 Erika Corretto1†
Erika Corretto1† Livio Antonielli1
Livio Antonielli1 Angela Sessitsch1
Angela Sessitsch1 Christoph Höfer2
Christoph Höfer2 Markus Puschenreiter2Siegrid Widhalm1Karivaradharajan Swarnalakshmi3
Markus Puschenreiter2Siegrid Widhalm1Karivaradharajan Swarnalakshmi3 Günter Brader1*
Günter Brader1*- 1Bioresouces Unit, Center for Health & Bioresources, AIT Austrian Institute of Technology GmbH, Tulln, Austria
- 2Institute of Soil Research, Department of Forest- and Soil Sciences, University of Natural Resources and Life Sciences, Vienna, Austria
- 3Division of Microbiology, Indian Agricultural Research Institute, New Delhi, India
Microbacterium species have been isolated from a wide range of hosts and environments, including heavy metal-contaminated sites. Here, we present a comprehensive analysis on the phylogenetic distribution and the genetic potential of 70 Microbacterium belonging to 20 different species isolated from heavy metal-contaminated and non-contaminated sites with particular attention to secondary metabolites gene clusters. The analyzed Microbacterium species are divided in three main functional clades. They share a small core genome (331 gene families covering basic functions) pointing to high genetic diversity. The most common secondary metabolite gene clusters encode pathways for the production of terpenoids, type III polyketide synthases and non-ribosomal peptide synthetases, potentially responsible of the synthesis of siderophore-like compounds. In vitro tests showed that many Microbacterium strains produce siderophores, ACC deaminase, auxins (IAA) and are able to solubilize phosphate. Microbacterium isolates from heavy metal contaminated sites are on average more resistant to heavy metals and harbor more genes related to metal homeostasis (e.g., metalloregulators). On the other hand, the ability to increase the metal mobility in a contaminated soil through the secretion of specific molecules seems to be widespread among all. Despite the widespread capacity of strains to mobilize several metals, plants inoculated with selected Microbacterium isolates showed only slightly increased iron concentrations, whereas concentrations of zinc, cadmium and lead were decreased.
Introduction
The genus Microbacterium belongs to the Microbacteriaceae family, a high GC actinobacterial taxon, and accounts for more than 90 recognized species that were isolated from a wide range of habitats and hosts (Behrendt et al., 2001; Park et al., 2008; Sharma et al., 2013; Wang et al., 2014; Hadjadj et al., 2016). Several studies associated these bacteria to metal contaminated sites. Several Microbacterium strains can survive in heavy metal contaminated environments (Brown et al., 2012; Fidalgo et al., 2016), reduce specific metals such as hexavalent chromium (Henson et al., 2015; Fierros-Romero et al., 2016; Kumar and Saini, 2019) and change the mobility of heavy metals in contaminated soils (Kuffner et al., 2010; Soni et al., 2013). Because of these interesting traits, they have already been used in phytoextraction trials for soil decontamination (Visioli et al., 2015).
Phytoextraction is meant to be an environment friendly remediation technique, which consists in the use of metal accumulating plants to remove metals from contaminated soils by concentrating them in the harvestable parts of the plant (Garbisu and Alkorta, 2001). The main limiting factors of this technique are the production of plant biomass and the plant metal uptake, which is influenced by the bioavailability of the metal in the soil (Wenzel, 2009). Therefore, bacteria, which can either promote plant growth/health, enhance the stress tolerance and mobilize metals, are considered as an interesting resource for the improvement of such clean-up techniques (Rajkumar et al., 2012; Sessitsch et al., 2013; Ma et al., 2016).
It is well known that members of the phylum Actinobacteria produce several secondary metabolites having diverse biological activities (e.g., siderophores, antibiotics, and pigments). Many are synthesized by polyketide synthases (PKS), non-ribosomal protein synthetases (NRPS) or hybrids of these (PKS/NRPS). PKS synthesize complex polyketide compounds and are divided in three categories based on their domain structures and synthesis mechanism (Shen, 2003). Non-ribosomal protein synthetases (NRPS) are large enzymes, which synthesize secondary metabolites of peptide origin using a wide range of building blocks. They are usually organized in modules, each containing a different catalytic domain (Marahiel, 2016; Winn et al., 2016). However, some non-modular enzymes have been characterized in biosynthetic pathways for the production of NRPS-siderophores such as enterobactin and vibriobactin (Crosa and Walsh, 2002). Siderophores and organic acids are the main compounds influencing metal availability in soils (Nett et al., 2009; Qin et al., 2010; Rajkumar et al., 2010). Beside the NRPS synthesized siderophores, several bacteria produce hydroxamate and α-hydroxyacid containing siderophores through NRPS-independent pathways as in the case of desferrioxamine (Challis, 2005).
In the last years, several reports linked Microbacterium species to heavy metal contaminated sites and described their potential as plant growth promoting bacteria (Lumactud et al., 2017; Sun et al., 2019). The increased interest in this taxon and the advance in genome sequencing methods resulted in a high number of Microbacterium genomes available in the NCBI database. For instance, the studies of Henson and Learman analyzed the genetic potential of Cr(VI) reducing Microbacterium spp. (Henson et al., 2015; Learman et al., 2019). However, to the best of our knowledge, a comprehensive investigation of the genus Microbacterium has not been published yet. In this study, we aimed to (i) perform a phylogenetic analysis and cluster the selected genomes in operational functional groups; (ii) explore the predicted secondary metabolite gene clusters; (iii) investigate their plant growth promoting traits, their ability to grow in the presence of heavy metals (zinc, lead, and cadmium) and to influence metal availability in contaminated soils.
Materials and Methods
Genomes, Phylogenetic Analysis and Tree Construction
All genomes classified as Microbacterium were downloaded from GenBank (on the 8th of May, 2020; total samples n = 366) and initially filtered according to a maximum number of contigs < 100 (n = 272), completeness (>90%) and contamination (<3%) analysis with CheckM (n = 240) (Parks et al., 2014). Of these, 70 Microbacterium genomes covering the branches of the 16S rRNA tree (Supplementary Figure S1) were considered for genome analysis. Supplementary Table S1 summarizes accession number and general features of these genomes, in Supplementary Table S2 the accession for all 240 are listed. Sequences of 16S rRNA gene were obtained using barrnap, and targeted extraction and orientation verified with Metaxa2. Fragmented short ribosomal gene sequences < 700 bp were filtered out using an ad hoc Python script. In addition, we included the 16S rRNA gene sequences of isolates considered in this study for which a genome sequence is not available (n = 240). Sequences were first aligned with MAFFT, then trimmed with TRIMal and a refined alignment was performed using MUSCLE. Finally, we used ModelTest-NG to identify the best model for the construction of the Maximum-Likelihood tree in RAxML using the Transfer Bootstrap method for branch support. The genome of Microbacterium sp. 1.5R (CP018151) was sequenced and assembled as described in Corretto et al. (2015).
Phylogenetic analysis was carried out with the following genes: (i) 16S rRNA gene sequences (Supplementary Figure S1 and Supplementary Table S2) and (ii) 38 unique marker genes identified using Phylosift v.1.0.1 (marker database v.4) (Darling et al., 2014) (Figure 1 and Supplementary Tables S2, S3) and (iii) core and accessory genes selected in pan-genome analysis (trees not shown). Sequences were aligned with MUSCLE v.3.8.31 (Edgar, 2004). Poorly aligned positions were removed using Gblocks v.0.91b with default parameters (Castresana, 2000), except for the 16S rRNA gene alignment. Maximum Likelihood trees were calculated in RAxML-NG v.0.8.0 (Kozlov et al., 2019) using the Transfer Bootstrap method for branch support (Lemoine et al., 2018) and according to the best-fit models selected with ModelTest-NG v.0.1.5 (Darriba et al., 2019). Tree were visualized using iTOL v4 (Letunic and Bork, 2019).
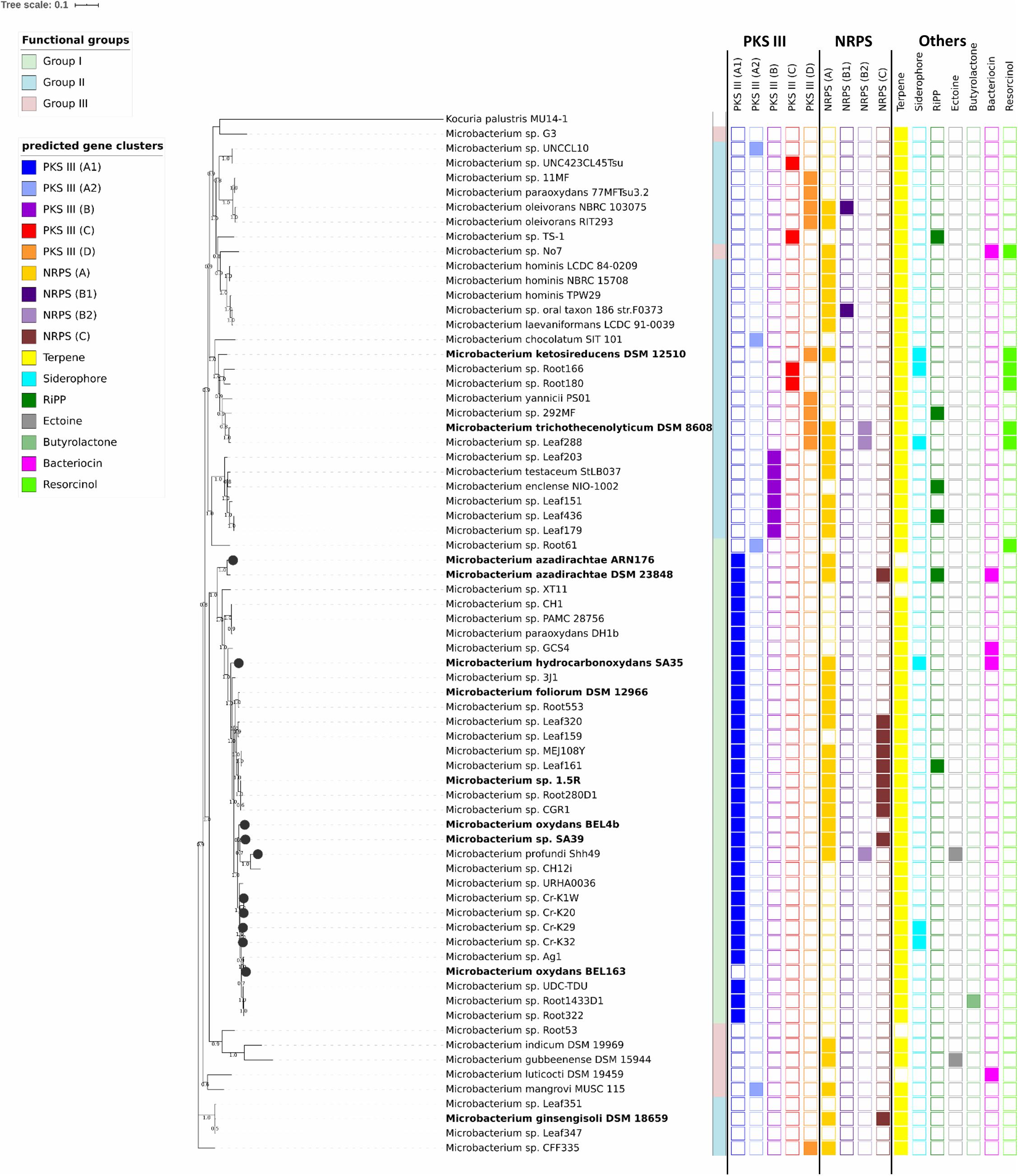
Figure 1. Maximum likelihood phylogenetic tree based on 38 concatenated unique marker genes identified using Phylosift (Darling et al., 2014). Kocuria palustris MU14/1 (CP012507) was used as outgroup. Bootstrap values > 50% are shown at branch points. Strains belonging to phylogenetic groups I, II and III are highlighted in green, blue, and red, respectively. Isolates in bold were tested in this study. Isolates marked with a black circle were isolated from heavy metal contaminated sites. The first column on the right side of the tree shows the different types of PKS gene clusters; whereas the second and third columns show the different types of NRPS gene clusters. The bars indicate the other predicted secondary metabolite gene clusters identified with antiSMASH 5.0.
