 Mei-Jie Yang1,2,3,4,5†
Mei-Jie Yang1,2,3,4,5† Hao Song1,2,3,4†Zheng-Lin Yu1,2,3,4Zhi Hu1,2,3,4,5Cong Zhou1,2,3,4,5Xiao-Long Wang1Tao Zhang1,2,3,4*
Hao Song1,2,3,4†Zheng-Lin Yu1,2,3,4Zhi Hu1,2,3,4,5Cong Zhou1,2,3,4,5Xiao-Long Wang1Tao Zhang1,2,3,4*- 1CAS Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 2Laboratory for Marine Science and Technology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 3Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, China
- 4CAS Engineering Laboratory for Marine Ranching, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 5University of Chinese Academy of Sciences, Beijing, China
The symbiotic microbiota can stimulate modulation of immune system, which also can promote immune system mature in critical developmental periods. In this study, we have investigated the symbiotic microbiota in Rapana venosa at five early development stages using Illumina high-throughput sequencing, and detected immune responses in larvae. Analysis of the symbiotic microbiota sequences identified that the most abundant phylum was Proteobacteria. Beta diversity analysis indicated that the structure of the symbiotic microbiota dramatically shifted in early development stages. The abundance of immune-related KEGG Orthologs (KOs) also increased in competent larval (J4, 30-day post-hatching) and postlarval after 3 days of metamorphosis (Y5, 33-day post-hatching) stages. Acid phosphatase activity decreased significantly in the Y5 stage, and alkaline phosphatase activity also at a lower level in Y5 stage, whereas lysozyme activities exhibited no remarkable change. Also, the activities of catalase and superoxide dismutase activities decreased dramatically during early development stages of R. venosa. Dramatic changes in the symbiotic microbiota and the immune response mainly occurred in the initially hatched veliger (C1), competent larval (J4) and postlarval (Y5) stages, during which the hosts might experience substantial environmental changes or changes in physiological structure and function. These findings expand our understanding of the stage-specific symbiotic microbiota in R. venosa and the close association between immune system and symbiotic microbiota in mollusks, however, the specific relationship may need more researches are needed to investigated in the future.
Introduction
The symbiotic microbiota is a complex ecosystem with multiple functions significant for the health of host (Ramirez and Romero, 2017), and the intricate interactions between the symbiotic microbiota and host start from early development stages in host (Nicholson et al., 2012). During host development, the symbiotic microbiota serves as important roles, including harvesting energy, against pathogens, and stimulating the development of the immune system (Al-Harbi and Uddin, 2005; Woodhams et al., 2007; Buchon et al., 2013; Ramirez and Romero, 2017). Furthermore, when the host undergoes a critical developmental event, corresponding changes also occur in its symbiotic microbiota that can reflect changes in the physiological functions of the host (Fraune and Bosch, 2010; Li et al., 2017), especially the immune system.
The veined rapa whelk (Rapana venosa) is an important fishery resource in China, and native to temperate Asian waters (Mann and Harding, 2003). Recently, the population of R. venosa has dramatically decreased because of the overexploitation and destruction of breeding grounds (Wei and Wang, 1999). Although efforts involving in the commercial aquaculture of R. venosa have been measured by enterprises since 1992 (Yuan, 1992), large-scale cultivation has been still severely restricted by the high mortality in larvae during metamorphosis (Song et al., 2017b). Metamorphosis is one of the most important developmental events in the life cycle of mollusks, usually accompanied by a high mortality (Harding, 2006). Furthermore, many morphological and behavioral variation occur during metamorphosis, including velum degradation and reabsorption, foot proliferation and elongation, and initiation of rapid secondary shell growth (Song et al., 2017b), all of which are regulated by the nervous, digestive and immune systems. Correspondingly, many changes occur in these systems at the molecular level during this important developmental process (He et al., 2014; Ueda and Degnan, 2014; Song et al., 2016b; Shen et al., 2018).
There are many studies about early development stages of R. venosa (Harding, 2006; Hacer and Ertug, 2007; Pan et al., 2013; Ban et al., 2014), and recent studies based on omics have paid more attention to the regulatory mechanism of its metamorphosis (Song et al., 2016a,b,c,d, 2017a,b). Additionally, a previous study explored the neuronal development of early larvae of R. venosa and investigated larval metamorphosis and feed conversion in R. venosa (Pan, 2014; Yang et al., 2016), suggesting that changes occur in the digestive system during the early development stages. Whereas, few studies investigated the development of immune system in R. venosa during metamorphosis, although the immune system is significant in early developmental stages, which has been extensively studied in other mollusks (Bao et al., 2015; Shen et al., 2018).
Accumulating evidence has shown that the symbiotic microbiota can stimulate the modulation of the immune system, resulting in mature and balanced immune responses (Mazmanian et al., 2005; Edelman et al., 2008). The effects are often reflected in the changes of immune enzymes [acid phosphatase (ACP), alkaline phosphatase (ALP), lysozyme (LSZ), superoxide dismutase (SOD), catalase (CAT) (Canicatti, 1990; Janeway and Medzhitov, 2002; Song et al., 2010)] and immune-related genes [Toll-like receptor 2, tumor necrosis factor, and defensin genes (Song et al., 2016b)]. Therefore, there is a close relationship between assembly of symbiotic microbiota and the development of host immune system. During early research, many studies examined the symbiotic microbiota of vertebrates such as rats and fishes (Mazmanian et al., 2005; Dhanasiri et al., 2011; Stephens et al., 2015; Li et al., 2017), while in recent years, due to experimental advantages, invertebrates have been used to help us understand the microbe–host interactions (Graf, 2016). However, few studies have focused on the symbiotic microbiota during the development of mollusks, and only Zhao et al. (2012) described the composition of intestinal symbiotic microbiota at different development stages of abalone. The changes that occur in the symbiotic microbiota composition during the development of mollusks may reflect the development of the immune system, which may play a regulatory role in metamorphosis.
In the present study, we aimed to elucidate the structure changes of the symbiotic microbiota during early development stages of R. venosa. and investigate the relationship between the development of the immune system and symbiotic microbiota. This information could help to expound the correlations between symbiotic microbiota and the development of the immune system, as well as further understand the mechanism of metamorphosis in R. venosa.
Materials and Methods
Sample Collection
Culturation of larval rearing was conducted according to Song et al. (2016b). Planktonic larvae were feed with a mixture of Isochrysis galbana, Chlorella vulgaris, and Platymonas subcordiformis three times a day. Samples were collected from five early developmental stages: the one-spiral whorl stage [C1, 1 day post-hatching (dph)], two-spiral whorl stage (D2, 5 dph), three-spiral whorl stage (F3, 12 dph), four-spiral whorl stage (competent larva, J4, 30 dph) and postlarval stage after 3 days of metamorphosis (Y5, 33 dph) (Pan et al., 2013; Song et al., 2016c). Each developmental stage had five replicates (30 larvae were used in each replicate). Larvae in each collected sample were euthanized and sterilized with 70% ethanol for 1 min and then washed three times using sterile water, snap-frozen and preserved at −80°C for DNA extraction. Environmental samples included five bait samples and five water samples. Five hundred milliliters of food samples and water samples were filtered through 0.22 μm cellulose nitrate filter, then subjected to bead beating and DNA extraction from the filter using the same method used for the other samples.
The samples from each development stage were homogenized and used to extract genomic DNA, total protein and total RNA by using the DNA/RNA/Protein Coextraction Kit (DP423, TIANGEN Biotech Co., Ltd., Beijing, China) according to the manufacturer’s protocol, and the extracted products were used for PCR amplification, analysis of immune enzyme activity and qRT-PCR, respectively. Subsequently, the DNA and RNA concentrations were detected by a NanoDrop 1000 (Thermo Fisher Scientific, United States), and the purities were analyzed according to the ratio of the absorbance values at 260 and 280 nm (OD 260/OD 280).
All procedures involved in the animal collection, rearing and dissection were conducted following the Guideline of Ethical Regulations of Animal Welfare of the Institute of Oceanology, Chinese Academy of Sciences (IOCAS 2013.3). Our study protocols were approved by the Animal Welfare Committee of the IOCAS.
PCR Amplification and Illumina MiSeq Platform Sequencing
PCR amplification of 16S rRNA gene and sequencing were performed by Majorbio Co., Ltd. (Shanghai, China). PCR amplification was carried out using the 338F/806R primer targeting V3-V4 regions of the bacterial 16S rRNA gene (338F 5′-ACTCCTACGGGAGGCAGCAG-3′ and 806R 5′-GGACTACHVGGGTWTCTAAT-3′) (Chu et al., 2015). PCR products were purified by the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States), and quantified by QuantiFluorTM-ST (Promega, United States), then sequenced (2 × 250 bp) on the MiSeq PE300 platform (Illumina, United States).
Analysis of Immune-Related Enzyme Activity
The soluble protein content of each samples was detected using Bradford (1976) method and expressed as mg mL–1 of larval serum. ACP and ALP were determined according to Barrett (1972) (A060-1-1, A059-1-1), and a unit of enzyme activity was defined as the amount of enzyme required to produce 1 μmol of phenol, and expressed as ACP/ALP King unit mL–1. LSZ activity was defined 7776 in terms of the change in absorbance of a Micrococcus luteus cell suspension per minute and expressed as LSZ unit mL–1. SOD activity was analyzed according to Ôyanagui (1984). A unit of SOD activity was defined as the amount of enzyme inhibited superoxide-induced oxidation by 50%, and expressed as SOD unit mL–1. CAT activity was determined according to Góth (1991) (A007-1-1). A unit of CAT activity was defined as the amount that can catalyze the conversion of 1 μmol H2O2 s–1, and expressed as CAT unit mL–1. All immune enzyme activities were measured following the manufacturer’s protocols of assay kits (Nanjing Jiancheng Bioengineering Institute, China).
Gene Expression Analysis
Immune-related gene expression was determined at different development stages. First-strand cDNAs for qRT-PCR were synthesized by a Prime ScriptTM RT Reagent Kit with gDNA Eraser (TaKaRa, Japan). Primers used for gene expression analysis were designed according to the full-length cDNA sequences of Toll-like receptor 2, tumor necrosis factor and defensin genes (Supplementary Table S1), the levels of which dramatically shifted during metamorphosis, which may be associated with the development of the immune system (Song et al., 2016b). The gene encoding the 60S ribosomal protein L28 (RL28-F, RL28-R) was selected as the housekeeping gene for signal normalization, which is the most stable gene in all developmental stages in R. venosa. (Song et al., 2017a). SYBR PrimeScriptTM RT-PCR Kit II (TaKaRa, Japan) was used with an Eppendorf Mastercycler® ep Realplex (Eppendorf, Hamburg, Germany) for qRT-PCR analysis following the manufacturer’s protocols (Yang et al., 2020). Standard curves were made with 10-, 102-, 103-, 104-, and 105-fold dilutions of each cDNA template, and qRT-PCR was carried out with this program: 95°C for 5 s and 40 cycles of 95°C for 15 s, then 60°C (Toll-like receptor 2, defensin) or 58°C (tumor necrosis factor) for 30 s. The 2–ΔΔCt method was used to analyzed the relative gene expression.
