4-fw and pgzrev508. It roughly encompasses the tac promoter, the lac operator and the multiple cloning site of the plasmid. FPY DNA (422 bp) was generated by PCR from E. coli XK1200 (pOX38-Km) and primers F_Py_fw and F_Py_rev. It contains DNA from −393 to +29 relative to the transcription start site of the F-plasmid tra operon. Atu DNA (395 bp) was generated by PCR from Agrobacterium tumefaciens C58 using primers GroEL_Atum_fw and GroEL_Atum_rev and covers a part of the groEL gene.
EMSA
A Cy-5 labeled DNA fragment (0.5 nM) was mixed by pipetting on ice with varying concentrations of purified protein in band shift buffer (12 mM HEPES pH 7.9, 30 mM Tris-Cl pH 7.5, 60 mM KCl, 10% glycerol, 1.7 mM EDTA, 26 mM boric acid, 5 mM TCEP). Samples were placed in 0.2 mL PCR tubes; total reaction volume was 15 μL. Tubes were then transferred to a thermocycler and incubated at 30°C for 15 min. DNA and protein–DNA complexes were then electrophoretically separated on non-denaturing 6% polyacrylamide gels (PAGs; acrylamide:bisacrylamide 80:1, 2.5% glycerol, 0.07% APS, and 0.07% TEMED). Vertical electrophoresis was performed in Hoefer Mighty Small II chambers with TBE buffer (100 mM Tris base, 100 mM boric acid, 2.5 mM EDTA pH 8.3) with 20 mA at 4°C for 30–45 min. After disassembly of the electrophoresis unit, the gel was scanned using a Typhoon 9400 scanner (GE Healthcare). For image analysis and estimation of the dissociation constant, the Quantity One software (Bio-Rad) was used. Each band shift experiment was performed at least twice.
DNase I Footprinting
For the DNase I footprint experiment, the same 319 bp R1PY DNA fragment as described for EMSA experiments was used with the exception that only one DNA strand was Cy5-labeled. Binding reaction mixtures (50 μL total volume) contained 50 mM Tris-Cl pH 7.2, 100 mM KCl, 10% glycerol, 30 ng of PY DNA, and varying concentrations of TraJ, ArcA-P, or H-NS. To allow protein–DNA complex formation, PY DNA and proteins were incubated for 20 min at 4°C and 2 min at 25°C. 50 μL of a 10 mM MgCl2, 5 mM CaCl2 solution were added and incubated for 1 min. To digest the DNA 0.1 U DNase I (1 U/μL) (Fermentas) were added directly to the protein-DNA mixes and incubated for 2 min. The reaction was terminated with 90 μL stop solution (20 mM EDTA, 200 mM NaCl, 1% SDS, 250 μg/mL herring sperm DNA). DNA was precipitated by adding 2.5 volumes of ethanol (96%) and incubation for 3 h at −70°C. Precipitates were sedimented by centrifugation (15,000 × g, 10 min, RT), washed with 70% ethanol, and centrifuged as before. After removal of the supernatant, the pellets were air-dried and resuspended in 10 μL formamide loading dye (Thermo Sequenase Primer Cycle Sequencing Kit, GE Healthcare), denatured for 10 min at 95°C, and separated on a 8% polyacrylamide sequencing gel (ReproGelTM, High resolution, GE Healthcare) in an Alf Express Sequencer (GE Healthcare). The sequencing reactions of the PY DNA fragment were done with the Thermo Sequenase fluorescent-labeled Primer Cycle Sequencing Kit (GE Healthcare) using 200 ng of DNA and Cy-5 labeled primer PYTransfw or PYTransrev1. Data were analyzed using the AlfWin Sequence Analyzer software (Amersham Pharmacia Biotech).
R1PY Promoter Activation Tests
For these tests, E. coli MC4100 co-transformed with the PY promoter test plasmid pRSYZ4 (carrying a R1PY-lacZ fusion) and traJ expressing plasmids (pJR1 or pJF or pJSLT2) or a vector control plasmid (pGZNSO2) were assayed for β-galactosidase activity. Promoter activation tests were similarly performed with derivatives of pRSYZ4 created by site specific mutagenesis. β-galactosidase assays were performed essentially as described previously (Wagner et al., 2013) and according to an Open Wetware protocol1 (last accessed 28/02/2020). Details of the procedure can be found in Supplementary Material.
Results
Binding of H-NS ArcA, and TraJ to R1PY DNA
In a previous study, we could show that H-NS is involved in silencing tra gene expression of plasmid R1 in vivo (Wagner et al., 2013). We therefore set up experiments to investigate direct H-NS binding to a fluorescently labeled DNA fragment containing the R1 PY promoter (R1PY DNA, 319 bp, 62% AT) as shown in Figure 1B. This DNA fragment was incubated with increasing concentrations of purified H-NS protein and the resulting DNA–protein complexes were separated electrophoretically on a non-denaturing PAG. As can be seen in Figure 2A, H-NS bound to R1PY with high affinity producing a distinct complex visible at protein concentrations between 30 and 250 nM H-NS. Higher H-NS concentrations shifted the DNA to the top of the gel. We estimated an apparent dissociation constant Kd of 15 nM. However, as is clearly obvious from Figure 2C, H-NS also bound to a control DNA fragment (GZ DNA, 261 bp, 49% AT). For a direct comparison in the DNA binding behavior, we performed binding studies using the same DNA fragments with phosphorylated ArcA protein (ArcA-P). The results are shown in Figures 2B,D. Binding of ArcA-P to R1PY with apparent Kd of 60 nM is highly specific. ArcA-P only bound to R1PY containing the abs site shown in Figure 1C but not to the GZ fragment which does not contain a binding motif for ArcA.
