 Congjuan Xu1
Congjuan Xu1 Dan Wang1Xinxin Zhang1Huimin Liu2Guangbo Zhu2Tong Wang3
Dan Wang1Xinxin Zhang1Huimin Liu2Guangbo Zhu2Tong Wang3 Zhihui Cheng1
Zhihui Cheng1 Weihui Wu1
Weihui Wu1 Fang Bai1*
Fang Bai1* Yongxin Jin1*
Yongxin Jin1*- 1State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Molecular Microbiology and Technology of the Ministry of Education, Department of Microbiology, College of Life Sciences, Nankai University, Tianjin, China
- 2Tianjin Union Medical Center, Nankai University Affiliated Hospital, Tianjin, China
- 3Department of Stomatology, Tianjin First Central Hospital, Tianjin, China
Infections by Pseudomonas aeruginosa are difficult to cure due to its high intrinsic and acquired antibiotic resistance. Once colonized the human host, and thanks to antibiotic treatment pressure, P. aeruginosa usually acquires genetic mutations which provide bacteria with antibiotic resistance as well as ability to better adapt to the host environment. Deciphering the evolutionary traits may provide important insights into the development of effective combinatory antibiotic therapy to treat P. aeruginosa infections. In this study, we investigated the molecular mechanisms by which a clinical isolate (ISP50) yields a carbapenem-resistant derivative (IRP41). RNAseq and genomic DNA reference mapping were conducted to compare the transcriptional profiles and in vivo evolutionary trajectories between the two isolates. Our results demonstrated that oprD mutation together with ampC hyper-expression contributed to the increased resistance to carbapenem in the isolate IRP41. Furthermore, a ldcA (PA5198) gene, encoding murein tetrapeptide carboxypeptidase, has been demonstrated for the first time to negatively influence the ampC expression in P. aeruginosa.
Introduction
Pseudomonas aeruginosa, as an opportunistic human pathogen, is one of the leading causes of nosocomial infections worldwide (Vincent et al., 1995). Infections by P. aeruginosa are difficult to treat due to its intrinsic and acquired resistance to a wide range of antibiotics, leaving limited number of effective antimicrobial agents. Carbapenems are used in clinical practice to treat P. aeruginosa infections. However, carbapenem resistance of clinical P. aeruginosa isolates has been increasingly reported (Davies et al., 2011). The mechanisms of carbapenem resistance are usually multifactorial which include: (i) acquisition of carbapenemase encoding genes through horizontal gene transfer (Poole, 2011; Potron et al., 2015), (ii) deficiency or repression of the porin (OprD) for carbapenem (Davies et al., 2011; Poole, 2011), (iii) overexpression of mexAB-oprM efflux pump (Poole, 2011; Liu et al., 2013; Choudhury et al., 2015), and (iv) overexpression of the chromosomal gene (ampC) encoding the P. aeruginosa intrinsic cephalosporinase (Poole, 2011; Mirsalehian et al., 2014). Although these and other studies have described the associated mechanisms of carbapenem resistance among clinical isolates of P. aeruginosa, there is little information on the detailed molecular mechanisms leading to the evolutionary dynamics of clinical P. aeruginosa isolates from carbapenems susceptibility to resistance and the impact of each of these resistance mechanisms.
In this study, we obtained two P. aeruginosa clinical isolates, later demonstrated to belong to the same clone, from sputum samples of the same patient with acute exacerbation of chronic bronchitis before and after treatment with biapenem. The first strain was obtained soon after the patient was admitted to the hospital while the second strain was obtained 4 days after the antibiotic treatment. The first isolate ISP50 was carbapenem susceptible, whereas the second one IRP41 was carbapenem resistant. Therefore, our goal was to decipher the molecular mechanisms by which the carbapenem resistance had been evolved so rapidly in the clinical setting. Our experimental results demonstrated that an oprD null mutation combined with an elevated ampC expression are the major contributory factors for the conversion. Furthermore, we have shown for the first time that LdcA functions as a repressor on the expression of ampC in P. aeruginosa. These findings provide novel insights into the regulatory mechanism of ampC expression in P. aeruginosa.
Materials and Methods
Basic Characterization of the Bacterial Strains
Bacterial strains and plasmids used in the study are listed in Table 1. Carbapenem-susceptible (ISP50) and resistant (IRP41) P. aeruginosa isolates characterized in this study were obtained from sputum samples of the same patient with acute exacerbation of chronic bronchitis before and after treatment with biapenem for 4 days (dosage at 0.3 g × 2/day) at the Nankai University Affiliated Hospital, Tianjin, China. The 16S rRNA encoding gene was amplified (primers listed in Supplementary Table S1) and sequenced to identify the species of these two isolates (Spilker et al., 2004). Random amplified polymorphic DNA (RAPD) typing was carried out using primer 272 as described previously (Mahenthiralingam et al., 1996). Antibiotic susceptibility test was conducted using the disk diffusion method according to the manufacturer’s recommendations, and minimum inhibitory concentration (MIC) of antibiotics was determined by the two-fold serial dilution method. Susceptibility was interpreted according to the Clinical and Laboratory Standards Institute guidelines (CLSI 2011–2018).
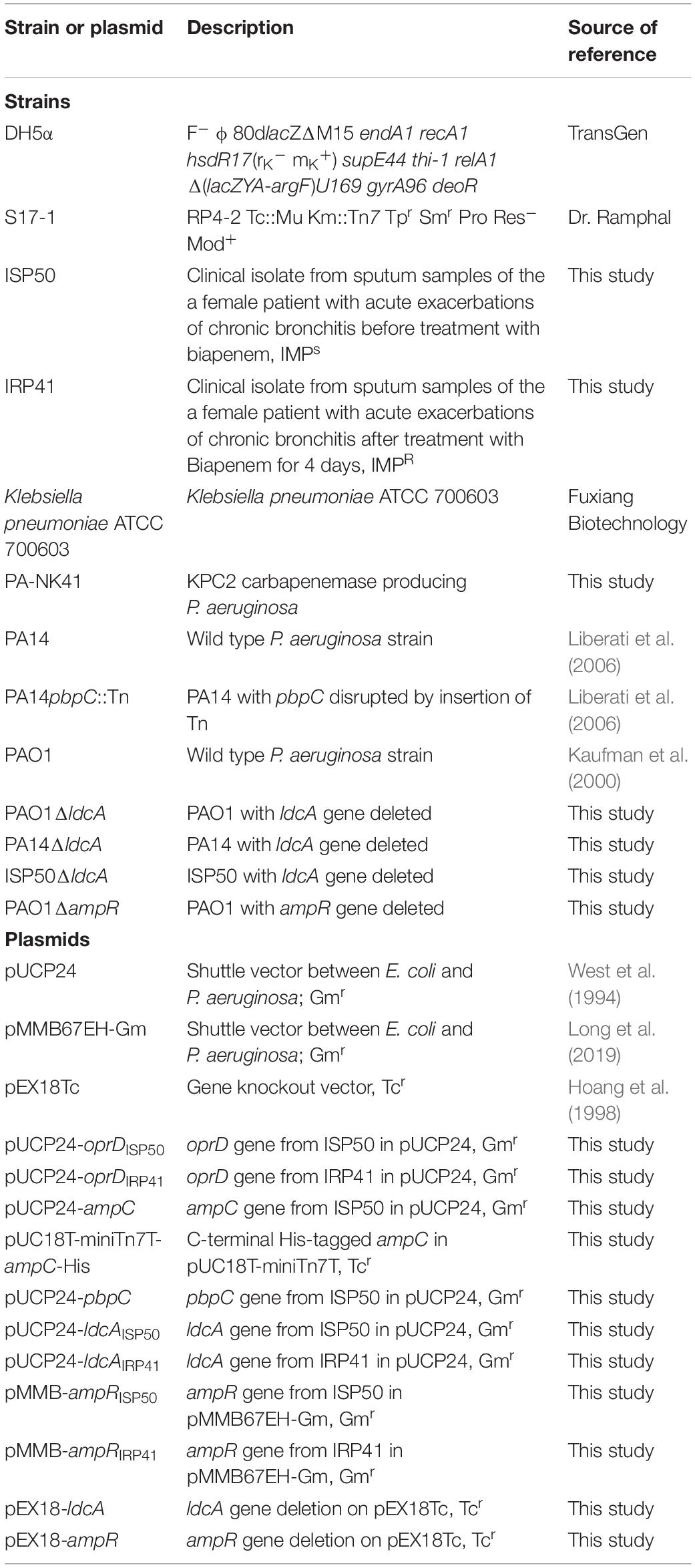
Table 1. Bacterial strains and plasmids used in this study.
Plasmid Construction
For overexpression of oprD, a 1,802 bp oprD-containing fragment with its putative Shine-Dalgarno (SD) sequence was PCR amplified using ISP50 and IRP41 genomic DNA as templates (the used primers are displayed in Supplementary Table S1). The PCR products were digested with BamHI and HindIII, and then ligated into a shuttle vector pUCP24, resulting in pUCP24-oprDISP50 and pUCP24-oprDIRP41, respectively. Constructs of pUCP24-ampC, pMMB-ampRISP50, pMMB-ampRIRP41, pUCP24-ldcAISP50, pUCP24-ldcAIRP41 and pUCP24-pbpC were all generated using similar procedures.
For deletion of the ldcA gene, a 836 bp fragment immediately upstream of the ldcA start codon and a 913 bp fragment downstream of the ldcA stop codon were PCR amplified, digested with EcoRI-BamHI and BamHI-HindIII, respectively. The two fragments were then ligated into pEX18Tc that was digested with EcoRI and HindIII, resulting in pEX18-ldcA. A pEX18-ampR was constructed by similar manipulations. Gene knockouts in P. aeruginosa were carried out by conjugal transfer of these plasmids followed by selection for single crossovers and then double crossovers as previously described (Schweizer, 1992).
Multilocus Sequence Typing
Multilocus sequence typing (MLST) was performed following a previous description (Curran et al., 2004) with minor modifications to confirm the allelic profiles of the two isolates. Briefly, genomic DNA was isolated from overnight bacterial culture with a DNA purification kit (Tiangen Biotech, Beijing, China) for use as PCR template. The internal fragments of seven housekeeping genes (acsA, aroE, guaA, mutL, nuoD, ppsA and trpE) were amplified by PCR and sequenced using primers described previously (Supplementary Table S1) (Curran et al., 2004). Gene sequences were then submitted to the P. aeruginosa MLST database1 for assignment of allelic numbers. A sequence type (ST) was assigned to each isolate by combining the seven allelic numbers.