Pan-Genome Construction
All genomes were re-annotated in Prokka v.1.13.3 (Seemann, 2014). The pan-genome was calculated with Roary v.3.11.2 (Page et al., 2015). We used default parameters and changed only the minimum blastp percentage identity to 80%. We divided the Microbacterium pan-genome in the following categories as calculated in Roary (Page et al., 2015): core (gene families present in 99–100% of the genomes, 69–70 genomes); soft-core (gene families present in 95–99% of the genomes, 67–69 genomes); shell (gene families present in 15–95% of the genomes, 11–67 genomes); cloud (gene families present in 0–15% of the genomes, 1–11 genomes).
Functional Diversity Analysis
Protein FASTA files of CDS sequences parsed and translated by Prokka (Seemann, 2014) were imported in a locally installed EggNOG v.1.0.3 mapper running in server mode (Huerta-Cepas et al., 2016). The functional annotation was carried out using the whole database based on bacterial orthologous groups (OG). The annotation outputs of each genome were then concatenated and formatted using an ad hoc shell script and imported in R for further statistical analysis. Unassigned proteins were removed. A contingency table was built counting the occurrences of each OG, resulting in a matrix were genomes are the samples and GOs the featuring variables. An exploratory multivariate analysis was conducted using a non-metric multidimensional scaling (NMDS) based on Bray–Curtis dissimilarity (vegan: Community Ecology Package. R package version 2.4-61) and a stress value reported (Figure 2). A statistical analysis was then performed on the same dissimilarity matrix to assess the significance of the origin (isolation) and the phylogenetic group via permutational multivariate analysis of variance (PERMANOVA; Anderson, 2001). A confirmatory analysis of deviance was then computed for a multivariate Generalized Linear Model (GLM) based on a negative binomial regression, with 999 iterations (mvabund: Statistical Methods for Analysing Multivariate Abundance Data. R package version 3.13.12).
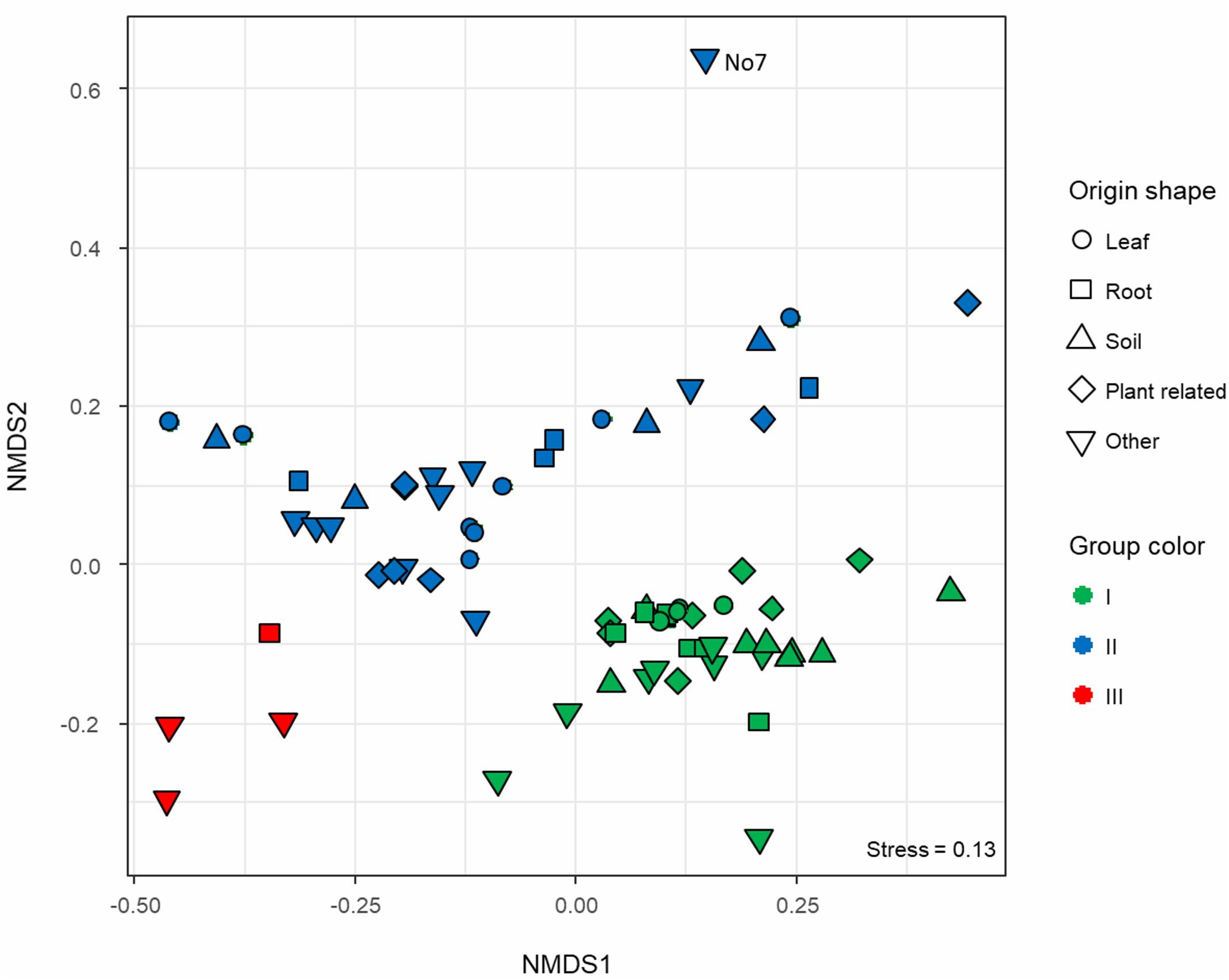
Figure 2. Non-metric multidimensional scaling (NMDS) ordination based on Bray–Curtis dissimilarity calculated on a contingency table where functionally annotated orthologous groups (eggNOG) were counted across all Microbacterium spp. genomes. Samples are displayed in different shapes according to the isolation origin, whereas the colors represent the functional group affiliation (green, blue, and red) as described in Figure 1.
Prediction of Secondary Metabolite Gene Clusters
The Genbank files obtained from the Prokka annotation (Seemann, 2014) were uploaded in antiSMASH 5.0 (Blin et al., 2019). The comparison of the terpenoid clusters was restricted to the Microbacterium strains that have a sequenced genome and were tested in this study, so that we could directly determine the color of the colonies.
Tested Isolates
A total of 29 bacteria belonging to the Microbacterium genus were obtained from different culture collections in order to assess their plant growth-promoting features, resistance to heavy metals and the ability to influence metal mobility in contaminated soil (Table 1). Experimental procedures are described in the next paragraphs. Strains SA5b, SA35, and SA39 were kindly provided by the Instituto de Investigaciones Agrobiológicas de Galicia (IIAG), Consejo Superior de Investigaciones Científicas (CSIC), Santiago de Compostela, Spain. Strains BEL125b, BEL156, and BEL163 were kindly provided by the Hasselt University, Centre for Environmental Sciences, Diepenbeek, Belgium. The DSM strains were obtained from the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures. The rest of the strains are part of the internal collection of the Austrian Institute of Technology. The corresponding papers on already published strains are provided in Table 1.
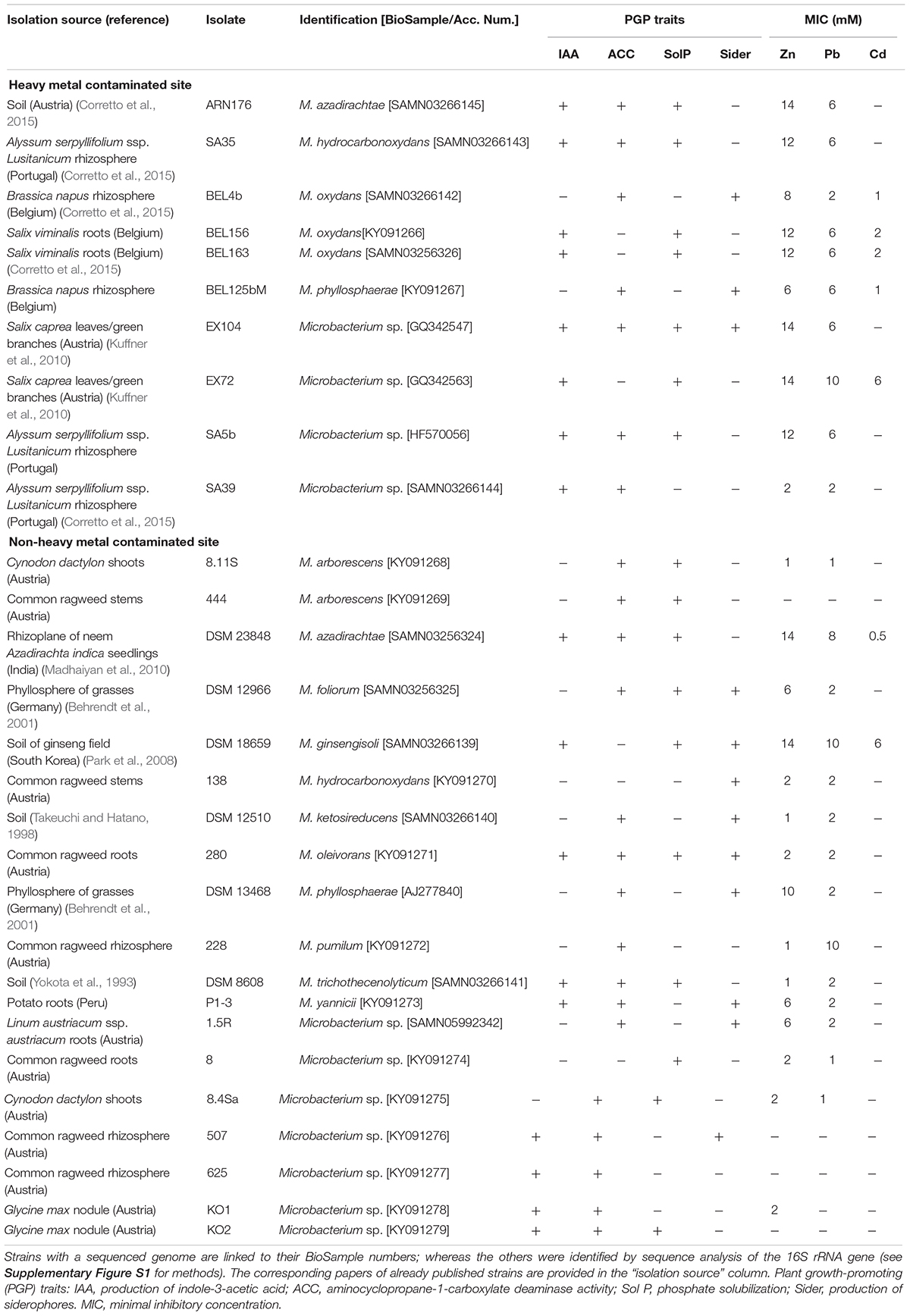
Table 1. Phylogenetic affiliation and features of 29 Microbacterium isolates tested in this study.