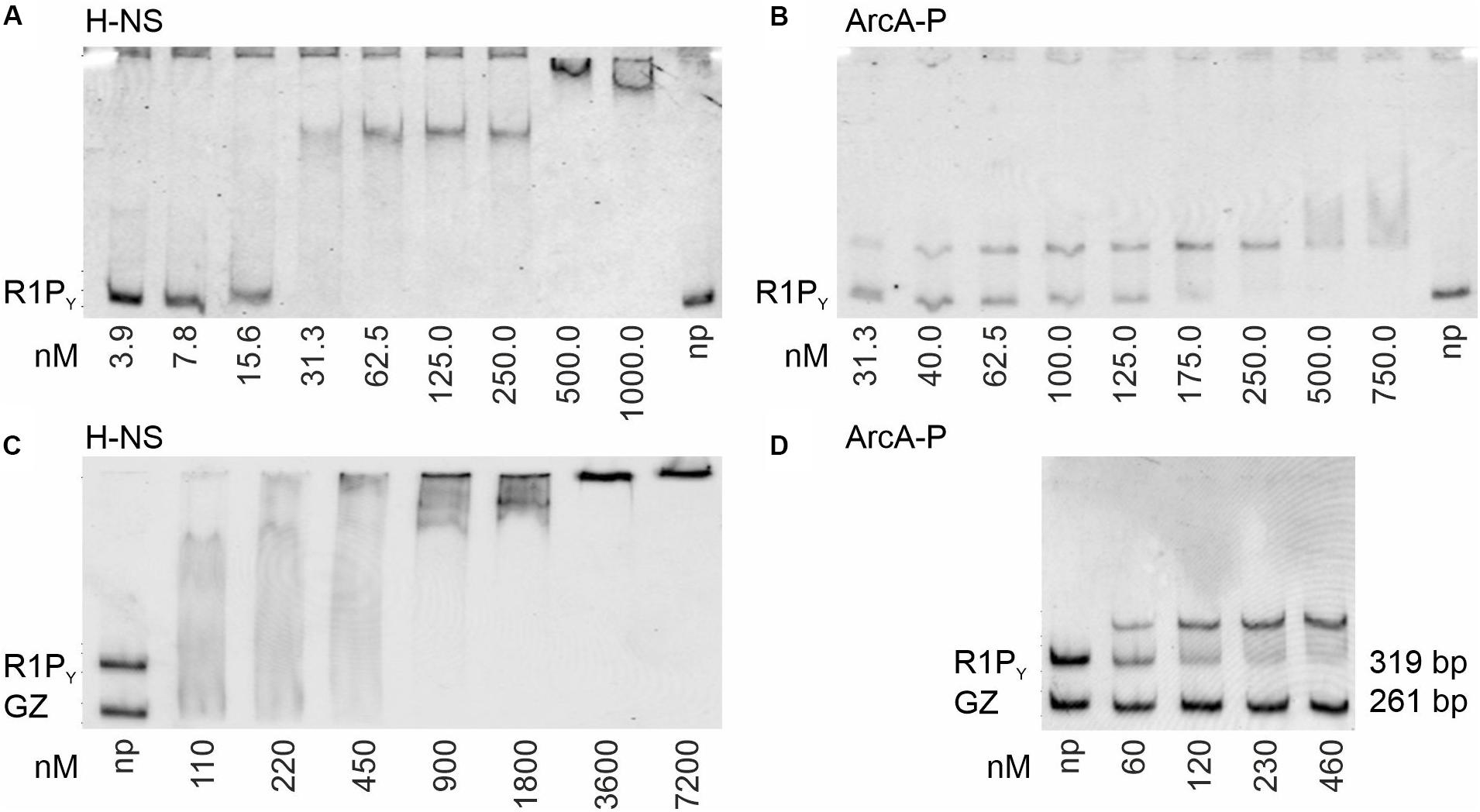
Figure 2. Binding of H-NS and ArcA-P (phosphorylated ArcA) to R1PY (319 bp, 62% AT, see Figure 1B) and control (GZ, 261 bp, 49% AT) DNA fragments. Electrophoretic mobility shift assays (EMSAs) were performed with the indicated fluorescently Cy5-labeled ds DNA fragments (0.5 nM) and purified proteins using the indicated protein concentrations (np: no protein present). Formed DNA–protein complexes were electrophoretically separated using non-denaturing polyacrylamide gels (PAGs) from free DNA and visualized by fluorescence imaging. Free DNA fragments are indicated at the bottom of the gels. Complexes appear as distinct bands, smear, or are retained in the gel slots. To increase perceptual contrast, images were inverted (gray fluorescence signal on a white background). (A) EMSA with purified H-NS protein and R1PY DNA. High affinity binding in with an apparent dissociation constant (Kd) of about 15 nM can be observed. (B) EMSA with ArcA-P protein demonstrates binding of ArcA-P to R1PY with Kd of 60 nM. (C) H-NS binds to both R1PY and GZ DNA. (D) ArcA-P is highly specific and only binds to R1PY which contains the abs tandem repeat sequence as depicted in Figure 1C.
In a second series of band shift experiments (Figure 3), we wished to investigate the DNA binding properties of TraJR1 which has been identified as an essential activator of the PY promoter and tra operon transcription of plasmid R1 (Wagner et al., 2013). As can be seen in Figure 3A, purified TraJR1 bound to R1PY DNA; however, no distinct DNA–protein complexes appeared except that at concentrations > 500 nM TraJR1 R1PY DNA is shifted to the top of the gel. The apparent Kd was estimated to be 250 nM for binding of TraJR1 to R1PY DNA. Importantly, the only protein found by mass spectroscopy in the top shifted band was TraJR1 (data not shown). Thus, we exclude the presence of contaminating DNA binding proteins in our TraJ protein preparations. We previously identified a loss-of-function mutant of TraJR1 (I187T); furthermore, ChIP experiments suggested that this mutant had severely disturbed the DNA binding capability (Wagner et al., 2013). Consistent with these findings, purified TraJR1 (I187T) displayed a considerably weaker DNA binding activity with an apparent Kd of 750 nM (Figure 3B). To investigate the specificity of TraJR1 DNA binding, we performed several DNA binding experiments. As can be seen in Figure 3C, TraJR1 also bound to FPY and Atu DNA fragments which do not contain the jbs recognition sequence (Figure 1C). However, quantitation of the remaining free DNA in relation to total DNA at 500 nM TraJR1 revealed only 17% in case of R1PY, whereas there were 34 or 28% in case of FPY or Atu fragments, respectively. This result suggested that TraJR1 bound with a higher affinity to R1PY (319 bp) when compared to FPY (422 bp) or a DNA fragment from A. tumefaciens (Atu, 395 bp). In another approach, TraJR1 binding was investigated using two different DNA fragments (R1PY and GZ) simultaneously. Although not a strikingly clear difference (compare ArcA-P binding to the same fragments, Figure 2D), there is a slight preference for the R1PY fragment (Figures 3D,E). We finally found that pre-incubation of the DNA fragments (R1PY and GZ) with 250 nM ArcA-P increased TraJR1 preference for R1PY DNA (Figures 3F,G). Taken together, these results suggest that TraJR1 has a DNA binding activity which is stronger when the cognate jbs site is present and that ArcA-P binding could direct TraJR1 toward the DNA containing jbs next to abs.
