 Chunsheng Li
Chunsheng Li Chi Li
Chi Li Laihao Li
Laihao Li Xianqing Yang
Xianqing Yang Shengjun Chen
Shengjun Chen Bo Qi
Bo Qi Yongqiang Zhao
Yongqiang Zhao- 1Key Laboratory of Aquatic Product Processing, Ministry of Agriculture and Rural Affairs, National R&D Center for Aquatic Product Processing, South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Guangzhou, China
- 2Co-Innovation Center of Jiangsu Marine Bio-industry Technology, Jiangsu Ocean University, Lianyungang, China
Agarose-oligosaccharide production from agar degradation by agarase exhibits lots of advantages and good application prospects. In this study, a novel agar-degrading bacterium Vibrio sp. A8 was isolated from a red algae in the South China Sea. The whole genome sequencing with comparative genomic and secretomic analysis were used to better understand its genetic components about agar degradation. This strain exhibited good agarase production in artificial seawater after culture optimization. The complete genome (4.88 Mb) of this strain comprised two circular chromosomes (3.19 and 1.69 Mb) containing 4,572 protein-coding genes, 108 tRNA genes and 31 rRNA genes. This strain was identified as Vibrio fluvialis A8 by comparative genomic analysis based on genome phylogenetic tree and average nucleotide identity (ANI) similarity. Different from other 20 similar strains including three strains of the same species, V. fluvialis A8 possessed unique agar degradation ability with four β-agarases (GH50) and one α-1,3-L-NA2 hydrolase (GH117) due to the horizontal gene transfer. Secretomic analysis showed that only β-agarase (gene 3152) was abundantly expressed in the secretome of V. fluvialis A8. This agarase had a good substrate specificity and wide work conditions in complex environments, suggesting its potential application for agarose-oligosaccharide production.
Introduction
Agar oligosaccharides (AOS) produced by agarase has a good application prospect. Agar, a marine polysaccharide producing from red algae, is widely used in food industry, pharmaceutical industry and biological engineering (Won-Jae et al., 2013). As the main component of agar, agarose is composed of 3,6-anhydro-L-galactose (L-AHG) and D-galactose (D-Gal) bonded alternately by α-1,3 and β-1,4 linkages (Araki, 1956). Agarose-oligosaccharides, including AOS and neoagaroligosacchrides (NAOS), are agarose degradation products, which exhibit various physiological functions, such as antioxidant (Kang et al., 2014), anti-inflammatory activity (Enoki et al., 2010), whitening effect (Kim et al., 2017), immune modulation (Mussatto and Mancilha, 2007), and antitumor activity (Bhattarai and Kashyap, 2016). There are two methods to produce agarose-oligosaccharides, including acid hydrolysis and agarase biodegradation. Compared with acid hydrolysis, agarase biodegradation can be easy to control agarose-oligosaccharide types, and produce the products with fixed polymerization degrees in an environmentally friendly way without damage of structure (Xu et al., 2018). Therefore, it is very important to develop agarases in the industrial production of agarose-oligosaccharides.
Most agarases are produced by marine gram-negative bacteria including Agarivorans (Zhang et al., 2019), Pseudoalteromonas (Wang et al., 2018; Li et al., 2019), Microbulbifer (Ma et al., 2019; Zhu et al., 2019), Alteromonas (Seo et al., 2014), etc., Vibrio is also an important genus of marine gram-negative bacteria to produce agarases, and various strains have been reported about their agarase production characteristics, such as Vibrio sp. JT0107 (Jam et al., 2005), Vibrio sp. CN41 (Liao et al., 2011), Vibrio sp. ZC-1 (Su et al., 2019), Vibrio sp. EJY3 (Yu et al., 2020), Vibrio sp. PO-303 (Dong et al., 2007), and Vibrio sp. V134 (Zhang and Sun, 2007). The agarases produced by these strains have been well characterized and classified. However, the genomic information and specific taxonomic status of these strains in Vibrio remain poorly understood. Here, we isolated and characterized a marine bacterium Vibrio sp. A8 with high agar degradation ability from a red algae in the South China Sea. This strain was identified as Vibrio fluvialis and showed 99.38% similarity with V. fluvialis NBRC103150 by 16S rRNA gene sequencing. To date, only three genome sequencing projects of V. fluvialis have been completed according to the NCBI Genome database. However, interestingly, no agarase genes have been found in these genomes based on the gene sequences and annotation data. Therefore, it is significantly useful and interesting to study whether Vibrio sp. A8 belongs to V. fluvialis, whether this strain can produce agarase, and why this strain possesses unique agar degradation ability.
In this study, the complete genome of novel agar-degrading bacterium Vibrio sp. A8 was sequenced using a combination of next-generation sequencing and third-generation sequencing technology. The microbial functions were annotated and analyzed against the gene databases to investigate the genetic elements related to agar degradation. Comparative genomic analysis between Vibrio sp. A8 and 20 similar strains in Vibrio was performed to ascertain the taxonomy position of this strain and to explain whether these bacteria had a common ability of agar degradation. Secretomic analysis using nanoLC-MS/MS was performed to study the molecular properties of agarase in the secretome of this strain, followed by the analysis of the biochemical properties of purified agarase. This study provides information about novel agarase in Vibrio and fills in the gap on the genomic information and taxonomy position of the agar-degrading strain in this genus, which is valuable for better understanding the agar degradation pathway in the biofilm of red algae surface, and providing insights into biotechnological applications of agarase for production of agarose-oligosaccharides.
Materials and Methods
Isolation and Biochemical Identification of Strain A8
Marine bacterium strain Vibrio sp. A8 with efficient agar degradation ability was isolated from the rotten red algae (Gracilaria lemaneiformis) in the South China Sea. Firstly, 5.0 g rotten algae was added into 50 mL sterilized saline water. After sufficient vortex oscillation, the solution was spread on LB ager plates (1% peptone, 0.5% yeast extract powder, 1% NaCl and 1.8% agar; pH 7.0) using gradient dilution. After cultivation at 37°C, the strains with concave around their colonies were selected and inoculated on fresh LB ager plates for 48 h at 37°C. The plates were then stained with Lugol’s iodine solution (5% I2 and 10% KI). Colonies with a clear zone formed by agar degradation were determined as agar-degrading strains. The strain with the highest agar degradation ability was maintained on LB agar slant (1% peptone, 0.5% yeast extract powder, 1% NaCl, and 1.8% agar; pH 7.0) and was used for further study.
Biochemical identification of Vibrio sp. A8 was carried out using the VITEK 2 GN test kit (BioMérieux) according to the manufacturer’s instructions.
Agarase-Producing Condition Optimization
Vibrio sp. A8 was first pre-cultured by transferring a loopful of slant culture to liquid LB medium (1% peptone, 0.5% yeast extract powder, and 1% NaCl; pH 7.0) and incubated at 37°C and 180 r/min for 24 h. Thereafter, the strain was transferred to 50 mL liquid agarase-producing medium (0.2% yeast extract powder and 0.2% agar in artificial seawater; pH 7.0) in 250 mL Erlenmeyer flask.
Unless otherwise stated in the agarase-producing experiments, the inoculation size of the strain after pre-culture was 2%, the pH value of the medium was 7.0, the culture temperature was 37°C, and the culture time was 24 h. Different carbon sources (0.2% galactose, 0.2% glucose, 0.2% sucrose, 0.2% agar, 0.2% agar + 0.2% galactose, 0.2% agar + 0.2% glucose, or 0.2% sucrose + 0.2% agar) were first added in the agarase-producing medium, and then the effects of different concentrations of the optimum carbon source on the agarase activity in the medium was studied. The optimum nitrogen source for the agarase production was selected by changing the yeast extract powder in the medium into other nitrogen sources (0.2% peptone, 0.2% NH4NO3, 0.2% (NH4)2SO4, 0.2% NH4Cl, or 0.2% NH4Cl + 0.2% yeast extract powder). The effects of NaCl concentrations (0–2.5%), pH value (4–9), temperature (15–40°C), and culture time (0–48 h) on the agarase activity in the medium were studied to determine the optimum culture conditions of Vibrio sp. A8.
The agarase activity was determined by measuring the release of the reducing sugars using the 3,5-dinitrosalicylic acid (DNS) method (Miller, 1959). Briefly, the medium (100 μL) after cultivation was incubated with 0.2% melted agar (2 mL) at 40°C for 30 min. The solution was then placed in boiling water for 5 min to stop the enzymatic reaction. Subsequently, the DNS reagent (2 mL) was added to the reaction solution which was heated in a boiling water bath for 5 min. When dropped to room temperature, the mixture was diluted with distilled water to 25 mL, and the released reducing sugars were quantified spectrophotometrically at 520 nm. One unit of agarase activity was defined as the amount of enzymes that released 1 μg of reducing sugars per minute under these experimental conditions.
Whole Genome Sequencing, Assembly and Annotation
The genomic DNA from Vibrio sp. A8 was extracted and purified using a Wizard Genomic DNA Purification Kit (Promega). The whole genome was sequenced using a combination of PacBio RS II SMRT and Illumina sequencing platforms. For Illumina sequencing, the genomic DNA sample (over 1 μg) was sheared into 400–500 bp fragments using a Covaris M220 Focused Acoustic Shearer (Covaris). Illumina sequencing libraries were got from these fragments using the NEXTflexTM Rapid DNA-Seq Kit (Bioo Scientific), and were then used for paired-end Illumina sequencing (2 × 150 bp) on an Illumina HiSeq × Ten machine (Illumina). For Pacific Biosciences sequencing, an aliquot of 15 μg DNA was spun in a Covaris g-TUBE (Covaris) at 6,000 r/min for 60 s. DNA fragments were then purified, end-repaired and ligated with SMRTbell sequencing adapters (Pacific Biosciences). The resulting sequencing library was purified three times using Agencourt AMPure XP beads (Beckman Coulter Genomics) and sequenced on one SMRT cell (Pacific Biosciences) using standard methods.
The complete genome was assembled using both the PacBio and Illumina reads (Feng et al., 2019). Briefly, the PacBio reads were assembled into a contig using HGAP and Canu. The circular step was checked and finished manually, generating a complete genome with seamless chromosomes and plasmids. The Illumina reads was used for the error correction of the PacBio assembly results using Pilon. The CDS, tRNA, and rRNA was, respectively predicated by Glimmer version 3.02, tRNA-scan-SE version 2.0 and Barrnap version 0.8. The predicted CDSs were annotated from NR, Swiss-Prot, Pfam, GO, COG, KEGG, and CAZyme database using sequence alignment tools such as BLAST, Blast2go, eggNOG, Diamond, and HMMER. Each set of query proteins were aligned with the databases, and annotations of the best-matched subject (E-value < 10–5) were used for gene annotation. The gene islands were predicted using the Island Viewer (Bertelli et al., 2017). The circular genomic plot of PMTB2.1 was plotted by Circos (Song H. et al., 2016).
The agarase and NAOS hydrolase gene sequences were compared with the gene sequences available from the NCBI database. Phylogenetic tree of these gene sequences were performed using MEGA based on the maximum-likelihood method after multiple alignments of the sequences by Clustal X (Larkin et al., 2007). Numbers represented the frequencies with which the tree topology was replicated after 1000 bootstrap iterations.
