 Tongda Li1,2,3*
Tongda Li1,2,3* Ross Mann1,2
Ross Mann1,2 Timothy Sawbridge1,2,3
Timothy Sawbridge1,2,3 Jatinder Kaur1,2
Jatinder Kaur1,2 Desmond Auer1
Desmond Auer1 German Spangenberg1,2,3
German Spangenberg1,2,3- 1Agriculture Victoria, AgriBio, Centre for AgriBioscience, Bundoora, VIC, Australia
- 2DairyBio, Bundoora, VIC, Australia
- 3School of Applied Systems Biology, La Trobe University, Bundoora, VIC, Australia
The productivity of the Australian dairy industry is underpinned by pasture grasses, and importantly perennial ryegrass. The performance of these pasture grasses is supported by the fungal endophyte Epichloë spp. that has bioprotection activities, however, the broader microbiome is not well characterized. In this study, we characterized a novel bioprotectant Xanthomonas species isolated from perennial ryegrass (Lolium perenne L. cv. Alto). In vitro and in planta bioassays against key fungal pathogens of grasses (Sclerotium rolfsii, Drechslera brizae and Microdochium nivale) indicated strong bioprotection activities. A complete circular chromosome of ∼5.2 Mb was generated for three strains of the novel Xanthomonas sp. Based on the 16S ribosomal RNA gene, the strains were closely related to the plant pathogen Xanthomonas translucens, however, comparative genomics of 22 closely related xanthomonad strains indicated that these strains were a novel species. The comparative genomics analysis also identified two unique gene clusters associated with the production of bioprotectant secondary metabolites including one associated with a novel nonribosomal peptide synthetase and another with a siderophore. The analysis also identified genes associated with an endophytic lifestyle (e.g., Type VI secretion system), while no genes associated with pathogenicity were identified (e.g., Type III secretion system and effectors). Overall, these results indicate that these strains represent a novel, bioactive, non-pathogenic species of the genus Xanthomonas. Strain GW was the designated type strain of this novel Xanthomonas sp.
Introduction
In Australia, the dairy industry has a farmgate value of $4.4 billion (2018 – 2019) and is ranked fourth for global market share (Dairy Australia, 2019). Despite its complexity in operation, the dairy industry can be summarized as the conversion of pastures grasses to milk and other dairy products. As such, the Australian dairy industry is underpinned by the performance of pasture grasses, and importantly perennial ryegrass.
The productivity of pasture grasses can be severely affected by plant pathogens. The major bacterial grass pathogen globally is Xanthomonas translucens pv. graminis, which causes bacterial wilt of pasture grasses (Egli and Schmidt, 1982), however, this pathovar is not present in Australia. According to the Victorian Plant Pathogen Herbarium (VPRI, Bundoora, Victoria, Australia), fungal grass pathogens are more common in Australia, including Pyrenophora spp., Sclerotium spp., Phoma spp., Bipolaris spp. and Microdochium nivale. The successful management of these pathogens is important for improving pasture productivity.
Biological controls (or biopesticides) are one management strategy that uses living organisms (e.g., microorganisms) to suppress deleterious or pathogenic organisms (Bulgarelli et al., 2013a). These bioprotection agents represent around 6.8% of the global pesticides market (2016) and are predicted to be worth $79.3 billion by 2022 (Chen, 2018). This growing area has seen more than 1320 bioprotection products registered in the US Environmental Protection Agency in 2014 (Mehrotra et al., 2017). For example, the fungal endophyte Epichloë spp. is a biological control that protects pasture grasses from herbivore via the production of bioactive compounds (Kauppinen et al., 2016). In addition, many bacteria have bioprotection activities, including Bacillus spp. and Pseudomonas spp. (Berg, 2009).
The plant microbiome provides an excellent reservoir where potential microbial bioprotection agents could be discovered. The diverse range of microorganisms associated with plant (plant microbiome) play a remarkable role in determining the health and productivity of the host (Berendsen et al., 2012). Therefore, substantial attention has been put on studying the bioprotection activities of these microorganisms (Bulgarelli et al., 2013b).
Next-generation sequencing technologies have led to fundamental changes to bacterial genomics by lowering cost and increasing throughput (Metzker, 2010). Recent advances in long read sequencing platforms like Oxford Nanopore Technologies (ONT) have made generating complete circular genomes for bacteria much easier (Koren et al., 2017). The availability of complete genome sequences underpins both the taxonomic identification and characterisation of novel microbial bioprotection agents, including the putative mode of action (i.e., identification of secondary metabolite gene clusters) and non-pathogenicity (i.e., absence of pathogenicity factors).
To gain insight into the broader microbiome of pasture grasses, we have profiled the microbiome of perennial ryegrass (Lolium perenne L. cv. Alto) and isolated bacterial strains (Tannenbaum et al., 2020), which were assessed for their beneficial activities (e.g., bioprotection). Three closely related strains (strain GW, seed-associated; strain SS and SI, mature-plant associated) exhibited excellent bioprotection activities against phytopathogens (in vitro and in planta). Complete genome assemblies were generated for these bacteria, and genome analysis showed that they represent a novel species of the genus Xanthomonas. Further bioinformatics analysis was conducted to determine the production of secondary metabolites that are putatively associated with bioprotection activities and to examine the presence/absence of pathogenicity related genes.
Materials and Methods
Bacterial Strain Isolation
Bacterial strains were isolated from perennial ryegrass (Lolium perenne L. cv. Alto, Barenbrug Agriseeds NZ). To isolate seed-associated bacteria, surface-sterilized seeds (3% NaOCl for 3 min, followed by 3 × sterile dH2O washes) were germinated under sterile conditions (on moistened sterile filter paper in sealed Petri dish). Germinated seedlings (5–7 days old) were harvested and sectioned into aerial and root tissue. Tissues were suspended in sterile Phosphate Buffered Saline (PBS), and ground using a Qiagen TissueLyser II (2 × 1 min at 30 Hz). Plant macerates were serial diluted (1:10, 100 μL in 900 μL), and plated onto Reasoners 2 Agar (R2A, Oxoid or Amyl Media, Australia) to isolate pure separated colonies. To isolate mature plant-associated bacteria, plants were grown in pots in a glasshouse for at least 60 days with standard potting mix and harvested for leaf and root tissues. Root tissues were washed in PBS to remove soil particles and then sonicated for 10 min to remove soil particulates and the rhizosphere. Tissue maceration, serial dilutions and bacterial isolations were prepared as above. All isolated bacterial strains were taxonomically classified using matrix assisted laser desorption ionization time-of-flight mass spectrometry (Bruker ultrafleXtreme MALDI-TOF/TOF MS and Biotyper System) (Tannenbaum et al., 2020), and stored in nutrient broth with 15% glycerol (v/v) at −80°C.
Bioprotection Assay (in vitro)
An assay was designed to assess the in vitro bioprotection activity of bacterial strains against fungal phytopathogens of Poaceae species. The bacterial strains assessed included three xanthomonads (GW, SS, SI) and one Paenibacillus sp. (BU). Six fungal phytopathogens of Poaceae species (Supplementary Table S1) were obtained from the Victorian Plant Pathogen Herbarium (VPRI, Bundoora, VIC, Australia). Each bacterial strain was cultured in Nutrient Broth (BD Bioscience) overnight (OD = 1.0) and drop-inoculated (20 μL) onto four equidistant points on a Nutrient Agar (BD Bioscience) plate, which was then incubated overnight at 28°C. Then, a 6 × 6 mm plug of the phytopathogen (actively growing hyphae) was placed at the center of the plate and incubated at 28°C in dark. The incubation time varied to accommodate the differences in growth rate of the fungal pathogens (Table 1). The diameter of the fungal colony on the plate was measured twice. One reading was taken from the straight line that was defined by two inoculation points and the center of the plate, and the other reading was taken after rotating the plate for 45 degrees. The average of the two readings was used for statistical analysis. For each treatment, three plates were prepared as biological replicates. For the blank control, sterile Nutrient Broth was used to replace the bacteria. Statistical analysis (One-way ANOVA and Tukey Test) was conducted using OriginPro 2018 (Version b9.5.1.195) for any significant difference (P < 0.05) between treatments.
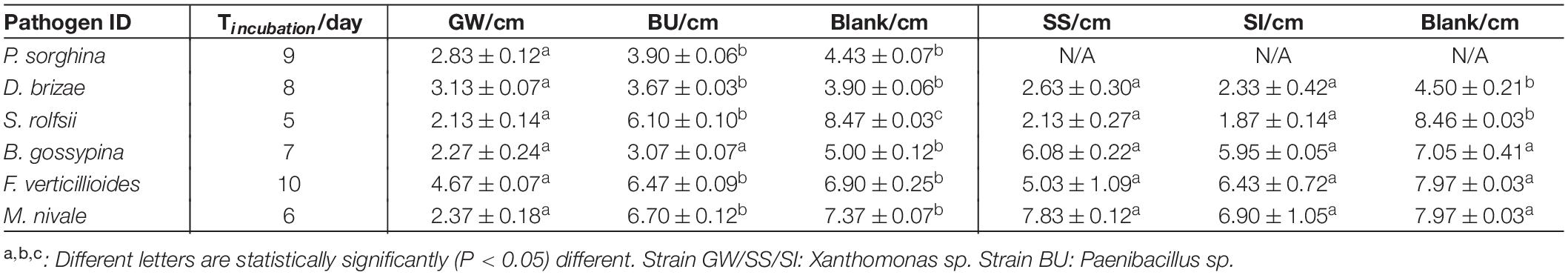
Table 1. The average colony diameter (±standard error) of fungal pathogens when exposed to the three xanthomonads in a bioprotection assay (in vitro).