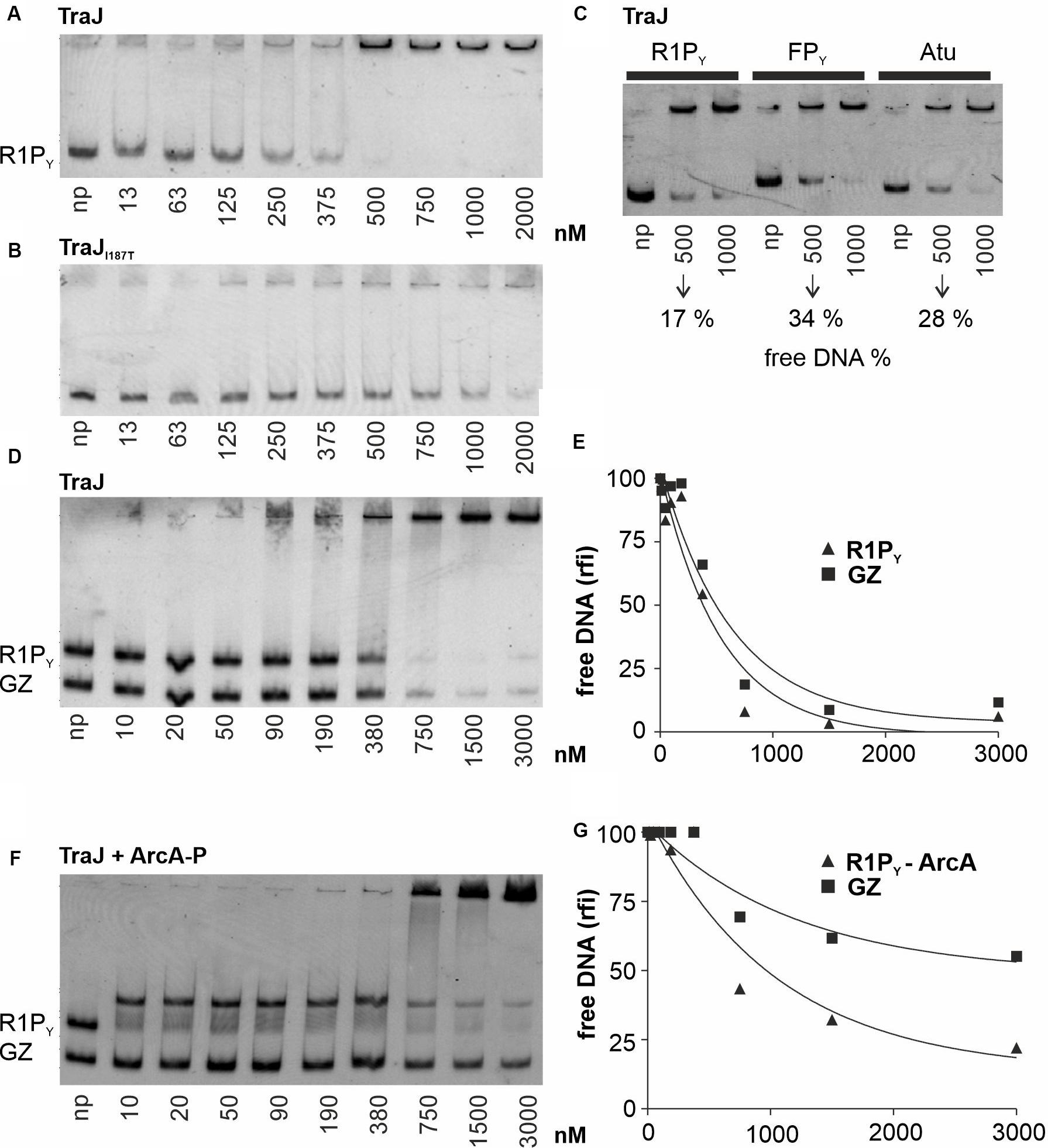
Figure 3. EMSAs with various proteins and DNA fragments to characterize TraJR1 binding to DNA. Binding studies were analyzed as described in Figure 2. np, no protein added. (A) TraJR1 binds to R1PY DNA. No distinct DNA–protein complexes can be seen. The Kd is estimated from this EMSA is 250 nM. At concentrations > 500 nM R1PY DNA is shifted to the gel slot. (B) An activation deficient TraJR1 mutant (I187T) displays a considerably weaker DNA binding activity with an apparent Kd of 750 nM. (C) Comparison of TraJR1 binding to different DNA fragments. TraJR1 binds with higher affinity to R1PY (319 bp) when compared to FPY (422 bp) or a DNA fragment from Agrobacterium tumefaciens (Atu, 395 bp). (D) Simultaneous binding of TraJR1 to R1PY and a control fragment (GZ). TraJR1 binds with higher affinity to R1PY when compared to a control DNA. (E) The percentage of free R1PY DNA and GZ DNA from D was calculated and graphically represented relative to the DNA fluorescence of the respective fragment without protein. (F) Pre-incubation of the DNA fragments (R1PY and GZ) with 250 nM ArcA-P increases TraJR1 preference for R1PY DNA. (G) The percentage of R1PY DNA bound to ArcA-P or free GZ DNA from F was calculated and graphically represented relative to the DNA fluorescence of the respective fragment without addition of TraJR1. The difference between TraJR1 binding to R1PY-ArcA-P versus binding to GZ when compared to E is clearly increased. rfi: relative fluorescence intensity.
H-NS and TraJR1 Binding Sites in the PY Promoter Region of Plasmid R1
Binding of ArcA-P to the R1 PY promoter region has been determined earlier by DNase I footprinting (Strohmaier et al., 1998) and clearly demonstrated that ArcA-P can bind to the abs site indicated in Figure 1C. To determine the binding sites for H-NS and TraJR1, we again performed DNase I protection assays using the 319 bp R1PY DNA fragment either fluorescently labeled on the top (non-coding) or bottom (coding) strand. The result of such an experiment is shown in Figure 4 where the binding regions for H-NS appear as three distinct regions with reduced band intensities when compared to the control DNase I digest without protein. These regions are labeled H-NS I (−184 to −157, 81% AT), H-NS II (−126 to −78, 77% AT), and H-NS III (−69 to + 4, 66% AT). The banding pattern changes when proteins ArcA-P and TraJR1 are additionally present (lane A + J + H in Figure 4), suggesting a replacement of H-NS by ArcA-P and TraJR1. The binding sites for ArcA-P and TraJR1 are indicated in Figure 4 and correspond to abs and jbs sequence motifs shown in Figure 1C.
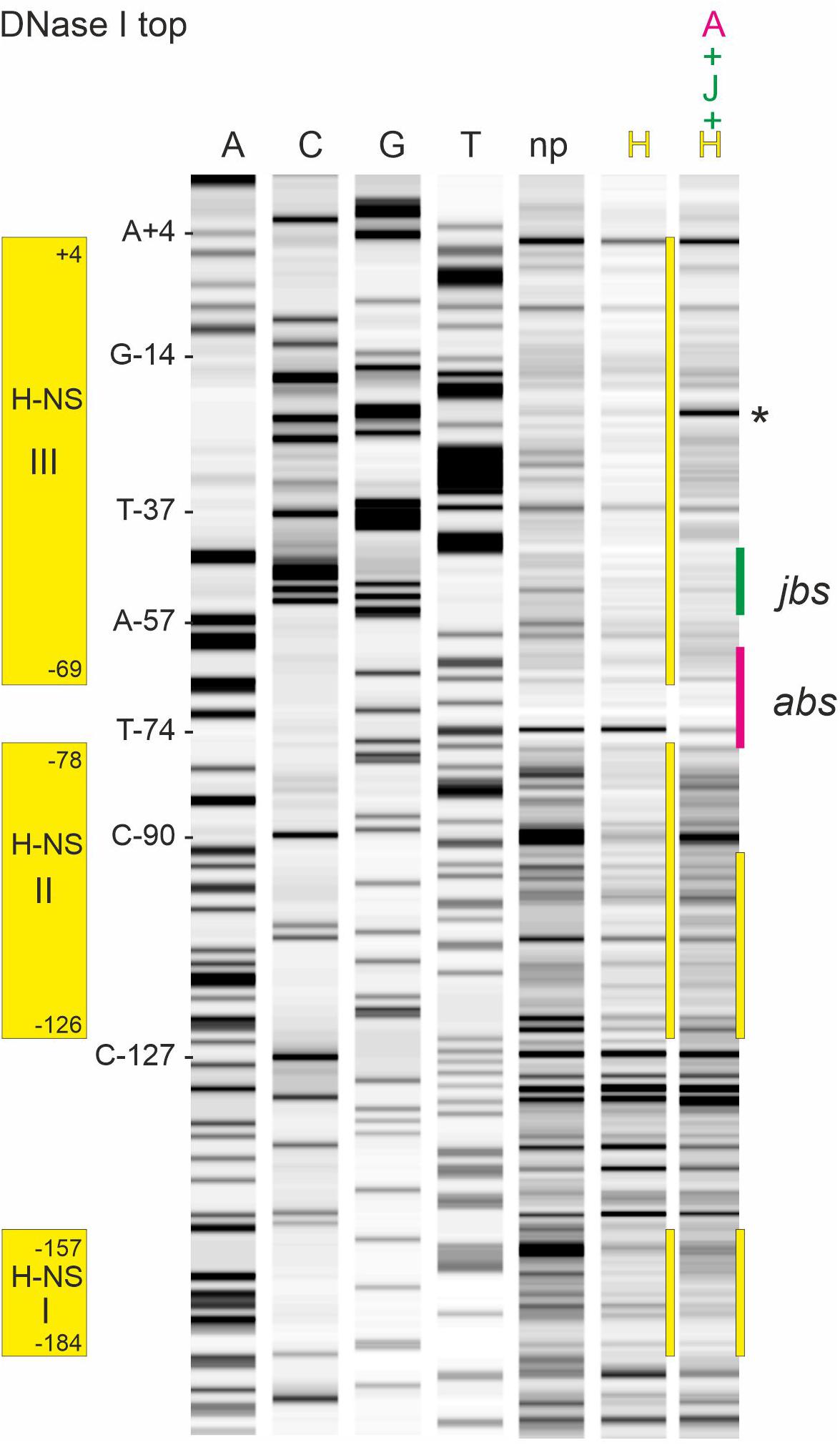
Figure 4. DNase I footprinting of the R1PY dsDNA fragment with the non-template strand fluorescently labeled reveals binding sites for H-NS, ArcA-P, and TraJR1 in the R1PY promoter region. Lanes A, C, G, and T are products of sequencing reactions with some of the bases indicated on the left side of lane A. np, DNA fragments obtained with DNase I digestion. H, DNA fragments obtained with 0.2 μM H-NS before DNase I digestion. Three distinct regions with reduced band intensities are indicated by yellow bars. H-NS protected regions are indicated on the left side and correspond to three AT rich regions as shown in Figure 1B. Last lane shows DNase I fragments obtained in the presence of ArcA-P (0.4 μM), TraJR1 (5 μM), and H-NS (0.2 μM). The resulting change in the banding pattern suggests a replacement of H-NS from binding site III and partially from binding site II. Note the appearance of an additional prominent band between −35 and −10 regions in the PY promoter (indicated by an asterisk). abs, binding site for ArcA-P; jbs, binding site for TraJR1.
