 Andrea Garretto1†
Andrea Garretto1† Taylor Miller-Ensminger1
Taylor Miller-Ensminger1 Adriana Ene1
Adriana Ene1 Zubia Merchant2Aashaka Shah1Athina Gerodias1Anthony Biancofiori2Stacey Canchola2Stephanie Canchola2Emanuel Castillo2Tasnim Chowdhury2Nikita Gandhi2Sarah Hamilton2Kyla Hatton2Syed Hyder2Koty Krull2Demetrios Lagios2Thinh Lam3Kennedy Mitchell2Christine Mortensen2Amber Murphy2Joseph Richburg2Meghan Rokas2Suzanne Ryclik2Pauline Sulit2Thomas Szwajnos2Manuel Widuch2Jessica Willis2Mary Woloszyn2
Zubia Merchant2Aashaka Shah1Athina Gerodias1Anthony Biancofiori2Stacey Canchola2Stephanie Canchola2Emanuel Castillo2Tasnim Chowdhury2Nikita Gandhi2Sarah Hamilton2Kyla Hatton2Syed Hyder2Koty Krull2Demetrios Lagios2Thinh Lam3Kennedy Mitchell2Christine Mortensen2Amber Murphy2Joseph Richburg2Meghan Rokas2Suzanne Ryclik2Pauline Sulit2Thomas Szwajnos2Manuel Widuch2Jessica Willis2Mary Woloszyn2 Bridget Brassil2Genevieve Johnson1Rita Mormando1
Bridget Brassil2Genevieve Johnson1Rita Mormando1 Laura Maskeri1
Laura Maskeri1 Mary Batrich4Nicole Stark2†
Mary Batrich4Nicole Stark2† Jason W. Shapiro2Cesar Montelongo Hernandez5
Jason W. Shapiro2Cesar Montelongo Hernandez5 Swarnali Banerjee6
Swarnali Banerjee6 Alan J. Wolfe5
Alan J. Wolfe5 Catherine Putonti1,2,5,7*
Catherine Putonti1,2,5,7*- 1Bioinformatics Program, Loyola University Chicago, Chicago, IL, United States
- 2Department of Biology, Loyola University Chicago, Chicago, IL, United States
- 3Neuroscience Program, Loyola University Chicago, Chicago, IL, United States
- 4Niehoff School of Nursing, Stritch School of Medicine, Loyola University Chicago, Maywood, IL, United States
- 5Department of Microbiology and Immunology, Stritch School of Medicine, Loyola University Chicago, Maywood, IL, United States
- 6Department of Mathematics and Statistics, Loyola University Chicago, Chicago, IL, United States
- 7Department of Computer Science, Loyola University Chicago, Chicago, IL, United States
Urinary tract infections (UTIs) are one of the most common human bacterial infections. While UTIs are commonly associated with colonization by Escherichia coli, members of this species also have been found within the bladder of individuals with no lower urinary tract symptoms (no LUTS), also known as asymptomatic bacteriuria. Prior studies have found that both uropathogenic E. coli (UPEC) strains and E. coli isolates that are not associated with UTIs encode for virulence factors. Thus, the reason(s) why E. coli sometimes causes UTI-like symptoms remain(s) elusive. In this study, the genomes of 66 E. coli isolates from adult female bladders were sequenced. These isolates were collected from four cohorts, including women: (1) without lower urinary tract symptoms, (2) overactive bladder symptoms, (3) urgency urinary incontinence, and (4) a clinical diagnosis of UTI. Comparative genomic analyses were conducted, including core and accessory genome analyses, virulence and motility gene analyses, and antibiotic resistance prediction and testing. We found that the genomic content of these 66 E. coli isolates does not correspond with the participant’s symptom status. We thus looked beyond the E. coli genomes to the composition of the entire urobiome and found that the presence of E. coli alone was not sufficient to distinguish between the urobiomes of individuals with UTI and those with no LUTS. Because E. coli presence, abundance, and genomic content appear to be weak predictors of UTI status, we hypothesize that UTI symptoms associated with detection of E. coli are more likely the result of urobiome composition.
