 Alfonso Benítez-Páez1*
Alfonso Benítez-Páez1* Marta Olivares1Hania Szajewska2Małgorzata Pieścik-Lech2
Marta Olivares1Hania Szajewska2Małgorzata Pieścik-Lech2 Isabel Polanco3Gemma Castillejo4Merce Nuñez4
Isabel Polanco3Gemma Castillejo4Merce Nuñez4 Carmen Ribes-Koninckx5Ilma R. Korponay-Szabó6
Carmen Ribes-Koninckx5Ilma R. Korponay-Szabó6 Sibylle Koletzko7,8
Sibylle Koletzko7,8 Caroline R. Meijer9M. Luisa Mearin9
Caroline R. Meijer9M. Luisa Mearin9 Yolanda Sanz1
Yolanda Sanz1- 1Microbial Ecology, Nutrition and Health Research Unit, Institute of Agrochemistry and Food Technology, Spanish National Research Council, Valencia, Spain
- 2Department of Pediatrics, The Medical University of Warsaw, Warsaw, Poland
- 3Department of Pediatric Gastroenterology and Nutrition, La Paz University Hospital, Madrid, Spain
- 4Gluten-Associated Disorder Unit, Institut d’Investigació Sanitària Pere Virgili, Universitat Rovira i Virgili, Reus, Spain
- 5Pediatric Gastroenterology Unit, La Fe University Hospital, Valencia, Spain
- 6Celiac Disease Center, Heim Pál Children’s Hospital, Budapest, Hungary
- 7Department of Pediatrics, Dr. von Hauner Children’s Hospital, University Hospital of Munich, Munich, Germany
- 8Department of Paediatrics, School of Medicine Collegium Medicum, University of Warmia and Mazury, Olsztyn, Poland
- 9Department of Pediatrics, Leiden University Medical Center, Leiden, Netherlands
Celiac disease (CeD) is an immune-mediated disorder triggered by exposure to dietary gluten proteins in genetically predisposed individuals. In addition to the host genome, the microbiome has recently been linked to CeD risk and pathogenesis. To progress in our understanding of the role of breast milk microbiota profiles in CeD, we have analyzed samples from a sub-set of mothers (n = 49) included in the PreventCD project, whose children did or did not develop CeD. The results of the microbiota data analysis indicated that neither the BMI, HLA-DQ genotype, the CeD condition nor the gluten-free diet of the mothers could explain the human milk microbiota profiles. Nevertheless, we found that origin country, the offspring’s birth date and, consequently, the milk sampling date influenced the abundance and prevalence of microbes in human milk, undergoing a transition from an anaerobic to a more aerobic microbiota, including potential pathogenic species. Furthermore, certain microbial species were more abundant in milk samples from mothers whose children went on to develop CeD compared to those that remained healthy. These included increases in facultative methylotrophs such as Methylobacterium komagatae and Methylocapsa palsarum as well as in species such as Bacteroides vulgatus, that consumes fucosylated-oligosaccharides present in human milk, and other breast-abscess associated species. Theoretically, these microbiota components could be vertically transmitted from mothers-to-infants during breastfeeding, thereby influencing CeD risk.
Introduction
Celiac disease (CeD) is an immune-mediated disorder triggered by gliadin proteins and related prolamines (defined as gluten) in genetically susceptible individuals. The disease is characterized by a variable combination of gluten-dependent clinical manifestations, CeD-specific antibodies, HLA-DQ2 or HLA-DQ8 haplotypes and enteropathy (Husby et al., 2012). The HLA-DQ2/8 genes predisposing to CeD and exposure to gluten are necessary but not sufficient to explain CeD onset. Emerging evidence suggests a role for the gut microbiota in CeD risk and pathogenesis (Galipeau et al., 2015; Sanz, 2015; Olivares et al., 2018a, b). The HLA-DQ2/8 genotype is associated with alterations in the early colonization pattern of infants at family risk of developing CeD (Palma et al., 2012; Olivares et al., 2015b). Early feeding patterns, including the type of milk-feeding (breastfeeding or formula) and the introduction of gluten into the infant’s diet are considered determinants of CeD onset (Silano et al., 2016). Both milk-feeding practices, particularly breastfeeding (Palma et al., 2012), and the amount of gluten in the diet (Sanz, 2010; Hansen et al., 2018) influence the gut microbiota composition and could, thus, act as moderators of CeD risk. Yet, little is known about how the host’s genetics and the environment interact with the gut microbiota, possibly contributing to the chain of causative mechanisms leading to CeD.
The first retrospective observational studies, which included control individuals from a general population, found a preventive effect of breastfeeding against later CeD development, particularly when breastfeeding was maintained parallel to introducing gluten into the infant diet (Auricchio et al., 1983; Stevens et al., 1987; Greco et al., 1988). Since then, several prospective and interventional studies including children with genetic risk predisposing to CeD or diabetes mellitus type I, have also tested this hypothesis but conclude that breastfeeding and its duration do not influence CeD risk (Szajewska et al., 2015; Silano et al., 2016). Some prospective studies report there is no association between CeD risk and gastrointestinal infections (Szajewska et al., 2015; Ziberna et al., 2016), however, our prospective gut microbiota analysis found an increased prevalence of pathogenic bacteria (enterotoxigenic Escherichia coli) in the feces of children with increased risk of developing CeD (Olivares et al., 2018a). Furthermore, findings from the prospective study TEDDY indicate that gastrointestinal infections increase the risk of CeD autoimmunity, and that the risk depends on other factors such as breastfeeding, the HLA genotype, infant gluten consumption, and rotavirus vaccination (Kemppainen et al., 2017). Therefore, the fact that previous prospective studies failed to control all interacting elements may well explain the inconsistency regarding the role of environmental variables like breastfeeding, which influence both gut microbiota and oral tolerance to allergens/gluten, in CeD onset.
A few studies have also suggested that breast-milk composition differs depending on the mother’s health status, which may influence its potential protective effect in CeD and partially explain the discrepant findings across studies. In particular, breast milk from control mothers and mothers with CeD was found to differ in microbiota composition (bifidobacteria) and immune mediators [secretory immunoglobulin A (sIgA) and transforming growth factor (TGFβ1)] and total IgA, which could be involved in the development of oral tolerance to gluten (Olivares et al., 2015a; Roca et al., 2018). However, this hypothesis has not been directly proven so far. Moreover, differences in mothers’ milk microbiota according to their CeD condition could lead to differences in the bacteria transmitted through breastfeeding and colonizing the infants’ gut, which could theoretically affect early immune development and disease risk (Olivares et al., 2015a; Demmelmair et al., 2020).
To progress in our understanding, the present pilot study aims to investigate the relationship between mother’s breast milk microbiota profiles and CeD development in their offspring, including the analysis of potential covariates. A strict methodology was applied to control for cross-contamination and accurately identify human-milk associated bacteria.
Materials and Methods
Study Participants and Sample Collection
The PreventCD cohort consists of 944 children with at least one first-degree relative with CeD and available genotype HLA-DQ2 or HLA-DQ8 enrolled at birth, between 2007 and 2010, in Croatia, Germany, Hungary, Israel, Italy, Netherlands, Poland, and Spain. The PreventCD project (prevention of celiac disease1) investigates the influence of genetic, immunologic and environmental factors on the risk of developing CeD. All the children were assessed regularly from birth onward for CeD development (monthly until the age of 1 year, bimonthly until the age of 2 years, and trimonthly onward). We periodically monitored parent-reported health status, anthropometrics, gluten consumption and specific CeD serology [IgA against tissue-transglutaminase (TGA)] as previously reported (Vriezinga et al., 2014). The parents of children with elevated TGA and/or CeD-associated symptoms suggestive of CeD, were offered small bowel biopsies to confirm the diagnosis or were diagnosed without biopsies according to the criteria of the European Society for Paediatric Gastroenterology Hepatology and Nutrition (ESPGHAN) (Husby et al., 2012). The disease onset in children of mothers providing CeD milk samples was diagnosed at 41.2 ± 6.38 months after birth (95% CI 34.8–47.6). Maternal milk samples were collected at 9 months after birth. Mothers expressed their milk manually or with a pump, but the preferred way for the collection was not recorded. No further specifications pre- or post-milk sampling or time of day were given to the mothers. The milk samples were frozen at −20°C at home, transferred to the hospital on ice to avoid thawing and stored at −80°C until processed for microbiota analyses. A total of 49 breast-milk samples from the mothers of the Dutch, Polish, Spanish, Hungarian, and German participants in PreventCD stored at the Leiden University Medical Center were used in this assessment. They corresponded to milk from mothers of 25 children who developed CeD and from mothers of 24 children who did not develop CeD, referred to as “controls.” The study was registered at ISRCTN (ISRCTN74582487) on February 26th, 2007. The first and last children were included between 26th of May 2007 and the 25th of September 2010, respectively. This study was conducted according to the guidelines laid down in the Declaration of Helsinki and it was approved by all medical ethics committees of the participating centers.
