 Li Wan1,2
Li Wan1,2 Haican Liu2
Haican Liu2 Machao Li2Yi Jiang2Xiuqin Zhao2Zhiguang Liu2Kanglin Wan2
Machao Li2Yi Jiang2Xiuqin Zhao2Zhiguang Liu2Kanglin Wan2 Guilian Li2*Cha-xiang Guan1*
Guilian Li2*Cha-xiang Guan1*- 1Department of Physiology, Xiangya School of Medicine, Central South University, Changsha, China
- 2State Key Laboratory for Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
Development of modern genomics provides us an effective method to understand the molecular mechanism of drug resistance and diagnose drug-resistant Mycobacterium tuberculosis. In this study, mutations in 18 genes or intergenic regions acquired by whole-genome sequencing (WGS) of 183 clinical M. tuberculosis strains, including 137 multidrug-resistant and 46 pan-susceptible isolates from China, were identified and used to analyze their associations with resistance of isoniazid, rifampin, ethambutol, and streptomycin. Using the proportional method as the gold standard method, the accuracy values of WGS to predict resistance were calculated. The association between synonymous or lineage definition mutations with different genotypes were also analyzed. The results show that, compared to the phenotypic proportional method, the sensitivity and specificity of WGS for resistance detection were 94.2 and 100.0% for rifampicin (based on mutations in rpoB), 90.5 and 97.8% for isoniazid (katG), 83.0 and 97.8% for streptomycin (rpsL combined with rrs 530 loop and 912 loop), and 90.9 and 65.1% for ethambutol (embB), respectively. WGS data also showed that mutations in the inhA promoter increased only 2.2% sensitivity for INH based on mutations in katG. Synonymous mutation rpoB A1075A was confirmed to be associated with the Beijing genotype. This study confirmed that mutations in rpoB, katG, rrs 530 loop and 912 loop, and rpsL were excellent biomarkers for predicting rifampicin, isoniazid, and streptomycin resistance, respectively, and provided clues in clarifying the drug-resistance mechanism of M. tuberculosis isolates from China.
Introduction
The World Health Organization’s (WHO’s) target is to end the tuberculosis epidemic by 2035 (Treatment Action Group and Stop Tb Partnership, 2018). The evolution and spread of rifampicin-/multidrug-resistant tuberculosis (RR-TB/MDR-TB) poses a major obstacle to success with an estimated half a million cases worldwide in 2018 alone (United Nations General Assembly, 2018). WHO estimated that only one in three of the approximately half a million RR-TB/MDR-TB cases were enrolled in treatment with a second-line regimen (World Health Organization, 2019). Closing the gap in detection and treatment of resistant TB cases requires much higher coverage of drug susceptibility testing among people diagnosed with TB and rapid, accurate, and sensitive susceptibility testing methods.
Generally, the culture-based conventional drug sensitivity test (DST) has long been considered as the gold standard for diagnosing drug-resistant Mycobacterium tuberculosis although it is time-consuming, labor-intensive, and lacking sensitivity. Therefore, nucleic acid–based antibiotic susceptibility tests, which can determine the isolate’s drug resistance profile within hours instead of culture-based diagnostics that require days or weeks, are increasingly considered as a diagnostic alternative. Acquired antibiotic resistance in M. tuberculosis mostly arises from the serial acquisition of point mutations in genes encoding drug targets or drug-activating enzymes. Many known mutations have been identified, e.g., katG315 and inhA(-15) for isoniazid (INH) resistance (Liu et al., 2018; Luo et al., 2019; Unissa et al., 2016), 81-bp of rifampicin (RMP) resistant determined region (RRDR) for RMP resistance (Li et al., 2010; Maningi et al., 2018; Luo et al., 2019), embB306 for ethambutol (EMB) resistance (Mokrousov et al., 2002; Campbell et al., 2011; Moure et al., 2014; Zhao et al., 2015a), and rpsL44 and rpsL88 for streptomycin (STR) resistance (Li et al., 2010; Maningi et al., 2018). Current molecular diagnostics amplify and detect known drug resistance–associated mutations, and their performance depends on the inclusion of a comprehensive catalog of these mutations. Although known mutations explain much resistance in M. tuberculosis, causative mutations have not been identified in 10–40% of clinically resistant isolates (Campbell et al., 2011; Zhang et al., 2014; Zhao et al., 2014a), implying the contribution of noncanonical mutations in known or unknown genes or other resistance mechanism(s). Mutations outside of RRDR in rpoB or mutations except the canonical mutation katG315 in katG have been reported to be associated with RMP or INH resistance, respectively (Zhao et al., 2014b; Torres et al., 2015). Maningi et al. (2018) reports that whole-genome sequencing (WGS) shows better concordance with the Lowenstein–Jensen (L-J) phenotypic assay than with Hain line probe assay in that many more mutations were found by WGS. Moreover, many genes, such as ndh, efpA, kasA, iniABC operon (for INH resistance) (Sandgren et al., 2009; Nguyen, 2016); rpoC (RMP) (Farhat et al., 2013; Perdigao et al., 2020); embA, embC, ubiA (EMB) (Plinke et al., 2010; Zhao et al., 2015a; Farhat et al., 2016); and gidB (STR) (Nhu et al., 2012; Spies et al., 2011; Perdigao et al., 2014), are reported to correlate with drug resistance.
Whole-genome sequencing enables the screening of known resistance-associated loci while also providing opportunities to characterize other loci as predictive of resistance or not (Farhat et al., 2013; Casali et al., 2014). In this study, we analyze the sequence polymorphisms in 18 chosen genes or regions of 183 M. tuberculosis isolates based on the whole-genome sequenced data. The sequences or regions were chosen on the basis of their demonstrated association with drug resistance and according to the TB Drug Resistance Database (Sandgren et al., 2009).
Materials and Methods
Strains
We used 183 M. tuberculosis isolates for WGS. H37Rv (ATCC 27294), which is susceptible to the four first-line anti-tuberculosis drugs INH, RMP, EMB, and pyrazinamide (i.e., pansusceptible), was used as a reference. The isolates used for WGS were obtained from 183 adult patients with pulmonary TB from 2005 to 2009 from institutes for tuberculosis control and prevention as well as tuberculosis hospitals distributed in 11 provincial-level administration divisions (PLADs) of China; the numbers isolated from each PLAD were as follows: Beijing, 13; Fujian, 24; Guangdong, 8; Guizhou, 21; Henan, 6; Inner Mongolia, 5; Liaoning, 20; Shaanxi, 25; Shanghai, 26; Tibet, 30; and Xinjiang, 5.
Drug Susceptibility Testing and Mycobacterium Species Identification
The isolate profiles of drug susceptibility were evaluated in our laboratory by the proportional method using L-J slants with the following drug concentrations: INH, 0.2 μg/mL; RMP, 40 μg/mL; STR, 4 μg/mL; EMB, 2 μg/mL; kanamycin (KAN), 30 μg/mL; ofloxacin (OFX), 2 μg/mL; capreomycin (CAP), 40 μg/mL (World Health Organization, 2008). L-J medium containing para-nitrobenzoic acid (500 μg/mL) was used to identify M. tuberculosis complex species from non-tuberculosis mycobacteria, and medium containing thiophen-2-carboxylic acid hydrazide (5 μg/mL) was used to exclude Mycobacterium bovis (M. bovis) from the M. tuberculosis complex. This study included the M. tuberculosis complex but did not include M. bovis clinical isolates. All the strains were stored in physiological saline containing 50% glycerol at −70°C. Prior to characterizing the drug susceptibility, the strains were recovered on L-J medium for 4 weeks at 37°C. DST, mycobacterium species identification, and inactivation of strains were all performed in a Biosafety Level 2 Laboratory.
Genome Sequencing
Genomic DNA was extracted from M. tuberculosis colonies on L-J medium using CTAB (van Embden et al., 1993). DNA libraries (350 bp insert) were constructed with genomic DNA using kits provided by Illumina according to the manufacturer’s instructions. DNA libraries were then selected to perform cluster growth and 90 bp paired-end sequencing on an Illumina HiSeq 2000 sequencer according to standard protocols. Raw reads with consecutive bases covered by fewer than five reads, duplicate reads, and the adapter were removed; then, the rest of the reads, called clean data, from each strain were mapped to the genome of H37Rv (GenBank accession number, NC_000962.2) using SOAP2 (Li et al., 2009). An average of 5.27 million sequence reads were acquired per genome at a depth of 112× and with coverage of 99.32%. The accuracy of the sequencing was assessed by sequencing rpoB in a random selection of 80 isolates on an ABI Prism 3730 automated DNA sequencer (ABI, Shirley, NY, United States) as described by Zhang et al. (2013), the results show 100% consensus between the Illumina HiSeq 2000 and ABI 3730 sequencing results.
Identify Mutations in Drug Resistance–Associated Genes and Promoter Regions
Identification of resistance-causing single-nucleotide polymorphisms (SNPs) from genome-wide sequence is challenging. We focused on putative or known resistance genes and promoter regions on the basis of their demonstrated association with drug resistance and according to the TB Drug Resistance Database (Sandgren et al., 2009; Table 1). All mutations in these genes and promoter regions were compared with the pan-susceptible reference genome (H37Rv, accession number: NC_000962.2) at the level of SNPs in promoter regions, amino acids in genes, or insertions and deletions. In this study, we first characterized the synonymous and lineage-defining mutations that did not cause resistance, and then, we characterized the mutations associated with drug resistance. The phenotypic and genotypic results were compared to determine the specificity and sensitivity for each gene or region studied.
