 Laura Biessy
Laura Biessy John K. Pearman
John K. Pearman Kirsty F. Smith
Kirsty F. Smith Ian Hawes
Ian Hawes Susanna A. Wood
Susanna A. Wood- 1Coastal and Freshwater, Cawthron Institute, Nelson, New Zealand
- 2Department of Biological Sciences, University of Waikato, Hamilton, New Zealand
- 3New Zealand Food Safety Science and Research Centre, Palmerston North, New Zealand
Tetrodotoxin (TTX) is one of the most potent naturally occurring compounds and is responsible for many human intoxications worldwide. Paphies australis are endemic clams to New Zealand which contain varying concentrations of TTX. Research suggests that P. australis accumulate the toxin exogenously, but the source remains uncertain. The aim of this study was to identify potential bacterial TTX-producers by exploring differences in bacterial communities in two organs of P. australis: the siphon and digestive gland. Samples from the digestive glands of a non-toxic bivalve Austrovenus stutchburyi that lives amongst toxic P. australis populations were also analyzed. Bacterial communities were characterized using 16S ribosomal RNA gene metabarcoding in P. australis sourced monthly from the Hokianga Harbor, a site known to have TTX-bearing clams, for 1 year, from ten sites with varying TTX concentrations around New Zealand, and in A. stutchburyi from the Hokianga Harbor. Tetrodotoxin was detected in P. australis from sites all around New Zealand and in all P. australis collected monthly from the Hokianga Harbor. The toxin averaged 150 μg kg–1 over the year of sampling in the Hokianga Harbor but no TTX was detected in the A. stutchburyi samples from the same site. Bacterial species diversity differed amongst sites (p < 0.001, F = 5.9) and the diversity in siphon samples was significantly higher than in digestive glands (p < 0.001, F = 65.8). Spirochaetaceae (4–60%) and Mycoplasmataceae (16–78%) were the most abundant families in the siphons and the digestive glands, respectively. The bacterial communities were compared between sites with the lowest TTX concentrations and the Hokianga Harbor (site with the highest TTX concentrations), and the core bacterial communities from TTX-bearing individuals were analyzed. The results from both spatial and temporal studies corroborate with previous hypotheses that Vibrio and Bacillus could be responsible for the source of TTX in bivalves. The results from this study also indicate that marine cyanobacteria, in particular picocyanobacteria (e.g., Cyanobium, Synechococcus, Pleurocapsa, and Prochlorococcus), should be investigated further as potential TTX producers.
Introduction
Tetrodotoxin (TTX) is one of the most potent naturally occurring neurotoxins in the world (Noguchi et al., 2011). This toxin was thought to occur only in tropical to sub-tropical climates but has now been reported from more temperate climates in the Pacific and Mediterranean (Lago et al., 2015; Biessy et al., 2019a). Tetrodotoxin is found in a wide variety of phylogenetically unrelated terrestrial, marine and freshwater organisms (Bane et al., 2014) and the consumption of seafood, primarily puffer fish and gastropods, contaminated with TTX has resulted in many human intoxications (Noguchi et al., 2011). It was first identified in marine bivalves (Japanese scallops, Pactinopecten yessoensis) in 1993 (Kodama et al., 1993), but recently there has been an increase in detection and published reports with TTX now identified in ten bivalve species from seven countries (Biessy et al., 2019a). The detection of TTX in harvested edible bivalves has led to concerns about health risks for human consumers and prompted further studies worldwide.
Despite substantial research over three decades, the origin of TTX remains uncertain (Chau et al., 2011). The two most common hypotheses regarding its origin are: (1) it is endogenously produced by symbiotic bacteria, or (2) it is exogenously accumulated through dietary sources (e.g., bacteria, micro-algae). Exogenous or symbiotic bacteria are commonly touted as the most likely TTX-producers, with a wide diversity of species and strains implicated. There are reports in the literature of at least 150 TTX-producing bacterial strains, with Vibrio species comprising more than 30% of records, followed by Bacillus, Pseudomonas, Actinomyces, and Micrococcus (Turner et al., 2018; Katikou, 2019). However, it is estimated that less than 1% of all bacteria are culturable, making isolation, culturing and confirmation of toxin production in bacteria a significant challenge (Chau et al., 2011). Studies have claimed to isolate bacteria that potentially produce TTX, but the concentration of toxin identified in laboratory cultures is usually low compared to amounts in the host animals. For example, 184 ng g–1 of TTX was reported from an isolated Vibrio sp. in comparison to 36 μg g–1 in the tissue of its host, the sea snail Nassarius semiplicatus (Wang et al., 2008). Additionally, there are indications that some of the methods (e.g., high performance-liquid chromatography and gas chromatography-mass spectrometry) used to analyze the TTX-producing bacteria in culture generate false positives (Matsumura, 1995).
Limited research has been undertaken on the source of TTX in marine bivalves but, as in other organisms, available evidence indicates an exogenous source (Biessy et al., 2018, 2019a). Gammaproteobacteria, particularly Vibrio and Pseudomonas species, have been linked to the accumulation of TTX in bivalves, with two recent studies finding correlations between the presence of Vibrio, Pseudomonas and TTX in shellfish (Turner et al., 2017b; Leão et al., 2018). However, these studies were unable to culture any TTX-producing bacteria from the shellfish samples. The hypothesis that bacteria or micro-algae are the source of TTX in these organisms is also fueled by reports of toxic episodes in bivalves being more prevalent during warmer months, particularly in late spring in Europe (Gerssen et al., 2018; Leão et al., 2018) and New Zealand (Biessy et al., 2019b). This may indicate seasonal changes in bacterial communities and an increase in warm-water adapted TTX-producing microorganisms.
The application of molecular genetics-based techniques to characterize entire communities (e.g., metabarcoding) is escalating rapidly (Taberlet et al., 2012; Deiner et al., 2017). Metabarcoding is a culture-independent technique that enables taxa in an environmental sample or organ to be identified simultaneously and has previously been used as a tool to investigate the source of TTX by characterizing the core bacterial communities of toxin-bearing organisms. Salvitti et al. (2017) used metabarcoding to investigate the foregut content of the highly toxic, carnivorous sea-slug Pleurobranchaea maculata from New Zealand. The authors found a high abundance of sequences taxonomically related to Cnidaria and Annelida, taxa known to contain neurotoxins that could have been ingested by P. maculata. Turner et al. (2018) studied the bacterial communities of the TTX-bearing nemertean worm Cephalothrix simula and showed the prevalence of a large number of bacterial genera previously associated with TTX production including Alteromonas, Vibrio and Pseudomonas. In a recent study, Melnikova and Magarlamov (2020) investigated the core bacterial communities of TTX-bearing and non-bearing nemertean worms from Japan and demonstrated that toxic individuals tend to have more potential TTX-producing bacterial strains (e.g., Pseudomonas, Vibrio and Bacillus) in their microflora but they were unable to identify a possible TTX producer. One issue with microbiome analyses of motile organisms is that it only provides a “snapshot” of what the organisms have ingested at one point in time and location. These organisms may move from the site of TTX-ingestion, making it challenging to identify TTX sources in the environment. Analysis of bacterial communities of sedentary animals, such as marine bivalves, would provide novel insights into the source of TTX, especially if investigated over spatial and temporal scales.
In 2011, McNabb et al. (2014) recorded high concentrations (800 μg kg–1) of TTX in an endemic marine clam Paphies australis in Whangapoua (Coromandel, New Zealand). Immunohistochemistry and chemical analyses of P. australis individuals from Hokianga Harbor (Northland, New Zealand) revealed that the siphons and organs involved in feeding contained the highest concentrations of TTX (means of 403 and 34 μg kg–1, respectively), providing evidence to support the hypothesis of an exogenous source (Biessy et al., 2018). This hypothesis was further strengthened during an experiment where P. australis individuals were maintained in captivity for 150 days and showed significant depuration of TTX. Tetrodotoxin present in the digestive glands at the start of the experiment depurated rapidly and only traces remained after 21 days (Biessy et al., 2019b). Furthermore, P. australis sourced from different locations around New Zealand have significant variations in TTX concentrations (Biessy et al., 2019b). The bivalves collected from warmer sites (at lower latitudes) contained higher TTX concentrations than the ones from the colder regions.
The overarching goal of the present study was to further investigate the source of TTX in P. australis. We used metabarcoding to characterize bacterial communities in the digestive glands and siphons sourced monthly from one site with high concentrations, for 1 year, and from single time points at ten sites with P. australis that contained varying TTX concentrations. The aims of the study were: (1) to explore differences in bacterial communities between P. australis populations with high and low TTX concentrations and a non-toxic bivalve species, the cockle Austrovenus stutchburyi, found in the same habitat at one site; (2) to evaluate the core bacterial communities of P. australis at one site where TTX concentrations were relatively stable over time; and (3) use the genetic information from bacterial amplicon sequence variants and corresponding toxin data to identify potential TTX-producing bacterial species. Intra- and inter-species variation in toxin concentration provides a unique opportunity to examine the differences in bacterial communities of these populations and identify any specific bacterial communities elements correlated with toxin concentrations in the host.
Materials and Methods
Sample Collection
Spatial Study
Paphies australis (n = 8 per site) were collected from ten sites spanning the length of New Zealand between September 2017 and February 2019 (Supplementary Figure S1). The P. australis were chilled (ca. 8°C) and sent overnight to the laboratory (Cawthron Institute, Nelson, New Zealand). Once in the laboratory, three of the eight individuals were aseptically dissected for genetic analysis: the siphons and digestive glands were placed in individual tubes and stored frozen (−20°C) until further analysis. The remaining five P. australis from each site were used for toxin extraction. Austrovenus stutchburyi were identified amongst the P. australis beds in the Hokianga Harbor and were also collected (n = 8) in September 2017 for further analysis. Five A. stutchburyi were used whole for toxin extraction and the remaining (n = 3) were aseptically dissected and the digestive glands were placed in individual tubes and stored frozen (−20°C) until DNA was extracted.
Temporal Study
Paphies australis were collected from the Hokianga Harbor (Northland, New Zealand, 35°28′S, 173°24′E; Supplementary Figure S1) monthly from April 2017 to March 2018 (except May 2017, n = 11 sampling events), chilled and sent to the laboratory overnight. P. australis from this location had previously been shown to contain TTX (Biessy et al., 2018). P. australis used for the temporal study were collected as part of a larger survey investigating TTX concentrations in recreationally harvested bivalves in New Zealand (Harwood et al. in preparation). The P. australis received each month were pooled together to follow the standard operating procedure which is used in the accredited laboratory that undertook the analysis of these samples. Three P. australis were put aside each month and were then dissected and stored for further genetic analysis as described above.