To better define the binding site for TraJR1 further DNase I footprinting experiments were performed with increasing TraJR1 concentrations, ArcA-P alone and TraJR1 together with ArcA-P. As shown in Figures 5A,D (top strand labeled) TraJR1 protected from DNase I cleavage at positions A-57 and C-90. Protection of C-90 by TraJR1 indicates a secondary binding site in R1PY which is located outside the proposed jbs recognition site. This can be attributed to the sequence independent DNA binding activity of TraJR1 which was also evident in the band shift experiments. ArcA-P protected T-74. Positions A-57 and T-74 are consistent with the proposed jbs and abs recognition sites, respectively. Addition of both proteins produced a protection pattern indicating simultaneous DNA binding of TraJR1 and ArcA-P. On the bottom strand TraJR1 protected from DNase I cleavage at position A-42. ArcA-P protected at T-72 and A-76 (Figures 5B,C). Again, these positions are consistent with the proposed jbs and abs motifs. From these results, we deduce that the recognition site for TraJR1 (jbs) in the R1PY promoter extends from −51 to −38 and includes the imperfect inverted repeat sequence CGCCAATTTTGGCG with two non-palindromic bases (TT) in the center. Incubation of R1PY DNA with ArcA-P alone resulted in the expected protection of the direct repeats TGTTAAGTAAATGTTAA from −76 to −60. Binding of ArcA-P to this site has been shown by our group previously (Strohmaier et al., 1998). This sequence closely resembles the proposed recognition matrix for ArcA-P in E. coli (Liu and De Wulf, 2004; Park et al., 2013). In addition, the sequence motif is highly conserved in PY promoter sequences of F-like plasmids.
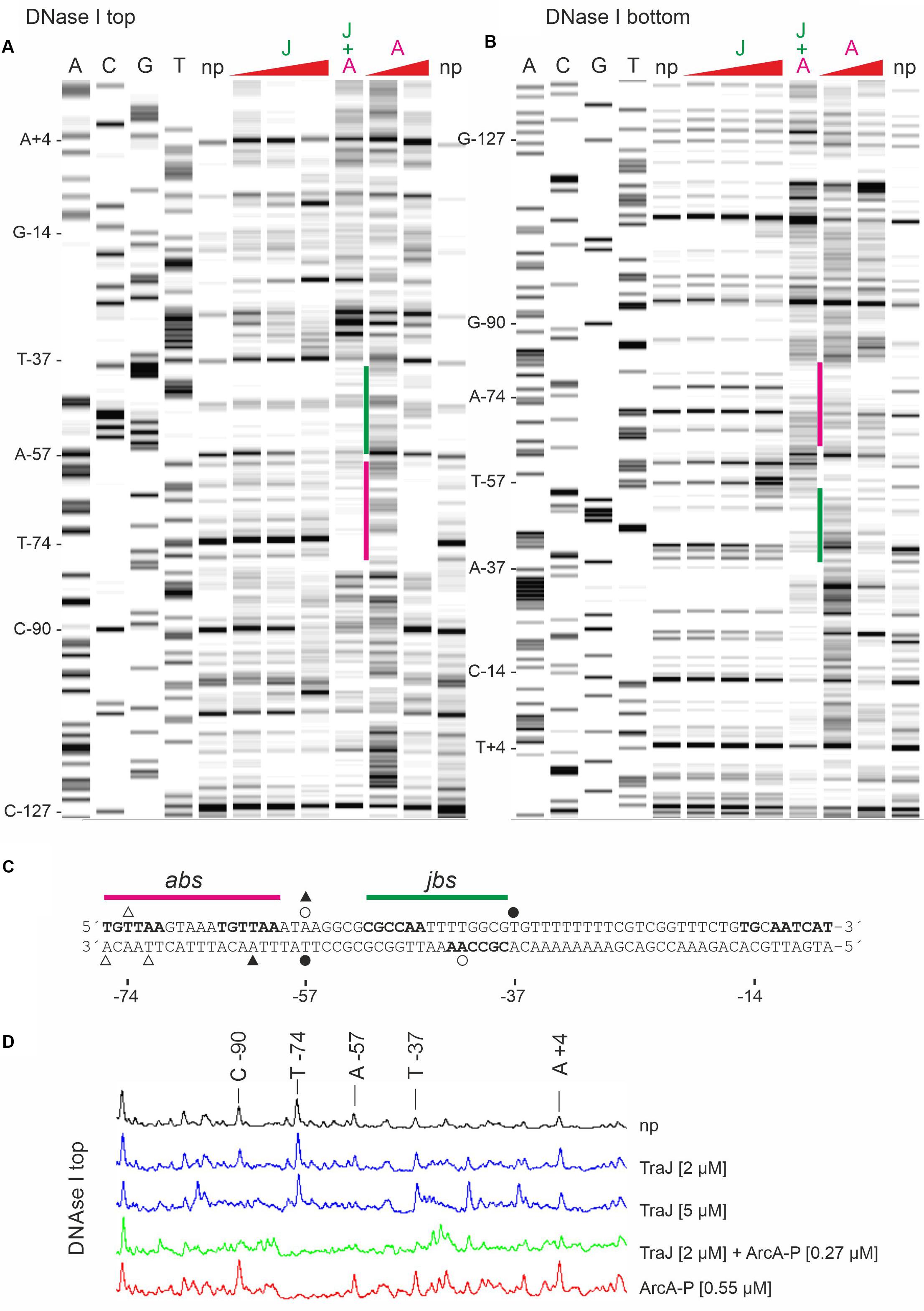
Figure 5. DNase I protection pattern by TraJR1 and ArcA-P obtained on R1PY DNA. R1PY DNA fluorescently 5′-labeled on the non-template (A) or the template (B) strand was incubated with proteins as indicated and subsequently digested with DNase I. The resulting DNA fragments were separated on a denaturing PAG. A gel-like image of the detected fluorescence signals is shown in A,B. Lanes A, C, G, and T are products of sequencing reactions. For orientation, some bases are indicated on the left side of lane A, respectively. Proteins were added before DNase I digestion as follows: J: TraJR1 0.5, 2, or 5 μM; A: ArcA-P 0.27 or 0.55 μM. J + A: TraJR1 + ArcA-P, 2 and 0.27 μM, respectively. np, no protein added before DNase I digestion. A: TraJR1 protects from DNase I cleavage at positions A-57 and C-90. ArcA-P protects T-74. Positions A-57 and T-74 are consistent with the proposed jbs and abs recognition sites as shown in C, respectively. Addition of both proteins produces a protection pattern indicating simultaneous DNA binding of TraJR1 (indicated by a green bar) and ArcA-P (magenta bar). (B) TraJR1 protects from DNase I cleavage at position A-42. ArcA-P protects T-72 and A-76. These positions are consistent with the proposed jbs and abs regions as shown in C, respectively. Addition of both proteins produces a protection pattern indicating simultaneous DNA binding. (C) DNA sequences of non-template (top) and template (bottom) DNA strands from the R1PY promoter region (−76 to −7). The proposed abs (−76 to −60) and jbs (−51 to −38) sites are indicated above the sequence. Open triangles indicate bases protected by ArcA-P and filled triangles represent ArcA-P induced DNase I cleavage sites. Open circles indicate bases protected by TraJR1 and filled circles represent TraJR1 induced DNase I cleavage sites. + 1: transcription start site as determined by reverse transcription of tra mRNA, −10: −10 region of the PY promoter, TG: extended −10 motif. (D) Curve view of DNase I footprinting experiment shown in A. Proteins were added as indicated. np, no protein added before DNase I treatment.