PAE-MHT Assay for Carbapenemase Test
The PAE-MHT assay was performed as described previously (Pasteran et al., 2011) with minor modifications. Briefly, an inoculum of Klebsiella pneumoniae ATCC 700603 was adjusted to a 0.5 McFarland turbidity standard (Mcfarland, 1907) followed by 10-fold dilution with sterile saline, and then inoculated onto the surfaces of Mueller-Hinton agar (Oxoid) plates (diameter, 10 cm) by swabbing. The plates were allowed to stand at room temperature. After 10 min, a disk of 10 μg imipenem/meropenem (Oxoid, United Kingdom) was placed in the center of each plate. Subsequently, one colony of each P. aeruginosa strain, grown overnight on LB agar plate, was inoculated onto the plate in a straight line from the edge of the disk to the periphery of the plate using a 1-μl loop (BOOPU Biological Technology, Changzhou, China). Presence of growth of the K. pneumoniae ATCC 700603 toward imipenem/meropenem disk was interpreted as carbapenemase positive.
Total RNA Isolation, RT-qPCR and RNAseq Analysis
Bacterial overnight culture was inoculated into fresh LB medium (1:50 dilution) and grown to an optical density of 1.0 (wavelength of 600 nm). Total RNA was extracted using an RNAprep Pure Cell/Bacteria Kit (Tiangen Biotech, Beijing, China) and reverse transcribed to cDNA with random primers and PrimeScript Reverse Transcriptase (Takara). The cDNA was mixed with indicated primers (Supplementary Table S1) and SYBR premix Ex Taq II (Takara), and then quantitatively amplified in a CFX Connect Real-Time system (Bio-Rad, United States). A 30S ribosomal protein encoding gene rpsL was used as an internal control.
For RNAseq, total RNA was extracted as described above, quantified and qualified by Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States), NanoDrop (Thermo Fisher Scientific Inc.) and 1% agarose gel. Then, the rRNA was depleted using Ribo-Zero rRNA Removal Kit (Bacteria, Illumina). The mRNA was then fragmented and reverse-transcribed. The double-strand cDNA was purified, ends repaired and ligated with adaptors. After 11 cycles of PCR amplification, the PCR products were cleaned, validated with an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States), and quantified by Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, United States). The resulting libraries were subjected to sequencing using an Illumina HiSeq 2500 platform with a 2 × 150 paired-end configuration.
Sequence reads were mapped onto PAO1 reference genome (NC_002516.2) via software Bowtie2 (v2.1.0). Gene expression levels were analyzed with HTSeq (v0.6.1p1). Differentially expressed genes were identified by the DESeq Bioconductor package, with the fold-change larger than 2 and P-value no more than 0.05 as cutoff values.
DNA Isolation and Reference Mapping
Bacterial genomic DNA was extracted with DNA purification kit (Tiangen Biotech, Beijing, China). Fragments smaller than 500 bp were obtained from 200 ng genomic DNA by sonication (Covaris S220), followed by end treatment and adaptors ligation. Adaptor-ligated DNA fragments of about 470 bp were recovered using beads and then PCR amplified for six cycles, and the PCR products were cleaned up using beads, validated using a Qsep100 (Bioptic, Taiwan, China), and quantified by Qubit3.0 Fluorometer (Invitrogen, Carlsbad, CA, United States). Sequencing was carried out using a 2 × 150 paired-end (PE) configuration on an Illumina Hiseq instrument according to manufacturer’s instructions (Illumina, San Diego, CA, United States). The data were aligned to the PAO1 reference genome (NC_002516.2) via software BW2 (version 0.7.12). Single nucleotide variation (SNV) or InDel mutation were detected using the software Samtools (version 1.1) and the Unified Genotyper module from GATK.
PCR and Sequencing of oprD, ampR and ldcA
The full-length oprD gene with its 105 bp upstream and 209 bp downstream region was amplified and sequenced using primers listed in Supplementary Table S1. The oprD gene sequence from each isolate was aligned with the oprD sequence of the reference strain PAO12. Analysis of ampR and ldcA were conducted with similar procedures.
Western Blot Assay
Subcultured samples from equivalent number of bacterial cells of an optical density of 1.0 (wavelength of 600 nm) were separated on a 12% SDS-PAGE. The proteins were then transferred onto a PVDF (polyvinylidene difluoride) membrane and probed with a mouse monoclonal antibody against His-tag (Cell Signaling Technology, United States) and the RNA polymerase alpha subunit RpoA (Biolegend). Signals were generated using the ECL-Plus kit (Millipore) and detected by a Biorad imager (ChemiDocXRS).
Quantification of Biofilm
Biofilm production was measured as described previously (Li et al., 2013). Overnight culture of each bacterium was diluted 50-fold in LB broth and incubated in each well of a 96-well plate at 37°C. After 24 h, each well was washed with phosphate buffered saline (PBS, 137 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, pH 7.4) for three times, stained with 0.1% crystal violet, and then washed three times with PBS. Then, 0.2 ml of decolorizing solution [methanol (4): acetic acid (1): water (5)] was added into each well and incubated 10 min at room temperature. Each sample was then subjected to measurement for OD590 using Varioskan Flash (Thermo Scientific, Netherlands).
Statistical Analysis
Graphpad software was used to perform the statistical analyses. The real-time qPCR results were analyzed by Student’s t-test (two-tailed).
Results
Clinical Isolates ISP50 and IRP41 Belong to the Same Clone
Sputum samples from a patient with acute exacerbation of chronic bronchitis were collected before and after treatment with biapenem for 4 days (dosage at 0.3 g × 2/day). Two mucoid isolates were selected, one before the biapenem treatment which was sensitive to imipenem (ISP50), and the other one after the biapenem treatment which showed resistance against the imipenem (IRP41). PCR amplification of the 16S rDNAs as well as their sequence analysis indicated that both of them were P. aeruginosa (Supplementary Figure S1A). The MLST analysis of both isolates revealed an allelic profile for acsA, aroE, guaA, mutL, nuoD, ppsA and trpE as 17, 5, 12, 3, 14, 4, 7, respectively, corresponding to the same sequence type, ST244. RAPD typing was further performed on the IRP41 and ISP50 isolates, which also suggested that they were of the same clone (Supplementary Figure S1B).
Isolate IRP41 Is Resistant to Carbapenem
Imipenem susceptibilities of IRP41 and ISP50 were found different based on the preliminary analysis with VITEK automatic microbe analysis instrument (data not shown). Therefore, we further examined the imipenem susceptibilities by both MIC and disk diffusion methods, with the results shown in Table 2 and Supplementary Figure S2. Initial isolate (ISP50) was found to be susceptible to the imipenem (53.3 mm/diameter of inhibition zone), while later isolate (IRP41) was resistant to the imipenem (10.7 mm/diameter of inhibition zone), with a 32-fold increase in MIC of imipenem. Although both strains were susceptible to meropenem (another carbapenem antibiotic) according to the Clinical and Laboratory Standards Institute guidelines (CLSI 2011–2018), IRP41 displayed a smaller diameter of inhibition zone (24 mm) compared to that of ISP50 (44 mm) (Supplementary Figures S2A–D). In addition, MIC of meropenem/biapenem in IRP41 showed two-fold/eight-fold increase compared to ISP50 (as displayed in Table 2).
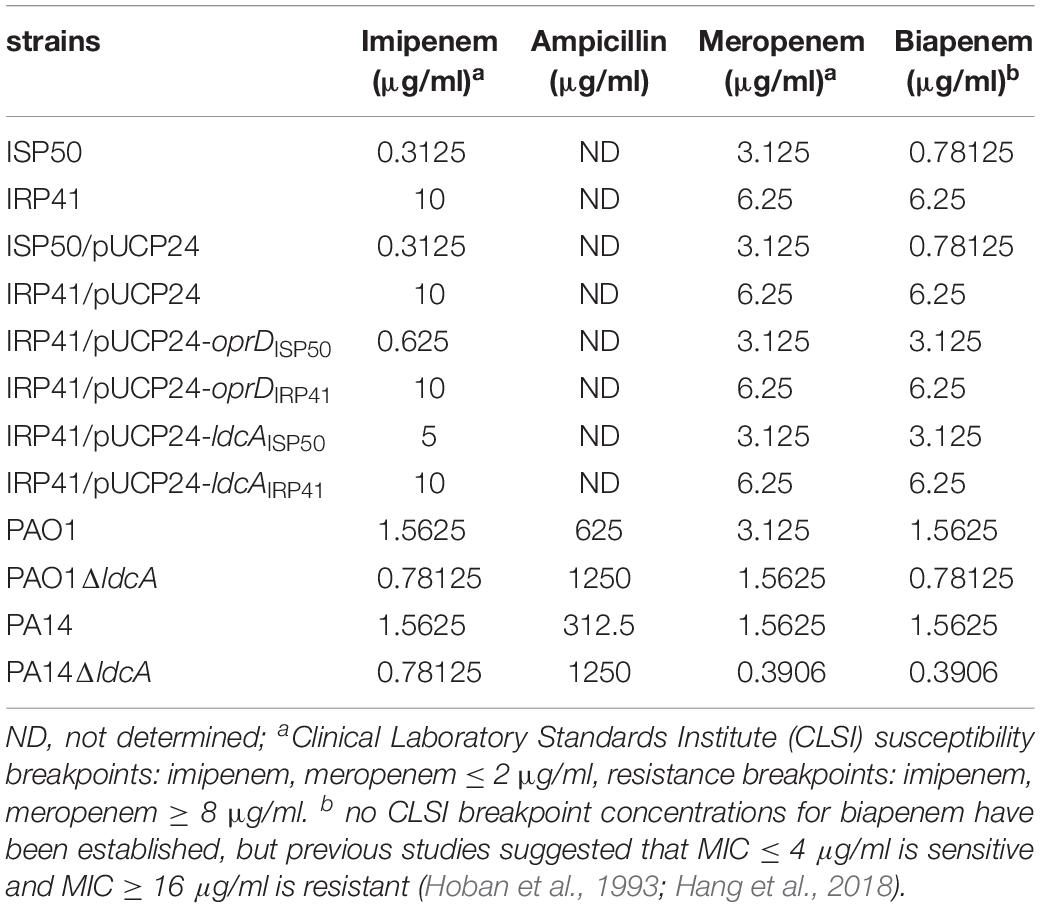
Table 2. MICs (μg/ml) of indicated P. aeruginosa strains.