Introduction
Urinary tract infections (UTIs) affect about 150 million people per year, mounting large medical costs for individuals and communities (Flores-Mireles et al., 2015). While several different bacterial species are known to cause UTIs, the most frequent culprit is uropathogenic E. coli (UPEC), which is thought to account for up to 74.4% of all community-acquired infections (Foxman, 2014). Phylogenomic studies have placed UPEC strains amongst multiple E. coli phylotypes (Moriel et al., 2016). Genome sequencing of UPEC isolates has identified numerous virulence factors and pathogenicity islands (Subashchandrabose and Mobley, 2015; Moriel et al., 2016; Terlizzi et al., 2017). Transcriptome analyses have revealed high expression of genes associated with copper efflux and potassium/nickel transport in patients with UTIs (see review; Subashchandrabose et al., 2014). Nevertheless, the UPEC genomic “signature” remains elusive (Johnson et al., 2001, 2015; Luo et al., 2012). At present, there is evidence that the pathogenic potential of E. coli cannot be predicted from the presence of specific genetic content, such as a distinct virulence factor. Instead, UTI may be a result of multiple variables (Eto et al., 2007; Ragnarsdóttir et al., 2011; Brubaker and Wolfe, 2017; Schreiber et al., 2017).
Previous evidence indicates that the gut of individuals can be a reservoir for E. coli and thus a source of UTI (Yamamoto et al., 1997; Nielsen et al., 2014). Evidence shows that flagellum-mediated motility is key during E. coli ascension into and colonization of the urinary tract (Lane et al., 2007). Furthermore, it has been shown that UPEC strains are able to colonize both the gut and urinary tract (Chen et al., 2013). E. coli strains isolated from fecal and urine samples of women with UTIs have been found to have similar genomes, and in the absence of symptoms, they cannot be distinguished (Nielsen et al., 2017). This supports the prevailing hypothesis that UTIs can be the result of colonization of the urinary tract by E. coli strains from the gut (Yamamoto et al., 1997; Hooton, 2001; Chen et al., 2013; Thänert et al., 2019).
However, the current UTI paradigm does not account for the human urinary microbiota (urobiome) – the newly discovered communities of microbes (bacteria, archaea, protists, fungi, and viruses) that inhabit the bladder. Consequently, traditional clinical care methods remain based on dogma that “urine is sterile.” High throughput DNA sequencing and improved urine culture techniques reveal bacterial DNA and live bacteria, respectively, in catheterized (bladder) urine deemed culture-negative by the standard urine culture method (Fouts et al., 2012; Wolfe et al., 2012; Khasriya et al., 2013; Brubaker et al., 2014; Hilt et al., 2014; Nienhouse et al., 2014; Pearce et al., 2014, 2015; Karstens et al., 2016; Price et al., 2016; Thomas-White et al., 2016, 2017; Coorevits et al., 2017). The urobiome of women without lower urinary tract symptoms (LUTS) often includes species of the genera Lactobacillus, Corynebacterium, Gardnerella, Staphylococcus, and Streptococcus (see review; Brubaker and Wolfe, 2017). Traditionally, it has been thought that the incidence of E. coli in the urinary tract necessitated infection and symptoms, but the discovery of the urobiome and isolation of E. coli in asymptomatic people indicates otherwise (Thomas-White et al., 2018b; Price et al., 2020). Moreover, the native microbiota of the bladder has been shown to be distinct from the gastrointestinal tract (Thomas-White et al., 2018a; Price et al., 2020). Thus, a UTI may not always be due to a uropathogen invasion; instead, the urinary tract could itself be the source of UPEC strains. This has been shown previously in individuals with recurrent UTIs (Mysorekar and Hultgren, 2006; Thänert et al., 2019). UTI also could be due to commensal E. coli being overly abundant and/or disproportionate to other commensals in the urinary tract.
Here, we present a study of 66 E. coli strains isolated from the bladders of women with or without a UTI diagnosis; the latter cohort was further subdivided into women with no lower urinary tract symptoms (no LUTS), overactive bladder (OAB), or urgency urinary incontinence (UUI). Genomes of each of these strains were sequenced. We then compared the evolutionary history, encoded virulence factors, and antibiotic sensitivity of each of these strains in order to identify any factors that distinguish strains associated with patient symptoms. Furthermore, we expanded our analyses to look not only at E. coli as the signature of a UTI, but rather the entire microbial community.
Materials and Methods
Patient Recruitment
Participant sampling for this study was through several different Institutional Review Board-approved studies [Loyola University Chicago (LU203986, LU205650, LU206449, LU206469, LU207102, and LU207152) and University of California San Diego (170077AW)]. Participants gave verbal and written consent for chart abstraction and urine collection with analysis for research purposes. Members of the Loyola Urinary Education and Research Collaborative (LUEREC), specifically clinicians from the Female Pelvic Medicine and Reconstructive Surgery Center at Loyola University Medical Campus (LUMC), performed recruitment and urine collection. Participants were recruited as a part of separate studies (Hilt et al., 2014; Pearce et al., 2014, 2015; Price et al., 2016, 2020; Thomas-White et al., 2016, 2018a; Dune et al., 2017).