Toxin Extractions
Tetrodotoxin extraction was performed as previously described in Biessy et al. (2018). Briefly, for the spatial samples, P. australis and A. stutchburyi were shucked and individually weighed (ca. 0.5–3.0 g) and placed in a sterile tube (50 mL) with a corresponding volume (ca. 500–3,000 μL) of Milli-Q water containing 1% acetic acid. Organisms were homogenized (Ultra-Turrax®, IKA®, NC, United States) for 45 s. The temporal samples were freshly opened, left to drain (5 min), pooled together for homogenisation (Ultra-Turrax®) for 5 min and a sub-sample was weighed (5 g) and placed in a sterile tube containing Milli-Q water and 1% acetic acid (5 mL). Subsequently extraction for both the spatial and temporal samples followed the same procedure. The tubes were boiled (5 min) and cooled in an ice bath (5 min) before briefly vortexing. Samples were centrifuged (3,200 × g, 10 min) and 0.5–1 mL of the supernatant transferred to a centrifuge tube (1.7 mL) containing 25% ammonia (2.5–5 μL; Honeywell, Seelze, Germany). Samples were then centrifuged (17,000 × g, 1 min) and the supernatant subjected to the graphitised carbon solid phase extraction (SPE) method as described in Boundy et al. (2015) using Supelclean ENVI-Carb 250 mg/3 mL cartridges (Sigma-Aldrich, MO, United States). Tetrodotoxin was analyzed and quantified by liquid chromatography tandem-mass spectrometry analysis as described by Turner et al. (2017a). The toxin results for samples collected in 2017 and 2018 were previously published by Biessy et al. (2019b) but are integrated with the molecular dataset in this study.
Statistical Analysis
Statistical analyses for toxin concentrations were performed using the R statistical package (R-Project, 2019). Normality was checked through inspection of Quantile-Quantile plots and conducting a Shapiro-Wilk test. As the data deviated from a normal distribution the spatial data was log transformed. Levene’s test was used to assess the equality of variance in TTX concentrations between sites (Carroll and Schneider, 1985). Statistical differences in TTX concentrations between sites was assessed using a one-way analysis of variance (ANOVA; Factor: Site; 10 levels). A Tukey’s honestly significant difference (HSD) post-hoc test was used to identify which sites were responsible for the significant differences.
Metabarcoding
DNA Extractions and PCR
Dissected organs from both studies were individually placed in the first tube containing bashing beads of the DNA extraction kit (DNeasy Powersoil Pro kit, Qiagen, MD, United States). The DNA was then extracted following manufacturer’s instructions using an automated homogenizer (1600 MiniG Automated Tissue Homogenizer and Cell Lyser, SPEX SamplePrep, NJ, United States) and a robotic workstation for DNA extraction (QIAcube, Qiagen). Negative extraction controls were performed every 23 samples. Amplification and sequencing PCR Amplicons were generated covering the V3 and V4 regions of the 16S ribosomal RNA (rRNA) gene using the primer sets as described in Klindworth et al. (2013) and were modified to include IlluminaTM overhang adaptors following the dual-indexing method from Kozich et al. (2013): Bact341F-5′–TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG–3’ and Bact785R-5′–GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C–3′. PCR reactions were undertaken in triplicate with 450 nM of each primer, 25 μL of 2X MyFiTM Mix (Bioline, United Kingdom), ca. 5 ng of DNA, and sterile water for a total reaction volume of 50 μL. Cycling conditions were: 95°C for 5 min, followed by 30 cycles of 94°C for 30 s, 54°C for 30 s, 72°C for 45 s, and a final extension of 72°C for 7 min. Triplicates of PCR products were pooled and visualized on 1.5% agarose gel with Red SafeTM DNA Loading Dye (Herogen Biotech, United States) and UV illumination. PCR negatives were run to assess for contamination during the PCR steps. The PCR products were purified, cleaned of primer dimers and normalized using SequalPrep Normalization plate (Thermo Fisher, MA, United States), and submitted to Auckland Genomics (University of Auckland, New Zealand) for library preparation. Sequencing adapters and sample-specific indices were added to each amplicon via a second round of PCR using the NexteraTM Index kit (Illumina Inc., United States). Amplicons were pooled into a single library and paired-end sequences (2 × 250 bp) generated on a MiSeq® instrument. The sequencing libraries were prepared following the Illumina 16S Metagenomics Library Prep manual with the exception that after the indexing, 5 μL of each sample (including three water samples acting as sequencing blank) was pooled and a single clean-up was undertaken on the pool instead of samples being individually cleaned. Quality control was undertaken using a bioanalyzer before the library was diluted to 4 nM and denatured. A 15% PhiX spike was used and the final loading concentration was 7 ρM. Sequence data were automatically demultiplexed using MiSeq® Reporter (version 2, Illumina Inc.), and forward and reverse reads assigned to samples. Raw sequence reads were deposited in the National Center for Biotechnology Information (NCBI) short read archive under the accession number PRJNA623388.
Bioinformatics
Amplicon sequence variant inference and taxonomic assignments
Raw reads were processed, subsequent to primers being removed with cutadapt (Martin, 2011), using the DADA2 package (Callahan et al., 2016) within R. Reads were truncated to 228 and 230 bp and filtered with a maxEE (maximum number of “expected errors”) of 2 and 4 for forward and reverse reads, respectively (reads not reaching this threshold were discarded). DADA2 constructs a parametric error matrix (based on the first 108 bps in the dataset), the samples are dereplicated and sequence variants for the forward and reverse reads are inferred based on the derived error profiles from the samples. Singletons observed in the inference step are discarded. Subsequently, paired-end reads were merged with a maximum mismatch of 1 bp and a required minimum overlap of 10 bp. Forward and reverse reads, which did not merge were not included in further analysis. Chimeras were removed using the function removeBimeraDenovo. The resulting chimera-checked, merged amplicon sequence variants (ASV) were used for taxonomic classification using the SILVA v132 database (Pruesse et al., 2007) within the DADA2 package, which is based on the rdp classifier (Wang et al., 2007) with a bootstrap of 50. The results were parsed into a table using the phyloseq package (Mcmurdie and Holmes, 2013), and reads assigned as eukaryotes, chloroplasts and mitochondria were removed. Negative controls were assessed and the sum of reads from contaminating ASVs was subtracted from the samples. Samples were subsampled to an even depth of 9,500 sequences for comparison.
Spatial study
Phylogenetically annotated 16S rRNA sequences were used to characterize bacterial community composition of each tissue type at the family level. Donut charts were generated using the package ggplot2 (Wickham, 2016) in R based on the average relative abundance of sequence reads attributed to a given bacterial family within each tissue type and for each site. A two-way permutational multivariate analysis of variance (PERMANOVA; Anderson, 2001) was used to examine the potential differences in community composition as a function of tissue types (digestive gland, siphon or cockle) and sampling sites, assessed at the ASV level. PERMANOVA was run on triangular similarity matrices derived from the fourth root transformed data calculating the Bray-Curtis. Visualization of the observed patterns was obtained by means of a principle coordinates analysis (PCoA) using a Bray-Curtis similarity matrix. Alpha diversity numbers were calculated using the estimate_richness script in R. Differences in alpha diversity between tissues were assessed by an analysis of variance (ANOVA) for the “Observed” diversity index. Comparative analysis of the bacterial communities from sites with low P. australis TTX concentrations (Bleinhem, Marahau and Riverton) with those from Hokianga Harbor which has P. australis with high TTX was undertaken to identify potential members of the bacterial community correlated with changes in TTX concentrations. The second replicate for the Hokianga siphons did not contain enough reads and was lost after rarefaction. The bacterial communities from the digestive glands of both P. australis and A. stutchburyi were compared using phyloseq. Relationships between TTX concentrations and the 20 most abundant ASVs in the 20 most abundant phyla were determined using linear regressions.
Temporal study
The core bacterial communities of digestive glands and siphons from P. australis over a one-year period was identified using the microbiome package in R (Lahti et al., 2017). ASVs present in at least 70% of samples were chosen as members of the core bacterial communities. A two-way PERMANOVA was used to identify potential differences in community composition as a function of organs and sampling months, assessed at the ASV level. PERMANOVA was run on triangular similarity matrices derived from the fourth root transformed data calculating the Bray-Curtis. Alpha diversity numbers were calculated using the estimate_richness script in R. Differences in alpha diversity between tissues were assessed by an analysis of variance (ANOVA) for the “Observed” diversity index Visualization of the observed patterns was plotted with principle coordinates analysis using the Bray-Curtis similarity matrix. The core bacterial communities of the digestive glands from A. stutchburyi was also identified using the microbiome package.
Results
Tetrodotoxin Analysis
Spatial Study
Tetrodotoxin was detected in P. australis from sites all around New Zealand (Figure 1A) with TTX as the main congener (>99%) in all samples. A Levene’s test showed that the degree of variance did not differ among sites (F = 0.59). One-way ANOVA showed a significant difference between TTX concentrations among all sites (F = 51.6, p < 0.001), with a Tukey’s HSD post-hoc test identifying a complex overlap between sites (Figure 1A). The highest concentrations were measured in P. australis from the northernmost site, the Hokianga Harbor from both years tested. Median toxin concentrations in P. australis from the South Island sites were significantly lower than the North Island sites. No TTX (<3 μg kg–1) was detected in the A. stutchburyi samples from the Hokianga Harbor site.
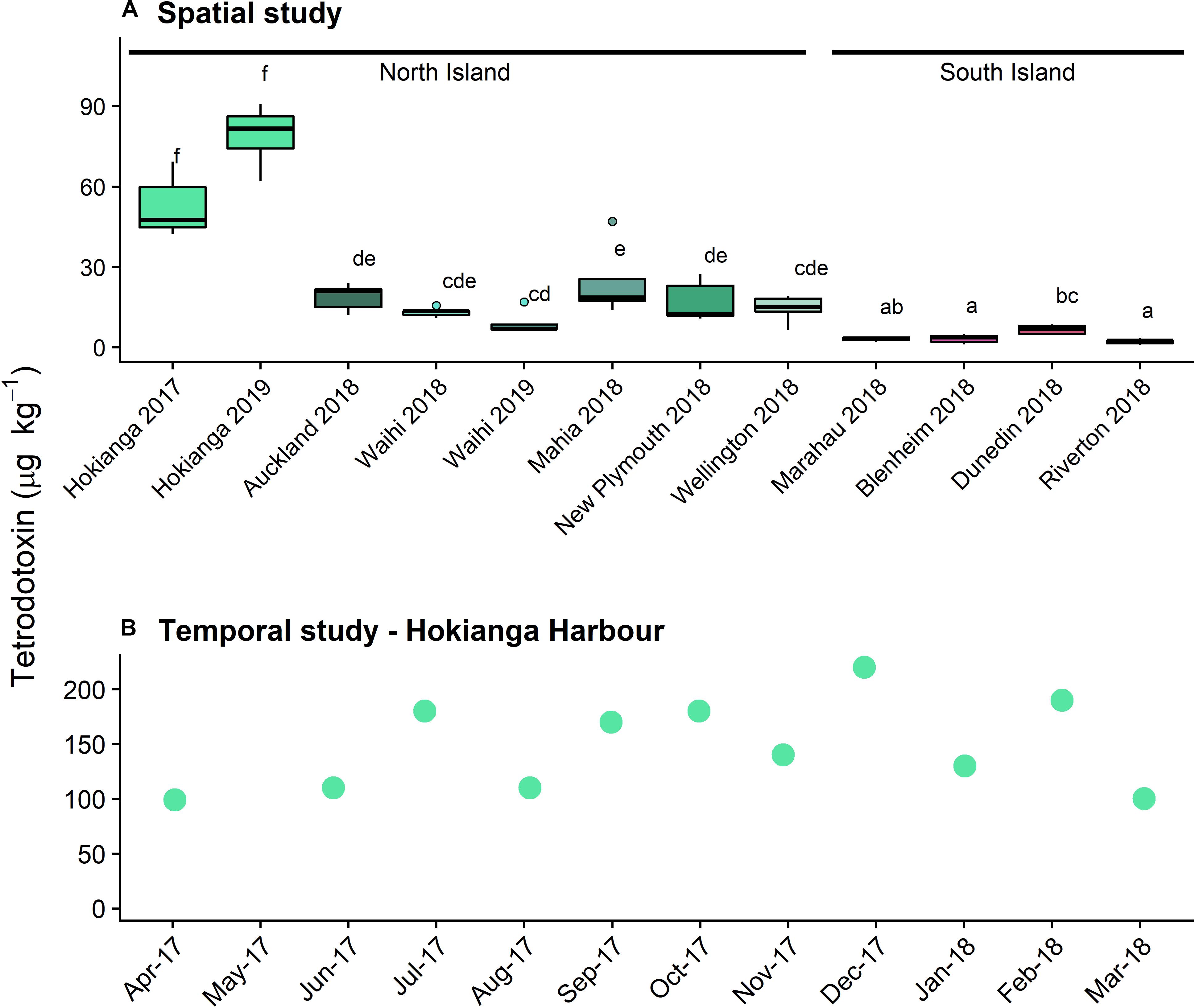
Figure 1. Tetrodotoxin concentrations in populations of Paphies australis, (A) collected around the New Zealand coastline, determined using liquid chromatography-mass spectrometry (LC/MS; n = 5). Solid black line shows median, box shows 1st and 3rd quartiles, whiskers extend to the last data point within 1.5 times the inter-quartile range. Dots outside the whiskers are considered as outliers, defined as beyond 1.5× interquartile range. Different letters indicate where significant differences occur between sites (one-way ANOVA with Tukey’s HSD post-hoc test, p < 0.001). Sites are ordered by increasing latitude for each island. Data from 2018 and earlier was previously reported in Biessy et al. (2019b). (B) Tetrodotoxin concentrations in populations of P. australis collected from the Hokianga Harbor site over a one-year period, determined using LCMS (n = 1 pooled sample).