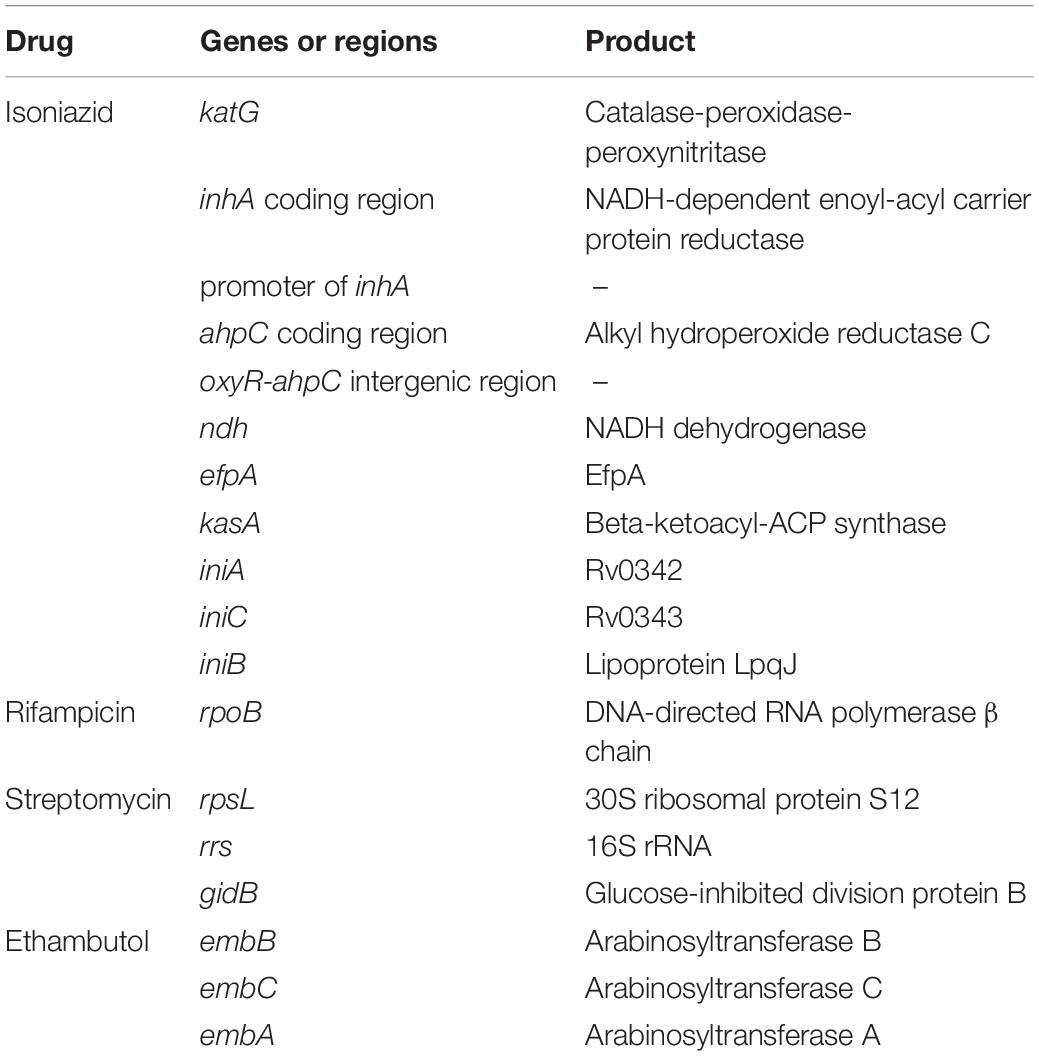
Table 1. Eighteen candidate genes and intergenic regions linked with acquisition of drug resistance.
Spoligotyping and Data Analysis
Spoligotyping was performed using 43 covalently bound oligonucleotides derived from the spacer sequences of M. tuberculosis H37Rv and M. bovis BCG P3 as previously described by Kamerbeek et al. (1997). The results in binary format were entered into an Excel spreadsheet and compared with the spoligotyping database SpolDB4.1
Statistical Analysis
SPSS 16.0 (SPSS Inc., Chicago, IL, Untied States) was used to perform Chi-square and Fisher’s exact analysis according to the sample number and multivariate regression analysis. The difference was considered to be statistically significant when P < 0.05.
Results
Drug Susceptibility Patterns
All isolates for WGS underwent culture-based DST to seven drugs: 46 were fully drug susceptible; 137 were resistant to both INH and RMP. Among 137 MDR strains, 100 (73.0%), 77 (56.2%), 60 (43.8%), 22 (16.1%), and 21 (15.3%) were resistant to STR, EMB, OFX, KAN, and CAP, respectively; 18% (24/137) of the MDR isolates were extensively drug resistant (XDR, MDR isolates are also resistant to both fluoroquinolone and an injectable drug). Detailed susceptibility profiles are shown in Supplementary Table 1.
Genotype Distribution of the M. tuberculosis Isolates
Among the M. tuberculosis isolates for WGS, 141 (77.0%) belonged to the Beijing genotype, and 42 (23.0%) were non-Beijing family, which included the T1 family (12), H3 family (3), T2 family (3), CAS family (2), Haarlem3 family (2), MANU2 family (2), CAS1-DELH1 family (1), LAM9 family (1), U family (1), and a new found genotype (12).
Synonymous Mutations in Chosen Genes and Regions
A total of 55 synonymous mutations were found and are shown in Supplementary Table 2. Although synonymous mutations were universally acknowledged to be unrelated with drug resistance, we found that the prevalence of rpoB 1075 GCT-GCC (Ala-Ala) in RMP-resistant M. tuberculosis was higher than that in RMP-susceptible isolates, and the prevalence of gidB 205 GCA-GCG (Ala-Ala) in STR-resistant isolates was higher than that in STR-susceptible isolates. Among 55 synonymous mutations, 17 were found only in interested drug–susceptible isolates, 27 were found only in interested drug–resistant isolates, and the remaining 11 were found in both interested drug–susceptible and –resistant isolates. Sixteen out of the 27 synonymous mutations were found in the 11 genes or regions related with INH resistance in INH-resistant isolates. None of the stand-alone synonymous mutations in the known genes katG (INH), rpoB (RMP), and rpsL (STR) was found only in isolates resistant to INH, RMP, and STR, respectively, and one stand-alone synonymous mutation 304 CTG-TTG (L-L) in the known gene embB was only found in one EMB-resistant isolate.
We further analyzed the associations between the 55 synonymous mutations and the Beijing genotype and found that the prevalence of iniA 178 GGT-GGC (Gly-Gly) and embA 1092 GCG-GCA (Ala-Ala) was much higher in the non-Beijing genotype than in the Beijing genotype M. tuberculosis, and the prevalence of rpoB 1075 GCT-GCC (Ala-Ala), embA 76 TGC-TGT (Cys-Cys), and gidB 205 GCA-GCG (Ala-Ala) was much higher in the Beijing genotype than in the non-Beijing genotype. Among 55 synonymous mutations, 31 were only found in Beijing genotype strains, 17 were only found in the non-Beijing genotype, and seven were found in both genotypes. The results are shown in Supplementary Table 3.
We then performed multivariate analysis toward the mutations of rpoB 1075 GCT-GCC (Ala-Ala) and gidB 205 GCA-GCG (Ala-Ala) based on the univariate analysis. As shown in Table 2, the analysis data revealed that the Beijing genotype is the high-risk factor for these two synonymous mutations, not the RMP or STR resistance.
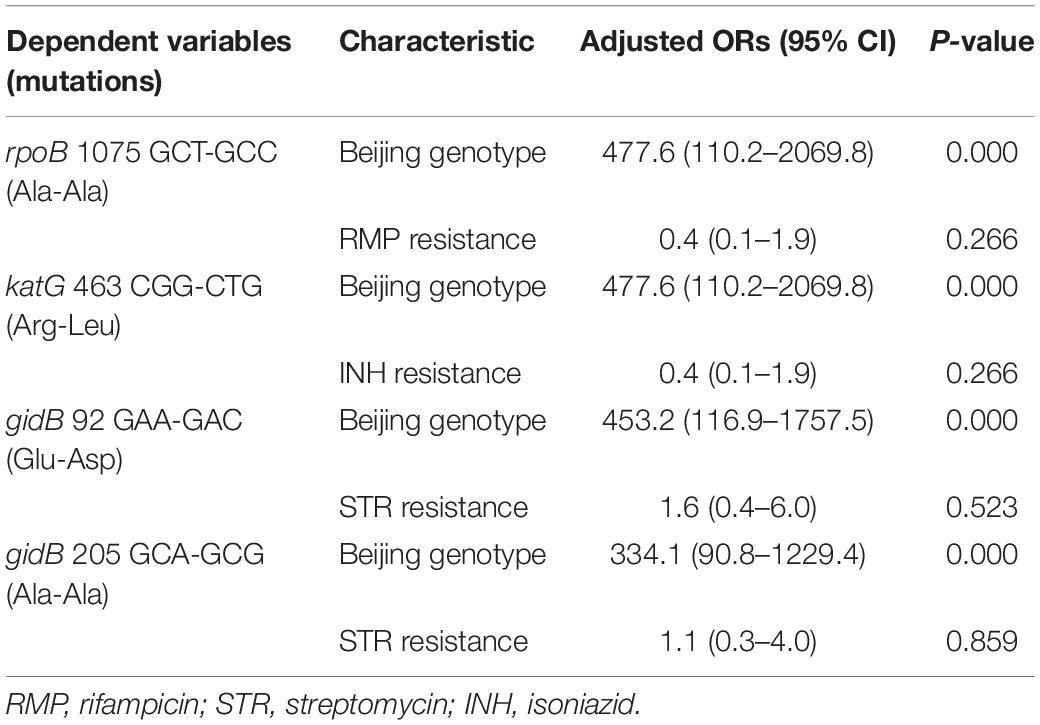
Table 2. Multivariate analysis of drug resistance and genotypes of M. tuberculosis according to the four mutations.
Analysis on Mutations katG R463L and gidB E92D Known Not to Code for Resistance
In the present study, 146 and 143 out of 183 isolates carried mutations katG 463 CGG-CTG (Arg-Leu) and gidB 92 GAA-GAC (Glu-Asp), respectively. According to previous studies, both mutations are known not to be associated with resistance (Meacci et al., 2005; Via et al., 2010; Feuerriegel et al., 2014; Nebenzahl-Guimaraes et al., 2014), and gidB92 polymorphism (276C allele) has been reported to be associated with the Beijing lineage (Spies et al., 2011). However, statistical analysis shows that the prevalence of katG 463 CGG-CTG (Arg-Leu) in INH-resistant isolates are higher than that in INH-susceptible isolates, and the prevalence of gidB 92 GAA-GAC (Glu-Asp) in STR-resistant isolates is higher than that in STR-susceptible isolates; both P-values were less than 0.05 (see Supplementary Table 4). We also found that the prevalence of katG 463 CGG-CTG (Arg-Leu) and gidB 92 GAA-GAC (Glu-Asp) in the Beijing genotype is much higher than in the non-Beijing genotype isolates; see Supplementary Table 5.
Multivariate analysis toward the mutations of katG 463 CGG-CTG (Arg-Leu) and gidB 92 GAA-GAC (Glu-Asp) based on the univariate analysis shows that the Beijing genotype is the high-risk factor for both mutations, not the INH resistance or STR resistance (Table 2).
Drug Resistance and Gene Mutations
Isoniazid Resistance and Mutations in Genes and Intergenic Regions
Whole-genome sequencing data shows that, respectively, among 137 INH-resistant and 46 INH-susceptible isolates, 124 and 1 carry mutations in katG, 28 and 0 in the inhA promoter, and 12 and 2 in the oxyR-ahpC intergenic region (Table 3). We found a rare high prevalence (90.5%) of the katG mutation in INH-resistant M. tuberculosis, which has never been reported in China. katG mutations combined with that in the inhA promoter only increased the sensitivity from 90.5 to 92.7% while there was no additional specificity (Figure 1). At the base of mutations in katG and the inhA promoter, adding the mutations in the oxyR-ahpC intergenic region, the sensitivity was not changed although the specificity fell from 97.8 to 93.5% as shown in Figure 1. The most frequent mutation site was katG315 (92/137, 67.2%); among the isolates that carried this mutation, only 14 combined mutations in the inhA promoter and/or the oxyR-ahpC intergenic region; among 32 INH-resistant isolates, which carried mutations in katG non-315, 21 combined mutations in the inhA promoter and/or the oxyR-ahpC intergenic region; among 13 INH-resistant isolates, which carried the wild-type of katG, three carried mutations in the inhA promoter and/or the oxyR-ahpC intergenic region, four carried mutations only in the other eight genes related to INH resistance, and there were still six INH-resistant strains with the wild-type of 11 sequenced genes that have been reported to be associated with INH resistance; see Figure 2. Among 28 INH-resistant isolates that carried mutations in the inhA promoter, 11 carried mutations of katG315, 14 carried mutations in katG but not in codon 315, and three carried wild-type katG.