Comparative Genomic Analysis
Genomic data of the similar Vibrio species analyzed in this study were obtained from the NCBI. The homologous genes were analyzed using OrthoMCL (Li et al., 2003) with the following parameters: E-Value, 1e–5; Percent Identity Cutoff, 0; Markov Inflation Index, 1.5. Phylogenetic analysis was performed by RA × ML (Stamatakis, 2006) after multiple alignments of single copy of homologous genes using Clustal X (Larkin et al., 2007). To compare the genetic relationship among Vibrio species, the average nucleotide identity (ANI) value was calculated using Jspecies (Richter and Rossello-Mora, 2009).
Secretomic Analysis
Label-free quantitative LC-MS/MS was performed to study the soluble extracellular proteins of Vibrio sp. A8. The extracellular proteins were isolated by ammonium sulfate fractionation (30∼60% ammonium sulfate saturation), redissolved in phosphate buffer solution (0.05 mol/L, pH 7.0), and then desalted by dialysis bag. The Bradford method (Bradford, 1976) was used to determine the protein concentration of sample. The protein sample (120 μg) was digested using trypsin (Promega) at 37°C overnight. Equal volume of 1% formic acid was mixed with digested sample and centrifuged at 12000 × g for 5 min at room temperature. The supernatant was slowly loaded to the C18 desalting column, washed by 1 mL of washing solution (0.1% formic acid and 4% acetonitrile) three times, and then eluted twice using 0.4 mL of elution buffer (0.1% formic acid and 75% acetonitrile). The eluents were lyophilized and reconstituted in 10 μL of 0.1% formic acid. Peptide samples were injected and analyzed using a Q Exactive HF-X mass spectrometer (Thermo) coupled with nanoEASY-nLC 1200 UHPLC (Thermo). Peptides were first trapped onto a home-made C18 Nano-Trap column (2 cm × 75 μm, 3 μm) and then eluted onto a home-made analytical column (15 cm × 150 μm, 1.9 μm) via a 60 min gradient of 2–80% acetonitrile with 0.1% formic acid at a flow rate of 600 nL/min (0–2 min: 4.8%; 2–49 min: 4.8–8%; 49–52 min: 4.8–24%; 52–54 min: 24–28%; 54–55 min: 28–40%; 55–60 min: 40–80%). MS full scan ranged from m/z 350 to 1500 with a target value of 3,000,000 at a resolution of 60,000 (at m/z 200). The top 40 precursors of the highest abundant in the full scan were fragmented using higher energy collisional dissociation, and analyzed in MS/MS with an automatic gain control target value of 50,000 and normalized collision energy of 27%. Raw MS/MS data were searched against the genome of Vibrio sp. A8 obtained in this study using Proteome Discoverer 2.2 (Thermo). For abundance calculation, mass spectrometric signal intensities of peptide precursor ions belonging to each protein were divided by the total abundance of all detected proteins. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD019302.
Phylogenetic tree of agarases based on their amino acid sequences was performed using MEGA based on the neighbor-joining method after multiple alignments of the sequences by Clustal X (Larkin et al., 2007). The sequences of agarases that were similar to that in Vibrio sp. A8 were obtained from UniProt database. Numbers represented the frequencies with which the tree topology was replicated after 1000 bootstrap iterations.
Agarase Purification and Properties
The extracellular proteins were isolated using ammonium sulfate fractionation (30∼60% ammonium sulfate saturation), redissolved in phosphate buffer solution (0.05 mol/L, pH 7.0), and then desalted by dialysis bag. The agarase purification was performed by DEAE Sephadex anion-exchange chromatography. The proteins were eluted by NaCl solution at 0.3 mol/L. The eluent collected from the protein peak was tested for the agarase activity. The eluent with the agarase activity was used for the study of agarase properties.
Unless otherwise stated in the study of agarase properties, the diluted enzyme solution (100 μL) was incubated with 0.2% melted agar (2 mL) at 40°C and pH 7.0 for 30 min, in which condition the agarase activity was set as 100%. The agarase solution was incubated with 2 mL of 2 g/L carrageenan, algin, chitosan, cellulose, or amylum to study the substrate specificity of agarase. One unit of enzyme activity was defined as the amount of enzymes that released 1 μg of reducing sugars per minute under these experimental conditions using DNS method as mentioned previously. To study the optimal reaction temperature, the agarase activity was measured at different temperatures (25, 30, 35, 40, 45, 50 and 55°C). The optimal pH of the agarase was assayed in a pH range of 4.0–9.0 in 20 mM Na2HPO4-citrate buffer (pH 4.0–6.0), 20 mM phosphate buffer (pH 7.0–8.0), and 20 mM glycine–NaOH buffer (pH 9.0). The effects of different metal ions (NaCl, FeCl2, FeCl3, CaCl2, CuSO4, KCl or MgCl2) at 1 mmol/L on the agarase activity were also studied. The same amount of agarase solution was placed in the water bath at 35, 40, 45, and 50°C, respectively for 0–60 min to study the agarase thermostability. The data were fitted to first order plots, and plots of ln(RA) (residual activity) versus time were constructed to calculate kd. The half-life of the enzyme (t1/2) was calculated using the following equation:
where t1/2 is the time required for the residual activity reducing to half, and kd is the deactivation rate constant.
Results and Discussion
Agarase-Producing Condition Optimization of Strain A8
The effects of medium components and culture conditions on the agarase production of Vibrio sp. A8 are shown in Figure 1. Different carbon sources showed obviously different effects on the agarase activity in the medium (Figure 1A). When glucose, galactose, sucrose or agar was added to the medium alone, the agarase activity was the highest in the presence of agar. However, the agarase activity decreased after glucose and sucrose were added to the medium, possibly because the co-existence of glucose or sucrose reduced the utilization of agar by this strain, thus inhibiting the secretion of agarase. The addition of galactose enhanced the agarase activity with the agar as the only carbon source, probably resulting from the improved strain growth by galactose. Therefore, the agar and galactose were chosen as the carbon sources of the agar-producing medium. The effects of agar and galactose concentrations on the agarase activity in the medium were, respectively studied. As shown in Figures 1B,C, the agarase activity in the medium first increased and then decreased with the increasing agar or galactose concentrations. The optimum agar and galactose concentration was respectively 0.2 and 0.3%.
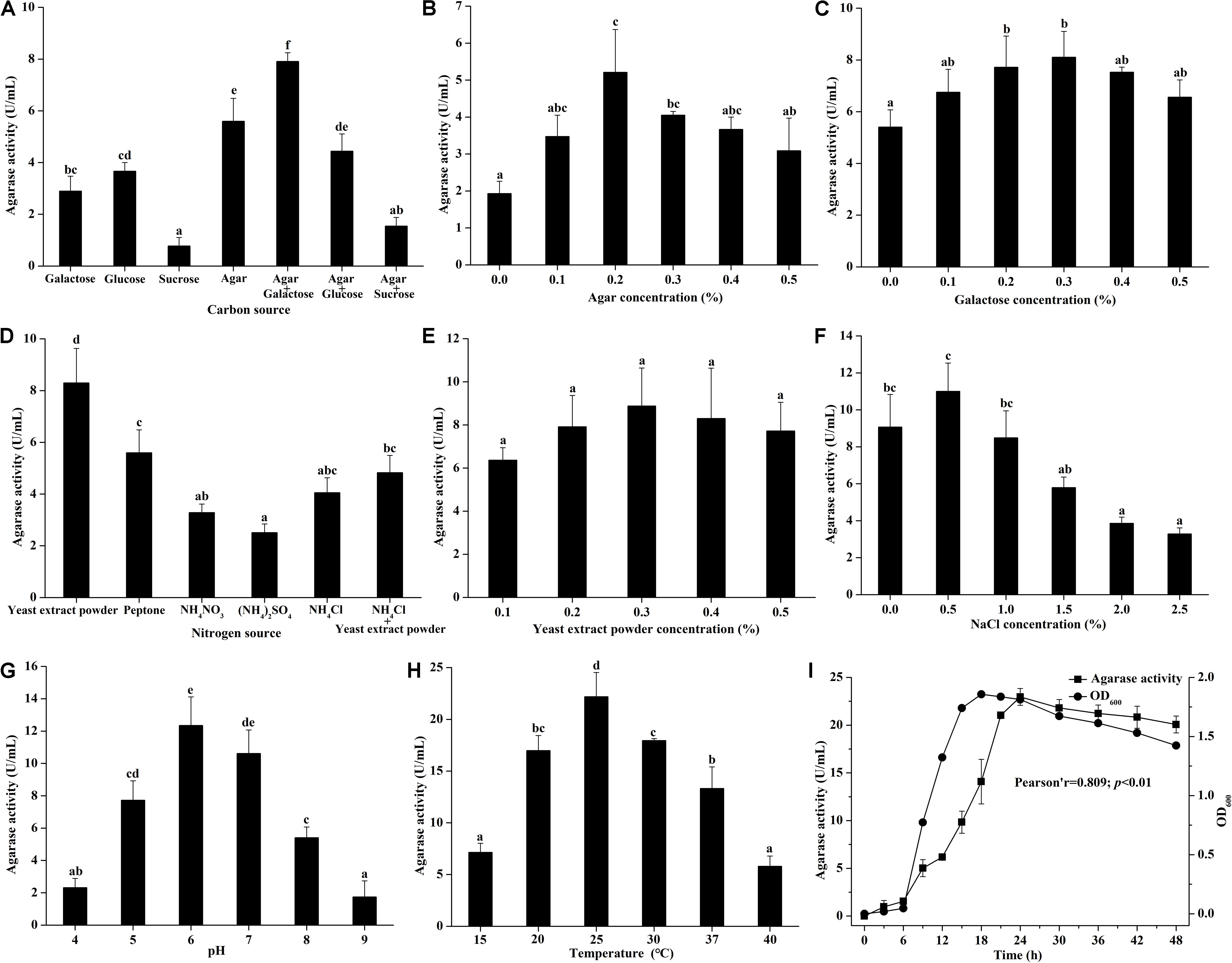
Figure 1. Effect of (A) carbon source (with 0.2% yeast extract powder), (B) agar concentration (with 0.2% yeast extract powder), (C) galactose concentration (with 0.2% yeast extract powder and 0.2% agar), (D) nitrogen source (with 0.2% agar and 0.3% galactose), (E) yeast extract powder concentration (with 0.2% agar and 0.3% galactose), (F) NaCl concentration (with 0.2% agar, 0.3% galactose, and 0.3% yeast extract powder), (G) pH (with 0.2% agar, 0.3% galactose, 0.3% yeast extract powder, and 0.5% NaCl), (H) temperature (with 0.2% agar, 0.3% galactose, 0.3% yeast extract powder, and 0.5% NaCl at pH 6.0), and (I) culture time (with 0.2% agar, 0.3% galactose, 0.3% yeast extract powder, and 0.5% NaCl at 25°C and pH 6.0) on the agarase activity in artificial seawater cultivation with strain A8. Bars labeled with the same letter are not statistically different (p < 0.05) tested by one-way ANOVA and multiple comparison Tukey test. Pearson correlation analysis was tested between agarase activity and OD600 at different culture time.
Different agarase activities was found in the medium with the addition of different nitrogen source (Figure 1D). Organic nitrogen source was more suitable for the agarase production of Vibrio sp. A8 than inorganic nitrogen source. The agarase activity was highest when yeast extract powder was the only nitrogen source in the medium. The agarase activity was not significantly different at various yeast extract powder concentrations (0.1–0.5%) (Figure 1E). The highest agarase activity was observed at 0.3% yeast extract powder.