Since the production of horizontally acquired carbapenem-hydrolyzing enzymes has been defined as a contributory factor in clinical P. aeruginosa isolates with carbapenem resistance (Potron et al., 2015), we tested if the IRP41 produced carbapenemase using a PAE-MHT assay. As the results shown in Supplementary Figures S2E,F, compared to the PA-NK41, which produces a KPC2 carbapenemase, no carbapenemase activity was observed in the IRP41 or ISP50 strain, suggesting that the carbapenem resistant phenotype of isolate IRP41 is not due to the presence of any horizontally acquired carbapenemases.
Differential Expression of Genes Relevant to β-Lactam Resistance
To elucidate the mechanism of reduced susceptibility to carbapenem in isolate IRP41, we compared the global gene expression profiles between strains ISP50 and IRP41. Expression levels of 284 genes were altered comparing the two strains (Table 3 and Supplementary Table S2). Among them, ampC gene, encoding the β-lactamase, displayed a 285-fold higher mRNA level in IRP41 than that in ISP50 (Table 3). To confirm the up-regulation, we further examined the mRNA levels by real-time qPCR. As shown in Figure 1A, consistent with the RNAseq result, the mRNA level of ampC was 413-fold higher in the strain IRP41 than that in ISP50. To further determine if the increased ampC expression contributed to the reduced susceptibility to carbapenem in IRP41, the ampC gene was overexpressed in the ISP50 strain background and the MIC of carbapenems was examined. As shown in Supplementary Table S3 and Supplementary Figure S3A, overexpression of the ampC (1,239-fold increase compared to that in ISP50/pUCP24) reduced the susceptibility of ISP50 to imipenem/meropenem/biapenem, with 16/2/4-fold increase in MIC. These results suggested that increased expression of the ampC indeed contributed to the decreased susceptibility of IRP41 to carbapenems, at least partially.
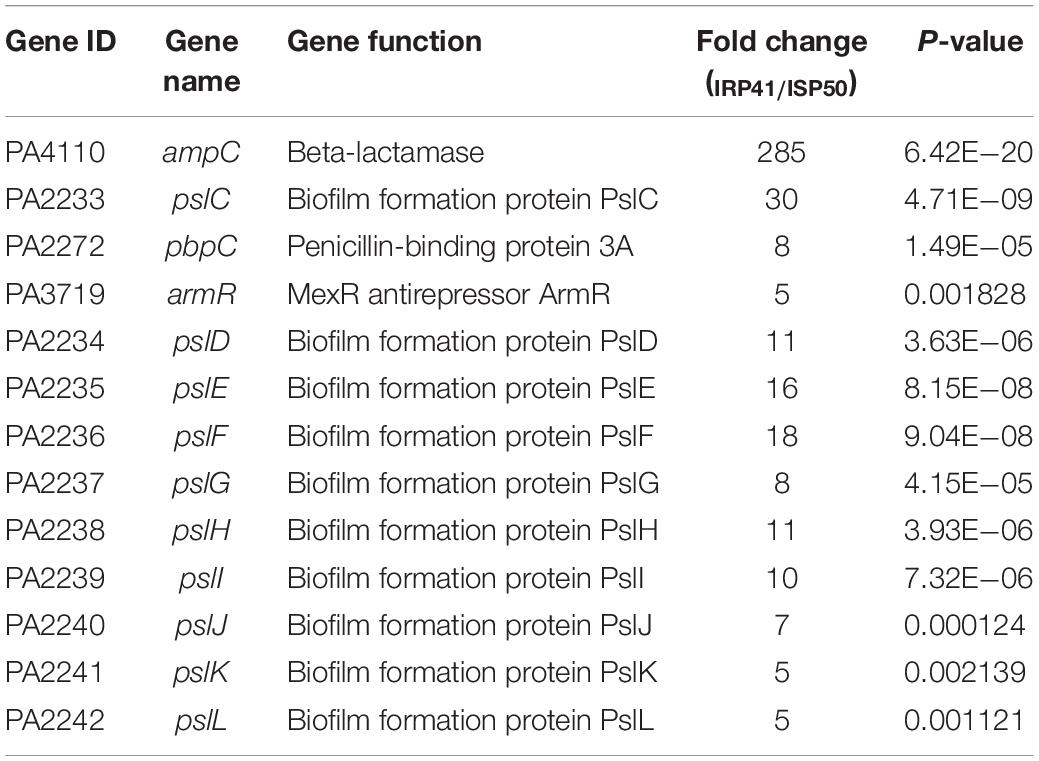
Table 3. mRNA levels of genes related to β-lactam resistance and biofilm in IRP41 compared to those in ISP50.
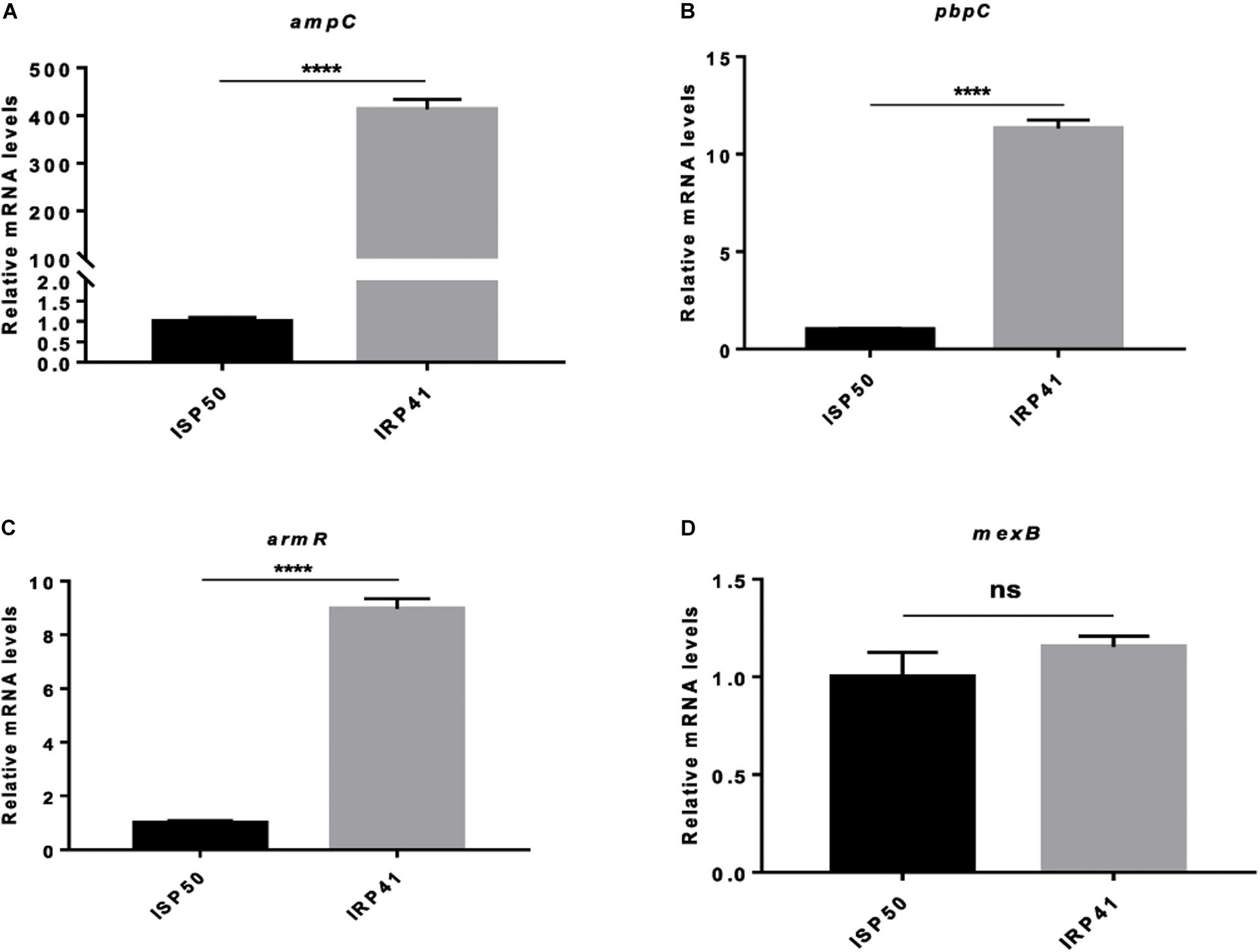
Figure 1. Relative mRNA levels of indicated genes in indicated strains. (A) Relative mRNA levels of ampC in indicated strains. (B) Relative mRNA levels of pbpC in indicated strains. (C) Relative mRNA levels of armR in ISP50 and IRP41. (D) Relative mRNA levels of mexB in ISP50 and IRP41. Total RNA was isolated from indicated strains at OD600 of 1.0, and the relative mRNA levels of indicated genes were determined by real-time qPCR using rpsL as an internal control. ns, not significant, ****P < 0.0001, by Student’s t-test.
Besides the ampC, a pbpC gene, encoding penicillin binding protein 3a, displayed an eight-fold up-regulation in IRP41 (Table 3). Real-time qPCR confirmed the increased expression of the pbpC in IRP41 (Figure 1B). However, overexpression of the pbpC (Supplementary Figure S3B) in ISP50 (ISP50/pUCP24-pbpC) had no effect on the bacterial MIC against imipenem/meropenem/biapenem (Supplementary Table S3), although it restored the MIC of carbenicillin in PA14pbpC::Tn to that of wild type PA14 strain (Supplementary Table S3). These results indicated that the elevated pbpC expression did not contribute to the carbapenem resistance in IRP41.
Our RNAseq analysis also revealed that armR, encoding MexR antirepressor (ArmR), was up-regulated five folds in IRP41. It had been demonstrated that MexR represses the expression of mexAB-oprM efflux pump (Daigle et al., 2007) whose over-expression confers meropenem resistance in P. aeruginosa (Okamoto et al., 2002). However, RNAseq results showed no significant difference in the transcriptional levels of the mexAB-oprM between the two strains, and this is confirmed by real-time qPCR (Figures 1C,D). Further sequence analysis showed that no mutation happened in the mexR, armR or mexR–mexA intergenic region of the IRP41 and ISP50 strains (data not shown).