Bioprotection Assay (in planta)
An assay was designed to assess the in planta bioprotection activity of the bacterial strains against the fungal phytopathogen Bipolaris sorokiniana (VPRI 42684). The xanthomonad strain GW was used in this assay. Wheat seeds were surface-sterilized as per section 2.1. The bacterial strain was cultured in Nutrient Broth (BD Bioscience) for 6 h (OD = 0.5). Sterile seeds were imbibed in the bacterial culture for 18 h, removed from the culture, dried under sterile conditions and then germinated in dark at room temperature (23°C) for 4 days for root and shoot development. Germinated seedlings were transferred into pots with standard potting mix (4 seeds per pot, 4 pots per treatment) in a glasshouse (Supplementary Table S2) for 39 days. A 7 cm segment of the lowest leaf that was green and fully extended from each plant was excised and placed on 0.5% water agar. A sterile sharp needle was used to create a wound at the center of each leaf, to which 1 μL of B. sorokiniana spore suspension (8.5 × 103 spores/mL) was added. Plates were then sealed and left at room temperature (23°C) for 3 days. To assess the bioprotection activity, the size (measured in mm2) of the lesion, chlorotic zones and fungal hyphal growth was recorded. For the blank control, sterile Nutrient Broth was used. Statistical analysis (One-way ANOVA and Tukey Test) was conducted using OriginPro 2018 (Version b9.5.1.195) for any significant difference (P < 0.05) between treatments.
Genome Sequencing
DNA was extracted from bacterial pellets of GW, SS and SI (overnight cultures) using a Wizard® Genomic DNA Purification Kit (A1120, Promega, Madison, WI, United States), and assessed for quality (average molecular weight ≥ 30 Kb) on an Agilent 2200 TapeStation (Agilent Technologies, Santa Clara, CA, United States).
Genomic sequencing libraries (short reads) were prepared from the DNA using the Illumina Nextera XT DNA library preparation kit (Cat# FC-131-1096) and sequenced on an Illumina HiSeq 3000 platform. Genomic sequence data (raw reads) were assessed for quality and filtered to remove any adapter and index sequence, and low-quality bases using fastp (Chen et al., 2018) with the following parameters: -w 8 -3 -5.
Genomic sequencing libraries (long reads) were prepared from the DNA using the Oxford Nanopore Technologies (ONT) transposases-based library preparation kit with minor modifications (SQK-RAD004, ONT, Oxford, United Kingdom) and sequenced on a MinION Mk1B platform (MIN-101B) with R9.4 flow cells (FLO-MIN106). Genomic sequence data (raw read signals) were basecalled using ONT’s Albacore software (Version 2.3.4), and assessed for quality using NanoPlot (De Coster et al., 2018). Basecalled data was filtered to remove adapter sequences using Porechop (Version 0.2.31), while reads shorter than 300 bp and the worst 5% of reads (based on quality) were discarded using Filtlong (Version 0.2.02).
Genome Assembly, Classification and Alignment
The whole genome of GW, SS, and SI were assembled with filtered long and short reads using Unicycler (Wick et al., 2017). Long reads were used for primary assembly and to resolve repeat regions in the genome, whereas short reads were used to correct small base-level errors. Assembly graphs were visualized using Bandage (Wick et al., 2015). Assembled genomes were taxonomically classified by Kraken2 (Wood and Salzberg, 2014) using a custom database containing all completed bacterial reference genomes in NCBI (20/03/2020). Genomes of GW, SS, and SI were aligned using LASTZ (Version 1.04.003), and visualized using AliTV (Ankenbrand et al., 2017).
Genome Annotation and Characterisation
The assembled genome of GW, SS and SI were annotated using Prokka (Seemann, 2014) with a custom Xanthomonas protein database (based on Kraken2 classification) to predict genes and corresponding functions. A further functional characterisation of annotated genomes was conducted using KEGG BlastKOALA (Kanehisa et al., 2016). Identification of secondary metabolite gene clusters from annotated genomes was conducted using antiSMASH (Weber et al., 2015) with the following options: –clusterblast –asf –knownclusterblast –subclusterblast –smcogs –full-hmmer. An evaluation of the presence of pathogenicity-related genes from the annotated genomes of all three strains (GW, SS, and SI) was conducted using BLAST (Camacho et al., 2009) (blastp and tblastn, e-value > 1e–10). Initially, pathogenicity-related genes previously reported in Xanthomonas spp. were targeted (133 genes), including secretion systems (Type I/II/III/VI), pili (Type IV), flagella, pathogenicity regulatory factors, xanthan biosynthesis and lipopolysaccharide biosynthesis. A further comparison of 36 genes involved in Type III secretion systems (T3SS) from six pathogenic strains, including X. translucens pv. translucens DSM18974, X. translucens pv. undulosa Xtu4699, X. translucens pv. cerealis CFBP2541, X. translucens DAR61454, X. translucens pv. graminis Xtg29 and X. translucens pv. graminis ICMP6431, and the three strains was conducted, including structural and regulatory genes, as well as conserved and variable Type III effectors (T3Es). Transcription activator-like effectors (TALEs) were predicted from the three strains (GW, SS and SI) and three pathogenic strains using annoTALE (Grau et al., 2016). Since TALE genes usually have multiple near-perfect repeats in the sequence and multiple copies of sequences in the genome (White et al., 2009), short reads often struggle to properly assemble the TALEs regions (Peng et al., 2016). Therefore, only pathogenic strains whose genome was completely assembled, i.e., X. translucens pv. translucens DSM18974, X. translucens pv. undulosa Xtu4699 and X. translucens pv. cerealis CFBP2541, were used in the prediction of TALE genes. The genome of strain GW and X. translucens pv. undulosa Xtu4699 were aligned using BLAST (Camacho et al., 2009). The alignment as well as the T3SS, T3Es and TALE genes that were detected on the genome of X. translucens pv. undulosa Xtu4699 were visualized using BRIG (Alikhan et al., 2011).
Phylogeny and Comparative Genomics
Eighteen Group 1 Xanthomonas spp. genomes and one X. campestris genome (Group 2 Xanthomonas) that were publicly available on NCBI (Supplementary Table S3) were downloaded and used for phylogenetic analysis (Young et al., 2008). These genomes were annotated de novo using the method above. Genes that were shared by all strains were identified using Roary and aligned (codon aware) using PRANK (Löytynoja, 2014). A maximum-likelihood phylogenetic tree was inferred using FastTree (Price et al., 2010) with Jukes-Cantor Joins distances, the Generalized Time-Reversible substitution model and the CAT approximation model. Local branch support values were calculated using 1000 resamples with the Shimodaira–Hasegawa test.
Results
Bioprotection Assay (in vitro)
Xanthomonas sp. strain GW significantly (P < 0.05) reduced the average colony diameter of all six fungal pathogens compared to the blank control, and four pathogens compared to Paenibacillus sp. strain BU (Table 1). Strain GW reduced the growth of S. rolfsii, M. nivae, D. brizae, P. sorghina, F. verticillioides and B. gossypina by 74.9, 67.8, 54.6, 36.1, 32.3, and 19.7%, respectively, compared to the blank control. Strain SI reduced the growth of S. rolfsii and D. brizae by 77.9 and 48.2%, respectively, and strain SS reduced the growth of S. rolfsii and D. brizae by 74.8 and 41.6%, respectively, when compared to the blank control. When comparing across the three xanthomonads, only strain GW significantly inhibited the growth of all pathogens, indicating its broad-spectrum bioprotection activity (Supplementary Figures S1, S2).
Bioprotection Assay (in planta)
Xanthomonas sp. strain GW significantly (P < 0.05) reduced the average size of lesion and fungal hyphal growth compared to the blank control (Figure 1 and Table 2). The lesion size was reduced by 96.7%, and the area of fungal hyphal growth was reduced by 94.7%.

Figure 1. Representative images of the in planta bioprotection assay for the blank control group (left) and the treatment group (inoculated with strain GW, right), with white arrows representing the point of inoculation of the pathogen B. sorokiniana (VPRI 42684) in wheat. Extensive leaf discoloration and white fungal hyphal growth are seen away from the point of inoculation in the blank control leaves, but not in the GW inoculated leaves.