Out of the 29 isolates, 11 strains have a sequenced genome. All isolates were plant- or soil-associated and were isolated from contaminated sites (10 isolates) and from non-contaminated sites (19 isolates) (Table 1). Cells were routinely grown at 27°C in Landy medium (20 g l–1 glucose, 5 g l–1 glutamate, 0.25 g l–1 MgSO4, 0.25 g l–1 KCl, 0.5 g l–1 KH2PO4, 150 μg l–1 FeSO4, 5 mg l–1 MnSO4, 160 μg l–1 CuSO4, 1 g l–1 yeast extract, pH 7.2) (Landy et al., 1948).
In vitro Tests
Resistance to heavy metals was tested on solid Landy medium supplemented with the following metals: zinc sulfate (1–14 mM), lead nitrate (1–10 mM) and cadmium nitrate (0.5–6 mM). Growth was assessed after 7 days of incubation at 27°C.
In addition, the isolates were tested for four common plant growth promotion activities: production of siderophores and auxins, phosphate mobilization and ACC deaminase activity. Siderophore production was analyzed using the modified CAS agar plate assay described in Milagres et al. (1999). Briefly, the agar plates were divided in two parts: one half containing solid Landy (without the addition of 150 μg l–1 FeSO4) as growth medium and the other half containing CAS agar prepared according to Schwyn and Neilands (1997). Plates were incubated for 5 days at 27°C. The ability to solubilize phosphate was tested on Pikovskaya agar plates (Pikovskaya, 1948): positive isolates produced a halo-zone around the colony after 5 days at 27°C. Production of indole acetic acid (IAA) was detected with the colorimetric assay described in Loper and Schroth (1986). Bacteria were inoculated in 5 ml Landy medium with the addition of 500 μg ml–1of tryptophan and were incubated at 27°C for 72 h at 200 rpm. Cells were removed by centrifugation (13,000 rpm, 10 min). To 2 ml of the supernatant, 40 μl orthophosphoric acid (85%) and 4 ml Salkowski reagent (50 ml perchloric acid 35%, 1 ml FeCl3 0.5 M) were added. After 10 min of incubation in the dark, absorbance at 530 nm was measured. Indole-3-acetic acid was used as standard for quantification. ACC deaminase activity was tested on a minimal medium containing 0.7 g l–1 ACC as sole nitrogen source (Brown and Dilworth, 1975). Minimal medium without nitrogen was used as negative control, whereas the positive control plates contained 0.7 g l–1 NH4Cl. Plates were incubated at 27°C for 2 weeks.
Detection of Genes Associated to Plant Growth Promotion Activity, Metal Resistance/Homeostasis and Iron Uptake Strategies
For this analysis, out of the 29 isolated, we selected the 10 Microbacterium genomes presented in Corretto et al. (2015) and Microbacterium sp. 1.5R (CP018151). These bacteria were selected for genome sequencing based on the following criteria: resistance to different concentrations of heavy metals (zinc, lead, and cadmium); different isolation sources. Five of the strains were isolated from contaminated sites and six from not-contaminated sites across Europe. These genomes were further annotated in RAST (Overbeek et al., 2014). The WebMGA server (Wu et al., 2011) was used to assign COG functional categories. Odds ratio (OR) of the different x categories (heavy metal relates genes, metallo-sensing regulators, arsR, genes belonging to the COG P category for inorganic ion transport and metabolism) were calculated as follows: OR = (A/B)/(C/D) with A being the number of genes in the genome(s) of interest assigned to a category x; B the number of genes in the genome(s) of interest assigned to all COG categories; C the number of genes in all the sequenced genomes assigned to a category x; D the number of genes in all the sequenced genomes assigned to all COG categories. Each contingency table was tested for significance using the one-tailed Fisher exact test (P ≤ 0.05). as described in Hemme et al. (2016).
Heavy Metal Mobilization Assay
A previous study (Kuffner et al., 2010) showed that bacterial exudates of Microbacterium spp. cultures mobilize metals in contaminated soils, particularly when collected during the stationary phase. Therefore, bacteria were grown in 50 ml of Landy medium at 27°C and 200 rpm until the stationary phase. Cells were removed by centrifugation (4,700 rpm, 20 min, 4°C) and the supernatants were filtrated through 0.2 μm filters (Millipore). Filtrates were stored at −20°C and pH values were measured prior starting the extraction assay. For each strain two cultures were prepared and from each culture three aliquots were analyzed as follows. Five ml of filtrates were shaken with 1 g of Zn, Cd, and Pb-contaminated soil for 2 h at room temperature. Soil characteristics can be found in Supplementary Table S11. As negative control, 1 g of soil was shaken with 5 ml of sterile Landy medium, which was adjusted to pH values between 6.5 and 3.7 to mimic the medium acidification caused by bacterial growth in some cultures. Soil particles were removed by centrifugation (7,000 rpm, 5 min), the supernatants were filtered through 0.45 μm filters and acidified to 2% HNO3 (p.a. grade, Sigma-Aldrich, Vienna, Austria). Concentration of Zn, Cd, Pb, Fe, Cu, and Mn were quantified by inductively coupled plasma mass spectrometry (Elan DRCe 9000, PerkinElmer).
Results
Phylogenetic and Functional Diversity Analysis of 70 Microbacterium Genomes
The analyzed genomes represent at least 20 different Microbacterium species that were isolated from a broad range of habitats, mainly plants and soils (Supplementary Table S1). We found an average genome size of more than 3.5 Mb, a GC% content of about 70% and more than 3,300 coding sequences per genome.
To elucidate the evolutionary relationships between the selected organisms, we performed a phylogenetic analysis using different sets of genes: the 16S rRNA gene as conventional marker (Supplementary Figure S1) and 38 unique marker genes (Figure 1 and Supplementary Table S3) that were identified by Phylosift. The 16S rRNA gene tree built with 240 genomes differs from the one based on the 38 unique marker genes and does not ensure sufficient phylogenetic resolution for a number of species. For instance, the strains showing high similarity to Microbacterium oxydans, Microbacterium hydrocarbonoxydans, Microbacterium phyllosphaerae and Microbacterium foliorum are grouped on one single branch (Supplementary Figure S1).
To characterize the functional potential of the selected Microbacterium strains, each genome was annotated using the eggNOG database. Functional diversity analysis shows that there are three main distinct operational functional groups. As exception, Microbacterium sp. No. 7 seems to be quite distinct from these groups (Figure 2), even though in the phylogenetic tree is positioned next to organisms belonging to the group II (Figure 1). Also, Microbacterium sp. G3 from group III seems to be functionally closer to group I, even though in the phylogenetic tree is located among organisms belonging to group II. Statistical analysis (PERMANOVA; Anderson, 2001) suggests that the taxonomic affiliation (pseudo-F = 10.41, R2 = 0.23, p < 0.001) has a higher influence than the isolation source (pseudo-F = 1.76, R2 = 0.08, p < 0.001) on the formation of the different functional clusters. Since the dispersion of variances resulted in not homoscedastic distribution for taxonomic affiliation, a confirmatory multivariate GLM followed by Likelihood-ratio test was applied. The model was validated with a residual plot and confirmed previous results (LRTtax.aff = 39920.88, LRTiso.sor = 34704.79, p = 0.001).
To further investigate the selected Microbacterium genomes, we performed a pan-genome analysis. The Microbacterium pan-genome has a total of 83,207 gene orthologs: 331 are shared between the core (165) and soft-core (166) genome; while 3,220 and 79,656 clusters form the shell and the cloud genome, respectively. Trees calculated with core and accessory genes show the presence of three main groups, which overlap with the functional groups described above. All COG categories are equally distributed among the analyzed Microbacterium genomes (data not shown). However, most of the core genes have housekeeping functions and belong to the COG categories of translation, ribosomal structure and biogenesis (J) or amino acid/nucleotide transport and metabolism (E/F). On the other hand, unique and accessory genes (cloud) have unknown function (S) or fall within more specific COG categories like carbohydrate transport and metabolism (G) and inorganic ion transport and metabolism (P).
Based on the nature (open/closed) and the size of the pan-genome, one can speculate on the lifestyle of an organism and its ability to acquire exogenous DNA, therefore gaining new advantageous functions (Rouli et al., 2015). Supplementary Figure S2 shows the effect on the total size of the pan-genome every time a new genome is added. The curve increases at each genome addition, but a plateau is not reached with the number of investigated genomes. This observation indicates that the Microbacterium pan-genome is open.
Secondary Metabolite Potential of Microbacterium Genomes
The analysis of secondary metabolite gene clusters with antiSMASH 5.0 (Blin et al., 2019) revealed the presence of several clusters for saccharides or fatty acids production (data not shown). These clusters showed low homology compared to the already characterized ones deposited in the MIBiG database3. In addition, we could detect and identify gene clusters for the production of terpenoids (in 96% of the analyzed genomes), type III polyketide synthase (PKS) genes (79%), non-ribosomal peptide synthetase (NRPS) genes (64%) and rarer clusters (<10%) involved in the production of siderophores, resorcinol, bacteriocins, ectoine, butyrolactone and ribosomally synthesized post-translationally modified peptides (RiPP) (Figure 1 and Supplementary Table S3).
Siderophore Gene Clusters: NRPS-Dependent and –Independent Pathways
Six Microbacterium genomes contain a cluster showing similarity to the one for the synthesis of desferrioxamine. All four biosynthetic genes (desABCD) are present, but not the related transporters (desEF) (Supplementary Table S5). Only few genes showed low amino acid identity (30–40%) with desF and cdtB, which is part of the ferric-siderophore transporter gene cluster (cdtABC) in Streptomyces species (Patel et al., 2010; Terrafria et al., 2011).
Several siderophores are synthesized by NRPS–dependent pathways. The NRPSs described here were identified in antiSMASH under the categories “nrps” and “other.” Within the “nrps” category, only four clusters were detected among the selected Microbacterium genomes (Supplementary Table S6). The clusters present in the genomes of Microbacterium yannicii PS01 and Microbacterium azadirachtae DSM 23848 contain genes harboring the characteristic NRPS domains: adenylation (A), peptidyl carrier protein (PCP) and condensation (C) domains. These genes did not show similarity to previously characterized NRPS gene clusters, impairing any prediction on the produced compounds. On the other hand, the clusters present in Microbacterium sp. No. 7 and Microbacterium gubbeenense DSM 15944 seem to be responsible for the synthesis of siderophores similar to enterobactin and griseobactin, respectively (Crosa and Walsh, 2002; Patzer and Braun, 2010) (Supplementary Table S6).
The antiSMASH category “other” is meant for a “cluster containing a secondary metabolite-related protein that does not fit into any other category.” However, in the case of the Microbacterium genomes, all the detected clusters contained NRPS genes. We divided these clusters in three types (A, B1/2, C) based on the predicted substrate used by the A domain of the NRPS gene (Figure 1 and Supplementary S6). Type A NRPSs use glycine, alanine or a hydrophilic compound as substrate and possess three essential domains: an adenylation (A) domain of siderophore synthetizing NRPSs (SidN3_like), a peptidyl carrier (PCP) domain and a terminal NRPS associated domain having acyltransferase activity and lipase-esterase activity only in the case of Microbacterium sp. No. 7 (Figure 1). In these clusters, right next to the NRPS gene, we could always identify an aminopeptidase N, which can function as a thioesterase domain (TE) releasing the product by hydrolysis. NRPS genes belonging to the cluster type B1/2 harbor the similar domains but can bind different substrates: polar (asn, gln, asp, glu, and 2-amino-adipate) (type B1) or hydrophobic/aliphatic amino acids (type B2) (Figure 1). The cluster type C NRPSs contain only the A domain (adenylate forming domain, AFD, class I superfamily). Apart from the NRPS genes identified in antiSMASH, we could find a group of genes belonging to the NRPS cluster type C and annotated as non-ribosomal protein synthetases, which are shorter than the ones described above (400–500 aa). These NRPSs have only a C domain of unknown function and are flanked by hydrolases (Figure 1 and Supplementary Figure S6).
The NRPS clusters are randomly distributed along the whole phylogenetic tree (Figure 1), suggesting they might be the result of horizontal gene transfers.