Data Analysis
USEARCH 7.11 (Caporaso et al., 2012) was used to demultiplex2 and quality filter raw sequences3 to discard singletons from 35 samples, and remove chimeric sequences4 to get clean reads (Haas et al., 2011). Operational taxonomic units (OTUs) were identified (equivalent to 97% sequence similarity) and clustered using UPARSE 7.1 software1 (Edgar, 2013). Meanwhile, effective sequences were aligned against the SILVA database5, and identified on phylum, class, order, family, genus and species levels by the Ribosomal Database Project Bayesian classifier6 with a 70% threshold. Partial data analysis was performed on the free online platform of the Majorbio Cloud Platform7.
OTU abundance information was normalized (Yang et al., 2019) for subsequent analysis. Alpha diversity indices used to assess the community diversity (Shannon and Simpson) and richness (Chao and ACE) were analyzed by Mothur8. Venn diagram analysis was performed using the Draw Venn Diagram online tool9 (Wang et al., 2019). Histograms of the community composition were conducted by the Vegan package in R 2.410. Beta diversity analyses, including non-metric multidimensional scaling (NMDS), typing analysis and hierarchical clustering analysis, were performed using QIIME 1.7.011 based on weighted UniFrac distances further investigated the similarity and differences of microbiota among samples from different development stages. In order to assess differences in the microbiota communities among groups, Adonis and ANOSIM were performed using the Vegan package in R 2.4. Permutational multivariate analysis of variance (PERMANOVA) was performed to compare dissimilarities in the structure of the microbiota (Anderson, 2001).
Microbial taxa that differed between every pair of groups at genera level were analyzed using linear discriminant analysis (LDA) effect size (LEfSe) method12, and Kruskal–Wallis test was used to identify the significant differences between two groups (Segata et al., 2011). Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was used to predict the functions of microbiota by predicting the Kyoto Encyclopedia of Genes and Genomes (KEGG) categories and functional enzymes (Langille et al., 2013).
In present study, Levene’s test was used to determine the homogeneity of variances in analysis of immune-related enzyme activity and gene expression, and mean differences were determined using Student’s t-test. SPSS 19.0 was used in all statistical analysis, and P-value < 0.05 indicated statistically significant difference.
Results
Composition of the Symbiotic Microbiota Population
A total of 1,432,056 high-quality sequences were produced from 32 samples (J4_4, Y5_4 and Y5_5 were excluded from sequencing because of the low quality DNA): 956,119 from the host corresponding to the five culture stages and 475,937 from 10 environmental water and bait samples, with an average of 44,752 reads (Table 1). The data has been submitted to the sequence read archive (SRA) with the project accession number PRJNA574861. Clustered at 3% distance, a total of 1,879 OTUs were obtained with an average of 535 OTUs, and 1744 OTUs were from the host (Table 1).
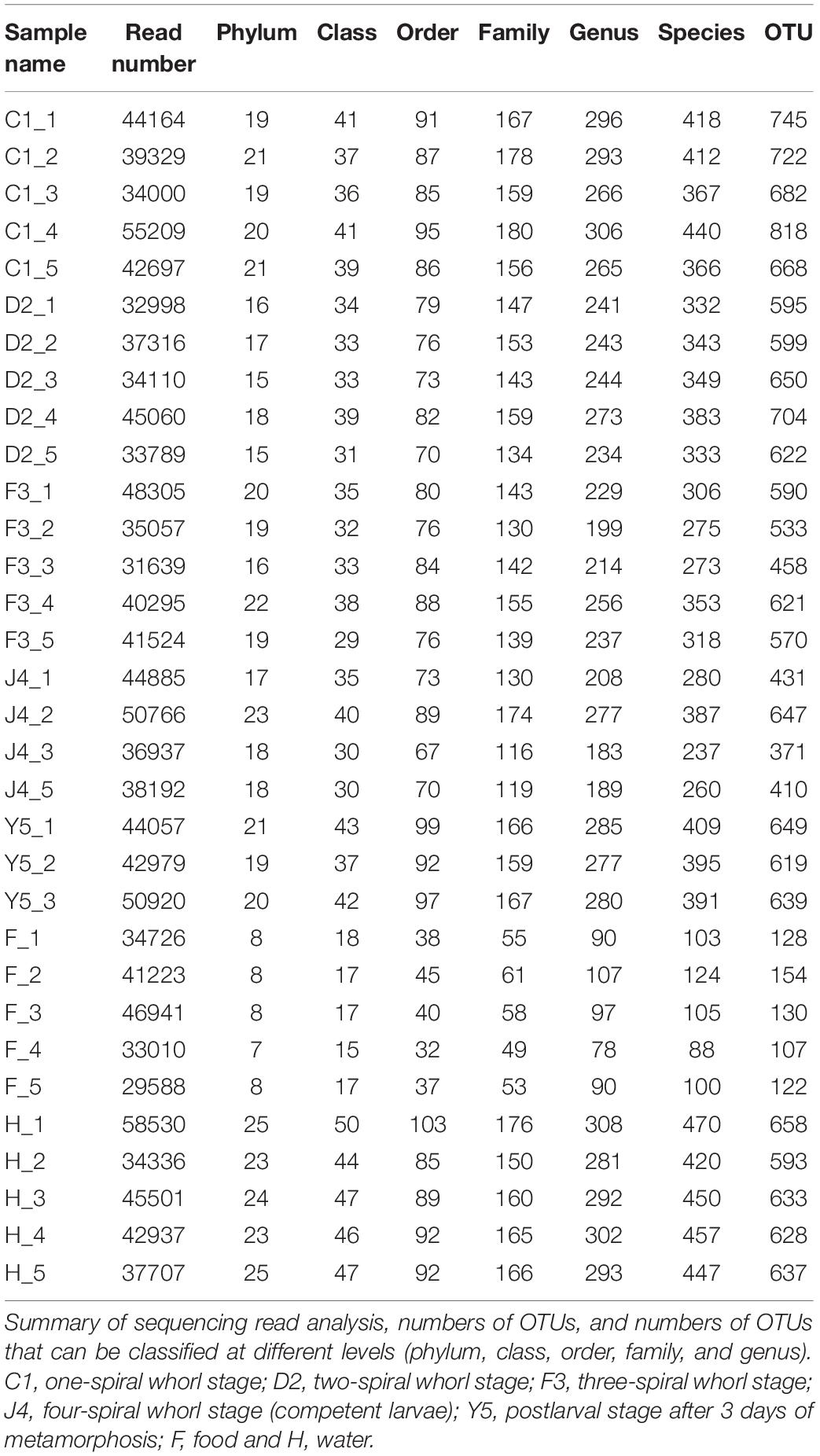
Table 1. Sequencing and OTU classification information.
The OTUs were classified into 32 phyla. Sequences that was not classified into any known groups were assigned as ‘unclassified’ (unclassified < 0.005). The phyla with the highest relative abundance in all samples were Proteobacteria (54.02%), Cyanobacteria (32.41%), Actinobacteria (4.86%), Bacteroidetes (5.29%), Firmicutes (1.28%), Verrucomicrobia (0.84%), Chlamydiae (0.36%), Planctomycetes (0.20%), TM6_Dependentiae_ (0.23%), WWE3 (0.09%), and Parcubacteria (0.08%) (Figure 1A). Proteobacteria was the most abundant phylum among all 22 samples from the host except samples J4_3 and J4_5, in which Cyanobacteria was the most abundant phylum, and Cyanobacteria was also the most abundant phylum among the 10 samples from the environment. At the genus level, a total of 289 taxa were identified, and the most abundant genera were norank_c__Cyanobacteria (29.26%), Vibrio (25.23%), Ruegeria (7.95%), unclassified_f__Rhodobacteraceae (4.75%), Synechococcus (2.91%), Marivita (2.68%), norank_o__PeM15 (1.82%), Nautella (1.74%), norank_f__Cryomorphaceae (1.24%), and Phaeodactylibacter (1.07%) (Figure 1B). Additionally, Venn diagram constructed to identify dominant OTUs present in samples suggested that 102 OTUs were shared among the five stages and environment samples, and 52, 13, 57, 33, 104, 2, and 141 OTUs were unique to groups C1, D2, F3, J4, Y5, F and H respectively (Figure 2).