Table 2. The average size of area showing disease symptoms (±standard error) of B. sorokiniana when exposed to strain GW in a bioprotection assay (in planta in wheat).
Genome Sequencing, Assembly and Annotation
A total of 9,674,929,775 bp short reads and 761,078,031 bp long reads were generated (Supplementary Table S4). Complete circular genome sequences were produced for all three strains. The genome size for strain GW, SS and SI were 5,233,349 bp (4358 CDSs), 5,185,085 bp (4227 CDSs) and 5,246,417 bp (4290 CDSs), respectively, (Table 3). The percent GC content ranged from 68.37% to 68.55%. There were no plasmids present in any strain.
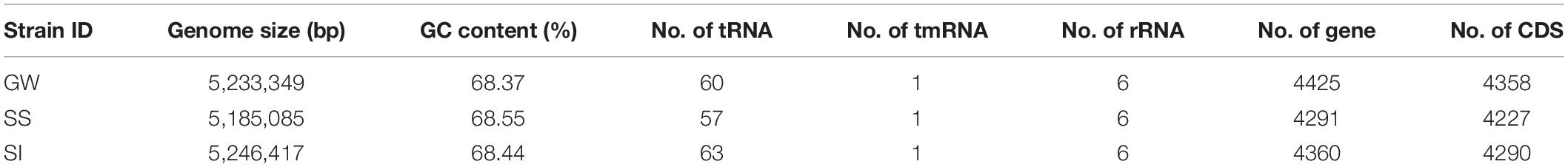
Table 3. General genomic characteristics of the three Xanthomonas strains.
Phylogeny and Comparative Genomics
The three Xanthomonas strains (GW, SS, and SI) were phylogenetically related to Xanthomonas translucens (strain XT2, Genbank Accession: NR_036968.1) with a sequence coverage of 100% and homology of 99.53 – 99.73% based on the 16S ribosomal RNA gene. The genomes of the three xanthomonads were also classified as X. translucens pv. cerealis (NCBI:txid 152263) by Kraken2, suggesting their close relationship with X. translucens.
A comparative genomics analysis indicated that the three Xanthomonas strains (GW, SS, SI) belonged to the Group 1 Xanthomonas based on a sequence homology comparison of 68 genes shared by all 22 strains (Figure 2). The topology of the tree was consistent with Young et al. (2008), with unique clades/branches apparent for X. albilineans, X. sacchari, X. theicola, X. hyacinthi and X. translucens, with the three Xanthomonas strains (GW, SS, SI) between X. hyacinthi and X. translucens. The tree showed the three Xanthomonas strains (GW, SS, SI) formed a unique clade adjacent to X. translucens pathovars and were separated with a strong local support value (100%). The X. translucens clade were divided into a subclade consisting of X. translucens pv. translucens DSM18974, X. translucens pv. undulosa Xtu4699, X. translucens pv. undulosa ICMP11055 and X. translucens DAR61454 (Figure 2, yellow, barley and wheat pathogens) and a subclade consisting of X. translucens pv. arrhenatheri LMG727, X. translucens pv. poae LMG728, X. translucens pv. phlei LMG730 and all X. translucens pv. graminis strains (Figure 2, blue, pasture grass pathogens).
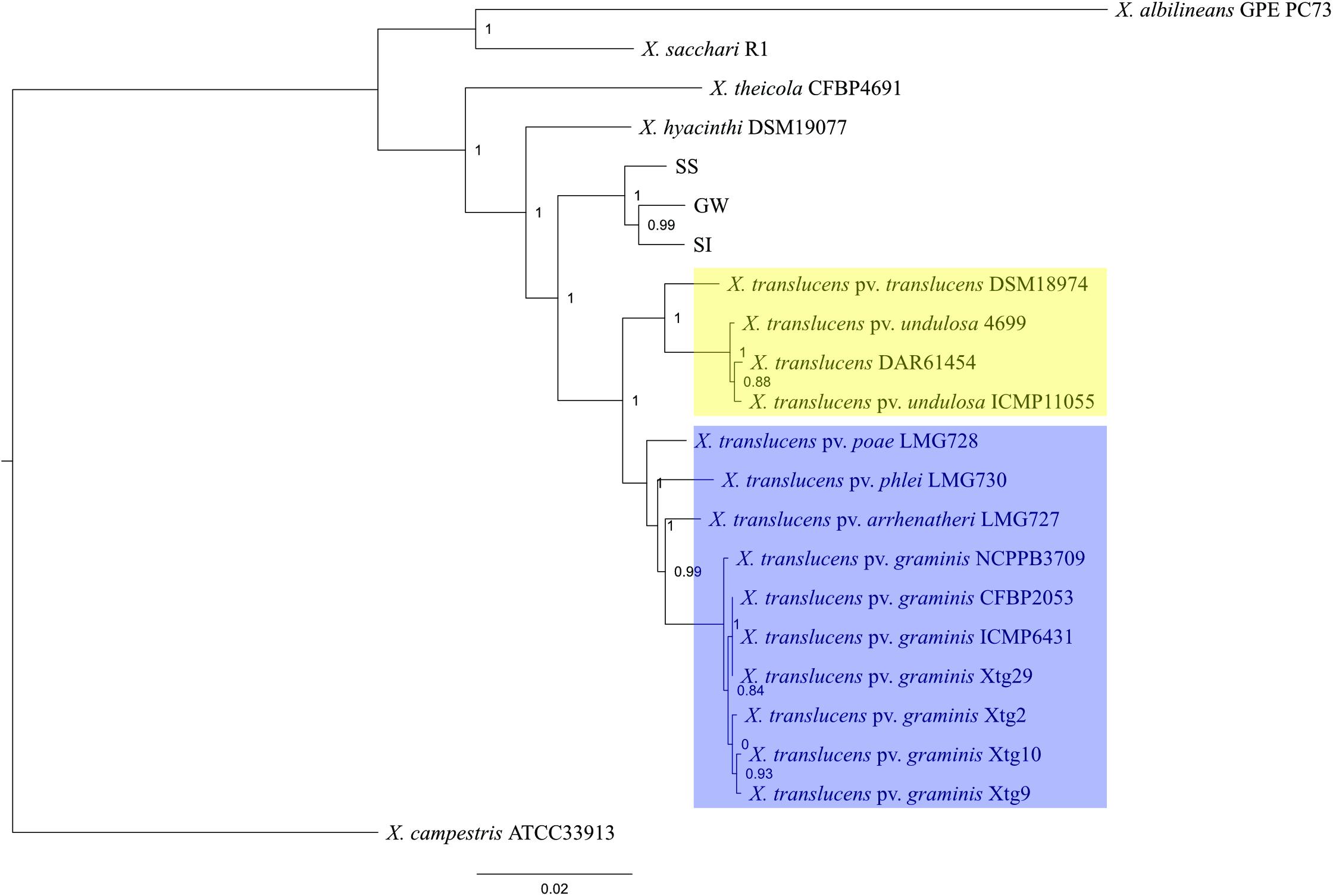
Figure 2. Phylogeny of Group 1 Xanthomonas species and strain GW, SS and SI. This maximum-likelihood tree was inferred based on 68 genes conserved among 22 genomes. Values shown next to branches were the local support values calculated using 1000 resamples with the Shimodaira–Hasegawa test. Strain GW, SS and SI formed a clade that was well separated from X. translucens pathovars that are pathogenic on crop species (yellow) and grass species (blue).
Average nucleotide identity (ANI) was calculated to further elucidate the relationship between the three Xanthomonas strains (GW, SS and SI) and X. translucens pathovars (Supplementary Table S5). The results showed 97.20 – 97.39% similarities between the three xanthomonads, and 92.97 – 94.07% similarities between the three xanthomonads and X. translucens pathovars.
Pathogenicity-Related Gene Analysis
The genomes of the three Xanthomonas strains (GW, SS and SI) were found to have a reduced complement of pathogenicity-related genes. The assessment of 133 pathogenicity-related genes identified that the three Xanthomonas strains (GW, SS and SI) was devoid of the T3SS that is critical for pathogenicity of most Xanthomonas species (White et al., 2009; Wichmann et al., 2013) (Supplementary Table S6). A comprehensive assessment of the T3SS structural and regulatory genes and T3Es across the three Xanthomonas strains (GW, SS and SI) and six pathogenic X. translucens strains identified that the three strains had 0 of 37 T3SS genes and T3Es (Table 4; Figure 3). This included an absence of the hrc genes, which encode the injectisome (Wagner et al., 2018), and the hrp genes, which are essential to suppress host plant defense responses for Xanthomonas species (Kay and Bonas, 2009). The hrpF gene, which encodes a translocon protein complex that is required to deliver T3Es (Chatterjee et al., 2013), was missing in all nine strains, which was supported by previous research (Pesce et al., 2017). However, the hpaT gene, which was described to encode an undescribed translocon protein complex of X. translucens strains (Pesce et al., 2017), was detected in all pathogenic strains but not in the three Xanthomonas strains (GW, SS and SI).