Type III Polyketide Synthases (PKS)
A total of 79% of the analyzed Microbacterium genomes harbor a type III PKS with a characteristic chalcone and stilbene synthase domain (Supplementary Table S4). We could define four different types of PKS clusters based on the genes flanking the pks11 or pks18 gene: types A (1/2), B, C and D (Figure 1 and Supplementary Figure S7). All the clusters harbor a long-chain fatty acid-CoA ligase (FadD15), that probably provides long-chain fatty acid starter units. The most common one is the type A, which can be divided in two subgroups based on the presence of different long-chain fatty acid-CoA ligase (FadD15). The ligases of PKS cluster types B, C, and D form a separate group and are positioned far away from the cluster or they are not included within the cluster due the genome fragmentation (Supplementary Figure S8). Organisms of PKS cluster type B contain a polyketide cyclase and separate clearly from the cluster type C, harboring genes with PKS/NRPS domains, and type D (Figure 1).
Overall, these results suggest that Microbacterium species might produce at least four different PKS types which are distributed in groups that largely correspond to the three main operational functional groups (Figure 1).
C50 Carotenoids Gene Clusters
Among the identified secondary metabolite gene clusters, the terpenoid cluster was the most commonly found. These compounds confer a yellow color to the colonies, whereas organisms lacking this cluster are usually white (Supplementary Table S3) (Qian et al., 2007; Vaz-Moreira et al., 2008; Corretto et al., 2016). The synthesis of these pigments can be influenced by the growth conditions like the presence/absence of light (Trutko et al., 2005; Meddeb-Mouelhi et al., 2016). In this study, the strains harboring the terpenoid cluster had pigmented colonies despite being grown in the dark.
The organization of the genes within the analyzed clusters resembles the one of C50 carotenoids like decaprenoxanthin, sarcinaxanthin or C.p. 450 (Heider et al., 2014), of which both the biosynthetic pathway and the chemical structure have already been characterized in Corynebacterium glutamicum ATCC 13032 (Heider et al., 2012), Micrococcus luteus NCTC 2665 (Netzer et al., 2010) and Dietzia sp. CQ4 (Tao et al., 2007), respectively. We performed a phylogenetic analysis using the main biosynthetic genes: lycopene elongase, geranylgeranyl-pyrophosphate synthase (GGPS), two cyclases and glycosyl transferase (Supplementary Figures S3, S4). The topology of the trees showed that the genes of Microbacterium species cluster in two main groups A, B (with the exception of Microbacterium hydrocarbonoxydans SA35 and M. azadirachtae DSM 23848) and separate well from the previously characterized genes of C. glutamicum ATCC 13032, Dietzia sp. CQ4 and M. luteus NCTC 2665. Isolates from both groups produce a methanol dissolvable yellow pigment, extractable from the water phase with chloroform only after hydrolysis. The pigment contains a nona-ene chromophore, pointing to a structure similar to glycosylated sarcinaxanthin or decaprenoxanthin (Supplementary Figure S5). Potential differences in the exact pigment structure due to differentiated biosynthetic genes require further investigation.
A specific glycosyl transferase catalyzes the addition of an activated sugar molecule via glycosylation. This gene can either flank the terpenoid cluster, as in M. luteus NCTC 2665 and Dietzia sp. CQ4, or can be separated by several kilobases as in C. glutamicum ATCC 13032 (Netzer et al., 2010). Blasting the glycosyl transferase of M. luteus NCTC 2665 against the analyzed Microbacterium genomes, we could identify glycosyl transferases that are located far away from the terpenoid cluster, but are possibly involved in the carotenoid glycosylation process (Supplementary Figure S4).
Other Secondary Metabolite Gene Clusters
Ribosomally synthesized post-translationally modified peptides (RiPP) are another huge group of structurally and functionally different metabolites (Ortega and van der Donk, 2016). Among the Microbacterium genomes, we could find only six gene clusters for RiPPs such as lantipeptides (Microbacterium sp. Leaf436 and TS-1), lassopeptides (Microbacterium sp. 292MF), ladderanes (Microbacterium enclense NIO-1002), thiopeptides (Microbacterium sp. Leaf161) and linaridin (M. azadirachtae DSM 23848).
Interestingly, only Microbacterium profundi Shh49, isolated from a deep-sea sediment, and Microbacterium gubbeenenense DSM 15944, isolated from cheese surface, possess a cluster for ectoine, which is an osmoprotectant widespread among halophilic and halotolerant microorganisms growing in saline environments (Reshetnikov et al., 2011).
Plant Growth Promoting Activities
Besides exploring the genetic potential of Microbacterium species, our next goal consisted in the functional characterization of selected strains by testing their ability to promote plant growth. For this purpose, we collected 29 Microbacterium strains isolated from contaminated and non-contaminated European sites, of which 11 have a sequenced genome (Table 1).
First, we screened the 29 isolates for the most prominent plant growth-promoting (PGP) traits such as aminocyclopropane-1-carboxylate (ACC) deaminase activity, phosphate solubilization, production of siderophores and indole-3-acetic acid (IAA). A high percentage (79%) of the tested Microbacterium isolates showed ACC deaminase activity (Table 1). However, in the 11 sequenced genomes, we were not able to identify annotated ACC deaminase genes.
More than half of the isolates (59%) could produce IAA and solubilize phosphate (Table 1). All the analyzed genomes, except M. oxydans BEL163, contain the genes for the synthesis of tryptophan, the IAA main precursor, as well as genes necessary for its conversion to IAA through the tryptamine pathway (Spaepen and Vanderleyden, 2011) (Supplementary Table S7).
Within the RAST subcategory dedicated to phosphate metabolism, several genes were found in all genomes (Supplementary Table S8). This support the fact that 59% of the tested isolates were able to solubilize phosphate in vitro. Among the detected genes, many encode phosphatases for the utilization of organic phosphorus and for transporters and regulatory elements belonging to the PHO regulon, which controls the use of inorganic phosphate (Pi) (Baek and Lee, 2006; Santos-Beneit, 2015).
Microbes adopt different strategies for iron uptake and homeostasis. Among the tested isolates, 41% were positive in the CAS assay for the detection of siderophores. However, it has to be mentioned, that not all types of siderophores are detected by the CAS assay (Schwyn and Neilands, 1997; Milagres et al., 1999). The RAST subsystem dedicated to “iron acquisition and metabolism” gives an overview on the different mechanisms and compounds involved (Supplementary Table S9). We detected two putative siderophore biosynthetic genes and confirmed the presence of defererrioxamine biosynthetic gene clusters. Despite the small number of siderophore biosynthetic genes detected, the 11 analyzed genomes contain several transporters for iron-siderophore complexes (unspecified siderophore interacting proteins, petrobactin- or aerobactin-like siderophore transporters) as well as for ferrous iron (low pH induced EfeUOB transporters, homologs to Streptococcus Pit iron transporters).
Interaction With Heavy Metals
Using the same 29 Microbacterium strains, we investigated other key features for the selection of strains employed in phytoextraction technologies. These include their resistance to heavy metals and their ability to change the metal mobility in contaminated soils. The investigation of these phenomena could help to understand the mechanisms that lead to adaptation to heavy metal-rich environments.
As expected, the number of resistant isolates decreased with the increase of metal concentration and its toxicity (Zn < Pb < Cd) (Figure 3A). Generally, Microbacterium isolated from metal contaminated sites were on average more resistant than the ones isolated from non-contaminated sites. Interestingly, all 11 sequenced genomes harbor genes related to metal homeostasis (transporters, permeases, and efflux pumps) and various metallo-regulators (ArsR, MerR, CsoR, Fur, Zur, and DtxR/IdeR) (Supplementary Table S10). However, in isolates from contaminated sites, genes related to heavy metal homeostasis and metallo-sensing regulators like arsR seem to be more abundant (Figures 3B,C).
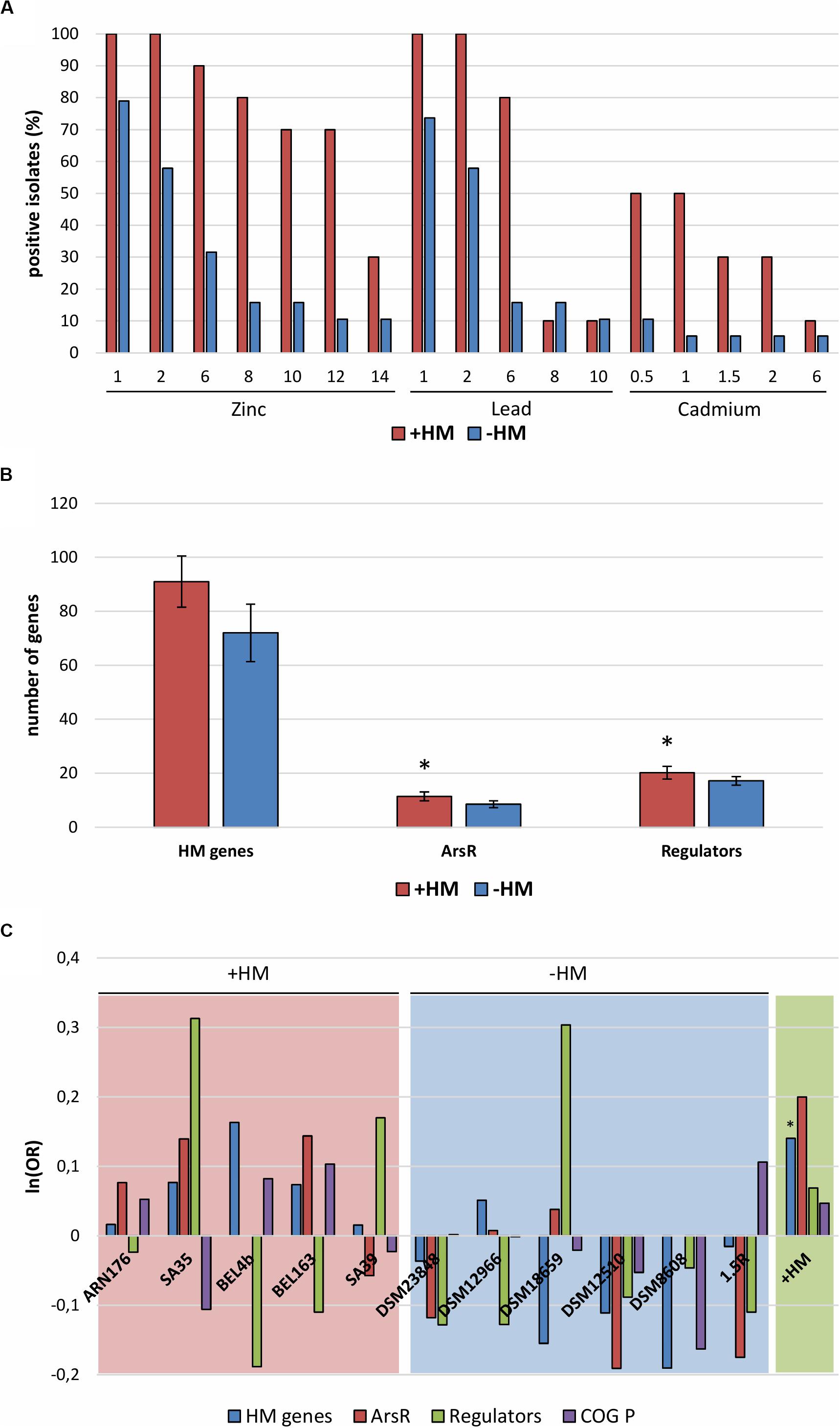
Figure 3. (A) Heavy metal resistance test: zinc sulfate (1–14 mM), lead nitrate (1–10 mM), and cadmium nitrate (0.5–6 mM). (B) Total number of genes related to heavy metal resistance and homeostasis (HM genes), of metallo-sensing regulators (regulators) and ArsR regulator family. Error bars show standard deviation. Asterisks indicate significantly different values of +HM compared to –HM (p < 0.05). (C) Odds ratio (OR) of genes belonging to single isolates compared to all sequenced bacteria (red and blue panels) and isolates from heavy metal contaminated sites compared from non-contaminated sites (green panel). Asterisks indicate significant deviation from the null hypothesis [ln(OR) = 0] at the 95% confidence level by one-tailed Fisher exact test. Gene categories: HM genes (blue), genes related to heavy metal resistance and homeostasis; ArsR (red), regulators belonging to the ArsR family; Regulators (green), metallo-sensing regulators; COG P (purple), genes assigned to the COG P category (Inorganic ion transport and metabolism). +HM, bacteria isolated from heavy metal contaminated sites (red); –HM, bacteria isolated from heavy metal non-contaminated sites (blue).
