 Shekhar Nagar1
Shekhar Nagar1 Chandni Talwar1
Chandni Talwar1 Shazia Haider2
Shazia Haider2 Akshita Puri1,3
Akshita Puri1,3 Kalaiarasan Ponnusamy4Madhuri Gupta1
Kalaiarasan Ponnusamy4Madhuri Gupta1 Utkarsh Sood1,5
Utkarsh Sood1,5 Abhay Bajaj1,6
Abhay Bajaj1,6 Rup Lal1,5*
Rup Lal1,5* Roshan Kumar1,7*
Roshan Kumar1,7*- 1Department of Zoology, University of Delhi, Delhi, India
- 2Department of Biotechnology, Jaypee Institute of Information Technology, Noida, India
- 3P.G.T.D, Zoology, R.T.M Nagpur University, Nagpur, India
- 4School of Biotechnology, Jawaharlal Nehru University, New Delhi, India
- 5The Energy and Resources Institute, New Delhi, India
- 6Environmental Biotechnology and Genomics Division, CSIR-National Environmental Engineering Research Institute, Nagpur, India
- 7P.G. Department of Zoology, Magadh University, Bodh Gaya, India
The genus Parapedobacter was established to describe a novel genus within the family Sphingobacteriaceae and derives its name from Pedobacter, with which it is shown to be evolutionarily related. Despite this, Parapedobacter and Pedobacter do not share very high 16S rRNA gene sequence similarities. Therefore, we hypothesized whether these substantial differences at the 16S rRNA gene level depict the true phylogeny or that these genomes have actually diverged. Thus, we performed genomic analysis of the four available genomes of Parapedobacter to better understand their phylogenomic position within family Sphingobacteriaceae. Our results demonstrated that Parapedobacter is more closely related to species of Olivibacter, as opposed to the genus Pedobacter. Further, we identified a significant class of enzymes called pectinases with potential industrial applications within the genomes of Parapedobacter luteus DSM 22899T and Parapedobacter composti DSM 22900T. These enzymes, specifically pectinesterases and pectate lyases, are presumed to have largely different catalytic activities based on very low sequence similarities to already known enzymes and thus may be exploited for industrial applications. We also determined the complete Bacteroides aerotolerance (Bat) operon (batA, batB, batC, batD, batE, hypothetical protein, moxR, and pa3071) within the genome of Parapedobacter indicus RK1T. This expands the definition of genus Parapedobacter to containing members that are able to tolerate oxygen stress using encoded oxidative stress responsive systems. By conducting a signal propagation network analysis, we determined that BatD, BatE, and hypothetical proteins are the major controlling hubs that drive the expression of Bat operon. As a key metabolic difference, we also annotated the complete iol operon within the P. indicus RK1T genome for utilization of all three stereoisomers of inositol, namely myo-inositol, scyllo-inositol, and 1D-chiro-inositol, which are abundant sources of organic phosphate found in soils. The results suggest that the genus Parapedobacter holds promising applications owing to its environmentally relevant genomic adaptations, which may be exploited in the future.
Introduction
The genus Parapedobacter was first described by Kim et al. (2007), under the family Sphingobacteriaceae of the phylum Bacteroidetes. Based on the highest gene sequence identity (<90%), Parapedobacter was found to be akin to the genus Pedobacter (Kim et al., 2007). Currently, nine validly published species that have been isolated from a wide range of ecological habitats represent this genus (www.bacterio.net/genus/parapedobacter). These are Parapedobacter koreensis (dried rice straw) (Kim et al., 2007), Parapedobacter soli (ginseng field soil) (Kim et al., 2008), Parapedobacter luteus and Parapedobacter composti (cotton waste compost) (Kim et al., 2010), Parapedobacter pyrenivorans (Zhao et al., 2013) and Parapedobacter indicus (polluted soils) (Kumar et al., 2015), Parapedobacter deserti (plant stem) (Liu et al., 2017), Parapedobacter lycopersici (rhizospheric soil) (Kim et al., 2017), and Parapedobacter defluvii (sewage treatment plant) (Yang et al., 2017). Altogether, the genomic understanding of this genus has been lacking since its identification more than a decade ago (Kim et al., 2007). Here, we uncover the evolutionary relationships and define the functional attributes of this genus through four available genome sequences: P. indicus RK1T [hexachlorocyclohexane (HCH) dumpsite), P. koreensis Jip14T (dried rice straw), P. composti DSM 22900T, and P. luteus DSM 22899T (cotton waste compost).
Our study provides a view of the phylogenomic relatedness of Parapedobacter with other genera of family Sphingobacteriaceae. To address this, we adopted an alignment-free method for computing pairwise dissimilarity values among the genomes that correspond to the number of substitution events separating two leaves over the evolutionary course (Criscuolo, 2019). These methods are becoming increasingly popular to address the challenges in computational requirements, runtime, and need for manual interventions with large genomic datasets, as in this study. We found Parapedobacter to be more closely related to the genus Olivibacter than to Pedobacter, which was previously reported based on 16S rRNA gene sequence similarities (Kim et al., 2007; Kumar et al., 2015). Through pangenome analyses, we clarified the genomic factors that shaped the unique distribution of Parapedobacter spp. while identifying functions that distinguished each species through their unique genomic contents. With the aim of finding the ecological relevance of Parapedobacter spp., we determined an industrially significant class of enzymes called pectinases encoded within the genomes of Parapedobacter spp. The pectinases identified within the genomes of Parapedobacter spp. shared low sequence similarity with the other known enzymes of this class and are likely to have important industrial applications. Our study also revealed the microaerophilic nature of one of the species, P. indicus RK1, as determined from the presence of the Bacteroides aerotolerance (Bat) operon genes within its genome and the expression of these genes under aerobic stress. This further widens the genomic definitions of the genus Parapedobacter, which was previously suggested to harbor strictly aerobic members (Kim et al., 2007). Through a systems biology approach, we conducted a signal propagation analysis of proteins encoded by the Bat operon genes to report the key proteins in their communication. We also report differences in the metabolic preferences of Parapedobacter spp., more specifically in the utilization of three stereo-isomers of inositol. Overall, the study resolved the peculiarities at the phylogenomic level as well as at the functional level in members of this novel genus.
Materials and Methods
Genome Sequencing and Assembly, Annotations and Genomic Features
The genome of P. indicus RK1 was sequenced by the Joint Genome Institute, CA, USA, under the Genomic Encyclopedia of Type Strains, Phase III (KMG-III), using the Illumina HiSeq 2500-1TB platform, and assembled using the SOAPdenovo assembler (Li et al., 2010). The sequence data for P. indicus RK1 have been submitted to the National Center for Biotechnology Information (NCBI) Genome database (GenBank: FOQO00000000). Subsequently, three other available genomes were obtained for comparative analyses: P. koreensis Jip14 (GenBank: FNZR00000000), P. composti DSM 22900 (GenBank: FOLL00000000), and P. luteus DSM 22899 (GenBank: FUYS00000000). The annotations were performed using the Rapid Annotation using Subsystems Technology (RAST) (Aziz et al., 2008) server with gene caller Glimmer-3 (Delcher et al., 2007). The presence of clustered regularly interspaced short palindromic repeats (CRISPR) elements was analyzed using the CRISPRFinder server (Grissa et al., 2007). The degree of genome completeness was predicted by analyzing 107 essential copy genes using hidden Markov models (Dupont et al., 2012).
Phylogenomic Analysis
The genome-based phylogeny of validly published members of family Sphingobacteriaceae (with available genomes on or before May 21, 2020) was inferred using an alignment-free distance-based method with JolyTree (Criscuolo, 2019). Briefly, for each pair of genomes, the pairwise p-distance (i.e., the proportion of aligned nucleotide differences) value was calculated using Mash v.2.1 (Ondov et al., 2016), followed by the correction of each value into a numerical quantity that is proportional to the evolutionary distance between the compared genomes. Further, the phylogeny was constructed using the balanced minimum-evolution (BME) method (Desper and Gascuel, 2002) based on an improved neighbor-joining principle using FastME v.2.1.5.1 (Lefort et al., 2015). Finally, the branch confidence values of the inferred tree were calculated using the REQ program (Guénoche and Garreta, 2001), which estimates the rate of elementary quartets (REQ) for each branch of the tree from the associated distance matrix. The program thus assigns confidence values from 0 to 1, where higher values denote branches fully supported by the pairwise evolutionary distances. To further decipher the genomic heterogeneity, the genome-wide average nucleotide identity (ANI) trend was explored using an ANIb (BLAST based) approach (Konstantinidis and Tiedje, 2005) and the heatmap was plotted using the pheatmap package in R (Kolde and Kolde, 2015).