Temporal Study
Tetrodotoxin was detected in all P. australis collected monthly from the Hokianga Harbor between April 2017 and March 2018 (Figure 1B). The lowest concentration was 99 μg k–1 in April 2017 and the highest was 220 μg kg–1 in December 2017 and the toxin averaged 150 μg kg–1 over the year of sampling. Only one sample was extracted for toxin analysis per time point and statistical analyses were not possible. The differences for the Hokianga Harbor toxin concentration between the spatial and temporal study are likely due to the minor methodological differences in toxin extraction.
Metabarcoding
Spatial Study
Across the 64 samples, encompassing the 10 sites and two tissues, the 16S rRNA gene sequencing yielded 6,865,560 reads sequences that resulted in a total of 17,188 distinct ASVs after processing and rarefaction. The number of reads for each sample at the various processing step is provided in Supplementary Table S1.
Species diversity significantly differed amongst the ten sites around New Zealand (p < 0.001, F = 5.9) but no differences between the two islands were observed (Supplementary Figure 2). The siphons had bacterial communities with significantly higher alpha diversity than the digestive glands (p < 0.001, F = 65.8). When examining the most abundant (top ten) families in term of read numbers (Figure 2), Spirochaetaceae (ranging from 4 to 60% across all samples) and Mycoplasmataceae (16–78%) accounted for the highest proportions of reads in the siphons and digestive glands, respectively, for both islands. Proteobacteria (purple and pink hues; Figure 2) were present at all sites and in both organs. Bacteroidetes (green hues, Figure 2) were present at all sites, except Waihi, and in higher abundances in the siphons. Cyanobiaceae were detected in two sites, Hokianga and Mahia (Figure 2).
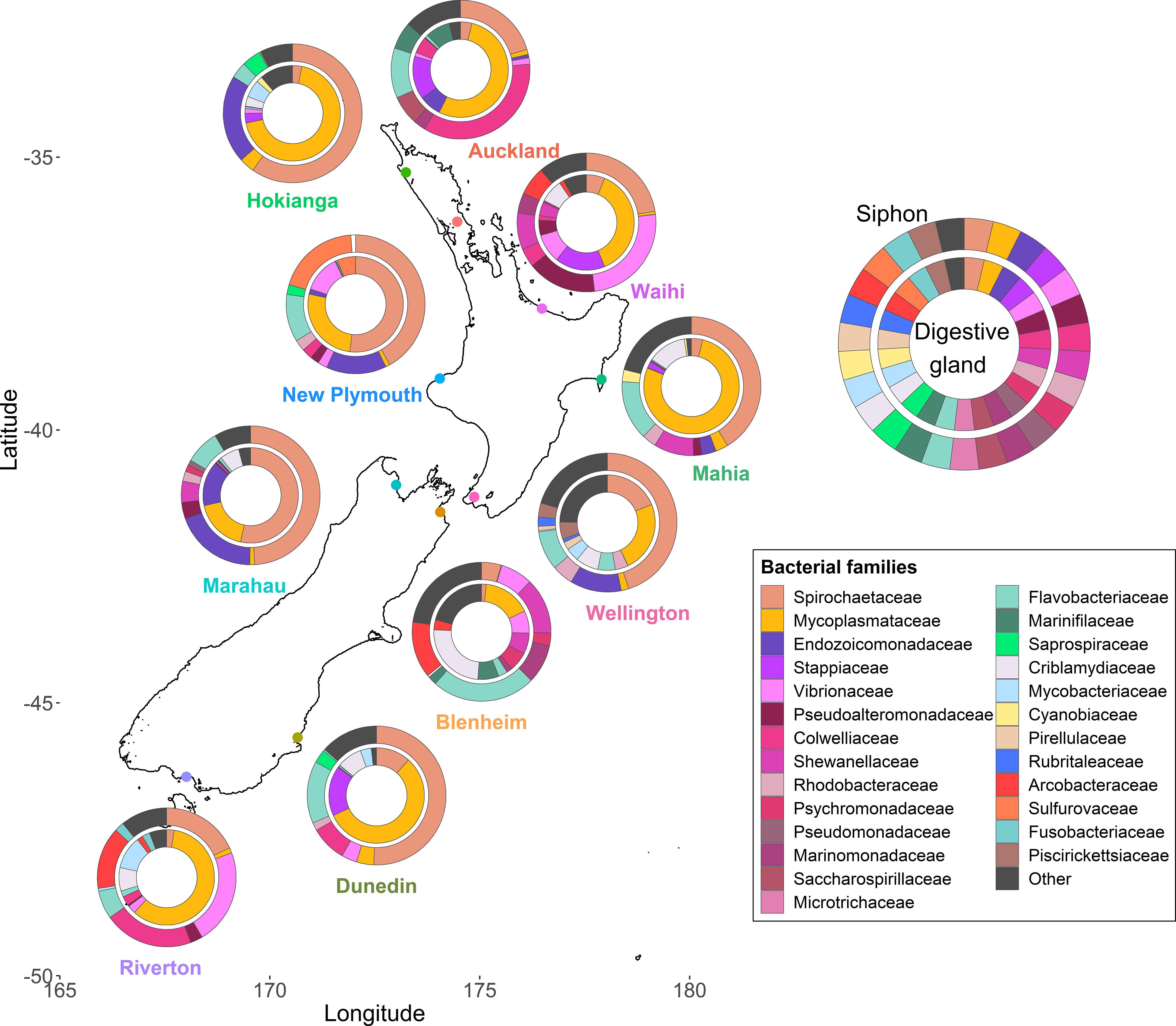
Figure 2. Bacterial community composition at the family level from the digestive glands and siphons of the New Zealand endemic clam Paphies australis collected from ten sites across the north and south islands. The relative abundance of the ten most abundant families in terms of sequence number for each site and organ are shown. Family are represented by different colors but the phylum Proteobacteria is represented by the purple-pink hues and the Bacteroidetes are represented by the green hues.
Multidimensional scaling based on Bray-Curtis similarities between bacterial communities (using ASVs), showed a clear separation between the two organs from P. australis and A. stutchburyi digestive gland (Figure 3). Comparison of bacterial communities across tissues and sites showed that the bacterial communities structure had a significant interaction term (PERMANOVA, p < 0.001, F = 1.61), suggesting inconsistent differences amongst the sites between organs.
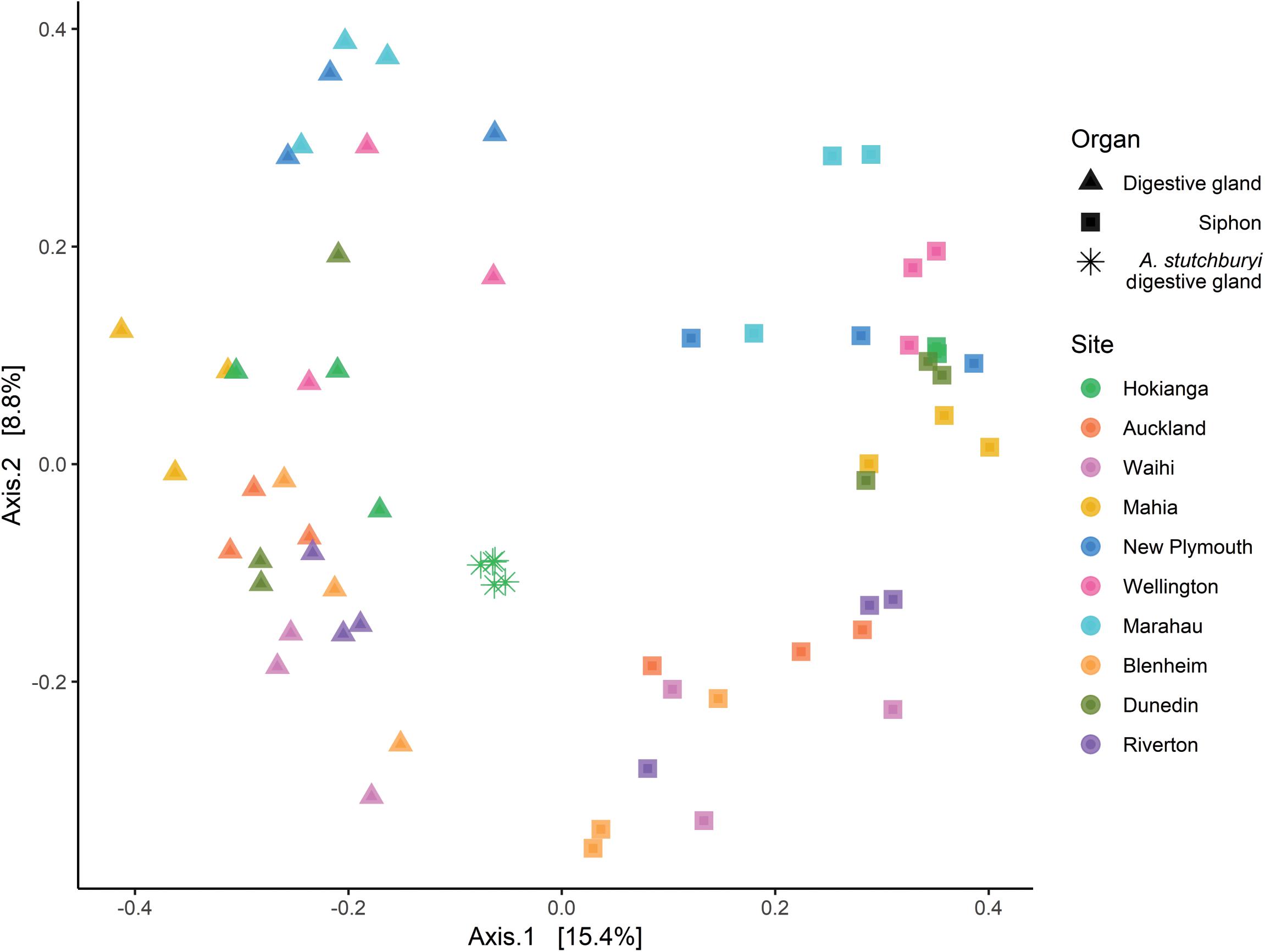
Figure 3. Principal Coordinates Analysis (PCoA) based on Bray-Curtis dissimilarities with 999 permutations of bacterial community composition of Paphies australis organs (unless indicated otherwise) at the Amplicon Sequence Variant level from ten different sites around New Zealand. For each axis, in square brackets, the percent of variation explained was reported. Sites are ordered by increasing latitude for New Zealand. A. stutchburyi = Austrovenus stutchburyi.