Based on artificial seawater, the different concentrations of NaCl were added to determine the optimum osmotic pressure for the agarase production of Vibrio sp. A8 (Figure 1F). The agarase activity was improved by 0.5% NaCl but was significantly inhibited by 2.0–2.5% NaCl. This might be because the excessive osmotic pressure inhibited the strain growth, leading to the decrease of its agarase production. Different pH values had great influence on the agarase activity in the medium (Figure 1G). The optimum pH was 6.0, at which the agarase activity was 12.35 U/mL. Similar to pH, the culture temperature showed significant influence on the agarase production of strain A8 (Figure 1H). The optimum culture temperature was 25°C, at which the agarase activity was as high as 22.19 U/mL.
The effect of culture time on the agarase production of Vibrio sp. A8 is shown in Figure 1I. The agarase activity increased slightly at 0–6 h, markedly rised at 6–24 h, and then reached equilibrium after 24 h. The growth curve was in accordance with the agarase-producing curve. Pearson correlation analysis showed that the growth of strain A8 was significantly correlated with agarase activity (p < 0.01), indicating that the positive influence of strain growth on the agarase production.
The optimum medium for agarase production of Vibrio sp. A8 was determined as 0.2% agar, 0.3% galactose, 0.3% yeast extract powder, and 0.5% NaCl in artificial seawater, and the optimum culture conditions were determined as temperature at 25°C, pH at 6.0 and incubation time for 24 h. Under these conditions, the agarase activity of the fermentation broth was 22.28 U/mL, which was 314% higher than that before optimization.
Genome Features of Strain A8
The genomic features of Vibrio sp. A8 are presented in Table 1. The complete genome of Vibrio sp. A8 was 4,882,363 bp (49.81% G+C) in length, composed of two circular chromosomes, Chr1 (3,190,163 bp, 49.89% G+C) (Figure 2A) and Chr2 (1,692,200 bp, 49.67% G+C) (Figure 2B), and no plasmid was detected. The genome contained 4,572 protein-coding genes, 108 tRNA genes and 31 rRNA genes, and all rRNA genes were located in Chr1. There were 3,401 genes (74.39%) assigned to the GO terms, including 2,633 genes in biological process, 2,737 genes in molecular function, and 1,823 genes in cellular component. The most abundant GO annotation included the metabolic process (1,929 genes), membrane (1,159 genes), and catalytic activity (1,845 genes) in respective category (Supplementary Figure S1). There were 3,975 genes (86.94%) assigned to the COG terms, and high percentage of genes was classified into the amino acid transport and metabolism (318 genes), transcription (305 genes), inorganic ion transport and metabolism (240 genes), energy production and conversion (238 genes), and carbohydrate transport and metabolism (230 genes), as well as the function unknown (1,112 genes) (Supplementary Figure S1). In addition, 2,645 genes (57.85%) were allocated to the KEGG pathways, in which the carbohydrate metabolism (242 genes), global and overview maps (237 genes), amino acid metabolism (224 genes) and membrane transport (222 genes) were the predominant pathways (Supplementary Figure S1).
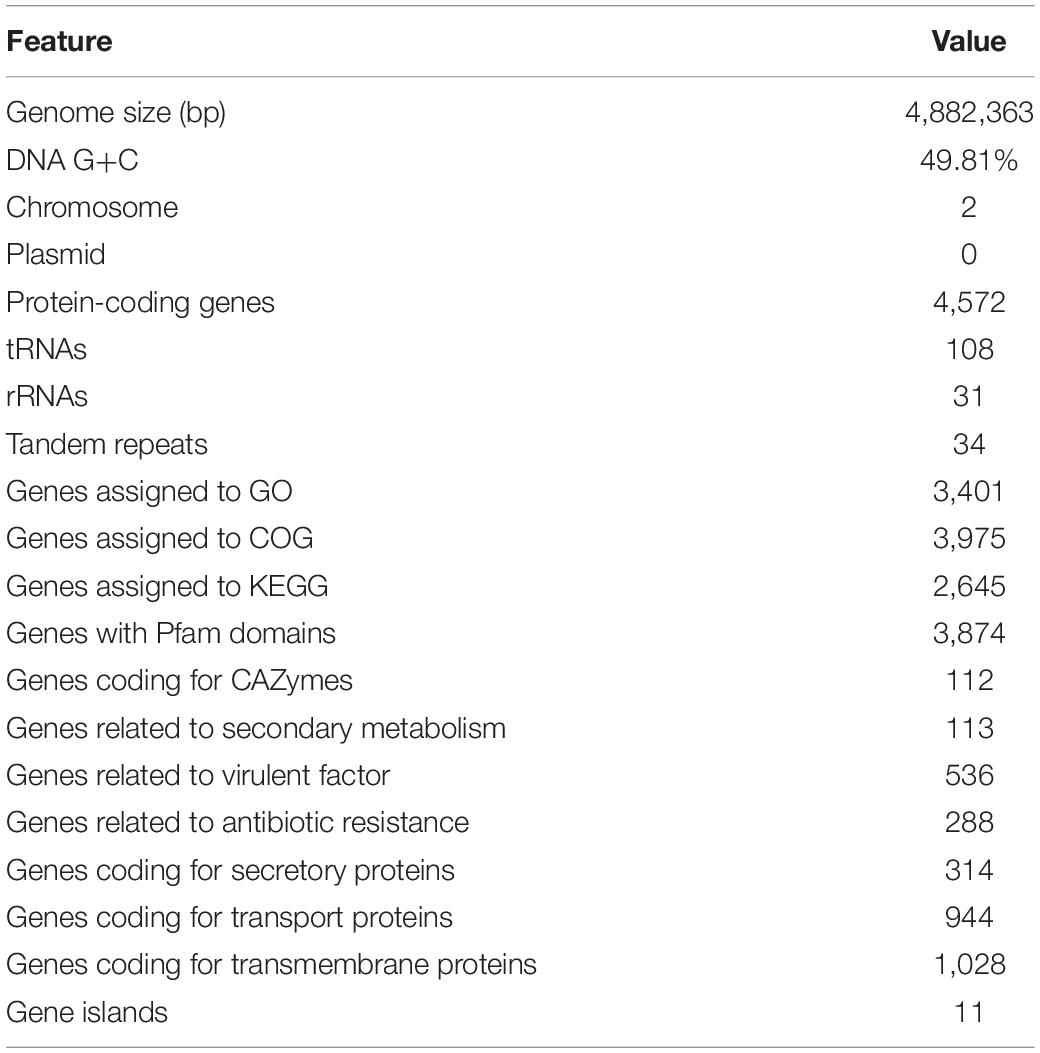
Table 1. Genomic features of strain A8.
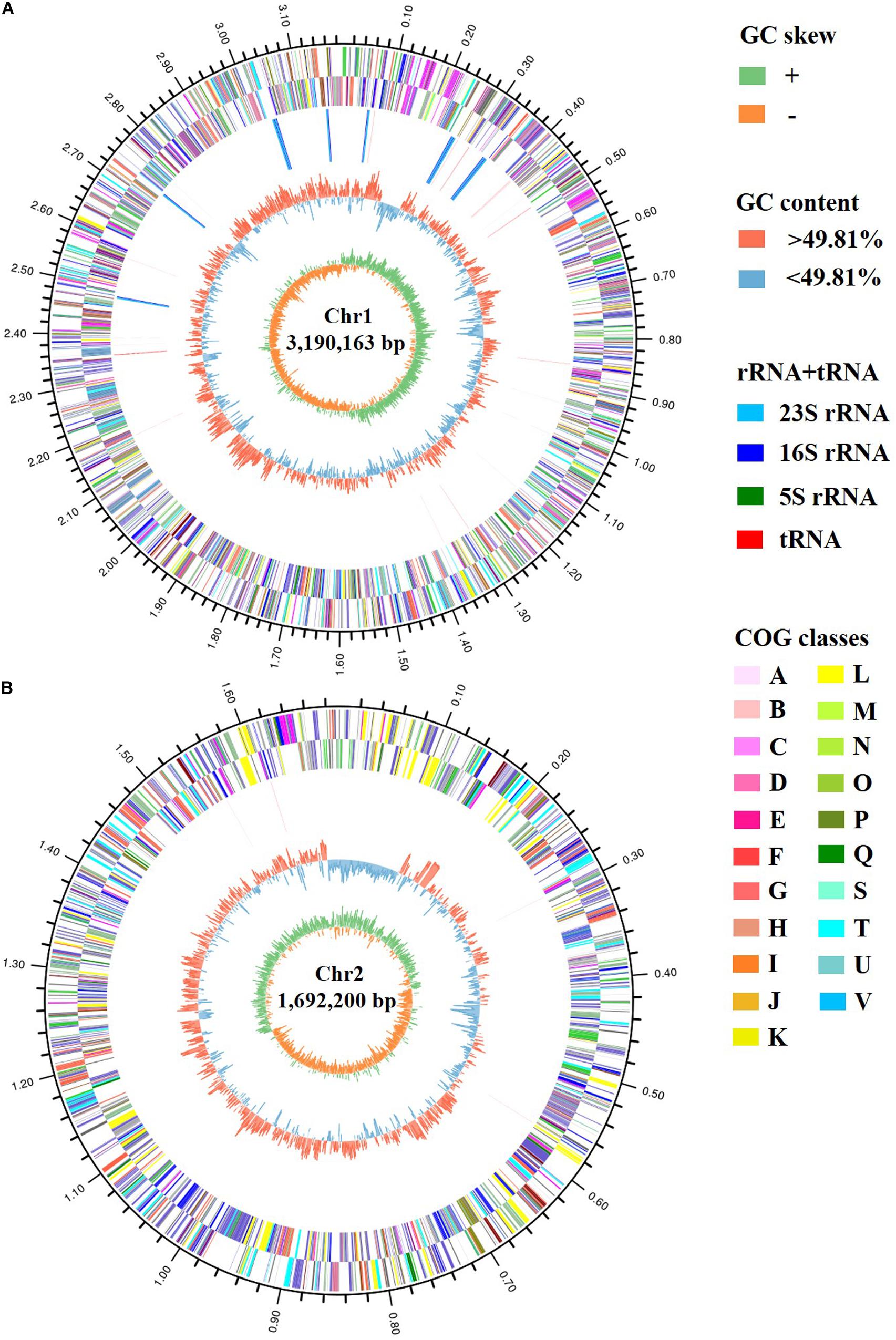
Figure 2. Circular maps of (A) Chr1 and (B) Chr2 of strain A8. The circles represent (from inner to outer): (1) GC skew; (2) GC content; (3) rRNA and tRNA; (4) COG assignments for CDSs on the reverse strand; (5) COG assignments for CDSs on the forward strand; (6) scale in Mb.