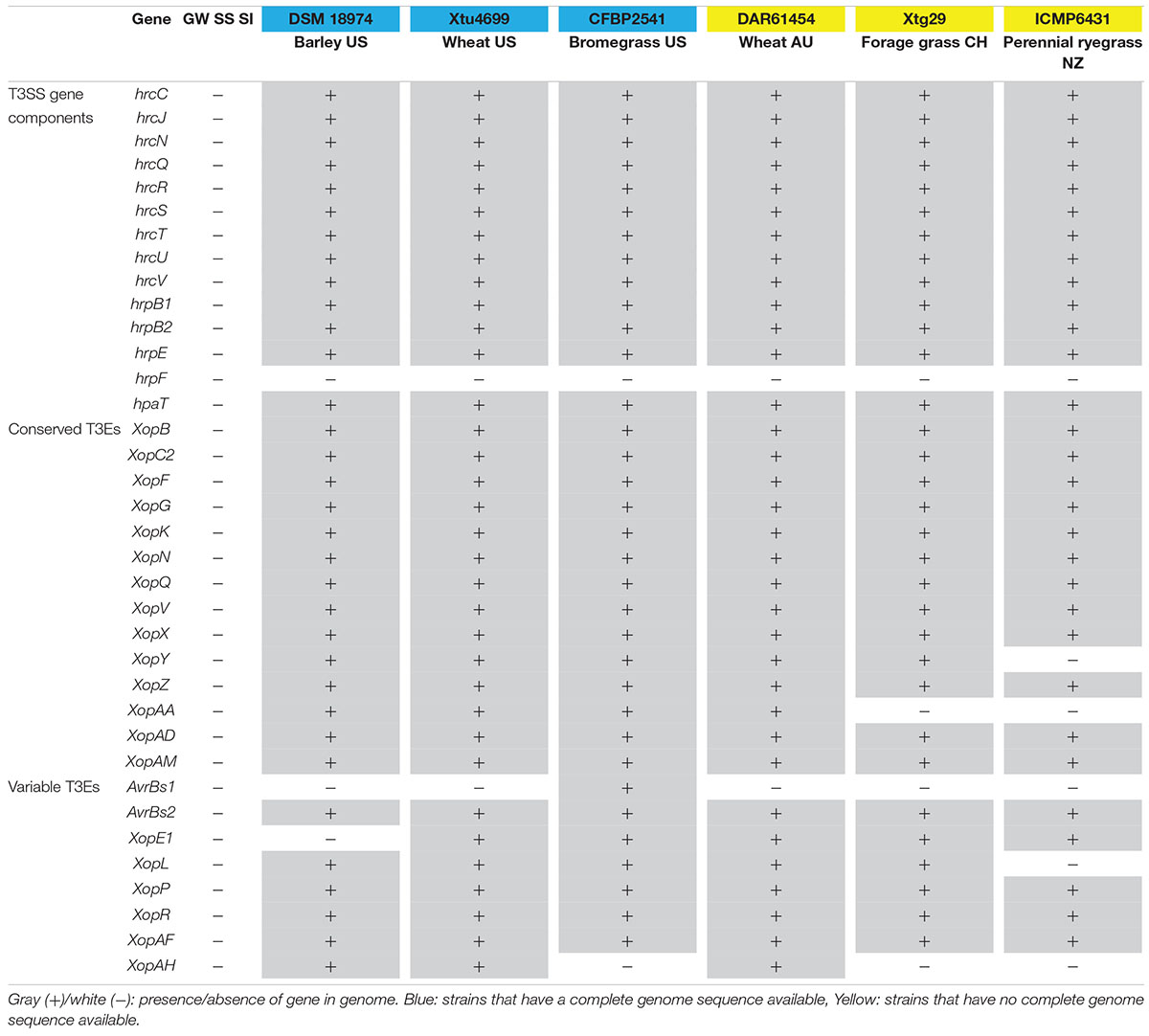
Table 4. T3SS and T3Es genes in the genome of the three Xanthomonas strains (GW, SS and SI) and other X. translucens strains.
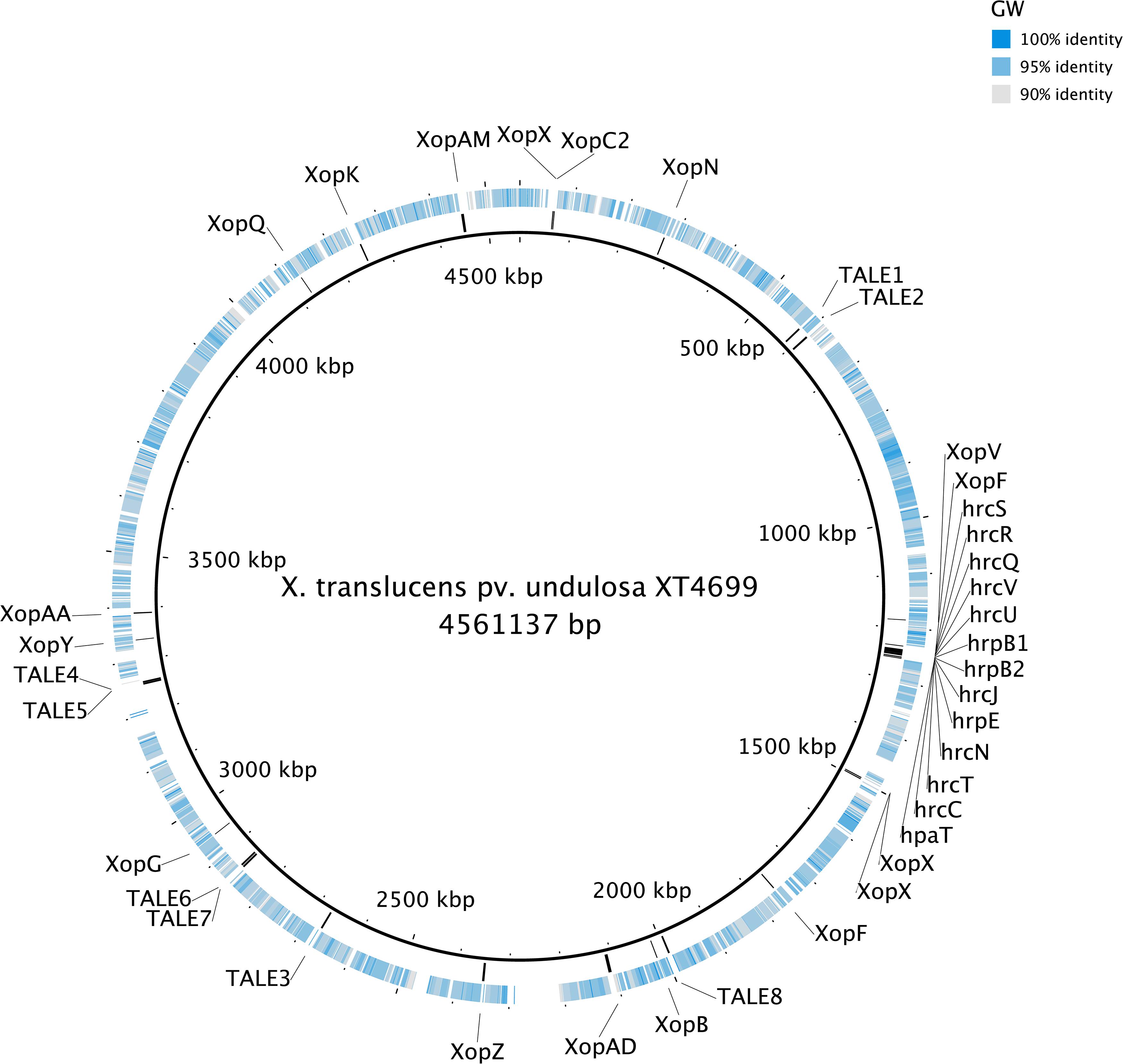
Figure 3. The genome alignment of the strain GW (the outer circle) and X. translucens pv. undulosa Xtu4699 (the inner circle, black). The color of the outer circle represented the sequence identity (gray to blue: 90–100%; white blocks: <90%) The locations of T3SS, T3Es and TALEs genes detected in the genome of X. translucens pv. undulosa Xtu4699 are also displayed.
Similar to the T3SS and T3Es genes, no TALE genes could be identified in the genome of the three Xanthomonas strains (GW, SS and SI). Eight TALE genes were predicted for strain X. translucens pv. undulosa Xtu4699 (Figure 3) and X. translucens pv. translucens DSM18974, and two TALE genes were predicted for strain X. translucens pv. cerealis CFBP2541.
Secondary Metabolite
The in vitro and in planta bioprotection activity of the three Xanthomonas strains (GW, SS and SI) indicated that they could produce biocidal secondary metabolites. Furthermore, it has been demonstrated that both live culture and cell-free extracts of strain GW have biocidal activity against fungal phytopathogens (unpublished data). Secondary metabolite gene analysis identified three clusters (Clusters 1 – 3), with strain GW having all three clusters, and strain SI and SS having two of the three clusters. These clusters contain all the genes (core/additional biosynthetic genes, regulatory genes, transport-related genes and other genes) required for complete function (Figures 4A–C).