DNA Isolation and Processing
Approximately 9 mL of milk sample was combined with 1 mL sterile 10× PBS + 0.1% Tween 20 (v/v) and mixed gently. The samples were then filtered through 5.0 μm membranes (Millipore) to eliminate eukaryotic cells and the flow-through was recovered for further processing. Filtered samples were centrifuged at 10,000 g for 10 min and pellets were stored in 1.5 mL nuclease-free tubes until processing for DNA extraction. Cell pellets were used for DNA isolation using the PowerFecal® DNA Isolation Kit (Mo Bio Laboratories) following manufacturer instructions and with a prior lysis treatment to improve DNA extraction from resistant species. Thus, pellets were resuspended in 300 μL sterile PBS, containing 20 U mutanolysin (Sigma) and 250 μg lysozyme (Sigma) and incubated during 60 min at 37°C. Given the potentially low bacterial load present in human milk samples, we included three negative controls corresponding to the different DNA extraction batches. These controls included a 1 mL aliquot of the 10× PBS + 0.1% Tween 20 buffer used to homogenized milk samples prior filtration and DNA extraction. These negative controls were equally processed during DNA extraction, PCR, and amplicon sequencing. The human-milk-derived genomic DNA was quantified by fluorescence-based methods such as Qubit 3.0 and the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States), and 1 μL DNA (0.5–5 ng) aliquot was used for microbiota analysis. The V4–V5 hypervariable regions of the bacterial 16S rRNA gene were amplified by 25-PCR cycles, including the following stages: 95°C for 20 s, 40°C for 30 s, and 72°C for 20 s. For PCR, Phusion High-Fidelity Taq Polymerase (Thermo Fisher Scientific) and the 6-mer barcoded primers S-D-Bact-0563-a-S-15 (AYTGGGYDTAAAGNG) and S-D-Bact-0907-a-A-20 (CCGTCAATTYMTTTRAGTTT), were used to targeting the bacterial 16S rRNA gene (Klindworth et al., 2012).
Sequencing and Data Processing
Dual barcoded PCR products, consisting of ∼400 bp, were purified from triplicate reactions by using the Illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare) and quantified through Qubit 3.0 and the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States). Samples were multiplexed by combining equimolar quantities of amplicon DNA (100 ng per sample) and sequenced in one lane of Illumina MiSeq platform (Eurofins Genomics GmbH, Ebersberg, Germany) with 2 × 300 PE configuration. The volume of purified PCR from negative controls (with no signal of amplified DNA) to be combined in the sequencing library was calculated as the median of the volume used for the entire set of samples (∼6 μL). Raw data were delivered in fastq files and paired-ends with quality filtering were assembled using Flash software (Magoc and Salzberg, 2011). Sample de-multiplexing was carried out using sequence information from respective DNA barcodes and Mothur v1.36.1 suite of analysis (Schloss et al., 2009). After assembly and barcodes/primers removal, the sequences were processed for chimera removal using Uchime algorithm (Edgar et al., 2011) and SILVA reference set of 16S rRNA gene sequences (Release 110) (Quast et al., 2013). Taxonomy assessment was performed using the RDP classifier v2.12 (Wang et al., 2007), using a high-quality and normalized subset of 5,000 sequences per sample, randomly selected after shuffling (10,000×) of the chimera removed dataset. The operational taxonomic unit (OTU)-picking approach was performed with the normalized subset of 5,000 sequences and the uclust algorithm (97% sequence identity) implemented in USEARCH v8.0.1623 (Edgar, 2010) discarding singletons and doubletons during the OTU clustering. Non-parametric linear discriminant analysis (LDA) among cases and controls was performed to measure differences among human milk microbial communities at different taxonomy level (Segata et al., 2011). Taxonomy identity of OTUs with differential abundance was assessed using a Blast-based approach and the NCBI non-redundant 16S rRNA gene database as well as the SINA aligner and SILVA database annotation (Pruesse et al., 2012). Common descriptors of the alpha diversity such as Chao’s index, observed OTUs, Shannon index, reciprocal Simpson’s index, dominance index, and phylogenetic distance were assessed using the OTU level information and the qiime v1.9.1 suite of analysis (Caporaso et al., 2010).
Potential Reagent Contaminants Identification and Removal
Given the very low bacterial load present in human milk, data analysis could be biased by the presence of foreign DNA coming from DNA extraction reagents, PCR, and sequencing approaches. To reduce this noise, we included negative controls that enable tracing of the bacterial species unpredictably present in reagents. In this way, we retrieved a few hundred DNA reads from respective negative controls, and identified dozens of bacterial groups present in those controls. The negative controls exhibited quite a different microbiota profiling compared to samples using multivariate analysis (Supplementary Figure S1). An abundance-based estimation of likely contaminant OTUs was performed by assessing predominant OTUs of negative control samples (>0.1% in average). Then, those OTUs were removed from control and cases samples if the abundance median was less than five reads (0.1%).
Statistical Analysis
The statistical assessment of individual parameters and comparison among groups was performed with Wilcoxon rank-sum test or t-test (Welch’s correction) according to Shapiro–Wilk normality test results, all the above implemented in R v 3.51. Qiime v1.9.1 was also used to analyze the global structure of microbial communities (beta diversity) through weighted and unweighted unifrac metrics as well as to perform principal coordinate analysis (PCoA) and statistical assessment with permutation-based test (Permanova) (Caporaso et al., 2010). Non-parametric correlations (Spearman’s rho) between principal coordinate (PC) values and OTU abundances were assessed and corrected for multiple comparisons, using the false discovery rate (FDR) post hoc test. Linear mixed models (LMM) were also used with log-transformed OTUs and clinical metadata to determine potential covariates influencing the human milk microbiota (hMM) by using the nlme R package. The metadata used to establish associations with hMM profiles include both children’s covariates (disease, gender, HLA-DQ genotype, birth year, and birth season) and mother’s covariates (disease, HLA-DQ genotype, birth year, maternity age, gluten-free diet, delivery mode, BMI, origin country). CeD genetic risk based on HLA-DQ genotyping (scale from 1 to 5) was calculated according to Vriezinga et al. (2014) for the PreventCD cohort, the lower the score the higher the DQ-associated genetic risk of CeD. Graphics and plots were designed in R v 3.5 and ggplot2 package2.
Results
Demographic and Clinical Characteristics
Summarized in Table 1 are the demographic and clinical variables taken into account in the present study, due to their potential relationship with CeD risk and the microbiota. We found no significant difference in distribution of such variables between the two groups compared (controls and CeD cases).