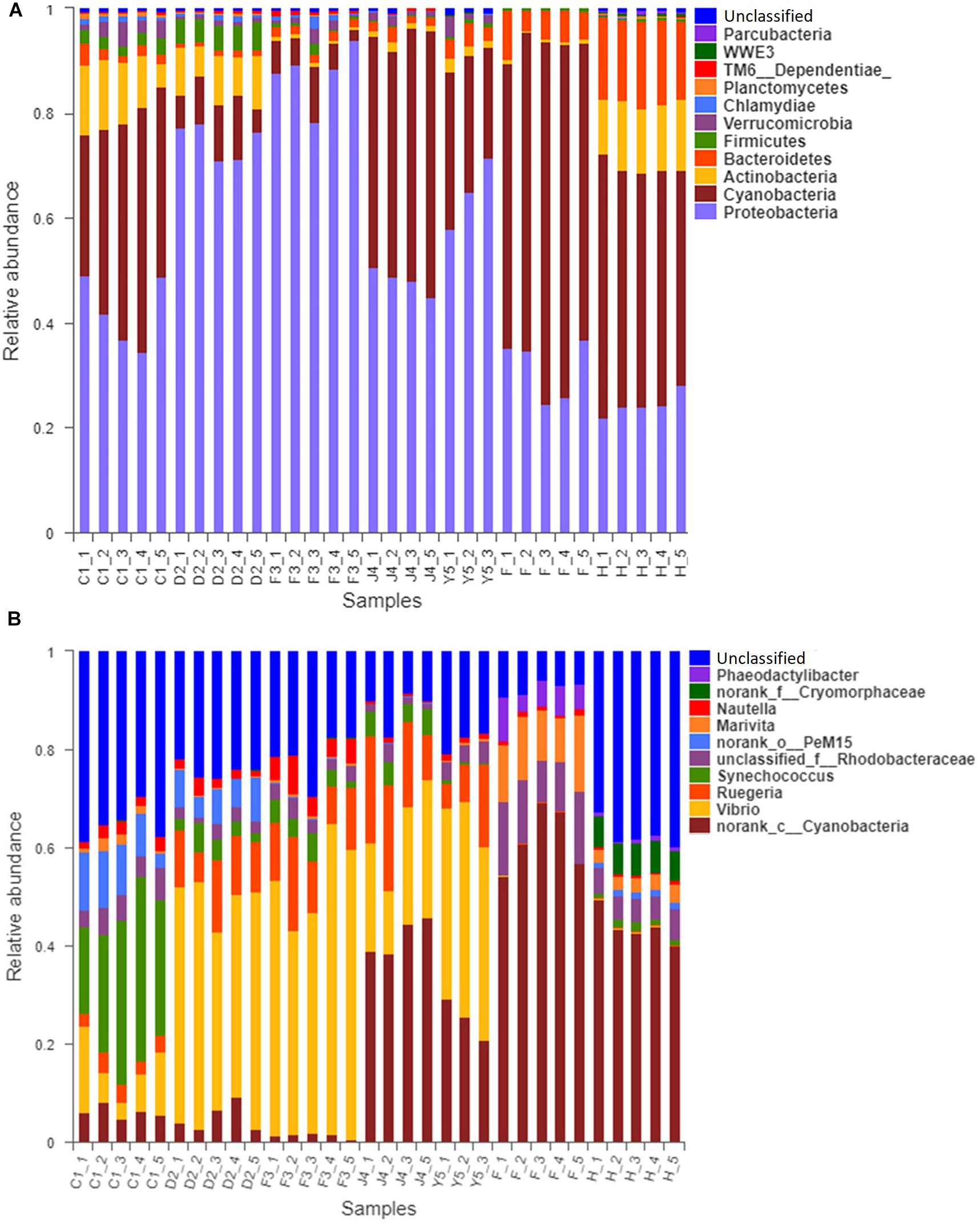
Figure 1. Relative read abundances of different bacterial phyla (A) and genera (B) within the different communities. Sequences that could not be classified into any known group are designated ‘unclassified.’ C1, one-spiral whorl stage; D2, two-spiral whorl stage; F3, three-spiral whorl stage; J4, four-spiral whorl stage (competent larvae); Y5, postlarval stage after 3 days of settling; F, food; H, water.
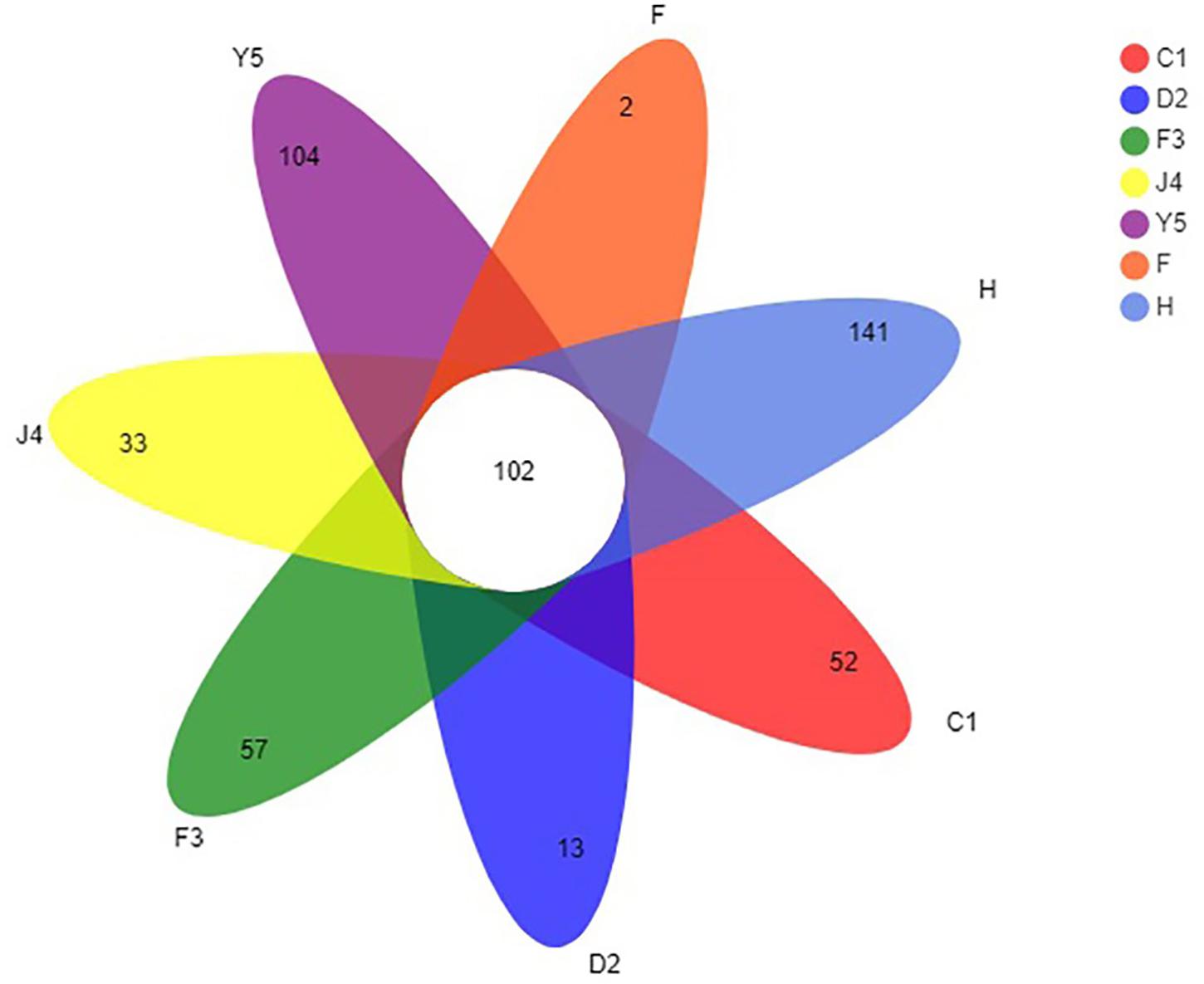
Figure 2. Venn diagram of OTUs in groups C1, D2, F3, J4, Y5, F, and H. The overlapping and separate sections represent the number of shared and unique OTUs, respectively. C1, one-spiral whorl stage; D2, two-spiral whorl stage; F3, three-spiral whorl stage; J4, four-spiral whorl stage (competent larvae); Y5, postlarval stage after 3 days of metamorphosis; F, food; H, water.
Diversity Analysis of the Five Development Stages
The alpha diversity of the microbiota community varied significantly in different development stages (Figure 3). The Shannon index ranged from 2.118 to 4.104 and significantly decreased with host development, while the Simpson index ranged from 0.052 to 0.494 and increased with host development, which suggests that the diversity of the symbiotic microbiota decreased with host development. The Chao index ranged from 155 to 929, while the ACE index ranged from 142 to 940, and both the Chao and ACE index did not show significant change with the development of the host.
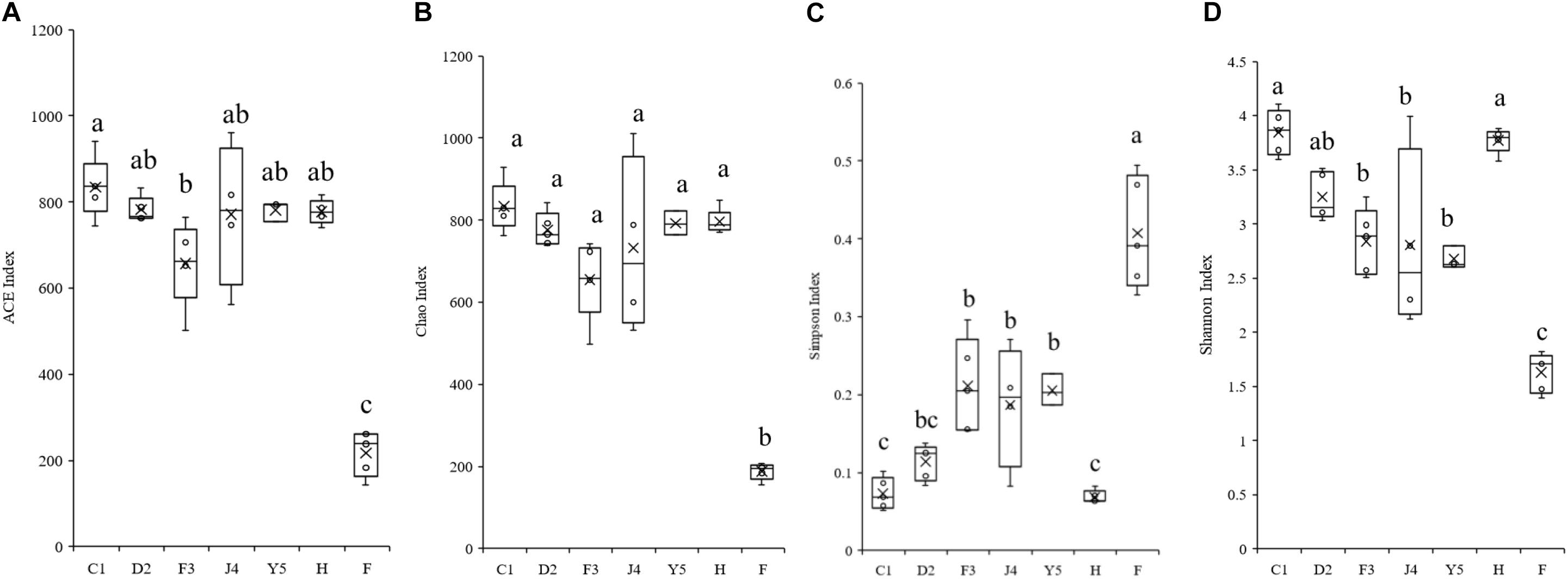
Figure 3. Boxplots showing the ranges of different alpha diversity indices. The boxplots show significant differences in the ACE index (A), Chao index (B), Simpson index (C), and Shannon index (D) at different culture stages. Different lower-case letters for each bar indicate significant differences (P < 0.05). C1, one-spiral whorl stage; D2, two-spiral whorl stage; F3, three-spiral whorl stage; J4, four-spiral whorl stage (competent larvae); Y5, postlarval stage after 3 days of metamorphosis; F, food; H, water.
The beta diversity of the microbiota community may suggest that structure of microbiota changes with development. NMDS analysis shown in Figure 4A and all the stages were clustered well. Meanwhile, typing analysis showed that stages D2 and F3 belonged to the same type, and stages J4 and Y5 belonged to the same type, while stage C1 alone belonged to a single type (Figure 4B). Meanwhile, unweighted pair group method with arithmetic mean (UPGMA) clustering showed that all the individual samples were clustered into groups according to the culture stage (Figure 4C). Groups D2 and F3 were clustered into the same branch, and groups J4 and Y5 were clustered into the same branch, while group C1 alone was clustered into a single branch, which was consistent with the results of the NMDS and the typing analysis. Adonis and ANOSIM showed that the symbiotic microbiota populations have significantly difference between any two of the compared stages (P-value < 0.05) (Table 2).