The symptoms of some of the women in the OAB and UUI groups were characterized with the validated Pelvic Floor Distress Inventory symptom questionnaire (Uebersax et al., 1995; Barber et al., 2001). The entire OAB cohort completed the Overactive Bladder Questionnaire (Coyne et al., 2002). For the OAB and UUI cohorts, women with a current UTI (based on urine dipstick test) or a history of recurrent UTI were excluded. For the UTI cohort, participants included women who responded affirmatively to the question “Do you feel you have a UTI?” and completed the UTI symptom assessment (UTISA) questionnaire (Clayson et al., 2005). All UTI samples were from women with cystitis (bladder infection). To be a member of the no LUTS cohort, women had to respond no to this question and be free of LUTS based on the Pelvic Floor Distress Inventory symptom and Overactive Bladder questionnaires. For all studies, exclusion criteria included age of <18 years, pregnancy, catheterization (indwelling or intermittent), or insufficient English skills to complete study measures.
Urine Collection and EQUC Bacterial Culturing
Urine was collected aseptically via transurethral catheter and was placed in a BD Vacutainer Plus C&S preservative tube for culturing. This technique bypasses the vulva, vagina, and urethra, thus resulting in samples of bacterial isolates specifically from the bladder niche (Wolfe et al., 2012). All samples underwent standard urine culture (SUC), as well as expanded quantitative urine culture (EQUC), as part of the aforementioned studies (Hilt et al., 2014; Pearce et al., 2014, 2015; Price et al., 2016, 2020; Thomas-White et al., 2016, 2018a; Dune et al., 2017). For SUC, 1 μL of urine was inoculated onto 5% sheep blood agar plate (BAP) and MacConkey agar plate (BD BBL prepared plated media), and incubated aerobically at 35°C for 24 h. The detection level was 1000 colony forming unit (CFU) per mL, represented by 1 colony of growth on either plate. EQUC was performed as described previously (Hilt et al., 2014). Briefly, 100 μL of urine was grown under five conditions with BD BBL prepared plated media: (1) BAP in CO2 for 48 h, (2) chocolate agar (CHOC) in CO2 for 48 h, (3) colistin and nalidixic acid (CNA) agar in CO2 for 48 h, (4) CDC anaerobe BAP in an anaerobic jar for 48 h, and (5) BAP in aerobic conditions (BD GasPak Anaerobe Sachets) for 48 h. The detection level was 10 CFU/mL, represented by 1 colony of growth on any of the plates.
Pure cultures were obtained by isolating each morphologically distinct colony type on a different plate of the same medium. Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrophotometry with the MALDI Biotyper 3.0 software program (Bruker Daltonics, Billerica, MA, United States) was used to identify the bacterial strains (see Hilt et al., 2014 for specific protocol, the same as is recommended by the Clinical Laboratory Standards Institute). For some of these isolates, 16S rRNA gene sequencing was performed to verify MALDI-TOF classifications. Previous work by our group showed a concordance between MALDI-TOF and 16S rRNA sequencing classification of urinary isolates (Pearce et al., 2014; Price et al., 2020). Strains identified as E. coli via MALDI-TOF were grown from single colonies in lysogeny (LB) broth (10.0 g/L tryptone, 5.0 g/L yeast, 10.0 g/L NaCl). Stocks of each strain were made using 900 μL 50% glycerol mixed with 90 μL E. coli cells, and stored at −80°C.
DNA Extraction and Genome Sequencing
Liquid cultures of each strain (Supplementary Table 1) were grown from freezer stock in LB at 37°C in a shaking (150 rpm) incubator for 12 h. DNA was extracted from liquid culture using the Qiagen DNeasy UltraClean Microbial Kit following the manufacturer’s protocol. DNA concentration was quantified using the Qubit fluorometer. To confirm the MALDI-TOF classification, the 16S rRNA gene sequence was amplified using the 63f and 1387r primers (Marchesi et al., 1998) and sequenced by Genewiz (New Brunswick, NJ, United States), using each primer individually for 2× coverage. The resulting sequences were manually trimmed and paired in Geneious v11.0.5 (Biomatters, Ltd., Auckland, New Zealand) and queried against the NCBI 16S rRNA sequences database via blastn to confirm that they were E. coli. DNA libraries were constructed using the Nextera XT DNA Library preparation kit and sequenced on the Illumina MiSeq platform using the MiSeq Reagent Kit v2 (500-cycles) at Loyola University Chicago’s Genomics Facility (Maywood, IL United States). Raw sequencing reads were deposited in NCBI’s SRA database.