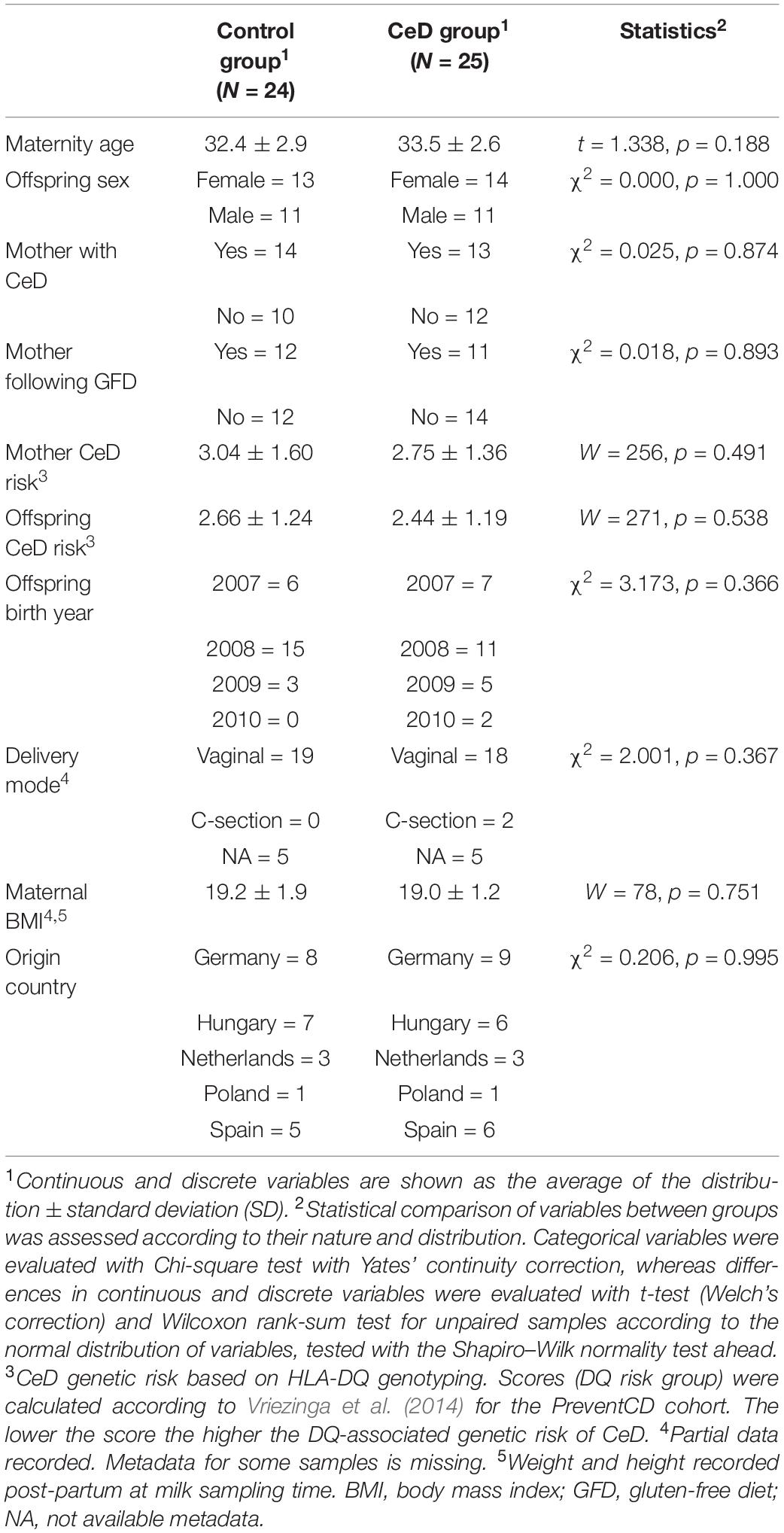
Table 1. Demographic and clinical variables of the mothers and children who developed celiac disease (CeD) and who did not (controls).
Alpha and Beta Diversities
Six common ecological descriptors were employed to assess the alpha diversity of the hMM in samples obtained from mothers enrolled in the PreventCD project included in this study. Globally, we found no differences in richness, entropy, dominance, or diversity in control and CeD-positive cases (mothers whose children later developed CeD), but we did retrieve an augmented phylogenetic distance among OTUs present in CeD samples (p < 0.001) (Figure 1). Regarding beta diversity, weighted and unweighted Unifrac metrics were calculated and a Permanova test was used to identify differences in hMM structure as a function of the different variables considered (see Table 1). As expected, we found that hMM profiles were influenced primarily by mothers’ origin country (unweighted unifrac based Permanova test = 1.53, p = 0.001, weighted unifrac based Permanova test = 3.20, p = 0.001) (Supplementary Figure S2A); however, we found that offspring’s year of birth, referred to henceforth as “offspring birth year” (unweighted unifrac based Permanova test = 1.40, p = 0.001, weighted unifrac based Permanova test = 1.66, p = 0.069) also resembled to shape the hMM (Supplementary Figure S2B). No other covariates expected to theoretically affect hMM, such as maternal BMI and delivery mode, were detected. Applying an exploratory multivariate PCoA, we found that distribution of the PC1 values of weighted unifrac correlates with birth year, suggesting that offspring birth year would be a covariate of hMM, likely influencing its composition. Additionally, we observed a gradient-like distribution of PC1 values across all samples dependent on children’s birth year (Supplementary Figure S2B).
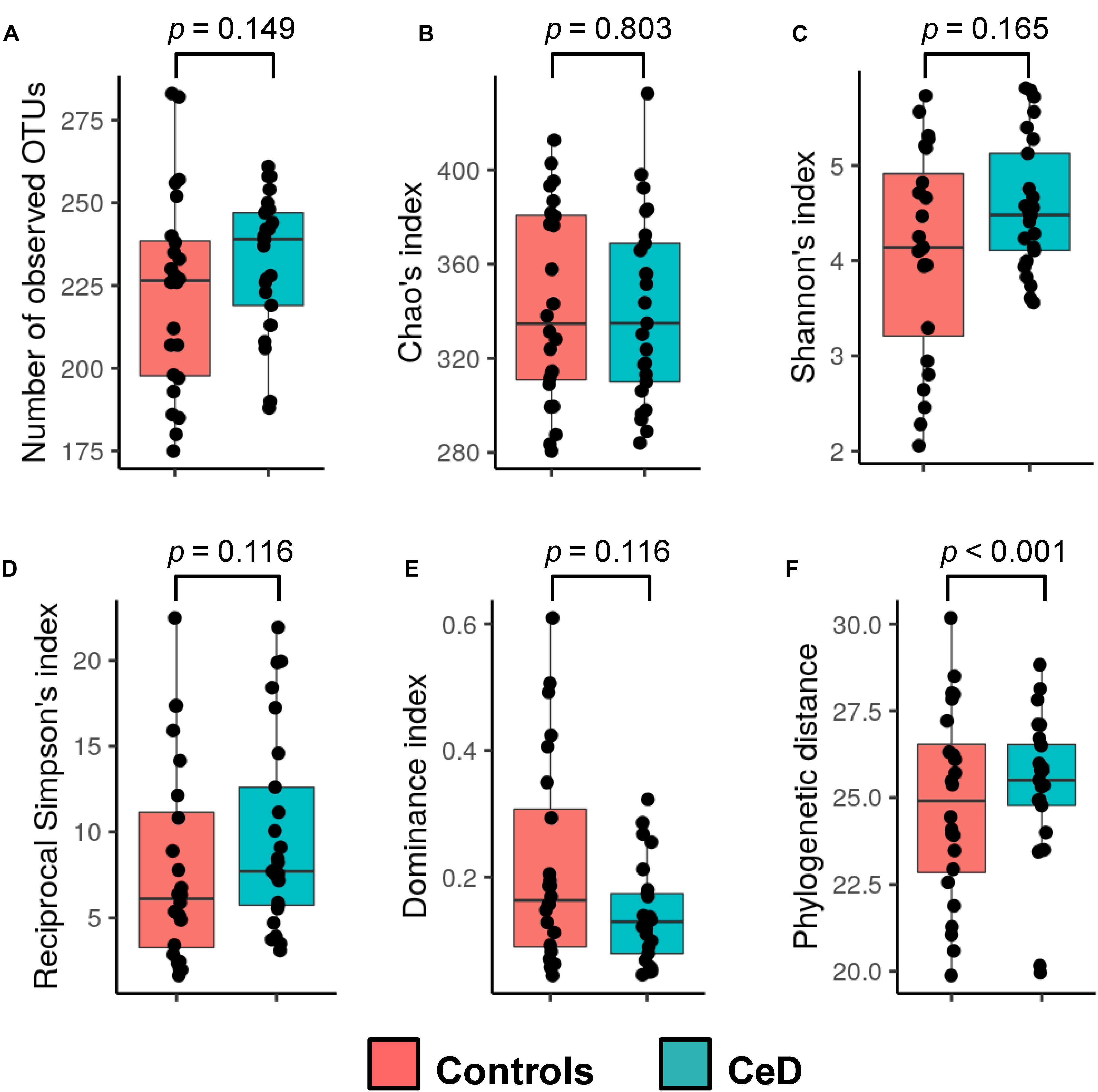
Figure 1. Alpha diversity of the human milk microbiota. The observed richness (A), Chao’s index (B), Shannon’s index (C), reciprocal Simpson’s index (D), dominance index (E), and phylogenetic distance (F) were calculated for samples and compared between the study groups (cases and controls; see legend color). Non-parametric Wilcoxon rank-sum test (unpaired samples) was used to calculate differences among groups. The p-values resulting from the statistical assessment are shown.