Identification of Strain A8
Strain A8 was initially identified as Vibrio sp. A8 based on 16S rRNA gene sequence. To determine the exact species of this strain, the comparative genomic analysis between A8 and 20 similar strains in Vibrio was studied. Among the 21 Vibrio strains, 1,419 homologous genes were found and constructed the core genome (Figure 3A). The genome phylogenetic tree was then constructed based on the core homologous genome (Figure 3B). The phylogenetic tree was capable of grouping various strains of a single species into a single cluster. Strain A8 clustered with V. fluvialis ATCC 33809, V. fluvialis 12605, and V. fluvialis AK 1296-A2-1, suggesting that these four strains might harbor similar biological properties. The biochemical identification and characterization observations for strain A8 were made on VITEK 2 GN system (Supplementary Table S1). Strain A8 was identified as V. fluvialis with 90% of probability. On the basis of above results, the strain A8 could be determined as V. fluvialis and was renamed as V. fluvialis A8. The ANI similarity analysis also proved that strain A8 belonged to V. fluvialis. It was reported that the strains could be considered as the same prokaryotic species when the ANI value was over 96% (Richter and Rossello-Mora, 2009). As shown in Table 2, intraspecies similarity of V. fluvialis was higher than 97%. The highest similarity with V. fluvialis A8 was V. fluvialis ATCC 33809, reaching 98.5%, which was consistent with the result of phylogenetic tree. Although 3,823 homologous genes were found among these V. fluvialis strains, there were still 448 unique genes in V. fluvialis A8 (Figure 3C).
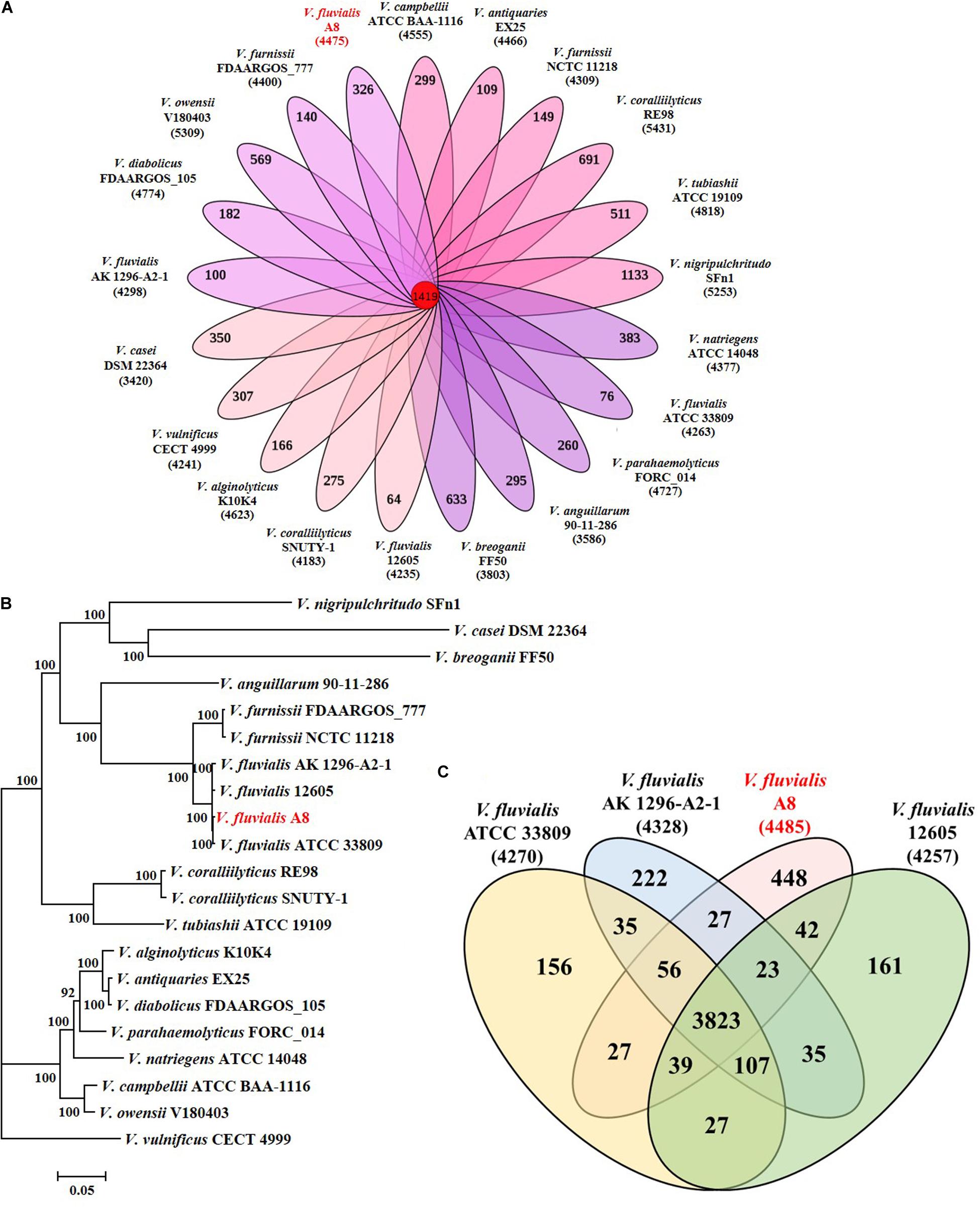
Figure 3. Phylogeny of Vibrio species based on single-copy core orthologs. (A) Core homologous genome (1419 genes) was computed by the genome of strain A8 along with 20 complete Vibrio genomes obtained from public database. (B) Phylogenetic analysis was performed by RA × ML after multiple alignments of single copy of homologous genes using Clustal X. (C) Venn diagram showing homologous and unique genes among V. fluvialis strains.
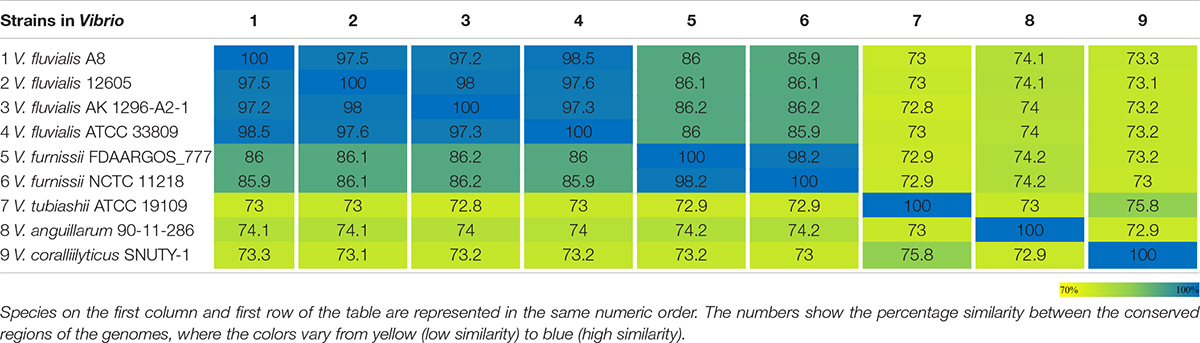
Table 2. Heatmap of the ANI value between the conserved regions in the genomes of strains in Vibrio.
Genes Related to Agar Degradation in Vibrio fluvialis A8
Carbohydrate-active enzymes (CAZymes) that are responsible for the complex carbohydrate metabolism, mainly include the recognition (carbohydrate-binding module, CBMs), synthesis (glycosyltransferases, GTs), and degradation (glycoside hydrolases, GHs; polysaccharide lyases, PLs; carbohydrate esterases, CEs; and enzymes of auxiliary activities, AAs) of the carbohydrates (Huang et al., 2018). There were 112 genes coding for CAZymes in the genome of V. fluvialis A8 (Table 1). This strain had a high fraction of carbohydrate-degrading enzymes, especially GHs (48 genes) which could hydrolyze various polysaccharides, including agar, agaro-oligosaccharides, amylum, cellulose, hemicellulose, and chitin (Figure 4).
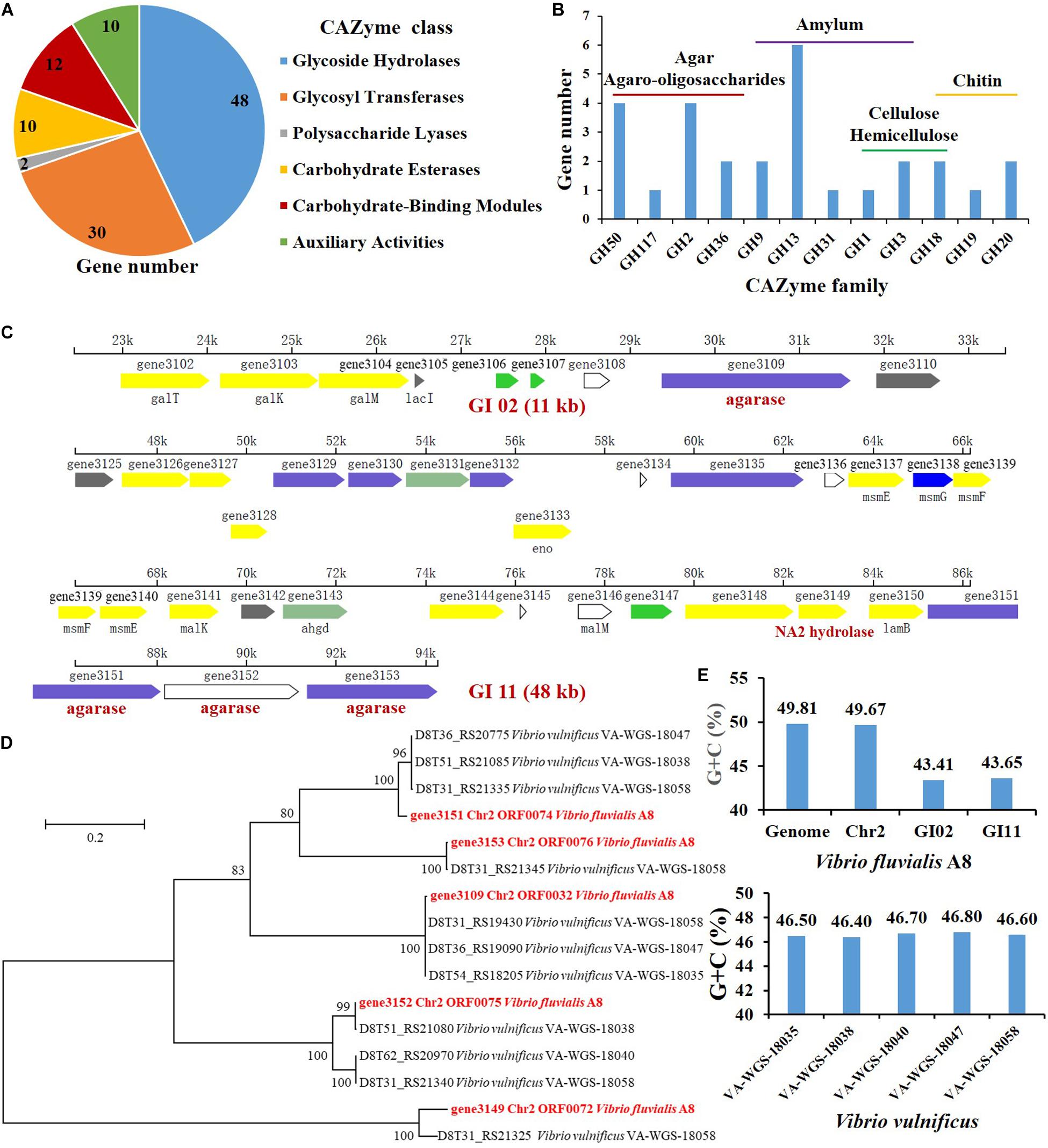
Figure 4. Genes related to agar degradation in the genome of V. fluvialis A8. (A) Number of genes in each CAZyme class. (B) Number of genes related to polysaccharide degradation in GHs. Genomic islands (GI) contained genes related to agar degradation: (C) gene3149 coding for α-1,3-L-NA2 hydrolase, and gene3151, gene3152, and gene3153 coding for β-agarase were in GI 11; (D) gene3109 coding for β-agarase was in GI 02. (E) The G+C content in the GI 02, GI 11, Chr2, and genome of V. fluvialis A8 as well as in the genomes of V. vulnificus strains.