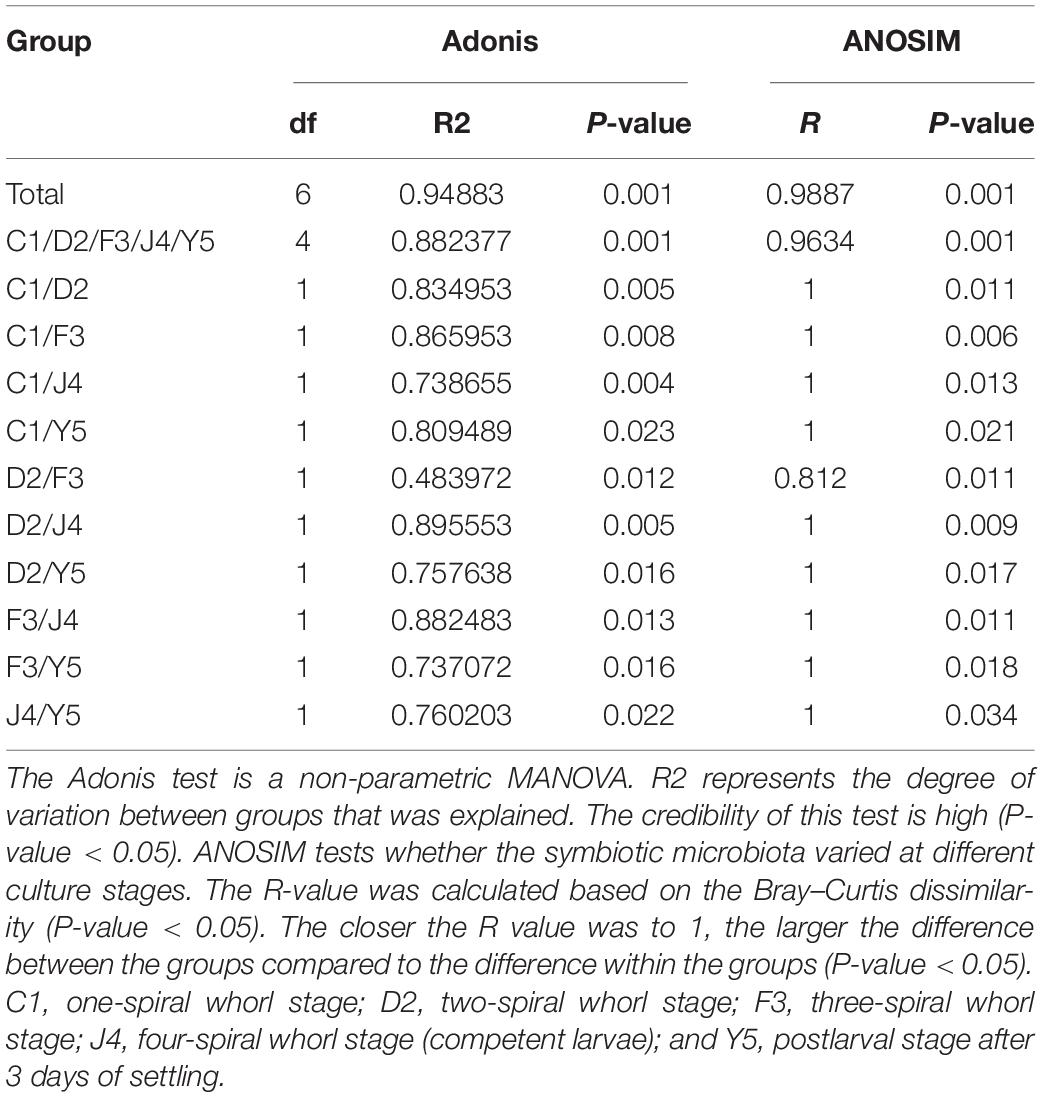
Table 2. Adonis and ANOSIM tests of significant differences between the five culture stages.
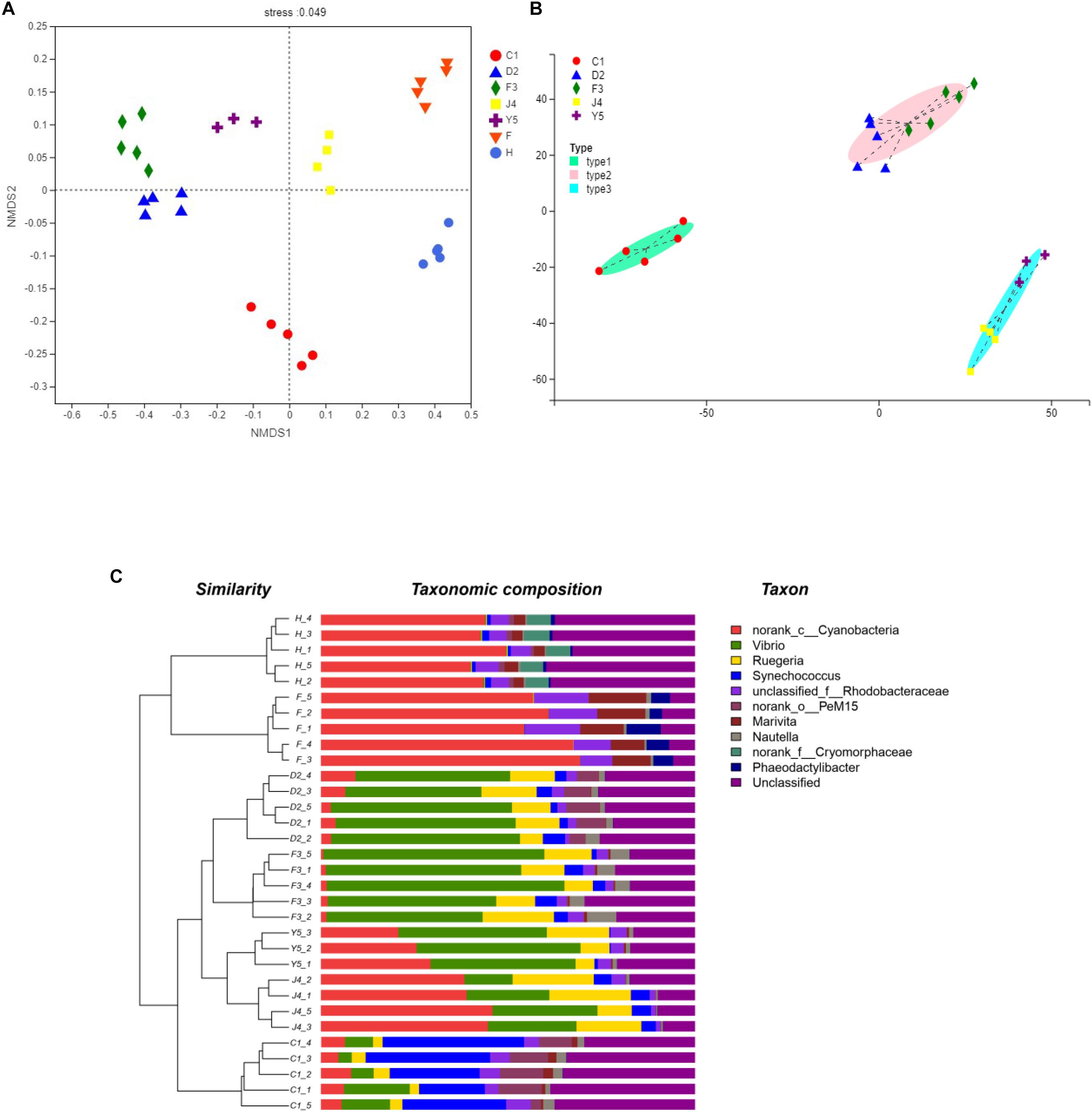
Figure 4. Beta diversity analysis. NMDS analysis (A), Typing analysis (B) at the genus level was based on the weighted UniFrac distances. A UPGMA tree (C) (based on the weighted UniFrac distances) and a genus-level relative abundance map are shown on the left and right, respectively. C1, one-spiral whorl stage; D2, two-spiral whorl stage; F3, three-spiral whorl stage; J4, four-spiral whorl stage (competent larvae); Y5, postlarval stage after 3 days of metamorphosis; F, food; H, water.
Changes in the Dominant Bacterial Community With Host Development
Among the top 10 phyla, the Kruskal–Wallis H test demonstrated that the abundances of Proteobacteria, Cyanobacteria, Actinobacteria, Bacteroidetes, Firmicutes, Verrucomicrobia, Chlamydiae, Planctomycetes, TM6_Dependentiae, and Chloroflexi changed significantly between different culture stages (P-value < 0.05) (Figure 5). Proteobacteria, the most dominant phylum, showed an increasing trend before the J4 stage and became dramatically depleted in the J4 stage. The abundances of Verrucomicrobia and Bacteroidetes increased from the D2 stage, while the abundances of Chlamydiae and Planctomycetes decreased with host development.