To discover the OTU abundance correlated with the PC1 values of weighted unifrac metrics, the Spearman’s rho parameter was calculated between the PC1 values and the OTUs, and the p-values obtained were corrected by multiple testing (FDR ≤ 0.05). As a result, we revealed eighteen OTUs (rho ≥ 0.60, p < 0.001) positively correlated with PC1 values indicating that such phylotypes tended to be more abundant in breast milk from mothers delivering their children during 2007 (with higher PC1 values) and reduced in the breast milk of mothers delivering their offspring later on (2010). This could also imply a reduction in the delivery of these phylotypes into the intestine of infants during breastfeeding throughout the study. When a Blast-based search was performed in order to unveil the bacterial identity of such DNA sequences, we found conclusive taxonomy identification for 13 out of 18 OTUs. In particular, the abundance of the following species appear to be in higher proportions in the breast-milk of mothers involved earlier in this study: Anaerostipes hadrus (OTU41), Alistipes shahii (OTU39), Parabacteroides golsteinii (OTU64), Prevotella copri (OTU7), Faecalibacterium spp. (OTU91), Bacteroides uniformis (OTU27), Bacteroides xylanisolvens (OTU2), Alistipes putredinis (OTU142), Butyrivibrio crossotus (OTU40), Alistipes obesi (OTU10), Parabacteroides johnsonii (OTU135), Bifidobacterium bifidum (OTU32), and Paraprevotella clara (OTU101). Conversely, negative correlations between PC1 values and OTU abundances (rho ≤ 0.56, p < 0.001) of the species Bacillus wiedmannii (OTU33), Enterococcus saigonensis (OTU197), and several species of the Staphylococcus genus (OTU193, OTU331, OTU84, and OTU2) were found. This indicates that the abundance of these bacterial species increased progressively in breast milk throughout the study.
Bacterial Taxonomic Composition of hMM
The inventory of the bacterial phyla and families present in breast milk samples was evaluated by assigning the respective taxonomy categories to DNA reads with the RDP classifier v2.12 as shown in Figure 2A. We found that Firmicutes and Proteobacteria were the most common bacterial phyla present in the entire set of samples followed by Bacteroidetes and Actinobacteria. In terms of family distribution, we observed that members of the Streptococcaceae and Staphylococcaceae families were predominantly present in human milk samples. Proteobacteria was mainly represented by members of Methylobacteriaceae and Comamonadaceae families, whereas Actinobacteria and Bacteroidetes were essentially represented by Propionibacteriaceae and Bacteroidaceae families, respectively. At first glance, there was a high inter-individual variability in the microbial composition of the samples and no abundance patterns could be visually associated with the human milk control and CeD samples. Applying a LDA we found that milk of mothers whose children later developed CeD contained a high proportion of Verrucomicrobia (LDA score = 3.18, p = 0.041) and Beijerinckiaceae species (LDA score = 3.36, p = 0.009) (Figure 2B). We also detected some differences in the abundance of specific OTUs in control mothers and mothers whose infants developed CeD, being OTU13 (Bacteroides vulgatus), OTU42 (Methylobacterium komagatae), OTU63 (Parabacteroides distasonis), and OTU54 (Methylocapsa palsarum) (Table 2).
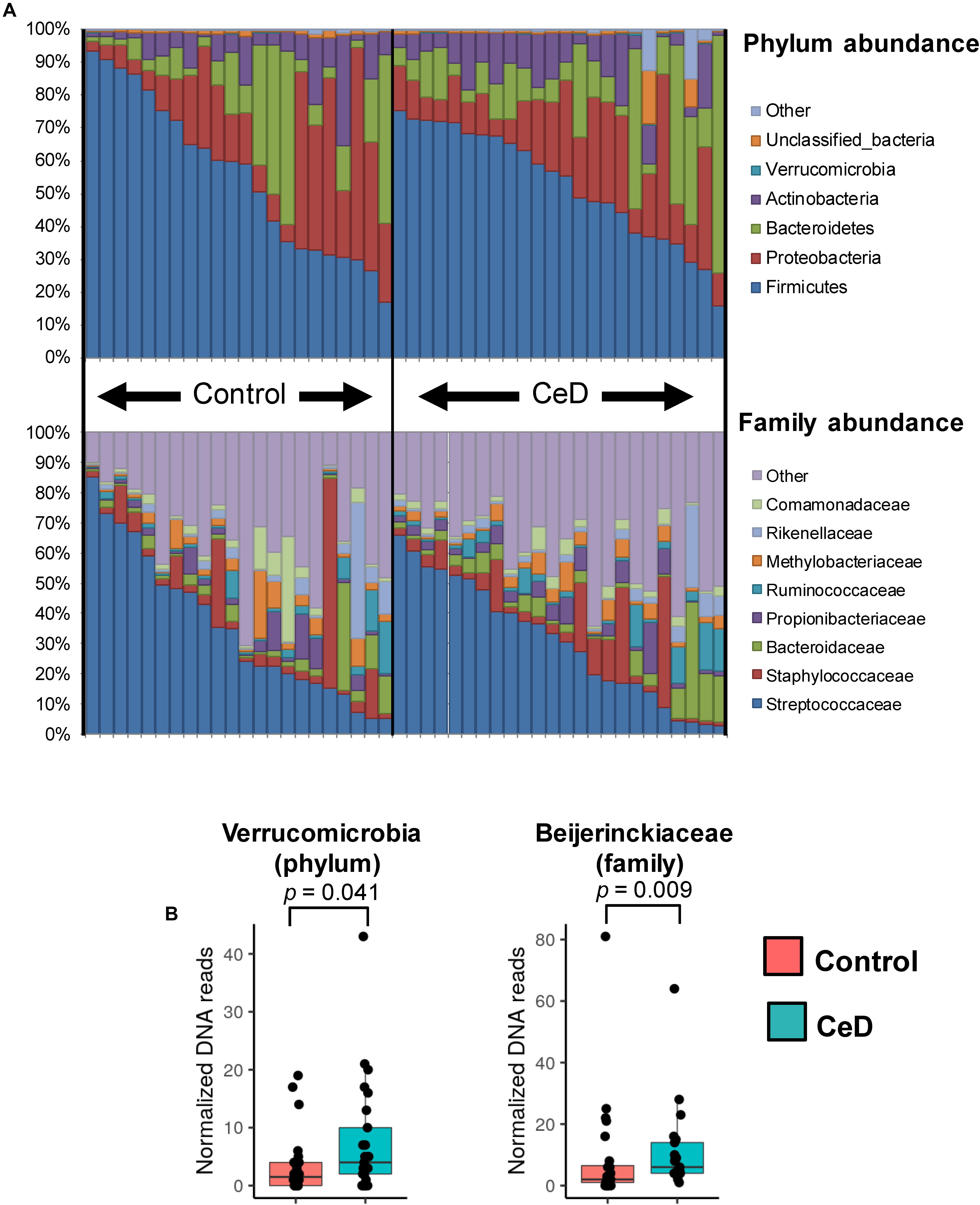
Figure 2. Bacterial phyla and families distribution in human milk microbiota. (A) The phylum (top bar plot) and family (bottom bar plot) distribution in the whole set of samples analyzed show the most predominant members present in different human milk samples. (B) Taxonomic categories with differential abundance in the study groups (cases and controls). The distribution of DNA reads assigned to Verrucomicrobia phylum and Beijerinckiaceae family are presented in boxplots. In both cases the abundance of reads is higher in case samples (CeD developed in the offspring) than in controls (p = 0.041 and p = 0.009, respectively).
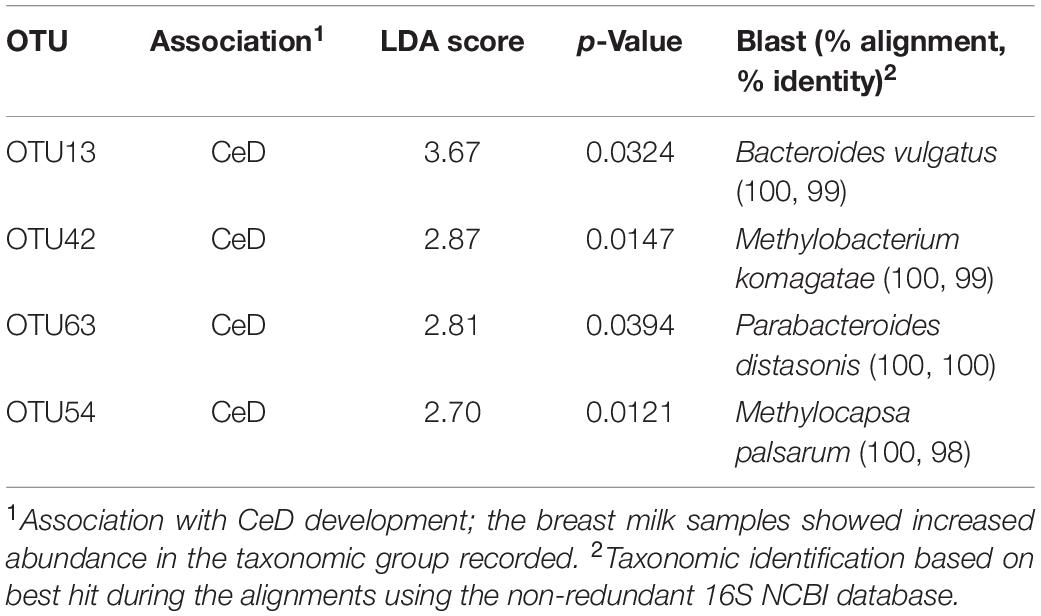
Table 2. OTUs with differential abundance in breast milk samples from mothers whose children later develop celiac disease (CeD) compared to those whose children remained healthy (controls).