Agarases are classified into α-agarase (E.C. 3.2.1.158) and β-agarase (E.C. 3.2.1.81) according to their cleavage position on agarose: α-agarase catalyzes the α-1,3 linkage to produce AOS, while β-Agarase produces NAOS by split of β-1,4 linkage (Fu and Kim, 2010). Agarases are grouped into GHs based on the sequence similarity, e.g., β-Agarase grouping into the GH16, GH50, GH86, and GH118 families and α-agarase only grouping into the GH96 family (Su et al., 2019). In this study, a total of four genes coding for β-agarase which all belonged to GH50 family, were found in the genome of V. fluvialis A8. Among the β-agarase genes, the gene3151, gene3152, and gene3153 arranged in order from 85221 to 94263 on the forward strand of Chr2, while the gene3109 was located on the reverse strand of Chr2 from 31608 to 29374 (Table 3). NAOS can be further catalyzed by α-NAOS hydrolase (EC 3.2.1.159) which has been classified into GH 117 family and can recognize neoagarobiose (NA2), neoagarotetraose (NA4), and neoagarohexaose (NA6) as substrate (Ariga et al., 2014; Watanabe et al., 2016). In the genome of V. fluvialis A8, the gene3149 coding for α-1,3-L-NA2 hydrolase was classified into GH 117 family. This enzyme can hydrolyze NA2 and produce L-AHG and D-Gal (Watanabe et al., 2016). It was reported that β-galactosidase in Vibrio sp. EJY3 could act on AOS to release D-Gal (Lee et al., 2014). There were four genes of β-galactosidase and two genes of α-galactosidase in the genome of V. fluvialis A8, which might contribute to its ability of agar degradation. The abundant genes of agarase, NAOS hydrolase and galactosidase in V. fluvialis A8 offer an enormous advantage in the saccharification of agar in red algae for its use as a biomass feedstock for the production of fermentable sugar.
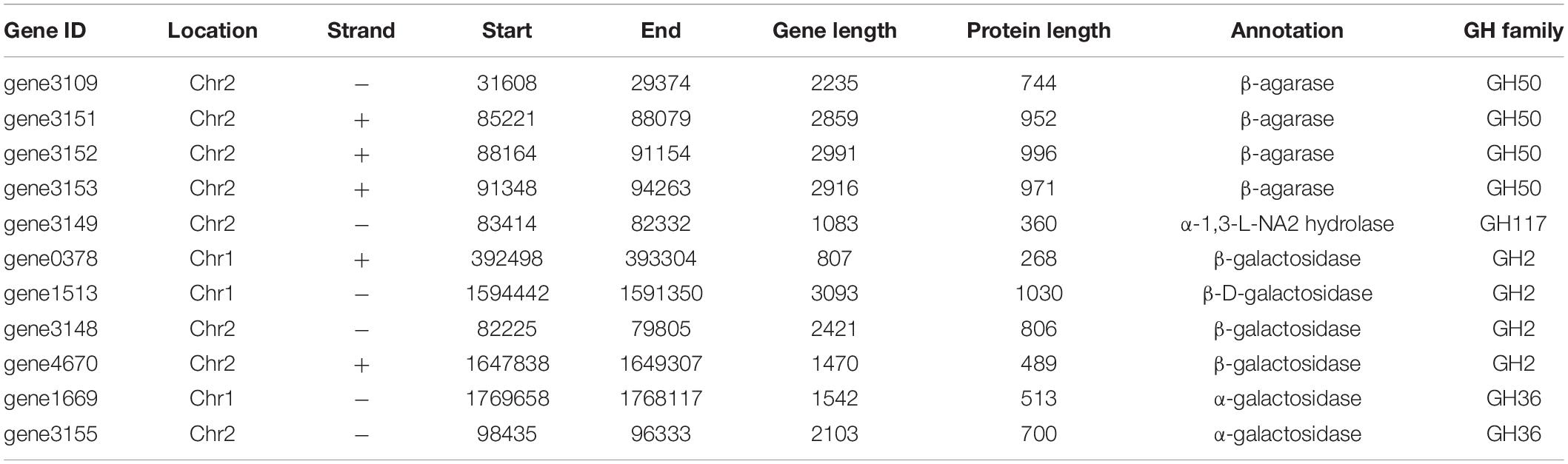
Table 3. Genes related to degradation of agar and relative compounds in the genome of V. fluvialis A8.
To study whether the strains closely related to V. fluvialis A8 shared the common ability of agar degradation, the agar-degrading genes of these strains were analyzed by comparative genomic analysis (Table 4). Interestingly, although the Vibrio strains exhibited the genes coding for galactosidase, the homologs for agarase and NA2 hydrolase were not observed in all other tested strains, even in the same species V. fluvialis 12605, V. fluvialis AK 1296-A2-1, and V. fluvialis ATCC 33809. This suggested that V. fluvialis A8 was probably different from other similar strains and possessed the unique agar degradation ability. Although the agar-degrading genes were not found in the tested genomes, another Vibrio sp. strain EJY3 exhibited similar agarolytic system with V. fluvialis A8 and also could produce four β-agarases (GH50) and one α-NAOS hydrolase (GH117) (Yu et al., 2020). Horizontal gene transfer, also known as lateral gene transfer, refers to nonsexual transmission of genetic material between unrelated genomes (Oliveira et al., 2017). As the most important form of horizontal gene transfer, genomic island (GI) contains numerous genes associated with a variety of biological functions (Godde et al., 2018). It has been reported that horizontal gene transfer in the bacteria affected the phenotypes such as thermotolerance and multidrug resistance of closely related strains (Hegstad et al., 2020; Matsutani et al., 2020). Horizontal gene transfer also gives bacteria unique polysaccharide degradation ability. Alteromonas macleodii 83-1 harbored an alginolytic system within a 24 kb GI, which comprised five putative alginate lyases and other CAZymes (Neumann et al., 2015). The marine bacterium Pseudoalteromonas haloplanktis ANT/505 acquired a GI with a functional pathway for pectin catabolism, including two multi-modular pectate lyases, PelA and PelB (Hehemann et al., 2017). A novel agarase gene aga1 in an inland soil agar-degrading bacterium Paenibacillus sp. SSG-1 might be horizontally transferred from marine bacteria via human microbiota (Song T. et al., 2016). In the genus Vibrio, horizontal gene transfer mainly contributes to the virulence of strains (Le Roux and Blokesch, 2018; Deng et al., 2019). However, few studies of horizontal gene transfer related to agar degradation were reported in the genus Vibrio. In the genome of V. fluvialis A8, there were 11 GIs, in which 474 genes located, accounting for over 10% of total protein-coding genes. Interestingly, all the GIs were located in the Chr2 of this strain. The genes coding for β-agarase and α-1,3-L-NA2 hydrolase were all contained in the GIs: the gene3151, gene3152, gene3153, and gene3149 were in the GI 11 (Figure 4C), while the gene3109 were in the GI 02 (Figure 4D). These results proved that these genes related to agar degradation were obtained from other species through horizontal gene transfer. The gene sequence similarity was further studied to determine the probable source using the phylogenetic tree analysis (Figure 4D). The gene3109, gene3151, gene3152, gene3153, and gene3149 were found most similar to that in Vibrio vulnificus VA-WGS-18058 (percent identity with 99.78%), V. vulnificus VA-WGS-18047 (percent identity with 96.08%), V. vulnificus VA-WGS-18038 (percent identity with 99.77%), V. vulnificus VA-WGS-18058 (percent identity with 99.90%), and V. vulnificus VA-WGS-18058 (percent identity with 91.20%), respectively. Furthermore, the G+C content in the GI 02 and GI 11 was 43.41 and 43.65%, respectively, which was obviously lower than that in the genome (49.81% G+C) and Chr2 (49.67% G+C) of V. fluvialis A8, but much closer to that in the genome of V. vulnificus strains (46.40–46.80% G+C) (Figure 4E). These results indicated that these genes might come from these V. vulnificus strains through horizontal gene transfer.
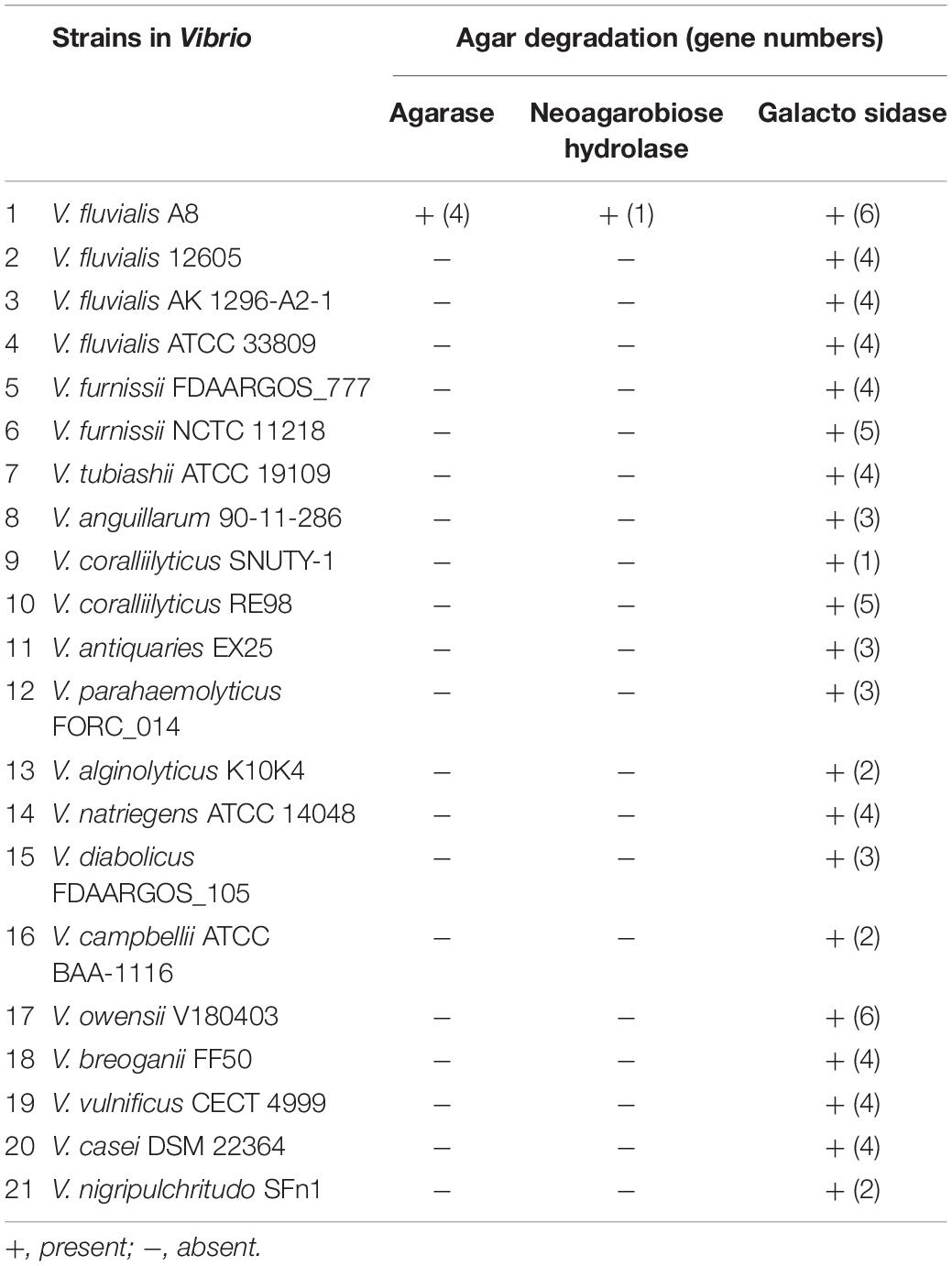
Table 4. Genes involved in degradation of agar and relative compounds in the genomes of strains in Vibrio based on homologous gene analysis.