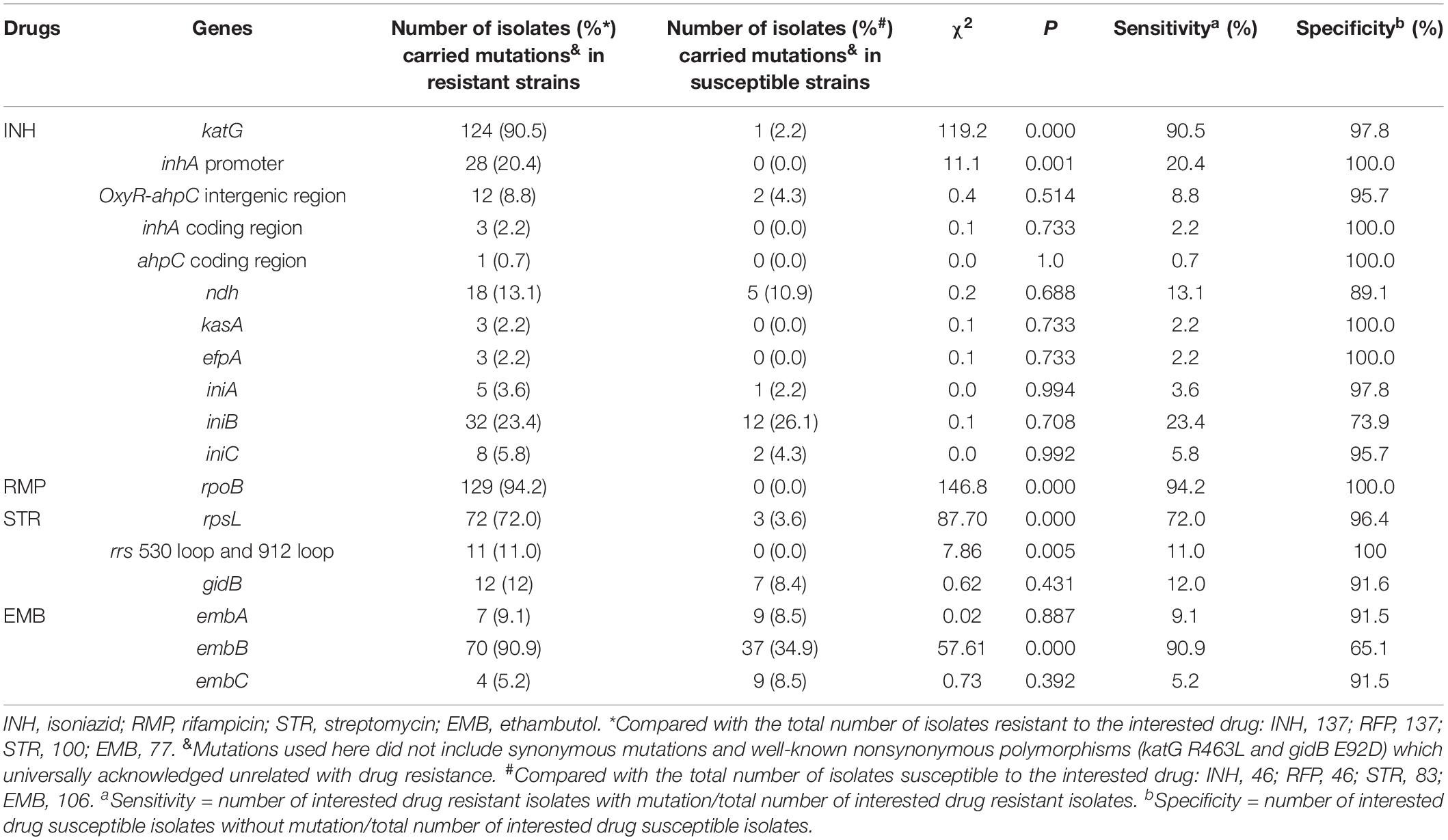
Table 3. The evaluation between whole-genome sequencing analysis of 18 drug-resistant associated genes or regions and the phenotypic drug susceptibility testing.
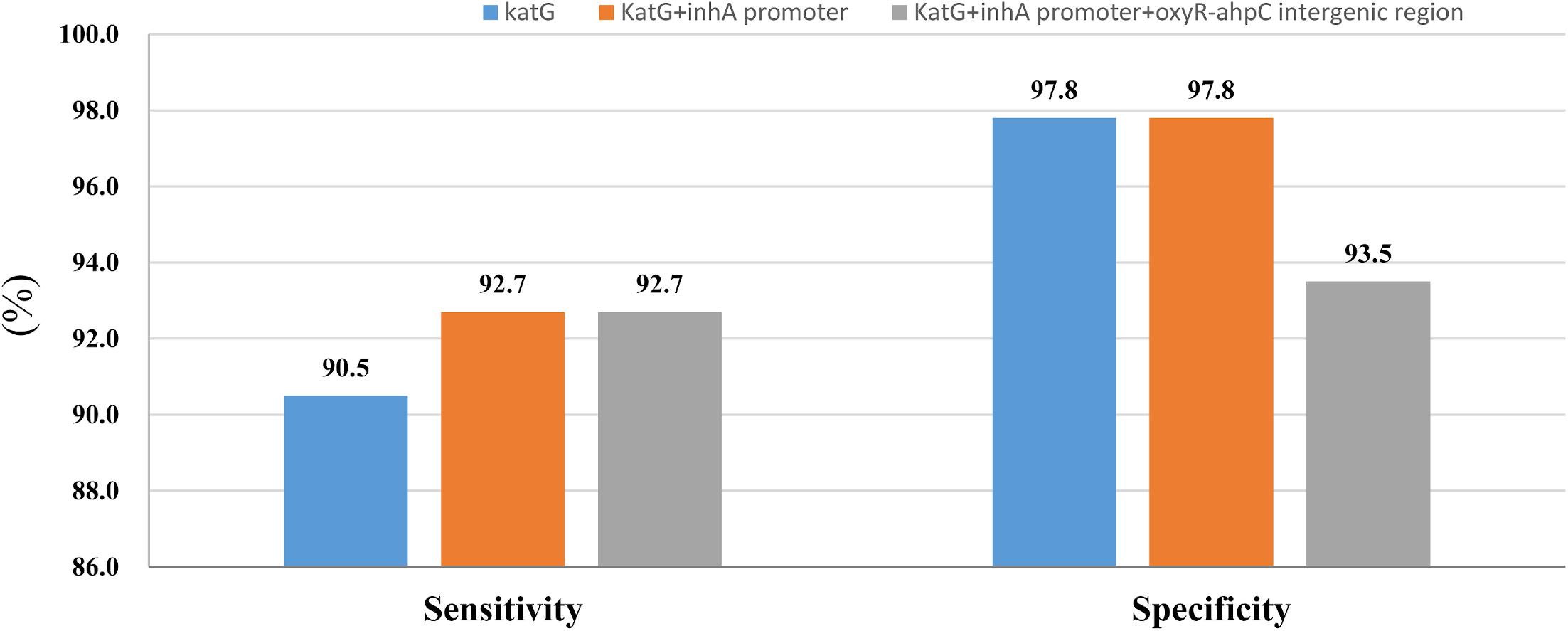
Figure 1. Sensitivity and specificity of sequencing of katG combined with inhA promoter and oxyR-ahpC intergenic region for isoniazid resistance and susceptibility diagnoses.
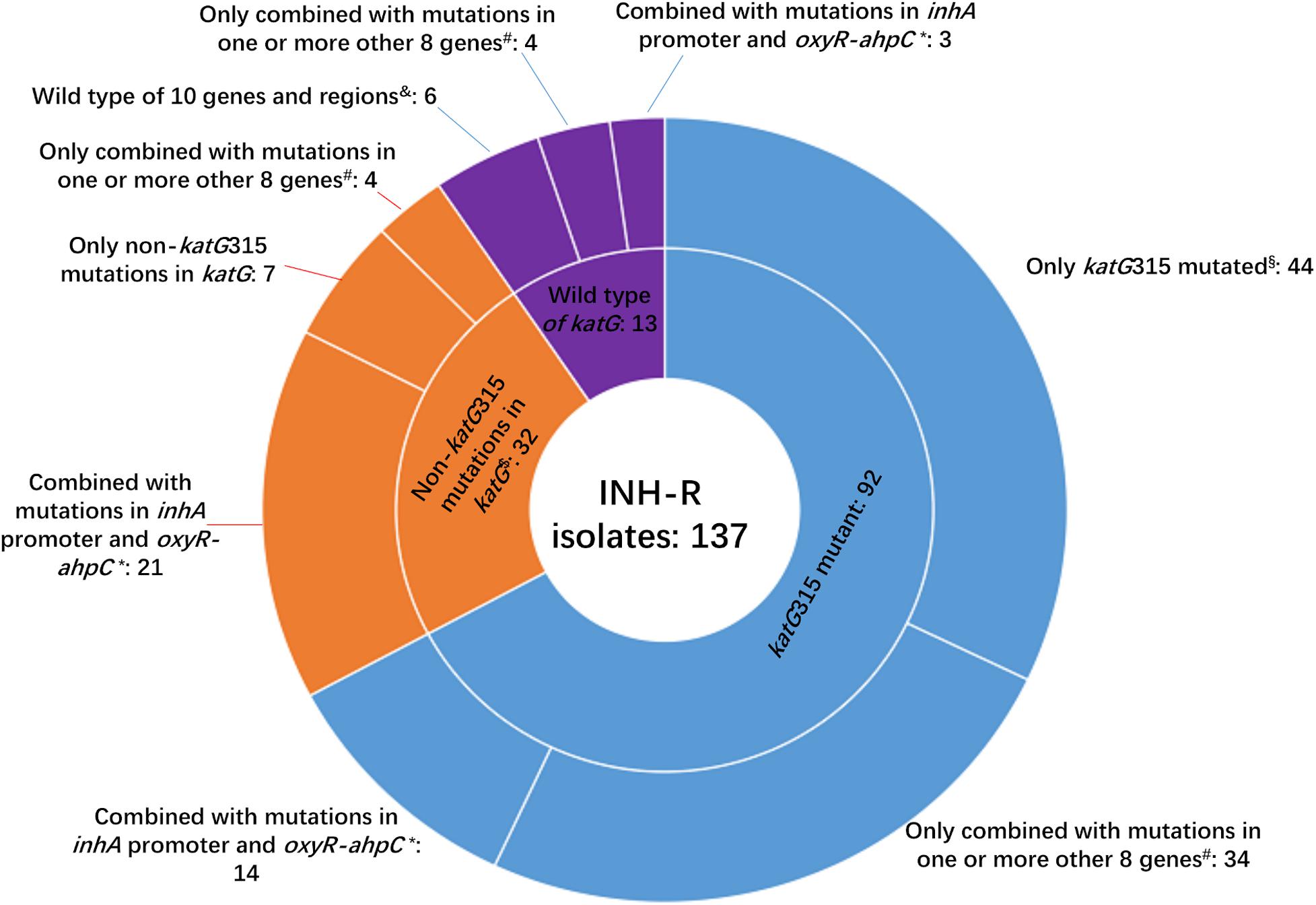
Figure 2. Number of isolates carrying katG mutations combined with mutations in another 10 genes or intergenic regions among 137 MDR M. tuberculosis. INH-R, isoniazid resistant; *this group included isoniazid-resistant isolates that also carried mutations in ahpC coding region, inhA coding region, ndh, efpA, kasA, iniA, iniB, and/or iniC. #Eight genes included ahpC coding region, inhA coding region, ndh, efpA, kasA, iniA, iniB, and iniC; $two isolates that had mutations of katG315 and other substitutes in katG were not included in this group. § This group included two isolates that had mutations of katG315 and other substitutes in katG; &10 genes and regions included inhA promoter, oxyR-ahpC intergenic region, ahpC coding region, inhA coding region, ndh, efpA, kasA, iniA, iniB, and iniC.