Associations Between the hMM and Different Covariates
A LMM analysis was performed to evaluate the possible influence of other clinical covariates on the hMM profiles. As previously detected by beta diversity analysis, this additional data analysis indicated that hMM structure was influenced only by the mothers’ origin country and offspring birth year, explaining approximately 52 and 11% of observed variability, respectively. The above was inferred from the observation that 71 OTU categories (p < 0.05), of the 247 most abundant identified (>0.01% of abundance), appeared to be differentially distributed across the offspring birth period between 2007 and 2010, whereas 93 OTUs appeared with differential abundance among the five origin countries (p < 0.05). The OTU categories and their most probable taxonomic identification with larger negative and positive associations according to offspring birth year are shown in Supplementary Figure S3. Of these, we highlight Pseudomonas spp. (OTU14) and Acinetobacter spp. (OTU22) (both from Pseudomonadales order), whose abundance increased progressively with offspring birth year irrespective of the origin country of samples (set as random effect on LMM). The remaining covariates contributed to explain the hMM profile to a lesser extent. Moreover, we identified 8 OTUs differentially present in the two groups of mothers, i.e., those whose children later developed CeD and those whose children remained healthy, after adjusting for all other covariates (Table 3). When comparing LDA and LMM data, we found four other OTUs associated with the microbiota of mothers whose infants developed CeD and two of them certainly correlated with specific species, including Peptoniphilus timonensis (OTU62) and Actinomyces turicencis (OTU483).
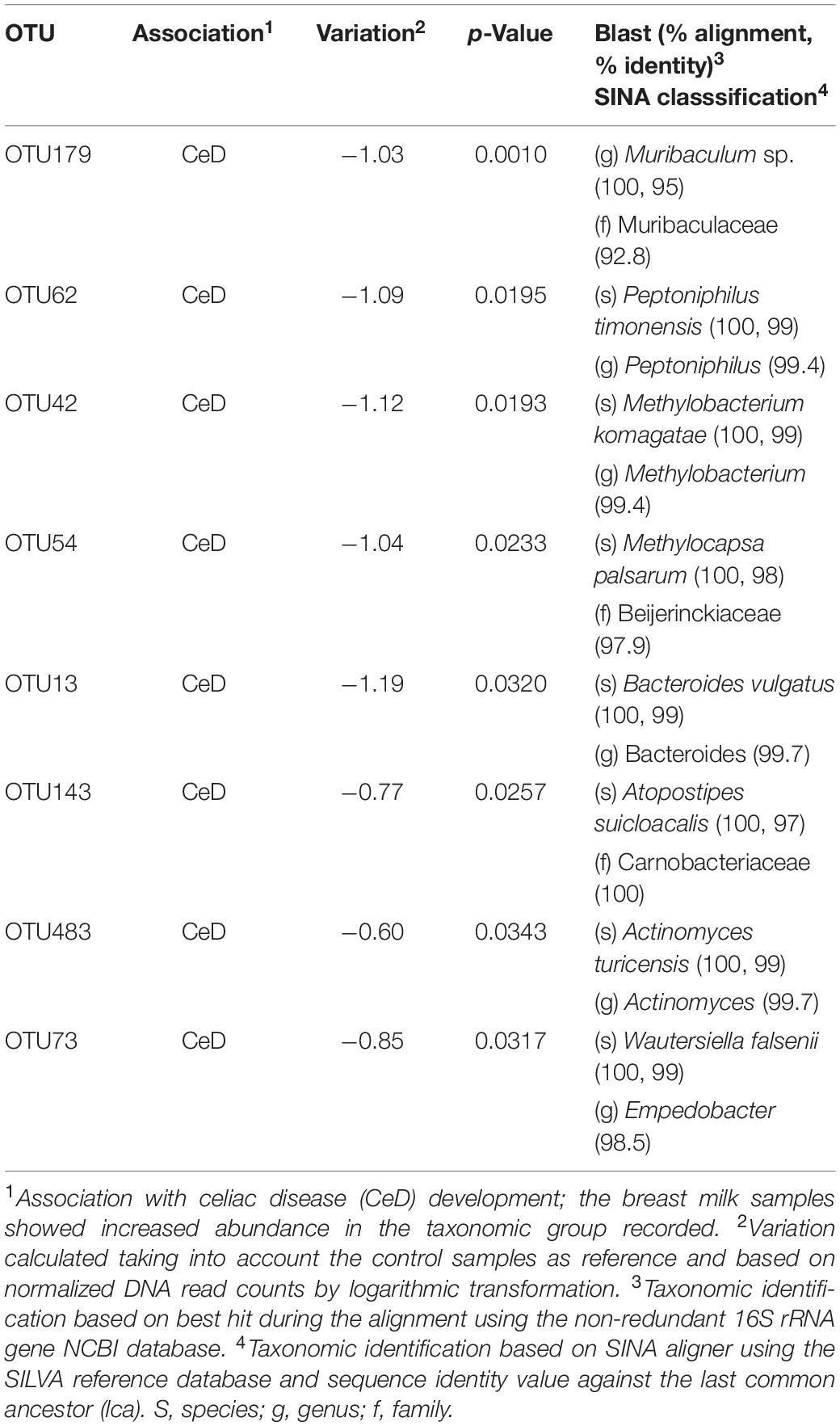
Table 3. OTUs with differential abundance in breast milk samples from mothers whose children later developed celiac disease (CeD) compared to those whose children remained healthy (controls) according to LMM analysis.
Discussion
Many recent hMM surveys report the microbiota profiles potentially involved in health and disease, not only of mothers but also of their offspring (Sakwinska and Bosco, 2019). Controversy exists on the most suitable methodology to accurately analyze the microorganisms present in human milk since this niche has a very low bacterial load. This makes it prone to misidentification of microbial profiles due to cross-contaminations of samples with foreign DNA from different processing stages and reagents used for DNA isolation, PCR amplification and sequencing (Salter et al., 2014). Therefore, the present study aimed to control contaminant DNA from reagents to minimize this source of bias when assessing the microbial signatures of different human milk samples (Sakwinska and Bosco, 2019), and to link specific microbial features with the development of CeD in children.
We found that the microbiota of CeD samples was more diverse than that of controls given the differences reflected in the phylogenetic distance metrics. Moreover, most of the primary variables and factors thought to modulate the hMM (genetic risk, CeD mother condition, mothers’ gluten-free diet, mothers’ age, delivery mode, and mothers’ BMI) exhibited no significant impact on its structure. Conversely, and apart from the well-known geographical influence on human microbiota assessments (Kumar et al., 2016; Deschasaux et al., 2018; He et al., 2018), replicated in this study, we found that hMM was primarily influenced by offspring birth year in our study population. As shown, human milk samples from 2007 to 2008 contained a higher abundance of strictly anaerobic and gut-associated microbes such as Anaerostipes spp., Parabacteroides spp., P. copri, Faecalibacterium spp., Bacteroides spp., Alistipes spp., and Bifidobacterium spp. among others, whereas later samples (2009–2010) were enriched in Bacillus, Enterococcus, and Staphylococcus species. Therefore, the strictly anaerobic bacteria found in the samples first collected seemed to be progressively replaced by aerobic or facultative bacteria in the samples of mothers collected afterward. This could be due to sample storage time and freeze-thaw cycles but, in our study, the samples stored for longer contained a higher abundance of strictly anaerobic bacteria. Therefore, we hypothesize that this shift might be the result of changes in lifestyle and environmental variables, which shaped the assembly of such bacterial communities in mothers’ milk. Moreover, given that abundance of any of the OTUs outlined above was not influenced by the mothers’ origin country, delivery mode, mothers’ BMI or mothers’ disease condition and gluten-free diet followed, the changes associated with the offspring birth year retrieved from this sample cohort of multi-national origin might be suggestive of a short-term switch of hMM signatures across the population. The above idea would be possible because of the lifestyles and environmental factors in urban settlements has been already associated with worsening human health, including human milk quality (Giles-Corti et al., 2016; Pajewska-Szmyt et al., 2019), and theoretically these factors could also negatively impact the breast milk microbiota. Nonetheless, we are conscious that the inspection of a greater sample size across a considerably longer sampling period will be mandatory to test such a hypothesis. Alternatively, we cannot discard that this hMM profile associated with offspring birth year may be a direct consequence of milk expression method for infant feeding, which has been demonstrated to induce meaningful changes in the human milk by reducing diversity and increasing potential pathogens (Moossavi et al., 2019). Despite the fact that our metadata lacks direct information of the milk expression method used, this hypothesis is plausible since there is an increased tendency to use expressed breast milk feeding as routine practice in developed countries, as those included in our study (reviewed by Johns et al., 2013).