Agarase Determination in Vibrio fluvialis A8 Based on Secretomic Analysis
Secretomic analysis using nanoLC-MS/MS was performed to study the agarase produced from V. fluvialis A8. A total of 1,078 peptides were identified and belonged to 207 proteins. The molecular weight of these proteins ranged from 6.6 to 212.2 kDa, and most of these proteins focused between 30 and 40 kDa (Supplementary Figure S2). The function of proteins with high abundance in the secretome of V. fluvialis A8 mainly included three major categories: macromolecule degradation (zinc protease, agarase, metalloprotease, and aminopeptidase), cell components (flagellin protein FlaD, FlaA, and FlaC, and membrane protein), and virulent factor (hemolysin HlyA, thermolabile hemolysin precursor) (Table 5). These proteins provided a great advantage for utilization the nutrients from red algae that helped V. fluvialis A8 grow. Among these extracellular proteins, zinc protease coded by gene3819 showed the highest relative abundance reaching 69.49%. A total of eight proteins were found in GHs, in which β-agarase coded by gene3152 had the highest relative abundance reaching 3.04% (Table 5). The 27 peptides of this agarase tested by Label-free quantitative LC-MS/MS were shown in Supplementary Table S2. Interestingly, there were four genes in the genome of V. fluvialis A8, among which the gene3152 and gene3153 coding for β-agarases were predicted as secretory proteins with signal peptide (Sec/SPI) using SignalP version 5.0, but only gene3152 was expressed extracellularly. It might be because there was little expression of the gene3153. In addition, the β-agarase encoded by gene3151 had a lipoprotein signal peptide (Sec/SPII), which was also called lipobox. Lipobox played an important role in anchoring the agarases onto the cell surface by acylation (Ekborg et al., 2006; Hutcheson et al., 2011). In this study, different from β-agarase (gene3152) that was secreted into the medium, β-agarase (gene3151) was predicted to be bound to the cell surface, which also contributed to the agar degradation and utilization of V. fluvialis A8. The β-agarase (gene3152) in the secretome was about 108.7 kDa with the pI 4.60 (Table 5).
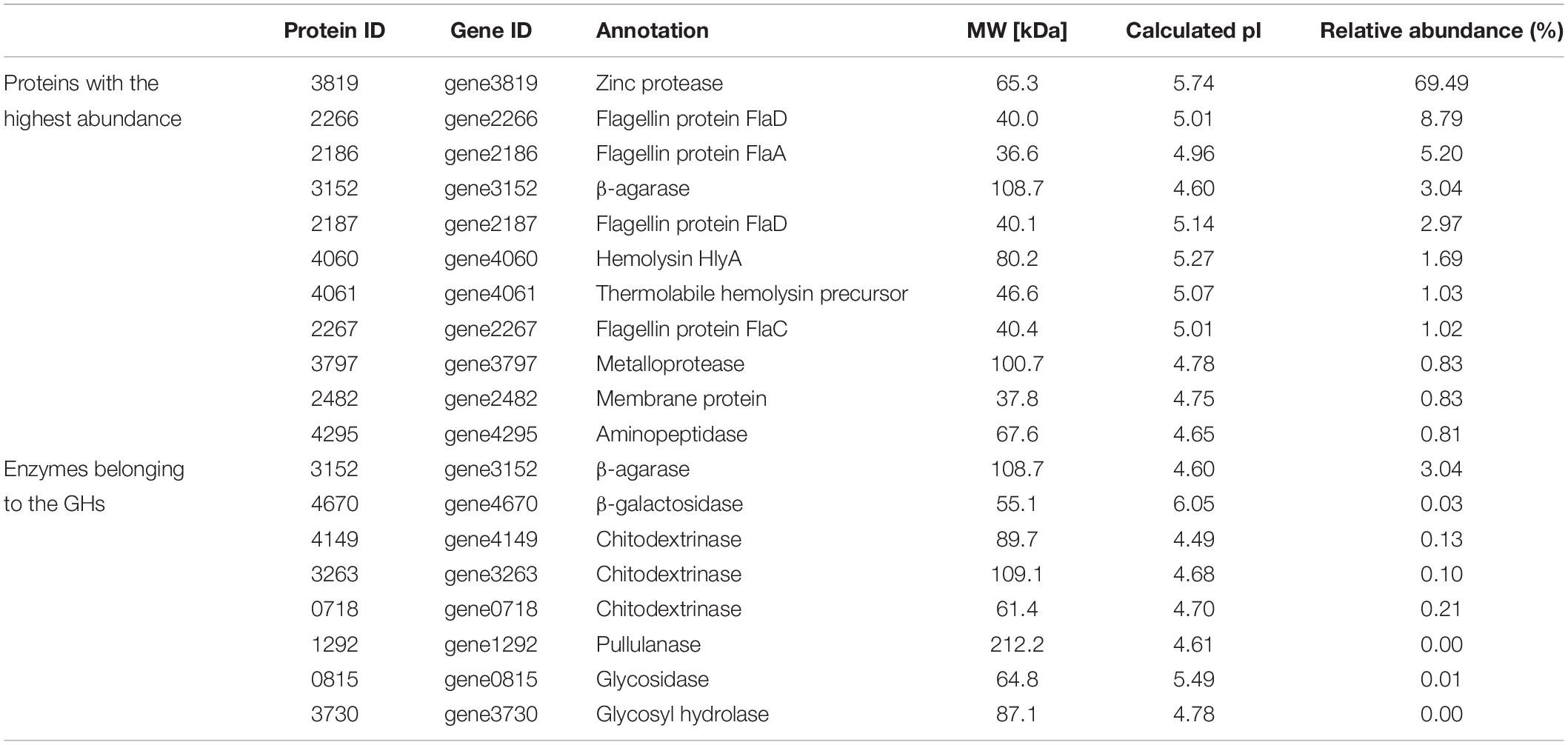
Table 5. Proteins with the highest abundance and enzymes belonging to the GHs in the secretome of V. fluvialis A8.
Two kinds of β-agarase have been determined in Vibrio sp. JT0107, β-agarase A (AgaA) which hydrolyzes agarose and also NA4 to yield NA2, and β-agarase B (AgaB) which hydrolyzes agarose to yield predominantly NA4 and NA6 (Jam et al., 2005). The phylogenetic tree consisting of β-agarase (gene3152) and known characterized agarases proved that the β-agarase secreted from V. fluvialis A8 was a member of GH50 (Figure 5A). The β-agarase (gene3152) showed most similar to AgaA (percent identity with 48.41%) from Vibrio sp. JT0107, suggesting that it might be classified as AgaA. Therefore, the major agar metabolic pathway in V. fluvialis A8 could be speculated: Agar or agarose was first hydrolyzed extracellularly by β-agarase (gene3152) to yield NA2, and after transport into the cells, NA2 was further catalyzed by α-1,3-L-NA2 hydrolase (gene3149) to produce L-AHG and D-GAL for use as a carbon source (Figure 5B).
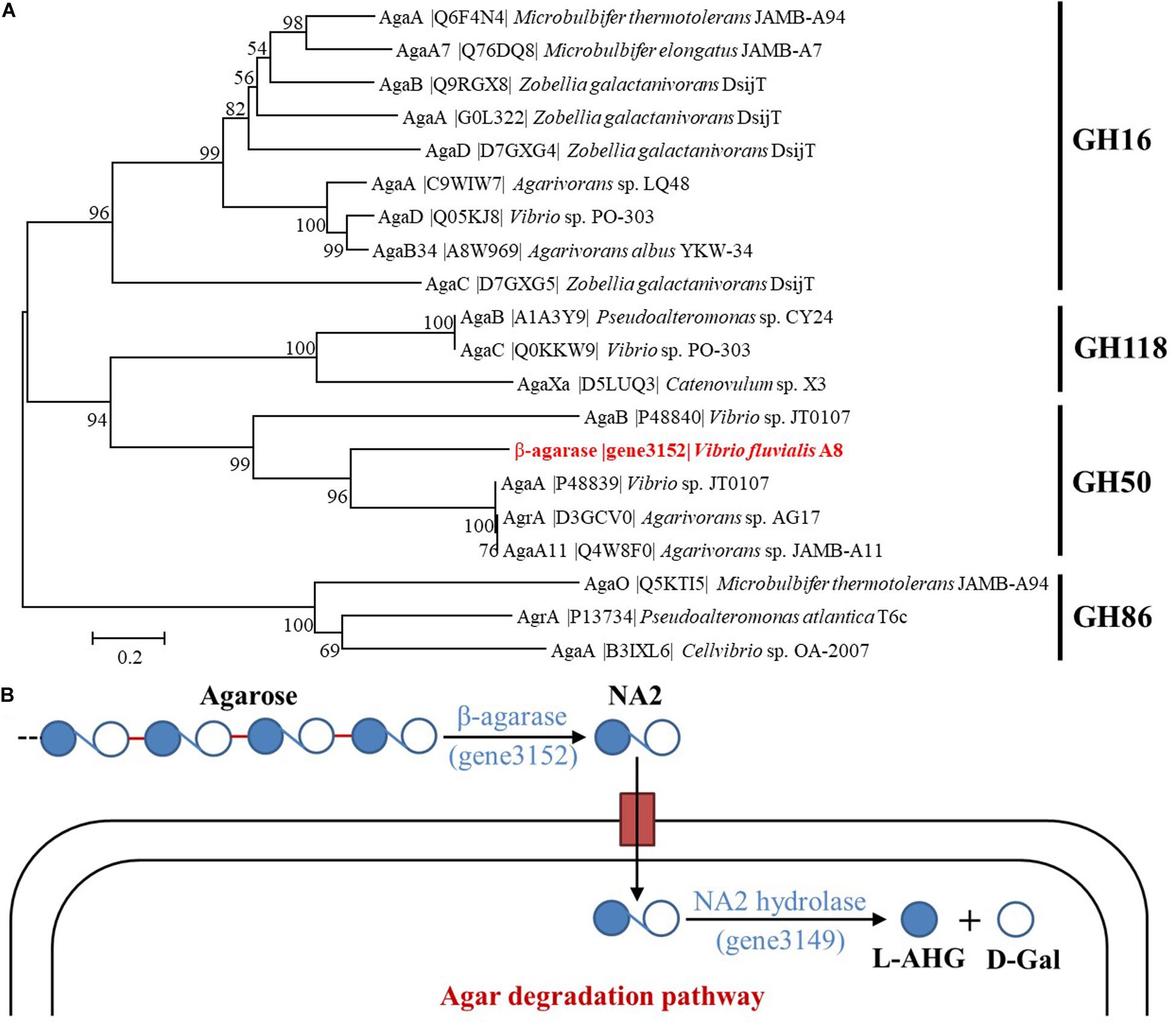
Figure 5. Classification of β-agarase and possible agar metabolic pathways in V. fluvialis A8. (A) Phylogenetic tree between β-agarase (gene3152) and the characterized agarases obtained from UniProt database. Phylogenetic tree of agarases based on their amino acid sequences was performed by MEGA based on the neighbor joining method after multiple alignments of the sequences by Clustal X. Bootstrap values (expressed as percentages) are given at the branching points. The bar corresponds to a genetic distance of 0.1 substitution per position (10% amino acid sequence difference). (B) Illustration for the possible agar metabolic pathways in V. fluvialis A8.