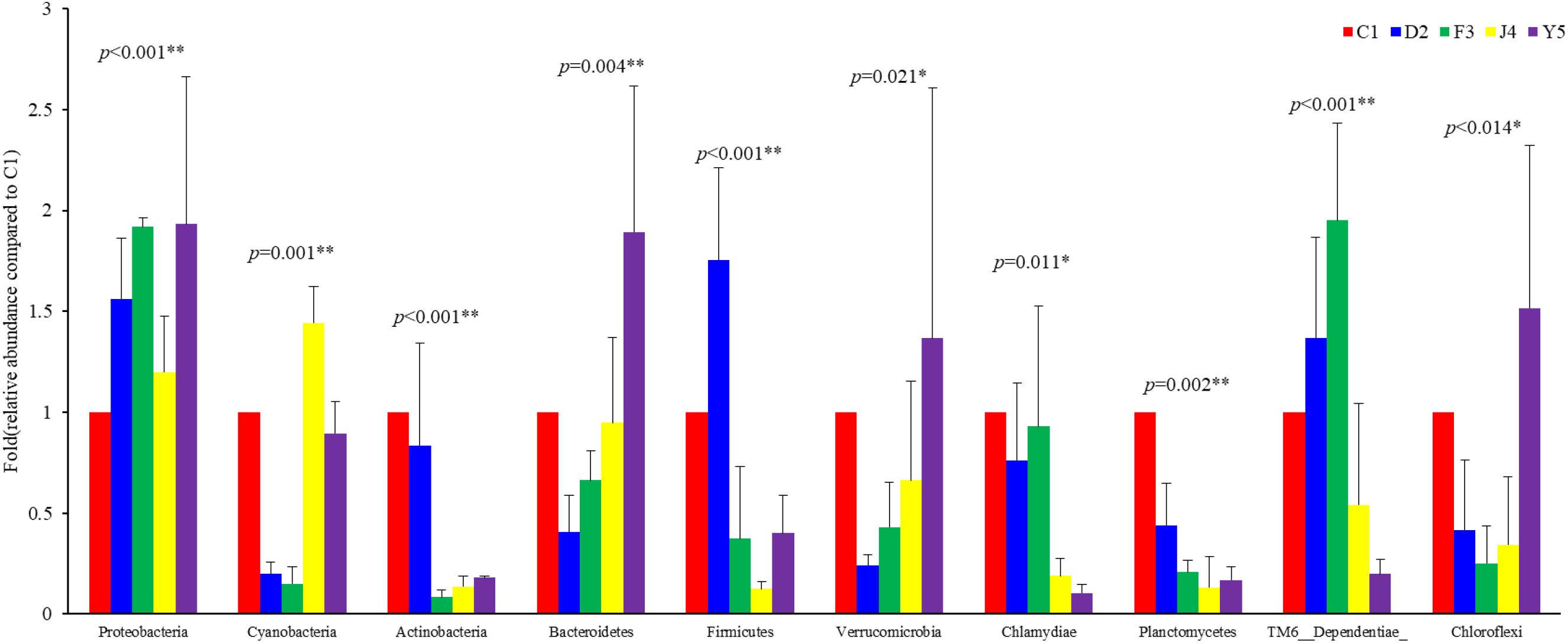
Figure 5. Abundances of the 10 most abundant phyla at different culture stages. The relative abundance of each phylum at the five culture stages is shown. The abundance at C1 was assigned a value of 1, and the relative abundances at other stages were calculated relative to this abundance. Asterisks indicate significant differences between groups by PERMANOVA (P < 0.05). Two asterisks indicate extremely significant differences between the groups (P < 0.01). C1, one-spiral whorl stage; D2, two-spiral whorl stage; F3, three-spiral whorl stage; J4, four-spiral whorl stage (competent larvae); Y5, postlarval stage after 3 days of metamorphosis.
To further investigate the different genera in the F3, J4, and Y5 stages, we conducted pairwise comparisons of relative abundances in the three stages using Student’s t-test. The results showed that the relative abundances of Vibrio, norank_c_Cyanobacteria, Nautella, norank_f_legionellaceae, Aquimarina, and Muricauda were significantly different in both F3 vs. J4 and J4 vs. Y5 and the relative abundance of Vibrio in F3 and Y5 was significantly higher than that in J4. In contrast, norank_c_Cyanobacteria, Sulfitobacter, and Gammaproteobacteria showed significant differences between F3 and J4, and Synechococcus, norank_f_Phyllobacteriaceae and unclassified_f_Flavobacteriaceae showed significant differences in J4 vs. Y5 (Figure 6).
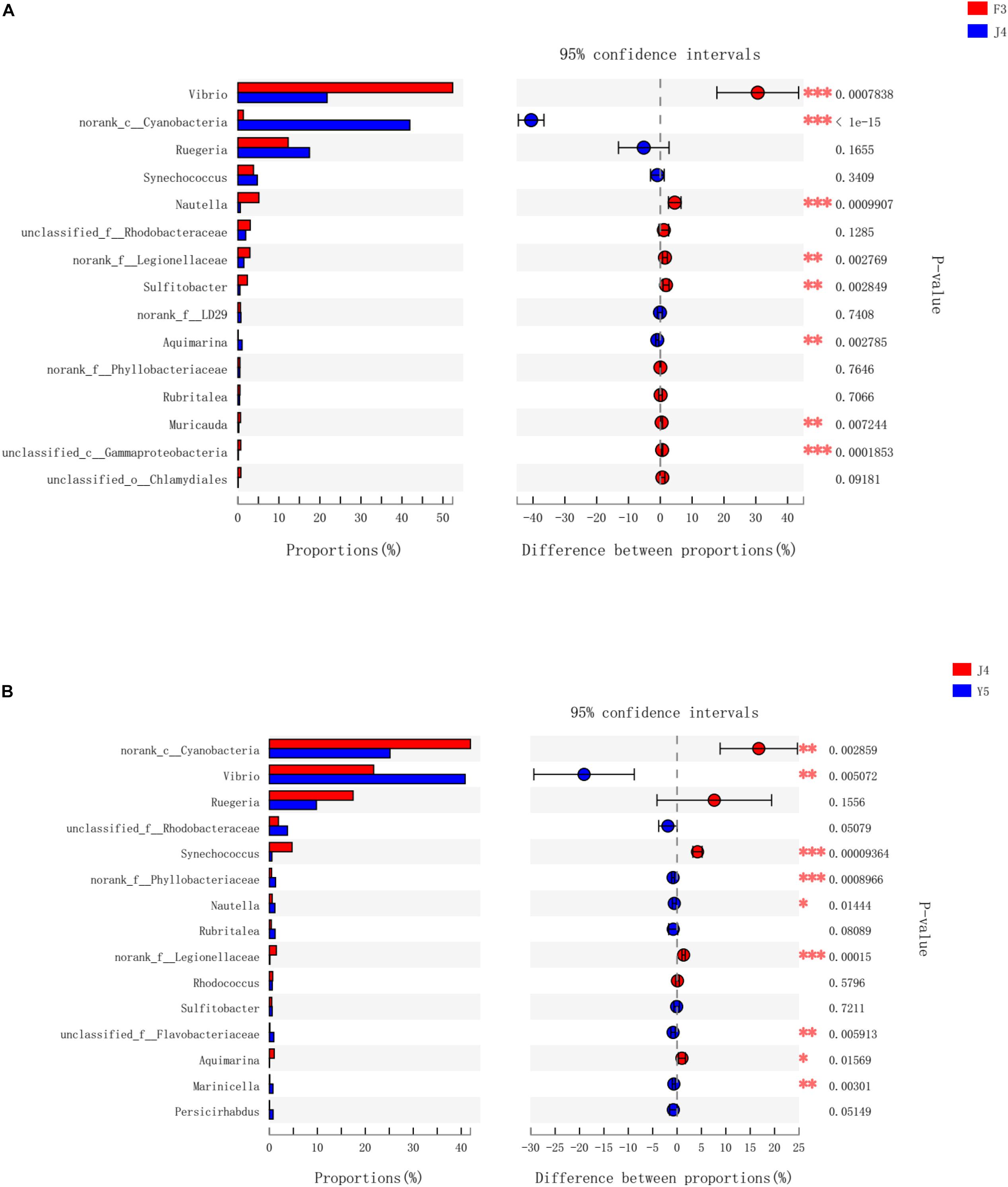
Figure 6. Bacterial taxa differentially represented between the three groups (F3, J4, and Y5) as determined by LEfSe using default parameters. F3 vs J4 (A), J4 vs Y5 (B). The asterisks indicate significant differences between the groups (P < 0.05). Two asterisks indicate extremely significant differences between the groups (P < 0.01). F3: three-spiral whorl stage, J4: four-spiral whorl stage (competent larvae) and Y5: postlarval stage after 3 days of metamorphosis.
Functional Prediction for the Symbiotic Microbiota
Changes in the putative functions of the symbiotic microbiota in R. venosa were analyzed by predicting metagenomes using PICRUSt, and the results showed that the symbiotic microbiota was mainly enriched in functions related to transporters, ATP-binding cassette (ABC) transporters, purine metabolism, ribosomes, peptidases, oxidative phosphorylation, bacterial motility proteins, porphyrin and chlorophyll metabolism, pyrimidine metabolism and amino acid-related enzymes (Supplementary Figure S1). We chose three KOs (KEGG Orthologs) at KEGG level 2 that are related to immune function (infectious diseases, immune system, immune system diseases) and analyzed their relative abundances among the five development stages. We found that all three KOs had high abundances in the J4 and Y5 stages, and their abundances increased with host development, peaking in the J4 stage (Figure 7).
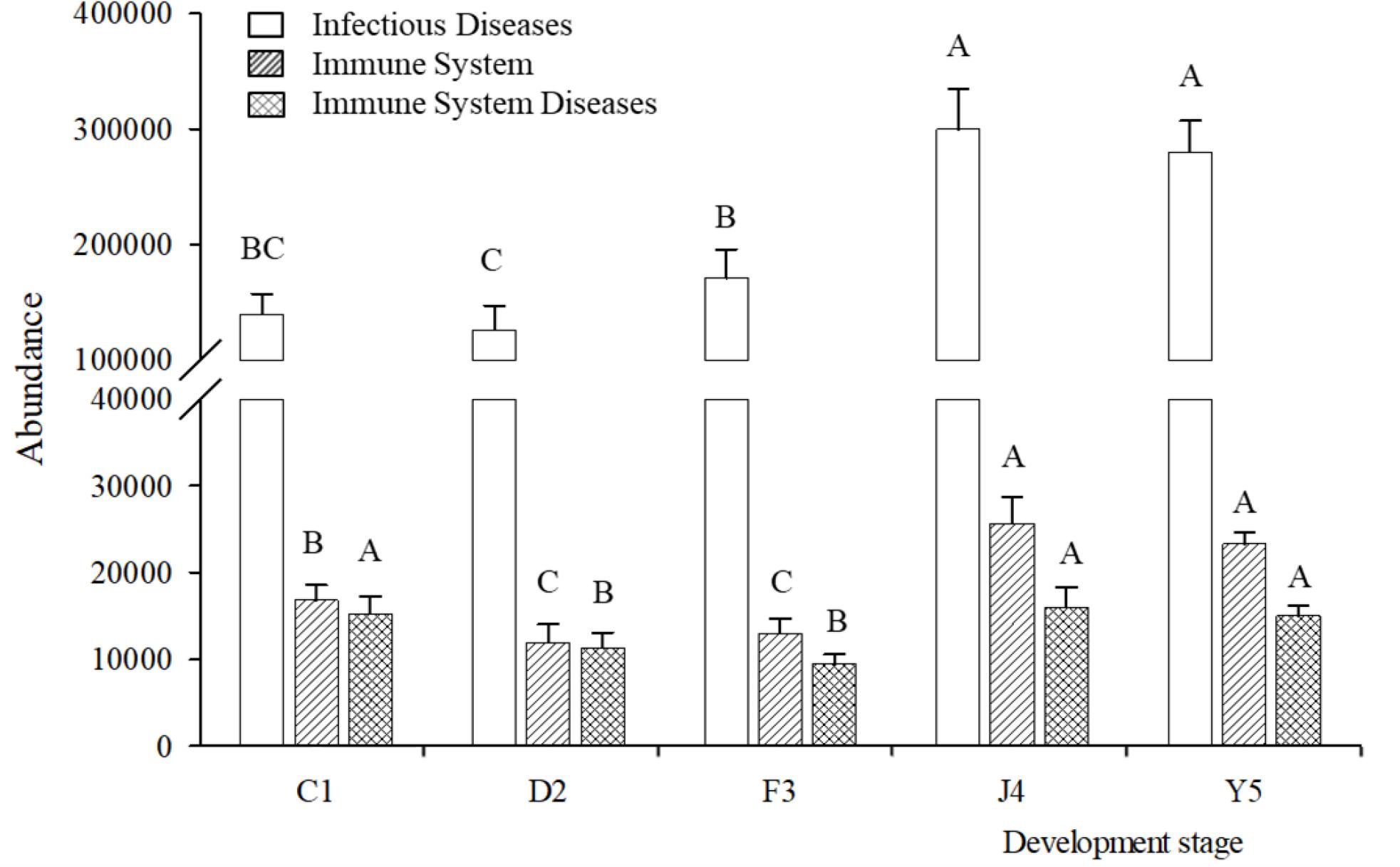
Figure 7. Relative abundances of predicted functions. Relative abundances of immune-related KOs at KEGG level 2 in different development stages. Different upper-case letters within each bar indicate significant differences (P < 0.05). C1, one-spiral whorl stage; D2, two-spiral whorl stage; F3, three-spiral whorl stage; J4, four-spiral whorl stage (competent larvae); Y5, postlarval stage after 3 days of metamorphosis.