The ability of enhancing the mobility of metals such as iron, zinc, cadmium, lead, and manganese seems to be widespread among the tested isolates, irrespective of their isolation source (Figure 4 and Supplementary Figure S9). Copper was the only metal that was immobilized by all isolates except Microbacterium sp. 625 (Supplementary Figure S9). The mobilization effect was partly caused by a decrease in pH (from 7.2 to 4.5) and partly due to other processes, such as the secretion of metabolites during the growth in Landy medium (Supplementary Figure S10).
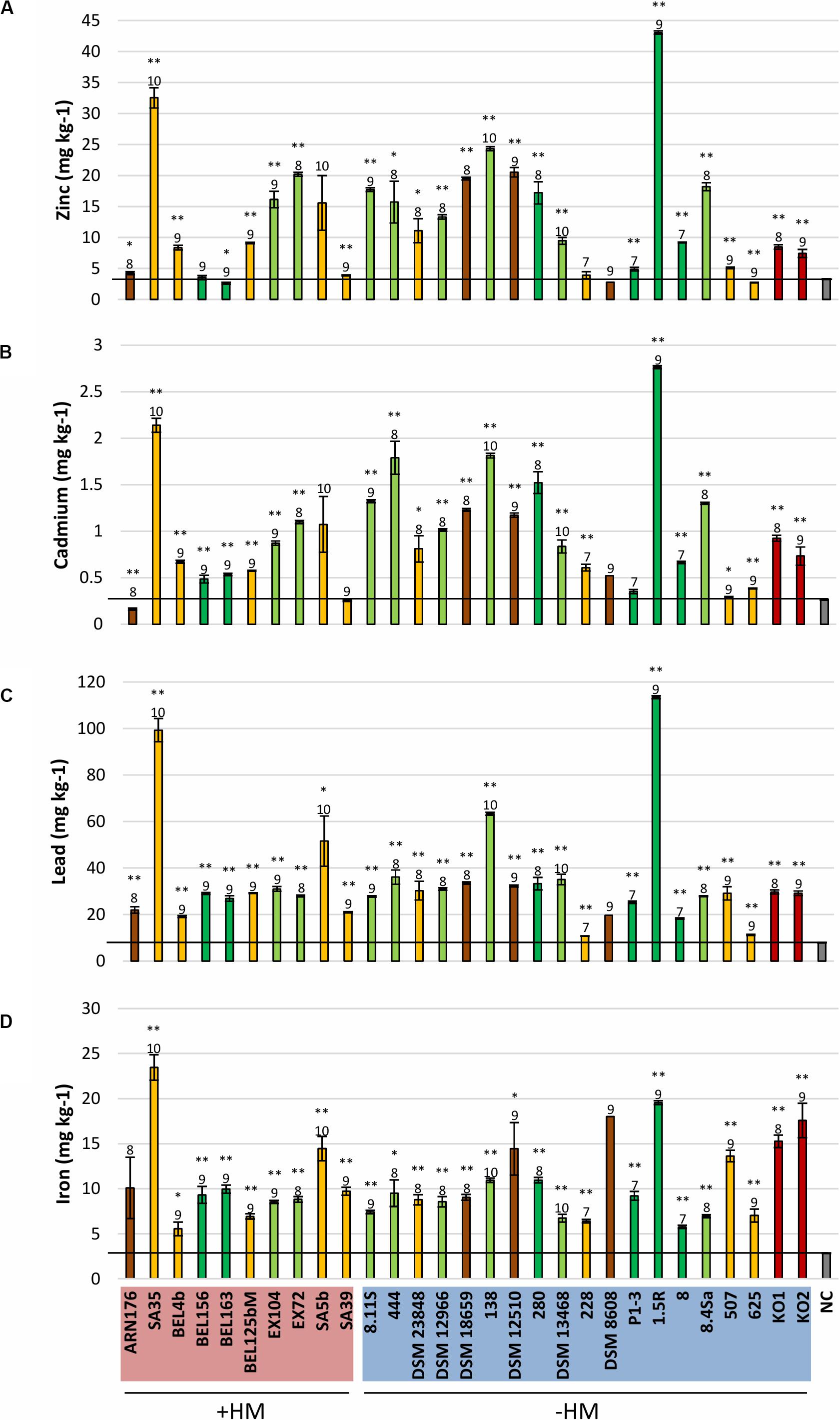
Figure 4. Mobilization of zinc (A), cadmium (B), lead (C), and iron (D) from contaminated soil by bacterial exudates. The different colors of the diagram bars represent different isolation sources: shoots in light green; roots in dark green; rhizosphere in orange; soil in brown; nodules in dark red. NC, negative control (gray). +HM, bacteria isolated from heavy metal contaminated sites; –HM, bacteria isolated from non-contaminated sited. Error bars show standard errors of the mean (n = 6, except for NC n = 3 and for bacteria producing a polysaccharide matrix difficult to filtrate: n = 5 for EX104 and 280; n = 3 for 228 and K02; n = 2 for K01; n = 1 for DSM 8608). Samples with extraction values differing significantly from the control (NC) are labeled with ∗(p < 0.05) and ∗∗(p < 0.01). The order of magnitude of CFU/mL prior to filtration is indicated above the diagram bars. The graphs representing the mobilization of copper and manganese can be found in the Supplementary Material, Supplementary Figure S9.
Such mobilization experiments provide information about the mobilization ability of the selected organisms. In order to verify if the same metabolites are produced not only from the substrates present in a lab growth medium, but also from the substrates available in a contaminated soil, we selected three Microbacterium strains with high (M. hydrocarbonoxydans SA35, Microbacterium sp. 1.5R) and low (M. azadirachtae ARN176) metal mobilization efficiency (see section “Materials and Methods” for the greenhouse assays can be found in the Supplementary Material). The inoculation of Brassica napus and Noccacea caerulescens grown on a contaminated soil did not cause an increase in the biomass production (Supplementary Figure S11). Surprisingly, the two best mobilizers, M. hydrocarbonoxydans SA35 and Microbacterium sp. 1.5R could decrease the cadmium concentration in B. napus shoots, but had no significant effect on the other metals. The weak mobilizer, M. azadirachtae ARN176 acted as an immobilizer causing a decrease in the concentration of zinc, cadmium, manganese, and copper in the shoots and manganese, cadmium and lead in the roots (Supplementary Figures S12, S13). In contrast, we observed no differences between the treated and untreated N. caerulescens plants (Supplementary Figure S12): only Microbacterium sp. 1.5R could increase the concentration of manganese and copper.
Discussion
Microbacterium species are Gram-positive bacteria, affiliated to the Actinobacteria phylum. In the last years, there has been an increased interest in Microbacterium strains isolated from heavy metal contaminated sites. Understanding their relationship with metals, the associated plants and exploring their genomes will provide insights on how these microbes adapted to such environments and how to exploit them for soil remediation processes. For this purpose, we analyzed 70 Microbacterium genomes and assessed the features of 29 Microbacterium strains as candidate organisms to speed up phytoremediation processes.
Previous phylogenetic studies showed that M. oxydans, M. hydrocarbonoxydans, M. phyllosphaerae, and M. foliorum are closely related species located on neighboring tree branches (Behrendt et al., 2001; Schippers et al., 2005). The Maximum Likelihood tree calculated with the 16S rRNA gene (Supplementary Figure S1) grouped these organisms in one big branch. By performing a multilocus sequence analysis, we could achieve a higher resolution resulting in a more robust phylogenetic analysis separating these species in different branches (Figure 1). The analyzed genomes can be grouped in three operational functional groups (Figure 2). Statistical analysis showed that taxonomy is the driving force defining these groups, whereas the isolation source has less influence.
The Microbacterium pan-genome is open. A wide and open pan-genome is usually associated with organisms living in a community with a high rate of horizontal gene transfer (Rouli et al., 2015). A total of 331 gene families are part of the core and soft-core of the pan-genome calculated with the 70 Microbacterium genomes. This extended core genome accounts for about 10% of the genes present in a typical Microbacterium genome (Table 2). The relatively small size of the extended core genome was obtained using a rather low blastp sequence identity threshold (80%), suggesting that the Microbacterium genus comprises species with high genetic diversity. Using 20 closely related strains and the An’vio pipeline, Learman et al. (2019) obtained a bigger core genome (26%), where the main COG categories were devoted to housekeeping functions as “Cellular processing and signaling” and “Information storage and processing.” As in this study, genes of the COG category “Inorganic ion transport and metabolism” or with unassigned function were mostly part of the accessory genome. They also registered a large genomic variability corresponding to different tolerance degrees to heavy metals and antibiotics (Learman et al., 2019). It is known that members of the Microbacteriaceae family have on average smaller core genomes compared to other Actinobacteria families like Micrococcaceae, Cellulomonadaceae, and Streptomycetaceae (Bai et al., 2015). In addition, the analyzed Microbacterium genomes have on average higher functional diversity compared to other members of the Microbacteriaceae family like Agromyces, Curtobacterium, and Plantibacter (data not shown). Taken together, Microbacterium genomes are characterized by a small number of conserved genes (core) covering basic functions (ribosomal proteins, elongation factors, transcriptional regulators, etc.) and by a high number of accessory genes having different sequences but similar or overlapping functions.

Table 2. Microbacterium pan-genome composition.
Compared to well-known Actinobacteria like Streptomyces, Microbacterium have smaller genomes (10 Mb vs. 3.5 Mb). Consequently, they host a lower number of secondary metabolite gene clusters (Jackson et al., 2019). Nonetheless, several secondary metabolite gene clusters were predicted by antiSMASH. They belong to different classes with NRPS, PKS III and terpenoids as the most common ones.
The presence of a desferrioxamine biosynthetic gene cluster, together with NRPS-like siderophore biosynthetic genes, suggest that Microbacterium species are able to synthesize siderophores via NRPS-dependent as well as -independent pathways. The NRPS genes are randomly distributed along the phylogenetic tree. This pattern suggests that these genes might be acquired via horizontal gene transfers. However, we have no information on the NRPS-siderophore type structures. In Streptomyces species, desferrioxamines have a role as regulatory or signaling molecules influencing cell growth and mediating ecological interactions with other bacteria and plants. The desferrioxamine biosynthetic pathway has been previously found in Streptomyces species and related genera like Salinispora and Agromyces (Terrafria et al., 2011; Roberts et al., 2012; Corretto et al., 2016). In Streptomyces coelicor A3(2), the gene cluster consists of four biosynthetic genes (desABCD) and two transporter genes (desEF) with high specificity to desferrioxamine E. Some Streptomyces species have a second ferric-siderophore transporter gene cluster (cdtABC) that binds desferrioxamine B and other hydroxamate-containing siderophores (Patel et al., 2010; Terrafria et al., 2011). Here, we were able to identify the four biosynthetic genes (desABCD), but only few genes showed similarity to the transporters. Eto et al. (2013) showed that three Microbacterium isolates (M. phyllosphaerae KW016, Microbacterium flavescens KW111 and KW080) grew only in the presence of desferrioxamine B, but to our knowledge this is the first detailed report about desferrioxamine biosynthetic genes in Microbacterium species. Only few Microbacterium tested in this study were positive in the CAS test for iron solubilization. This result might be explained by the fact that many bacteria harbor transporters with a broader specificity range to exploit siderophores produced by other microbes (D’Onofrio et al., 2010; Terrafria et al., 2011; Mitter et al., 2013) or they might as well adopt other strategies for iron uptake such as acidification (Miethke et al., 2013; Mathew et al., 2014).