The bacterial community was compared between the pooled sites with the lowest TTX concentrations (Blenheim, Marahau, and Riverton) and the ones from the Hokianga Harbor containing the highest toxin concentration. The most abundant bacterial genera in P. australis’ siphons from the Hokianga Harbor (Figure 4A) that were not present in low TTX sites were Kistimonas and Spirochaeta_2 followed by Pleurocapsa and Roseibacillus. For the digestive glands, Mycoplasma was the main genus remaining in the P. australis from the Hokianga Harbor after ASVs from the low TTX sites had been subtracted (Figure 4B). Other genera present included Bacillus, Clostridium, Prochlorococcus and Vibrio. Finally, when comparing the bacterial communities present in the digestive glands of the TTX-bearing P. australis to those in the non-toxic A. stutchburyi, Mycoplasma was also the most abundant genus remaining. Bacillus, Mycobacterium, Spirochaeta, and Vibrio were also present (Figure 4C).
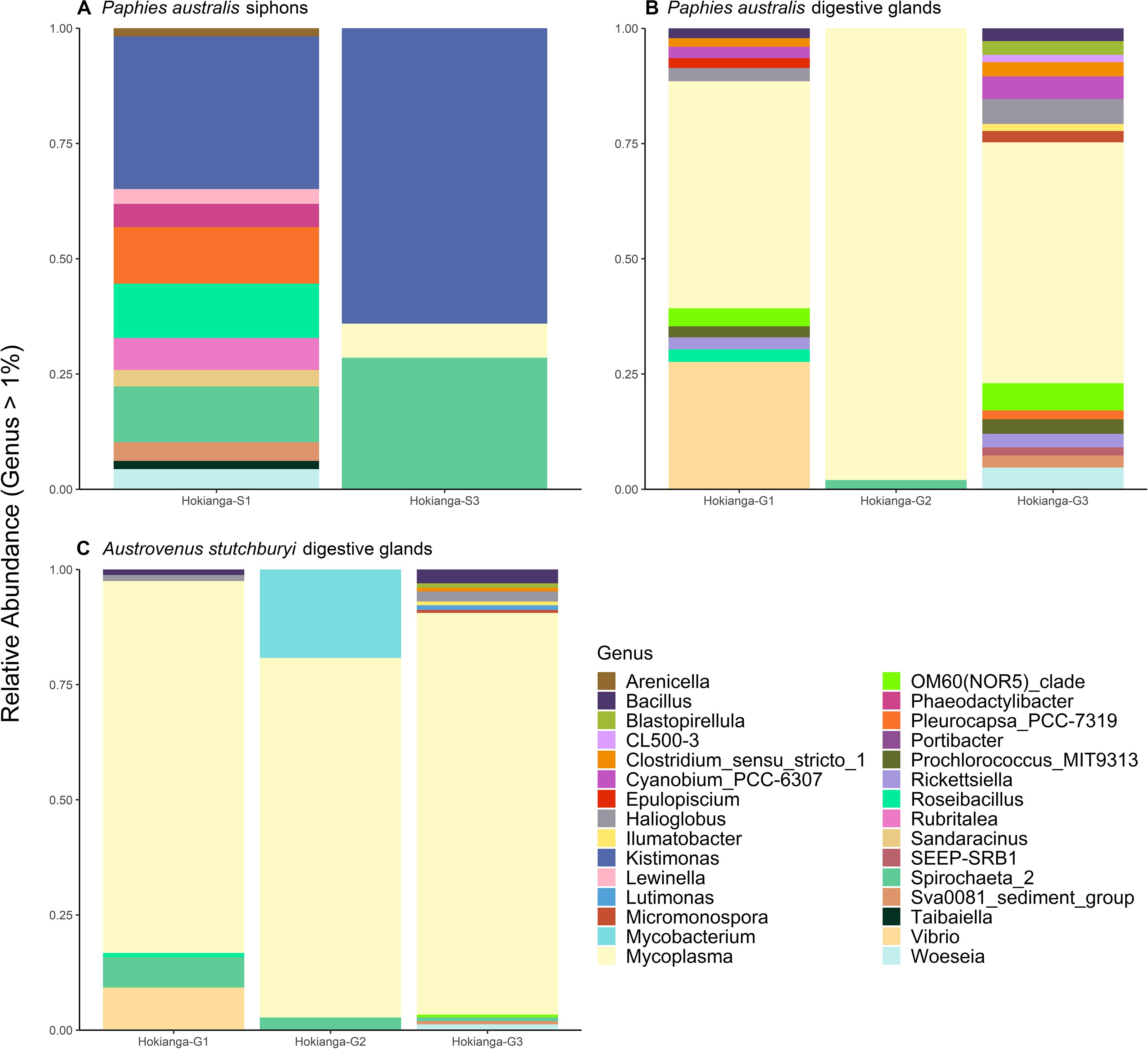
Figure 4. Stacked barplots showing the relative abundance of bacterial 16S ribosomal RNA sequences, at the genus level (>1% of total abundance), that are present in tetrodotoxin (TTX)-bearing Paphies australis from the Hokianga Harbor samples but not in tissue samples from sites with low TTX. (A,B) show communities present in P. australis’ siphons and digestive glands, respectively, at the Hokianga site but not in similar tissues from the three lowest TTX sites. (C) shows relative abundance of bacteria communities present in TTX-bearing P. australis’ digestive glands and the non-toxic Austrovenus stutchburyi’s digestive gland from the same bed in the Hokianga Harbor. S1: Siphon replicate 1, S3: Siphon replicate 3, G1: digestive gland replicate 1, G2: digestive gland replicate 2, G3: digestive gland replicate 3.
The results from the linear regression analysis identified 17 ASV that were significantly and positively correlated to TTX concentrations, however, all relationships were weak (R2 < 0.35; Supplementary Table S2). The phyla present were: Verrucomicrobia (4 ASVs from the Rubritaleaceae family), Tenericutes (4 ASVs from the Mycoplasmataceae family), Acidobacteria (2 ASVs), Cyanobacteria (2 ASVs including the genus Synechococcus_CC9902), Marinimicrobia (2 ASVs), and one ASVs from each Firmicutes (Romboutsia genus), Planctomycetes and Proteobacteria phyla.
Temporal Study
Bacterial composition
Bacterial DNA was isolated from P. australis siphons and digestive glands every month for 11 months (n = 3 per month), totaling 62 samples after rarefaction and contamination removal. The 16S rRNA gene sequencing yielded 589,000 reads that resulted in 14,782 distinct ASVs. The number of reads for each sample at the various processing steps is provided in Supplementary Table S1.
Taxonomic community composition of the dominant families (abundance > 5%) by tissue type revealed that similar numbers were present in both P. australis’ tissues (Figure 5). There were high relative abundances of Mycoplasmataceae (65–95%) and Spirochaetaceae (60–90%) in the digestive glands and siphons, respectively. In the siphons, Flavobacteriaceae (0–14%) and Mycoplasmataceae (0–12%) were also substantial components of the community occurring in samples from most months. Endozoicomonadaceae (0–18%) were also present half of the year, mostly in winter (July–September). In the digestive glands, Spirochaetaceae (4–37%) and Criblamydiaceae (0–25%) were present in samples from the majority of the sampling period. Stappiaceae (0–8%), Endozoicomonadaceae (0–4%), and Vibrionaceae (0–2%; Figure 5) were less frequently observed in the digestive gland communities (Figure 5).
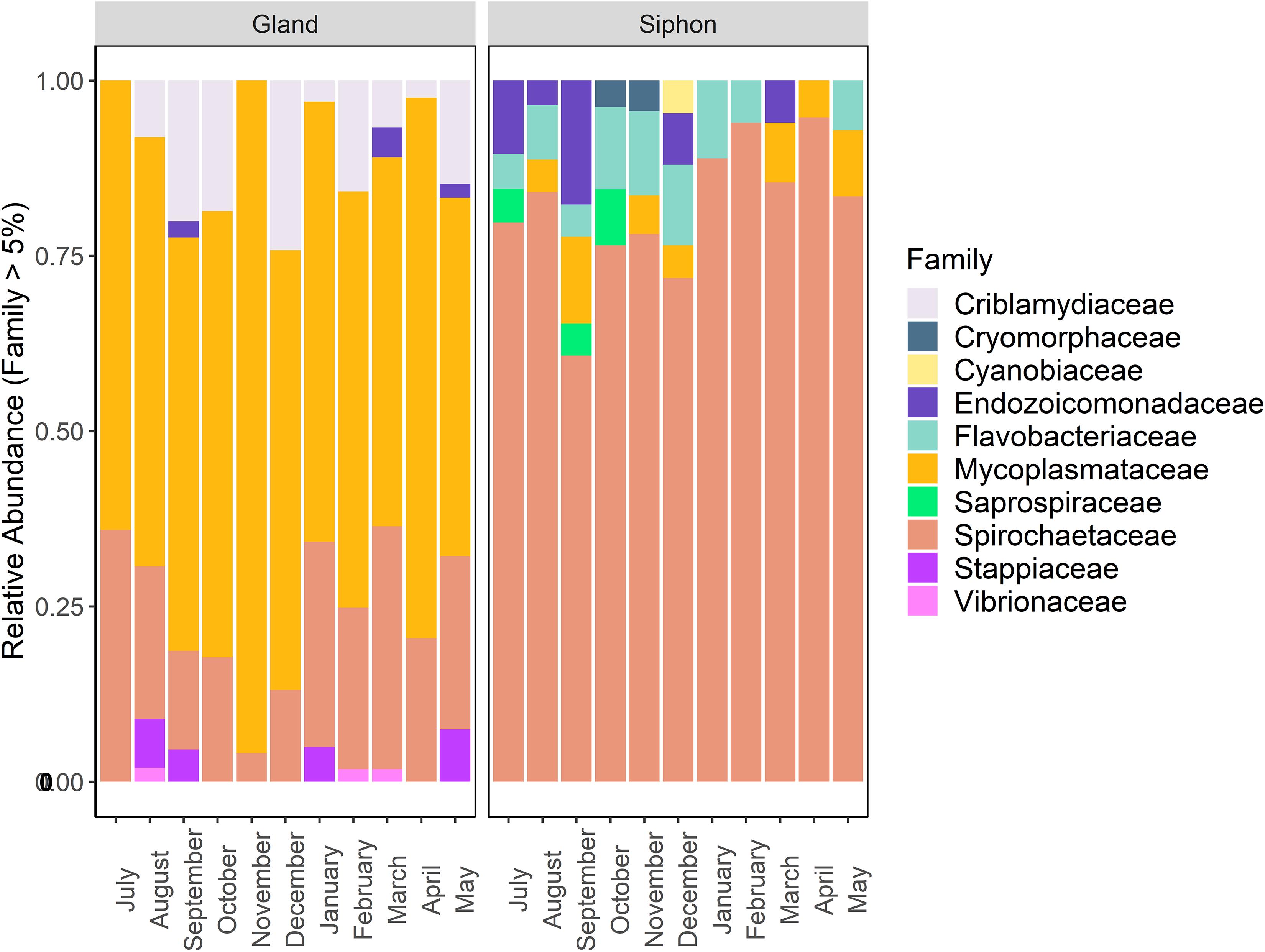
Figure 5. Stacked bar plot showing the relative abundance of bacterial 16S ribosomal RNA sequences at the family level (contributing to >5% of total abundance) in Paphies australis digestive glands and siphons from the Hokianga Harbor (Northland, New Zealand), sampled every month from July 2017 to May 2018.