IRP41 Encodes an Inactive OprD
To further elucidate the molecular mechanisms of the decreased susceptibility to carbapenem in IRP41, genome reference mapping was performed to identify the accumulated mutations in the genomes of IRP41 and ISP50 in comparison with PAO1 reference genome (see text footnote 2). Between strains ISP50 and IRP41, there are 84 frameshifts (deletion/insertion) and 377 nonsynonymous single nucleotide variations (SNV) (including 17 early stops) (Supplementary Table S4). Among them, a G831A substitution in the oprD gene resulted in a premature termination of the OprD in the IRP41 strain. To confirm the result, oprD genes were PCR amplified from the genomic DNA of strains ISP50 and IRP41. Sequence analysis of the amplicons revealed that oprD in ISP50 was the same as that of PAO1, while the oprD from IRP41 showed a G831A substitution, which confirmed the reference mapping results. To assess if the G831A substitution in oprD contributed to the reduced susceptibility of the IRP41 to carbapenems, the oprD gene from both ISP50 and IRP41 were expressed in the IRP41 strain background. As can be observed in Table 2, introduction of the oprDISP50 reduced the MIC of imipenem/meropenem/biapenem in IRP41, with 16/2/2-fold decrease, while the oprDIRP41 did not. These results indicated that the G831A substitution in oprD indeed disrupted its function and lead to the reduced susceptibility to carbapenem in strain IRP41.
Mechanisms of ampC Hyper-Expression in IRP41
AmpR is a transcriptional regulator encoded immediately upstream of the ampC, regulating the ampC expression (Kong et al., 2005). The genome reference mapping result revealed that the ampR sequence of IRP41 is identical to that of PAO1, while in strain ISP50 an “A” was substituted by “G” at 821th position of the ampR gene, resulting in an E274G substitution. These results were further confirmed by sequencing of the PCR amplicons. To test if the nonsynonymous mutation in ampR of ISP50 resulted in a lower level of ampC expression, both ampR genes from IRP41 and ISP50 were cloned and expressed in the background of ISP50. As shown in Supplementary Figure S3C and Supplementary Table S3, expression of the ampRISP50 conferred ISP50 strain a higher ampC mRNA level and increased MIC against ampicillin compared to that of ampRIRP41, although it had no observable influence on the MIC of imipenem/meropenem/biapenem. The ampRISP50 and ampRIRP41 were further introduced into PAO1ΔampR, and their effects on MICs to imipenem, meropenem, biapenem and ampicillin in PAO1ΔampR were examined. As shown in Supplementary Table S3, both ampRISP50 and ampRIRP41 restored the MIC against ampicillin and biapenem in PAO1ΔampR to that in wild type PAO1 strain, while in contrast to ampRIRP41, the ampRISP50 conferred PAO1ΔampR a repeatable two-fold increase in MIC against imipenem compared to that of wild type PAO1. Unexpectedly, no influence on MIC of meropenem was observed in PAO1ΔampR. These results suggested that A821G SNV of ampR in fact confers slightly higher ampC expression-activating capacity than the wild type AmpR, thus resulting in higher, rather than lower ampC in ISP50.
ldcA (PA5198) encodes a cytosolic LD-carboxypeptidase which removes the terminal amino acid D-alanine from the tetra peptide of the peptidoglycan (Korza and Bochtler, 2005). Our genome reference mapping and PCR amplicons sequencing results revealed that a C was deleted at 445 bp of the ldcA gene in strain IRP41, leading to a frameshift mutation after R149 of the LdcA. To test if this frameshift mutation in ldcA contributed to the elevated expression of the ampC in IRP41, we expressed the ldcAIRP41 or ldcAISP50 (same as ldcA of PAO1) in the IRP41 background. As shown in Figure 2A, the relative mRNA level of ampC in IRP41/pUCP24-ldcAISP50, but not in IRP41/pUCP24-ldcAIRP41, was restored to that in ISP50/pUCP24, and the MIC to imipenem/meropenem/biapenem in IRP41/pUCP24-ldcAISP50 was decreased by two folds (Table 2). To confirm the expression level of ampC gene, a C-terminal His-tagged ampC driven by its native promoter was integrated into the genomes of ISP50 and IRP41. As shown in Figure 2B, the AmpC-His level was much higher in IRP41 than that in ISP50, and overexpression of ldcAISP50, but not ldcAIRP41, reduced the amount of AmpC-His in the IRP41 to almost that of the ISP50. These results demonstrated that frameshift-mutation of the ldcA in IRP41 contributes to the increased expression of the ampC in IRP41.
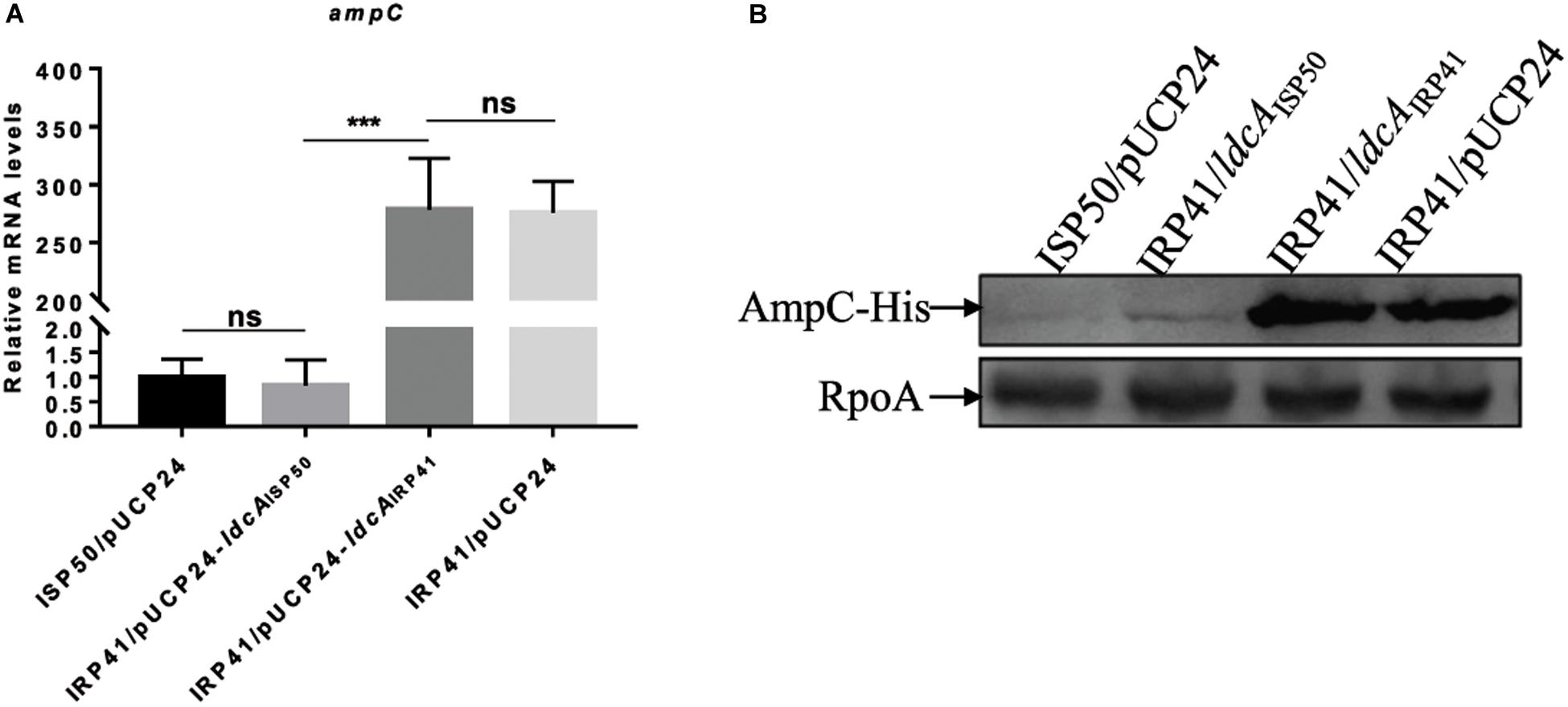
Figure 2. Mechanism of increased mRNA levels of ampC in IRP41. (A) Relative mRNA levels of ampC gene in indicated strains. Total RNA was isolated from indicated strains at OD600 of 1.0, and the relative mRNA levels of ampC were determined by real-time qPCR using rpsL as an internal control. ns, not significant, ***P < 0.001, by Student’s t-test. (B) Levels of AmpC-His in indicated strains detected with Western blot assay. Strains with an ampC-His in their chromosomes were cultured to OD600 of 1.0. Samples from equal number of bacterial cells were loaded onto 12% SDS-PAGE gels and probed with an antibody against His-tag or RpoA. The RNA polymerase alpha subunit RpoA served as a control.
To further confirm the effect of ldcA on ampC repression, the whole ldcA open reading frame was deleted in PA14 and PAO1 backgrounds, resulting in PA14ΔldcA and PAO1ΔldcA, respectively. Similar to previous report in E. coli (Templin et al., 1999), both of the ldcA deletion mutants showed a reduced growth rate and proneness to lysis during stationary growth phase (Supplementary Figure S4). Interestingly, the expression levels of ampC displayed 2.6-fold and 4.0-fold increase in the ΔldcA mutants compared to their parental strains PAO1 and PA14, respectively (Figure 3A). Accordingly, the MIC of ampicillin in PAO1ΔldcA and PA14ΔldcA displayed a two-fold and four-fold increase compared to their respective parental strains (Table 2). We further deleted the ldcA gene from the ISP50 strain. Consistent with the above observations, the expression level of ampC in the ISP50ΔldcA increased 1.8-fold compared to that in the ISP50 strain (Figure 3B). The difference in fold increase of ampC mRNA levels between ISP50 and IRP41 (413 folds) vs. between ISP50 and ISP50ΔldcA (1.8 folds) suggested that other factors also contributed to the increased expression of the ampC in IRP41.