Assembly and Annotation
Raw reads were first trimmed for quality using the tool Sickle v1.331 and then assembled with SPAdes v3.10.1 using the assembly-only option for k values of 55, 77, 99, and 127 (Bankevich et al., 2012). Contigs less than 500 nucleotides in length were removed from further consideration. Assembled contigs were then manually inspected and individually queried against the nr/nt database via megablast, confirming that no contaminants were included in the sequence data. The genome coverage of each assembly was calculated using BBMap’s bbwrap and pileup scripts2. Each assembly was annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) v4.7 (Tatusova et al., 2016).
Assemblies were evaluated for additional genomic features. The E. coli serotype was determined using SerotypeFinder v2.0 using default parameters (threshold ID = 85%; minimum length = 60%) (Joensen et al., 2015). The EzClermont web application was used to phylotype each genome (Waters et al., 2020). Genome assemblies were scanned for plasmid replicon sequences using PlasmidFinder v2.1 with default settings (threshold ID = 95%; minimum% coverage = 60%) (Carattoli et al., 2014). All genomes are publicly available through NCBI’s Assembly database (Supplementary Table 2). Virulence factors present within each genome were identified using the virulence factor database (VFDB) using VFanalyzer (Liu et al., 2019). Genes for flagellar synthesis, flagellar rotation, chemotactic signal transduction, and chemotactic membrane receptors were identified within the genome sequences using reciprocal blasts and manual curation. Local blast databases were made using the PGAP annotation files and flg (flgA-flgN), flh (flhA-flhE), fli (fliA, fliC-fliT, and fliZ), mot (motA, motB) and che (cheA, cheB, cheR, cheW, cheY, and cheZ) genes as well as chemotactic receptors tap, tar, trg, tsr, and aer were identified. These sequences were manually inspected. Antibiotic resistance prediction was conducted using ResFinder v3.2 using the default settings (threshold ID = 90%; minimum length = 60%) (Zankari et al., 2012).
Core Genome and Phylogenomic Analysis
Amino acid sequences for each genome’s RefSeq annotation derived by PGAP were concatenated into a single FASTA format file, sorted by length, and clustered using usearch via the cluster_fast method (Edgar, 2010). Sequence identity thresholds of 70, 80, and 90% were tested; the clusters generated for 80% were selected for further analysis. After manual inspection to confirm homologs were properly identified, the average sequence identity was confirmed to be >90%. The final set of clusters were next parsed using Python to generate a gene presence/absence matrix for the genomes. Using this matrix, hierarchical clustering was performed with SciPy3 and the Ward variance minimization algorithm. The resulting tree was converted to Newick format and visualized using iTOL (Letunic and Bork, 2016). This matrix also was used to identify genes belonging to the core and accessory genomes for the 66 E. coli. For each gene within the core genome set, the sequences were aligned using MAFFT and alignments were manually inspected (Katoh and Standley, 2013). A consensus tree was derived by first concatenating the alignments followed by derivation using FastTree (Price et al., 2010). The resulting tree was visualized using iTOL (Letunic and Bork, 2016).
Antibiotic Susceptibility Testing
Antibiotics frequently prescribed for UTI treatment were tested (Chu and Lowder, 2018). Using BBLTM Sensi-Discs (Becton, Dickinson and Company; Franklin Lakes, NJ), each strain was tested for its susceptibility to fosfomycin (200 μg, BD 231709), ciprofloxacin (5 μg, BD 231657), amoxicillin with clavulanic acid (20/10 μg; BD 231628), sulfamethoxazole-trimethoprim (23.75/1.25 μg; BD 231536) and cefpodoxime (10 μg; BD 231673). Testing was conducted as follows. Each E. coli strain was incubated in 3 mL of LB broth overnight aerobically, shaking at 37°C. 1 mL was spread on a 1.7% LB agar plate using a cell spreader and left to dry for 5 m. Sensi-Discs were then placed on the E. coli lawn using sterile forceps and the plates were incubated overnight at 37°C. The clearance diameter was measured for each Sensi-Disc and each antibiotic was tested in triplicate for each E. coli strain. Based upon the diameter measured, the manufacturer’s instructions were used to classify strains as resistant, intermediate, or susceptible.