Multidimensional scaling based on Bray-Curtis similarities between bacterial communities (using ASVs), showed a clear separation between the two organs (Figure 6). Analysis of overall bacterial community structure revealed a significant difference between siphons and digestive glands (PERMANOVA: p < 0.0001, F = 11.5). No significant interaction was found between sampling months and tissue type (PERMANOVA, p = 0.12, F = 1.26). The digestive glands did not display a seasonal pattern in species diversity but had a significantly lower diversity than the siphons (p < 0.001, F = 181.85; Figure 6). In contrast, the siphons had a higher diversity in warmer months (October to January; Supplementary Figure S3).
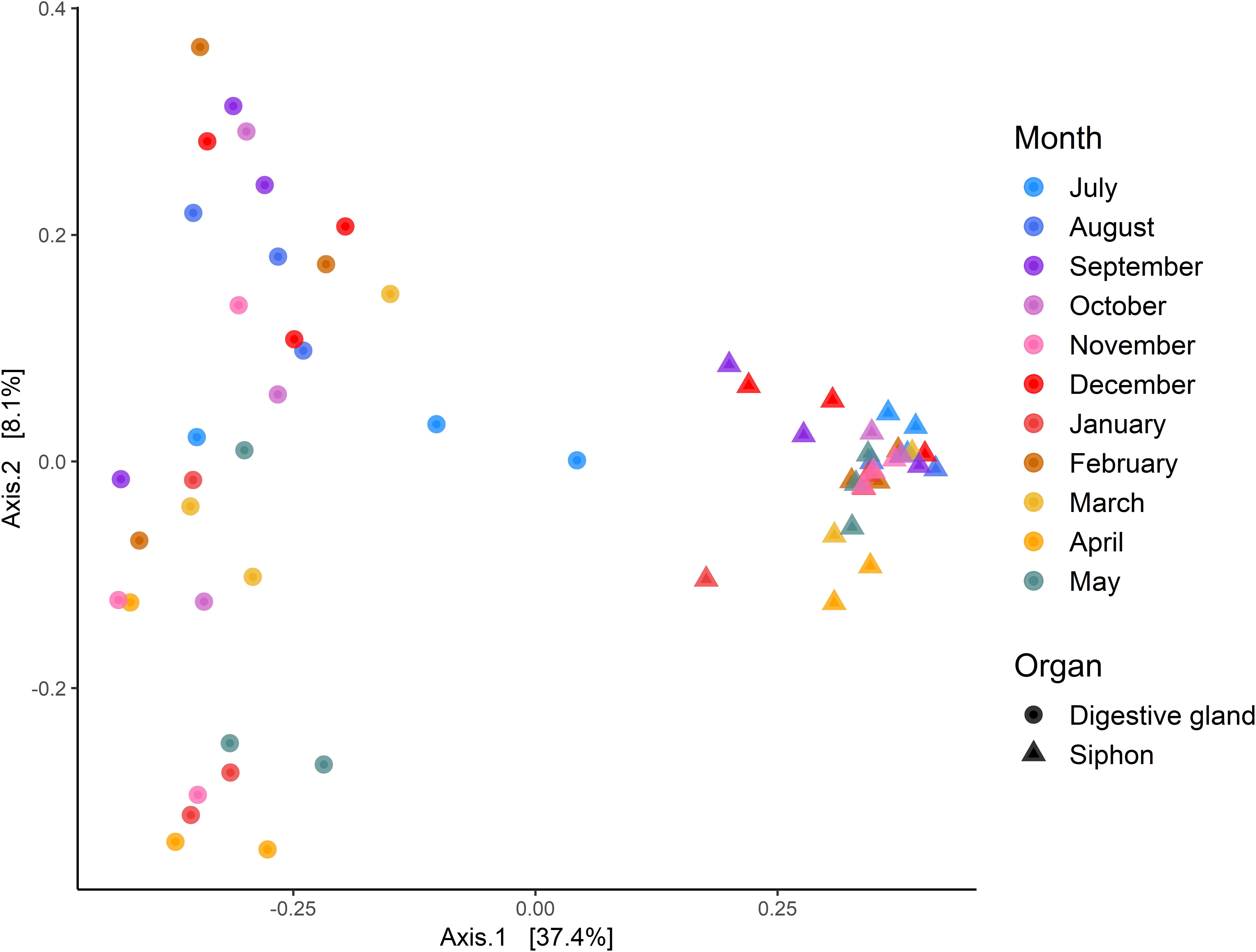
Figure 6. Principal Coordinates Analysis (PCoA) based on Bray-Curtis dissimilarities with 999 permutations of bacterial community composition of Paphies australis’ organs at the Amplicon Sequence Variant level, sampled every month from July 2017 to May 2018 from the Hokianga Harbor (Northland, New Zealand). For each axis, in square brackets, the percent of variation explained is reported.
Core bacterial communities
A total of 38 and 74 bacterial ASVs present in at least 70% of all samples were identified as members of the core bacterial communities of the P. australis’ digestive glands and siphons, respectively (Tables 1, 2). Mycoplasmatales (15 ASVs total) was the most dominant order in the digestive gland bacterial communities, followed by Spirochaetales (9 ASVs total). Many more orders were present in the core bacterial communities of the siphons from P. australis with the Flavobacteriales the most abundant (12 ASVs total), followed closely by Chitinophagales (7 ASVs), Microtrichales (7 ASVs) and Steroidobacterales (5 ASVs; Table 2). Only 12 ASVs were present in the core bacterial communities of A. stutchburyi’s digestive glands (Supplementary Table S3): Clostridiales was the most dominant order (4 ASVs), followed by Mycoplasmatales (3 ASVs).
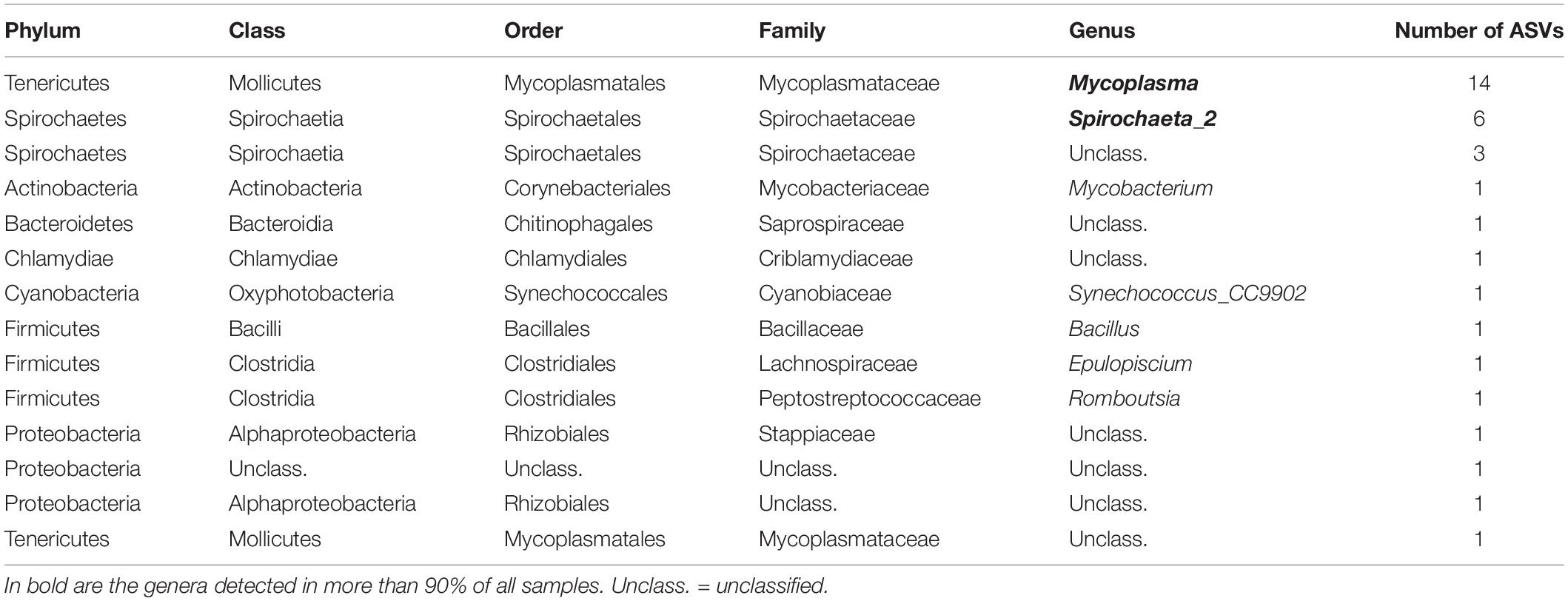
Table 1. Summary of bacterial amplicon sequence variants (ASV) corresponding to the core bacterial communities of Paphies australis’ digestive glands, define as present in at least 70% of all samples.