In the present study, a total of 29 mutation sites in katG except katG315 were found in 34 INH-resistant isolates of which two were combined with the mutation of katG315 (Figure 2 and Supplementary Table 6). We also found that 23 out of 45 isolates possessed katG non-315 mutations, or wild-type katG carried inhA C(-15)T and/or mutations in the region of ahpC from −84 to −48, which occupied 16.8% of the INH-resistant isolates. So the inhA(-15) and ahpC−84 to −48 combined with katG315 can make a preferable set for INH-resistance diagnoses, and the mutation sites in katG except 315 were scattered, which made these codons have less diagnostic value when used in gene chip, line/dot-blot hybridization or multiplex fluorescence melting curve analyses.
Besides katG, the inhA promoter, and oxyR-ahpC intergenic region, eight genes (coding regions) were also included to clarify the mechanism of INH resistance. The strain numbers that carried mutations in eight studied genes (coding regions) in INH-resistant and -susceptible isolates were as follows, respectively: inhA coding region, 3 and 0; ahpC coding region, 1 and 0; ndh, 18 and 5; kasA, 3 and 0; efpA, 3 and 0; iniA, 5 and 1; iniB, 32 and 12; iniC, 8 and 2 (Table 3). Low mutation prevalence of these genes in INH-resistant strains and mutations has even been found in INH-susceptible strains, which made the associations confused between these genes and INH resistance.
As shown in Table 4, we found 18 novel mutations, which included 10 in the gene katG, three in the oxyR-ahpC intergenic region, one in the ahpC coding region, two in the kasA gene, and four in the efpA gene.
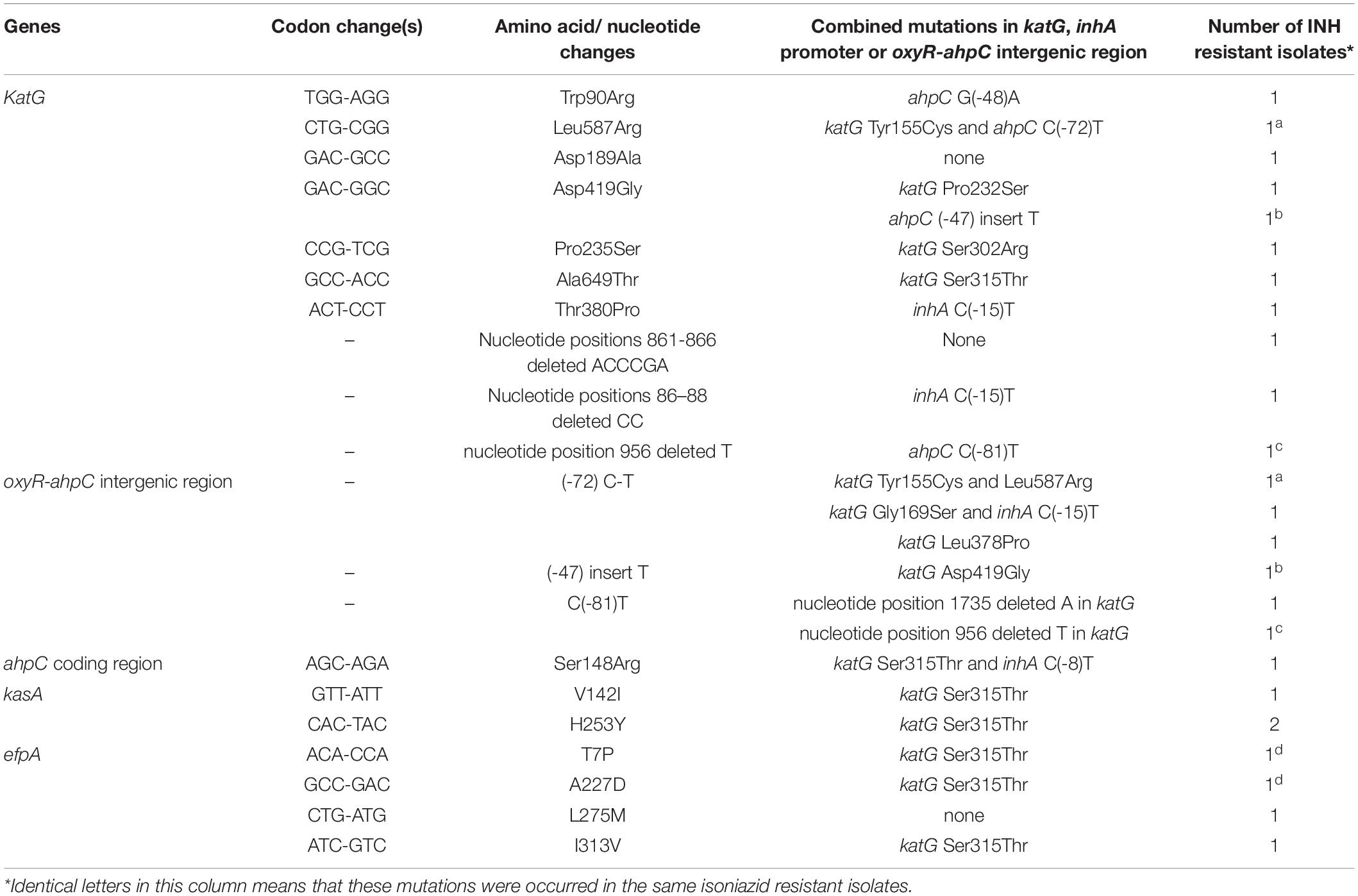
Table 4. Novel mutations occurred in genes and regions associated with isoniazid resistance and found only in phenotypic isoniazid resistant M. tuberculosis.
Rifampicin Resistance and rpoB Mutations
The whole rpoB sequence of 183 M. tuberculosis was analyzed. Altogether, 94.2% (129/137) of the RMP-resistant isolates harbored at least one mutation within rpoB, and other eight isolates lacked such a mutation (Supplementary Table 7). Eighty-nine isolates (65%) had a single mutation, and 48 (35%) had two or more mutations each. When all of the mutations were considered, regardless of single, double, or more, a total of 55 genotype patterns were identified, and 127 out of 137 RMP-resistant M. tuberculosis isolates carried mutations in the 81-bp RRDR of the rpoB gene. The most frequently mutated codons were 450 (Escherichia coli 531), 445 (E. coli 526), and 435 (E. coli 516) with mutation frequencies of 51.1% (70/137 isolates), 23.4% (32/137 isolates), and 12.4% (17/137 isolates) (Table 5). An independent and novel mutation was detected in rpoB: 675 GGC-GAC (Gly-Glu), which was only found in an RMP-resistant isolate. In contrast, none of 46 susceptible isolates possessed a nonsynonymous mutation within the whole sequence of the rpoB gene.
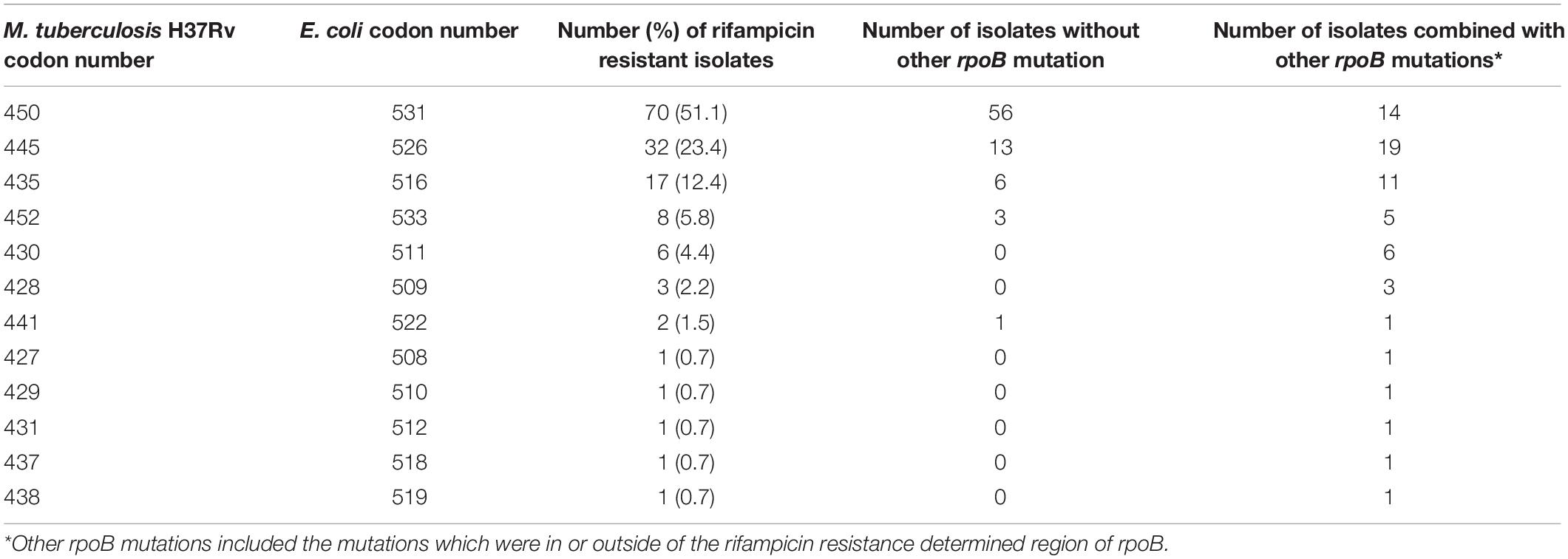
Table 5. Mutation frequency of codons of rpoB 81-bp rifampicin resistance determined region among 137 MDR isolates from China.