Pangenome Analyses
The pangenome of Parapedobacter was studied by means of the anvi'o workflow (Eren et al., 2015). Proteins were predicted within each genome using Prodigal (Hyatt et al., 2010). Each protein in every genome was compared with every other protein using the NCBI BLASTP program (–use-ncbi-blast). The sequence similarity and algorithm sensitivity were estimated using bitscore and MCL inflation (van Dongen and Abreu-Goodger, 2012) parameters while identifying clusters in a protein similarity search. The program was run at –min-bit = 0.5, which denotes the sequences to be at least 50% identical in length for clustering together, and –min-inflation was set to 10 for maximum sensitivity (from a scale of 2–10). The genomes were subsequently clustered based on their core and accessory gene clusters using the Euclidean distance (Gower, 1982) and Ward linkage (Ward, 1963). The genes were annotated for Clusters of Orthologous Groups (COG) functional classes implemented in the anvi'o pangenomics workflow (Eren et al., 2015). The strain-specific or unique gene clusters of each genome were distributed into different functional COG categories and a heatmap was generated using the pheatmap package in R (Kolde and Kolde, 2015). Further, the core genome was also estimated using the GET_HOMOLOGUES pipeline using an 80% cutoff value for both query coverage (QC) and percentage identity (PI).
Comparative Functional Analysis and Genome Orthology
For each genome, proteins were annotated to be placed in COG categories by using RPS-Blast against the NCBI COG database (Tatusov et al., 2001) and a heatmap was plotted using average clustering. The genomes were searched for similar gene contents using the orthoMCL algorithm that is available in GET_HOMOLOGUES (Contreras-Moreira and Vinuesa, 2013). As the genomes were highly variable, we determined their core contents using QC and percentage identity at regular intervals of 5%, starting from 100% and going down to 50% (for both parameters), to classify the protein families that are widely shared among the genomes. The shared sequences within each interval were subsequently annotated into COG categories using RPS-Blast against the NCBI COG database (Tatusov et al., 2001). Further, the amino acid sequences of each genome were searched against Pfam (Finn et al., 2016) and the profiles of unique and shared protein families among the genomes were compared. The protein sequences of pectinesterases and pectate lyases annotated as unique Pfam families in P. luteus DSM 22899 and P. composti DSM 22900 were compared by using the BLASTP program on the NCBI database based on PI and QC with the top 10 hits. Annotations of genes involved in inositol utilization and those coding for pectate lyase and pectinesterase identified by Glimmer 3 in RAST were checked manually on UniProt (Pundir et al., 2016).
Computational Systems Biology Analysis of Bat Proteins
To date, the protein–protein interaction (PPIs) studies of Parapedobacter spp. have not been performed and thus no PPI data for this genus are available. We followed the systems biology approach to analyze the proteins of the Bat operon cluster (n = 8) to identify the importance of each of these proteins. For this, the eight protein (BatA, BatB, BatC, BatD, BatE, hypothetical protein, MoxR, and PA3071) sequences of P. indicus RK1 were searched against those of the closest phylogenetic neighbor, Pedobacter heparinus (Steyn et al., 1998), using BLASTP to find homologous proteins. PPI data for P. heparinus were retrieved from the STRING database v.10 (Szklarczyk et al., 2015), which consists of known and predicted PPIs including direct (physical) and indirect (functional) interactions among sets of proteins. The interactions with a confidence score equal to or >0.4 (default parameters) were considered. The PPI networks were visualized in Cytoscape v.3.5 (Shannon et al., 2003) and analyzed using the Network analyzer to find the important protein. Further, to explore the signal communications of these important proteins in the cell, we carried out signal propagation analysis. For this, each protein was separately analyzed for direct interaction with other proteins, which form the shells of nodes at a distance r. These interacting partners (r = 1) were searched in the STRING database and network growth was constructed up to r = 8 while taking care of the repeated proteins. Perl script v.5.18.2.2 was used to construct the propagation of network growth.
The average degree of nodes in the shell at distance r was calculated as , and the average residual degree as . So, the probability that the nodes at a distance r interlink with the nodes in the shell, |D (r)|, is given by , where is the rate of network expansion (Barzel and Barabási, 2013).
Bacteroides Aerotolerance (Bat) Gene Expression in Parapedobacter indicus RK1
P. indicus RK1, previously reported to be isolated from HCH-contaminated soil by our laboratory (Kumar et al., 2015), was grown to mid-exponential phase under aerobic as well as under microaerophilic conditions (CO2 incubator containing 5% CO2) at 30°C in Luria–Bertani medium. Growth was monitored at regular intervals by measuring OD values at 600 nm by means of a spectrophotometer. Primers for the eight Bat operon genes, namely batA, batB, batC, batD, batE, pa3071, hypothetical protein, and moxR, were designed (Supplementary File S1) and the genes were amplified to confirm their presence within the genome. The amplified products were cleaned using a NucleoSpin Gel and PCR Clean-up Kit according to the manufacturer's instructions. The eluted products were further Sanger sequenced and reconstructed using Sequencing Analysis v.5.1.1 (Applied Biosystems) and confirmed as Bat operon genes through BLASTn analysis. The expression of these genes under aerobic as well as microaerophilic conditions was studied by harvesting the cells after 24 h of growth under these conditions. Total RNA was isolated using an RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized by using a Revert Aid First Strand cDNA synthesis kit (Thermo Scientific) according to the manufacturer's instructions. The expression of Bat operon genes was confirmed through amplification using cDNA.
Results and Discussion
General Genomic Attributes and Distribution of Parapedobacter Across the Phylogenetic Clade
In this study, we analyzed four draft genomes of genus Parapedobacter; namely, P. indicus RK1, P. koreensis Jip14, P. luteus DSM 22899, and P. composti DSM 22900. As they belong to a novel genus, so far no genomic studies have been reported. The genome size and %G+C content of these Parapedobacter strains varied between 4.6–6.1 Mbp and 48–50%, respectively (Supplementary Table 1). These features can be attributed to their evolution in different ecological settings, as the complexity of the environment differs with their respective habitats. For example, strain RK1 was isolated from an HCH dumpsite (Kumar et al., 2015) and its genome was found to harbor genes for aromatic compound degradation pathways, such as the beta-ketoadipate pathway (muconate cyclo-isomerase, succinyl-CoA:3-ketoacid-coenzyme A transferase subunit A and B, and mandelate racemase) and salicylate and gentisate catabolism (salicylate esterase and fumarylacetoacetate hydrolase) (Sharma et al., 2015; Talwar et al., 2020). Further, the abundance of membrane transport genes (n = 208) and genes related to the stress response contributing toward osmotic stress (n = 54) and oxidative stress (n = 51) were significantly abundant within the genome of strain RK1. The abundance of these genes has been suggested as an adaptation in other organisms under toxic soil environments and might contribute toward the adaptability of P. indicus RK1 in an HCH-contaminated environment (Sangwan et al., 2012; Talwar et al., 2020).
Additionally, we aimed to study the evolutionary relationships of Parapedobacter spp. among other genera of the family Sphingobacteriaceae. For this, we carried out parallel whole genome-based phylogeny assessments of all validly published species of the family Sphingobacteriaceae using pairwise genomic dissimilarity measures based on substitution events and BLAST-based whole-genome average nucleotide identities (ANIb) analyses. The former method performs alignment-free, pairwise whole-genome comparisons and has been used previously for resolving phylogenies from large genomic datasets (Parks et al., 2018; Badell et al., 2020; Tarlachkov et al., 2020). Although the genus Parapedobacter derives its name from Pedobacter based on 16S rRNA gene sequence similarity (Kim et al., 2007), the inferred trees from both the described methods revealed a close association of Parapedobacter with the genus Olivibacter (Figures 1A,B). This provides evidence for the greater significance of whole genome-based evaluation of the phylogeny over a single 16S rRNA gene-based approach in bacterial taxonomy (Mahato et al., 2017). However, the clustering of Parapedobacter with Olivibacter was supported by low confidence scores (Figure 1A). Thus, sequencing and addition of more genomes of Olivibacter spp. would reveal their phylogenetic association with Parapedobacter more clearly. In addition, ANIb trends also revealed the Parapedobacter spp. to be related to Arcticibacter and Pararcticibacter; however, additional genomes of recognized species will unveil the genomic relatedness of these genera more clearly (Figure 1B). Within the genus Parapedobacter, a whole genome-based dissimilarity matrix clustered P. composti DSM 22900 and P. luteus DSM 22899 together and P. koreensis Jip14 and P. indicus RK1 joined the clade distantly. These results were consistent with the 16S rRNA gene sequence analysis based on which P. indicus RK1 was reported to be closest to P. koreensis Jip14 (Kumar et al., 2015). P. composti DSM 22900 and P. luteus DSM 22899 are isolates from same habitat, i.e., cotton waste compost, which may account for their high evolutionary relatedness inferred based on the substitution events. However, ANIb-based phylogeny placed P. luteus DSM 22899 with P. koreensis Jip14 (ANI = 79.08%), while P. luteus DSM 22899 and P. composti DSM 22900 shared 76.3% ANIb. The pairwise similarities between all Parapedobacter genomes ranged between 73.9 and 79.0%, clearly separating them from other members of the family Sphingobacteriaceae (Figure 1B). Therefore, although the Parapedobacter were originally identified and named based on their 16S rRNA gene sequence relatedness with Pedobacter spp., sequencing and addition of whole genomes in the family Sphingobacteriaceae has resolved their taxonomic position with other genera in the family.