Host Immune Response
The SOD and CAT activities in the larvae exhibited a decreasing trend with the development of R. venosa (Figure 8A) and reached their lowest levels at the Y5 stage (P < 0.05). The ALP and ACP activities were also at their lowest levels at the Y5 stage, while the LSZ activity did not change significantly during the early development stages (P < 0.05) (Figure 8B).
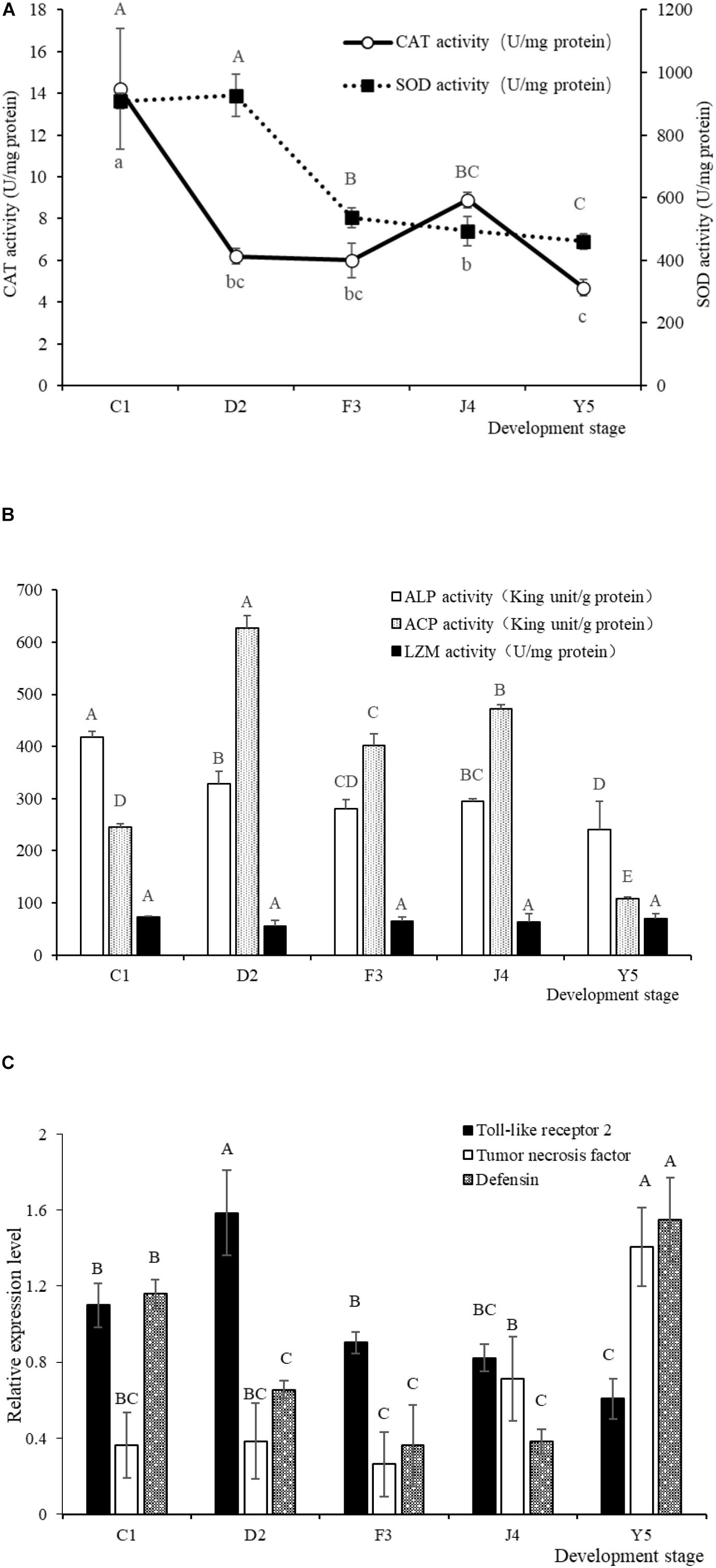
Figure 8. SOD and CAT activities (A) and LSZ, ACP and ALP activities (B) in larvae of R. venosa. Relative mRNA expression of Toll-like receptor 2, tumor necrosis factor and defensin (C). Values are expressed as the mean ± SD (n = 5). Different upper-case letters within each bar indicate significant differences (P < 0.05). C1, one-spiral whorl stage; D2, two-spiral whorl stage; F3, three-spiral whorl stage; J4, four-spiral whorl stage (competent larva); Y5, postlarval stage after 3 days of metamorphosis.
The relative expression of Toll-like receptor 2, tumor necrosis factor and defensin significantly changed during host development (Figure 8C). The relative expression of Toll-like receptor 2 peaked at the D2 stage, after which the expression of this gene decreased significantly with host development, becoming lowest at the Y5 stage. The relative expression of tumor necrosis factor increased with host development and peaked at the Y5 stage, which is similar to the observed trend for defensin.
Discussion
Increased attention is being paid to the relationship between aquatic hosts and their symbiotic microbiota. Previous researches have indicated that symbiotic microbiota is important for the maturation of immune system (Mazmanian et al., 2005; Edelman et al., 2008), so there may be some changes in the symbiotic microbiota during host development. Present results showed that significant shifts in the structure of symbiotic microbiota and immune response of R. venosa occurred at early development stages, which may reflect a substantial change in the physiological functions of the larvae.
The core microbiota consists of the OTUs shared among all samples (Turnbaugh et al., 2009). In our study, 102 OTUs were shared by all samples from the host, accounting for 5.85% of the total OTUs (Figure 2). The core microbiota could reflect the impact of diet, development, stocking density and environment on microbiota (Wong et al., 2013) and is usually determined based on the genetic background. We found that most of the shared OTUs belonged to Proteobacteria and Cyanobacteria, which were also the dominant phyla in R. venosa in this study. Proteobacteria are commonly found in the digestive tract of adult R. venosa (Yang et al., 2019), Pacific white shrimp (Zeng et al., 2017) and gibel carp (Li et al., 2017) and seem to be the dominant phylum among aquacultured animals, which may result from the widespread distribution of Proteobacteria in environmental water and the ability of these bacteria to interact with host. However, Cyanobacteria are not abundant in adult whelks (Song et al., 2018; Yang et al., 2019) and have a low abundance (less than 0.01%) in most aquaculture animals (Dhanasiri et al., 2011; Rungrassamee et al., 2014; Li et al., 2017), while they were detected at a high abundance (4.5–37.0%) in this study, which might be associated with the water environment and feed because Cyanobacteria were also the dominant phylum in water and feed samples (Figure 1A).
A previous study has indicated that neutral processes are able to dramatically affect the gut microbiota during development in zebrafish (Stephens et al., 2015). Li et al. (2017) also indicated that the structure of gut microbiota in gibel carp mainly depended on by host-associated processes instead of stochastic processes, and they found that the diversity of gut microbiota community increased with developing, which may be related to the morphological differentiation of the gastrointestinal tract (Li, 2007). In present study, the diversity of symbiotic microbiota showed a decreasing trend during the development stages, while the richness indices showed no significant change, which suggests that the role of the dominant population becomes increasingly prominent as the host develops. Moreover, UPGMA clustering, NMDS and typing analysis also showed differences in the symbiotic microbiota between development stages. The microbiota compositions of stages D2 and F3 are highly similar, while those of J4 and Y5 belong to the same type. Metamorphosis occurs in the late phase of the J4 stage, so there should be a considerable difference in the symbiotic microbiota between the J4 and Y5 stages. However, in our study, significant changes were observed earlier than expected, which may suggest that a series of changes in the internal physiological functions of the host during metamorphosis precede the changes in apparent features, including the further development of the digestive and immune system. This shift in the symbiotic microbiota in R. venosa occurs because the conditions of host (e.g., physicochemical environments, niche availability, and microbial interactions) change dramatically during host development.
Many studies have reported that the balance of the symbiotic microbiota could make difference on immune system, especially in invertebrates lacking specific immune system, which is crucial for disease resistance (Gómez and Balcázar, 2008; Jaenike et al., 2010). Therefore, changes in the abundance of some pathogenic bacteria may reflect changes in the immune function of the host. Chlamydiae is an obligate intracellular bacterium that can cause many diseases in the host (Moulder, 1991). In the present study, the abundance of Chlamydiae decreased significantly with host development, which may indicate the strengthening of host immune function. Additionally, the genus Vibrio, which is widely distributed in aquaculture, contains many pathogenic species, such as Vibrio anguillarum, Vibrio ordalii, and Vibrio parahaemolyticus, which can cause disease in fish, shrimp and shellfish. Previous studies have indicated that V. anguillarum has a noticeable impact on the phosphatase activity of Cyclina sinensis and a significant stimulatory effect on the immune system of clams (Song et al., 2010). We found that the abundance of Vibrio decreased dramatically in the J4 stage in the present study, which may be another indicator of changes in the immune system that occur during metamorphosis in the host. All three KOs predicted by PICRUSt are related to immune function, namely, infectious diseases, immune system and immune system diseases, had significantly higher abundances in the J4 and Y5 stages than in the other stages, indicating the involvement of many microbes in the host immune system. However, the applicability of PICRUSt prediction in R. venosa needs further validation, although PICRUSt prediction in humans shows consistency across all body tissues.