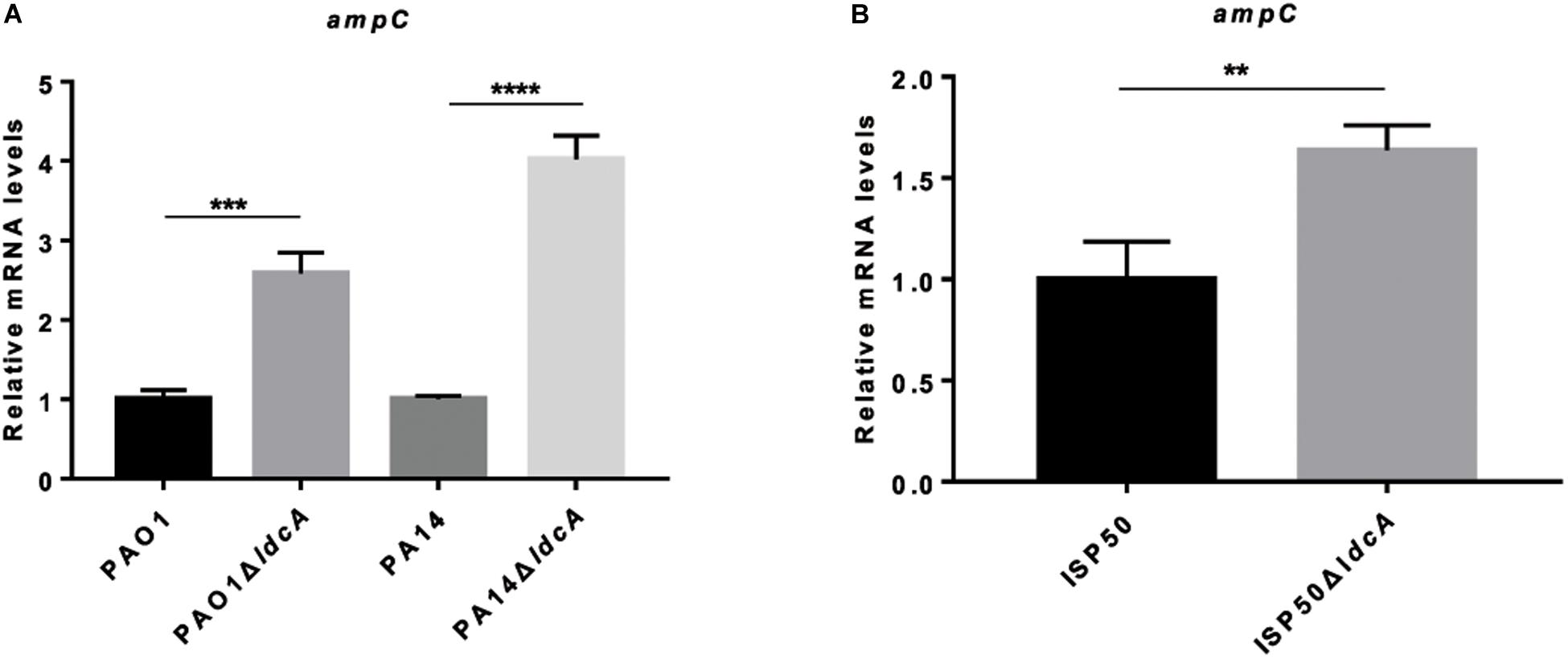
Figure 3. Relative mRNA levels of ampC gene in indicated strains (A,B). Total RNA was isolated from indicated strains at OD600 of 1.0, and the relative mRNA levels of ampC gene were determined by real-time qPCR using rpsL as an internal control. **P < 0.01, ***P < 0.001, ****P < 0.0001, by student’s t-test.
In addition, we also examined the effect of ldcA mutation on MICs against piperacillin, ceftazidime, aztreonam, ciprofloxacin and tobramycin. Except for a two-fold decrease against ceftazidime and meropenem in PAO1ΔldcA, no MICs change was observed for piperacillin, aztreonam, ciprofloxacin and tobramycin in ldcA mutants (Table 4). However, we didn’t obtain the IRP41ΔldcA, although deletion of ldcA in IRP41 strain had been tried for several times. We assume that it may be due to presence of some unknown factors to prevent the recombination in IRP41.
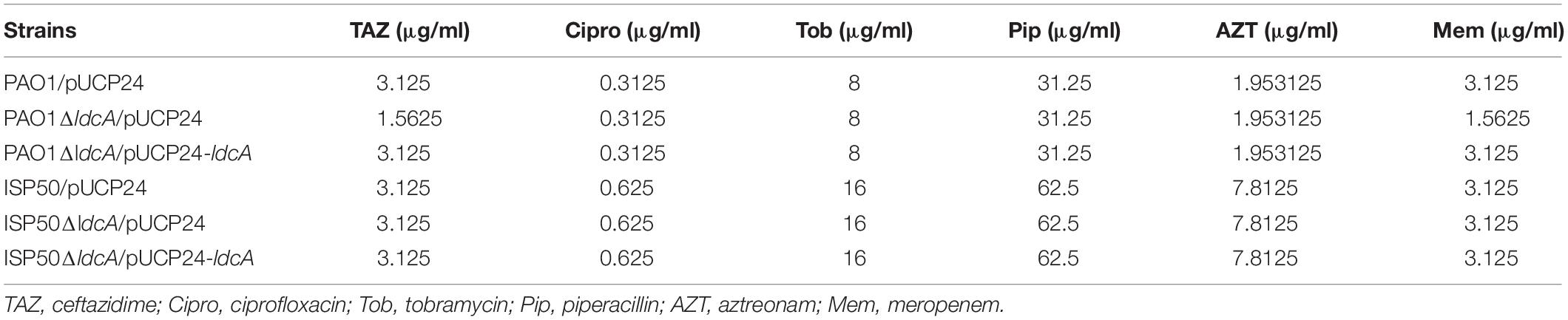
Table 4. MICs (μg/ml) of indicated P. aeruginosa strains.
Since AmpR is the regulator of ampC, and its 274th amino acid is different between IRP41 and ISP50, we further compared the ampC expression levels and the MIC to ampicillin/imipenem/meropenem/biapenem between ISP50ΔldcA expressing AmpRISP50 and AmpRIRP41. As shown in Supplementary Figure S3D, consistent with that in the ISP50 strain, expression of AmpRISP50 in ISP50ΔldcA (ISP50ΔldcA/pMMB-ampRISP50) resulted in a higher mRNA level of ampC compared to that of ISP50ΔldcA/pMMB-ampRIRP41, as well as a two-fold increase in MIC of ampicillin (Supplementary Table S3). Similar to that in ISP50 strain, overexpression of both AmpRIRP41 and AmpRISP50 showed no influence on MIC of imipenem/meropenem/biapenem in the ISP50ΔldcA strain. Again, these are consistent with the earlier observation that the AmpR point mutation in ISP50 confers a higher transcriptional activator activity on ampC.
Furthermore, as the expression level of pbpC is much higher in IRP41 than that in ISP50, we overexpressed pbpC in the ISP50ΔldcA background to see if the elevated pbpC expression had any effect on the ampC mRNA levels. However, the mRNA level of ampC was not affected by the overexpression of pbpC in ISP50ΔldcA (data not shown), consistent with such null effect observed in the ISP50 background (Supplementary Table S3 and Supplementary Figure S3E).
IRP41 Is a Better Biofilm Former Than ISP50
Our RNA-seq result also revealed an increased expression of pslD-pslL operon (Table 3) which is involved in biofilm formation. Accordingly, we examined and compared the biofilm formation between IRP41 and ISP50 strains. As the results shown in Figure 4, the IRP41 strain produced more biofilm than the ISP50 strain.
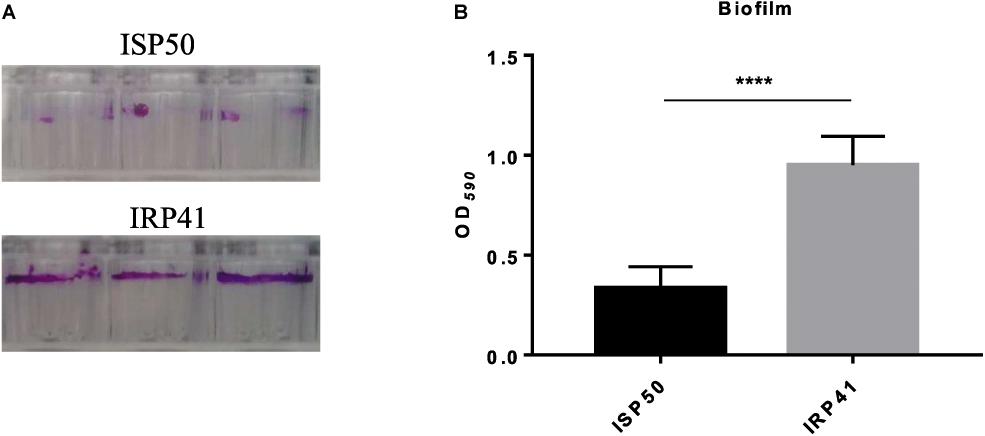
Figure 4. Biofilm formation by ISP50 and IRP41 strains. Overnight cultures of ISP50 and IRP41 were 50-fold diluted in LB and incubated in each well of a 96-well plate for 24 h at 37°C. Then the biofilm was stained with 0.1% crystal violet (A), dissolved by 0.2 ml of decolorizing solution and subjected to measurement at OD590 (B). ****P < 0.0001 by Student’s t-test.
Discussion
In this study, we examined MICs of imipenem, meropenem and biapenem, three carbapenem antibiotics, for all the strains in Table 2 and Supplementary Table S3. Compared to meropenem, the contribution of AmpC overproduction and OprD deficiency to biapenem susceptibility in IRP41 resembles to that of imipenem. The difference among the susceptibility to imipenem, meropenem and biapenem between ISP50 and IRP41 may reflect their difference in penetration rate, stability to the hydrolysis by β-lactamase, PBP protein binding profile (Yang et al., 1995; Bonfiglio et al., 2002), β-lactamase deactivating capability and export by the efflux pumps (Chen and Livermore, 1994; Okamoto et al., 2002).