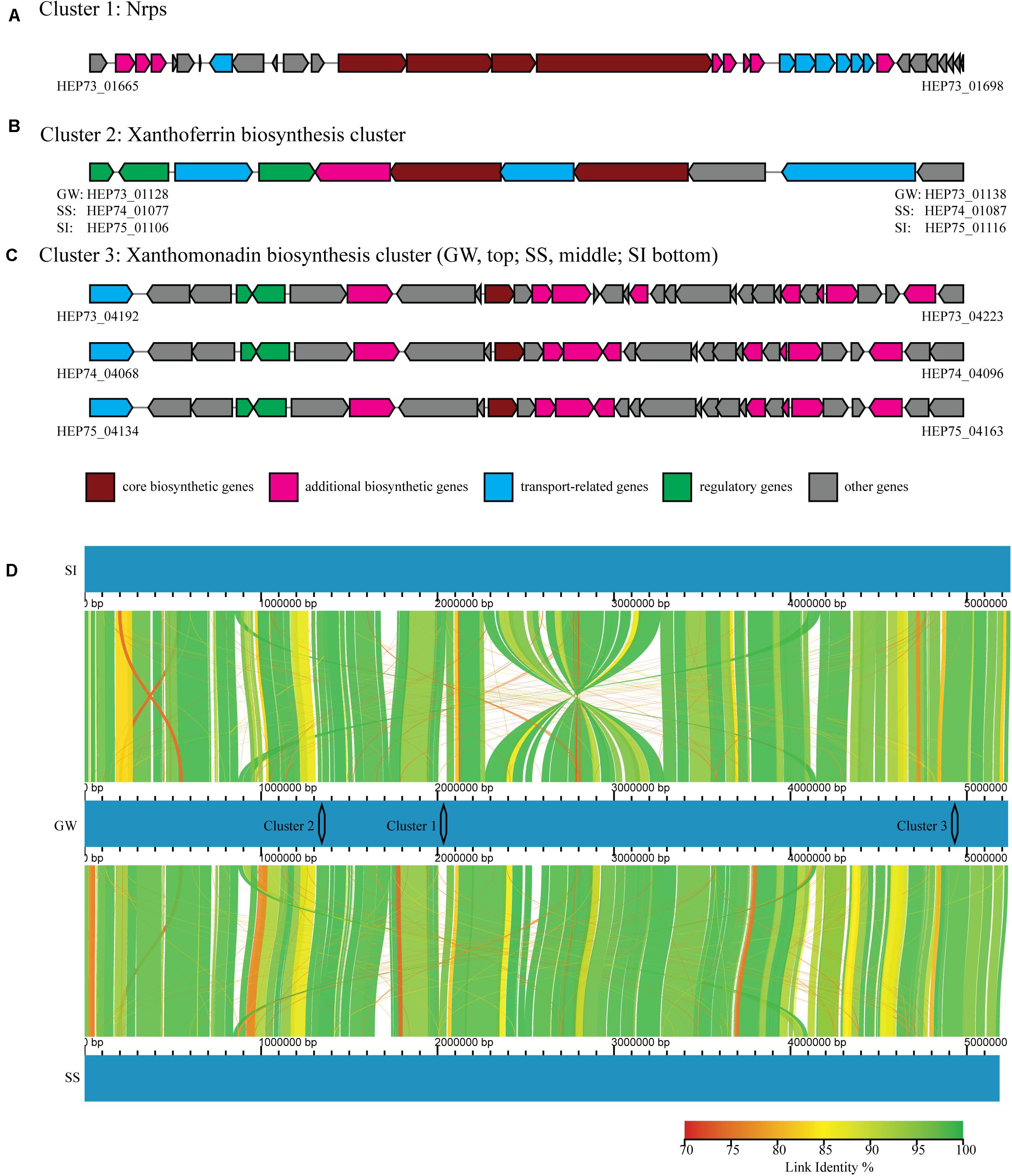
Figure 4. Three secondary metabolite gene clusters (A–C) identified by antiSMASH, including core biosynthetic genes (maroon), additional biosynthetic genes (pink), regulatory genes (green), transport-related genes (blue) and other genes (gray). The NCBI identifiers were shown for genes located at both ends of each cluster. (D) Whole genome comparison of strain GW (middle), SI (top) and SS (bottom), with color graduation representing nucleotide percentage similarity between genomes (from 70 to 100%, red to green). The locations of the three clusters were also represented.
Cluster 1 contained a nonribosomal peptide synthetase (Nrps), and the entire cluster was unique to strain GW. Cluster 1 was located between bases 1,997,794 and 2,067,075 in the genome of strain GW, while this region was absent from strain SS and SI (Figure 4D). Cluster 1 appears novel based on sequence homology searching against the antiSMASH gene clusters database. Cluster 2 contained a siderophore synthetase and the entire cluster was present in all three strains. Cluster 2 was located between bases 1,300,000 and 1,380,000 in the genomes of strain GW, SS and SI (Figure 4D). Cluster 2 has sequence homology to the xanthoferrin biosynthesis gene cluster. Cluster 3 contained an aryl polyene synthase and the entire cluster was present in all three strains, however, slight variations in the cluster structure were observed (Figure 4C). Cluster 3 was located between bases 4,860,000 and 4,980,000 in the genomes of strain GW, SS and SI (Figure 4D). Cluster 3 has sequence homology to the xanthomonadin biosynthesis gene cluster.
Discussion
Plant microbiomes are a repository from which plant beneficial bacteria can be isolated and identified. In this study, we compared three related Xanthomonas strains from the L. perenne microbiome. These had differing in vitro bioprotection activities, and the strain with the strongest activities against a wide range of phytopathogens (GW) became the focus of this study. Based on the complete genome assembly, strain GW possesses a novel Nrps cluster compared to the other two strains. All three strains lack many of the genes that are essential for pathogenicity in pathogenic Xanthomonas strains. Such characteristics made strain GW a promising candidate to be developed as a bioprotection agent for crops and grasses.
Identification of a Novel Xanthomonas Species
Taxonomic identification of bacterial species often uses 16S ribosomal RNA, whole genome sequence homology and ANI, with each technique providing varying degrees of taxonomic resolution. Taxonomic assignment based on the 16S ribosomal RNA gene provides genus level resolution, whereas whole genome techniques provide species or sub-species resolution. In this study, the initial classification based on 16S ribosomal RNA and whole genome sequence against the NCBI RefSeq database suggested that strain GW, SS and SI were most likely representatives of the plant pathogenic X. translucens. However, comparative genome analysis demonstrated the three strains formed a cluster that was separated from other X. translucens pathovars. Most importantly, the ANI between these three strains and X. translucens pathovars was lower than the species boundary, which is 95 – 96% ANI (Richter and Rosselló-Móra, 2009; Chun et al., 2018). Therefore, the three xanthomonads used in this study represent a novel species of the genus Xanthomonas. This clearly demonstrated the limitations of 16S ribosomal RNA-based classification (Klenk and Goker, 2010). Due to the technical limitations of the short-read sequencing platforms, most microbiome studies only used a variable region of the 16S ribosomal RNA (Pollock et al., 2018). It was likely that such novel, bioactive Xanthomonas species were present in the samples but were overlooked since they were classified as the known pathogenic Xanthomonas species. Moreover, this study also emphasized the importance of available whole genome sequences. The hybrid assembly approach used here combined the advantages of both short reads and long reads to produce high quality genome sequences for all three strains, which underpinned the downstream analysis including taxonomic identification and functional characterisation of the genomic resources.
Absence of Pathogenicity-Related Genes in the Three Xanthomonads
An analysis of pathogenicity-related genes clarified that the three xanthomonads were highly likely non-pathogenic. The T3SS and T3SS-related effector proteins (T3Es and TALEs) were completely absent from the genome of the three xanthomonads. The T3SS is a needle and syringe-like system that delivers (i) T3Es that suppress plant innate immunity and modulate plant cellular pathways to enhance bacterial infection (Büttner, 2016), and (ii) TALEs that induce host susceptibility genes to enhance virulence (Cernadas et al., 2014; Hu et al., 2014), both of which are important for pathogenicity in Xanthomonas species (Green and Mecsas, 2016). For instance, deletion mutations of the T3SS structural genes hrpE or hrcR showed significantly reduced symptoms of X. translucens pv. graminis Xtg29 when compared with the wildtype strain (Wichmann et al., 2013). Furthermore, complete loss of symptoms was observed for a X. translucens pv. undulosa Xtu4699 strain with an insertion mutation in the T3SS structure gene hrcC (Peng et al., 2016). Complete absence of the T3SS and T3Es has been reported in other Xanthomonas species, such as X. arboricola strains (Group 2 Xanthomonas) which were referred to as non-pathogenic (Garita-Cambronero et al., 2017).