PY Promoter Sequence Requirements for TraJ Activation
The in vitro results demonstrated that TraJR1 can bind to DNA. Based on the above described preference for jbs containing DNA and the DNAse I footprinting data, we concluded that TraJR1 specifically recognizes an inverted repeat sequence in the PY promoter including bases from −51 to −38 in the R1PY promoter region. This is consistent with previous results of ChIP experiments where we could demonstrate specific in vivo binding of TraJR1 to the R1PY DNA (Wagner et al., 2013). We also noted that sequences in the PY promoter region between different subclasses of F-like plasmids differ as do the cognate TraJ proteins (Wagner et al., 2013; Koraimann, 2018). To verify the jbs recognition sequence and to demonstrate subclass-specific activation of the PY promoter of F-like plasmids, we systematically mutagenized the jbs site in the R1PY promoter and tested its activation by TraJ proteins from plasmid R1, pSLT, or F. The mutations shown in Figure 6A were introduced in the promoter test plasmid pRSYZ4 in which the R1PY promoter is fused to the lacZ reporter gene. β-Galactosidases assays were performed to evaluate basal promoter activity (with a vector control plasmid) and its activation by the above mentioned TraJ proteins. As can be seen in Figure 6B, there was a very low basal expression level from the R1PY promoter which could be induced by both TraJR1 and TraJpSLT but not by TraJF. In the fully stimulated promoter expression was about 6.5-fold to 8-fold higher than the basal expression. The mutation of three bases (m1) in jbs did not change the basal expression level but completely abolished activation by TraJR1 showing that these bases in jbs are absolutely essential for recognition by TraJR1. There was no activation by TraJF. Further sequential mutations toward the F jbs did not change this phenotype dramatically (m4, m41). However, finally, mutants with an almost completely changed jbs sequence (m42, m43) displayed a specificity switch. The PY promoter in these mutants was activated by TraJF and not by TraJR1. We also observed an approximately twofold increase in the basal PY promoter activity in mutant m43 which could be activated by TraJF fourfold (Figure 6).
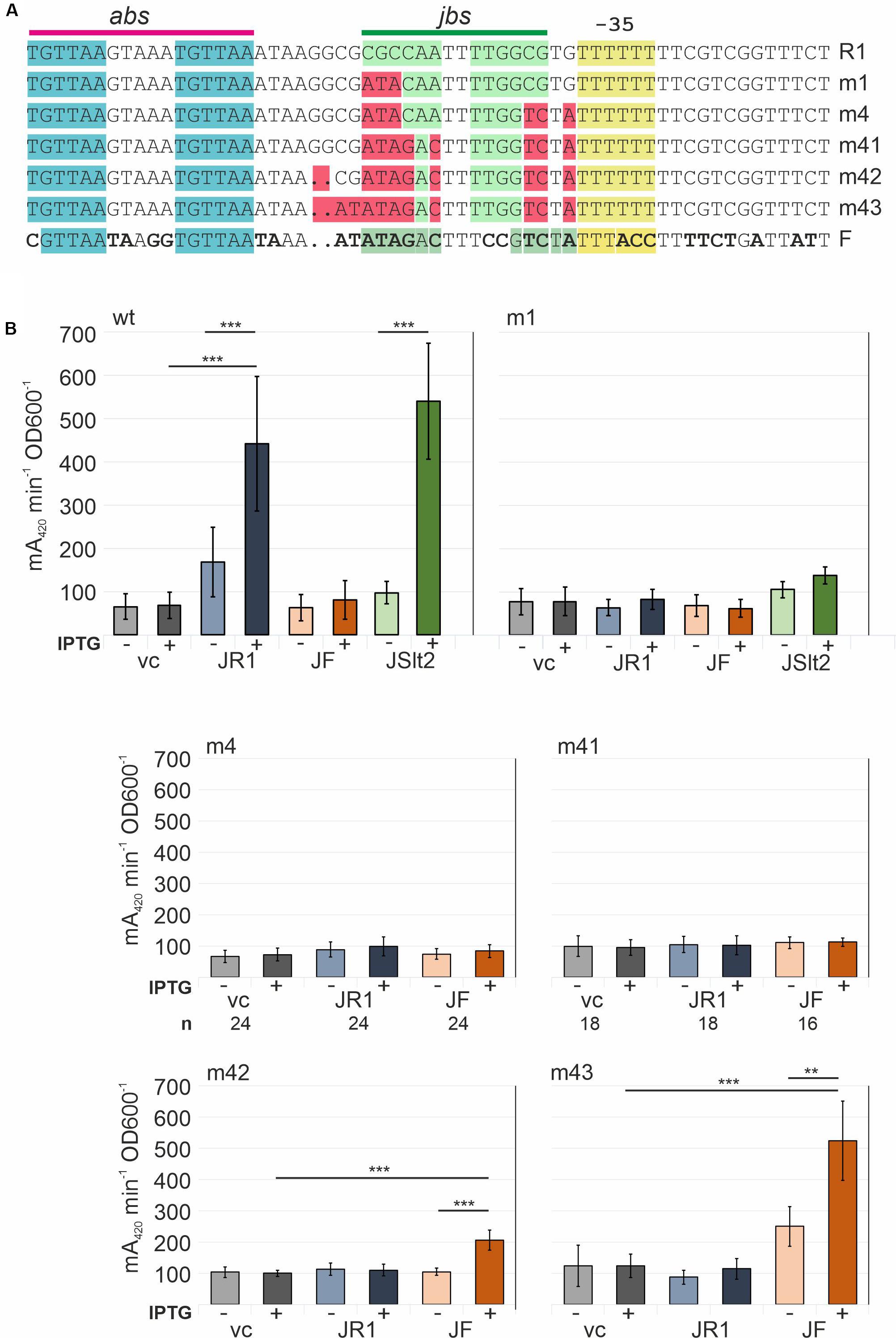
Figure 6. Determination of important sequence elements in the R1PY promoter for activation by TraJR1. Successively introduced mutations switch recognition specificity to TraJF. (A) Sequences showing sequentially created site specific mutations in R1PY. Top line shows the R1PY wt sequence with the abs and jbs sites indicated above. Bottom line shows FPY wt sequence with bases that differ from R1PY in bold letters. (B) β-Galactosidase assays with bacterial cells harboring a R1PY-lacZ promoter-test plasmid and a second compatible plasmid IPTG-inducible for expression of traJ from plasmids R1 (JR1), F (JF), or pSLT (JSlt2); vc, vector control (no traJ). IPTG (inductor): +(added), −(not added). Mean values and standard deviations were calculated from at least three independently carried out experiments with two technical replicates for each experiment. P-values (paired t-test): **P < 0.01; ***P < 0.001.