Biochemical Properties of Agarase in Vibrio fluvialis A8
After preliminary purification by DEAE Sephadex anion-exchange chromatography (Supplementary Figure S3), the biochemical properties of agarase in the secretome of V. fluvialis A8 was studied (Figure 6). The agarase could hardly degrade other polysaccharides such as carrageenan, algin, chitosan, cellulose, and amylum into small-molecule reducing sugars, indicating its good substrate specificity (Figure 6A). The agarase could work over a wide temperature range that its relative activity at 25–50°C maintained over 60% (Figure 6B). The optimum temperature of the agarase was 45°C. This result is consistent with the optimum reaction temperature of agarase from Cellulophaga omnivescoria W5C (Ramos et al., 2018) and Pseudoalteromonas hodoensis H7 (Chi et al., 2014). The optimum pH of the agarase was 7.0 (Figure 6C). The agarase activity was relatively stable to pH, and the activity could remain over 47% when the pH value was in the range of 4.0–9.0. Such performance could hardly reached by agarase from other bacteria that the activity was significantly inhibited at low pH (Tawara et al., 2015; Chen et al., 2019; Su et al., 2019). The agarase possessed stable activity at high concentration of various metal ions (Figure 6D). Na+ significantly enhanced the agarase activity by 8.0%. Mg2+, Ca2+, and K+ had little effect on the agarase activity, while Cu2+, Fe2+, and Fe3+ manifested a strong inhibition of agarase activity. Similar results were found that the activity of agarase was significantly suppressed by Cu2+ and Fe2+ (Chen et al., 2019; Li et al., 2019). The agarase in V. fluvialis A8 showed good thermostability at 35–45°C (Figure 6E). The t1/2 of agarase at 35, 40, and 45°C reached 866.4, 169.1, and 44.4 min, respectively. The agarase in V. fluvialis A8 was easily inactivated over 50°C and its activity completely lost after 30 min at this temperature. The agarase in V. fluvialis A8 possessed more thermostability than agarase rAgaZC-1 and D622G in Vibrio sp. ZC-1 whose t1/2 was only 3.1 and 6.3 min at 45°C, respectively (Su et al., 2019). In this study, the agarase in V. fluvialis A8 had a good substrate specificity, possessed stable activity in the conditions of temperature, pH and various metal ions, and exhibited good thermostability, which suggests that it could be developed as a potential agarase preperation for agarose-oligosaccharide production in future.
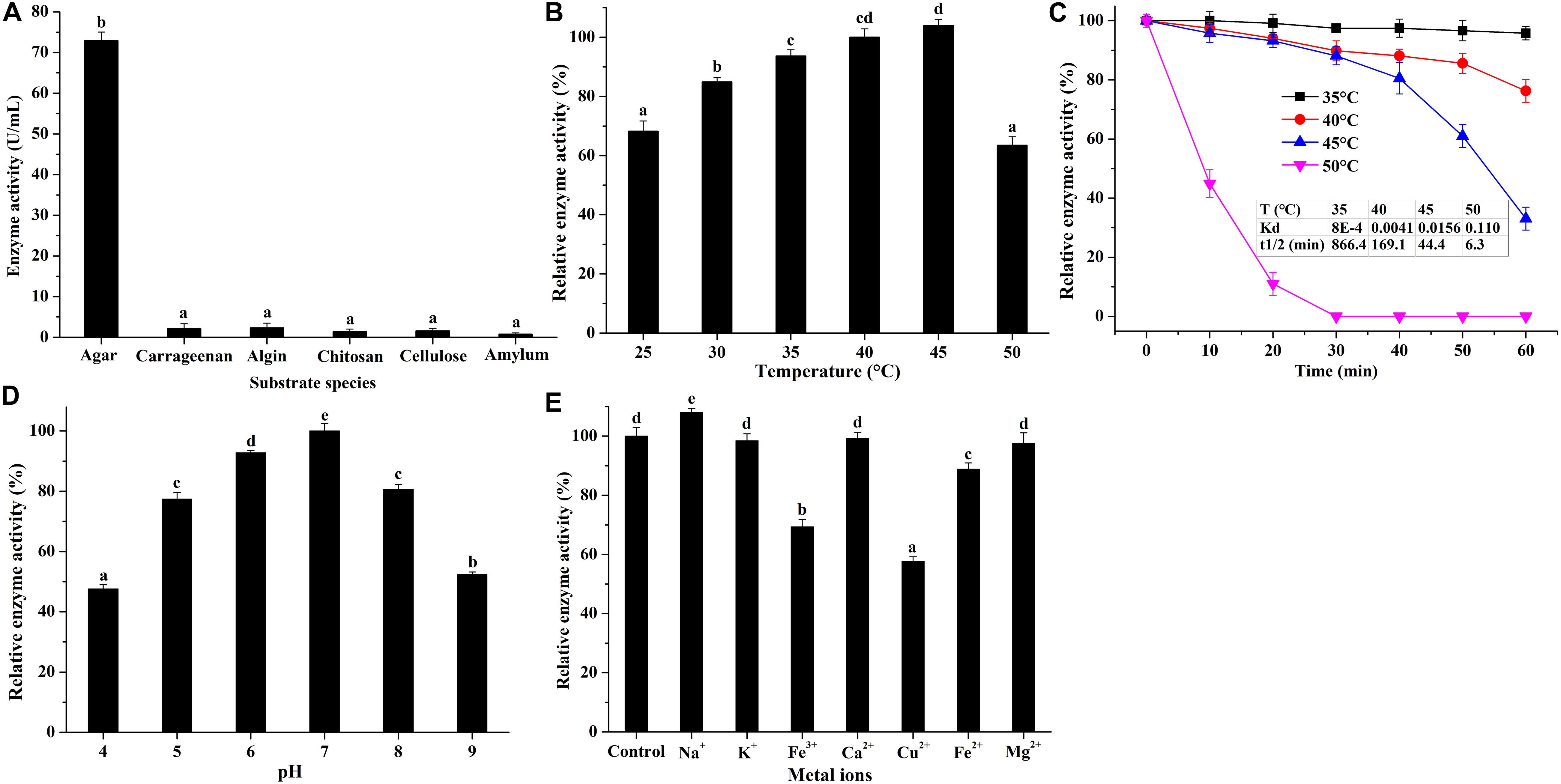
Figure 6. Biochemical properties of purified agarase in V. fluvialis A8. (A) Substrate specificity of agarase was studied using various substrates, including carrageenan, algin, chitosan, cellulose, and amylum. (B) Effects of temperature, (C) pH, and (D) metal ions on the agarase activity. Bars labeled with the same letter are not statistically different (p < 0.05) tested by one-way ANOVA and multiple comparison Tukey test. (E) Thermostability of agarase was studied at different temperatures.
Conclusion
Complete genome of novel agar-degrading bacterium Vibrio sp. A8 was sequenced to investigate the genetic elements related to agar degradation. The complete genome was 4.88 Mb with two circular chromosomes (3.19 and 1.69 Mb) and no plasmid. This strain was identified as V. fluvialis A8 by genome phylogenetic tree and ANI similarity with biochemical identification. Different from other similar strains including the same species, V. fluvialis A8 possessed unique agar degradation ability with 4 β-agarases (GH50) and 1 α-1,3-L-NA2 hydrolase (GH117) due to horizontal gene transfer. Only β-agarase coding by gene3152 was expressed extracellularly based on secretomic analysis. This agarase had a good substrate specificity and stability in complex environments, suggesting its potential application for agarose-oligosaccharide production.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
CSL and CL conceived, designed, and performed the experiments. CSL analyzed the data, prepared all tables and figures, and wrote the manuscript. SC and BQ provided assistance with analysis tools. LL, SC, BQ, and YZ critically reviewed and curated the manuscript. CSL and XY were responsible for the project. All authors contributed to the article and approved the submitted version.