It must be stated that some xanthomonads that lacked the Hrp T3SS were found to be associated with diseased plants including X. cannabis NCPPB3735 and X. cannabis NCPPB2877 strains (Group 2 Xanthomonas) that could cause symptoms on hemp, barley and tobacco (Jacobs et al., 2015). While they lacked the Hrp T3SS, they had HrpG and HrpX, which are two key Hrp pathogenicity regulator genes (Büttner and Bonas, 2010), that were absent from the genome of the three xanthomonads. Moreover, there is another pathogenic strain of the same species, X. cannabis pv. phaseoli (Nyagatare strain), that has been reported to have both regulator genes, the full Hrp T3SS and T3Es (Aritua et al., 2015). In Group 1 Xanthomonas, X. albilineans GPE PC73, which is a xylem-limited pathogen, also lacked the Hrp T3SS (Pieretti et al., 2009). However, this strain had a Salmonella pathogenicity island-1 (SPI-1) containing an alternate T3SS and a gene cluster that encodes the phytotoxic albicidin, neither of which was detected in the three xanthomonads used in this study. X. sacchari, which is also a Group 1 Xanthomonas, lacked the Hrp T3SS and the SPI-1 T3SS (Studholme et al., 2011). However, the strain was isolated from an insect from a diseased banana plant and there was no evidence of plant pathogenicity, which could be explained by the missing T3SS. T3SS has been proven to be essential for pathogenicity for X. translucens (Wichmann et al., 2013), which has the closest phylogenetic relationship amoug all known Xanthonomas species to the three xanthomonads in this study. Therefore, without any known type of T3SS, T3Es and TALEs the three xanthomonads are highly likely non-pathogenic, and no symptoms have been seen in inoculated wheat, barley and ryegrass plants. Furthermore, given the fact that these genes are widely distributed across the whole chromosome (Figure 3), they are highly unlikely to acquire all the genes necessary to become pathogenic through horizontal gene transfer.
The three xanthomonads contained gene clusters (T1SS, T2SS, T6SS, type 4 pilus, flagella) linked to pathogenicity of X. translucens pathovars (Supplementary Table S6), however, these clusters have also been reported to possess functions associated with an endophytic lifestyle. For example, the T1SS was associated with biofilm formation (Tseng et al., 2009), the T2SS was used to secrete enzymes that facilitate environmental adaptation (Green and Mecsas, 2016), and the T6SS was involved in communication between bacteria or bacteria and the symbiotic host plant (Boyer et al., 2009). The three xanthomonads also had a type IV pilus cluster and a flagellar gene cluster that are associated with adherence and motility (Dunger et al., 2016; Hersemann et al., 2017). The presence of these gene clusters is supportive of the endophytic lifestyle proposed for the three xanthomonads.
Bioprotection Activity and Putative Mode of Action
Biological controls agents (e.g., bioprotectant bacteria) have been widely adopted globally for managing plant diseases as they are an effective and environmentally sustainable alternative to agrochemicals (Glare et al., 2012). For instance, biological control agents offer unique, complex modes of action, whereas agrichemicals have specific mode of action that can more easily lead to the development of resistance (Grimmer et al., 2015). Furthermore, there is less regulatory burden associated with biological control agents, in contrast to some agrichemicals that are under increased regulatory scrutiny as they have increasing environmental and public health concerns (Bach et al., 2016; Droby et al., 2016). Many Bacillus- and Pseudomonas- based biological control products have been commercialized globally for controlling bacterial and fungal phytopathogens (e.g., Bacillus subtilis for controlling Fusarium spp., and Pseudomonas fluorescens for controlling Erwinia amylovora) (Berg, 2009). These types of bacteria protect plants from phytopathogens directly via microbial antagonism, either endophytically (within the plant) or on the rhizosphere and phyllosphere (on the plant surface) (Eljounaidi et al., 2016; O’brien, 2017). Such antagonism can be carried out by competing for nutrients and spaces for colonization on the plant surface (Kamilova et al., 2005), and/or synthesizing allelochemicals such as antibiotics and siderophores to suppress pathogens (Beneduzi et al., 2012).
Bioprotection activity against fungi has not been reported to be associated with xanthomonads. Xanthomonas spp. are commonly associated with plants as either endophytes (Bouffaud et al., 2014; Bulgarelli et al., 2015; Zarraonaindia et al., 2015; Mitter et al., 2017) or as phytopathogens (An et al., 2019). In this study, we isolated three xanthomonads that had bioprotection activity, providing inhibitory activity against fungal pathogens from a broad taxonomic range (2 Phyla, 5 Families) in in vitro and in planta assays. The activity observed in the assays was a reduction in growth of the pathogen, and while no complete control was observed the xanthomonad strains restricted growth up to 77.9% of some pathogens. Strain GW also provided prolonged protection (up to 39 days) against the pathogen in the in planta assay. This suggests two methods of plant protection including (1) localized microbial colonization of a plant tissue from which antibiotic compounds are produced that are translocated systemically throughout the plant, or (2) systemic microbial colonization of the plant from which the bacteria either competes for nutrients or produces antibiotic compounds. Method 1 is utilized by Epichloë spp. endophytes to protect Poaceae species against pests and pathogens (Johnson et al., 2013), whereas method 2 is utilized by Erwinia and Pantoea species (Born et al., 2016; Smits et al., 2019). Given the in vitro bioprotection assay indicated production of a bioactive suppressant, we propose that the in planta activity is analogous. Further experiments including in planta assays and glasshouse and field assays have been planned to explore the potential bioactivity of strain GW.
Xanthomonas spp. have been shown to produce an array of bioactive secondary metabolites including the siderophore xanthoferrin, the pigment xanthomonadin, and the polysaccharide xanthan gum (Poplawsky et al., 2000; He et al., 2011; Palaniraj and Jayaraman, 2011; Huang et al., 2015; Pandey et al., 2017; Madden et al., 2019). A genomics-based assessment identified two secondary metabolite gene clusters that could be linked to the bioactivity of strain GW, SS and SI. A xanthoferrin siderophore synthesis cluster was detected in all three strains. First described in X. campestris pv. campestris, xanthoferrin is a vibrioferrin-type siderophore which facilitate iron uptake of bacteria by binding ferric iron from the environment (Andrews et al., 2003). Bacterial siderophores have higher affinity to iron compared to fungal siderophores (Compant et al., 2005), and therefore they can act as bioprotection agents under iron-limiting environments by depriving fungi of this essential element. This has been observed in fluorescent pseudomonads against the fungal pathogen Fusarium oxysporum (Kloepper et al., 1980; Dwivedi and Johri, 2003). Therefore, xanthoferrin could be responsible for the in vitro bioprotection activity that was observed from the three xanthomonads. Given that siderophores are predominantly produced locally (Saha et al., 2016), the mode of action of such bioprotection activity could be explained by method 2 described above. Moreover, strain GW showed a stronger and broader spectrum bioprotection activity against phytopathogens compared to strain SS and SI. Given that a novel Nrps cluster that was unique to strain GW but was missing from strain SS and SI, we proposed a hypothesis that the product of this novel Nrps cluster was responsible for the broad-spectrum of bioprotection activity of strain GW. Further research is needed to prove this hypothesis, including creating mutants of the Nrps cluster and evaluate the bioprotection activity (in vitro), along with identifying, purifying and characterizing the active compound(s). The mode of action of the bioprotection activity that provide by this Nrps could be either method described above.
Data Availability Statement
Annotated genome sequences of all strains were deposited in the NCBI GenBank with the accession numbers: CP051189 for GW, CP051190 for SS, and CP051261 for SI.
Author Contributions
TS conceptualized the study. TL prepared the manuscript. TL, TS, and RM designed the experiment. TL, JK, and DA contributed to the laboratory work. RM, TS, DA, and GS reviewed and edited the manuscript. TS and RM supervised the study. GS contributed to the funding acquisition. All authors have read and agreed to the submitted version of the manuscript.
Funding
This research was supported by the Agriculture Victoria, Dairy Australia, and Gardiner Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
TL received La Trobe University Full-Fee Research Scholarship, La Trobe University Postgraduate Research Scholarship and DairyBio Scholarship. The authors wish to thank Dr. Jacqueline Edwards for access to the Victorian Plant Pathogen Herbarium.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.01991/full#supplementary-material
FIGURE S1 | Representative images of the in vitro bioprotection assay when challenging strain GW and BU with Microdochium nivale.
FIGURE S2 | Representative images of the in vitro bioprotection assay when challenging strain SS and SI with Microdochium nivale.
TABLE S1 | Pathogens used in the in vitro bioprotection assay.
TABLE S2 | Programmed conditions of the glasshouse used in this study.
TABLE S3 | Xanthomonas spp. genomes used in phylogeny and comparative genomics.
TABLE S4 | Summary of reads available for genome assembly.
TABLE S5 | The average nucleotide identity (ANI) between Xanthomonas spp. genomes used in comparative genomics.