Analyses of the taxonomic features present in hMM at the phylum and family level indicate that Streptococcaceae and Staphylococcaceae species are predominant members of the hMM. These data are consistent with previous inventories of microbial species identified in human milk, where the predominance of Proteobacteria and Firmicutes seems to be a common feature (Biagi et al., 2017; Li et al., 2017; Murphy et al., 2017). Moreover, presence of certain bacterial groups associated with skin microbiota (like Propionibacteriaceae species) also appears to be frequent in this type of sample.
We found increased abundance of B. vulgatus in breast-milk samples of mothers whose children developed CeD. This species is capable of efficiently fermenting fucosylated oligosaccharides present in human milk (Marcobal et al., 2010) and, therefore, human milk oligosaccharides could support their prevalence in breast milk samples. Furthermore, B. vulgatus has been previously linked to CeD as infants with a high genetic risk showed an increased prevalence of this genus (Sanchez et al., 2011). Moreover, P. distasonis was also present in higher proportions in such samples. Although there are no previous reports identifying the presence of these common human gut microbes in breast milk, the abundance of B. vulgatus has been associated with irritable bowel disease (IBD) severity (Schirmer et al., 2018), and that of P. distasonis with Crohn’s disease (Lopetuso et al., 2018). This evidence could hint as to the potentially harmful effects of the transmission of these bacterial species from mothers to their infants. Future studies to unveil potential relationships between human milk oligosaccharides and the abundance of microbial species found in hMM associated with CeD children should be warranted. This would provide further insights on the mother-child vertical microbiota transmission since the abundance of some species in fecal microbiota of breast-fed infants seem to correlate to oligosaccharides present in human milk (Wang et al., 2015). Consequently, precise mechanistic studies are necessary to test such hypothetical associations.
Interestingly, we also found environmental-associated microbes to be present in human milk, showing differential abundance between study groups with higher abundance in CeD samples. This included, in particular, Methylobacterium species of the Rhizobiales order (phylum Proteobacteria), which are commonly known as one-carbon compound reducer microorganisms widely found in soil and plant surfaces. Moreover, species of the Methylocapsa genera, members of the Beijerinckiaceae family, formed part of the hMM after minimizing the confounding effects of contaminant DNA coming from reagents. Species belonging to the Methylobacterium genus are prevalent in water pipes and reservoirs (Barbeau et al., 1996; Vaz-Moreira et al., 2017) and as contaminants of industrial milk samples (Bracke et al., 2014). In addition, some studies also indicate that Methylobacterium species are part of the human oral and skin microbiota (Hung et al., 2011; Alekseyenko et al., 2013) and, therefore, their presence in breast milk microbiota could partly be due to their transmission via the infant’s oral cavity, an event that has been recently hypothesized as important modulator of the hMM (Moossavi and Azad, 2019; Moossavi et al., 2019). Interestingly, a recent microbiota analysis of human breast tissue through 16S rRNA gene amplicon sequencing has also reported the presence of Methylobacterium (among other environmental microbes such as Ralstonia) in healthy and cancerous breast biopsies (Costantini et al., 2018), supporting the presence of these type of microbes in human breast-related samples. Although the presence of such methylotrophs in hMM is corroborated by our study, their particular role in CeD onset needs further evaluation. In this regard, recent reports of the ability of Methylobacterium species to utilize organic acids and ethanolamines, and to produce central metabolic compounds should be investigated further to understand their possible functional role in health and disease (Yang et al., 2009).
A LMM-based analysis to control several covariates of the microbiota enabled us to distinguish additional microbial species potentially associated with the development of CeD in children due to its increased abundance in the milk of their respective mothers. As a result and, in addition to those species described in Table 2, we found that the abundance of P. timonensis and Actinomyces turicensis related OTUs were increased in milk samples from mothers whose children developed CeD. In this regard, Peptoniphilus species have been considered as potential inducers of pro-inflammatory signals associated with an increased risk of suffering from human immunodeficiency virus (HIV) infection and wound severity in skin ulcers (Liu et al., 2017; Park et al., 2019), which could also confer higher susceptibility to other inflammatory conditions such as CeD. Moreover, breast abscesses caused by A. turicensis and Peptoniphilus harei also indicate these bacterial taxa are potentially pathogenic (Le Bihan et al., 2019). On the other hand, the LMM analysis also revealed several species potentially associated with offspring birth year, which could play an important role in the colonization of the infant’s gut during breastfeeding. We found that P. copri, a species recurrently associated with traditional lifestyle and non-Westernized populations (Fragiadakis et al., 2019), was more abundant in milk samples of mothers who delivered their children in 2007, regardless their origin’s country, BMI, and mothers’ disease status, and the abundance seemed to decline in samples from mothers who delivered their children in subsequent years. Interestingly, a similar pattern of evolution has been found for the abundance of Prevotella species, which seems to have been reduced in the human gut microbiota, particularly as a result of the transition from traditional to westernized lifestyles (Smits et al., 2017; Fragiadakis et al., 2019). In turn, the abundance of potential pathogenic species such as Acinetobacter and Pseudomonas exhibited the opposite pattern. Interestingly, a recent study has reported that Pseudomonas aeruginosa, an opportunistic pathogen from CeD patients, may have proteolytic activity and could trigger a severe intestinal inflammation synergistically with gluten in experimental CeD models (Caminero et al., 2019). All in all, our study provides new insights into the human milk bacterial profile and its potential role in CeD onset in children in a prospective study for the first time. This shows it could serve as a vehicle for vertical transmission of protective or harmful bacteria, whose pathogenic role should be investigated further.
Limitations of our study include the low sample size, the sample heterogeneity (country origin) and the lack of records of relevant clinical data recently demonstrated to impact the hMM such as milk expression method (Moossavi et al., 2019). Moreover, milk sampling for analysis (9 months after birth) could have been completed at an earlier point, given that infants’ gut microbiota modulation by breastfeeding also starts earlier. However, this analysis could only be done at 9 months because: earlier samples were processed for macronutrient and endocrine profiling (Grunewald et al., 2019), all available milk sample from mothers included in this pilot study were uniform at this time point, and because all were still breastfeeding their children, together with the introduction of complementary food. Accordingly, we assumed that hMM would still act as a modulator of the infants’ gut microbiota. In spite of the limitations, this pilot study holds promise to identify new factors that could affect hMM and impact on CeD onset in larger cohorts and, thus, provide a more comprehensive understanding of moderators and mediators of the disease etiology. Such a robust design would also be necessary to clarify if the hMM profiles could be informative to trace short-term lifestyles changes in Western societies.
Data Availability Statement
The datasets generated for this study can be found in the MG-RAST server (Meyer et al., 2008), accession number mgp88214.
Ethics Statement
The studies involving human participants were reviewed and approved by the Medical University of Warsaw, Hospital La Paz, Institut d’Investigació Sanitària Pere Virgili, Hospital La Fe, Heim Pál Children’s Hospital, Dr. von Hauner Children’s Hospital, University of Warmia and Mazury, and Leiden University Medical Center ethics committees. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MM and YS designed and coordinated the study. HS, MP-L, IP, GC, MN, CR-K, IK-S, SK, and CM recruited participants in their respective clinical centers and recorded samples-associated metadata. AB-P and MO processed human milk samples and performed amplicon sequencing analyses. AB-P executed massive DNA data analysis. AB-P, MO, MM, and YS drafted the manuscript. All authors contributed to the critical review of the manuscript and approved the final version submitted to this journal.