Type III PKS genes were first described in plants and later discovered in bacteria as well (Funa et al., 1999; Yu et al., 2012). These are homodimeric enzymes that iteratively catalyze condensation reactions to produce different polyketides like chalcones, pyrones and stilbenes (Shen, 2003; Yu et al., 2012). The identified PKS genes are annotated as “α-pyrone synthesis polyketide synthase-like Pks11 or 18.” Such genes have been previously characterized in other Gram-positive bacteria like Streptomyces coelicolor (Yu et al., 2012), Mycobacterium tuberculosis (Saxena et al., 2003) and Bacillus subtilis (Nakano et al., 2009). These studies showed that PKS11 and PKS18 usually accept long-chain aliphatic acid CoA-esters (C6–C20) as starter units rather than small-chain substrates to produce alkylpyrones and alkylresorcinols. The different types of PKS III clusters defined in our study roughly correspond to the phylogenetic distribution of the analyzed organisms. Thus, we can speculate that PKS III are vertically transmitted, whereas NRPS were acquired horizontally.
In nature, carotenoids are synthetized by plants, algae, fungi, and bacteria to carry out essential functions involved in nutrition, photosynthesis and protection against oxidative stress. In particular, C50 carotenoids have higher antioxidative properties compared to carotenoids with shorter hydrophobic backbones (C25, C30, and C40) and are known only from a small number of Gram-positive bacteria (Pfander, 1994; Heider et al., 2014). The presence of terpenoids clusters in Microbacterium strains has been recently documented (Han et al., 2016). However, there are only few studies describing the chemical structure of the produced carotenoids (Trutko et al., 2005; Godinho and Bhosle, 2008; Meddeb-Mouelhi et al., 2016). For instance, Microbacterium sp. LEMMJ01 produces a mix of carotenoids including neurosporene, which confer protection against UV-A and UV-B radiation (Reis-Mansur et al., 2019). Another key function of carotenoids is the stabilization of membranes. The presence of glycosylated groups at the molecule ends enhances the association of the carotenoids to the membrane, therefore increasing its rigidity and consequently the resistance to toxic compounds and osmotic stress (Britton, 2008). C50 carotenoids can be glycosylated through the addition of an activated sugar molecule catalyzed by a specific glycosyl transferase that has been characterized in M. luteus NCTC 2665 (Netzer et al., 2010). In the analyzed Microbacterium genomes, we could identify glycosyl transferases that are located far away from the terpenoid cluster, but are possibly involved in the carotenoid glycosylation process.
In heavy metal contaminated environments both bacteria and plants are highly sensitive to abiotic/biotic stresses. It is recognized that PGP bacteria can help the plant to cope with metal toxicity. For example, the production of growth hormones can increase the plant biomass and the root surface area or improve the uptake of essential nutrients like iron and phosphorus. Moreover, PGP traits confer the bacteria selective advantages for niche colonization against other competitors or pathogens (Rajkumar et al., 2012). Although several Microbacterium strains were positive in the ACC deaminase test, we could detect only genes coding for tryptophan synthase beta subunit. ACC deaminase is responsible for the degradation of ethylene, a plant hormone that is produced under various stress conditions including the presence of metals, other inorganic and organic chemicals (Arshad et al., 2007; Glick, 2014). ACC deaminase and tryptophan synthase belong to the same family, pyridoxal 5′-phosphate (PLP)-dependent enzymes. Even though ACC is the only supplied nitrogen source, traces of nitrogen might still support the growth of some strains and the results might be interpreted as false positives. Alternatively, the tested bacteria might have a PLP-dependent deaminase with non-specific ACC deaminase activity or use alternative ACC degradation pathways (Li et al., 2015). Even though, we could detect genes related to phosphorus metabolism in all genomes, not all Microbacterium strains were able to solubilize phosphate in vitro. The discrepancy between the results of the in vitro test and the in silico analysis might be due to the fact that phosphorus solubilization can be carried out using different strategies and pathways, which might not be activated under these specific conditions. The mechanisms leading to phosphorus mobilization influence also the mobility of other metals present in the soil. Bacteria can alter the absorption equilibria through the secretion of protons, organic anions, phosphatases and chelating molecules including siderophores (Richardson and Simpson, 2011). Some of them possess the PHO regulon, which controls the use of inorganic phosphate (Pi). This is a two-component regulatory system that is activated during Pi starvation and seems to be involved in the expression of genes related to metal stress as well (Baek and Lee, 2006; Santos-Beneit, 2015).
Heavy metal resistance tests showed that on average Microbacterium isolated from metal contaminated sites were more resistant to Zn, Pb, and Cd than the one isolated from “clean” sites. Moreover, the Microbacterium from contaminated sites possess more genes related to heavy metal homeostasis and resistance. The overabundance of genes conferring resistance to heavy metals and other stresses (e.g., nitrate, organic solvents, and pH) was also reported in a heavy metal contaminated groundwater microbial community (Hemme et al., 2010). Metallo-sensors function as co-repressors leading to the downregulation of genes for metal uptake and they stimulate the activation of genes related to metal efflux and storage. For instance, they control the activation of membrane transporters, channels and efflux pumps to reduce the overall uptake of potentially toxic metals. In addition, metallo-sensing regulators influence the production of secondary metabolites as siderophores and other chelators (Nies, 2003). The presence of resistance genes and metallo-sensing regulators seem to be essential factors for the adaptation to heavy metal contaminated environments (Hemme et al., 2016; Locatelli et al., 2016).
On the other hand, the ability to change the metal mobility in a contaminated soil seems to be widespread among all tested Microbacterium spp. The mobilization effect seems to be caused by a modification of the pH and by the production of specific compounds. Although the nature of such metabolites is still unclear, they could either be organic acids, specific ligands or siderophores with a broader metal specificity and able to chelate metals other than iron (Sessitsch et al., 2013; Johnstone and Nolan, 2015). Despite the promising results of the in vitro mobilization tests, the phytoremediation experiments did not provide the expected outcome. The inoculated microbes did not cause a significant change in the metal accumulation in N. caerulescens shoots. The weak mobilizer M. azadirachtae ARN176 induced an immobilization effect causing the decrease of some metals both in the shoots and roots of B. napus. On the other hand, the two best mobilizers, M. hydrocarbonoxydans SA35 and Microbacterium sp. 1.5R had limited or no influence on the accumulation of metals in B. napus.). The differences between the two plants might be due to the fact that N. caerulescens has a highly efficient regulation system that allows the plant to tolerate and accumulate very high metal concentrations (Halimaa et al., 2014; Mourato et al., 2015). Another important point to consider is the colonization efficiency of the inoculated Microbacterium strains. Since we used a non-sterile soil, these bacteria have to compete with the already existing microbial communities and find a way to successfully establish themselves in the new environment. Microbacterium strains were often isolated from plants grown in contaminated and non-contaminated soils (Behrendt et al., 2001; Kuffner et al., 2010). Nevertheless, the actual concentration and the metal-mobilizing effects of these bacteria was perhaps too small to provide excess amounts of metals readily available for plant uptake and translocation into leaf tissues. In this experimental set up, the concentration of the strains in planta as well as the mobilization of metals might be too small to be observed. Further experiments are required to assess the survival rate of these Microbacterium strains in the chosen contaminated soil and their ability to colonize N. caerulescens and B. napus plants.
The selection of bacterial isolates based on their genetic potential and their performance under laboratory conditions is unfortunately not always sufficient to predict their behavior under natural conditions. In fact, this process is influenced by several biotic (e.g., colonization ability and plant genotype) and abiotic factors (e.g., contamination levels) that should be considered. The investigation of the genomic repertoire and the chemical analysis of selected metabolites combined with microbial community studies may contribute to a better understanding of plant-microbe interactions and their adaptation mechanisms to harsh environments.
Data Availability Statement
The datasets generated for this study can be found in the NCBI with accession number CP018151.