TABLE S6 | Pathogenicity-related gene clusters identified in Xanthomonas spp.
Footnotes
- ^ https://github.com/rrwick/Porechop
- ^ https://github.com/rrwick/Filtlong
- ^ http://www.bx.psu.edu/~rsharris/lastz/
References
Alikhan, N.-F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
An, S. Q., Potnis, N., Dow, M., Vorhölter, F. J., He, Y. Q., Becker, A., et al. (2019). Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas. FEMS Microbiol. Rev. 44, 1–32.
Andrews, S. C., Robinson, A. K., and Rodríguez-Quiñones, F. (2003). Bacterial iron homeostasis. FEMS Microbiol. Rev. 27, 215–237. doi: 10.1016/s0168-6445(03)00055-x
Ankenbrand, M. J., Hohlfeld, S., Hackl, T., and Förster, F. (2017). AliTV—interactive visualization of whole genome comparisons. PeerJ Comput. Sci. 3:e116. doi: 10.7717/peerj-cs.116
Aritua, V., Musoni, A., Kabeja, A., Butare, L., Mukamuhirwa, F., Gahakwa, D., et al. (2015). The draft genome sequence of Xanthomonas species strain Nyagatare, isolated from diseased bean in Rwanda. FEMS Microbiol. Rev. 362, 1–4.
Bach, E., Seger, G. D. D. S., Fernandes, G. D. C., Lisboa, B. B., and Passaglia, L. M. P. (2016). Evaluation of biological control and rhizosphere competence of plant growth promoting bacteria. Appl. Soil Ecol. 99, 141–149. doi: 10.1016/j.apsoil.2015.11.002
Beneduzi, A., Ambrosini, A., and Passaglia, L. M. (2012). Plant growth-promoting rhizobacteria (PGPR): their potential as antagonists and biocontrol agents. Genet. Mol. Biol. 35, 1044–1051. doi: 10.1590/s1415-47572012000600020
Berendsen, R. L., Pieterse, C. M., and Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Berg, G. (2009). Plant-microbe interactions promoting plant growth and health: perspectives for controlled use of microorganisms in agriculture. Appl. Microbiol. Biotechnol. 84, 11–18. doi: 10.1007/s00253-009-2092-7
Born, Y., Remus-Emsermann, M. N., Bieri, M., Kamber, T., Piel, J., and Pelludat, C. (2016). Fe2+ chelator proferrorosamine A: a gene cluster of Erwinia rhapontici P45 involved in its synthesis and its impact on growth of Erwinia amylovora CFBP1430. Microbiology 162, 236–245. doi: 10.1099/mic.0.000231
Bouffaud, M. L., Poirier, M. A., Muller, D., and Moenne-Loccoz, Y. (2014). Root microbiome relates to plant host evolution in maize and other Poaceae. Environ. Microbiol. 16, 2804–2814. doi: 10.1111/1462-2920.12442
Boyer, F., Fichant, G., Berthod, J., Vandenbrouck, Y., and Attree, I. (2009). Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genomics 10:104. doi: 10.1186/1471-2164-12-104
Bulgarelli, D., Garrido-Oter, R., Munch, P. C., Weiman, A., Droge, J., Pan, Y., et al. (2015). Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microb. 17, 392–403. doi: 10.1016/j.chom.2015.01.011
Bulgarelli, D., Rott, M., Schlaeppi, K., Van Themaat, E. V. L., Ahmadinejad, N., Assenza, F., et al. (2013a). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 501:S25.
Bulgarelli, D., Schlaeppi, K., Spaepen, S., Van Themaat, E. V. L., and Schulze-Lefert, P. (2013b). Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838. doi: 10.1146/annurev-arplant-050312-120106
Büttner, D. (2016). Behind the lines-actions of bacterial type III effector proteins in plant cells. FEMS Microbiol. Rev. 40, 894–937. doi: 10.1093/femsre/fuw026
Büttner, D., and Bonas, U. (2010). Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol. Rev. 34, 107–133.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2164-12-421
Cernadas, R. A., Doyle, E. L., Nino-Liu, D. O., Wilkins, K. E., Bancroft, T., Wang, L., et al. (2014). Code-assisted discovery of TAL effector targets in bacterial leaf streak of rice reveals contrast with bacterial blight and a novel susceptibility gene. PLoS Pathog. 10:e1003972. doi: 10.1371/journal.ppat.1004126
Chatterjee, S., Chaudhury, S., Mcshan, A. C., Kaur, K., and De Guzman, R. N. (2013). Structure and biophysics of type III secretion in bacteria. Biochemistry 52, 2508–2517. doi: 10.1021/bi400160a
Chen, J. (2018). Biopesticides: GLOBAL MARKETs to 2022. BCC Research. Available: https://www.bccresearch.com/market-research/chemicals/biopesticides-global-markets-report.html (accessed September 9, 2019).
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., Da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Compant, S., Duffy, B., Nowak, J., Clément, C., and Barka, E. A. (2005). Use of plant growth-promoting bacteria for biocontrol of plant diseases: principles, mechanisms of action, and future prospects. Appl. Environ. Microbiol. 71, 4951–4959. doi: 10.1128/aem.71.9.4951-4959.2005
Dairy Australia, (2019). The Australian Dairy Industry In Focus 2019. Available: https://www.dairyaustralia.com.au/industry/farm-facts/in-focus (accessed January 22, 2020).
De Coster, W., D’hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Droby, S., Wisniewski, M., Teixidó, N., Spadaro, D., and Jijakli, M. H. (2016). The science, development, and commercialization of postharvest biocontrol products. Postharvest Biol. Technol. 122, 22–29. doi: 10.1016/j.postharvbio.2016.04.006
Dunger, G., Llontop, E., Guzzo, C. R., and Farah, C. S. (2016). The Xanthomonas type IV pilus. Curr. Opin. Microbiol. 30, 88–97. doi: 10.1016/j.mib.2016.01.007
Dwivedi, D., and Johri, B. (2003). Antifungals from fluorescent pseudomonads: biosynthesis and regulation. Curr. Sci. 85, 1693–1703.
Egli, T., and Schmidt, D. (1982). Pathogenic variation among the causal agents of bacterial wilt of forage grasses. J. Phytopathol. 104, 138–150. doi: 10.1111/j.1439-0434.1982.tb00520.x
Eljounaidi, K., Lee, S. K., and Bae, H. (2016). Bacterial endophytes as potential biocontrol agents of vascular wilt diseases - review and future prospects. Biol. Control 103, 62–68. doi: 10.1016/j.biocontrol.2016.07.013
Garita-Cambronero, J., Palacio-Bielsa, A., Lopez, M. M., and Cubero, J. (2017). Pan-genomic analysis permits differentiation of virulent and non-virulent strains of Xanthomonas arboricola that cohabit Prunus spp. and elucidate bacterial virulence factors. Front. Microbiol. 8:573. doi: 10.3389/fmicb.2017.00573
Glare, T., Caradus, J., Gelernter, W., Jackson, T., Keyhani, N., Kohl, J., et al. (2012). Have biopesticides come of age? Trends Biotechnol. 30, 250–258. doi: 10.1016/j.tibtech.2012.01.003
Grau, J., Reschke, M., Erkes, A., Streubel, J., Morgan, R. D., Wilson, G. G., et al. (2016). AnnoTALE: bioinformatics tools for identification, annotation, and nomenclature of TALEs from Xanthomonas genomic sequences. Sci. Rep. 6:21077.
Green, E. R., and Mecsas, J. (2016). Bacterial secretion systems: an overview. Microbiol. Spectr. 4:10.1128/microbiolsec.VMBF-0012-2015. doi: 10.1128/microbiolspec.VMBF-0012-2015
Grimmer, M. K., Van Den Bosch, F., Powers, S. J., and Paveley, N. D. (2015). Fungicide resistance risk assessment based on traits associated with the rate of pathogen evolution. Pest Manag. Sci. 71, 207–215. doi: 10.1002/ps.3781
He, Y.-W., Wu, J. E., Zhou, L., Yang, F., He, Y.-Q., Jiang, B.-L., et al. (2011). Xanthomonas campestris diffusible factor is 3-hydroxybenzoic acid and is associated with xanthomonadin biosynthesis, cell viability, antioxidant activity, and systemic invasion. Mol. Plant Microb. Interact. 24, 948–957. doi: 10.1094/mpmi-02-11-0031
Hersemann, L., Wibberg, D., Blom, J., Goesmann, A., Widmer, F., Vorhölter, F. J., et al. (2017). Comparative genomics of host adaptive traits in Xanthomonas translucens pv. graminis. BMC Genomics 18:35. doi: 10.1186/1471-2164-12-35
Hu, Y., Zhang, J., Jia, H., Sosso, D., Li, T., Frommer, W. B., et al. (2014). Lateral organ boundaries 1 is a disease susceptibility gene for citrus bacterial canker disease. Proc. Natl. Acad. Sci. U.S.A. 111, E521–E529.