Funding
This work was supported by grant AGL2017-88801-P from Ministerio de Ciencia, Innovación y Universidades (MCIU, Spain). The postdoctoral contract of Marta Olivares from “Juan de la Cierva” program (MCIU, Spain) is fully acknowledged. PreventCD consortium is supported by grants from the European Commission (FP6-2005-FOOD-4B-36383–PREVENTCD), the Azrieli Foundation, Deutsche Zöliakie Gesellschaft, Eurospital, Fondazione Celiachia, Fria Bröd, Instituto de Salud Carlos III, Spanish Society for Pediatric Gastroenterology, Hepatology, and Nutrition, Komitet Badañ Naukowych (1715/B/P01/2008/34), Fundacja Nutricia (1W44/FNUT3/2013), Hungarian Scientific Research Funds (OTKA101788 and TAMOP 2.2.11/1/KONV-20 12-0023), Stichting Coeliakie Onderzoek Nederland (STICOON), Thermo Fisher Scientific, and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01335/full#supplementary-material
FIGURE S1 | Principal Coordinate Analysis (PCoA) of samples and negative controls. Bray–Curtis dissimilarity index and distance between samples and negative controls was evaluated with OTU abundance information through multidimensional analysis. Controls (turquoise filled circles), cases/celiac disease (CeD, red filled circles), and negative controls (black filled circles) are represented in this scatter plot, containing information of two main principal coordinates explaining the largest variation among samples.
FIGURE S2 | Beta diversity analysis of the human milk microbiota. Multivariate analysis (Principal Coordinate Analysis –PCoA) of OTUs associated with the origin country (A) and offspring birth year (B). Distribution of PC1 and PC2 values as a function of the both covariates are shown upon unweighted and weighted unifrac metrics, respectively. Permanova test and p-values are shown accordingly. Color legend is shown below respective scatter plots.
FIGURE S3 | OTUs most significantly associated with offspring birth year. The OTUs showing the extreme variation between groups (p < 0.05) according to LMM analysis were taxonomically identified using the SINA aligner (Pruesse et al., 2012) and SILVA database (Quast et al., 2013). When taxonomic assignment at genus level reached ≥99% sequence identity, identification at the species level using a BlastN-based search against the NCBI 16S rRNA gene non-redundant database (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was completed. The distribution of offspring delivery dates between 2007 and 2010 is presented in the plot at the bottom. The OTUs negatively associated (yearly time-course decrease, in light blue) and positively associated (yearly time-course increase, light red), together with respective taxonomic identification are shown at the top. The association slope is directly correlated to the inter-group differences detected by LMM analysis and the respective p-values are shown within square brackets. The abundance (Log) in terms of log10 normalized DNA reads counts and prevalence (Prev) discriminated per birth year subgroups are disclosed, respectively.
Footnotes
References
Alekseyenko, A. V., Perez-Perez, G. I., De Souza, A., Strober, B., Gao, Z., Bihan, M., et al. (2013). Community differentiation of the cutaneous microbiota in psoriasis. Microbiome 1:31.
Auricchio, S., Follo, D., De Ritis, G., Giunta, A., Marzorati, D., Prampolini, L., et al. (1983). Does breast feeding protect against the development of clinical symptoms of celiac disease in children? J. Pediatr. Gastroenterol. Nutr. 2, 428–433. doi: 10.1097/00005176-198302030-00006
Barbeau, J., Tanguay, R., Faucher, E., Avezard, C., Trudel, L., Cote, L., et al. (1996). Multiparametric analysis of waterline contamination in dental units. Appl. Environ. Microbiol. 62, 3954–3959. doi: 10.1128/aem.62.11.3954-3959.1996
Biagi, E., Quercia, S., Aceti, A., Beghetti, I., Rampelli, S., Turroni, S., et al. (2017). The bacterial ecosystem of mother’s milk and infant’s mouth and gut. Front. Microbiol. 8:1214. doi: 10.3389/fmicb.2017.01214
Bracke, N., Van Poucke, M., Baert, B., Wynendaele, E., De Bels, L., Den Broeck, W. V., et al. (2014). Identification of a microscopically selected microorganism in milk samples. J. Dairy Sci. 97, 609–615. doi: 10.3168/jds.2013-6932
Caminero, A., Mccarville, J. L., Galipeau, H. J., Deraison, C., Bernier, S. P., Constante, M., et al. (2019). Duodenal bacterial proteolytic activity determines sensitivity to dietary antigen through protease-activated receptor-2. Nat. Commun. 10:1198.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336.
Costantini, L., Magno, S., Albanese, D., Donati, C., Molinari, R., Filippone, A., et al. (2018). Characterization of human breast tissue microbiota from core needle biopsies through the analysis of multi hypervariable 16S-rRNA gene regions. Sci. Rep. 8:16893.
Demmelmair, H., Jimenez, E., Collado, M. C., Salminen, S., and Mcguire, M. K. (2020). Maternal and perinatal factors associated with the human milk microbiome. Curr. Dev. Nutr. 4:nzaa027.
Deschasaux, M., Bouter, K. E., Prodan, A., Levin, E., Groen, A. K., Herrema, H., et al. (2018). Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 24, 1526–1531. doi: 10.1038/s41591-018-0160-1
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fragiadakis, G. K., Smits, S. A., Sonnenburg, E. D., Van Treuren, W., Reid, G., Knight, R., et al. (2019). Links between environment, diet, and the hunter-gatherer microbiome. Gut Microbes 10, 216–227. doi: 10.1080/19490976.2018.1494103
Galipeau, H. J., Mccarville, J. L., Huebener, S., Litwin, O., Meisel, M., Jabri, B., et al. (2015). Intestinal microbiota modulates gluten-induced immunopathology in humanized mice. Am. J. Pathol. 185, 2969–2982. doi: 10.1016/j.ajpath.2015.07.018
Giles-Corti, B., Vernez-Moudon, A., Reis, R., Turrell, G., Dannenberg, A. L., Badland, H., et al. (2016). City planning and population health: a global challenge. Lancet 388, 2912–2924.
Greco, L., Auricchio, S., Mayer, M., and Grimaldi, M. (1988). Case control study on nutritional risk factors in celiac disease. J. Pediatr. Gastroenterol. Nutr. 7, 395–399. doi: 10.1097/00005176-198805000-00013
Grunewald, M., Hellmuth, C., Kirchberg, F. F., Mearin, M. L., Auricchio, R., Castillejo, G., et al. (2019). Variation and interdependencies of human milk macronutrients, fatty acids, adiponectin, insulin, and IGF-II in the European PreventCD Cohort. Nutrients 11:2034. doi: 10.3390/nu11092034
Hansen, L. B. S., Roager, H. M., Sondertoft, N. B., Gobel, R. J., Kristensen, M., Valles-Colomer, M., et al. (2018). A low-gluten diet induces changes in the intestinal microbiome of healthy danish adults. Nat. Commun. 9:4630.
He, Y., Wu, W., Zheng, H. M., Li, P., Mcdonald, D., Sheng, H. F., et al. (2018). Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 24, 1532–1535. doi: 10.1038/s41591-018-0164-x
Hung, W. L., Wade, W. G., Boden, R., Kelly, D. P., and Wood, A. P. (2011). Facultative methylotrophs from the human oral cavity and methylotrophy in strains of Gordonia, Leifsonia, and Microbacterium. Arch. Microbiol. 193, 407–417. doi: 10.1007/s00203-011-0689-6
Husby, S., Koletzko, S., Korponay-Szabo, I. R., Mearin, M. L., Phillips, A., Shamir, R., et al. (2012). European society for pediatric gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J. Pediatr. Gastroenterol. Nutr. 54, 136–160. doi: 10.1097/mpg.0b013e31821a23d0
Johns, H. M., Forster, D. A., Amir, L. H., and Mclachlan, H. L. (2013). Prevalence and outcomes of breast milk expressing in women with healthy term infants: a systematic review. BMC Pregnancy Childbirth 13:212. doi: 10.1186/1471-2393-13-212
Kemppainen, K. M., Lynch, K. F., Liu, E., Lonnrot, M., Simell, V., Briese, T., et al. (2017). Factors that increase risk of celiac disease autoimmunity after a gastrointestinal infection in early life. Clin. Gastroenterol. Hepatol. 15, 694.e5–702.e5.