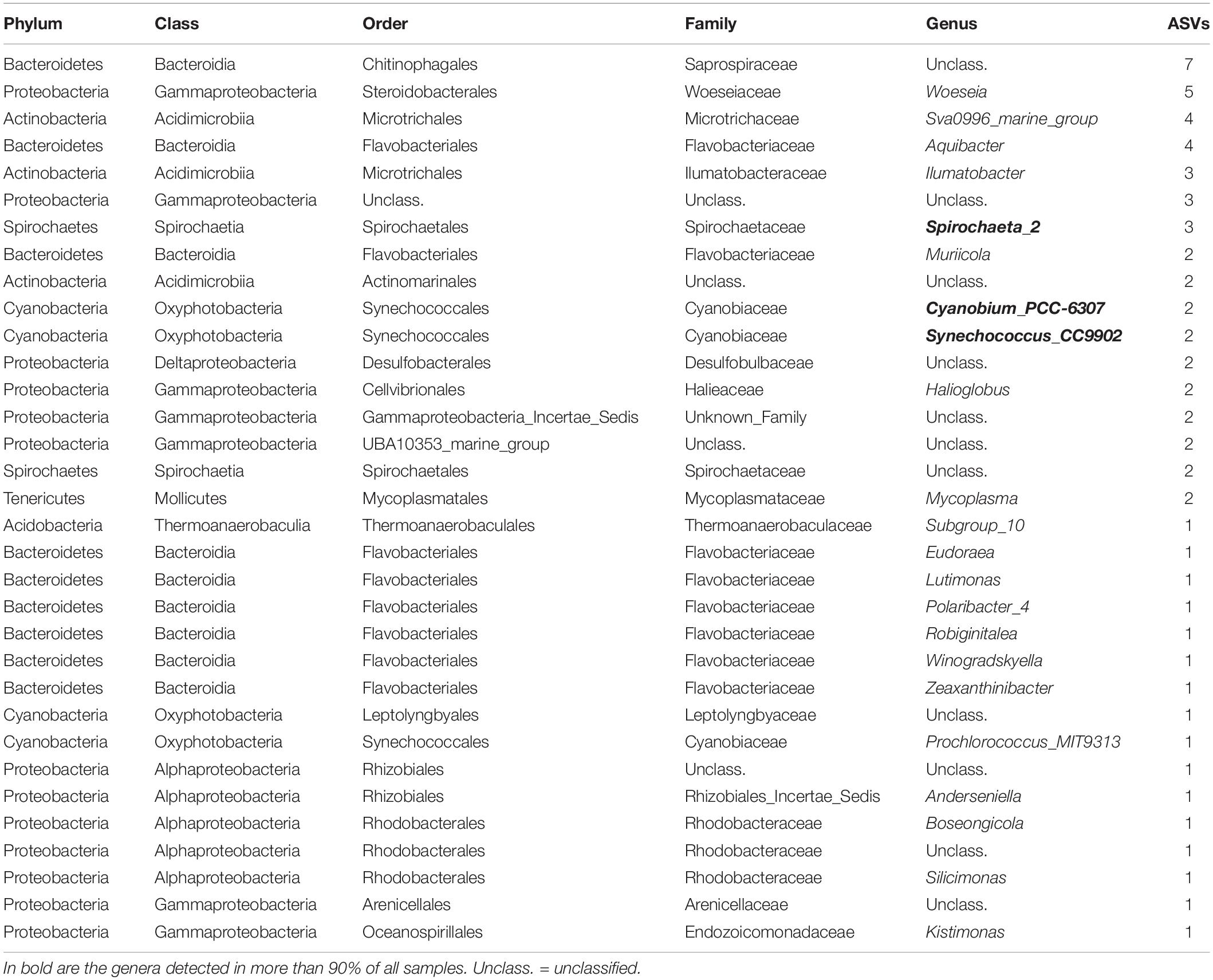
Table 2. Summary of bacterial amplicon sequence variants (ASVs) corresponding to the core bacterial communities of the clam Paphies australis’ siphons, present in at least 70% of all samples.
Discussion
The endemic New Zealand clam P. australis is an excellent study organism to investigate the source of TTX. Individual clams are mostly stationary, and samples can be repeatedly collected from the same location over time, reducing the effects of seasons, tides and weather conditions. This study explored seasonal and spatial differences in bacterial communities’ present in the siphons and digestive glands of P. australis and aimed to identify any relationships with TTX-production. To achieve this, the bacterial communities were characterized using metabarcoding in P. australis sourced monthly from the Hokianga Harbor for 1 year and the bacterial communities of P. australis from 10 sites with varying TTX concentrations spanning the length of New Zealand were compared. Previous studies have shown that P. australis from the Hokianga Harbor contained relatively high levels of TTX and that toxin concentrations in clam populations were variable around the country (Biessy et al., 2019b).
The bacterial community present in the siphons was more diverse than the digestive glands, potentially due to being in direct contact with the external environment (Pierce and Ward, 2018). Previous studies on other bivalves have shown similar patterns with the gills of oysters Crassostrea gigas containing a higher diversity of bacteria than digestive glands (Hernández-Zárate and Olmos-Soto, 2006). Kueh and Chan (1985) also found that total bacterial counts were comparable between seawater and organs in contact with the external environment like the gills, mantle and adductor muscles of the oyster C. gigas. The most abundant phylum in the siphon was Spirochetes. Species from this phylum have previously been found associated with the crystalline style or stomach of bivalves (Gunnar et al., 2010) but are also free-living bacteria that are abundant in marine environments, explaining their abundance in the siphons of shellfish (Paster and Dewhirst, 2000). The bacterial communities of the digestive glands were dominated by ASVs from the genus Mycoplasma, a genus that is also consistently associated with bivalves, often in high abundances (Lokmer et al., 2016; Aceves et al., 2018; Pierce and Ward, 2018). Proteobacteria such as Vibrionaceae, Stappiaceae or Endozoicomonadaceae were also abundant in both organs, which corroborates previous studies of TTX-bearing bivalves, for example in the clams Ruditapes philippinarum (Milan et al., 2018).
Many bacterial genera have been reported as potential producers of TTX, the most common being Bacillus, Pseudomonas, Aeromonas, Vibrio, Actinomyces, and Shewanella (Pratheepa and Vasconcelos, 2013), and over 150 TTX-producing strains are described in the literature (Magarlamov et al., 2017; Katikou, 2019). With less than 1% of bacteria being culturable there are challenges with isolating, culturing and thus characterizing TTX producers and it is likely that many other strains may be TTX producers (Chau et al., 2011). Many marine bacteria are capable of producing toxic secondary metabolites including; cyanobacteria (Williamson et al., 2004; Mejean et al., 2010), Actinomycetes (Fiedler et al., 2005), and Bacilli (Teasdale et al., 2009). The development of molecular techniques, such as metabarcoding, have revolutionized environmental surveys and are increasingly used worldwide for biodiversity monitoring (Taberlet et al., 2012; Valentini et al., 2016; Stat et al., 2017). Metabarcoding has previously been used for detecting rare, toxin-producing dinoflagellates (Smith et al., 2017), to first report harmful algal species in new territories (Grzebyk et al., 2017) and to monitor toxic cyanobacterial blooms in freshwater reservoirs (Casero et al., 2019). Salvitti et al. (2017) used metabarcoding to investigate the diet and gut content of the TTX-containing sea slug P. maculata and observed that most TTX-bearing individuals contained Cnidarian and Annelids sequences, two phyla known to contain toxic species (Miyazawa and Noguchi, 2001; Salvitti et al., 2017). However, the bacterial content of the slug’s foregut was not analyzed and blocking primers to remove the background sequences of the sea slugs themselves were not used, reducing the number of taxa that could be identified (Vestheim and Jarman, 2008). Another study investigating the microbiome of TTX-containing marine nemertean Cephalothrix simula showed the prevalence of a large number of bacterial genera previously associated with TTX production including Alteromonas, Vibrio and Pseudomonas but could not conclusively identify a potential producer (Turner et al., 2018).
The present study comprized of a spatial and temporal component. When analyzing the spatial study data, we subtracted the bacterial ASVs from sites containing very low TTX concentrations (approx. limit of detection; 2–3 μg kg−1) from the Hokianga Harbor bacterial ASVs. In the siphons, the first genus of interest was Phaeodactylibacter, a gram-negative bacteria associated with the diatoms Phaeodactylum spp. (Chen et al., 2014) that have been shown to produce the neurotoxin non-protein amino acid β-methylamino-L-alanine (BMAA) (Réveillon et al., 2016). Bacillus (phylum: Firmicutes) was detected in the digestive glands from P. australis from high TTX sites and has previously been described as a potential TTX producer (Chau, 2013) and TTX was recently detected in the strain Bacillus sp. 1839 tested after 5 years of cultivation in the laboratory (Melnikova et al., 2019). However, the concentrations detected in the culture were low and would not likely account for the extremely high concentrations in higher trophic organisms. The last genus of interest in the differential analysis was Vibrio, which comprise more than 30% of all the suggested 150 TTX-producing strains reported in the literature (Turner et al., 2018). When the bacteria present in the digestive glands from the non-toxic A. stutchburyi were subtracted from those in the glands of toxic P. australis, the genera Bacillus and Vibrio were also present indicating that these taxa remain candidates for further investigation as potential TTX producers.
In the temporal study, the focus was on the core bacterial communities of TTX-bearing P. australis over an entire year. The core microbiome of a species is defined as the group of microbes present in all individuals regardless of the environment (Turnbaugh et al., 2007). Given that TTX was present in all samples in the temporal study at Hokianga Harbor, we hypothesized that the TTX producer would be present in the core bacterial communities of P. australis. The siphons and the digestive glands had some similarities: two genera, Mycoplasma and Spirochaeta, were present in both organs’ bacterial communities. Spirochaeta and Mycoplasma are common in the core bacterial communities of bivalves (Pierce and Ward, 2018) and were also present from all sites in the spatial study but have never been previously associated with toxin production. In the digestive glands, Bacillus was present in 70% of all samples.
When analyzing the spatial study data, three cyanobacterial genera remained when subtracting the bacterial ASVs from sites containing very low TTX concentrations from the Hokianga Harbor bacterial ASVs. The first was Pleurocapsa strain PCC-7319, a cyanobacterium that was found to be abundant in a toxic bacterial mat, although the toxin was not identified (Aboal et al., 2002). Two other cyanobacterial genera, Cyanobium strain PCC-6307 and Prochlorococcus strain MIT-9313, were also detected. Cyanobium have been shown to produce toxins similar to microcystins (Jakubowska and Szeląg-Wasielewska, 2015) and one strain (in addition to other cyanobacteria) was toxic to brine shrimp, but the analysis did not reveal the production of known toxins (Frazão et al., 2010). Prochlorococcus is one of the smallest and most abundant photosynthetic organisms in the ocean (Partensky et al., 1999) and at least one strain has been shown to produce BMAA (Jakubowska and Szeląg-Wasielewska, 2015). Prochlorococcus and Cyanobium are picocyanobacteria (i.e., have a cell size between 0.2 and 2 μm) and are common in freshwater and marine ecosystems but remain poorly studied due to their small size and difficulty to isolate (Jakubowska and Szeląg-Wasielewska, 2015). Cyanobacterial ecotoxicology is particularly well documented in freshwater habitats where they have been found to produce a wide range of toxins (Jakubowska and Szeląg-Wasielewska, 2015; Rastogi et al., 2015; Cirés et al., 2017) including saxitoxin. Saxitoxin is a potent neurotoxin produced by cyanobacteria and marine dinoflagellates and is known to exert the same toxic effect as TTX through an interaction with voltage gated sodium channels resulting in inhibition of neuromuscular transmission (Narahashi, 1988). Both toxins are active on the α-subunit of the sodium channels (Walker et al., 2012) and have been detected simultaneously in several aquatic species (Bane et al., 2014). However, toxicological studies on cyanobacteria have not been performed to the same extent in marine environments (Frazão et al., 2010), and while no specific links have previously been reported between cyanobacteria and TTX, they are an interesting new lead for future studies with the aim to identify TTX-producing bacterial species.