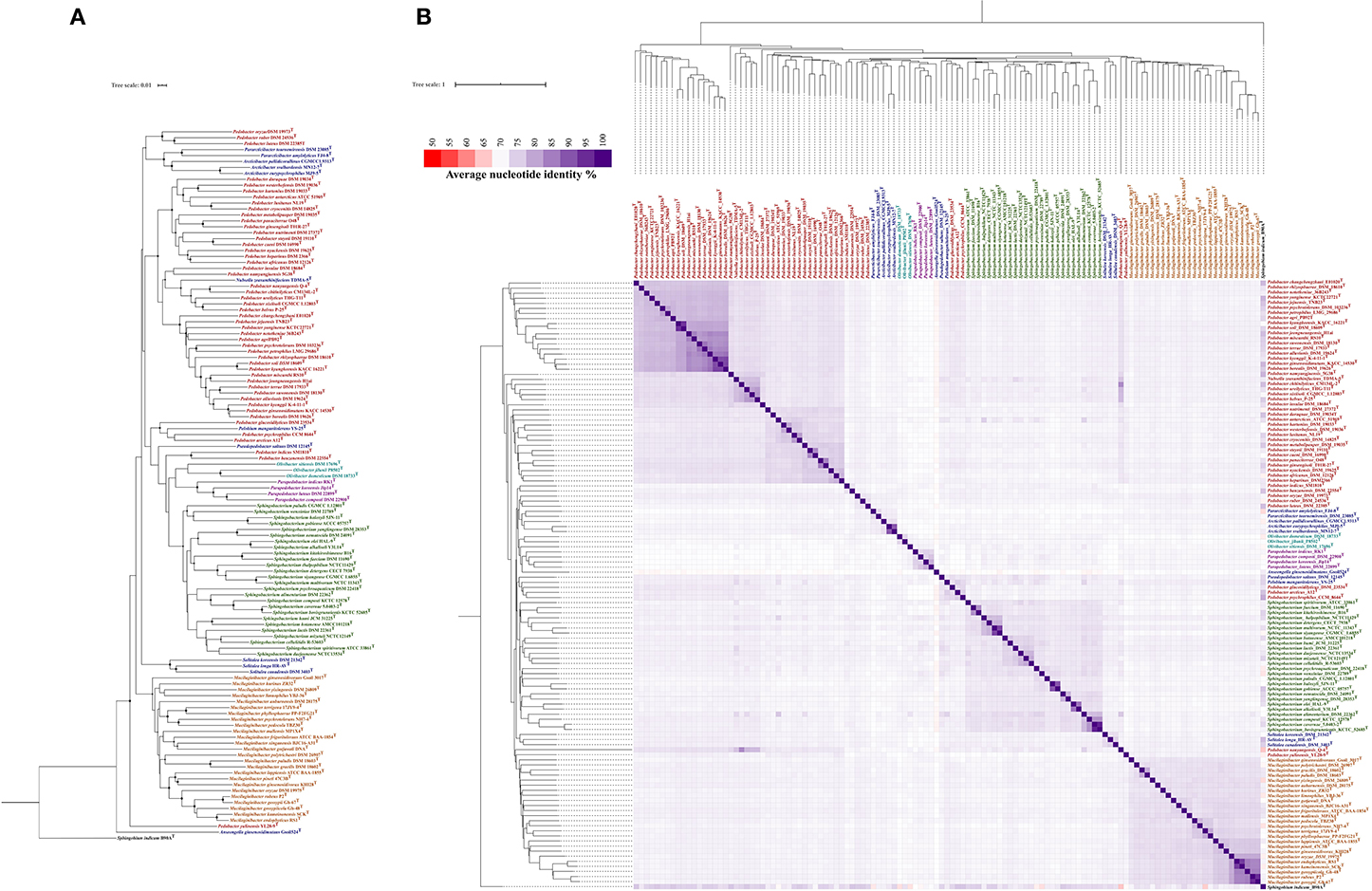
Figure 1. Phylogenetic analysis of genus Parapedobacter. (A) Phylogenetic tree constructed from the pairwise distance estimates from unaligned genomes of the validly published species of family Sphingobacteriaceae. Sphingobium indicum B90A was used as an outgroup. The tree was constructed in JolyTree (Criscuolo, 2019) using the balanced minimum-evolution method, based on an improved Neighbor Joining principle using FastME v.2.1.5.1 (Lefort et al., 2015). The branch confidence values were computed using the REQ program (Guénoche and Garreta, 2001) and values in the range 0.7–1 are shown with circles of corresponding sizes on branches of the inferred tree. (B) Phylogeny reconstructed from BLAST-based pairwise average nucleotide identity (ANIb) using available genomes of validly published species of the family Sphingobacteriaceae and Sphingobium indicum B90A as an outgroup. The pairwise %ANIb values among genomes were compared using the Manhattan distance and hierarchical clustering and a heatmap was constructed.
Pangenomic Attributes of Parapedobacter
The genomes were more than 98% complete, as estimated from the presence of essential single-copy genes (Dupont et al., 2012; Kumar et al., 2017) (Supplementary Table 1). This removed bias from the results of whole-genome comparisons based on core and accessory gene contents from subsequent analyses. The pangenome distributed into 17,761 gene clusters (minbit = 0.5; min-inflation = 10; distance: Euclidean; linkage: Ward). Further, the core genome analysis revealed a total of 1,084 genes present in four Parapedobacter spp. genomes (QC = 80% and PI = 80%). The genomes were also revealed to be highly non-redundant (98.6–99.3%) (Figure 2). This reflects that the Parapedobacter spp. have a tendency to reduce genomic paralogy yet they maintain functional diversity, which might have implications in niche-specific adaptations (Mendonça et al., 2011).
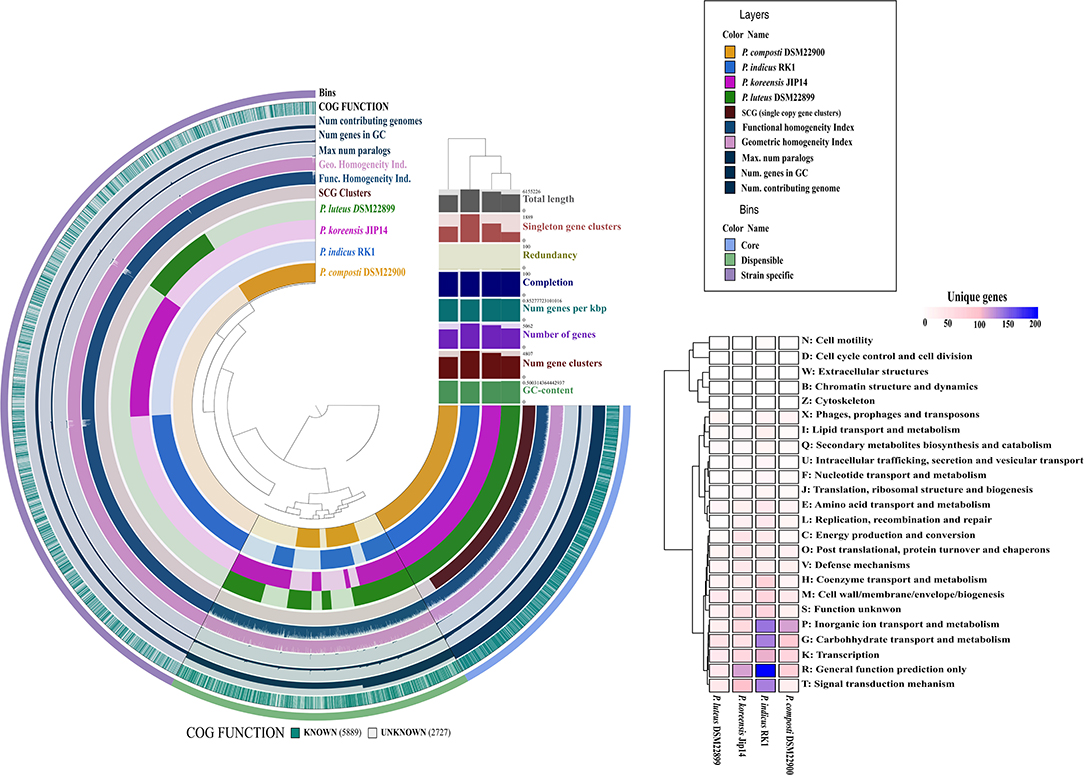
Figure 2. Comparative circular phylogram of Parapedobacter genomes based on common genetic attributes and gene frequencies. The genomes are arranged in radial layers with their genomic contents organized into core and accessory gene contents computed using the Euclidean distance and the Ward linkage method. The clustering of genomes is shown as a dendrogram based on core and accessory genomic contents at the top right corner of the circular phylogram. The strain-specific contents of each of the genomes were annotated into different COG functional categories and plotted as a heatmap, shown on the right.