Huang, C.-L., Pu, P.-H., Huang, H.-J., Sung, H.-M., Liaw, H.-J., Chen, Y.-M., et al. (2015). Ecological genomics in Xanthomonas: the nature of genetic adaptation with homologous recombination and host shifts. BMC Genom. 16:188. doi: 10.1186/1471-2164-12-188
Jacobs, J. M., Pesce, C., Lefeuvre, P., and Koebnik, R. (2015). Comparative genomics of a cannabis pathogen reveals insight into the evolution of pathogenicity in Xanthomonas. Front. Plant Sci. 6:431. doi: 10.3389/fmicb.2017.00431
Johnson, L. J., De Bonth, A. C. M., Briggs, L. R., Caradus, J. R., Finch, S. C., Fleetwood, D. J., et al. (2013). The exploitation of epichloae endophytes for agricultural benefit. Fungal Divers. 60, 171–188. doi: 10.1007/s13225-013-0239-4
Kamilova, F., Validov, S., Azarova, T., Mulders, I., and Lugtenberg, B. (2005). Enrichment for enhanced competitive plant root tip colonizers selects for a new class of biocontrol bacteria. Environ. Microbiol. 7, 1809–1817. doi: 10.1111/j.1462-2920.2005.00889.x
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kauppinen, M., Saikkonen, K., Helander, M., Pirttila, A. M., and Wali, P. R. (2016). Epichloë grass endophytes in sustainable agriculture. Nat. Plants 2:15224.
Kay, S., and Bonas, U. (2009). How Xanthomonas type III effectors manipulate the host plant. Curr. Opin. Microbiol. 12, 37–43. doi: 10.1016/j.mib.2008.12.006
Klenk, H. P., and Goker, M. (2010). En route to a genome-based classification of Archaea and Bacteria? Syst. Appl. Microbiol. 33, 175–182. doi: 10.1016/j.syapm.2010.03.003
Kloepper, J. W., Leong, J., Teintze, M., and Schroth, M. N. (1980). Pseudomonas siderophores: a mechanism explaining disease-suppressive soils. Curr. Microbiol. 4, 317–320. doi: 10.1007/bf02602840
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Löytynoja, A. (2014). “Phylogeny-aware alignment with PRANK,” in Multiple Sequence Alignment Methods, ed. D. J. Russell, (Totowa, NJ: Humana Press), 155–170. doi: 10.1007/978-1-62703-646-7_10
Madden, K. S., Jokhoo, H., Conradi, F., Knowles, J., Mullineaux, C., Whiting, A. J. O., et al. (2019). Using nature’s polyenes as templates: studies of synthetic xanthomonadin analogues and realising their potential as antioxidants. Organic Biomol. Chem. 17, 3752–3759. doi: 10.1039/c9ob00275h
Mehrotra, S., Kumar, S., Zahid, M., and Garg, M. (2017). “Biopesticides,” in Principles and Applications of Environmental Biotechnology for a Sustainable Future, ed. R. L. Singh, (Berlin: Springer), 273–292.
Mitter, E. K., De Freitas, J. R., and Germida, J. J. (2017). Bacterial root microbiome of plants growing in oil sands reclamation covers. Front. Microbiol. 8:849. doi: 10.3389/fmicb.2017.00849
Palaniraj, A., and Jayaraman, V. (2011). Production, recovery and applications of xanthan gum by Xanthomonas campestris. J. Food Eng. 106, 1–12. doi: 10.1016/j.jfoodeng.2011.03.035
Pandey, S. S., Patnana, P. K., Rai, R., and Chatterjee, S. (2017). Xanthoferrin, the alpha-hydroxycarboxylate-type siderophore of Xanthomonas campestris pv. campestris, is required for optimum virulence and growth inside cabbage. Mol. Plant Pathol. 18, 949–962. doi: 10.1111/mpp.12451
Peng, Z., Hu, Y., Xie, J., Potnis, N., Akhunova, A., Jones, J., et al. (2016). Long read and single molecule DNA sequencing simplifies genome assembly and TAL effector gene analysis of Xanthomonas translucens. BMC Genom. 17:21. doi: 10.1186/1471-2164-12-21
Pesce, C., Jacobs, J. M., Berthelot, E., Perret, M., Vancheva, T., Bragard, C., et al. (2017). Comparative genomics identifies a novel conserved protein, HpaT, in proteobacterial Type III secretion systems that do not possess the putative translocon protein HrpF. Front. Microbiol. 8:1177. doi: 10.3389/fmicb.2017.01177
Pieretti, I., Royer, M., Barbe, V., Carrere, S., Koebnik, R., Cociancich, S., et al. (2009). The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics 10:616. doi: 10.1186/1471-2164-12-616
Pollock, J., Glendinning, L., Wisedchanwet, T., and Watson, M. (2018). The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 84:e02627-17.
Poplawsky, A. R., Urban, S. C., and Chun, W. (2000). Biological role of xanthomonadin pigments in Xanthomonas campestris pv. campestris. Appl. Environ. Microbiol. 66:5123. doi: 10.1128/aem.66.12.5123-5127.2000
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.ppat.009490
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Saha, M., Sarkar, S., Sarkar, B., Sharma, B. K., Bhattacharjee, S., and Tribedi, P. (2016). Microbial siderophores and their potential applications: a review. Environ. Sci. Pollut. Res. 23, 3984–3999. doi: 10.1007/s11356-015-4294-0
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Smits, T. H. M., Duffy, B., Blom, J., Ishimaru, C. A., and Stockwell, V. O. (2019). Pantocin A, a peptide-derived antibiotic involved in biological control by plant-associated Pantoea species. Archiv. Microbiol. 201, 713–722. doi: 10.1007/s00203-019-01647-7
Studholme, D. J., Wasukira, A., Paszkiewicz, K., Aritua, V., Thwaites, R., Smith, J., et al. (2011). Draft Genome Sequences of Xanthomonas sacchari and two banana-associated Xanthomonads reveal insights into the Xanthomonas Group 1 Clade. Genes 2, 1050–1065. doi: 10.3390/genes2041050
Tannenbaum, I., Kaur, J., Mann, R., Sawbridge, T., Rodoni, B., and Spangenberg, H. (2020). Profiling the Lolium perenne microbiome: from seed to seed. Phytobiome J. 4, 281–289. doi: 10.1094/PBIOMES-03-20-0026-R
Tseng, T. T., Tyler, B. M., and Setubal, J. C. (2009). Protein secretion systems in bacterial-host associations, and their description in the gene ontology. BMC Microbiol. 9(Suppl. 1):S2. doi: 10.1186/1471-2180-9-S1-S2
Wagner, S., Grin, I., Malmsheimer, S., Singh, N., Torres-Vargas, C. E., and Westerhausen, S. (2018). Bacterial type III secretion systems: a complex device for the delivery of bacterial effector proteins into eukaryotic host cells. FEMS Microbiol. Lett. 365:fny201.
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., and Bruccoleri, R. (2015). antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243.
White, F. F., Potnis, N., Jones, J. B., and Koebnik, R. (2009). The type III effectors of Xanthomonas. Mol. Plant Pathol. 10, 749–766.
Wichmann, F., Vorhölter, F. J., Hersemann, L., Widmer, F., Blom, J., Niehaus, K., et al. (2013). The noncanonical type III secretion system of Xanthomonas translucens pv. graminis is essential for forage grass infection. Mol. Plant Pathol. 14, 576–588. doi: 10.1111/mpp.12030
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computat. Biol. 13:e1005595. doi: 10.1371/journal.ppat.1005595
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:R46.
Young, J. M., Park, D. C., Shearman, H. M., and Fargier, E. (2008). A multilocus sequence analysis of the genus Xanthomonas. Syst. Appl. Microbiol. 31, 366–377. doi: 10.1016/j.syapm.2008.06.004
Keywords: bioprotection, Xanthomonas, perennial ryegrass, microbiome, non-pathogenic, secondary metabolite
Citation: Li T, Mann R, Sawbridge T, Kaur J, Auer D and Spangenberg G (2020) Novel Xanthomonas Species From the Perennial Ryegrass Seed Microbiome – Assessing the Bioprotection Activity of Non-pathogenic Relatives of Pathogens. Front. Microbiol. 11:1991. doi: 10.3389/fmicb.2020.01991
Received: 18 April 2020; Accepted: 28 July 2020;
Published: 26 August 2020.
Edited by:
Christina Cowger, Plant Science Research Unit (USDA-ARS), United StatesReviewed by:
Jeffrey Jones, University of Florida, United StatesXianjun Yuan, Nanjing Agricultural University, China
Alison Jean Popay, AgResearch Ltd., New Zealand
Ralf Koebnik, Institut de Recherche pour le D veloppement (IRD), France
Copyright © 2020 Li, Mann, Sawbridge, Kaur, Auer and Spangenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tongda Li, dG9uZ2RhLmxpQGFncmljdWx0dXJlLnZpYy5nb3YuYXU=