Streptomycin Resistance and Mutations in rpsL, rrs, and gidB
Previous studies show that mutations in loop 530 and loop 912 of rrs are associated with STR resistance (Finken et al., 1993; Sreevatsan et al., 1996; Li et al., 2010), so we only analyzed the mutations in these two regions of rrs and the whole sequence of rpsL and gidB in this study.
The WGS data shows that 72, 11, and 12 out of 100 STR-resistant isolates carried mutations in rpsL, rrs 530 loop, or 912 loop and gidB, respectively; in contrast, 3, 0, and 7 out of 83 STR-susceptible isolates carried, respectively (Table 3). None of the STR-resistant isolates were found to carry mutations simultaneously in both rpsL and rrs 530 loop or 912 loop. The sensitivity and susceptibility of mutations in rpsL combined with rrs 530 loop and 912 loop were 83.0% (83/100) and 92.8% (77/83). We also found that, among 83 STR-susceptible isolates, eight carried mutations in rrs outside of the 530 loop or 912 loop: one carried a mutation with nucleotides in positions 334–344 deleted, four carried mutations with nucleotides in positions 388–394 deleted, one carried a mutation of 555 A-T, and two carried mutations with 846 C-T and 1017 G-C. Among these eight isolates, seven were susceptible to INH, RMP, STR, EMB, CPM, KAN, and OFX.
As shown in Table 6, for rpsL, the most frequently mutated codons were 43 and 88 with mutation frequencies of 50.0% (50/100 isolates) and 19.0% (19/100 isolates), respectively. For the 530 loop and 912 loop, the most frequently mutated positions were 515, 518, and 888 with mutation frequencies of 8.0% (8/100 isolates), 2.0% (2/100 isolates), and 1.0% (1/100 isolates), respectively. In the present study, no isolate was found to carry mutations in rpsL combined with rrs 530 loop or 912 loop although one STR-resistant isolate was found to carry mutations of rpsL Lys88Arg and rrs 38 G-A. The mutation results of gidB were confused as shown in Table 3 and Supplementary Table 8; both STR-resistant and susceptible isolates carried mutations, and the mutated codons were scattered throughout the gene.
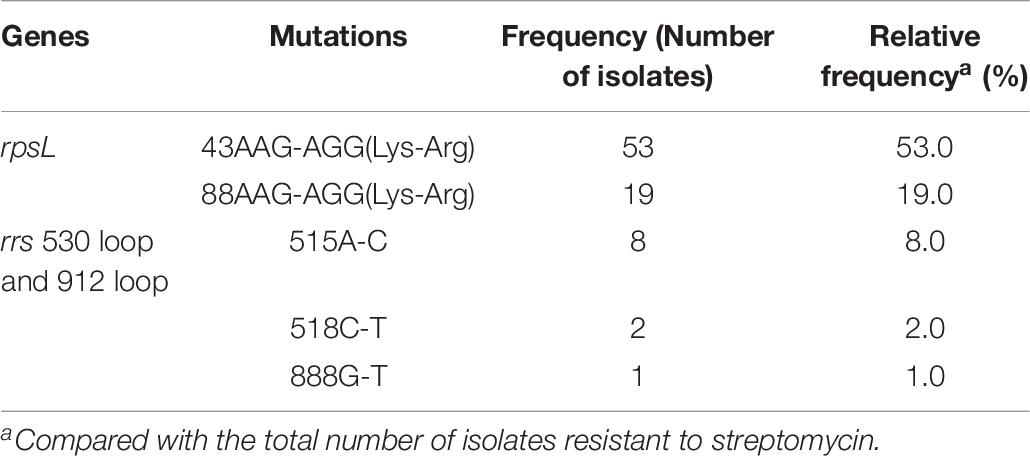
Table 6. Mutation characterizations of rpsL and rrs 530 loop and 912 loop among 100 streptomycin resistant M. tuberculosis from China.
Ethambutol Resistance and Mutations in embB, embA, and embC
Known mechanisms of EMB resistance are caused by mutations in the embCAB operon, especially in embB. The whole sequences of embA, embB, and embC were analyzed in this study. The results show that, among 77 EMB-resistant isolates, there were 7, 70, and 4 carried mutations in embA, embB, and embC, respectively, and among 106 EMB-susceptible isolates, there were 9, 37, and 9 carried mutations in embA, embB, and embC, respectively (Table 3).
The most predominant mutation in embB occurred at codon 306 (45, 58.4%), where the codon ATG (Met) was replaced with GTG (Val, 26, 33.8%), ATA (Ile, 11, 14.3%), ATC (Ile, 4, 5.2%), CTG (Leu, 2, 2.6%), ATT (Ile, 1, 1.3%), and GTA (Val,1, 1.3%), respectively. Mutation of embB Gly406 was the next most predominant mutation (9, 11.7%). Among the EMB-susceptible isolates, mutations in Met306 and Gly406 were also the most predominant in embB. As shown in Supplementary Table 9, among the EMB-resistant isolates, all of the mutations in embA or embC combined with mutations in embB and the mutated codons scattered in embA and embC.
Discussion
The present study is the first to comprehensively analyze the whole sequences of 18 drug resistance–associated genes or intergenic regions of 183 M. tuberculosis isolates, which include MDR-TB and XDR-TB isolates from 11 provinces of China. The results suggest that sequencing the whole sequence of four genes can now characterize profiles of resistance to INH, RMP, and STR with an acceptable degree of accuracy sufficient for clinical use. Furthermore, more drug resistance–associated loci and Beijing genotype–associated loci were found in our research.
Compared to the molecular assays of DST, such as line-probe arrays, PCR methods based on TaqMan probes, or melting curves (Boehme et al., 2010; Chen et al., 2014; Zhao et al., 2015b; World Health Organization, 2016; Makhado et al., 2018; Cao et al., 2019), WGS displays better performance to predict drug resistance according to the limited known mutations as well as a big catalog of mutations in various genes. However, among the mutations found by WGS, synonymous mutations, mutations of lineage markers, and some well-known nonsynonymous polymorphisms (e.g., katG R463L, gyrA S95T) (Feuerriegel et al., 2014; Zhao et al., 2014b), which are universally acknowledged to be unrelated with drug resistance, must be excluded before predicting resistance. In the present study, we found that both gidB E92D and katG R463L occurred in almost all of the Beijing genotype strains although they sparsely occurred in strains with CAS, MANU, and new genotypes. As reported by previous studies (Jagielski et al., 2014; Sun et al., 2016), we also found that the nonsynonymous mutations gidB E92D and the synonymous mutation gidB A205A were associated with the Beijing genotype. The synonymous mutations rpoB A1075A and embA C76C were associated with the Beijing genotype, and iniA G178G and embB A1092A were associated with the non-Beijing genotype, which were first reported by us. The remaining 50 synonymous mutations were not found to have statistics association with drug resistance or genotypes.
It is well known that the mechanism of action of INH, which has a simple chemical structure, is very complex, and several bactericidal strategies have been reported (Timmins and Deretic, 2006). Consequently, several genes in multiple biosynthetic networks and pathways involved in INH action have been reported to play a role in INH resistance (Unissa et al., 2016). Mutations in the katG gene are the major contributors for INH resistance, followed by inhA, ahpC, kasA, ndh, iniABC, efpA, fadE, furA, Rv1592c, and Rv1772 (Unissa et al., 2016). In the present study, There were 90.5% INH-resistant isolates that carried mutations in katG, which was higher than the 86.2% reported by Liu et al. (2018) and the 74.5% reported by Luo et al. (2019). Liu et al. (2018) reported that 47.1% (16/34) INH-resistant isolates had inhA promoter mutations combined with a mutation in katG, which was far lower than the 89.3% (25/28) found in our study. One explanation may be that they only sequenced 518 bp of katG while we analyzed the whole sequence of katG. In the present study, 29 additional mutations except katG315 were found in katG, of which 10 were novel mutations, and only three novel mutations combined with katG315 or inhA(-15) mutations. All of these novel mutations were found only in phenotypic INH-resistant isolates, suggesting that these mutations were resistance-associated but needed to be further verified by site-directed mutagenesis or other experiments. Many non-katG315 mutations in the katG gene, e.g., Y95C, P131T, D142G, A162V, T306P, Y64S, F483L, and A541D, have been confirmed causing INH resistance, and katG R385W and D387G did not play a role in INH resistance by in vitro mutagenesis experiments (Torres et al., 2015).
In the present study, the mutation rates in inhA and ahpC coding regions, kasA and efpA in INH-resistant isolates were low. To make sure these mutations are associated with INH resistance, more studies for the phenotypic effect of these mutations are required. Another four genes (ndh, iniA, iniB, and iniC) show similar mutation frequency in both INH-resistant and -susceptible M. tuberculosis. The total mutation frequency of ndh, iniA, iniB, and iniC were 12.6% (23/183), 3.3% (6/183), 24% (44/183), and 5.5% (10/183), respectively. The mutation rate of ndh in INH-resistant isolates was higher than that reported by Islam et al. (13.1 vs. 2.9%) (Islam et al., 2019). Many reports have shown that isolates with mutations in iniABC had mutations in other genes as well (Ramaswamy et al., 2003), which was similar to our findings. However, the high mutation frequency of iniABC among clinical isolates in our study has never been reported. Previous studies have shown that IniA, IniB, and IniC were proteins that can be induced by isoniazid (Colangeli et al., 2005; Unissa et al., 2016). We speculated that there was a joint mechanism between efflux pumps and acquired mutations, e.g., iniABC and katG, and the accumulation of mutations under the pressure of drug selection may contribute to the appearance of INH resistance. Such knowledge of other genes (apart from katG) aids in developing better means to diagnose and prevent the transmission of INH-resistant tuberculosis.
Previous studies show that mutations in RRDR of rpoB account for 90% or higher RMP resistance (Li et al., 2010; Zhao et al., 2014b; Luo et al., 2019). We found 92.7% of RMP-resistant isolates carried mutations in this area, which is concordant with previous studies (Li et al., 2010; Zhao et al., 2014b; Luo et al., 2019). Only two RMP isolates carried mutations outside of RRDR: one carried a novel independent mutation of 675 GGC-GAC (Gly-Glu); the other isolate carried two substitutions of 170 GTC-TTC (Val-Phe) and 920 ATG-GTG (Met-Val). The most common mutations of the rpoB gene were in codons 450 (E. coli 531), 435 (E. coli 526), and 445 (E. coli 516) (Table 5); the results are consistent with other studies performed in China and other countries that reported the same trends (Li et al., 2010; Maningi et al., 2018; Luo et al., 2019). However, data from WGS in the present study reveal that 40 additional mutations outside of the RRDR region of rpoB are found in RMP-resistant phenotypes, and most of them are shown in the form of joint mutations with codons in RRDR, which may explain why isolates carrying the same mutation patterns in RRDR show different minimum inhibitory concentrations against RMP. Mutations in rpoB cannot answer the 5.8% RMP resistance in the present study. Previous studies show that substitutions in rpoC were frequent in RMP-resistant isolates; however, most rpoC substitutions combined with mutations in rpoB and are recognized as a modification or compensation for the phenotypes of mutations in the rpoB (Farhat et al., 2013; Perdigao et al., 2020).