More than one-fourth (28.0%) of the pangenome was formed from the strain-specific gene contents for which high abundance was noted in the COG classes: general function prediction (R), inorganic ion transport and metabolism (P), carbohydrate transport and metabolism (G), signal transduction mechanisms (T), and transcription (K). Thus, genomic fractions that greatly distinguished strain-specific contents from core contents were assigned to inorganic ion transport and metabolism (P) and signal transduction mechanisms (T). Specifically, these functions were abundant in the strain-specific contents of the P. indicus RK1, P. composti DSM 22900, and P. koreensis Jip14 genomes. Being an isolate from HCH-polluted soil, the abundance of unique genes involved in signal transduction and inorganic ion transport and metabolism within P. indicus RK1 might enable the organism to sense environmental signals and use chemotaxis; this is consistent with the previous assertions in species of different genera isolated from this stressed niche (Verma et al., 2014; Sharma et al., 2015; Talwar et al., 2020). P. indicus RK1 harbored the highest unique gene content (10.9%) followed by P. koreensis Jip-14 (7.4%), P. composti DSM 22900 (6.1%), and P. luteus DSM 22899 (3.9%) (Figure 2). Thus, the large strain-specific content might account for the largest of the Parapedobacter genomes, represented by P. indicus RK1 at 6.2 Mbp. Since, P. composti DSM 22900 and P. luteus DSM 22899 both inhabited the same habitat of cotton waste compost, it was interesting to outline the major functional differences among them that placed them at a greater distance at the pangenomic level. P. composti DSM 22900 harbored 20.1% of its unique gene content involved in inorganic ion transport and metabolism, compared with that in P. luteus DSM 22899 (7.9%) (Figure 2). The unique contents of the two genomes were also highly diverged for functions of transcription (K), carbohydrate transport and metabolism (G), and general function prediction only (R), which were all greatly abundant (more than 2-fold) in P. composti DSM 22900 (Figure 2). Thus, although evolving in the same ecological niche, the two bacterial species might have adapted differently with respect to their key functions, as has been described previously (de la Haba et al., 2019). Since strain RK1 has large strain-specific genomic attributes that may be phage induced, as has already been documented for many rapidly evolving bacterial genomes (Lopez et al., 2012; Blesa et al., 2018), we wanted to look for CRISPR elements. While genomes of all three strains except P. composti DSM 22900 harbored CRISPR elements (Supplementary Table 1), only one CRISPR from P. koreensis Jip14 showed significant similarity to Caldilinea aerophila DSM 14535 (QC = 76%, PI = 75%, E = 2e-14). It was found to code for gliding motility-associated C-terminal domain-containing protein, suggesting the phage-mediated acquisition of this protein in the strain. CRISPRs from the two other genomes, P. indicus RK1 and P. luteus DSM 22899, were not found to code for any known protein. The low number of CRISPRs detected within Parapedobacter spp. suggested their non-frequent encounters with the phages. To uncover the important functions of the genus, we further analyzed Parapedobacter genomes to compare them based on broad COG functional categories and further delved into key functions under these COG categories.
Comparative Account of Potential Functional Attributes
Based on whole-genome-encoded functions, P. indicus RK1 separated out from the rest of the genomes as opposed to core- and accessory genome-based clustering (Figure 3A). The strain had an abundance of genes involved in unknown functions, carbohydrate transport and metabolism, and transcription. In addition, inorganic ion transport and metabolism was highest in P. indicus RK1, which was also revealed from strain-specific contents analysis as discussed above. Thus, the strain possessed mechanisms for the utilization of resources as well as for adapting to environmental pressures imposed by HCH pollution.
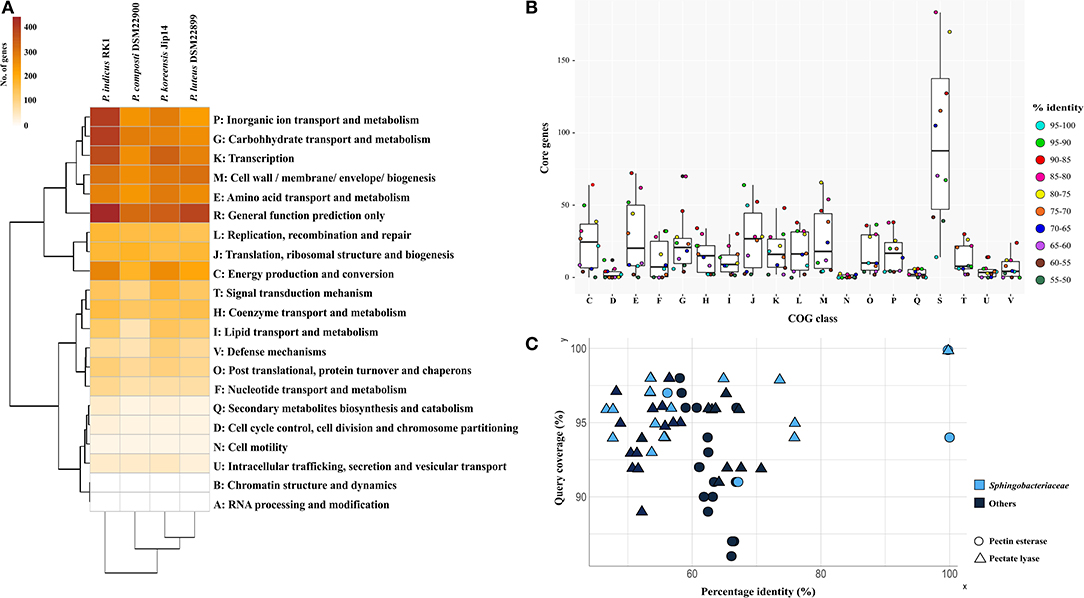
Figure 3. Comparative functional analysis of genus Parapedobacter. (A) Annotation of proteins using RPS-Blast against the COG database. Pearson correlation and complete clustering were used to generate the heatmap. (B) The distribution of proteins shared at discrete intervals of percentage identity and coverage cutoff in different COG categories showing the relative similarities of proteins under each functional category. (C) Top 10 hits of pectinesterase and pectate lyase proteins identified within genomes of strains DSM 22899 and DSM 22900 showing sequence identities plotted as a function of percentage identity (x-axis) vs. the coverage cutoff (y-axis). The shape and color of each protein denote the protein type and whether the protein belongs to the family Sphingobacteriaceae, respectively.
To identify the level of COG functional similarity among the genomes, we further determined the core genes using both QC and PI at intervals of 5%, starting from 100 to 95% and going down to 55 to 50%, and annotated these genes from the genomes to reveal the relative abundance of COG functions at these discrete windows (Figure 3B). This was done with aim of identifying the percentage similarity between core functional genes to provide an estimate of the functional relatedness of Parapedobacter genomes. The maximum gene content of each of these genomes was found to be shared at above 55% identity and QC. The annotations revealed that the majority of these core functional genes shared highest similarity in the range 90% to 85% under each COG category, with the exceptions of translation, ribosomal structure, and biogenesis (J; 95–90%) and cell wall/membrane/envelope biogenesis (M; 80–75%) (Figure 3B). This revealed the high similarity of the functional core genes of Parapedobacter genomes.
We observed P. luteus DSM 22899 and P. composti DSM 22900 to be least abundant for all Pfam categories compared with the two other species (data not shown). This can be justified as these are comparatively smaller among the four members in terms of coding potential (Figure 2). In spite of having the smaller coding potential, these strains possessed unique protein families under carbohydrate transport and metabolism. Most importantly, the strains possessed pectate lyase (pfam09492) and pectinesterase (pfam01095) as unique enzymes. Since, they form an important class of industrial enzymes, we were interested in studying their diversity within Parapedobacter. In DSM 22899, two variants of pectinesterases and one pectate lyase were annotated while strain DSM 22900 maintained one and three copies of these genes, respectively. This suggested the important functional significance of these enzymes in the two strains in the context of their habitat of cotton waste compost. Pectin is one of the major polysaccharides in plant cell walls. It is modified and degraded by a group of naturally occurring pectinases that are essential for cell wall extension and the recycling of plant materials (Voragen et al., 2009). Pectinesterase breaks down pectin into pectate and methanol (Fries et al., 2007), while pectate lyase catalyzes the eliminative cleavage of de-esterified pectin and thus mediates cell wall degradation and fruit softening (Marín-Rodríguez et al., 2002). Pectate lyases of plant-associated bacteria enable them to utilize the pectin from dead or living plants as a carbon source for growth (Hugouvieux-Cotte-Pattat et al., 2014). As strains DSM 22899 and DSM 22900 are isolates from cotton waste composts used for the cultivation of the oyster mushroom (Pleurotus ostreatus) (Kim et al., 2010), they might be assumed to naturally help in bio-scouring of cotton fiber using pectinases encoded within their genomes. To determine whether this is an acquired phenotype in these strains, we compared the percentage identities of each of these proteins with the top 10 hits obtained through BLASTP on NCBI (Supplementary File S2). The analysis revealed that the two copies of pectinesterase annotated within the genome of DSM 22899 were 100% identical at 94% QC while these proteins were only 66.8% similar to those of DSM 22900 (91% QC), displaying the differences between them despite being isolated from the same habitat (Figure 3C). Moreover, the two pectinesterase proteins of DSM 22899 shared similarity in the range of 63–66% with their top 10 neighbors, none of which belonged to the family Sphingobacteriaceae. The topmost hit of pectinesterase protein of DSM 22900 after DSM 22899 was that from Pedobacter glucosidilyticus (56.1% PI at 97% QC), which belongs to the family Sphingobacteriaceae. Percentage identities with all other hits ranged between 59 and 62% at low QCs with sequences from genera not belonging to the family Sphingobacteriaceae (Figure 3C). Similarly, pectate lyase proteins from the two strains were also checked for their closest hits. The only copy of pectate lyase within DSM 22899 showed similarity to that of DSM 22900 (76% PI; 95% QC). It showed similarities in the range 48–53% to the rest of its neighbors, most of which were not from Sphingobacteriaceae (Figure 3C). Out of the three copies of pectate lyase present in DSM 22900, one showed maximum similarity to proteins of genus Sphingobacterium (55.98% PI; 94% QC); however, the two other copies showed maximum similarity to proteins from Catalinimonas (67.22% PI; 96% QC) and Rufibacter (56.49% PI; 98% QC) (Figure 3C; Supplementary File S2). Pectinases find several other industrial applications, including clarification and stabilization of fruit juices; softening of pickles; fermentation of coffee, cocoa, and tea; preparation of jams and jellies; as supplements in animal feed for optimal absorption of nutrients; for increasing bioethanol production; and in paper and wine manufacturing (Kubra et al., 2018). Microbial pectinases are preferred over other chemical methods as they are ecofriendly and energy efficient (Maldonado and Strasser de Saad, 1998; Kashyap et al., 2001; Garg et al., 2016). Thus, these enzymes from DSM 22899 and DSM 22900 can be further explored for their industrial implications.