Author Contributions
AS, GB, and EC designed the study. LA, EC, and GB contributed to the bioinformatics analysis. EC, CH, and MP performed the experiments. SW and KS isolated some of the strains used in this study. EC, GB, and LA wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Austrian Science Fund FWF, project P 24569-B25 and LS12-016 of the NFB, NÖ Forschungs- und Bildungsges.m.b.H. The funder bodies were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge Dr. Petra Kidd [Instituto de Investigaciones Agrobiológicas de Galicia (IIAG), Consejo Superior de Investigaciones Científicas (CSIC), Santiago de Compostela, Spain] and Dr. Nele Weyens (Hasselt University, Centre for Environmental Sciences, Diepenbeek, Belgium) for providing some of the Microbacterium strains used in this study. We would also like to acknowledge Dr. Stefan Pfeiffer (Bioresouces Unit, Center for Health & Bioresources, AIT Austrian Institute of Technology GmbH, Tulln, Austria) and Dr. Andreas Oswald [International Potato Center (CIP), Integrated Crop Management Division, Peru; current address: Agroforestry and Sustainable Agriculture Program, CATIE, Turrialba, Costa Rica] for isolating Microbacterium sp. P1-3. We thank Prof. Dr. Jörn Piel (Institut für Mikrobiologie, ETH Zürich, Switzerland) for the fruitful discussion about secondary metabolite gene clusters.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01869/full#supplementary-material
Footnotes
- ^ https://CRAN.R-project.org/package=vegan
- ^ https://rdrr.io/cran/mvabund/
- ^ http://mibig.secondarymetabolites.org/
References
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Aust. Ecol. 26, 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
Arshad, M., Saleem, M., and Hussain, S. (2007). Perspectives of bacterial ACC deaminase in phytoremediation. Trends Biotechnol. 8, 356–362. doi: 10.1016/j.tibtech.2007.05.005
Baek, J. H., and Lee, S. Y. (2006). Novel gene members in the Pho regulon of Escherichia coli. FEMS Microbiol. Lett. 264, 104–109. doi: 10.1111/j.1574-6968.2006.00440.x
Bai, Y., Müller, D. B., Srinivas, G., Garrido-Oter, R., Potthoff, E., Rott, M., et al. (2015). Functional overlap of the Arabidopsis leaf and root microbiota. Nature 528, 364–369. doi: 10.1038/nature16192
Behrendt, U., Ulrich, A., and Schumann, P. (2001). Description of Microbacterium foliorum sp. nov. and Microbacterium phyllosphaerae sp. nov., isolated from the phyllosphere of grasses and the surface litter after mulching the sward, and reclassification of Aureobacterium resistens (Funke et al. 1998) as Microbacterium resistens comb. nov. Int. J. Syst. Evol. Microbiol. 51(Pt 4), 1267–1276. doi: 10.1099/00207713-51-4-1267
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). AntiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Britton, G. (2008). “Functions of intact carotenoids,” in Carotenoids: Natural Functions, eds G. Britton, S. Liaaen-Jensen, and H. Pfader (Basel: Birkhäuser Verlag), 189–212. doi: 10.1007/978-3-7643-7499-0_10
Brown, C. M., and Dilworth, M. J. (1975). Ammonia assimilation by Rhizobium cultures and bacteroids. J. Gen. Microbiol. 122, 61–67. doi: 10.1099/00221287-86-1-39
Brown, S. D., Palumbo, A. V., Panikov, N., Ariyawansa, T., Klingeman, D. M., Johnson, C. M., et al. (2012). Draft genome sequence for Microbacterium laevaniformans strain OR221, a bacterium tolerant to metals, nitrate, and low pH. J. Bacteriol. 194, 3279–3280. doi: 10.1128/JB.00474-12
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Challis, G. L. (2005). A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. ChemBioChem 6, 601–611. doi: 10.1002/cbic.200400283
Corretto, E., Antonielli, L., Sessitsch, A., Compant, S., Höfer, C., Puschenreiter, M., et al. (2016). Complete genome sequence of the heavy metal resistant bacterium Agromyces aureus AR33 and comparison with related Actinobacteria. Stand Genomic Sci. 12:2. doi: 10.1186/s40793-016-0217-z
Corretto, E., Antonielli, L., Sessitsch, A., Kidd, P., Weyens, N., and Brader, G. (2015). Draft genome of 10 Microbacterium sp., with emphasis on heavy metal-contaminated environments. Genome Announc. 3:e00432-15. doi: 10.1128/genomeA.00432-15
Crosa, J. H., and Walsh, C. T. (2002). Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol. Mol. Biol. Rev. 66, 223–249. doi: 10.1128/mmbr.66.2.223-249.2002
Darling, A. E., Jospin, G., Lowe, E., Matsen, F. A., Bik, H. M., and Eisen, J. A. (2014). PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 2:e243. doi: 10.7717/peerj.243
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2019). ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 18, 291–294. doi: 10.1093/molbev/msz189
D’Onofrio, A., Crawford, J. M., Stewart, E. J., Witt, K., Gavrish, E., Epstein, S., et al. (2010). Siderophores from neighboring organisms promote the growth of uncultured bacteria. Cell Chem. Biol. 17, 254–264. doi: 10.1016/j.chembiol.2010.02.010
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eto, D., Watanabe, K., Saeki, H., Oinuma, K., Otani, K., Nobukuni, M., et al. (2013). Divergent effects of desferrioxamine on bacterial growth and characteristics. J. Antibiot. 66, 199–203. doi: 10.1038/ja.2012.111
Fidalgo, C., Henriques, I., Rocha, J., Tacão, M., and Aves, A. (2016). Culturable endophytic bacteria from the salt marsh plant Halimione portulacoides. Environ. Sci. Pollut. Res. 23, 10200–10214. doi: 10.1007/s11356-016-6208-1
Fierros-Romero, G., Gómez-Ramírez, M., Arenas-Isaac, G. E., Pless, R. C., and Rojas-Avelizapa, N. (2016). Identification of Bacillus megaterium and Microbacterium liquefaciens genes involved in metal resistance and metal removal. Can. J. Microbiol. 62, 505–513. doi: 10.1139/cjm-2015-0507
Funa, N., Ohnishi, Y., Fujii, I., Shibuya, M., Ebizuka, Y., and Horinouchi, S. (1999). A new pathway for polyketide synthesis in microorganisms. Nature 400, 897–899. doi: 10.1038/23748
Garbisu, C., and Alkorta, I. (2001). Phytoextraction: a cost-effective plant-based technology for the removal of metals from the environment. Biores. Technol. 77, 229–236. doi: 10.1016/s0960-8524(00)00108-5
Glick, B. R. (2014). Bacteria with ACC deaminase can promote plant growth and help to feed the world. Microbiol. Res. 1, 30–39. doi: 10.1016/j.micres.2013.09.009
Godinho, A., and Bhosle, S. (2008). Carotenes produced by alkaliphilic orange-pigmented strain of Microbacterium arborescens AGSB isolated from coastal sand dunes. Indian J. Marine Sci. 37, 307–312.
Hadjadj, L., Rathored, J., Keita, M. B., Michelle, C., Levasseur, A., Raoult, D., et al. (2016). Non contiguous-finished genome sequence and description of Microbacterium gorillae sp. nov. Stand. Gen. Sci. 11:32. doi: 10.1186/s40793-016-0152-z
Halimaa, P., Blande, D., Aarts, M. G. M., Tuomainen, M., Tervahauta, A., and Kärenlampi, S. (2014). Comparative transcriptome analysis of the metal hyperaccumulator Noccaea caerulescens. Front. Plant Sci. 5:213. doi: 10.3389/fpls.2014.00213
Han, S., Kim, K., Ahn, D., Park, H., and Oh, T. (2016). Complete genome sequence of carotenoid-producing Microbacterium sp. strain PAMC28756 isolated from an Antarctic lichen. J. Biotechnol. 226, 18–19. doi: 10.1016/j.jbiotec.2016.03.034
Heider, S. A. E., Peters-Wendisch, P., and Wendisch, V. F. (2012). Carotenoid biosynthesis and overproduction in Corynebacterium glutamicum. BMC Microbiol. 12:198. doi: 10.1186/1471-2180-12-198
Heider, S. A. E., Peters-Wendisch, P., Wendisch, V. F., Beekwilder, J., and Brautaset, T. (2014). Metabolic engineering for the microbial production of carotenoids and related products with a focus on the rare C50 carotenoids. Appl. Microbiol. Biotechnol. 98, 4355–4368.
Hemme, C. L., Deng, Y., Gentry, T. J., Fields, M. W., Wu, L., Barua, S., et al. (2010). Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J. 4, 660–672. doi: 10.1007/s00253-014-5693-8
Hemme, C. L., Green, S. J., Rishishwar, L., Prakash, O., Pettenato, A., Chakraborty, R., et al. (2016). Lateral gene transfer in a heavy metal-contaminated-groundwater microbial community. mBio 7:e02234-15. doi: 10.1128/mBio.02234-15
Henson, M. W., Santo Domingo, J. W., Kourtev, P. S., Jensen, R. V., Dunn, J. A., and Learman, D. R. (2015). Metabolic and genomic analysis elucidates strain-level variation in Microbacterium spp. isolated from chromate contaminated sediment. PeerJ 3:e1395. doi: 10.7717/peerj.1395
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucl. Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Jackson, S. A., Crossman, L., Almeida, E. L., Margassery, L. M., Kennedy, J., and Dobson, A. D. W. (2019). Diverse and abundant secondary metabolism biosynthetic gene clusters in the genomes of marine sponge derived Streptomyces spp. isolates. Mar. Drugs 16:67. doi: 10.3390/md16020067
Johnstone, T., and Nolan, E. M. (2015). Beyond iron: non-classical biological functions of bacterial siderophores. Dalton Trans. 44, 6320–6339. doi: 10.1039/c4dt03559c
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Kuffner, M., De Maria, S., Puschenreiter, M., Fallmann, K., Wieshammer, G., Gorfer, M., et al. (2010). Culturable bacteria from Zn- and Cd-accumulating Salix caprea with differential effects on plant growth and heavy metal availability. J. Appl. Microbiol. 108, 1471–1484. doi: 10.1111/j.1365-2672.2010.04670.x
Kumar, M., and Saini, H. S. (2019). Reduction of hexavalent chromium (VI) by indigenous alkaliphilic and halotolerant Microbacterium sp. M5: comparative studies under growth and nongrowth conditions. J. Appl. Microbiol. 127, 1057–1068. doi: 10.1111/jam.14366
Landy, M., Warren, G. H., Roseman, S. B., and Colio, L. G. (1948). Bacillomycin: an antibiotic from Bacillus subtilis active against pathogenic fungi. Proc. Soc. Exp. Biol. Med. 67, 539–541. doi: 10.3181/00379727-67-16367
Learman, D. R., Ahmad, Z., Brookshier, A., Henson, M. W., Hewitt, V., Lis, A., et al. (2019). Comparative genomics of 16 Microbacterium spp. that tolerate multiple heavy metals and antibiotics. PeerJ 6:e6258. doi: 10.7717/peerj.6258
Lemoine, F., Domelevo Entfellner, J.-B., Wilkinson, E., Correia, D., Dávila Felipe, M., De Oliveira, T., et al. (2018). Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 556, 452–456. doi: 10.1038/s41586-018-0043-0
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (ITOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, Z., Chang, S., Ye, S., Chen, M., Lin, L., Li, Y., et al. (2015). Differentiation of 1-aminocyclopropane-1-carboxylate (ACC) deaminase from its homologs is the key for identifying bacteria containing ACC deaminase. FEMS Microbiol. Ecol. 10:fiv112. doi: 10.1093/femsec/fiv112
Locatelli, F. M., Goo, K., and Ulanova, D. (2016). Effects of trace metal ions on secondary metabolism and the morphological development of streptomycetes. Metallomics 8, 469–480. doi: 10.1039/C5MT00324E
Loper, J. E., and Schroth, M. N. (1986). Influence of bacterial sources of indole-3-acetic acid on root elongation of sugar beet. Phytopathology 76, 386–389. doi: 10.1094/Phyto-76-386
Lumactud, R., Fulthorpe, R., Sentchilo, V., and van der Meer, J. R. (2017). Draft genome sequence of Microbacterium foliorum strain 122 isolated from a plant growing in a chronically hydrocarbon-contaminated site. Genome Announc. 5:e00434-17. doi: 10.1128/genomeA.00434-17
Ma, Y., Rajkumar, M., Zhang, C., and Freitas, H. (2016). Beneficial role of bacterial endophytes in heavy metal phytoremediation. J. Environ. Manag. 174, 14–25. doi: 10.1016/j.jenvman.2016.02.047
Madhaiyan, M., Poonguzhali, S., Lee, J.-S., Lee, K.-C., Saravanan, V. S., and Santhanakrishnan, P. (2010). Microbacterium azadirachtae sp. nov., a plant-growth-promoting actinobacterium isolated from the rhizoplane of neem seedlings. Int. J. Syst. Evol. Microbiol. 60, 1687–1692. doi: 10.1099/ijs.0.015800-0
Marahiel, M. A. (2016). A structural model for multimodular NRPS assembly lines. Nat. Prod. Rep. 33, 136–140. doi: 10.1039/c5np00082c
Mathew, A., Eberl, L., and Carlier, A. L. (2014). A novel siderophore-independent strategy of iron uptake in the genus Burkholderia. Mol. Microbiol. 91, 805–820. doi: 10.1111/mmi.12499
Meddeb-Mouelhi, F., Moisan, J. K., Bergeron, J., Daoust, B., and Beauregard, M. (2016). Structural characterization of a novel antioxidant pigment produced by a photochromogenic Microbacterium oxydans strain. Appl. Biochem. Biotechnol. 180, 1286–1300. doi: 10.1007/s12010-016-2167-8
Miethke, M., Montefeörante, C., Marahiel, M. A., and van Dijl, J. M. (2013). The Bacillus subtilis EfeUOB transporter is essential for high-affinity acquisition of ferrous and ferric iron. J. Bacteriol. 191, 4916–4923. doi: 10.1016/j.bbamcr.2013.05.027
Milagres, A. M. F., Machuca, A., and Napoleao, D. (1999). Detection of siderophore production from several fungi and bacteria by modification of chromeazurol S (CAS) agar plate assay. J. Microbiol. Meth. 37, 1–6. doi: 10.1016/s0167-7012(99)00028-7
Mitter, B., Petric, A., Shin, M. W., Chain, P. S. G., Hauberg-Lotte, L., Reinhold-Hurek, B., et al. (2013). Comparative genome analysis of Burkholderia phytofirmans PsJN reveals a wide spectrum of endophytic lifestyles based on interaction strategies with host plants. Front. Plant Sci. 4:120. doi: 10.3389/fpls.2013.00120
Mourato, M. P., Moreira, I. N., Leitão, I., Pinto, F. R., Sales, J. R., and Louro Martins, L. (2015). Effect of heavy metals in plants of the Genus Brassica. Int. J. Mol. Sci. 16, 17975–17998. doi: 10.3390/ijms160817975
Nakano, C., Ozawa, H., Akanuma, G., Funa, N., and Horinouchi, S. (2009). Biosynthesis of aliphatic polyketides by type III polyketide synthase and methyltransferase in Bacillus subtilis. J. Bacteriol. 191, 4916–4923. doi: 10.1128/JB.00407-09
Nett, M., Ikeda, H., and Moore, B. S. (2009). Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 26, 1362–1384. doi: 10.1039/B817069J
Netzer, R., Stafsnes, M. H., Andreassen, T., Goksøyr, A., Bruheim, P., and Brautaset, T. (2010). Biosynthetic pathway for γ-cyclic sarcinaxanthin in Micrococcus luteus: heterologous expression and evidence for diverse and multiple catalytic functions of C50 carotenoid cyclases. J. Bacteriol. 192, 5688–5699. doi: 10.1128/JB.00724-10
Nies, D. H. (2003). Efflux-mediated heavy metal resistance in Prokaryotes. FEMS Microbiol. Rev. 27, 313–339. doi: 10.1016/S0168-6445(03)00048-2
Ortega, M. A., and van der Donk, W. A. (2016). New insights into the biosynthetic logic of ribosomally synthesized and post-translationally modified peptide natural products. Cell Chem. Biol. 23, 31–44. doi: 10.1016/j.chembiol.2015.11.012
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Park, M., Kim, M. K., Kim, H., Im, W., Yi, T., Kim, S., et al. (2008). Microbacterium ginsengisoli sp. nov., a β-glucosidase-producing bacterium isolated from soil of a ginseng field. Int. J. Syst. Evol. Microbiol. 58, 429–433. doi: 10.1099/ijs.0.65226-0
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2014). Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055.