AmpC expression is regulated by AmpR, a LysR family transcriptional regulator (Kong et al., 2005). Previous studies reported that ampR gene point mutations were associated with increased ampC expression in both Enterobacter cloacae and P. aeruginosa clinical isolates (Kuga et al., 2000; Bagge et al., 2002). Our study presents E274G substitution in AmpR actually lead to a higher level of ampC expression. Besides the expression of ampC, AmpR regulates expression of hundreds of genes involved in diverse pathways such as physiological processes and metabolism (Balasubramanian et al., 2015). It has been demonstrated that ampR mutant displays an impaired growth under iron-limiting conditions and sensitive to many agents that affect cell growth (Balasubramanian et al., 2014, 2015). Thus, it is possible that restoration to wild type AmpR from E274G substituted AmpR of ISP50 provided the IRP41 strain a better adaption to the host environment. Also, an increased expression of the pbpC is neither responsible for the higher ampC expression nor the decreased susceptibility to carbapenem in IRP41 strain. The regulatory mechanism for the elevated pbpC expression in IRP41 and its functional roles remain to be investigated.
AmpD, a cytosolic peptidoglycan amidase, cleaves the peptide chain from N-acetyl-anhydromuramic acid peptides which are the inducer molecules for the ampC expression via binding to the AmpR (Langaee et al., 1998; Lister et al., 2009; Dhar et al., 2018). Mutations in AmpD and its two homologous proteins, AmpDh2 and AmpDh3, contribute to a stepwise de-repression of the ampC in the wild-type strain PAO1 (Juan et al., 2006). Similarly, ampD mutations leading to ampC de-repression have been reported in clinical isolates (Schmidtke and Hanson, 2008). However, some AmpC over-expressing P. aeruginosa strains do not exhibit mutation in ampD, ampDh2, ampDh3, ampR, or the ampR–ampC intergenic region (Wolter et al., 2007; Schmidtke and Hanson, 2008), suggesting that other undiscovered factors or pathways likely contribute to the upregulation of ampC expression. Besides, mutational inactivation of dacB, encoding penicillin-binding protein 4 (PBP4), has been reported to trigger a stable AmpC overproduction in P. aeruginosa (Moya et al., 2009). Mutation of the lytic transglycosylases encoding genes sltB1 and mltB1 in PAO1 resulted in a stable AmpC hyperproduction in the presence of β-lactam antibiotic (Cavallari et al., 2013; Juan et al., 2017). Inactivation of mpl (encoding cytosolic UDP-N-acetylmuramate:L-alanyl-γ-D-glutamyl-meso-diaminopimelate ligase) and nuoN (encoding NADH dehydrogenase I chain N) led to an increase in the expression of ampC (Tsutsumi et al., 2013; Juan et al., 2017). In the two isolates used in this study, no mutation was found in the above genes, except for the AmpR with E274G substitution in ISP50 which had no effect on the increased ampC expression in IRP41. These suggested the presence of unknown molecular basis driving AmpC hyperproduction in the IRP41 strain.
In the present study, inactivation of the ldcA contributes to the elevated expression of the ampC in P. aeruginosa. LdcA, encoding an LD carboxypeptidase, cleaves the D-alanine from N-acetyl-anhydromuramic acid tetrapeptides (anhNAM-P4) in P. aeruginosa (Korza and Bochtler, 2005), generating N-acetyl-anhydromuramic acid tripeptide (anhNAM-P3). Functional loss of the LdcA leads to the accumulation of anhNAM-P4, thus our observation suggests that the anhNAM-P3 is less potent inducer for the ampC expression than the anhNAM-P4. This is consistent with a previous study in which the anhNAM-P4 was shown to be a critical activator ligand for β-lactamase expression in Stenotrophomonas maltophilia (Huang et al., 2017). Results from previous studies implied anhNAM-P5 as the genuine AmpR-binding signal (Dietz et al., 1997; Lee et al., 2016) and further suggested that only muropeptides containing a terminal d-Ala-d-Ala motif (i.e., muropentapeptides) is capable of binding to the AmpR for ampC induction (Vadlamani et al., 2015; Dik et al., 2017). However, in a most recent report, anhNAM-P3 was demonstrated to function as an ampC inducer, albeit much less potent than the anhNAM-P5 (Torrens et al., 2019). As these studies were carried out under different conditions (with or without antibiotic induction) using different mutant backgrounds, it is possible that different signaling pathways may have been involved in the AmpR mediated regulation of the ampC in the clinical strains analyzed in this study.
This is the first report linking ldcA to the regulation of ampC expression in P. aeruginosa. Interestingly, compared to the 400-fold increase of ampC mRNA in IRP41, the ΔldcA mutants of PA14, PAO1 and ISP50 showed only 1.8–4 folds increases in the ampC mRNA compared to their parental strains. Such minor changes in the ampC expression, in addition to the growth defect of the ldcA mutant at stationary phase, may explain why LdcA was not identified as a repressor for the ampC in previous studies. One may argue that the observed 2–4 folds increase in MIC to ampicillin may be due to the reduced growth of the ΔldcA mutant. However, we do not feel this is the case because the proneness to lysis happens during the stationary growth phase (OD600 about 2.0). In the case of IRP41, we postulate that there are other factors contributing to the increased ampC expression in addition to the ldcA. The mechanism underlying this observation is yet to be elucidated.
OprD has been reported to be the most prevalent cause of imipenem resistance among clinical isolates of P. aeruginosa. Insertion of various IS elements and point mutations with premature stops on the oprD gene had been associated with the carbapenem resistance among clinical isolates of P. aeruginosa (Hirabayashi et al., 2017). In this study, a G831A substitution in oprD resulted in loss of the OprD function. With the OprD inactivation and elevated ampC expression, the clinical isolate IRP41 displayed resistance to carbapenem. It has been demonstrated that derepression of AmpC in PAO1 has no obvious impact on the MIC of imipenem (Juan et al., 2006). While in another study, the loss of AmpC from PAO1 displayed a four-fold increase in susceptibility to imipenem (Masuda et al., 1999). Derepression of AmpC was associated with 4–8/8–64-fold increase in MIC of imipenem in an OprD+/OprD– background, respectively (Livermore, 1992; Mushtaq et al., 2004). And also, mutational variants of the AmpC cephalosporinase provide P. aeruginosa with imipenem resistance (Rodriguez-Martinez et al., 2009). In our study, overexpression of ampC conferred ISP50 a 16-fold increase in MIC of imipenem. However, both AmpC and OprD are of PAO1-type in ISP50, which suggests that other unknown factors may contribute to the change in susceptibility to imipenem by the overexpression of AmpC.
MexAB-OprM is able to expel a wide variety of antibiotics (Kohler et al., 1999; Lister et al., 2009) and its expression is repressed by MexR (Poole et al., 1996). A 53-amino-acid long antirepressor, ArmR, could interact with and inhibit the MexR function, upregulating the mexAB-oprM expression (Wilke et al., 2008). However, in the present study, increased expression of armR in IRP41 did not cause altered expression level of mexAB-oprM. In fact, mexAB-oprM is expressed and contributes to antimicrobial resistance in wild type Pseudomonas aeruginosa, it is more difficult to detect mexAB-oprM overexpression in mutant cells than the other efflux pump encoding genes (e.g., mexCD-oprJ, mexEF-oprN). In addition, previous study has revealed MexR is a redox regulator which senses oxidative stress inside bacterial cells to regulate mexAB-oprM expression (Chen et al., 2008). nalD encodes a second repressor of the mexAB-oprM (Morita et al., 2006). Mutations in nalD resulted in increased MexAB-OprM expression in lab and clinical isolates of P. aeruginosa (Sobel et al., 2005). However, no mutation occurred in the nalD gene of the IRP41 or ISP50 isolates. The mechanisms underlying these observations are under active investigation. It has been demonstrated that nalC (PA3721), encoding a TetR family repressor, negatively regulated armR, and S127P substitution in NalC impaired its armR-repressing capacity (Cao et al., 2004). Compared to ISP50, no mutations were observed in the intergenic region between nalC and PA3720, while a S127P substitution in NalC happened in IRP41 (Supplementary Table S4), which may be the cause to result in the increased armR transcriptional level in IRP41.
IRP41 is a better biofilm former than ISP50. However, the MICs against ciprofloxacin and levofloxacin in IRP41 are the same as ISP50 (data not shown), and the MICs against gentamicin and tobramycin display a two-fold decrease in IRP41 than that in ISP50 (data not shown). A previous study had shown that the Psl did not affect bacterial MIC to biapenem for planktonic cells (Murakami et al., 2017). Therefore, we assumed that the increased biofilm production did not contribute to the increased MIC to carbapenem in IRP41.
It is not necessary for clinical isolates processing a major resistance determinant against a certain antibiotic category to accumulate the other resistance mechanisms. However, the concomitant presence of multiple carbapenem resistance mechanisms has been observed in P. aeruginosa (Meletis et al., 2014). In our study, the overproduction of AmpC β-lactamase together with OprD deficiency leads imipenem-susceptible ISP50 to imipenem-resistant IRP41 derivative, as well as a reduced susceptibility to meropenem and biapenem.
Data Availability Statement
The datasets generated for this study can be found in NCBI, under accession PRJNA635437.
Ethics Statement
We have a waiver from the ethics committee, exempting this study from the requirement to have ethics approval and written informed as the clinical strains used in the study come from the routine procedures of the clinical laboratory rather than the clinical trials.
Author Contributions
YJ and FB conceived and designed the experiments. CX, DW, XZ, HL, GZ, and TW performed the experiments. WW, FB, ZC, and YJ analyzed the data. YJ wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Science Foundation of China (31600110, 31870130, 31670130, 81670766, and 31970680), Science and Technology Committee of Tianjin (17JCQNJC09200 and 19JCYBJC24700), National Key Research and Development Project of China (2017YFE0125600), Science and Technology Program of Sichuan Province (2018JZ0069), and the Fundamental Research Funds for the Central Universities, Nankai University (63191121, 63191117, and 63191521).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01390/full#supplementary-material
FIGURE S1 | PCRresults of ISP50 and IRP41 strains. (A) 16S rDNA gene amplification; (B) RAPD typing.
FIGURE S2 | Carbapenem susceptibility and carbapenemase production of indicated strains. (A,B) Imipenem inhibition zones of indicated strains on a 6-cm disk. (C,D) Meropenem inhibition zones of indicated strains on a 6-cm disk. (E,F) PAE-MHT assay using K. pneumoniae ATCC 700603 as indicator. Carbapenemase production test on imipenem (E) and meropenem (F) on a 10-cm disk, 0: ISP50, 1: IRP41, 2: PA-NK41, a producer of KPC2 carbapenemase.