We further annotated the complete cluster of Bat operon genes within the genome of P. indicus RK1, which were lacking in the three other genomes. The Bat proteins were first identified within the genome of Bacteroides fragilis to have a role in aerotolerance (Tang et al., 1999) and have been reported to be involved in the bacterial response to oxidative stress in other species (Meuric et al., 2008). Despite their identification more than two decades ago, the literature on Bat proteins has been very limited. Through this study, we performed genome-based analysis of these proteins for the first time by analyzing the signal propagation in their PPI network using a systems biology approach. We further studied the expression of these genes qualitatively under oxidative stress to understand this responsive mechanism.
Analysis of Signal Propagation in Protein–Protein Interactions of Bat Proteins and Their Expression Under Microaerophilic Stress
PPIs of Bat proteins of P. indicus RK1 were studied through those of its closest neighbor, P. heparinus, with available datasets on the STRING database (Figure 4). The genome was used as a reference following a preliminary BLASTP analysis of Bat proteins of P. indicus RK1 (BatA, BatB, BatC, BatD, BatE, hypothetical protein, MoxR, and PA3071), which revealed those from P. heparinus as the topmost hits. The network comprised 78 proteins involved in 216 interactions, as visualized in Cytoscape v.3.5. The network analysis identified highly interacting proteins (hubs) among these eight proteins (Figure 4A). BatD showed maximum interaction with degree 55 followed by hypothetical protein (Hypo; degree: 53), BatE (degree: 39), BatB (degree: 30), BatA (degree: 29), BatC (degree: 23), MoxR (degree: 22), and PA3071 (degree: 21). These results suggested that BatD is the most important protein of the Bat operon. In order to understand the expression of the other proteins, we carried out computational signal propagation, which could suggest the communication of these proteins in P. indicus RK1. The propagation of a signal from a specific node in a large network to other nodes at a distance r was measured using the probability that the nodes at a distance r link with the specific node, |D (r)|. This parameter indicates how perturbation at a certain node affects the stability in the network and its topological characteristics. We calculated |D (r)| for each of the proteins (BatA, BatB, BatC, BatD, BatE, hypothetical protein, MoxR, and PA3071) (Figure 4B), and found that |D (r)| first increased exponentially with r. This indicated that the signal processing from the respective hubs was active (propagation of perturbation from the respective hubs increased) and efficient. Then this propagation of signal was found to saturate at a particular value of r, denoted by rs. The saturation value of rs for all eight proteins was rs = 4. The peak value of |D (r)| was maximum for BatD, BatE, and hypothetical protein when rs < 4, whereas MoxR and PA3071 had maximum propagation when rs > 4. Thus, the |D (r)| decreased as a function of r owing to the contraction of the shells of nodes, which allowed a decrease in signal propagation. The analysis indicated that BatD, BatE, and hypothetical protein had the maximum impact on the regulatory network of aerotolerance, and the remaining hubs (BatB, BatC, and BatA) had similar controlling capability (Figure 4B).
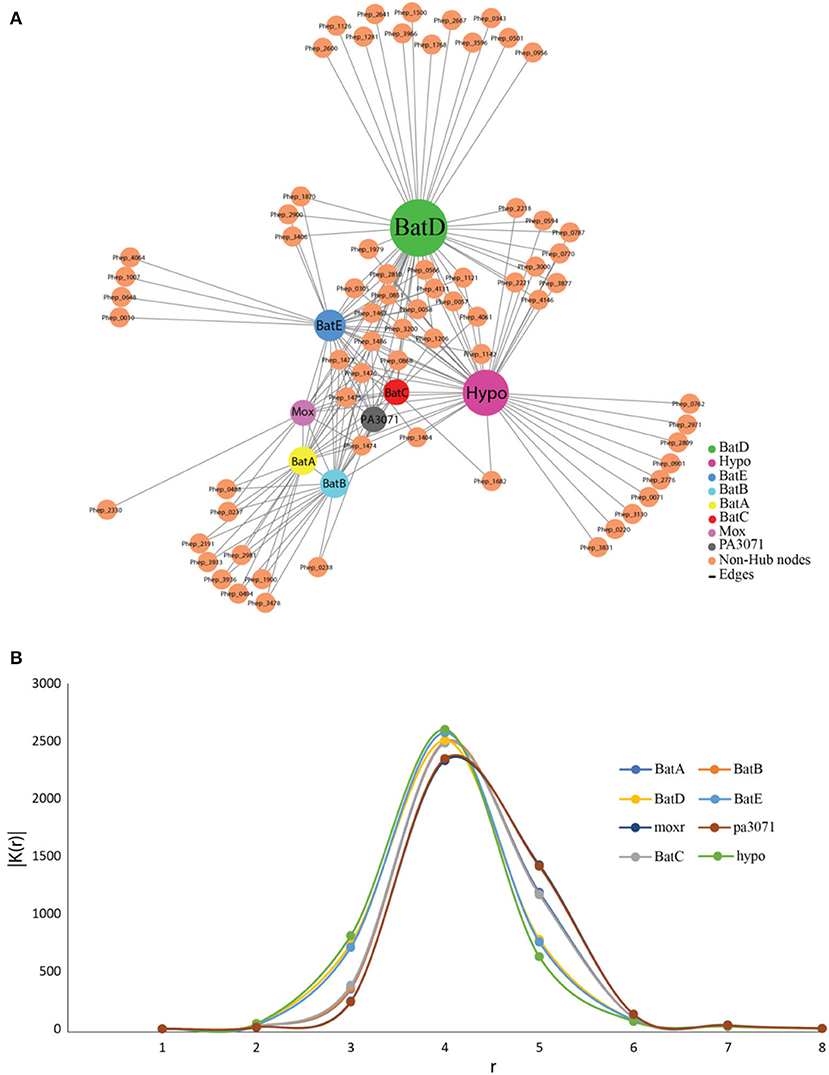
Figure 4. The protein–protein interaction network and signal propagation of Bat operon genes of P. indicus RK1. (A) Expanded view of the network imported from Cytoscape, where nodes represent proteins and edges represent their physical interactions. All the nodes and edges are in orange and gray, respectively. The existence of the few sparsely distributed main gene hubs, namely BatD, hypothetical protein, BatE, BatB, BatA, BatC, MoxR, and PA3071, in the network is indicated by a color other than orange. (B) Propagation of signal in the protein–protein interaction network growth of BatD, Hypo, BatE, BatB, BatA, BatC, MoxR, and PA3071 is highlighted by colored lines.
The presence of genes associated with the Bat operon was confirmed within the genome of P. indicus RK1 through the amplification of the eight genes (BatA, BatB, BatC, BatD, BatE, hypothetical protein, MoxR, and PA3071) using custom-designed primers for each gene (Supplementary Figure S1; Supplementary File S1). The expression of these Bat genes in strain RK1 was analyzed by growing the culture under aerobic as well as microaerophilic conditions. All eight Bat-associated genes could be amplified from the cDNA obtained from cells of P. indicus RK1 following 24 h of growth under aerobic and microaerophilic conditions (data shown for microaerophilic conditions; Supplementary Figure S1). This supports our hypothesis based on comparative genomic analysis that the genus might harbor microaerophilic species that have not yet been identified. The study provides preliminary insights based on qualitative data and further studies focused independently on the nature of growth of Parapedobacter spp. under different oxic conditions would reveal the quantitative expression of each of these Bat operon-associated genes and their underlying mechanisms in Parapedobacter indicus RK1, which is beyond the scope of this genome-based study. However, to gain more genome-based insights, we further performed comparative genomic analysis of Parapedobacter spp. for oxidative stress response-related genes and proteins.