Another series of mutations was introduced into the R1PY promoter to investigate the effects of exchanges in abs and the −35 region (Figure 7). In the m6 mutation in which the second TGTTAA ArcA recognition motif in abs is changed to TCATAA, we observed a reduced basal expression level (approximately 50% of the wt promoter) and a complete loss of its activation by TraJR1. This is consistent with earlier R1PY promoter activity determinations using arcA mutant strains (Wagner et al., 2013). Interestingly, there is some promoter activation by TraJpSLT which is probably due to a somewhat higher expression of this protein variant (data not shown). The −35-region mutant (TTTTTT to TTGATT) unexpectedly led to a complete different of the R1PY promoter behavior. First, the basal expression level was higher than from the wt promoter in the fully induced state, second, there was no observable induction by TraJ, and third, there was no more dependence on ArcA since the abs site mutation had no effect on the high expression level. Thus, by introducing just two bases making the −35 sequence more similar to the canonical −35 recognition sequence (TTGACA) which is contacted by region 4 of the σ70 subunit of the RNA polymerase converted the R1PY promoter into a constitutive and completely deregulated promoter.
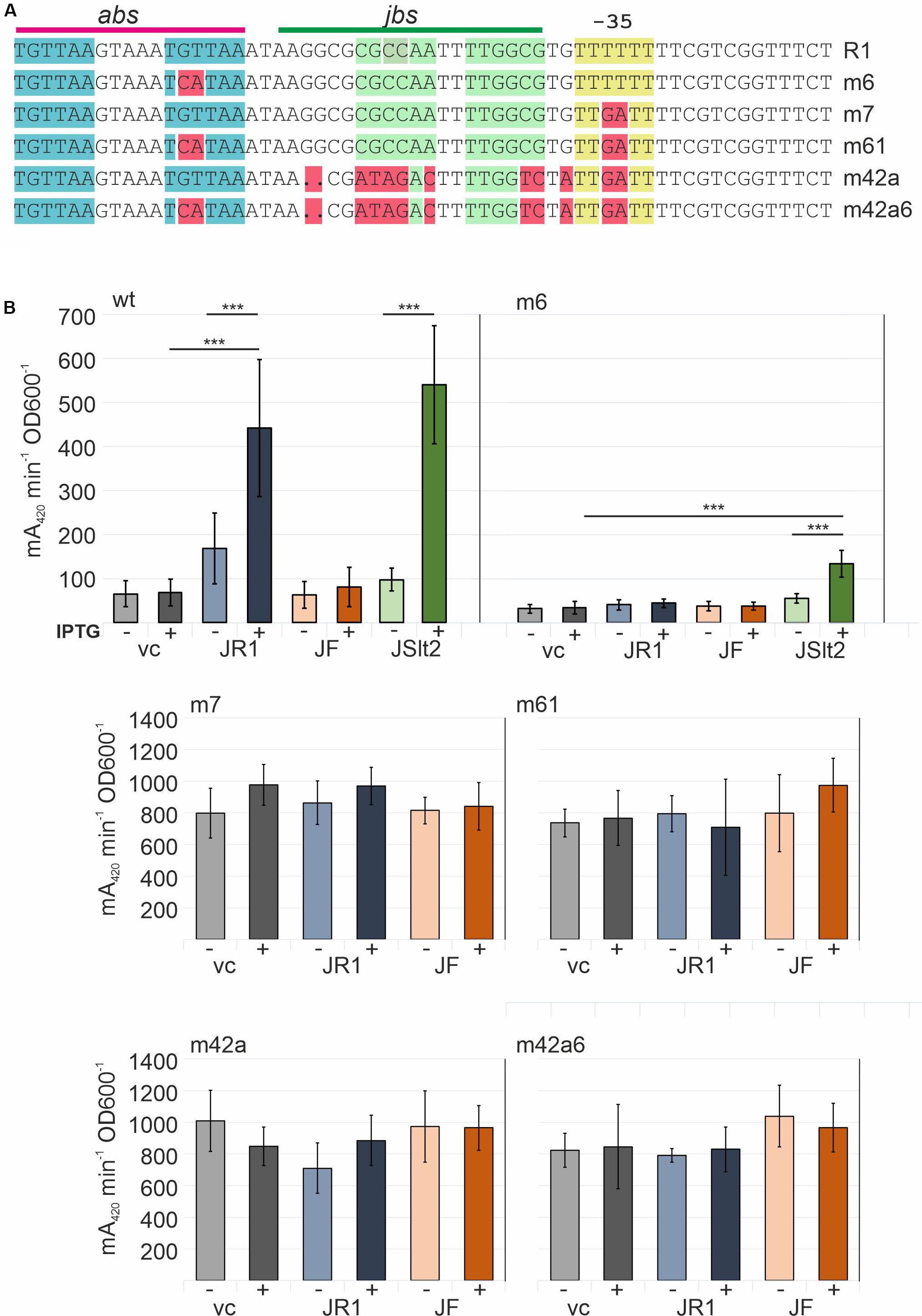
Figure 7. Determination of the role of abs and the non-canonical −35 region in the R1PY promoter. (A) Top line shows the R1PY wt sequence with the abs and jbs sites indicated above. Mutations tested in β-galactosidase assays are shown in the R1PY sequence context and highlighted in red. (B) β-Galactosidase assays with bacterial cells harboring a R1PY-lacZ promoter-test plasmid and a second compatible plasmid IPTG-inducible for expression of traJ from plasmids R1 (JR1), F (JF), or pSLT (JSlt2). vc: vector control (no traJ). IPTG (inductor): +(added), -(not added). Mean values and standard deviations were calculated from at least three independently carried out experiments with two technical replicates for each experiment. P-values (paired t-test): ***P < 0.001.