The unicellular cyanobacterial genus Synechococcus was significantly correlated to higher TTX concentrations in the spatial study and was also present in the bacterial communities of both P. australis’ organs in the temporal study. Marine Synechococcus are abundant throughout the world’s oceans and are significant primary producers in coastal environments but very little is known about their toxic effects on other marine organisms (Hamilton et al., 2014). Some strains produce substances with neurotoxic effects in mice and negative effects on invertebrates (Martins et al., 2005, 2007). Although no toxin production has been associated with the strain CC9902 (the strain whose 16S rRNA sequences were most closely related to the ASV in this study; Genbank accession number CP000097.1), it has been shown to alter fish behavior after a 3-day exposure (Hamilton et al., 2014). More toxicological studies should be undertaken on Synechococcus using analytical methods. When investigating the core bacterial communities of the siphons, Cyanobium PCC-6307 and Synechococcus CC9902 were again present in 90% of all samples and Prochlorococcus MIT9313 in 70% of all samples, strengthening the hypothesis that cyanobacteria (especially picocyanobacteria) could be responsible for the presence of TTX in P. australis. This is further reinforced by the fact that no cyanobacteria were present in the core bacterial communities of the non-TTX bearing A. stutchburyi.
Conclusion and Future Research
This is the first study to evaluate the bacterial communities of TTX-bearing and non-TTX bearing bivalves using high-throughput sequencing. Our results from both spatial and temporal studies correlate with some previous hypotheses that Vibrio and Bacillus could be responsible for the source of TTX in the New Zealand endemic clams P. australis. This study highlights that further investigation of marine cyanobacteria as potential TTX producers is warranted, especially Synechococcus, Cyanobium, and Prochlorococcus that were present in both spatial and temporal studies. Culturing, isolating and testing these bacterial species, in particular picocyanobacteria, for toxin production would be a logical next step in the search to identify the source of TTX in marine organisms. It has recently been shown that DNA from toxic flatworms were detected in the intestinal content of juvenile pufferfish (Okabe et al., 2019). So alternatively, it is also feasible that P. australis could source TTX from ingesting larvae, eggs, or pieces of the highly toxic sea slug P. maculata and flatworm Stylochoplana sp., which are common throughout New Zealand, Japan and Europe (Miyazawa et al., 1987; Salvitti et al., 2015; Turner et al., 2018). Other eukaryotes communities, such as dinoflagellates, should also be investigated as many of them produce other marine biotoxins. Molecular techniques such as qPCR, droplet digital PCR or metabarcoding with the use of blocking primers could be used to assess whether these toxic species are present in the digestive system of P. australis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material.
Author Contributions
LB, KS, IH, and SW conceived and designed the experiments. LB performed the experiments. LB and JP undertook the metabarcoding. KS, JP, and SW helped with the data interpretation and experiments. All authors contributed to data analysis and the editing and writing of the manuscript.
Funding
This work was supported by; the MBIE-funded Safe New Zealand Seafood Research Programe (contract no.: CAWX1801), a Ph.D. scholarship from the New Zealand Food Safety Science and Research Center to Laura Biessy, and the Cawthron Institute Internal Capability Investment Fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the following who assisted with P. australis collections: Dana Clark, David White, Julie Hope, Sam Thomas, Phil Bremer, Martin Black, Geoff and Raewynn Foreman, Jan and Roy Smith, and Elizabeth and John Wood. They also thank and acknowledge Jonathan Puddick for help with interpreting possible TTX biosynthesis pathways.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01860/full#supplementary-material
References
Aboal, M., Puig, M. A., Mateo, P., and Perona, E. (2002). Implications of cyanophyte toxicity on biological monitoring of calcareous streams in north-east Spain. J. Appl. Phycol. 14, 49–56.
Aceves, A. K., Johnson, P., Bullard, S. A., Lafrentz, S., and Arias, C. R. (2018). Description and characterization of the digestive gland microbiome in the freshwater mussel Villosa nebulosa (Bivalvia: Unionidae). J. Molluscan Stud. 84, 240–246. doi: 10.1093/mollus/eyy014
Anderson, M. J. (2001). Permutation tests for univariate or multivariate analysis of variance and regression. Can. J. Fish. Aqu. Sci. 58, 626–639. doi: 10.1139/f01-004
Bane, V., Lehane, M., Dikshit, M., O’riordan, A., and Furey, A. (2014). Tetrodotoxin: chemistry, toxicity, source, distribution and detection. Toxins 6, 693–755. doi: 10.3390/toxins6020693
Biessy, L., Boundy, M. J., Smith, K. F., Harwood, D. T., Hawes, I., and Wood, S. A. (2019a). Tetrodotoxin in marine bivalves and edible gastropods: a mini-review. Chemosphere 236:124404. doi: 10.1016/j.chemosphere.2019.124404
Biessy, L., Smith, K. F., Harwood, D. T., Boundy, M. J., Hawes, I., and Wood, S. A. (2019b). Spatial variability and depuration of tetrodotoxin in the bivalve Paphies australis from New Zealand. Toxicon: X 2:100008. doi: 10.1016/j.toxcx.2019.100008
Biessy, L., Smith, K. F., Boundy, M. J., Webb, S., Hawes, I., and Wood, S. A. (2018). Distribution of tetrodotoxin in the New Zealand clam, Paphies australis, established using immunohistochemistry and liquid chromatography-tandem quadrupole mass spectrometry. Toxins 10:282. doi: 10.3390/toxins10070282
Boundy, M. J., Selwood, A. I., Harwood, D. T., McNabb, P. S., and Turner, A. D. (2015). Development of a sensitive and selective liquid chromatography–mass spectrometry method for high throughput analysis of paralytic shellfish toxins using graphitised carbon solid phase extraction. J. Chromatogr. A 1387, 1–12. doi: 10.1016/j.chroma.2015.01.086
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Carroll, R. J., and Schneider, H. (1985). A note on Levene’s tests for equality of variances. Stat. Probab. Lett. 3, 191–194. doi: 10.1016/0167-7152(85)90016-1
Casero, M. C., Velázquez, D., Medina-Cobo, M., Quesada, A., and Cirés, S. (2019). Unmasking the identity of toxigenic cyanobacteria driving a multi-toxin bloom by high-throughput sequencing of cyanotoxins genes and 16S rRNA metabarcoding. Sci. Total Environ. 665, 367–378. doi: 10.1016/j.scitotenv.2019.02.083
Chau, R. (2013). Microbial and Chemical Diversity of Tetrodotoxin-Producing Marine Animals. Doctorate. Sydney, NSW: The University of New South Wales.
Chau, R., Kalaitzis, J. A., and Neilan, B. A. (2011). On the origins and biosynthesis of tetrodotoxin. Aqu. Toxicol. 104, 61–72. doi: 10.1016/j.aquatox.2011.04.001
Chen, Z., Lei, X., Lai, Q., Li, Y., Zhang, B., Zhang, J., et al. (2014). Phaeodactylibacter xiamenensis gen. nov., sp. nov., a member of the family Saprospiraceae isolated from the marine alga Phaeodactylum tricornutum. Int. J. Syst. Evol. Microbiol. 64, 3496–3502. doi: 10.1099/ijs.0.063909-0
Cirés, S., Casero, M. C., and Quesada, A. (2017). Toxicity at the edge of life: a review on cyanobacterial toxins from extreme environments. Mar. Drugs 15:233. doi: 10.3390/md15070233
Deiner, K., Bik, H. M., Mächler, E., Seymour, M., Lacoursière-Roussel, A., Altermatt, F., et al. (2017). Environmental DNA metabarcoding: transforming how we survey animal and plant communities. Mol. Ecol. 26, 5872–5895. doi: 10.1111/mec.14350
Fiedler, H.-P., Bruntner, C., Bull, A. T., Ward, A. C., Goodfellow, M., Potterat, O., et al. (2005). Marine actinomycetes as a source of novel secondary metabolites. Ant. Van Leeuwenhoek 87, 37–42. doi: 10.1007/s10482-004-6538-8
Frazão, B., Martins, R., and Vasconcelos, V. (2010). Are known cyanotoxins involved in the toxicity of picoplanktonic and filamentous North Atlantic marine cyanobacteria? Mar. Drugs 8, 1908–1919. doi: 10.3390/md8061908
Gerssen, A., Bovee, T., Klijnstra, M., Poelman, M., Portier, L., and Hoogenboom, R. (2018). First report on the occurrence of tetrodotoxins in bivalve mollusks in The Netherlands. Toxins 10:450. doi: 10.3390/toxins10110450
Grzebyk, D., Audic, S., Lasserre, B., Abadie, E., De Vargas, C., and Bec, B. (2017). Insights into the harmful algal flora in northwestern Mediterranean coastal lagoons revealed by pyrosequencing metabarcodes of the 28S rRNA gene. Harmful Algae 68, 1–16. doi: 10.1016/j.hal.2017.06.003
Gunnar, H., Gunnar, G., and Antje, W. (2010). Spirochetes in crystalline styles of marine bivalves: group-specific PCR detection and 16S rRNA sequence analysis. J. Shellfish Res. 29, 1069–1075. doi: 10.2983/035.029.0409
Hamilton, T. J., Paz-Yepes, J., Morrison, R. A., Palenik, B., and Tresguerres, M. (2014). Exposure to bloom-like concentrations of two marine Synechococcus cyanobacteria (strains CC9311 and CC9902) differentially alters fish behaviour. Conserv. Physiol. 2, 20–24.
Hernández-Zárate, G., and Olmos-Soto, J. (2006). Identification of bacterial diversity in the oyster Crassostrea gigas by fluorescent in situ hybridization and polymerase chain reaction. J. Appl. Microbiol. 100, 664–672. doi: 10.1111/j.1365-2672.2005.02800.x
Jakubowska, N., and Szeląg-Wasielewska, E. (2015). Toxic picoplanktonic cyanobacteria. Mar. Drugs 13, 1497–1518. doi: 10.3390/md13031497
Katikou, P. (2019). Public health risks associated with tetrodotoxin and its analogues in European waters: recent advances after the EFSA scientific opinion. Toxins 11:240. doi: 10.3390/toxins11050240
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl. Acids Res. 41:e1. doi: 10.1093/nar/gks808
Kodama, M., Sato, S., and Ogata, T. (1993). “Alexandrium tamarense as a source of tetrodotoxin in the scallop Patinopecten yessoensis,” in Toxic Phytoplankton Blooms in the Sea, eds T. J. Smayda and Y. Shimizu (New York, NY: Elsevier).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/aem.01043-13
Kueh, C. S. W., and Chan, K. Y. (1985). Bacteria in bivalve shellfish with special reference to the oyster. J. Appl. Bacteriol. 59, 41–47. doi: 10.1111/j.1365-2672.1985.tb01773.x
Lago, J., Rodríguez, L. P., Blanco, L., Vieites, J. M., and Cabado, A. G. (2015). Tetrodotoxin, an extremely potent marine neurotoxin: distribution, toxicity, origin and therapeutical uses. Mar. Drugs 13, 6384–6406. doi: 10.3390/md13106384
Lahti, L., Shetty, S., Blake, T., and Salojarvi, J. (2017). Tools for Microbiome Analysis in R. Microbiome Package Version. Available online at: http://microbiome.github.io/microbiome (accessed June 26, 2020).