Resistance to STR is due mostly to mutations in the rpsL, followed by mutations in rrs 530 loop or 912 loop. Only two mutations, Lys43Arg and Lys88Arg in rpsL, were found in STR-resistant isolates, similar to the data from other areas in China (Li et al., 2010) and South Africa (Maningi et al., 2018). The most common mutations of the rrs were A515C, C518T, and G888T, which are located in the rrs 530 loop or 912 loop. Eight out of 83 STR-susceptible isolates carried mutations outside of the rrs 530 loop or 912 loop, suggesting that these mutations are not related to STR resistance. Recently, mutations in gidB were reported to cause STR resistance (Perdigao et al., 2014; Verma et al., 2014). gidB mutations were found in both STR-resistant and -susceptible strains in the present study, consistent with data from previous studies (Nhu et al., 2012; Spies et al., 2011). Among 17 STR-resistant isolates without mutations in the rpsL or rrs 530 loop or 912 loop, seven carried gidB mutations found in six codons, of which one mutation (K163stop codon) was found in two STR-resistant isolates, and one mutation (nucleotide 120 C deleted) was also found in three STR-susceptible isolates. The results suggest that mutations in gidB do not help to explain STR resistance in isolates without the rpsL or rrs 530 loop and 912 loop mutations. The overexpressed proteins in STR-resistant isolates identified by Sharma and Bisht (2017b) are assumed to be responsible for STR resistance; however, the corresponding genes were not analyzed in the present study.
Resistance to EMB is mostly attributed to mutations in codon 306 in embB, accounting for 48.3–70.6% resistant isolates (Mokrousov et al., 2002; Campbell et al., 2011; Moure et al., 2014; Zhao et al., 2015a). In the present study, 58.4% EMB-resistant isolates carried mutations of embB306, in line with previous studies (Mokrousov et al., 2002; Campbell et al., 2011; Moure et al., 2014; Zhao et al., 2015a). Mutations of embB406 were found in 11.7% EMB-resistant isolates. WGS data from the present study found 19 more mutations besides the two canonical mutations in 16 EMB-resistant isolates, of which four isolates had double noncanonical mutations and one combined with embB M306I. embB mutations were found in 90.9% EMB-resistant isolates while previous studies ranged from 38.2 to 89.9% (Li et al., 2010; Campbell et al., 2011; Moure et al., 2014; Zhao et al., 2015a). However, 34.9% of EMB-susceptible isolates also carried mutations in embB, which is much higher that a previous study (6.5%) (CRyPTIC Consortium and the 100, 000 Genomes Project et al., 2018). One explanation may be that previous studies showed that the conventional DST method for EMB resistance was an imperfect standard, particularly for isolates with embB mutations (Zhang et al., 2009; Walker et al., 2015; Schon et al., 2017). A previous study in our laboratory shows that, using the EMB concentration with 1.6 μg/mL instead of 2.0 μg/mL in L-J slants by the proportional method, 23 out of 28 EMB-susceptible isolates that carried emb306 mutations could be successfully recognized as EMB-resistant isolates while the susceptibility patterns of 26 EMB susceptible isolates with wild-type embB were not affected (Zhang et al., 2009). A recent study shows that embB mutations are also associated with INH resistance in EMB-susceptible isolates (Wan et al., 2020) and another study shows that the embB M306I and M306V mutations are significantly associated with INH resistance in both EMB-resistant and -susceptible strains (Farhat et al., 2016). We speculate that the ambiguous relationship between mutations in embB and EMB or INH resistance may also lower the specificity of embB for predicting EMB resistance.
Mutations in embC and embA, which are suggested to be involved in EMB resistance development (Plinke et al., 2010; Zhao et al., 2015a; Farhat et al., 2016) were also analyzed in the present study; all of the isolates carried mutations in embC or embA combined with mutations in embB, and the prevalence of mutations in these two genes among EMB-resistant and -susceptible isolates were comparable. Therefore, 9.1% EMB resistance cannot be explained by embC, embA and embB in the present study. Farhat et al. (2016) finds univariate associations between embA N54D or iniB A70T and EMB resistance; however, no isolates carried these two mutations in the present study. Zhao et al. (2015a) reports that the mutations in the embA upstream region showed significant correlation with EMB resistance; however, this region was not included in the present study. Mutations in ubiA except a lineage-specific mutation E149D are reported to correlate with high-level EMB resistance and responsible for 3.2–6.4% EMB resistance (He et al., 2015; Xu et al., 2015; Tulyaprawat et al., 2019).
The WHO target product profiles for new molecular assays for M. tuberculosis require more than 90% sensitivity and 95% specificity (World health organization, 2014). Our findings show the predicted resistance to rifampicin and isoniazid exceeded 90% sensitivity and 95% specificity by WGS and analyzing rpoB (RMP) and katG (INH) (Table 3). In the present study, the additional mutation loci found in katG except in codon 315 made the mutations in the inhA promoter and oxyR-ahpC less meaningful for predicting INH resistance for that mutation in the latter two regions increased only 2.7% sensitivity by WGS. Although several molecular DSTs for INH resistance testing are recommended by WHO, several reports show that the calculated sensitivity among clinical isolates is far lower 90% (Li et al., 2015; Javed et al., 2016, 2018; World Health Organization, 2016; Maningi et al., 2018). According to the standard (World health organization, 2014) and the actual situations in clinical practice of new molecular DST assays (Li et al., 2015; Javed et al., 2016, 2018; World Health Organization, 2016; Maningi et al., 2018), the sensitivity for STR by WGS in the present study was recognized to achieve to an acceptable degree (using rpsL and rrs 530 loop and 912 loop, 83%), and the specificity was excellent (97.8%) while the sensitivity of embB for predicting EMB resistance was excellent (90.9%), but the specificity (65.1%) was far lower than the standard, which requires more than 95% (World health organization, 2014).
The results in the present study show that the sensitivity of drug resistance-associated genes or intergenic regions, whether alone or combined, could not predict 100% of the interested drug resistance, which is similar to that reported by most studies (Li et al., 2010; Farhat et al., 2016; Miotto et al., 2017; Shea et al., 2017). For the gap between genotypic and phenotypic resistance, one most fundamental cause may be that current sequencing technologies have varying capabilities to detect low frequencies (<20%) of resistant strains mixed with susceptible strains relative to phenotypic testing that can detect resistant strains making up only 1% of the total population (Canetti et al., 1969; Do and Dobrovic, 2009; Oh et al., 2010; Miotto et al., 2017). Second, breakpoint artifacts (i.e., inappropriately high critical concentrations) can be a major source of misclassification of phenotypes (Miotto et al., 2017), e.g., the threshold value for EMB resistance used in L-J medium mentioned in Zhang et al.’s (2009) report. Third, synonymous mutations are universally acknowledged to be unrelated with drug resistance as they do not cause any change in the structure of the protein (Torres et al., 2015), so synonymous mutations are excluded in the present study; however, drug-resistant strains are more likely to carry synonymous mutations although no statistical differences were found for most of loci (Supplementary Table 5), which cut down the sensitivity of sequencing. Previous studies show that these mutations can sometimes confer resistance (Safi et al., 2013; Van Deun et al., 2013). Fourth, predicting drug resistance based on WGS data relies on the knowledge of drug-resistance mechanisms; however, drug-resistance mechanisms have not been understood clearly, which results in WGS mispredicting resistance or susceptibility according to including mutations or without mutations, respectively. Previous studies (Li et al., 2010; Farhat et al., 2016; Miotto et al., 2017; Shea et al., 2017) as well as the present study report certain phenotypically susceptible isolates carried mutations while phenotypically resistant isolates show wild types in interested genes, especially in noncanonical genes associated with resistance. Applied proteomics and bioinformatics analysis (such as molecular docking, pupylation, and protein–protein interaction) on uncharacterized and hypothetical proteins in M. tuberculosis might give a clue for the novel mechanism of drug resistance (Sharma and Bisht, 2017a, b; Sharma et al., 2018).
Conclusion
The data in this study raise our understanding of the molecular determinants of resistance to INH, RMP, EMB, and STR; high sensitivities and specificities of mutations in the genes of katG (INH), rpoB (RMP), rpsL (STR), and rrs 530 loop and 912 loop (STR) provide us a good choice to predict INH, RMP, and STR resistance by WGS or target region sequencing in the future. Further, the results provide clues in clarifying the drug-resistance mechanisms of M. tuberculosis isolates from China.
Data Availability Statement
The datasets generated for this study can be found in the NCBI with accession numbers SRA065095 and PRJNA633954.
Ethics Statement
This study was approved by the ethics committee of the National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention. The patients with TB were included in the present research only after we received informed written consent from themselves or from their parents/guardians in cases in which the patient was a child (18 years of age). An assent was also obtained from participants between 14 and 18 years of age.
Author Contributions
GL designed and conducted this study. LW and GL wrote the first drafts of the manuscript. LW, HL, and ML sequenced the isolates and performed molecular characterization. YJ, XZ, and ZL performed drug susceptibility testing and spoligotyping. KW collected M. tuberculosis strains and provided oversight for sequencing and bioinformatics support. C-XG provided key manuscript edits. All authors commented on the manuscript draft and read and approved the final manuscript.
Funding
This work was financially supported by projects (2018ZX10103001-003-012) of the National Key Programs of Mega Infectious Diseases from the Ministry of Science and Technology, People’s Republic of China, grant (2017zzts069) from Fundamental Research Funds for the Central Universities of Central South University, and the grant (81670014) from National Natural Science Foundation of China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the staff of Beijing, Fujian, Guangdong, Guizhou, Heilongjiang, Henan, Inner Mongolia, Liaoning, Shaanxi, Shanghai, Tibet, and Xinjiang for supplying strains.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01444/full#supplementary-material
TABLE S1 | Drug susceptibility patterns of 183 clinical M. tuberculosis for whole-genome sequencing.
TABLE S2 | Synonymous mutations in 183 M. tuberculosis and their associations with drug resistance.
TABLE S3 | Associations between synonymous mutations and Beijing genotype of 183 M. tuberculosis clinical isolates.
TABLE S4 | Associations between mutations katG463 or gidB92 and drug resistance in 183 M. Tuberculosis.
TABLE S5 | Associations between mutations katG463 or gidB92 and Beijing genotype in 183 M. Tuberculosis.
TABLE S6 | Characterizations of katG mutations and its combination mutations with other 10 genes or regions associated with isoniazid resistance among 137 multi-drug resistant isolates.
TABLE S7 | Mutation characterizations of rpoB among 137 MDR isolates from China.
TABLE S8 | Mutation characterizations of gidB among 100 streptomycin resistant isolates and 83 streptomycin susceptible isolates from China.
TABLE S9 | Mutation characterizations of embABC among 77 ethambutol resistant isolates and 106 ethambutol susceptible isolates from China.