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2012). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Kumar, H., Du Toit, E., Kulkarni, A., Aakko, J., Linderborg, K. M., Zhang, Y., et al. (2016). Distinct patterns in human milk microbiota and fatty acid profiles across specific geographic locations. Front. Microbiol. 7:1619. doi: 10.3389/fmicb.2016.01619
Le Bihan, A., Ahmed, F., and O’driscoll, J. (2019). An uncommon cause for a breast abscess: actinomyces turicensis with Peptoniphilus harei. BMJ Case Rep. 12:e231194. doi: 10.1136/bcr-2019-231194
Li, S. W., Watanabe, K., Hsu, C. C., Chao, S. H., Yang, Z. H., Lin, Y. J., et al. (2017). Bacterial composition and diversity in breast milk samples from mothers living in taiwan and Mainland China. Front. Microbiol. 8:965. doi: 10.3389/fmicb.2017.00965
Liu, C. M., Prodger, J. L., Tobian, A. A. R., Abraham, A. G., Kigozi, G., Hungate, B. A., et al. (2017). Penile anaerobic dysbiosis as a risk factor for HIV infection. MBio 8e00996-17. doi: 10.1128/mBio.00996-17
Lopetuso, L. R., Petito, V., Graziani, C., Schiavoni, E., Paroni Sterbini, F., Poscia, A., et al. (2018). Gut microbiota in health, diverticular disease, irritable bowel syndrome, and inflammatory bowel diseases: time for microbial marker of gastrointestinal disorders. Dig Dis. 36, 56–65. doi: 10.1159/000477205
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Marcobal, A., Barboza, M., Froehlich, J. W., Block, D. E., German, J. B., Lebrilla, C. B., et al. (2010). Consumption of human milk oligosaccharides by gut-related microbes. J. Agric. Food Chem. 58, 5334–5340. doi: 10.1021/jf9044205
Meyer, F., Paarmann, D., D’souza, M., Olson, R., Glass, E. M., Kubal, M., et al. (2008). The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386
Moossavi, S., and Azad, M. B. (2019). Origins of human milk microbiota: new evidence and arising questions. Gut Microbes 4, 1–10. doi: 10.1080/19490976.2019.1667722
Moossavi, S., Sepehri, S., Robertson, B., Bode, L., Goruk, S., Field, C. J., et al. (2019). Composition and variation of the human milk microbiota are influenced by maternal and early-life factors. Cell Host Microbe 25, 324–335e324.
Murphy, K., Curley, D., O’callaghan, T. F., O’shea, C. A., Dempsey, E. M., O’toole, P. W., et al. (2017). The composition of human milk and infant faecal microbiota over the first three months of life: a pilot study. Sci. Rep. 7:40597.
Olivares, M., Albrecht, S., De Palma, G., Ferrer, M. D., Castillejo, G., Schols, H. A., et al. (2015a). Human milk composition differs in healthy mothers and mothers with celiac disease. Eur. J. Nutr. 54, 119–128. doi: 10.1007/s00394-014-0692-1
Olivares, M., Neef, A., Castillejo, G., Palma, G. D., Varea, V., Capilla, A., et al. (2015b). The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut 64, 406–417. doi: 10.1136/gutjnl-2014-306931
Olivares, M., Benitez-Paez, A., De Palma, G., Capilla, A., Nova, E., Castillejo, G., et al. (2018a). Increased prevalence of pathogenic bacteria in the gut microbiota of infants at risk of developing celiac disease: The PROFICEL study. Gut Microbes 9, 551–558.
Olivares, M., Walker, A. W., Capilla, A., Benitez-Paez, A., Palau, F., Parkhill, J., et al. (2018b). Gut microbiota trajectory in early life may predict development of celiac disease. Microbiome 6:36.
Pajewska-Szmyt, M., Sinkiewicz-Darol, E., and Gadzala-Kopciuch, R. (2019). The impact of environmental pollution on the quality of mother’s milk. Environ. Sci. Pollut. Res. Int. 26, 7405–7427. doi: 10.1007/s11356-019-04141-1
Palma, G. D., Capilla, A., Nova, E., Castillejo, G., Varea, V., Pozo, T., et al. (2012). Influence of milk-feeding type and genetic risk of developing coeliac disease on intestinal microbiota of infants: the PROFICEL study. PLoS One 7:e30791. doi: 10.1371/journal.pone.0030791
Park, J. U., Oh, B., Lee, J. P., Choi, M. H., Lee, M. J., and Kim, B. S. (2019). Influence of microbiota on diabetic foot wound in comparison with adjacent normal skin based on the clinical features. Biomed. Res. Int. 2019:7459236.
Pruesse, E., Peplies, J., and Glockner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596.
Roca, M., Vriezinga, S. L., Crespo-Escobar, P., Auricchio, R., Hervas, D., Castillejo, G., et al. (2018). Anti-gliadin antibodies in breast milk from celiac mothers on a gluten-free diet. Eur. J. Nutr. 57, 1947–1955. doi: 10.1007/s00394-017-1476-1
Sakwinska, O., and Bosco, N. (2019). Host microbe interactions in the lactating mammary gland. Front. Microbiol. 10:1863. doi: 10.3389/fmicb.2019.01863
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Sanchez, E., De Palma, G., Capilla, A., Nova, E., Pozo, T., Castillejo, G., et al. (2011). Influence of environmental and genetic factors linked to celiac disease risk on infant gut colonization by Bacteroides species. Appl. Environ. Microbiol. 77, 5316–5323. doi: 10.1128/aem.00365-11
Sanz, Y. (2010). Effects of a gluten-free diet on gut microbiota and immune function in healthy adult humans. Gut Microbes 1, 135–137. doi: 10.4161/gmic.1.3.11868
Schirmer, M., Franzosa, E. A., Lloyd-Price, J., Mciver, L. J., Schwager, R., Poon, T. W., et al. (2018). Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 3, 337–346.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60.
Silano, M., Agostoni, C., Sanz, Y., and Guandalini, S. (2016). Infant feeding and risk of developing celiac disease: a systematic review. BMJ Open 6:e009163. doi: 10.1136/bmjopen-2015-009163
Smits, S. A., Leach, J., Sonnenburg, E. D., Gonzalez, C. G., Lichtman, J. S., Reid, G., et al. (2017). Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 357, 802–806. doi: 10.1126/science.aan4834
Stevens, F. M., Egan-Mitchell, B., Cryan, E., Mccarthy, C. F., and Mcnicholl, B. (1987). Decreasing incidence of coeliac disease. Arch. Dis. Child. 62, 465–468. doi: 10.1136/adc.62.5.465
Szajewska, H., Shamir, R., Chmielewska, A., Piescik-Lech, M., Auricchio, R., Ivarsson, A., et al. (2015). Systematic review with meta-analysis: early infant feeding and coeliac disease–update 2015. Aliment Pharmacol. Ther. 41, 1038–1054. doi: 10.1111/apt.13163
Vaz-Moreira, I., Nunes, O. C., and Manaia, C. M. (2017). Ubiquitous and persistent Proteobacteria and other Gram-negative bacteria in drinking water. Sci. Total Environ. 586, 1141–1149. doi: 10.1016/j.scitotenv.2017.02.104
Vriezinga, S. L., Auricchio, R., Bravi, E., Castillejo, G., Chmielewska, A., Crespo Escobar, P., et al. (2014). Randomized feeding intervention in infants at high risk for celiac disease. N. Engl. J. Med. 371, 1304–1315.
Wang, M., Li, M., Wu, S., Lebrilla, C. B., Chapkin, R. S., Ivanov, I., et al. (2015). Fecal microbiota composition of breast-fed infants is correlated with human milk oligosaccharides consumed. J. Pediatr. Gastroenterol. Nutr. 60, 825–833. doi: 10.1097/mpg.0000000000000752
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-07
Yang, S., Sadilek, M., Synovec, R. E., and Lidstrom, M. E. (2009). Liquid chromatography-tandem quadrupole mass spectrometry and comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry measurement of targeted metabolites of Methylobacterium extorquens AM1 grown on two different carbon sources. J. Chromatogr. A 1216, 3280–3289. doi: 10.1016/j.chroma.2009.02.030
Keywords: celiac disease, children, mothers, human milk microbiota, HLA genotype
Citation: Benítez-Páez A, Olivares M, Szajewska H, Pieścik-Lech M, Polanco I, Castillejo G, Nuñez M, Ribes-Koninckx C, Korponay-Szabó IR, Koletzko S, Meijer CR, Mearin ML and Sanz Y (2020) Breast-Milk Microbiota Linked to Celiac Disease Development in Children: A Pilot Study From the PreventCD Cohort. Front. Microbiol. 11:1335. doi: 10.3389/fmicb.2020.01335
Received: 21 February 2020; Accepted: 25 May 2020;
Published: 23 June 2020.
Edited by:
Francesca Bottacini, University College Cork, IrelandReviewed by:
Kieran James, University College Cork, IrelandShirin Moossavi, University of Manitoba, Canada
Copyright © 2020 Benítez-Páez, Olivares, Szajewska, Pieścik-Lech, Polanco, Castillejo, Nuñez, Ribes-Koninckx, Korponay-Szabó, Koletzko, Meijer, Mearin and Sanz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alfonso Benítez-Páez, YWJlbml0ZXpAaWF0YS5jc2ljLmVz; YWJlbml0ZXpAY2lwZi5lcw==