Discussion
Expression of the tra operon is an absolute requirement for bacteria carrying CPs to develop transfer competence. Only transfer competent cells which usually only represent a minor subpopulation of plasmid harboring cells can actively transfer DNA into recipients and thereby spread genes horizontally in bacterial populations. Intricate genetic regulatory networks ensure that transfer genes are expressed only under optimal conditions (Frost and Koraimann, 2010; Koraimann and Wagner, 2014; Stingl and Koraimann, 2017). Here, we investigated the molecular basis of tra gene activation of the classical antibiotic resistance plasmid R1 which belongs to a large family of F-like plasmids, also categorized as MOBF12A (Fernandez-Lopez et al., 2017). The performed experiments are based on a previous study where we genetically characterized the contribution of hns, arcA, and traJ on silencing and activating DNA transfer and type IV secretion genes of plasmid R1 (Wagner et al., 2013). H-NS binding to the R1PY promoter DNA could be directly shown by EMSA and DNase I footprinting experiments. As expected, DNA binding by H-NS was not specific for the R1PY promoter but also bound to a control DNA fragment in the EMSA with a lower AT content. However, the observed apparent Kd with the R1PY fragment was very low (15 nM) and comparable to DNA fragments containing nucleation sites for H-NS such as csgD or proU promoters (Bouffartigues et al., 2007; Gulvady et al., 2018). In the DNase I protection experiment, we identified three AT binding regions in the R1PY promoter region (H-NS I, H-NS II, and H-NS III). Due to their high AT content of 81 and 77%, the first two could serve as nucleation sites since this high AT content represents the documented preferred composition of H-NS targets in vivo (Navarre et al., 2006). Our results are fully consistent with the proposed silencing role of hns in tra gene expression of plasmid R1 (Wagner et al., 2013).
Counter-silencing functions of the known activators of tra gene expression ArcA (the response regulator of the ArcAB two component system) and TraJ, requires displacement of H-NS from at least the H-NS III binding region which could facilitate RNA polymerase access to −35 and −10 regions of the PY promoter. In vitro binding of ArcA-P and in vivo activation functions have been established earlier suggesting that these proteins could help clearing the promoter from H-NS and recruiting RNA polymerase (Strohmaier et al., 1998; Wagner et al., 2013). The EMSA results confirmed specific ArcA-P binding the R1PY promoter, in addition TraJR1 bound to DNA but only with a slight preference for R1PY DNA. DNase I footprinting analysis nevertheless revealed a protection pattern for TraJR1 that is consistent with the proposed jbs site with the inverted repeat sequence located between −51 and −38 and confirmed ArcA-P binding immediately next to TraJR1 to two direct repeats from −76 to −60. The TGTTAA hexamer represents the core of the ArcA sequence motif with the highest conservation. In most E. coli ArcA regulated promoters, two or three direct repeats of these ArcA boxes are found (Park et al., 2013). Since the simultaneous addition of both proteins can change the protection pattern of H-NS in a way that suggests replacement of H-NS by ArcA and TraJ from binding site III and partially from binding site II (Figure 4), we propose that such a mechanism is responsible for the observed in vivo activation of R1PY by ArcA and TraJ. Phosphorylated ArcA may be responsible for a first counter-silencing activity and opening the PY promoter for TraJ activator binding in a second step. It has similarly been shown that the SsrB response regulator of S. enterica can replace H-NS from type III effector gene promoters in SPI-2 required for intracellular growth and maintenance of this pathogen (Walthers et al., 2011). A cooperative activity of TraJF and ArcA has also recently been proposed to mediate activation of the FPY promoter which is also silenced by H-NS (Will and Frost, 2006; Rodriguez-Maillard et al., 2010; Lu et al., 2018). The results of our promoter mutagenesis studies fully supported the idea that counter-silencing in the R1PY promoter requires ArcA since when only two bases in the second ArcA box hexamer were exchanged (from TGTTAA to TCATAA), together with a lower basal activity, induction of the R1PY promoter was virtually abolished. On the other hand, silencing required a non-canonical −35 hexamer (TTTTTT) which when mutated to TTGATT completely disrupted silencing an converted the mutant promoter into a constitutive promoter independent of both ArcA and TraJ. We envision that this mutation disrupts binding of H-NS in H-NS III of the R1PY promoter and at the same time allows binding of the RNA polymerase holoenzyme (with σ70) to productively initiate transcription from R1 PY.
Although the role of the plasmid encoded activator protein TraJ is well established, the exact mechanism how TraJ acts at the molecular level is still unclear. Whereas the EMSA experiments showed DNA binding with an apparent dissociation constant Kd of about 250 nM only revealed a weak preference for R1PY fragment which was somewhat enhanced by ArcA. Clearly, as shown in the DNase I footprints, TraJ was able to bind next to ArcA in the R1PY promoter. However, there was also a prominent protected band outside the TraJ recognition motif jbs suggesting binding to DNA outside of jbs (C-90 in Figure 5). Unspecific DNA binding was also observed in the band shift experiments and is possibly due to our in vitro conditions which do not reflect the true in vivo situation. Weak DNA binding and low specificity have also been observed for TraJF binding to the FPY promoter; in addition, the authors of that study suggested a cooperative binding mode without a direct protein-protein interaction (Lu et al., 2018). Our findings that ArcA-P binding to R1PY DNA somehow enhanced TraJR1 binding to jbs containing DNA is consistent with this notion. Although we observed no direct protein-protein interaction between TraJR1 and ArcA (data not shown), cooperativity between ArcA and TraJR1 could also be an important aspect of R1PY activation. Furthermore, results of several experiments suggest a direct interaction of TraJ with RNA polymerase (G. Koraimann, unpublished observations). Such a complex may strongly enhance affinity and sequence specificity. Investigations to better understand the interaction and complex formation between TraJ and the RNA polymerase of E. coli are currently ongoing in our laboratory.
In stark contrast to the in vitro observations are the results of R1PY promoter mutations in jbs and the effects of these mutations on the activation potential by TraJR1 or TraJF. The sequence identity between these two TraJ variants is only 18%—explaining the observed specificity of these activators, which means that TraJR1 activates only the R1PY promoter whereas TraJF only functions to activate the FPY promoter containing the cognate jbs site (Wagner et al., 2013; Lu et al., 2014). Based on TraJ sequence variations within the MOBF12A family of F-like plasmids, nine subgroups have been recently proposed, presumably resulting in a subgroup-specific activation of the PY promoter (Koraimann, 2018). Two groups that share a rather high identity in the TraJ protein sequence (73%) are the R1 subgroup and the pSLT subgroup. As we demonstrate here, TraJR1 and TraJpSLT are exchangeable. Furthermore, there are only a few bases that differ in the respective promoter sequences (Figure 1). Exchanges of these bases, mostly outside of jbs did not affect silencing nor activation by TraJR1 or TraJpSLT (Supplementary Figure S1). However, when jbs in R1PY was mutated in only three positions of the inverted repeat sequence, we immediately observed a loss of activation by TraJR1 indicating that recognition by TraJ in vivo is extremely specific. After a series of mutations toward the jbs sequence in FPY, we finally observed a specificity switch, as the promoter was activated by TraJF and not by TraJR1. This result shows that the sole determinant for recognition by TraJ resides in the identified jbs sequence.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
KB, DS, ST, TH, AH, and GR performed and analyzed the experiments. GK designed, evaluated, and analyzed the data and wrote the manuscript.