Comparative Genomic Basis of the Oxidative Stress Response in Parapedobacter spp
One of the key classes of enzymes that are inherently associated with growth under different oxic conditions in bacteria are the ribonucleotidereductases (RNR), which catalyze the synthesis of deoxyribonucleotide triphosphates (dNTPs). RNRs of classes Ib (nrdE, nrdF, nrdI) and class III (nrdD, nrdG), which are reported to be active under aerobic and anaerobic conditions, respectively (Jordan and Reichard, 1998), were annotated within the genome of RK1 (Supplementary Table 2). BLASTN analysis of nrdEF genes encoding class Ib RNR and cofactor assembly protein nrdI showed the highest similarities to proteins of P. koreensis Jip14 (Supplementary Table 2). The genes of anaerobic class III RNR nrdDG also showed the highest similarity to the proteins of P. koreensis Jip14. RNRs are categorized as class I, II, or III depending upon radical generation, cofactor requirement, and oxygen dependence (Jordan and Reichard, 1998). Class I represents aerobic enzymes generating a tyrosyl radical with an iron–oxygen center and class II functions under both aerobic and anaerobic conditions generating adenosylcobalamin. Class III are anaerobic enzymes generating a glycyl radical from S-adenosylmethionine and an iron–sulfur cluster. Class I RNRs are further divided into class Ia, Ib, and Ic, depending upon the metal center required for protein radical generation (Jordan and Reichard, 1998). The presence of both aerobic and anaerobic classes of RNRs in P. indicus RK1 strengthened the prediction for its survival at different oxic conditions.
Further, we studied the presence/absence patterns of five osr elements, which are expressed during oxidative stress in bacteria (Smalley et al., 2002; Supplementary Table 3). Three genes – namely, class Ia RNR-encoding gene nrdA; an aspartate decarboxylase-encoding gene, asdA; and a putative outer-membrane protein similar to susC of Bacteroides thetaiotaomicron – which are known to be highly induced during periods of oxidative stress in bacteria were annotated within all Parapedobacter spp. (Smalley et al., 2002). The two other genes – namely, cation efflux pump-encoding gene czcD and a heat shock protein, htpG – were absent in all the strains. To note, Smalley et al. (2002) reported that these two genes were not as highly induced under oxidative stress. In addition, several other genes coding for enzymes participating in the oxidative stress response were annotated within Parapedobacter genomes, including manganese superoxide dismutase, alkyl hydroperoxide reductase (ahp), rubredoxin, catalase, glutathione peroxidase, and non-specific DNA binding proteins which are responsible for scavenging superoxide radicals and peroxides, thereby combating oxidative stress (Rocha and Smith, 1999; Fu et al., 2007; Hagelueken et al., 2007). Thus, the annotation of these oxidative stress response systems hinted toward possible aerotolerance mechanisms used by Parapedobacter spp. for protection from cellular reactive oxygen species.
We were further interested in the comparative metabolic profiles of the Parapedobacter spp. Since the comparative functional profiling of genomes revealed the abundance of carbohydrate metabolism genes within the P. indicus RK1 genome, we looked into specific functional genes that were enriched under this category. As one of the distinguishing metabolic characteristics, P. indicus RK1 was found to harbor a complete gene cluster for degradation of all three stereo-isomers of inositol, namely myo-inositol, scyllo-inositol and 1D-chiro-inositol, which were studied in detail.
Inositol Utilization in P. indicus RK1
The carbohydrate myo-inositol commonly exists as the mono- or phosphorylated form (commonly known as phytic acid) and corresponds to more than 80% of organic phosphate in the soil (Turner et al., 2002). Many soil microbes utilize these forms as an energy source. However, the utilization differs among organisms based on the genetic organization of these gene clusters. The molecular genetics of inositol metabolism is best studied in Bacillus subtilis, which is shown to metabolize all three stereo-isomers using the iol operon (Yoshida et al., 1997; Yamaoka et al., 2011). We annotated the complete iol operon only in strain RK1, which is an isolate of HCH-contaminated soil, as the other strains lacked its complete gene cluster (Figure 5A). For uptake of myo-inositol from the extracellular environment strain RK1 possessed a sodium/myo-inositol co-transporter iolT. Upon entering into the bacterial cell, myo-inositol is converted to intermediates that enter glycolysis and subsequently the tricarboxylic acid cycle in successive reactions catalyzed by enzymes encoded by the iol operon genes (Yoshida et al., 1997; Yamaoka et al., 2011) (Figure 5B). Inside the bacterial cell, inositol-2-dehydrogenase encoded by iolG initiates the catabolism of inositol isomers. The strain RK1 harbored two variants of this gene, iolG and iolG1. The presence of iolI, which encodes an inosose isomerase, suggested the potential interconversion of scyllo-inositol into 1D-chiro-inositol via an intermediate, namely 1-keto-D-chiro-inositol. The successive steps in the catabolism of inositol are catalyzed by the enzymes inosose dehydratase (iolE), epi-inositol hydrolase (iolD), 5-deoxy-glucuronate isomerase (iolB), 5-dehydro-2-deoxygluconokinase (iolC), fructose-bisphosphate aldolase (iolJ), and malonate-semialdehyde dehydrogenase (iolA), as illustrated in Figure 5B (Krings et al., 2006). The catabolic products glyceraldehyde-3-phosphate and acetyl coenzyme A then enter the glycolytic and tricarboxylic acid cycle, respectively, for energy production. In addition to the presence of a complete gene cluster for inositol catabolism, a large copy number of iolG (n = 23) and the presence of variant form iolG1 within the genome of strain RK1 suggest extensive utilization of inositol in this organism as an acquired mechanism for utilization of myo-inositol reserves in soil.
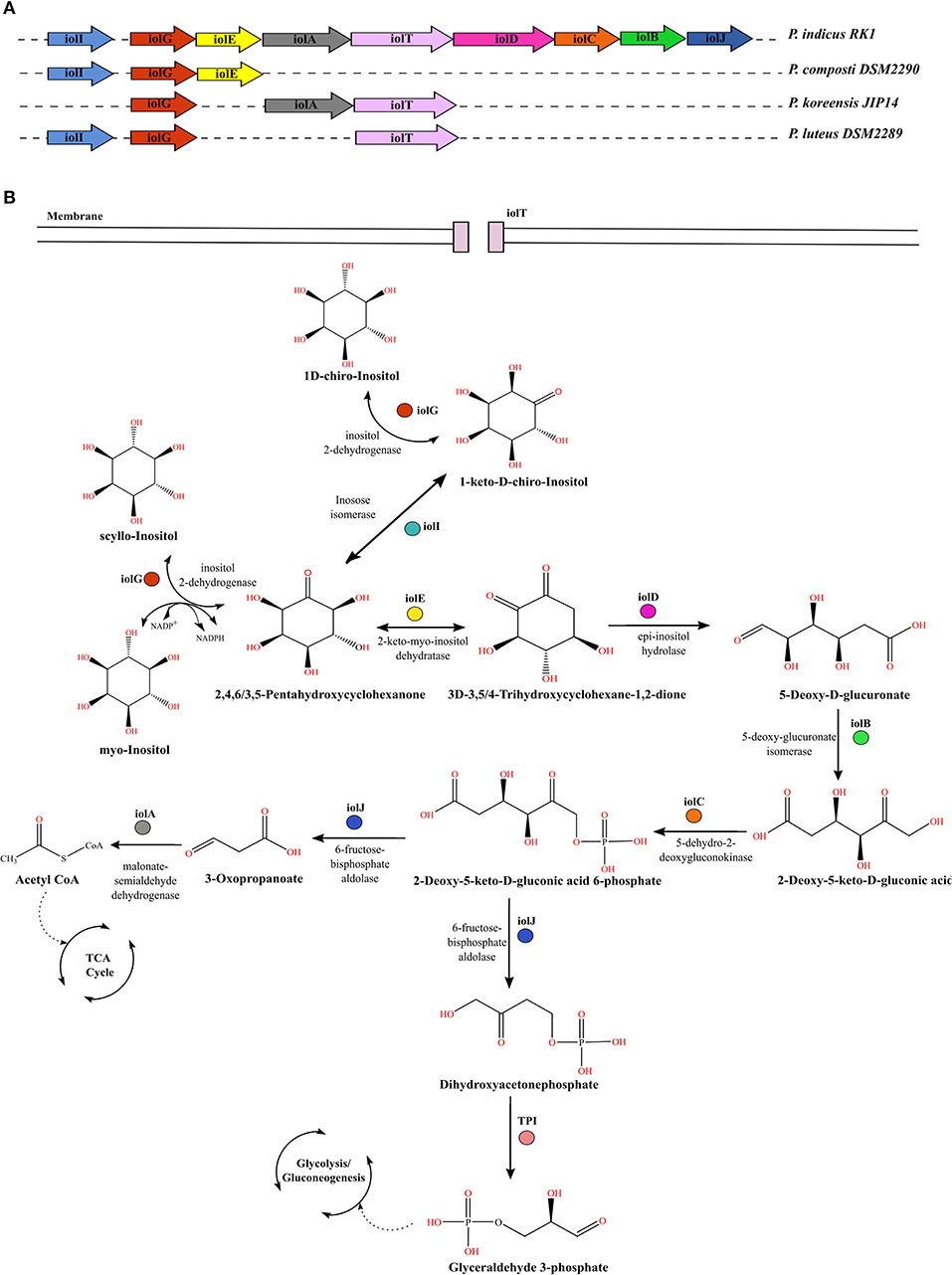
Figure 5. Comparative genomic profiles for utilization of inositol stereo-isomers by Parapedobacter spp. (A) Organization of iol genes in the operon within the genome of strain RK1 as compared with other strains that lack the complete iol gene cluster. (B) Pathway for catabolism of three stereo-isomers of inositol, namely myo-inositol, scyllo-inositol, and chiro-inositol, in bacteria. Each gene encoding the enzyme responsible for catalyzing a reaction is denoted by the same color as in panel (A).