Footnotes
References
Boehme, C. C., Nabeta, P., Hillemann, D., Nicol, M. P., Shenai, S., Krapp, F., et al. (2010). Rapid molecular detection of tuberculosis and rifampin resistance. N. Engl. J. Med. 363, 1005–1015. doi: 10.1056/NEJMoa0907847
Campbell, P. J., Morlock, G. P., Sikes, R. D., Dalton, T. L., Metchock, B., Starks, A. M., et al. (2011). Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 55, 2032–2041. doi: 10.1128/AAC.01550-10
Canetti, G., Fox, W., Khomenko, A., Mahler, H. T., Menon, N. K., Mitchison, D. A., et al. (1969). Advances in techniques of testing mycobacterial drug sensitivity, and the use of sensitivity tests in tuberculosis control programmes. Bull. World Health Organ. 41, 21–43.
Cao, Y., Parmar, H., Simmons, A. M., Kale, D., Tong, K., Lieu, D., et al. (2019). Automatic identification of individual rpoB gene mutations responsible for rifampin resistance in mycobacterium tuberculosis by use of melting temperature signatures generated by the Xpert MTB/RIF ultra assay. J. Clin. Microbiol. 58, 907–919. doi: 10.1128/JCM.00907-19
Casali, N., Nikolayevskyy, V., Balabanova, Y., Harris, S. R., Ignatyeva, O., Kontsevaya, I., et al. (2014). Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat. Genet. 46, 279–286. doi: 10.1038/ng.2878
Chen, C., Kong, W., Zhu, L., Zhou, Y., Peng, H., Shao, Y., et al. (2014). Evaluation of the GenoType((R)) MTBDRplus line probe assay on sputum-positive samples in routine settings in China. Int. J. Tuberc. Lung. Dis. 18, 1034–1039. doi: 10.5588/ijtld.13.0857
Colangeli, R., Helb, D., Sridharan, S., Sun, J., Varma-Basil, M., Hazbon, M. H., et al. (2005). The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol. Microbiol. 55, 1829–1840. doi: 10.1111/j.1365-2958.2005.04510.x
CRyPTIC Consortium and the 100,000 Genomes Project. Allix-Beguec, C., Arandjelovic, I., Bi, L., Beckert, P., Bonnet, M., et al. (2018). Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N. Engl. J. Med. 379, 1403–1415. doi: 10.1056/NEJMoa1800474
Do, H., and Dobrovic, A. (2009). Limited copy number-high resolution melting (LCN-HRM) enables the detection and identification by sequencing of low level mutations in cancer biopsies. Mol. Cancer 8:82. doi: 10.1186/1476-4598-8-82
Farhat, M. R., Shapiro, B. J., Kieser, K. J., Sultana, R., Jacobson, K. R., Victor, T. C., et al. (2013). Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat. Genet. 45, 1183–1189. doi: 10.1038/ng.2747
Farhat, M. R., Sultana, R., Iartchouk, O., Bozeman, S., Galagan, J., Sisk, P., et al. (2016). Genetic determinants of drug resistance in Mycobacterium tuberculosis and their diagnostic value. Am. J. Respir. Crit. Care Med. 194, 621–630. doi: 10.1164/rccm.201510-2091OC
Feuerriegel, S., Koser, C. U., and Niemann, S. (2014). Phylogenetic polymorphisms in antibiotic resistance genes of the Mycobacterium tuberculosis complex. J. Antimicrob. Chemother. 69, 1205–1210. doi: 10.1093/jac/dkt535
Finken, M., Kirschner, P., Meier, A., Wrede, A., and Bottger, E. C. (1993). Molecular basis of streptomycin resistance in Mycobacterium tuberculosis: alterations of the ribosomal protein S12 gene and point mutations within a functional 16S ribosomal RNA pseudoknot. Mol. Microbiol. 9, 1239–1246. doi: 10.1111/j.1365-2958.1993.tb01253.x
He, L., Wang, X., Cui, P., Jin, J., Chen, J., Zhang, W., et al. (2015). ubiA (Rv3806c) encoding DPPR synthase involved in cell wall synthesis is associated with ethambutol resistance in Mycobacterium tuberculosis. Tuberculosis (Edinb) 95, 149–154. doi: 10.1016/j.tube.2014.12.002
Islam, M. M., Tan, Y., Hameed, H. M. A., Liu, Z., Chhotaray, C., Liu, Y., et al. (2019). Detection of novel mutations associated with independent resistance and cross-resistance to isoniazid and prothionamide in Mycobacterium tuberculosis clinical isolates. Clin. Microbiol. Infect. 25, 1041.e1–1041.e7. doi: 10.1016/j.cmi.2018.12.008
Jagielski, T., Ignatowska, H., Bakula, Z., Dziewit, L., Napiorkowska, A., Augustynowicz-Kopec, E., et al. (2014). Screening for streptomycin resistance-conferring mutations in Mycobacterium tuberculosis clinical isolates from Poland. PLoS One 9:e100078. doi: 10.1371/journal.pone.0100078
Javed, H., Bakula, Z., Plen, M., Hashmi, H. J., Tahir, Z., Jamil, N., et al. (2018). Evaluation of genotype MTBDRplus and MTBDRsl assays for rapid detection of drug resistance in extensively drug-resistant Mycobacterium tuberculosis isolates in Pakistan. Front. Microbiol. 9:2265. doi: 10.3389/fmicb.2018.02265
Javed, H., Jamil, N., Jagielski, T., Bakula, Z., and Tahir, Z. (2016). Evaluation of genotype MTBDRplus assay for rapid detection of isoniazid and rifampicin resistance in Mycobacterium tuberculosis clinical isolates from Pakistan. Int. J. Mycobacteriol. 5(Suppl 1), S147–S148. doi: 10.1016/j.ijmyco.2016.11.010
Kamerbeek, J., Schouls, L., Kolk, A., van Agterveld, M., van Soolingen, D., Kuijper, S., et al. (1997). Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 35, 907–914. doi: 10.1128/jcm.35.4.907-914.1997
Li, G. L., Zhao, D. F., Xie, T., Ju, H. F., Mu, C., Zhao, H., et al. (2010). Molecular characterization of drug-resistant Beijing family isolates of Mycobacterium tuberculosis from Tianjin, China. Biomed. Environ. Sci. 23, 188–193. doi: 10.1016/S0895-3988(10)60051-7
Li, Q., Dong, H. Y., Pang, Y., Xia, H., Ou, X. C., Zhang, Z. Y., et al. (2015). Multicenter evaluation of the molecular line probe assay for multidrug resistant Mycobacterium Tuberculosis detection in China. Biomed. Environ. Sci. 28, 464–467. doi: 10.3967/bes2015.066
Li, R., Yu, C., Li, Y., Lam, T. W., Yiu, S. M., Kristiansen, K., et al. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Liu, L., Jiang, F., Chen, L., Zhao, B., Dong, J., Sun, L., et al. (2018). The impact of combined gene mutations in inhA and ahpC genes on high levels of isoniazid resistance amongst katG non-315 in multidrug-resistant tuberculosis isolates from China. Emerg. Microbes Infect. 7:183. doi: 10.1038/s41426-018-0184-0
Luo, D., Chen, Q., Xiong, G., Peng, Y., Liu, T., Chen, X., et al. (2019). Prevalence and molecular characterization of multidrug-resistant M. tuberculosis in Jiangxi province, China. Sci. Rep. 9:7315. doi: 10.1038/s41598-019-43547-2
Makhado, N. A., Matabane, E., Faccin, M., Pincon, C., Jouet, A., Boutachkourt, F., et al. (2018). Outbreak of multidrug-resistant tuberculosis in South Africa undetected by WHO-endorsed commercial tests: an observational study. Lancet Infect. Dis. 18, 1350–1359. doi: 10.1016/S1473-3099(18)30496-1
Maningi, N. E., Daum, L. T., Rodriguez, J. D., Said, H. M., Peters, R. P. H., Sekyere, J. O., et al. (2018). Multi- and extensively drug resistant Mycobacterium tuberculosis in South Africa: a molecular analysis of historical isolates. J. Clin. Microbiol. 56:e01214-17. doi: 10.1128/JCM.01214-17
Meacci, F., Orru, G., Iona, E., Giannoni, F., Piersimoni, C., Pozzi, G., et al. (2005). Drug resistance evolution of a Mycobacterium tuberculosis strain from a noncompliant patient. J. Clin. Microbiol. 43, 3114–3120. doi: 10.1128/JCM.43.7.3114-3120.2005
Miotto, P., Tessema, B., Tagliani, E., Chindelevitch, L., Starks, A. M., Emerson, C., et al. (2017). A standardised method for interpreting the association between mutations and phenotypic drug resistance in Mycobacterium tuberculosis. Eur. Respir. J. 50, 1701354. doi: 10.1183/13993003.01354-2017
Mokrousov, I., Otten, T., Vyshnevskiy, B., and Narvskaya, O. (2002). Detection of embB306 mutations in ethambutol-susceptible clinical isolates of Mycobacterium tuberculosis from Northwestern Russia: implications for genotypic resistance testing. J. Clin. Microbiol. 40, 3810–3813. doi: 10.1128/jcm.40.10.3810-3813.2002
Moure, R., Espanol, M., Tudo, G., Vicente, E., Coll, P., Gonzalez-Martin, J., et al. (2014). Characterization of the embB gene in Mycobacterium tuberculosis isolates from Barcelona and rapid detection of main mutations related to ethambutol resistance using a low-density DNA array. J. Antimicrob. Chemother. 69, 947–954. doi: 10.1093/jac/dkt448
Nebenzahl-Guimaraes, H., Jacobson, K. R., Farhat, M. R., and Murray, M. B. (2014). Systematic review of allelic exchange experiments aimed at identifying mutations that confer drug resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 69, 331–342. doi: 10.1093/jac/dkt358
Nguyen, L. (2016). Antibiotic resistance mechanisms in M. tuberculosis: an update. Arch. Toxicol. 90, 1585–1604. doi: 10.1007/s00204-016-1727-6
Nhu, N. T., Lan, N. T., Phuong, N. T., Chau, N., Farrar, J., and Caws, M. (2012). Association of streptomycin resistance mutations with level of drug resistance and Mycobacterium tuberculosis genotypes. Int. J. Tuberc. Lung. Dis. 16, 527–531. doi: 10.5588/ijtld.11.0202
Oh, J. E., Lim, H. S., An, C. H., Jeong, E. G., Han, J. Y., Lee, S. H., et al. (2010). Detection of low-level KRAS mutations using PNA-mediated asymmetric PCR clamping and melting curve analysis with unlabeled probes. J. Mol. Diagn. 12, 418–424. doi: 10.2353/jmoldx.2010.090146
Perdigao, J., Gomes, P., Miranda, A., Maltez, F., Machado, D., Silva, C., et al. (2020). Using genomics to understand the origin and dispersion of multidrug and extensively drug resistant tuberculosis in Portugal. Sci. Rep. 10:2600. doi: 10.1038/s41598-020-59558-3