Funding
This research was financed by the National Key R&D Program of China (No. 2019YFD0901905), the China Agriculture Research System (No. CARS-50), the Central Public-interest Scientific Institution Basal Research Fund, South China Sea Fisheries Research Institute, CAFS (No. 2020XK01), the National Natural Science Foundation of China (No. 31801520), and the Special Scientific Research Funds for Central Non-profit Institutes, Chinese Academy of Fishery Sciences (No. 2020TD69).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Weizhou Chen from the Shantou University for the help with the isolation of V. fluvialis A8.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01934/full#supplementary-material
References
Araki, C. (1956). Structure of the agarose constituent of agar-agar. Bull. Chem. Soc. Jpn. 29, 543–544. doi: 10.1246/bcsj.29.543
Ariga, O., Okamoto, N., Harimoto, N., and Nakasaki, K. (2014). Purification and characterization of alpha-neoagarooligosaccharide hydrolase from Cellvibrio sp OA-2007. J. Microbiol. Biotechnol. 24, 48–51. doi: 10.4014/jmb.1307.07018
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bhattarai, Y., and Kashyap, P. C. (2016). Agaro-oligosaccharides: a new frontier in the fight against colon cancer? Am. J. Physiol. Gastrointest. Liver Physiol. 310, G335–G336. doi: 10.1152/ajpgi.00049.2016
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Chen, X., Lin, H., Jin, M., Zeng, R., and Lin, M. (2019). Characterization of a novel alkaline beta-agarase and its hydrolysates of agar. Food Chem. 295, 311–319. doi: 10.1016/j.foodchem.2019.05.132
Chi, W. J., Park, J. S., Kang, D. K., and Hong, S. K. (2014). Production and characterization of a novel thermostable extracellular agarase from Pseudoalteromonas hodoensis newly isolated from the West Sea of South Korea. Appl. Biochem. Biotechnol. 173, 1703–1716. doi: 10.1007/s12010-014-0958-3
Deng, Y., Xu, H., Su, Y., Liu, S., Xu, L., Guo, Z., et al. (2019). Horizontal gene transfer contributes to virulence and antibiotic resistance of Vibrio harveyi 345 based on complete genome sequence analysis. BMC Genomics 20:761. doi: 10.1186/s12864-019-6137-8
Dong, J., Tamaru, Y., and Araki, T. (2007). A unique beta-agarase, AgaA, from a marine bacterium, Vibrio sp strain PO-303. Appl. Microbiol. Biotechnol. 74, 1248–1255. doi: 10.1007/s00253-006-0781-z
Ekborg, N. A., Taylor, L. E., Longmire, A. G., Henrissat, B., Weiner, R. M., and Hutcheson, S. W. (2006). Genomic and proteomic analyses of the agarolytic system expressed by Saccharophagus degradans 2-40. Appl. Environ. Microbiol. 72, 3396–3405. doi: 10.1128/AEM.72.5.3396-3405.2006
Enoki, T., Okuda, S., Kudo, Y., Takashima, F., Sagawa, H., and Kato, I. (2010). Oligosaccharides from agar inhibit pro-Inflammatory mediator release by inducing heme oxygenase 1. Biosci. Biotechnol. Biochem. 74, 766–770. doi: 10.1271/bbb.90803
Feng, Z., Chen, G., Zhang, J., Zhu, H., Yu, X., Yin, Y., et al. (2019). Characterization and complete genome analysis of the carbazomycin B-producing strain Streptomyces luteoverticillatus SZJ61. Curr. Microbiol. 76, 982–987. doi: 10.1007/s00284-019-01711-x
Fu, X. T., and Kim, S. M. (2010). Agarase: review of major sources, categories, purification method, enzyme characteristics and applications. Mar. Drugs 8, 200–218. doi: 10.3390/md8010200
Godde, J. S., Baichoo, S., Mungloo-Dilmohamud, Z., and Jaufeerally-Fakim, Y. (2018). Comparison of genomic islands in cyanobacteria: evidence of bacteriophage-mediated horizontal gene transfer from eukaryotes. Microbiol. Res. 211, 31–46. doi: 10.1016/j.micres.2018.03.005
Hegstad, K., Mylvaganam, H., Janice, T., Josefsen, E., Sivertsen, A., and Skaare, D. (2020). Role of horizontal gene transfer in the development of multidrug resistance in Haemophilus influenzae. mSphere 5:e00969-19. doi: 10.1128/mSphere.00969-19
Hehemann, J. H., Truong, L. V., Unfried, F., Welsch, N., Kabisch, J., Heiden, S. E., et al. (2017). Aquatic adaptation of a laterally acquired pectin degradation pathway in marine gammaproteobacteria. Environ. Microbiol. 19, 2320–2333. doi: 10.1111/1462-2920.13726
Huang, L., Zhang, H., Wu, P., Entwistle, S., Li, X., Yohe, T., et al. (2018). dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 46, D516–D521. doi: 10.1093/nar/gkx894
Hutcheson, S. W., Zhang, H., and Suvorov, M. (2011). Carbohydrase systems of Saccharophagus degradans degrading marine complex polysaccharides. Mar. Drugs 9, 645–665. doi: 10.3390/md9040645
Jam, M., Flament, D., Allouch, J., Potin, P., Thion, L., Kloareg, B., et al. (2005). The endo-beta-agarases AgaA and AgaB from the marine bacterium Zobellia galactanivorans: two paralogue enzymes with different molecular organizations and catalytic behaviours. Biochem. J. 385, 703–713. doi: 10.1042/BJ20041044
Kang, O. L., Ghani, M., Hassan, O., Rahmati, S., and Ramli, N. (2014). Novel agaro-oligosaccharide production through enzymatic hydrolysis: physicochemical properties and antioxidant activities. Food Hydrocoll. 42, 304–308. doi: 10.1016/j.foodhyd.2014.04.031
Kim, J. H., Yun, E. J., Yu, S., Kim, K. H., and Kang, N. J. (2017). Different levels of skin whitening activity among 3,6-anhydro-l-galactose, agarooligosaccharides, and neoagarooligosaccharides. Mar. Drugs 15:321. doi: 10.3390/md15100321
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Le Roux, F., and Blokesch, M. (2018). Eco-evolutionary dynamics linked to horizontal gene transfer in Vibrios. Annu. Rev. Microbiol. 72, 89–110. doi: 10.1146/annurev-micro-090817-062148
Lee, C. H., Kim, H. T., Yun, E. J., Lee, A. R., Kim, S. R., Kim, J. H., et al. (2014). A novel agarolytic beta-galactosidase acts on agarooligosaccharides for complete hydrolysis of agarose into monomers. Appl. Environ. Microbiol. 80, 5965–5973. doi: 10.1128/AEM.01577-14
Li, J., Xie, M., and Gao, Y. (2019). Identification and biochemical characterization of a novel exo-type beta-agarase Aga3463 from an Antarctic Pseudoalteromonas sp. strain. Int. J. Biol. Macromol. 129, 162–170. doi: 10.1016/j.ijbiomac.2019.01.204
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Liao, L., Xu, X. W., Jiang, X. W., Cao, Y., Yi, N., Huo, Y. Y., et al. (2011). Cloning, expression, and characterization of a new beta-agarase from Vibrio sp strain CN41. Appl. Environ. Microbiol. 77, 7077–7079. doi: 10.1128/AEM.05364-11
Ma, J., Yan, Q., Yi, P., Yang, S., Liu, H., and Jiang, Z. (2019). Biochemical characterization of a truncated beta-agarase from Microbulbifer sp. suitable for efficient production of neoagarotetraose. Process Biochem. 87, 119–127. doi: 10.1016/j.procbio.2019.08.021
Matsutani, M., Matsumoto, N., Hirakawa, H., Shiwa, Y., Yoshikawa, H., Okamoto-Kainuma, A., et al. (2020). Comparative genomic analysis of closely related Acetobacter pasteurianus strains provides evidence of horizontal gene transfer and reveals factors necessary for thermotolerance. J. Bacteriol. 202:e00553-19. doi: 10.1128/JB.00553-19
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing Sugar. Anal. Chem. 31, 426–428. doi: 10.1021/ac60147a030
Mussatto, S. I., and Mancilha, I. M. (2007). Non-digestible oligosaccharides: a review. Carbohydr. Polym. 68, 587–597. doi: 10.1016/j.carbpol.2006.12.011
Neumann, A. M., Balmonte, J. P., Berger, M., Giebel, H. A., Arnosti, C., Voget, S., et al. (2015). Different utilization of alginate and other algal polysaccharides by marine Alteromonas macleodii ecotypes. Environ. Microbiol. 17, 3857–3868. doi: 10.1111/1462-2920.12862
Oliveira, P. H., Touchon, M., Cury, J., and Rocha, E. P. C. (2017). The chromosomal organization of horizontal gene transfer in bacteria. Nat. Commun. 8:841. doi: 10.1038/s41467-017-00808-w
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Ramos, K. R. M., Valdehuesa, K. N. G., Nisola, G. M., Lee, W. K., and Chung, W. J. (2018). Identification and characterization of a thermostable endolytic beta-agarase Aga2 from a newly isolated marine agarolytic bacteria Cellulophaga omnivescoria W5C. New Biotechnol. 40, 261–267. doi: 10.1016/j.nbt.2017.09.006
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Seo, Y. B., Lu, Y., Chi, W. J., Park, H. R., Jeong, K. J., Hong, S. K., et al. (2014). Heterologous expression of a newly screened beta-agarase from Alteromonas sp.GNUM1 in Escherichia coli and its application for agarose degradation. Process Biochem. 49, 430–436. doi: 10.1016/j.procbio.2013.12.014
Song, H., Wang, P., Li, C., Han, S., Lopez-Baltazar, J., Zhang, X., et al. (2016). Identification of lipoxygenase (LOX) genes from legumes and their responses in wild type and cultivated peanut upon Aspergillus flavus infection. Sci. Rep. 6:35245. doi: 10.1038/srep35245
Song, T., Xu, H., Wei, C., Jiang, T., Qin, S., Zhang, W., et al. (2016). Horizontal transfer of a novel soil agarase gene from marine bacteria to soil bacteria via human microbiota. Sci. Rep. 6:34103. doi: 10.1038/srep34103
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Su, B. M., Xu, X. Q., Yan, R. X., Xie, Y., and Lin, J. (2019). Mutagenesis on the surface of a beta-agarase from Vibrio sp. ZC-1 increased its thermo-stability. Enzyme Microb. Technol. 127, 22–31. doi: 10.1016/j.enzmictec.2019.04.006
Tawara, M., Sakatoku, A., Tiodjio, R. E., Tanaka, D., and Nakamura, S. (2015). Cloning and characterization of a novel agarase from a newly isolated bacterium Simiduia sp strain TM-2 able to degrade various seaweeds. Appl. Biochem. Biotechnol. 177, 610–623. doi: 10.1007/s12010-015-1765-1
Wang, Y., Liu, T., Guo, S., Zhang, P., Sun, P., Chen, M., et al. (2018). Characterization and overexpression of a glycosyl hydrolase family 16 beta-agarase Aga0917 from Pseudoalteromonas fuliginea YTW1-15-1. J. Gen. Appl. Microbiol. 64, 276–283. doi: 10.2323/jgam.2018.02.002
Watanabe, T., Kashimura, K., and Kirimura, K. (2016). Purification, characterization and gene identification of a α-Neoagarooligosaccharide hydrolase from an alkaliphilic bacterium Cellvibrio sp. WU-0601. J. Mol. Catal. B Enzym. 133, S328–S336. doi: 10.1016/j.molcatb.2017.02.003
Won-Jae, C., Park, J. S., Kwak, M. J., Kim, J. F., Chang, Y. K., and Hong, S. K. (2013). Isolation and characterization of a novel agar-degrading marine bacterium, Gayadomonas joobiniege gen, nov, sp nov., from the Southern Sea, Korea. J. Microbiol. Biotechnol. 23, 1509–1518. doi: 10.4014/jmb.1308.08007
Xu, X. Q., Su, B. M., Xie, J. S., Li, R. K., Yang, J., Lin, J., et al. (2018). Preparation of bioactive neoagaroligosaccharides through hydrolysis of Gracilaria lemaneiformis agar: a comparative study. Food Chem. 240, 330–337. doi: 10.1016/j.foodchem.2017.07.036
Yu, S., Yun, E. J., Kim, D. H., Park, S. Y., and Kim, K. H. (2020). Dual agarolytic pathways in a marine bacterium, Vibrio sp. strain EJY3: molecular and enzymatic verification. Appl. Environ. Microbiol. 86:e02724-19. doi: 10.1128/AEM.02724-19
Zhang, P., Zhang, J., Zhang, L., Sun, J., Li, Y., Wu, L., et al. (2019). Structure-based design of agarase AgWH50C from Agarivorans gilvus WH0801 to enhance thermostability. Appl. Microbiol. Biotechnol. 103, 1289–1298. doi: 10.1007/s00253-018-9540-1
Zhang, W., and Sun, L. (2007). Cloning, characterization, and molecular application of a beta-agarase gene from Vibrio sp strain V134. Appl. Environ. Microbiol. 73, 2825–2831. doi: 10.1128/AEM.02872-06
Keywords: complete genome, secretome, agarase, horizontal gene transfer, Vibrio
Citation: Li C, Li C, Li L, Yang X, Chen S, Qi B and Zhao Y (2020) Comparative Genomic and Secretomic Analysis Provide Insights Into Unique Agar Degradation Function of Marine Bacterium Vibrio fluvialis A8 Through Horizontal Gene Transfer. Front. Microbiol. 11:1934. doi: 10.3389/fmicb.2020.01934
Received: 25 May 2020; Accepted: 22 July 2020;
Published: 11 August 2020.
Edited by:
Baolei Jia, Chung-Ang University, South KoreaReviewed by:
Chang-Ro Lee, Myongji University, South KoreaGo Furusawa, Universiti Sains Malaysia (USM), Malaysia
Copyright © 2020 Li, Li, Li, Yang, Chen, Qi and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunsheng Li, bGljaHVuc2hlbmdAc2NzZnJpLmFjLmNu; Xianqing Yang, eWFuZ3hxQHNjc2ZyaS5hYy5jbg==; eXhxZ2RAMTYzLmNvbQ==