Leão, J., Lozano-Leon, A., Giráldez, J., Vilariño, Ó, and Gago-Martínez, A. (2018). Preliminary results on the evaluation of the occurrence of tetrodotoxin associated to marine Vibrio spp. in bivalves from the Galician Rias (Northwest of Spain). Mar. Drugs 16:81. doi: 10.3390/md16030081
Lokmer, A., Goedknegt, M. A., Thieltges, D. W., Fiorentino, D., Kuenzel, S., Baines, J. F., et al. (2016). Spatial and temporal dynamics of Pacific oyster hemolymph microbiota across multiple scales. Front. Microbiol. 7:1367. doi: 10.3389/fmicb.2016.01367
Magarlamov, T. Y., Melnikova, D. I., and Chernyshev, A. V. (2017). Tetrodotoxin-producing bacteria: detection, distribution and migration of the toxin in aquatic systems. Toxins 9:166. doi: 10.3390/toxins9050166
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12.
Martins, R., Fernandez, N., Beiras, R., and Vasconcelos, V. (2007). Toxicity assessment of crude and partially purified extracts of marine Synechocystis and Synechococcus cyanobacterial strains in marine invertebrates. Toxicon 50, 791–799. doi: 10.1016/j.toxicon.2007.06.020
Martins, R., Pereira, P., Welker, M., Fastner, J., and Vasconcelos, V. M. (2005). Toxicity of culturable cyanobacteria strains isolated from the Portuguese coast. Toxicon 46, 454–464. doi: 10.1016/j.toxicon.2005.06.010
Matsumura, K. (1995). Reexamination of tetrodotoxin production by bacteria. Appl. Environ. Microbiol. 61, 3468–3470. doi: 10.1128/aem.61.9.3468-3470.1995
Mcmurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
McNabb, P. S., Taylor, D. I., Ogilvie, S. C., Wilkinson, L., Anderson, A., Hamon, D., et al. (2014). First detection of tetrodotoxin in the bivalve Paphies australis by liquid chromatography coupled to triple quadrupole mass spectrometry with and without precolumn reaction. J. AOAC Int. 97, 325–333. doi: 10.5740/jaoacint.sgemcnabb
Mejean, A., Peyraud-Thomas, C., Kerbrat, A. S., Golubic, S., Pauillac, S., Chinain, M., et al. (2010). First identification of the neurotoxin homoanatoxin-a from mats of Hydrocoleum lyngbyaceum (marine cyanobacterium) possibly linked to giant clam poisoning in New Caledonia. Toxicon 56, 829–835. doi: 10.1016/j.toxicon.2009.10.029
Melnikova, D. I., Vlasenko, A. E., and Magarlamov, T. Y. (2019). Stable tetrodotoxin production by Bacillus sp. strain 1839. Mar. Drugs 17:704. doi: 10.3390/md17120704
Melnikova, I. D., and Magarlamov, Y. T. (2020). The microbial community of tetrodotoxin-bearing and non-tetrodotoxin-bearing ribbon worms (Nemertea) from the Sea of Japan. Mar. Drugs 18:177. doi: 10.3390/md18030177
Milan, M., Carraro, L., Fariselli, P., Martino, M. E., Cavalieri, D., Vitali, F., et al. (2018). Microbiota and environmental stress: how pollution affects microbial communities in Manila clams. Aqu. Toxicol. 194, 195–207. doi: 10.1016/j.aquatox.2017.11.019
Miyazawa, K., Jeon, J. K., Noguchi, T., Ito, K., and Hashimoto, K. (1987). Distribution of tetrodotoxin in the tissues of the flatworm Planocera multitentaculata (Platyhelminthes). Toxicon 25, 975–980. doi: 10.1016/0041-0101(87)90160-7
Miyazawa, K., and Noguchi, T. (2001). Distribution and origin of tetrodotoxin. J. Toxicol. Toxin Revi. 20, 11–33. doi: 10.1081/txr-100103081
Narahashi, T. (1988). “Mechanism of tetrodotoxin and saxitoxin action,” in Handbook of Natural Toxins, Marine toxins and Venoms, ed. A. T. Tu (New York, NY: Mercel Dekker Inc.).
Noguchi, T., Onuki, K., and Arakawa, O. (2011). Tetrodotoxin poisoning due to pufferfish and gastropods, and their intoxication mechanism. Int. Schol. Res. Netw. Toxicol. 2011:276939.
Okabe, T., Oyama, H., Kashitani, M., Ishimaru, Y., Suo, R., Sugita, H., et al. (2019). Toxic flatworm egg plates serve as a possible source of tetrodotoxin for Pufferfish. Toxins 11:402. doi: 10.3390/toxins11070402
Partensky, F., Hess, W. R., and Vaulot, D. (1999). Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 63, 106–127. doi: 10.1128/mmbr.63.1.106-127.1999
Paster, B. J., and Dewhirst, F. E. (2000). Phylogenetic foundation of spirochetes. J. Mol. Microbiol. Biotechnol. 2, 341–344.
Pierce, M. L., and Ward, J. E. (2018). Microbial ecology of the bivalvia, with an emphasis on the family Ostreidae. J. Shellfish Res. 37, 793–806. doi: 10.2983/035.037.0410
Pratheepa, V., and Vasconcelos, V. (2013). Microbial diversity associated with tetrodotoxin production in marine organisms. Environ. Toxicol. Pharmacol. 36, 1046–1054. doi: 10.1016/j.etap.2013.08.013
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Rastogi, R. P., Madamwar, D., and Incharoensakdi, A. (2015). Bloom dynamics of cyanobacteria and their toxins: environmental health impacts and mitigation strategies. Front. Microbiol. 6:1254. doi: 10.3389/fmicb.2015.01254
Réveillon, D., Séchet, V., Hess, P., and Amzil, Z. (2016). Production of BMAA and DAB by diatoms (Phaeodactylum tricornutum, Chaetoceros sp., Chaetoceros calcitrans and, Thalassiosira pseudonana) and bacteria isolated from a diatom culture. Harmful Algae 58, 45–50. doi: 10.1016/j.hal.2016.07.008
R-Project (2019). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Salvitti, L., Wood, S. A., Fairweather, R., Culliford, D., McNabb, P., and Cary, S. C. (2017). In situ accumulation of tetrodotoxin in non-toxic Pleurobranchaea maculata (Opisthobranchia). Aqu. Sci. 79, 335–344. doi: 10.1007/s00027-016-0500-5
Salvitti, L., Wood, S. A., Taylor, D. I., McNabb, P., and Cary, S. C. (2015). First identification of tetrodotoxin (TTX) in the flatworm Stylochoplana sp.; a source of TTX for the sea slug Pleurobranchaea maculata. Toxicon 95, 23–29. doi: 10.1016/j.toxicon.2014.12.006
Smith, K. F., Kohli, G. S., Murray, S. A., and Rhodes, L. L. (2017). Assessment of the metabarcoding approach for community analysis of benthic-epiphytic dinoflagellates using mock communities. N. Z. J. Mar. Freshw. Res. 51, 555–576. doi: 10.1080/00288330.2017.1298632
Stat, M., Huggett, M. J., Bernasconi, R., Dibattista, J. D., Berry, T. E., Newman, S. J., et al. (2017). Ecosystem biomonitoring with eDNA: metabarcoding across the tree of life in a tropical marine environment. Sci. Rep. 7, 1–11.
Taberlet, P., Coissac, E., Pompanon, F., Brochmann, C., and Willerslev, E. (2012). Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 21, 2045–2050. doi: 10.1111/j.1365-294x.2012.05470.x
Teasdale, M. E., Liu, J., Wallace, J., Akhlaghi, F., and Rowley, D. C. (2009). Secondary metabolites produced by the marine bacterium Halobacillus salinus that inhibit quorum sensing-controlled phenotypes in gram-negative bacteria. Appl. Environ. Microbiol. 75, 567–572. doi: 10.1128/aem.00632-08
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449, 804–810. doi: 10.1038/nature06244
Turner, A., Fenwick, D., Powell, A., Dhanji-Rapkova, M., Ford, C., Hatfield, R., et al. (2018). New invasive Nemertean species (Cephalothrix Simula) in England with high levels of tetrodotoxin and a microbiome linked to toxin metabolism. Mar. Drugs 16:452. doi: 10.3390/md16110452
Turner, A. D., Boundy, M. J., and Rapkova, M. D. (2017a). Development and single-laboratory validation of a liquid chromatography tandem mass spectrometry method for quantitation of Tetrodotoxin in mussels and oysters. J. AOAC Int. 100, 1469–1482. doi: 10.5740/jaoacint.17-0017
Turner, A. D., Dhanji-Rapkova, M., Coates, L., Bickerstaff, L., Milligan, S., O’neill, A., et al. (2017b). Detection of tetrodotoxin shellfish poisoning (TSP) toxins and causative factors in bivalve molluscs from the UK. Mar. Drugs 15:277. doi: 10.3390/md15090277
Valentini, A., Taberlet, P., Miaud, C., Civade, R., Herder, J., Thomsen, P. F., et al. (2016). Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol. Ecol. 25, 929–942. doi: 10.1111/mec.13428
Vestheim, H., and Jarman, S. N. (2008). Blocking primers to enhance PCR amplification of rare sequences in mixed samples–a case study on prey DNA in Antarctic krill stomachs. Front. Zool. 5:12. doi: 10.1186/1742-9994-5-12
Walker, J. R., Novick, P. A., Parsons, W. H., Mcgregor, M., Zablocki, J., Pande, V. S., et al. (2012). Marked difference in saxitoxin and tetrodotoxin affinity for the human nociceptive voltage-gated sodium channel (Nav1. 7). Proc. Natl. Acad. Sci. 109, 18102–18107. doi: 10.1073/pnas.1206952109
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-07
Wang, X.-J., Yu, R.-C., Luo, X., Zhou, M.-J., and Lin, X.-T. (2008). Toxin-screening and identification of bacteria isolated from highly toxic marine gastropod Nassarius semiplicatus. Toxicon 52, 55–61. doi: 10.1016/j.toxicon.2008.04.170
Keywords: bacteria, biotoxin, bivalves, cyanobacteria, metabarcoding, microbiome, high-throughput sequencing, Paphies australis
Citation: Biessy L, Pearman JK, Smith KF, Hawes I and Wood SA (2020) Seasonal and Spatial Variations in Bacterial Communities From Tetrodotoxin-Bearing and Non-tetrodotoxin-Bearing Clams. Front. Microbiol. 11:1860. doi: 10.3389/fmicb.2020.01860
Received: 27 April 2020; Accepted: 15 July 2020;
Published: 05 August 2020.
Edited by:
Chris Dupont, J. Craig Venter Institute, United StatesReviewed by:
Maria Fátima Carvalho, Universidade do Porto, PortugalDetmer Sipkema, Wageningen University & Research, Netherlands
Copyright © 2020 Biessy, Pearman, Smith, Hawes and Wood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Biessy, bGF1cmEuYmllc3N5QGNhd3Rocm9uLm9yZy5ueg==