Perdigao, J., Macedo, R., Machado, D., Silva, C., Jordao, L., Couto, I., et al. (2014). GidB mutation as a phylogenetic marker for Q1 cluster Mycobacterium tuberculosis isolates and intermediate-level streptomycin resistance determinant in Lisbon, Portugal. Clin. Microbiol. Infect. 20, O278–O284. doi: 10.1111/1469-0691.12392
Plinke, C., Cox, H. S., Zarkua, N., Karimovich, H. A., Braker, K., Diel, R., et al. (2010). embCAB sequence variation among ethambutol-resistant Mycobacterium tuberculosis isolates without embB306 mutation. J. Antimicrob. Chemother. 65, 1359–1367. doi: 10.1093/jac/dkq120
Ramaswamy, S. V., Reich, R., Dou, S. J., Jasperse, L., Pan, X., Wanger, A., et al. (2003). Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 47, 1241–1250. doi: 10.1128/aac.47.4.1241-1250.2003
Safi, H., Lingaraju, S., Amin, A., Kim, S., Jones, M., Holmes, M., et al. (2013). Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-beta-D-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 45, 1190–1197. doi: 10.1038/ng.2743
Sandgren, A., Strong, M., Muthukrishnan, P., Weiner, B. K., Church, G. M., and Murray, M. B. (2009). Tuberculosis drug resistance mutation database. PLoS Med. 6:e2. doi: 10.1371/journal.pmed.1000002
Schon, T., Miotto, P., Koser, C. U., Viveiros, M., Bottger, E., and Cambau, E. (2017). Mycobacterium tuberculosis drug-resistance testing: challenges, recent developments and perspectives. Clin. Microbiol. Infect. 23, 154–160. doi: 10.1016/j.cmi.2016.10.022
Sharma, D., and Bisht, D. (2017a). M. tuberculosis hypothetical proteins and proteins of unknown function: hope for exploring novel resistance mechanisms as well as future target of drug resistance. Front. Microbiol. 8:465. doi: 10.3389/fmicb.2017.00465
Sharma, D., and Bisht, D. (2017b). Secretory proteome analysis of streptomycin-resistant Mycobacterium tuberculosis clinical isolates. SLAS Discov. 22, 1229–1238. doi: 10.1177/2472555217698428
Sharma, D., Bisht, D., and Khan, A. U. (2018). Potential alternative strategy against drug resistant tuberculosis: a proteomics prospect. Proteomes 6:26. doi: 10.3390/proteomes6020026
Shea, J., Halse, T. A., Lapierre, P., Shudt, M., Kohlerschmidt, D., Van Roey, P., et al. (2017). Comprehensive whole-genome sequencing and reporting of drug resistance profiles on clinical cases of Mycobacterium tuberculosis in New York state. J. Clin. Microbiol. 55, 1871–1882. doi: 10.1128/JCM.00298-17
Spies, F. S., Ribeiro, A. W., Ramos, D. F., Ribeiro, M. O., Martin, A., Palomino, J. C., et al. (2011). Streptomycin resistance and lineage-specific polymorphisms in Mycobacterium tuberculosis gidB gene. J. Clin. Microbiol. 49, 2625–2630. doi: 10.1128/JCM.00168-11
Sreevatsan, S., Pan, X., Stockbauer, K. E., Williams, D. L., Kreiswirth, B. N., and Musser, J. M. (1996). Characterization of rpsL and rrs mutations in streptomycin-resistant Mycobacterium tuberculosis isolates from diverse geographic localities. Antimicrob. Agents Chemother. 40, 1024–1026. doi: 10.1128/aac.40.4.1024
Sun, H., Zhang, C., Xiang, L., Pi, R., Guo, Z., Zheng, C., et al. (2016). Characterization of mutations in streptomycin-resistant Mycobacterium tuberculosis isolates in Sichuan, China and the association between Beijing-lineage and dual-mutation in gidB. Tuberculosis (Edinb) 96, 102–106. doi: 10.1016/j.tube.2015.09.004
Timmins, G. S., and Deretic, V. (2006). Mechanisms of action of isoniazid. Mol. Microbiol. 62, 1220–1227. doi: 10.1111/j.1365-2958.2006.05467.x
Torres, J. N., Paul, L. V., Rodwell, T. C., Victor, T. C., Amallraja, A. M., Elghraoui, A., et al. (2015). Novel katG mutations causing isoniazid resistance in clinical M. tuberculosis isolates. Emerg. Microbes Infect. 4:e42. doi: 10.1038/emi.2015.42
Treatment Action Group and Stop Tb Partnership (2018). Tuberculosis Research Funding Trends 2005–2017. New York, NY: Treatment Action Group.
Tulyaprawat, O., Chaiprasert, A., Chongtrakool, P., Suwannakarn, K., and Ngamskulrungroj, P. (2019). Association of ubiA mutations and high-level of ethambutol resistance among Mycobacterium tuberculosis Thai clinical isolates. Tuberculosis (Edinb) 114, 42–46. doi: 10.1016/j.tube.2018.11.006
Unissa, A. N., Subbian, S., Hanna, L. E., and Selvakumar, N. (2016). Overview on mechanisms of isoniazid action and resistance in Mycobacterium tuberculosis. Infect. Genet. Evol. 45, 474–492. doi: 10.1016/j.meegid.2016.09.004
United Nations General Assembly (2018). Resolution 73/3: Political Declaration of the High-Level Meeting of the General Assembly on the Fight Against Tuberculosis. New York, NY: United Nations General Assembly.
Van Deun, A., Aung, K. J., Bola, V., Lebeke, R., Hossain, M. A., de Rijk, W. B., et al. (2013). Rifampin drug resistance tests for tuberculosis: challenging the gold standard. J. Clin. Microbiol. 51, 2633–2640. doi: 10.1128/JCM.00553-13
van Embden, J. D., Cave, M. D., Crawford, J. T., Dale, J. W., Eisenach, K. D., Gicquel, B., et al. (1993). Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J. Clin. Microbiol. 31, 406–409. doi: 10.1128/jcm.31.2.406-409.1993
Verma, J. S., Gupta, Y., Nair, D., Manzoor, N., Rautela, R. S., Rai, A., et al. (2014). Evaluation of gidB alterations responsible for streptomycin resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 69, 2935–2941. doi: 10.1093/jac/dku273
Via, L. E., Cho, S. N., Hwang, S., Bang, H., Park, S. K., Kang, H. S., et al. (2010). Polymorphisms associated with resistance and cross-resistance to aminoglycosides and capreomycin in Mycobacterium tuberculosis isolates from South Korean Patients with drug-resistant tuberculosis. J. Clin. Microbiol. 48, 402–411. doi: 10.1128/JCM.01476-09
Walker, T. M., Kohl, T. A., Omar, S. V., Hedge, J., Del Ojo, Elias, C., et al. (2015). Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect. Dis. 15, 1193–1202. doi: 10.1016/S1473-3099(15)00062-6
Wan, L., Guo, Q., Wei, J.-H., Liu, H.-C., Li, M.-C., Jiang, Y., et al. (2020). Accuracy of a reverse dot blot hybridization assay for simultaneous detection of the resistance of four anti-tuberculosis drugs in Mycobacterium tuberculosis isolated from China. Infect. Dis. Poverty 9:38. doi: 10.1186/s40249-020-00652-z
World Health Organization (2008). Policy Guidance on Drug-Susceptibility Testing (DST) of Second-line Antituberculosis Drugs (WHO/HTM/TB/2008.392). Geneva: World Health Organization.
World health organization (2014). High-Priority Target Product Profiles for New Tuberculosis Diagnostics: Report of a Consensus Meeting. Geneva: World Health Organization.
World Health Organization (2016). The Use of Molecular Line Probe Assays for the Detection of Resistance to Isoniazid and Rifampicin, Policy Update. Geneva: World Health Organization.
World Health Organization (2019). Global Tuberculosis Report 2019. Geneva: World Health Organization.
Xu, Y., Jia, H., Huang, H., Sun, Z., and Zhang, Z. (2015). Mutations found in embCAB, embR, and ubiA genes of ethambutol-sensitive and -resistant Mycobacterium tuberculosis clinical isolates from China. Biomed. Res. Int. 2015:951706. doi: 10.1155/2015/951706
Zhang, H., Li, D., Zhao, L., Fleming, J., Lin, N., Wang, T., et al. (2013). Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat. Genet. 45, 1255–1260. doi: 10.1038/ng.2735
Zhang, N., Zhao, X. Q., and Wan, K. L. (2009). Preliminary study on the appropriate concentrations of drug used in the drug-susceptibility test to detect the Ethambutol-resistant isolates of Mycobacterium tuberculosis. Chin. J. Zoonoses 25, 1049–1053. doi: 10.3969/j.issn.1002-2694.2009.11.005
Zhang, Z., Liu, M., Wang, Y., Pang, Y., Kam, K. M., and Zhao, Y. (2014). Molecular and phenotypic characterization of multidrug-resistant Mycobacterium tuberculosis isolates resistant to kanamycin, amikacin, and capreomycin in China. Eur. J. Clin. Microbiol. Infect. Dis. 33, 1959–1966. doi: 10.1007/s10096-014-2144-5
Zhao, L. L., Chen, Y., Chen, Z. N., Liu, H. C., Hu, P. L., Sun, Q., et al. (2014a). Prevalence and molecular characteristics of drug-resistant Mycobacterium tuberculosis in Hunan, China. Antimicrob. Agents Chemother. 58, 3475–3480. doi: 10.1128/AAC.02426-14
Zhao, L. L., Chen, Y., Liu, H. C., Xia, Q., Wu, X. C., Sun, Q., et al. (2014b). Molecular characterization of multidrug-resistant Mycobacterium tuberculosis isolates from China. Antimicrob. Agents Chemother. 58, 1997–2005. doi: 10.1128/AAC.01792-13
Zhao, L. L., Sun, Q., Liu, H. C., Wu, X. C., Xiao, T. Y., Zhao, X. Q., et al. (2015a). Analysis of embCAB mutations associated with ethambutol resistance in multidrug-resistant Mycobacterium tuberculosis isolates from China. Antimicrob. Agents Chemother. 59, 2045–2050. doi: 10.1128/AAC.04933-14
Keywords: Mycobacterium tuberculosis, resistance, whole-genome sequencing, mutation, Beijing genotype
Citation: Wan L, Liu H, Li M, Jiang Y, Zhao X, Liu Z, Wan K, Li G and Guan C-x (2020) Genomic Analysis Identifies Mutations Concerning Drug-Resistance and Beijing Genotype in Multidrug-Resistant Mycobacterium tuberculosis Isolated From China. Front. Microbiol. 11:1444. doi: 10.3389/fmicb.2020.01444
Received: 07 January 2020; Accepted: 04 June 2020;
Published: 15 July 2020.
Edited by:
Onya Opota, University of Lausanne, SwitzerlandReviewed by:
Divakar Sharma, Indian Institute of Technology Delhi, IndiaGonzalo Greif, Institut Pasteur de Montevideo, Uruguay
Copyright © 2020 Wan, Liu, Li, Jiang, Zhao, Liu, Wan, Li and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guilian Li, bGlndWlsaWFuQGljZGMuY24=; Cha-xiang Guan, Z3VhbmNoYXhpYW5nQGNzdS5lZHUuY24=