Funding
This work was supported by the Austrian Science Foundation (FWF), grant number 17857-B12. We acknowledge the support of the field of excellence BioHealth, NAWI Graz, and the University of Graz.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01254/full#supplementary-material
Footnotes
References
Bouffartigues, E., Buckle, M., Badaut, C., Travers, A., and Rimsky, S. (2007). H-NS cooperative binding to high-affinity sites in a regulatory element results in transcriptional silencing. Nat. Struct. Mol. Biol. 14, 441–448. doi: 10.1038/nsmb1233
Christie, P. J. (2016). The mosaic type IV secretion systems. EcoSal Plus 7. doi: 10.1128/ecosalplus.ESP-0020-2015
Cox, K. E. L., and Schildbach, J. F. (2017). Sequence of the R1 plasmid and comparison to F and R100. Plasmid 91, 53–60. doi: 10.1016/j.plasmid.2017.03.007
Datta, N., and Kontomichalou, P. (1965). Penicillinase synthesis controlled by infectious R factors in Enterobacteriaceae. Nature 208, 239–241. doi: 10.1038/208239a0
Fernandez-Lopez, R., de Toro, M., Moncalian, G., Garcillan-Barcia, M. P., and de la Cruz, F. (2016). Comparative genomics of the conjugation region of F-like plasmids: five shades of F. Front. Mol. Biosci. 3:71. doi: 10.3389/fmolb.2016.00071
Fernandez-Lopez, R., Redondo, S., Garcillan-Barcia, M. P., and de la Cruz, F. (2017). Towards a taxonomy of conjugative plasmids. Curr. Opin. Microbiol. 38, 106–113. doi: 10.1016/j.mib.2017.05.005
Frost, L. S., and Koraimann, G. (2010). Regulation of bacterial conjugation: balancing opportunity with adversity. Future Microbiol. 5, 1057–1071. doi: 10.2217/fmb.10.70
Gulvady, R., Gao, Y., Kenney, L. J., and Yan, J. (2018). A single molecule analysis of H-NS uncouples DNA binding affinity from DNA specificity. Nucleic Acids Res. 46, 10216–10224. doi: 10.1093/nar/gky826
Koraimann, G. (2018). Spread and persistence of virulence and antibiotic resistance genes: a ride on the F plasmid conjugation module. EcoSal Plus 8. doi: 10.1128/ecosalplus.ESP-0003-2018
Koraimann, G., and Wagner, M. A. (2014). Social behavior and decision making in bacterial conjugation. Front. Cell. Infect. Microbiol. 4:54. doi: 10.3389/fcimb.2014.00054
Laible, M., and Boonrod, K. (2009). Homemade site directed mutagenesis of whole plasmids. J. Vis. Exp. 27, 2–4. doi: 10.3791/1135
Liu, H., and Naismith, J. H. (2008). An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8:91. doi: 10.1186/1472-6750-8-91
Liu, X., and De Wulf, P. (2004). Probing the ArcA-P modulon of Escherichia Coli by whole genome transcriptional analysis and sequence recognition profiling. J. Biol. Chem. 279, 12588–12597. doi: 10.1074/jbc.m313454200
Lu, J., Peng, Y., Wan, S., Frost, L. S., Raivio, T., and Glover, J. N. M. (2018). Cooperative function of TraJ and ArcA in regulating the F plasmid Tra operon. J. Bacteriol. 201:e00448-18. doi: 10.1128/JB.00448-18
Lu, J., Wu, R., Adkins, J. N., Joachimiak, A., and Glover, J. N. (2014). Crystal structures of the F and PSLT plasmid TraJ N-terminal regions reveal similar homodimeric PAS folds with functional interchangeability. Biochemistry 53, 5810–5819. doi: 10.1021/bi500244m
Meynell, E., and Datta, N. (1966). The relation of resistance transfer factors to the F-Factor (Sex-Factor) of Escherichia Coli K12. Genet. Res. 7, 134–140. doi: 10.1017/S0016672300009538
Miller, J. H. (1972). Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Navarre, W. W., Porwollik, S., Wang, Y., McClelland, M., Rosen, H., Libby, S. J., et al. (2006). Selective silencing of foreign DNA with Low GC content by the H-NS protein in Salmonella. Science 313, 236–238. doi: 10.1126/science.1128794
Park, D. M., Akhtar, M. S., Ansari, A. Z., Landick, R., and Kiley, P. J. (2013). The bacterial response regulator ArcA uses a diverse binding site architecture to regulate carbon oxidation globally. PLoS Genet. 9:e1003839. doi: 10.1371/journal.pgen.1003839
Rodriguez-Maillard, J. M., Arutyunov, D., and Frost, L. S. (2010). The F plasmid transfer activator TraJ is a dimeric Helix-Turn-Helix DNA-binding protein. FEMS Microbiol. Lett. 310, 112–119. doi: 10.1111/j.1574-6968.2010.02064.x
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Stingl, K., and Koraimann, G. (2017). “Prokaryotic information games: how and when to take up and secrete DNA,” in Type IV Secretion in Gram-Negative and Gram-Positive Bacteria, eds S. Backert and E. Grohmann (Cham: Springer), 61–92. doi: 10.1007/978-3-319-75241-9_3
Strohmaier, H., Noiges, R., Kotschan, S., Sawers, G., Högenauer, G., Zechner, E. L. L., et al. (1998). Signal transduction and bacterial conjugation: characterization of the role of ArcA in regulating conjugative transfer of the resistance plasmid R1. J. Mol. Biol. 277, 309–316. doi: 10.1006/jmbi.1997.1598
Wagner, M. A., Bischof, K., Kati, D., and Koraimann, G. (2013). Silencing and activating type IV secretion genes of the F-like conjugative resistance plasmid R1. Microbiology 159(Pt 12), 2481–2491. doi: 10.1099/mic.0.071738-0
Waksman, G. (2019). From conjugation to T4S systems in gram−negative bacteria: a mechanistic biology perspective. EMBO Rep. 20:e47012. doi: 10.15252/embr.201847012
Walthers, D., Li, Y., Liu, Y., Anand, G., Yan, J., and Kenney, L. J. (2011). Salmonella Enterica response regulator SsrB relieves H-NS silencing by displacing H-NS bound in polymerization mode and directly activates transcription. J. Biol. Chem. 286, 1895–1902. doi: 10.1074/jbc.M110.164962
Will, W. R., and Frost, L. S. (2006). Characterization of the opposing roles of H-NS and TraJ in transcriptional regulation of the F-plasmid Tra operon. J. Bacteriol. 188, 507–514. doi: 10.1128/JB.188.2.507-514.2006
Wong, J. J., Lu, J., and Glover, J. N. (2012). Relaxosome function and conjugation regulation in F-like plasmids – a structural biology perspective. Mol. Microbiol. 85, 602–617. doi: 10.1111/j.1365-2958.2012.08131.x
Keywords: F-like plasmids, horizontal gene transfer, bacterial conjugation, type IV secretion, antibiotic resistance, gene silencing
Citation: Bischof K, Schiffer D, Trunk S, Höfler T, Hopfer A, Rechberger G and Koraimann G (2020) Regulation of R1 Plasmid Transfer by H-NS, ArcA, TraJ, and DNA Sequence Elements. Front. Microbiol. 11:1254. doi: 10.3389/fmicb.2020.01254
Received: 07 March 2020; Accepted: 18 May 2020;
Published: 11 June 2020.
Edited by:
Eva M. Top, University of Idaho, United StatesReviewed by:
Hideaki Nojiri, The University of Tokyo, JapanBeth Traxler, University of Washington, United States
Copyright © 2020 Bischof, Schiffer, Trunk, Höfler, Hopfer, Rechberger and Koraimann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Günther Koraimann, Z3VlbnRoZXIua29yYWltYW5uQHVuaS1ncmF6LmF0
†These authors share first authorship