There is no specific immune system in mollusks, which mainly rely on non-specific immunity to prevent infections and maintain good health. The phagocytic process is the first line of internal defense in the body (Janeway and Medzhitov, 2002), and mollusk phagocytes can completely degrade exogenous bacteria via the activities of LSZ, ACP, and ALP (Canicatti, 1990; Ellis, 1999). Therefore, the activity of these immune-related enzymes can reflect the body’s immune status. ACP and ALP are important hydrolases that participate in immune defense (Xiao et al., 2007). ACP is widely distributed in animal tissues and can destroy foreign bodies through hydrolysis to prevent pathogenic infection (Cheng, 1978; Lackie, 1980). ALP is mainly located on the plasma membranes of cells and is an important metabolic regulatory enzyme in animals. ALP is also related to calcium absorption, membrane absorption and transport, and keratin secretion in shellfish. Moreover, ALP as a significant component of animal lysosomal enzymes, plays an important role in the immune response (Xie et al., 2000). In our study, ACP activity was significantly decreased in the Y5 stage, while ALP and LZM activities exhibited no substantial change. CAT and SOD are the primary components of the antioxidant defense system of organisms, and the activities of these enzymes could indicate the ability of body to resist oxidative stress though reducing the H2O2 and O2– content and indirectly reflect the immunity levels (Bao et al., 2017). The activities of CAT and SOD were dramatically decreased during the early development of R. venosa, which might be related to the decreases in the abundances of pathogens, such as Chlamydiae and Vibrio. On the other hand, ALP, CAT and SOD all exhibited high activities in the C1 stage, which may be because larvae hatching from the oocysts first enter the water environment with an abundance of microorganisms and initial colonization by the symbiotic microbiota in larvae stimulates the immune system of the host.
Previous research has indicated that Toll-like receptor 2 can identify the capsular polysaccharide antigens of commensal bacteria, stimulating the innate immune response (Edelman et al., 2008). We found that the expression of the Toll-like receptor 2 gene was not significantly increased in the Y5 stage, which was not in line with the results in previous study (Song et al., 2016b), and we speculated that this difference is related to the differences in the composition of the sampled microbiota in different environments. Furthermore, tumor necrosis factor and defensin are two other important genes related to the symbiotic microbiota in the host (Jordans and Misra, 2012; Yuzhakova et al., 2016). Tumor necrosis factor exhibited a high expression level in the Y5 stage, which may suggest that this gene is related to the shift in symbiotic microbiota during metamorphosis. On the other hand, defensin also exhibited relatively high expression in the C1 stage, which indicates that defensin is associated with not only the shift in symbiotic microbiota during metamorphosis but also the initial colonization in host by the symbiotic microbiota.
The host and symbiotic microbiota are components of an indivisible whole that interact with each other. On the one hand, the development of immune system in host affects the structure of the symbiotic microbiota. On the other hand, the colonization by and shift in the symbiotic microbiota stimulate the development of the immune system. Our present study comprehensively examined the symbiotic microbiota and the immune response in R. venosa during the early development stage and investigated the relationship between the symbiotic microbiota and the immune system of the host. We found that dramatic changes in the symbiotic microbiota and the immune response mainly occurred in the initially hatched veliger (C1), competent larval (J4) and postlarval (Y5) stages, during which the hosts experienced substantial environmental changes or changes in physiological structure and function. These findings expand our knowledge of the stage-specific symbiotic microbiota in R. venosa and the close association between symbiotic microbiota and immune system in mollusks. However, additional studies are needed to explore the specific relationship between microbiota and immune system during the early development stages, especially during metamorphosis.
Data Availability Statement
The datasets generated for this study can be found in the SRA under accession number PRJNA574861.
Author Contributions
TZ conceived and designed the experiments. M-JY conducted the experiments. M-JY and HS analyzed the data. Z-LY, ZH, CZ, and X-LW contributed reagents, materials, and analytical tools. M-JY wrote the manuscript.
Funding
This research was supported by the National Key Research and Development Program of China (2019YFD0900800), the National Natural Science Foundation of China (Grant No. 31972814), the Natural Science Foundation of Shandong Province (Grant No. ZR2019BD003), the China Postdoctoral Science Foundation (Grant No. 2019M652498), the Major Scientific and Technological Innovation Project of Shandong Provincial Key Research and Development Program (2019JZZY020708), the Earmarked Fund for Modern Agro-industry Technology Research System (CARS-49), the Industry Leading Talents Project of Taishan Scholars (Recipient: TZ), the ‘Double Hundred’ Blue Industry Leader Team of Yantai (Recipient: TZ), the National Natural Science Foundation of China (Grant No. 31572636), and the Creative Team Project of the Laboratory for Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology (No. LMEES-CTSP-2018-1). The funders had no role in the study design, data collection and analysis, decision to publish and preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Mashan Group Co., Ltd. for providing support for the experimental material.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01265/full#supplementary-material
FIGURE S1 | Relative abundances of predicted functions. KOs at KEGG level 3 are listed according to the relative average abundances. C1, one-spiral whorl stage; D2, two-spiral whorl stage; F3, three-spiral whorl stage; J4, four-spiral whorl stage (competent larvae); Y5, postlarval stage after 3 days of metamorphosis.
TABLE S1 | Immune-related genes analyzed in the present work and primer sequences used for qPCR.
Footnotes
- ^ http://drive5.com/uparse/
- ^ http://drive5.com/usearch/manual/dereplication.html
- ^ http://drive5.com/usearch/manual/singletons.html
- ^ https://drive5.com/usearch/manual/chimeras.html
- ^ http://www.arb-silva.de
- ^ http://rdp.cme.msu.edu/
- ^ www.majorbio.com
- ^ http://www.mothur.org/wiki
- ^ http://bioinformatics.psb.ugent.be/webtools/Venn/
- ^ https://cran.r-project.org/web/packages/vegan/
- ^ http://qiime.org/index.html
- ^ http://huttenhower.sph.harvard.edu/galaxy/
References
Al-Harbi, A. H., and Uddin, N. (2005). Bacterial diversity of tilapia (Oreochromis niloticus) cultured in brackish water in Saudi Arabia. Aquaculture 250, 566–572. doi: 10.1016/j.aquaculture.2005.01.026
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
Ban, S., Zhang, T., Pan, H. Q., Pan, Y., Wang, P. C., and Xue, D. X. (2014). Effects of temperature and salinity on the development of embryos and larvae of the veined rapa whelk Rapana venosa (valenciennes, 1846). J. Oceanol. Limnol. 32, 773–782. doi: 10.1007/s00343-014-3264-6
Bao, N., Ren, T., Han, Y., Wang, F., Chen, F., and Jiang, Z. (2017). Alteration of growth, intestinal lactobacillus, selected immune and digestive enzyme activities in juvenile sea cucumber apostichopus japonicus, fed dietary multiple probiotics. Aquacult. Int. 25, 1721–1731. doi: 10.1007/s10499-017-0148-8
Bao, X. B., He, C. B., Fu, C. D., Wang, B., and Liu, W. D. (2015). A c-type lectin fold gene from japanese scallop mizuhopecten yessoensis, involved with immunity and metamorphosis. Genet. Mol. Res. 14, 2253–2267. doi: 10.4238/2015.March.27.11
Barrett, A. J. (1972). “Lysosomal enzymes,” in Lysosomes: A Laboratory Handbook, ed. J. T. Dingle (Amsterdam: North-Holland), 46–135.
Bradford, M. M. (1976). A rapid sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1006/abio.1976.9999
Buchon, N., Broderick, N. A., and Lemaitre, B. (2013). Gut homeostasis in a microbial world: insights from Drosophila melanogaster. Nat. Rev. Microbiol. 11, 615–626. doi: 10.1038/nrmicro3074
Canicatti, C. (1990). Lysosomal enzyme pattern in Holothuria polii coelomocytes. J. Invertebr. Pathol. 56, 70–74. doi: 10.1016/0022-2011(90)90146-w
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Cheng, T. C. (1978). The role of lysosmal hydrolases in molluscan cellar response to immunologic challenge. Comp. Pathbiol. 4, 59–71. doi: 10.1007/978-1-4757-1278-0_4
Chu, Z., Wang, K., Li, X., Zhu, M., Yang, L., and Zhang, J. (2015). Microbial characterization of aggregates within a one-stage nitritation-anammox system using high-throughput amplicon sequencing. Chem. Eng. J. 262, 41–48. doi: 10.1016/j.cej.2014.09.067
Dhanasiri, A. K., Brunvold, L., Brinchmann, M. F., Korsnes, K., Bergh, O., and Kiron, V. (2011). Changes in the intestinal microbiota of wild atlantic cod Gadus morhua L. upon captive rearing. Microb. Ecol. 61, 20–30. doi: 10.1007/s00248-010-9673-y
Edelman, M., Sanna, K., and Dennis, L. (2008). Symbiotic commensal bacteria direct maturation of the host immune system. Curr. Opin. Gastroen. 24, 720–724. doi: 10.1097/MOG.0b013e32830c4355
Edgar, R. C. (2013). Uparse: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods. 10, 996–998. doi: 10.1038/nmeth.2604
Ellis, A. E. (1999). Immunity to bacteria in fish. Fish Shellfish Immunol. 9, 291–308. doi: 10.1006/fsim.1998.0192
Fraune, S., and Bosch, T. C. G. (2010). Why bacteria matter in animal development and evolution. BioEssays 32, 571–580. doi: 10.1002/bies.200900192
Gómez, G. D., and Balcázar, J. L. (2008). A review on the interactions between gut microbiota and innate immunity of fish. Fems Immunol. Med. Mic. 52, 145–154. doi: 10.1111/j.1574-695X.2007.00343.x
Góth, L. (1991). A simple method for determination of serum catalase activity and revision of reference range. Clin. Chim. Acta. 196, 143–151. doi: 10.1016/0009-8981(91)90067-m
Graf, J. (2016). Lessons from digestive-tract symbiosis between bacteria and invertebrates. Annu. Rev. Microbiol. 70, 75–93. doi: 10.1146/annurev-micro-091014-104258
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., et al. (2011). Chimeric 16S rRNA sequence formation and detection in sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504. doi: 10.1101/gr.112730.110
Hacer, S., and Ertug, D. (2007). Deposition of egg capsule and larval development of Rapana venosa (Gastropoda: Muricidae) from the south-eastern black sea. J. Mar. Biol. Assoc. U..K. 87, 953–957. doi: 10.1017/s0025315407056330
Harding, J. M. (2006). Growth and development of veined rapa whelk Rapana venosa veligers. J. Shellfish Res. 25, 941–946. doi: 10.2983/0730-8000(2006)25[941:gadovr]2.0.co;2
He, T. F., Chen, J., Zhang, J., Ke, C. H., and You, W. W. (2014). Sarp 19 and vdg3 gene families are functionally related during abalone metamorphosis. Dev. Genes Evol. 224, 197–207. doi: 10.1007/s00427-014-0478-8
Jaenike, J., Unckless, R., Cockburn, S. N., Boelio, L. M., and Perlman, S. J. (2010). Adaptation via symbiosis: recent spread of a Drosophila defensive symbiont. Science 329, 212–215. doi: 10.1126/science.1188235
Janeway, C. A., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216.