FIGURE S3 | Relative mRNA levels of indicated genes in indicated strains. (A,C–E) Relative mRNA levels of ampC in indicated strains. (B) Relative mRNA levels of pbpC in indicated strains. Total RNA was isolated from indicated strains at OD600 of 1.0, and the relative mRNA levels of ampC or pbpC gene were determined by real-time qPCR using rpsL as an internal control. ns, not significant, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ****P < 0.0001, by Student’s t-test.
FIGURE S4 | Growth curves of indicated strains.
TABLE S1 | Primers used in this study.
TABLE S2 | Transcriptome analysis: differentially expressed genes by RNAseq without those listed in Table 2.
TABLE S3 | MICs (μg/ml) of indicated P. aeruginosa strains.
TABLE S4 | SNVs identified by genome reference mapping with elimination of SNVs with synonymous mutation and the same mutation between ISP50 and IRP41.
Footnotes
References
Bagge, N., Ciofu, O., Hentzer, M., Campbell, J. I., Givskov, M., and Hoiby, N. (2002). Constitutive high expression of chromosomal beta-lactamase in Pseudomonas aeruginosa caused by a new insertion sequence (IS1669) located in ampD. Antimicrob. Agents Chemother. 46, 3406–3411. doi: 10.1128/aac.46.11.3406-3411.2002
Balasubramanian, D., Kumari, H., Jaric, M., Fernandez, M., Turner, K. H., Dove, S. L., et al. (2014). Deep sequencing analyses expands the Pseudomonas aeruginosa AmpR regulon to include small RNA-mediated regulation of iron acquisition, heat shock and oxidative stress response. Nucleic Acids Res. 42, 979–998. doi: 10.1093/nar/gkt942
Balasubramanian, D., Kumari, H., and Mathee, K. (2015). Pseudomonas aeruginosa AmpR: an acute-chronic switch regulator. Pathog. Dis. 73, 1–14. doi: 10.1111/2049-632x.12208
Bonfiglio, G., Russo, G., and Nicoletti, G. (2002). Recent developments in carbapenems. Expert Opin. Investig. Drugs 11, 529–544. doi: 10.1517/13543784.11.4.529
Cao, L., Srikumar, R., and Poole, K. (2004). MexAB-OprM hyperexpression in NalC-type multidrug-resistant Pseudomonas aeruginosa: identification and characterization of the nalC gene encoding a repressor of PA3720-PA3719. Mol. Microbiol. 53, 1423–1436. doi: 10.1111/j.1365-2958.2004.04210.x
Cavallari, J. F., Lamers, R. P., Scheurwater, E. M., Matos, A. L., and Burrows, L. L. (2013). Changes to its peptidoglycan-remodeling enzyme repertoire modulate beta-lactam resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 57, 3078–3084. doi: 10.1128/aac.00268-13
Chen, H., Hu, J., Chen, P. R., Lan, L., Li, Z., Hicks, L. M., et al. (2008). The Pseudomonas aeruginosa multidrug efflux regulator MexR uses an oxidation-sensing mechanism. Proc. Natl. Acad. Sci. U.S.A. 105, 13586–13591. doi: 10.1073/pnas.0803391105
Chen, H. Y., and Livermore, D. M. (1994). In-vitro activity of biapenem, compared with imipenem and meropenem, against Pseudomonas aeruginosa strains and mutants with known resistance mechanisms. J. Antimicrob. Chemother. 33, 949–958. doi: 10.1093/jac/33.5.949
Choudhury, D., Das Talukdar, A., Dutta Choudhury, M., Maurya, A. P., Paul, D., Dhar Chanda, D., et al. (2015). Transcriptional analysis of MexAB-OprM efflux pumps system of Pseudomonas aeruginosa and its role in carbapenem resistance in a tertiary referral hospital in India. PLoS One 10:e0133842. doi: 10.1371/journal.pone.0133842
Curran, B., Jonas, D., Grundmann, H., Pitt, T., and Dowson, C. G. (2004). Development of a multilocus sequence typing scheme for the opportunistic pathogen Pseudomonas aeruginosa. J. Clin. Microbiol. 42, 5644–5649. doi: 10.1128/jcm.42.12.5644-5649.2004
Daigle, D. M., Cao, L., Fraud, S., Wilke, M. S., Pacey, A., Klinoski, R., et al. (2007). Protein modulator of multidrug efflux gene expression in Pseudomonas aeruginosa. J. Bacteriol. 189, 5441–5451. doi: 10.1128/jb.00543-07
Davies, T. A., Marie Queenan, A., Morrow, B. J., Shang, W., Amsler, K., He, W., et al. (2011). Longitudinal survey of carbapenem resistance and resistance mechanisms in Enterobacteriaceae and non-fermenters from the USA in 2007-09. J. Antimicrob. Chemother. 66, 2298–2307. doi: 10.1093/jac/dkr290
Dhar, S., Kumari, H., Balasubramanian, D., and Mathee, K. (2018). Cell-wall recycling and synthesis in Escherichia coli and Pseudomonas aeruginosa - their role in the development of resistance. J. Med. Microbiol. 67, 1–21. doi: 10.1099/jmm.0.000636
Dietz, H., Pfeifle, D., and Wiedemann, B. (1997). The signal molecule for beta-lactamase induction in Enterobacter cloacae is the anhydromuramyl-pentapeptide. Antimicrob. Agents Chemother. 41, 2113–2120. doi: 10.1128/aac.41.10.2113
Dik, D. A., Dominguez-Gil, T., Lee, M., Hesek, D., Byun, B., Fishovitz, J., et al. (2017). Muropeptide binding and the X-ray structure of the effector domain of the transcriptional regulator AmpR of Pseudomonas aeruginosa. J. Am. Chem. Soc. 139, 1448–1451. doi: 10.1021/jacs.6b12819
Hang, Y., Chen, Y., Xue, L., Sun, S., Liu, L., Gao, J., et al. (2018). Evaluating biapenem dosage regimens in intensive care unit patients with Pseudomonas aeruginosa infections: a pharmacokinetic/pharmacodynamic analysis using Monte Carlo simulation. Int. J. Antimicrob. Agents 51, 484–487. doi: 10.1016/j.ijantimicag.2017.07.005
Hirabayashi, A., Kato, D., Tomita, Y., Iguchi, M., Yamada, K., Kouyama, Y., et al. (2017). Risk factors for and role of OprD protein in increasing minimal inhibitory concentrations of carbapenems in clinical isolates of Pseudomonas aeruginosa. J. Med. Microbiol. 66, 1562–1572. doi: 10.1099/jmm.0.000601
Hoang, T. T., Karkhoff-Schweizer, R. R., Kutchma, A. J., and Schweizer, H. P. (1998). A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86. doi: 10.1016/s0378-1119(98)00130-9
Hoban, D. J., Jones, R. N., Yamane, N., Frei, R., Trilla, A., and Pignatari, A. C. (1993). In vitro activity of three carbapenem antibiotics. Comparative studies with biapenem (L-627), imipenem, and meropenem against aerobic pathogens isolated worldwide. Diagn. Microbiol. Infect. Dis. 17, 299–305. doi: 10.1016/0732-8893(93)90039-a
Huang, Y. W., Wang, Y., Lin, Y., Lin, C., Lin, Y. T., Hsu, C. C., et al. (2017). Impacts of penicillin binding protein 2 Inactivation on beta-Lactamase expression and muropeptide profile in Stenotrophomonas maltophilia. mSystems 2:17. doi: 10.1128/mSystems.00077-17
Juan, C., Moya, B., Perez, J. L., and Oliver, A. (2006). Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level beta-lactam resistance involves three AmpD homologues. Antimicrob. Agents Chemother. 50, 1780–1787. doi: 10.1128/aac.50.5.1780-1787.2006
Juan, C., Torrens, G., Gonzalez-Nicolau, M., and Oliver, A. (2017). Diversity and regulation of intrinsic beta-lactamases from non-fermenting and other Gram-negative opportunistic pathogens. FEMS Microbiol. Rev. 41, 781–815. doi: 10.1093/femsre/fux043
Kaufman, M. R., Jia, J., Zeng, L., Ha, U., Chow, M., and Jin, S. (2000). Pseudomonas aeruginosa mediated apoptosis requires the ADP-ribosylating activity of exoS. Microbiology 146(Pt 10), 2531–2541. doi: 10.1099/00221287-146-10-2531
Kohler, T., Michea-Hamzehpour, M., Epp, S. F., and Pechere, J. C. (1999). Carbapenem activities against Pseudomonas aeruginosa: respective contributions of OprD and efflux systems. Antimicrob. Agents Chemother. 43, 424–427. doi: 10.1128/aac.43.2.424
Kong, K. F., Jayawardena, S. R., Indulkar, S. D., Del Puerto, A., Koh, C. L., Hoiby, N., et al. (2005). Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB beta-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob. Agents Chemother. 49, 4567–4575. doi: 10.1128/aac.49.11.4567-4575.2005
Korza, H. J., and Bochtler, M. (2005). Pseudomonas aeruginosa LD-carboxypeptidase, a serine peptidase with a Ser-His-Glu triad and a nucleophilic elbow. J. Biol. Chem. 280, 40802–40812. doi: 10.1074/jbc.M506328200
Kuga, A., Okamoto, R., and Inoue, M. (2000). ampR gene mutations that greatly increase class C beta-lactamase activity in Enterobacter cloacae. Antimicrob. Agents Chemother. 44, 561–567. doi: 10.1128/aac.44.3.561-567.2000
Langaee, T. Y., Dargis, M., and Huletsky, A. (1998). An ampD gene in Pseudomonas aeruginosa encodes a negative regulator of AmpC beta-lactamase expression. Antimicrob. Agents Chemother. 42, 3296–3300. doi: 10.1128/aac.42.12.3296
Lee, M., Dhar, S., De Benedetti, S., Hesek, D., Boggess, B., Blazquez, B., et al. (2016). Muropeptides in Pseudomonas aeruginosa and their role as elicitors of beta-lactam-antibiotic resistance. Angew. Chem. Int. Ed. Engl. 55, 6882–6886. doi: 10.1002/anie.201601693
Li, K., Xu, C., Jin, Y., Sun, Z., Liu, C., Shi, J., et al. (2013). SuhB is a regulator of multiple virulence genes and essential for pathogenesis of Pseudomonas aeruginosa. mBio 4:e419-13. doi: 10.1128/mBio.00419-13