Conclusion
The present study defines the evolutionary relationships of the genus Parapedobacter with members of the family Sphingobacteriaceae. Further, this comparative study uncovered the microaerobic nature of P. indicus RK1, which harbors the Bat operon as well as other genes, such as the osr and rnr group, that participate in the response to oxidative stress. The expression of Bat operon-associated genes in P. indicus RK1 was confirmed when grown under aerobic and microaerophilic conditions. Further, for the first time using a systems biology approach, signal propagation analysis of the proteins encoded by the Bat operon genes was performed. The results suggest that BatD, BatE, and hypothetical protein had the maximum impact on the regulatory network of aerotolerance and thus these are the major players of the Bat operon. The study also identified industrially important enzymes of the classes pectinesterases and pectate lyases within two bacterial strains, namely P. composti DSM 22900 and P. luteus DSM 22899. This opens up new avenues to study their enzymatic activity both in vitro and in vivo for their use as industrial enzymes in the future.
Data Availability Statement
All datasets presented in this study are included in the article/Supplementary Material.
Author Contributions
SN, RL, AB, and RK planned the study. SN, RK, CT, SH, AP, KP, MG, US, and AB performed the experiments and analysis. SN, CT, SH, AP, and RK wrote the manuscript. RL and RK critically reviewed the manuscript and improved it. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SN and CT acknowledge the Council of Scientific and Industrial Research (CSIR), New Delhi, for providing doctoral fellowships. SH would like to thank Jaypee Institute of Information Technology, Noida India for providing support. AP acknowledges UGC for providing fellowship and P.G.T.D, Zoology, R.T.M Nagpur University for support. KP thanks Hub of Bioinformatics for providing support. US and RL thank The National Academy of Sciences, India, for support under the NASI-Senior Scientist Platinum Jubilee Fellowship Scheme. RK acknowledges the Post Graduate Department of Zoology, Magadh University, Bodh Gaya, for support. All authors gratefully acknowledge Dr. Ram Krishan Negi, Associate Professor, University of Delhi, for providing guidance that shaped the final manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01725/full#supplementary-material
Supplementary Figure S1. Electrophoretogram (0.8% agarose gel matrix) of Bat operon genes of Parapedobacter indicus RK1. (A1,A2) Lane 1, Gene ruler (Fermentas no. SM0331, USA); lanes 2–7 in (A1) and lanes 2–3 in (A2), amplified Bat operon genes from strain RK1. Sizes (in bp) of the different genes are labeled on each band. (B) Bat operon genes amplified from cDNA of strain RK1 following 24 h of microaerophilic growth.
References
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Badell, E., Hennart, M., Rodrigues, C., Passet, V., Dazas, M., Panunzi, L., et al. (2020). Corynebacterium rouxii sp. nov., a novel member of the diphtheriae species complex. Res. Microbiol. 171, 122–127. doi: 10.1016/j.resmic.2020.02.003
Barzel, B., and Barabási, A.-L. (2013). Universality in network dynamics. Nat. Phys. 9, 673–681. doi: 10.1038/nphys2741
Blesa, A., Averhoff, B., and Berenguer, J. (2018). Horizontal gene transfer in Thermus spp. Curr Issues Mol. Biol. 29, 23–36. doi: 10.21775/cimb.029.023
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Criscuolo, A. (2019). A fast alignment-free bioinformatics procedure to infer accurate distance-based phylogenetic trees from genome assemblies. Res. Ideas Outcomes 5:e36178. doi: 10.3897/rio.5.e36178
de la Haba, R. R., López-Hermoso, C., Sánchez-Porro, C., Konstantinidis, K. T., and Ventosa, A. (2019). Comparative genomics and phylogenomic analysis of the genus Salinivibrio. Front. Microbiol. 10:2104. doi: 10.3389/fmicb.2019.02104
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
Desper, R., and Gascuel, O. (2002). Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle. J. Comput. Biol. 9, 687–705. doi: 10.1089/106652702761034136
Dupont, C. L., Rusch, D. B., Yooseph, S., Lombardo, M. J., Richter, R. A., Valas, R., et al. (2012). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J. 6, 1186–1199. doi: 10.1038/ismej.2011.189
Eren, A. M., Esen, O. C., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi'o: an advanced analysis and visualization platform for 'omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–285. doi: 10.1093/nar/gkv1344
Fries, M., Ihrig, J., Brocklehurst, K., Shevchik, V. E., and Pickersgill, R. W. (2007). Molecular basis of the activity of the phytopathogen pectin methylesterase. EMBO J. 26, 3879–3887. doi: 10.1038/sj.emboj.7601816
Fu, R. Y., Chen, J., and Li, Y. (2007). The function of glutathione/glutathione peroxidase system in the oxidative stress resistance systems of microbial cells. Chin. J. Biotechnol. 23, 770–775. doi: 10.1016/s1872-2075(07)60048-x
Garg, G., Singh, A., Kaur, A., Singh, R., Kaur, J., and Mahajan, R. (2016). Microbial pectinases: an ecofriendly tool of nature for industries. Biotech 6:47. doi: 10.1007/s13205-016-0371-4
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 8:172. doi: 10.1186/1471-2105-8-172
Guénoche, A., and Garreta, H. (2001). “Can we have confidence in a tree representation?” in Computational Biology, eds O. Gascuel and M.-F. Sagot (Berlin: Springer), 45–56.
Hagelueken, G., Wiehlmann, L., Adams, T. M., Kolmar, H., Heinz, D. W., Tümmler, B., et al. (2007). Crystal structure of the electron transfer complex rubredoxin rubredoxin reductase of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 104, 12276–12281. doi: 10.1073/pnas.0702919104
Hugouvieux-Cotte-Pattat, N., Condemine, G., and Shevchik, V. E. (2014). Bacterial pectate lyases, structural and functional diversity. Environ. Microbiol. Rep. 6, 427–440. doi: 10.1111/1758-2229.12166
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Jordan, A., and Reichard, P. (1998). Ribonucleotide reductases. Annu. Rev. Biochem. 67, 71–98. doi: 10.1146/annurev.biochem.67.1.71
Kashyap, D. R., Vohra, P. K., Chopra, S., and Tewari, R. (2001). Applications of pectinases in the commercial sector: a review. Bioresour. Technol. 77, 215–227. doi: 10.1016/s0960-8524(00)00118-8
Kim, J. M., Lee, S. A., Cho, H., Kim, S. J., Joa, J. H., Kwon, S. W., et al. (2017). Parapedobacter lycopersici sp. nov., isolated from the rhizosphere soil of tomato plants (Solanum lycopersicum L.). Int. J. Syst. Evol. Microbiol. 67, 3728–3732. doi: 10.1099/ijsem.0.002162
Kim, M. K., Kim, Y. A., Kim, Y. J., Soung, N. K., Yi, T. H., Kim, S. Y., et al. (2008). Parapedobacter soli sp. nov., isolated from soil of a ginseng field. Int. J. Syst. Evol. Microbiol. 58, 337–340. doi: 10.1099/ijs.0.65249-0
Kim, M. K., Na, J. R., Cho, D. H., Soung, N. K., and Yang, D. C. (2007). Parapedobacter koreensis gen. nov., sp. nov. Int. J. Syst. Evol. Microbiol. 57, 1336–1341. doi: 10.1099/ijs.0.64677-0
Kim, S. J., Weon, H. Y., Kim, Y. S., Yoo, S. H., Kim, B. Y., Anandham, R., et al. (2010). Parapedobacter luteus sp. nov. and Parapedobacter composti sp. nov., isolated from cotton waste compost. Int. J. Syst. Evol. Microbiol. 60, 1849–1853. doi: 10.1099/ijs.0.013318-0
Kolde, R., and Kolde, M. R. (2015). Package “pheatmap.” Version 1.0.12. Avaialble online at: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed December 26, 2018).
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A.102, 2567–2572. doi: 10.1073/pnas.0409727102
Krings, E., Krumbach, K., Bathe, B., Kelle, R., Wendisch, V. F., Sahm, H., et al. (2006). Characterization of myo-inositol utilization by Corynebacterium glutamicum: the stimulon, identification of transporters, and influence on L-lysine formation. J. Bacteriol. 188, 8054–8061. doi: 10.1128/JB.00935-06
Kubra, K. T., Ali, S., Walait, M., and Sundus, H. (2018). Potential applications of pectinases in food, agricultural and environmental sectors. J. Pharm. Chem. Biol. Sci. 6, 23–34.