Jordans, R., and Misra, J. (2012). Down regulation of enteric defensins is responsible for alteration of gut microbiota and development of visceral hypersensitivity in chronically stressed rats, American. Gastroenterology 142, S–98.
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Li, Q. (2007). The Study on Intestinal Histology and Developing Regulation of Nutrients of Allogynogenetic Crucian. Nanjing: Nanjing Agricultural University.
Li, X., Zhou, L., Yu, Y., Ni, J., Xu, W., and Yan, Q. (2017). Composition of gut microbiota in the gibel carp (Carassius auratus gibelio) varies with host development. Micro. Ecol. 74, 239–250. doi: 10.1007/s00248-016-0924-4
Mann, R., and Harding, J. M. (2003). Salinity tolerance of larval Rapana venosa: implications for dispersal and establishment of an invading predatory gastropod on the North American Atlantic coast. Biol. Bull. 204, 96–103. doi: 10.2307/1543499
Mazmanian, S. K., Liu, C. H., Tzianabos, A. O., and Kasper, D. L. (2005). An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118. doi: 10.1016/j.cell.2005.05.007
Moulder, J. W. (1991). Interaction of chlamydiae and host cells in vitro. Microbiol. Rev. 55, 143–190. doi: 10.1128/mmbr.55.1.143-190.1991
Nicholson, J. K., Holmes, E., Kinross, J., Burcelin, R., Gibson, G., and Jia, W. (2012). Pettersson. Host-gut microbiota metabolic interactions. Science 336, 1262–1267. doi: 10.1126/science.1223813
Ôyanagui, Y. (1984). Reevaluation of assay methods and establishment of kit for superoxide dismutase activity. Anal. Biochem. 142, 290–296. doi: 10.1016/0003-2697(84)90467-6
Pan, H. Q. (2014). Study on Neuronal Development of the Embryo and Early Larvae of the Gastropod Mollusc Rapana venosa. Qingdao: Ocean University of China.
Pan, Y., Qiu, T. L., Zhang, T., Wang, P., and Ban, S. J. (2013). Morphological studies on the early development of Rapana venosa. J. Fish. China 37, 1503– 1510.
Ramirez, C., and Romero, J. (2017). Fine Flounder (Paralichthys adspersus) microbiome showed important differences between wild and reared specimens. Front. Microbiol. 8:271. doi: 10.3389/fmicb.2017.00271
Rungrassamee, W., Klanchui, A., Maibunkaew, S., Chaiyapechara, S., Jiravanichpaisal, P., and Karoonuthaisiri, N. (2014). Characterization of intestinal bacteria in wild and domesticated adult black tiger shrimp (Penaeus monodon). PLoS One 9:e91853. doi: 10.1371/journal.pone.0091853
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60.
Shen, M., Di, G. L., Li, M., Fu, J. Q., Dai, Q., and Miao, X. L. (2018). Proteomics studies on the three larval stages of development and metamorphosis of babylonia areolate. Sci. Rep. U. K. 8, 62–69. doi: 10.1038/s41598-018-28807-x
Song, H., Dang, X., He, Y. Q., Zhang, T., and Wang, H. Y. (2017a). Selection of housekeeping genes as internal controls for quantitative rt-pcr analysis of the veined rapa whelk (Rapana venosa). PeerJ 5:e3398. doi: 10.7717/peerj.3398
Song, H., Qi, L., Zhang, T., and Wang, H. Y. (2017b). Understanding microrna regulation involved in the metamorphosis of the veined rapa whelk (Rapana venosa). G3Gene. Genom. Genet. 7, 3999–4008. doi: 10.1534/g3.117.300210
Song, H., Sun, L. Y., Yu, Z. L., Sun, L. N., Xue, D. X., and Zhang, T. (2016a). Metabolomic analysis of competent larvae and juvenile veined rapa whelks (Rapana venosa). Mar. Biol. 163:145.
Song, H., Yu, Z. L., Sun, L. N., Xue, D. X., Zhang, T., and Wang, H. Y. (2016b). Transcriptomic analysis of differentially expressed genes during larval development of Rapana venosa, by digital gene expression profiling. G3 Genes Genom. Genet. 6, 2181–2193. doi: 10.1534/g3.116.029314
Song, H., Yu, Z. L., Sun, L. N., Zhang, T., and Wang, H. Y. (2016c). De novo transcriptome sequencing and analysis of Rapana venosa from six different development stages using hi-seq 2500. Comp. Biochem. Phys. D 17, 48–57. doi: 10.1016/j.cbd.2016.01.006
Song, H., Wang, H. Y., and Zhang, T. (2016d). Comprehensive and quantitative proteomic analysis of metamorphosis-related proteins in the veined rapa whelk, Rapana venosa. Int. J. Mol. Sci. 17:924. doi: 10.3390/ijms17060924
Song, H., Yu, Z. L., Yang, M. J., Zhang, T., and Wang, H. Y. (2018). Analysis of microbial abundance and community composition in esophagus and intestinal tract of wild veined rapa whelk (Rapana venosa) by 16s rRNA gene sequencing. J. Gen. Appl. Microbiol. 64, 158–166. doi: 10.2323/jgam.2017.11.003
Song, X., Zhang, L. Y., Gao, W. W., and Pan, B. P. (2010). Effect of vibrio anguillarum on activity of phosphatase in cyclina sinensis. Oceanol. Limnol. Sini. 41, 254–258.
Stephens, W. Z., Burns, A. R., Stagaman, K., Wong, S., Rawls, J. F., Guillemin, K., et al. (2015). The composition of the zebrafish intestinal microbial community varies across development. ISME J. 10, 644–654. doi: 10.1038/ismej.2015.140
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Ueda, N., and Degnan, S. M. (2014). Nitric oxide is not a negative regulator of metamorphic induction in the abalone Haliotis asinine. Front. Mar. Sci. 1:21. doi: 10.3389/fmars.2014.00021
Wang, J., Huang, Y. J., Xu, K. H., Zhang, X., Sun, H., Fan, L., et al. (2019). White spot syndrome virus (wssv) infection impacts intestinal microbiota composition and function in Litopenaeus vannamei. Fish Shellfish Immun. 84, 130–137. doi: 10.1016/j.fsi.2018.09.076
Wei, L. P., and Wang, B. G. (1999). Study on the reproductive biology of Rapana venosa. J. Fish. China 2, 150–155.
Wong, S., Waldrop, T., Summerfelt, S., Davidson, J., Barrows, F., Kenney, P. B., et al. (2013). Aquacultured rainbow trout (Oncorhynchus mykiss) possess a large core intestinal microbiota that is resistant to variation in diet and rearing density. Appl. Environ. Microb. 79, 4974–4984. doi: 10.1128/AEM.00924-13
Woodhams, D. C., Vredenburg, V. T., Simon, M. A., Billheimer, D., Shakhtour, B., and Shyr, Y. (2007). Symbiotic bacteria contribute to innate immune defenses of the threatened mountain yellow-legged frog, rana muscosa. Bio. Conserv. 138, 390–398. doi: 10.1016/j.biocon.2007.05.004
Xiao, K. Y., Deng, S. M., and Xiang, J. G. (2007). Immunity and Application of Aquatic Animals. Beijing: Science Press, 89–96.
Xie, L. P., Lin, J. Y., Xiao, R., and Zhang, R. Q. (2000). Purification and characterization of alkaline phosphatase from Pinctada fucata. Mar. Sci. 24, 37–40.
Yang, M. J., Song, H., Sun, L. N., Yu, Z. L., Hu, Z., Wang, X. L., et al. (2019). Effect of temperature on the microflora community composition in the digestive tract of the veined rapa whelk (Rapana venosa) revealed by 16S rRNA gene sequencing. Comp. Biochem. Phys. D. 29, 145–153. doi: 10.1016/j.cbd.2018.10.006
Yang, M. J., Song, H., Yu, Z. L., Bai, Y. C., and Zhang, T. (2020). Expression and activity of critical digestive enzymes during early larval development of the veined rapa whelk, Rapana venosa (Valenciennes, 1846). Aquaculture 519:734722. doi: 10.1016/j.aquaculture.2019.734722
Yang, Z., Yu, H., Yu, R. H., Kong, L. F., and Li, Q. (2016). Studies on larval metamorphosis and feed conversion of Rapana venosa. J. Fish. China 38, 249–259.
Yuzhakova, D. V., Shirmanova, M. V., Bocharov, A. A., Astrakhantseva, I. V., Vasilenko, E. A., Gorshkova, E. N., et al. (2016). Microbiota induces expression of tumor necrosis factor in postnatal mouse skin. Biochemistry 81, 1303–1308. doi: 10.1134/S0006297916110080
Zeng, S., Huang, Z., Hou, D., Liu, J., and He, J. (2017). Composition, diversity and function of intestinal microbiota in pacific white shrimp (Litopenaeus vannamei) at different culture stages. PeerJ 5:e3986. doi: 10.7717/peerj.3986
Keywords: Rapana venosa, symbiotic microbiota, immune system, metamorphosis, early development stages
Citation: Yang M-J, Song H, Yu Z-L, Hu Z, Zhou C, Wang X-L and Zhang T (2020) Changes in Symbiotic Microbiota and Immune Responses in Early Development Stages of Rapana venosa (Valenciennes, 1846) Provide Insights Into Immune System Development in Gastropods. Front. Microbiol. 11:1265. doi: 10.3389/fmicb.2020.01265
Received: 05 January 2020; Accepted: 18 May 2020;
Published: 16 June 2020.
Edited by:
Jaime Romero, University of Chile, ChileReviewed by:
Ricardo Vazquez-Juarez, Centro de Investigaciones Biológicas del Noroeste (CIBNOR), MexicoMorten Schiøtt, Technical University of Denmark, Denmark
Copyright © 2020 Yang, Song, Yu, Hu, Zhou, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Zhang, dHpoYW5nQHFkaW8uYWMuY24=
†These authors share first authorship