Liberati, N. T., Urbach, J. M., Miyata, S., Lee, D. G., Drenkard, E., Wu, G., et al. (2006). An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U.S.A. 103, 2833–2838. doi: 10.1073/pnas.0511100103
Lister, P. D., Wolter, D. J., and Hanson, N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22, 582–610. doi: 10.1128/cmr.00040-09
Liu, Y., Li, X. Y., Wan, L. G., Jiang, W. Y., Li, F. Q., and Yang, J. H. (2013). Efflux system overexpression and decreased OprD contribute to the carbapenem resistance among extended-spectrum beta-lactamase-producing Pseudomonas aeruginosa isolates from a Chinese university hospital. Microb. Drug Resist. 19, 463–468. doi: 10.1089/mdr.2013.0010
Livermore, D. M. (1992). Interplay of impermeability and chromosomal beta-lactamase activity in imipenem-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 36, 2046–2048. doi: 10.1128/aac.36.9.2046
Long, Y., Fu, W., Li, S., Ren, H., Li, M., Liu, C., et al. (2019). Identification of novel genes that promote persister formation by repressing transcription and cell division in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 74, 2575–2587. doi: 10.1093/jac/dkz214
Mahenthiralingam, E., Campbell, M. E., Foster, J., Lam, J. S., and Speert, D. P. (1996). Random amplified polymorphic DNA typing of Pseudomonas aeruginosa isolates recovered from patients with cystic fibrosis. J. Clin. Microbiol. 34, 1129–1135. doi: 10.1128/jcm.34.5.1129-1135.1996
Masuda, N., Gotoh, N., Ishii, C., Sakagawa, E., Ohya, S., and Nishino, T. (1999). Interplay between chromosomal beta-lactamase and the MexAB-OprM efflux system in intrinsic resistance to beta-lactams in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43, 400–402. doi: 10.1128/aac.43.2.400
Mcfarland, J. (1907). The nephelometer: an instrument for estimating the number of bacteria in suspensions used for calculating the opsonic index and for vaccines. J. Am. Med. Assoc. 49, 1176–1178. doi: 10.1001/jama.1907.25320140022001f
Meletis, G., Vavatsi, N., Exindari, M., Protonotariou, E., Sianou, E., Haitoglou, C., et al. (2014). Accumulation of carbapenem resistance mechanisms in VIM-2-producing Pseudomonas aeruginosa under selective pressure. Eur. J. Clin. Microbiol. Infect. Dis. 33, 253–258. doi: 10.1007/s10096-013-1952-3
Mirsalehian, A., Kalantar-Neyestanaki, D., Nourijelyani, K., Asadollahi, K., Taherikalani, M., Emaneini, M., et al. (2014). Detection of AmpC-beta-lactamases producing isolates among carbapenem resistant P. aeruginosa isolated from burn patient. Iran J. Microbiol. 6, 306–310.
Morita, Y., Cao, L., Gould, V. C., Avison, M. B., and Poole, K. (2006). nalD encodes a second repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Bacteriol. 188, 8649–8654. doi: 10.1128/jb.01342-06
Moya, B., Dotsch, A., Juan, C., Blazquez, J., Zamorano, L., Haussler, S., et al. (2009). Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 5:e1000353. doi: 10.1371/journal.ppat.1000353
Murakami, K., Ono, T., Viducic, D., Somiya, Y., Kariyama, R., Hori, K., et al. (2017). Role of psl Genes in Antibiotic Tolerance of Adherent Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 61:e002587-16. doi: 10.1128/aac.02587-16
Mushtaq, S., Ge, Y., and Livermore, D. M. (2004). Doripenem versus Pseudomonas aeruginosa in vitro: activity against characterized isolates, mutants, and transconjugants and resistance selection potential. Antimicrob. Agents Chemother. 48, 3086–3092. doi: 10.1128/aac.48.8.3086-3092.2004
Okamoto, K., Gotoh, N., and Nishino, T. (2002). Alterations of susceptibility of Pseudomonas aeruginosa by overproduction of multidrug efflux systems, MexAB-OprM, MexCD-OprJ, and MexXY/OprM to carbapenems: substrate specificities of the efflux systems. J. Infect. Chemother. 8, 371–373. doi: 10.1007/s10156-002-0193-7
Pasteran, F., Veliz, O., Rapoport, M., Guerriero, L., and Corso, A. (2011). Sensitive and specific modified Hodge test for KPC and metallo-beta- lactamase detection in Pseudomonas aeruginosa by use of a novel indicator strain, Klebsiella pneumoniae ATCC 700603. J. Clin. Microbiol. 49, 4301–4303. doi: 10.1128/jcm.05602-11
Poole, K. (2011). Pseudomonas aeruginosa: resistance to the max. Front. Microbiol. 2:65. doi: 10.3389/fmicb.2011.00065
Poole, K., Tetro, K., Zhao, Q., Neshat, S., Heinrichs, D. E., and Bianco, N. (1996). Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob. Agents Chemother. 40, 2021–2028. doi: 10.1128/aac.40.9.2021
Potron, A., Poirel, L., and Nordmann, P. (2015). Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int. J. Antimicrob. Agents 45, 568–585. doi: 10.1016/j.ijantimicag.2015.03.001
Rodriguez-Martinez, J. M., Poirel, L., and Nordmann, P. (2009). Extended-spectrum cephalosporinases in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 1766–1771. doi: 10.1128/aac.01410-08
Schmidtke, A. J., and Hanson, N. D. (2008). Role of ampD homologs in overproduction of AmpC in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 52, 3922–3927. doi: 10.1128/aac.00341-08
Schweizer, H. P. (1992). Allelic exchange in Pseudomonas aeruginosa using novel ColE1-type vectors and a family of cassettes containing a portable oriT and the counter-selectable Bacillus subtilis sacB marker. Mol. Microbiol. 6, 1195–1204. doi: 10.1111/j.1365-2958.1992.tb01558.x
Sobel, M. L., Hocquet, D., Cao, L., Plesiat, P., and Poole, K. (2005). Mutations in PA3574 (nalD) lead to increased MexAB-OprM expression and multidrug resistance in laboratory and clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49, 1782–1786. doi: 10.1128/aac.49.5.1782-1786.2005
Spilker, T., Coenye, T., Vandamme, P., and LiPuma, J. J. (2004). PCR-based assay for differentiation of Pseudomonas aeruginosa from other Pseudomonas species recovered from cystic fibrosis patients. J. Clin. Microbiol. 42, 2074–2079. doi: 10.1128/jcm.42.5.2074-2079.2004
Templin, M. F., Ursinus, A., and Holtje, J. V. (1999). A defect in cell wall recycling triggers autolysis during the stationary growth phase of Escherichia coli. EMBO J. 18, 4108–4117. doi: 10.1093/emboj/18.15.4108
Torrens, G., Hernandez, S. B., Ayala, J. A., Moya, B., Juan, C., Cava, F., et al. (2019). Regulation of AmpC-Driven beta-Lactam resistance in Pseudomonas aeruginosa: different pathways, different signaling. mSystems 4:e00524-19. doi: 10.1128/mSystems.00524-19
Tsutsumi, Y., Tomita, H., and Tanimoto, K. (2013). Identification of novel genes responsible for overexpression of ampC in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 57, 5987–5993. doi: 10.1128/aac.01291-13
Vadlamani, G., Thomas, M. D., Patel, T. R., Donald, L. J., Reeve, T. M., Stetefeld, J., et al. (2015). The beta-lactamase gene regulator AmpR is a tetramer that recognizes and binds the D-Ala-D-Ala motif of its repressor UDP-N-acetylmuramic acid (MurNAc)-pentapeptide. J. Biol. Chem. 290, 2630–2643. doi: 10.1074/jbc.M114.618199
Vincent, J. L., Bihari, D. J., Suter, P. M., Bruining, H. A., White, J., Nicolas-Chanoin, M. H., et al. (1995). The prevalence of nosocomial infection in intensive care units in Europe. results of the european prevalence of infection in intensive care (EPIC) study. EPIC international advisory committee. JAMA 274, 639–644. doi: 10.1001/jama.274.8.639
West, S. E., Schweizer, H. P., Dall, C., Sample, A. K., and Runyen-Janecky, L. J. (1994). Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 148, 81–86. doi: 10.1016/0378-1119(94)90237-2
Wilke, M. S., Heller, M., Creagh, A. L., Haynes, C. A., McIntosh, L. P., Poole, K., et al. (2008). The crystal structure of MexR from Pseudomonas aeruginosa in complex with its antirepressor ArmR. Proc. Natl. Acad. Sci. U.S.A. 105, 14832–14837. doi: 10.1073/pnas.0805489105
Wolter, D. J., Schmidtke, A. J., Hanson, N. D., and Lister, P. D. (2007). Increased expression of ampC in Pseudomonas aeruginosa mutants selected with ciprofloxacin. Antimicrob. Agents Chemother. 51, 2997–3000. doi: 10.1128/aac.00111-07
Keywords: P. aeruginosa, carbapenem resistance, oprD, ampC, ldcA
Citation: Xu C, Wang D, Zhang X, Liu H, Zhu G, Wang T, Cheng Z, Wu W, Bai F and Jin Y (2020) Mechanisms for Rapid Evolution of Carbapenem Resistance in a Clinical Isolate of Pseudomonas aeruginosa. Front. Microbiol. 11:1390. doi: 10.3389/fmicb.2020.01390
Received: 31 January 2020; Accepted: 29 May 2020;
Published: 19 June 2020.
Edited by:
Yuji Morita, Meiji Pharmaceutical University, JapanReviewed by:
Carlos Juan, Instituto de Investigación Sanitaria de Palma (IdISPa), SpainErwin Bohn, University Hospital Tübingen, Germany
Copyright © 2020 Xu, Wang, Zhang, Liu, Zhu, Wang, Cheng, Wu, Bai and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Bai, YmFpZmFuZzExMjJAbmFua2FpLmVkdS5jbg==; Yongxin Jin, eXhqaW5AbmFua2FpLmVkdS5jbg==