Kumar, R., Dwivedi, V., Nayyar, N., Verma, H., Singh, A. K., Rani, P., et al. (2015). Parapedobacter indicus sp. nov., isolated from hexachlorocyclohexane-contaminated soil. Int. J. Syst. Evol. Microbiol. 65, 129–134. doi: 10.1099/ijs.0.069104-0
Kumar, R., Verma, H., Haider, S., Bajaj, A., Sood, U., Ponnusamy, K., et al. (2017). Comparative genomic analysis reveals habitat-specific genes and regulatory hubs within the genus Novosphingobium. mSystems 2:e00020-17. doi: 10.1128/mSystems.00020-17
Lefort, V., Desper, R., and Gascuel, O. (2015). FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800. doi: 10.1093/molbev/msv150
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., et al. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Liu, L., Li, L., Song, Z., Wang, S., Zhang, J., Zhang, X., et al. (2017). Parapedobacter deserti sp. nov., an endophytic bacterium isolated from Haloxylon ammodendron stems. Int. J. Syst. Evol. Microbiol. 67, 2148–2152. doi: 10.1099/ijsem.0.001911
Lopez, C. A., Winter, S. E., Rivera-Chávez, F., Xavier, M. N., Poon, V., Nuccio, S. P., et al. (2012). Phage-mediated acquisition of a type III secreted effector protein boosts growth of Salmonella by nitrate respiration. mBio 3, e00143–e00112. doi: 10.1128/mBio.00143-12
Mahato, N. K., Gupta, V., Singh, P., Kumari, R., Verma, H., Tripathi, C., et al. (2017). Microbial taxonomy in the era of OMICS: application of DNA sequences, computational tools and techniques. Antonie Van Leeuwenhoek 110, 1357–1371. doi: 10.1007/s10482-017-0928-1
Maldonado, M. C., and Strasser de Saad, A. M. (1998). Production of pectinesterase and polygalacturonase by Aspergillus niger in submerged and solid state systems. J. Ind. Microbiol. Biotechnol. 20, 34–38. doi: 10.1038/sj.jim.2900470
Marín-Rodríguez, M. C., Orchard, J., and Seymour, G. B. (2002). Pectate lyases, cell wall degradation and fruit softening. J. Exp. Bot. 53, 2115–2119. doi: 10.1093/jxb/erf089
Mendonça, A. G., Alves, R. J., and Pereira-Leal, J. B. (2011). Loss of genetic redundancy in reductive genome evolution. PLoS Comput. Biol. 7:e1001082. doi: 10.1371/journal.pcbi.1001082
Meuric, V., Gracieux, P., Tamanai-Shacoori, Z., Perez-Chaparro, J., and Bonnaure-Mallet, M. (2008). Expression patterns of genes induced by oxidative stress in Porphyromonas gingivalis. Oral Microbiol. Immunol. 23, 308–314. doi: 10.1111/j.1399-302X.2007.00429.x
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonnee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17:132. doi: 10.1186/s13059-016-0997-x
Parks, D., Chuvochina, M., Waite, D., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Pundir, S., Martin, M. J., O'Donovan, C., and UniProt Consortium. (2016). UniProt tools. Curr Protoc. Bioinform. 53, 1–1.29. doi: 10.1002/0471250953.bi0129s53
Rocha, E. R., and Smith, C. J. (1999). Role of the alkyl hydroperoxide reductase (ahpCF) gene in oxidative stress defense of the obligate anaerobe Bacteroides fragilis. J. Bacteriol. 181, 5701–5710.
Sangwan, N., Lata, P., Dwivedi, V., Singh, A., Niharika, N., Kaur, J., et al. (2012). Comparative metagenomic analysis of soil microbial communities across three hexachlorocyclohexane contamination levels. PLoS ONE 7:e46219. doi: 10.1371/journal.pone.0046219
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sharma, A., Sangwan, N., Negi, V., Kohli, P., Khurana, J. P., Rao, D. L., et al. (2015). Pan-genome dynamics of Pseudomonas gene complements enriched across hexachlorocyclohexane dumpsite. BMC Genomics 16:313. doi: 10.1186/s12864-015-1488-2
Smalley, D., Rocha, E. R., and Smith, C. J. (2002). Aerobic-type ribonucleotide reductase in the anaerobe Bacteroides fragilis. J. Bacteriol. 184, 895–903. doi: 10.1128/jb.184.4.895-903.2002
Steyn, P. L., Segers, P., Vancanneyt, M., Sandra, P., Kersters, K., and Joubert, J. J. (1998). Classification of heparinolytic bacteria into a new genus, Pedobacter, comprising four species: Pedobacter heparinus comb. nov., Pedobacter piscium comb. nov., Pedobacter africanus sp. nov. and Pedobacter saltans sp. nov. proposal of the family Sphingobacteriaceae fam. nov. Int. J. Syst. Bacteriol. 48, 165–177. doi: 10.1099/00207713-48-1-165
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. doi: 10.1093/nar/gku1003
Talwar, C., Nagar, S., Kumar, R., Scaria, J., Lal, R., and Negi, R. K. (2020). Defining the environmental adaptations of genus Devosia: Insights into its expansive short peptide transport system and positively selected genes. Sci Rep. 10:1151. doi: 10.1038/s41598-020-58163-8
Tang, Y. P., Dallas, M. M., and Malamy, M. H. (1999). Characterization of the Batl (Bacteroides aerotolerance) operon in Bacteroides fragilis: isolation of a B. fragilis mutant with reduced aerotolerance and impaired growth in in vivo model systems. Mol. Microbiol. 32, 139–149.
Tarlachkov, S. V., Shevchuk, T. V., Montero-Calasanz, M. C., and Starodumova, I. P. (2020). Diversity of rhodopsins in cultivated bacteria of the family Geodermatophilaceae associated with non-aquatic environments. Bioinformatics 36, 1668–1672. doi: 10.1093/bioinformatics/btz840
Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., Rao, B. S., et al. (2001). The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29, 22–28. doi: 10.1093/nar/29.1.22
Turner, B. L., Papházy, M. J., Haygarth, P. M., and McKelvie, I. D. (2002). Inositol phosphates in the environment. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 357, 449–469. doi: 10.1098/rstb.2001.0837
van Dongen, S., and Abreu-Goodger, C. (2012). Using MCL to extract clusters from networks. Methods Mol Biol. 804, 281–295. doi: 10.1007/978-1-61779-361-5_15
Verma, H., Kumar, R., Oldach, P., Sangwan, N., Khurana, J. P., Gilbert, J. A., et al. (2014). Comparative genomic analysis of nine Sphingobium strains: insights into their evolution and hexachlorocyclohexane (HCH) degradation pathways. BMC Genomics 15:1014. doi: 10.1186/1471-2164-15-1014
Voragen, A. G. J., Coenen, G.-J., Verhoef, R. P., and Schols, H. A. (2009). Pectin, a versatile polysaccharide present in plant cell walls. Struct. Chem. 20:263. doi: 10.1007/s11224-009-9442-z
Ward, J. H. (1963). Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58, 236–244. doi: 10.1080/01621459.1963.10500845
Yamaoka, M., Osawa, S., Morinaga, T., Takenaka, S., and Yoshida, K. (2011). A cell factory of Bacillus subtilis engineered for the simple bioconversion of myo-inositol to scyllo-inositol, a potential therapeutic agent for Alzheimer's disease. Microb. Cell Fact. 10:69. doi: 10.1186/1475-2859-10-69
Yang, L., Wang, Y. H., Zhu, H. Z., Muhadesi, J. B., Wang, B. J., Liu, S. J., et al. (2017). Parapedobacter defluvii sp. nov., isolated from the sewage treatment packing of a coking chemical plant. Int. J. Syst. Evol. Microbiol. 67, 4698–4703. doi: 10.1099/ijsem.0.002360
Yoshida, K. I., Aoyama, D., Ishio, I., Shibayama, T., and Fujita, Y. (1997). Organization and transcription of the myo-inositol operon, iol, of Bacillus subtilis. J. Bacteriol. 179, 4591–4598. doi: 10.1128/jb.179.14.4591-4598.1997
Zhao, J. K., Li, X. M., Zhang, M. J., Jin, J. H., Jiang, C. Y., and Liu, S. J. (2013). Parapedobacter pyrenivoranssp. nov., isolated from a pyrene-degrading microbial enrichment, and emended description of the genus Parapedobacter. Int. J. Syst. Evol. Microbiol. 63, 3994–3999. doi: 10.1099/ijs.0.051938-0
Keywords: Parapedobacter, Bat operon, pectinases, inositol, ortholog analysis
Citation: Nagar S, Talwar C, Haider S, Puri A, Ponnusamy K, Gupta M, Sood U, Bajaj A, Lal R and Kumar R (2020) Phylogenetic Relationships and Potential Functional Attributes of the Genus Parapedobacter: A Member of Family Sphingobacteriaceae. Front. Microbiol. 11:1725. doi: 10.3389/fmicb.2020.01725
Received: 19 March 2020; Accepted: 30 June 2020;
Published: 04 September 2020.
Edited by:
John R. Battista, Louisiana State University, United StatesReviewed by:
Pradipta Saha, University of Burdwan, IndiaAnandham Rangasamy, Tamil Nadu Agricultural University, India
Copyright © 2020 Nagar, Talwar, Haider, Puri, Ponnusamy, Gupta, Sood, Bajaj, Lal and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roshan Kumar, cm9zaGFuemhjQGdtYWlsLmNvbQ==; Rup Lal, cnVwbGFsQGdtYWlsLmNvbQ==