Patel, P., Song, L., and Challis, G. L. (2010). Distinct extracytoplasmic siderophore binding proteins recognize ferrioxamines and ferricoelichelin in Streptomyces coelicolor A3(2). Biochemistry 49, 8033–8042. doi: 10.1021/bi100451k
Patzer, S. I., and Braun, V. (2010). Gene cluster involved in the biosynthesis of griseobactin, a catechol-peptide siderophore of Streptomyces sp. ATCC 700974. J. Bacteriol. 192, 426–435. doi: 10.1128/JB.01250-09
Pikovskaya, R. I. (1948). Mobilization of phosphorus in soil in connection with the vital activity of some microbial species. Mikrobiologiya 17, 362–370.
Qian, F., An, L., Wang, M., Li, M., and Li, X. (2007). Isolation and characterization of a xanthan-degrading Microbacterium sp. strain XT11 from garden soil. J. Appl. Microbiol. 102, 1362–1371. doi: 10.1111/j.1365-2672.2006.03215.x
Qin, S., Xing, K., Jiang, J., Xu, L., and Li, W. (2010). Biodiversity, bioactive natural products and biotechnological potential of plant-associated endophytic actinobacteria. Appl. Microbiol. Biotechnol. 89, 457–473. doi: 10.1007/s00253-010-2923-6
Rajkumar, M., Ae, N., Prasad, M. N. V., and Freitas, H. (2010). Potential of siderophore-producing bacteria for improving heavy metal phytoextraction. Trends Biotechnol. 28, 142–149. doi: 10.1016/j.tibtech.2009.12.002
Rajkumar, M., Sandhya, S., Prasad, M. N. V., and Freitas, H. (2012). Perspectives of plant-associated microbes in heavy metal phytoremediation. Biotechnol. Adv. 30, 1562–1574. doi: 10.1016/j.biotechadv.2012.04.011
Reis-Mansur, M. C. P. P., Cardoso-Rurr, J. S., Silva, J. V. M. A., de Souza, G. R., da Silva Cardoso, V., et al. (2019). Carotenoids from UV-resistant antarctic Microbacterium sp. LEMMJ01. Sci. Rep. 9:9554. doi: 10.1038/s41598-019-45840-6
Reshetnikov, A. S., Khmelenina, V. N., Mustakhimov, I. I., and Trotsenko, Y. A. (2011). “Genes and enzymes of ectoine biosynthesis in halotolerant methanotrophs,” in Methods in Enzymology, eds C. R. Amy and W. R. Stephen (Cambridge, MA: Academic Press), 15–30. doi: 10.1016/B978-0-12-386905-0.00002-4
Richardson, A. E., and Simpson, R. J. (2011). Soil microorganisms mediating phosphorus availability update on microbial phosphorus. Plant Physiol. 156, 989–996. doi: 10.1104/pp.111.175448
Roberts, A. A., Schultz, A. W., Kersten, R. D., Dorrestein, P. C., and Moore, B. S. (2012). Iron acquisition in the marine actinomycete genus Salinispora is controlled by the desferrioxamine family of siderophores. FEMS Microbiol. Lett. 335, 95–103. doi: 10.1111/j.1574-6968.2012.02641.x
Rouli, L., Merhej, V., Fournier, P. E., and Raoult, D. (2015). The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 7, 72–85. doi: 10.1016/j.nmni.2015.06.005
Santos-Beneit, F. (2015). The Pho regulon: a huge regulatory network in bacteria. Front. Microbiol. 6:402. doi: 10.3389/fmicb.2015.00402
Saxena, P., Yadav, G., Mohanty, D., and Gokhale, R. S. (2003). A new family of type III polyketide synthases in Mycobacterium tuberculosis. J. Biol. Chem. 278, 44780–44790. doi: 10.1074/jbc.M306714200
Schippers, A., Bosecker, K., Spröer, C., and Schumann, P. (2005). Microbacterium oleivorans sp. nov. and Microbacterium hydrocarbonoxydans sp. nov., novel crude-oil-degrading Gram-positive bacteria. IJSEM 2, 655–660. doi: 10.1099/ijs.0.63305-0
Schwyn, B., and Neilands, J. B. (1997). Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 160, 46–56. doi: 10.1016/0003-2697(87)90612-9
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sessitsch, A., Kuffner, M., Kidd, P., Vangronsveld, J., Wenzel, W. W., Fallmann, K., et al. (2013). The role of plant-associated bacteria in the mobilization and phytoextraction of trace elements in contaminated soils. Soil Biol. Biochem. 60, 182–194. doi: 10.1016/j.soilbio.2013.01.012
Sharma, P., Diene, S. M., Thibeaut, S., Bittar, F., Roux, V., Gomez, C., et al. (2013). Phenotypic and genotypic properties of Microbacterium yannicii, a recently described multidrug resistant bacterium isolated from a lung transplanted patient with cystic fibrosis in France. BMC Microbiol. 13:97. doi: 10.1186/1471-2180-13-97
Shen, B. (2003). Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 7, 285–295. doi: 10.1016/s1367-5931(03)00020-6
Soni, S. K., Singh, R., Awasthi, A., and Kalra, A. (2013). A Cr(VI)-reducing Microbacterium sp. strain SUCR140 enhances growth and yield of Zea mays in Cr(VI) amended soil through reduced chromium toxicity and improves colonization of arbuscular mycorrhizal fungi. Environ. Sci. Pollut. Res. 21, 1971–1979. doi: 10.1007/s11356-013-2098-7
Spaepen, S., and Vanderleyden, J. (2011). Auxin and plant-microbe interactions. Cold Spring Harb. Perspect. Biol. 3:a001438. doi: 10.1101/cshperspect.a001438
Sun, W., Xiong, Z., Chu, L., Li, W., Soares, M. A., White, J. F., et al. (2019). Bacterial communities of three plant species from Pb-Zn contaminated sites and plant-growth promotional benefits of endophytic Microbacterium sp. (Strain BXGe71). J. Hazard. Mater. 370, 225–231. doi: 10.1016/j.jhazmat.2018.02.003
Takeuchi, M., and Hatano, K. (1998). Proposal of six new species in the genus Microbacterium and transfer of Flavobacterium marinotypicum ZoBell and Upham to the genus Microbacterium as Microbacterium maritypicum comb. nov. Int. J. Syst. Bacteriol. 48, 973–982. doi: 10.1099/00207713-48-3-973
Tao, L., Yao, H., and Cheng, Q. (2007). Genes from a Dietzia sp. for synthesis of C40 and C50 β-cyclic carotenoids. Gene 386, 90–97. doi: 10.1016/j.gene.2006.08.006
Terrafria, V. H., Ramos-Aboites, H. E., Gosset, G., and Barona-Gomez, F. (2011). Disruption of the siderophore−binding desE receptor gene in Streptomyces coelicolor A3(2) results in impaired growth in spite of multiple iron–siderophore transport systems. Microb Biotechnol. 4, 275–285. doi: 10.1111/j.1751-7915.2010.00240.x
Trutko, S. M., Dorofeeva, L. V., Evtushenko, L. I., Ostrovskii, D. N., Hintz, M., Wiesner, J., et al. (2005). Isoprenoid pigments in representatives of the family Microbacteriaceae. Mikrobiologiia 74, 335–341.
Vaz-Moreira, I., Lopes, A. R., Falsen, E., Schumann, P., Nunes, O. C., and Manaia, C. M. (2008). Microbacterium luticocti sp. nov., isolated from sewage sludge compost. Int. J. Syst. Evol. Microbiol. 58, 1700–1704. doi: 10.1099/ijs.0.65494-0
Visioli, G., Vamerali, T., Mattarozzi, M., Dramis, L., and Sanangelantoni, A. M. (2015). Combined endophytic inoculants enhance nickel phytoextraction from serpentine soil in the hyperaccumulator Noccaea caerulescens. Front. Plant Sci. 6:638. doi: 10.3389/fpls.2015.00638
Wang, H., Xiang, T., Wang, Y., Song, J., Zhai, Y., Chen, X., et al. (2014). Microbacterium petrolearium sp. nov., isolated from an oil-contaminated water sample. Int. J. Syst. Evol. Microbiol. 64, 4168–4172. doi: 10.1099/ijs.0.061119-0
Wenzel, W. W. (2009). Rhizosphere processes and management in plant-assisted bioremediation (phytoremediation) of soils. Plant Soil 321, 385–408. doi: 10.1007/s11104-008-9686-1
Winn, M., Fyans, J. K., Zhuo, Y., and Micklefield, J. (2016). Recent advances in engineering nonribosomal peptide assembly lines. Nat. Prod. Rep. 33, 317–347. doi: 10.1039/C5NP00099H
Wu, S., Zhu, Z., Fu, L., Niu, B., and Li, W. (2011). WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12:444. doi: 10.1186/1471-2164-12-444
Yokota, A., Takeuchi, M., Sakane, T., and Weiss, N. (1993). Proposal of six new species in the genus Aureobacterium and transfer of Flavobacterium esteraromaticum Omelianski to the genus Aureobacterium as Aureobacterium esteraromaticum comb. nov. Int. J. Syst. Bacteriol. 43, 555–564. doi: 10.1099/00207713-43-3-555
Keywords: heavy metals, plant associated bacteria, comparative genomics, polyketide synthases, siderophore, terpenoids, secondary metabolites
Citation: Corretto E, Antonielli L, Sessitsch A, Höfer C, Puschenreiter M, Widhalm S, Swarnalakshmi K and Brader G (2020) Comparative Genomics of Microbacterium Species to Reveal Diversity, Potential for Secondary Metabolites and Heavy Metal Resistance. Front. Microbiol. 11:1869. doi: 10.3389/fmicb.2020.01869
Received: 03 February 2020; Accepted: 16 July 2020;
Published: 04 August 2020.
Edited by:
Qiaoyun Huang, Huazhong Agricultural University, ChinaReviewed by:
Virginie Chapon, Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA), FranceJunhui Li, Vanderbilt University, United States
Copyright © 2020 Corretto, Antonielli, Sessitsch, Höfer, Puschenreiter, Widhalm, Swarnalakshmi and Brader. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Günter Brader, Z3VlbnRlci5icmFkZXJAYWl0LmFjLmF0
†Present address: Erika Corretto, Faculty of Science and Technology, Free University of Bozen-Bolzano, Bolzano, Italy