Comparing Bladder E. coli Isolates to Publicly Available Genomes
Each of the 66 bladder E. coli genome assemblies was compared to NCBI’s nr/nt database via megablast. The assembled contigs for each genome assembly were concatenated such that the entire genome sequence was queried. The top hit was recorded for each and the source for the top hit was determined from the GenBank record and/or the literature.
Statistical Analysis of Communities
The CFU/mL count for all bacteria in each of the 63 independent samples containing one of the sequenced E. coli genomes were examined. The individual contributing 3 isolates to our collection of E. coli genomes (UMB6653, UMB6721, and UMB7431) was removed from consideration as they are not independent samples. In addition to these data, we included CFU/mL counts for the urobiomes that we have previously characterized from samples of women with UTI symptoms and women without LUTS. In total, this resulted in 109 participants with UTI symptoms and 90 participants with no LUTS. CFU/mL data was recorded at the genus level rather than the species level for our analysis. To analyze prevalence of UTI among participants, a multiple logistic regression model (Cramer, 2002) was used. We treated the no LUTS group as our baseline category for the UTI group. For each participant, abundances of 27 different bacteria were measured; genera appearing in only one sample were removed from consideration. Abundance of these 27 species of bacteria were treated as covariates in our model. Akaike Information Criterion (AIC) (Akaike, 1974; Aho et al., 2014) was used to reduce the full model. All analyses were conducted in R4.
Results
Sixty-six E. coli isolates were investigated here, each isolated using the EQUC method from catheterized urine samples from women with (n = 42) or without (n = 24) a clinical diagnosis of UTI. These isolates were collected from 64 different women as one individual with a recurrent UTI was sampled three times [E. coli strains UMB6653 (first visit), UMB6721 (second visit), and UMB7431 (third visit)]. The non-UTI group included asymptomatic ‘healthy’ women [the “no LUTS” group (n = 6)] as well as women with OAB (n = 5) or UUI (n = 13). Supplementary Table 1 lists each isolate and participant symptom diagnosis, as well as the abundance of E. coli relative to isolates of other species within the catheterized urine specimens. These data revealed that the abundance of E. coli does not strictly relate to diagnostic group, as the UTI group included 6 individuals with low abundances (<10,000 CFU/mL) of E. coli within their bladder microbiota, whereas the non-UTI group included 9 individuals with high E. coli abundances (≥100,000 CFU/mL).
Whole genome sequencing, genome assembly, and annotation were performed for each isolate (see section “Materials and Methods”). Supplementary Table 2 lists the genome assembly statistics. The genomes ranged in size from 4.6 to 5.6 Mbp. Each genome is represented by long, high quality contig sequences. The average assembly consisted of 73 contigs, ranging from E. coli isolate UMB6454 (33 contigs) to E. coli isolate UMB2019 (161 contigs). The serotype and phylotypes were determined for each strain (Supplementary Table 1). The genomes represent 39 different serotypes. While most (n = 36) isolates belong to the B2 phylotype, 20 isolates belong to phylotype D, 5 to phylotype A, 4 to phylotype B1, and 1 to phylotype F. 59 of the 66 genomes included a contig(s) with a replicon sequence, suggesting that many isolates included plasmid sequences (Supplementary Table 2). Contigs showing similarity to the Col156, IncFII, and IncFIB plasmid replicons were the most abundant, with 28, 45, and 41 isolates harboring these plasmids, respectively.
The core genome of these isolates contained 2,814 genes. This is significantly smaller than the pangenome, which contained an additional 10,182 genes present in one or more of the isolates. However, 28% of the pangenome consists of genes that were only found within one of the 66 E. coli genomes, reinforcing prior claims that E. coli has an open genome (Rasko et al., 2008).
From the core genome, we constructed a phylogenomic tree (Figure 1). The 66 isolates belong to two distinct clades: the isolates belonging to the B2 phylotype and the isolates belonging to the A, B1, D, and F phylotypes. Symptom status was not associated with these clades; there is no evolutionary distinction between E. coli isolates associated with UTI symptoms and those that are not. While the no LUTS isolates were only found within the bottom clade, the sample size is too small to draw conclusions from this observation. In addition to the clear distinction between phylotypes, members of the same serotype often had the most similar core genome sequences.
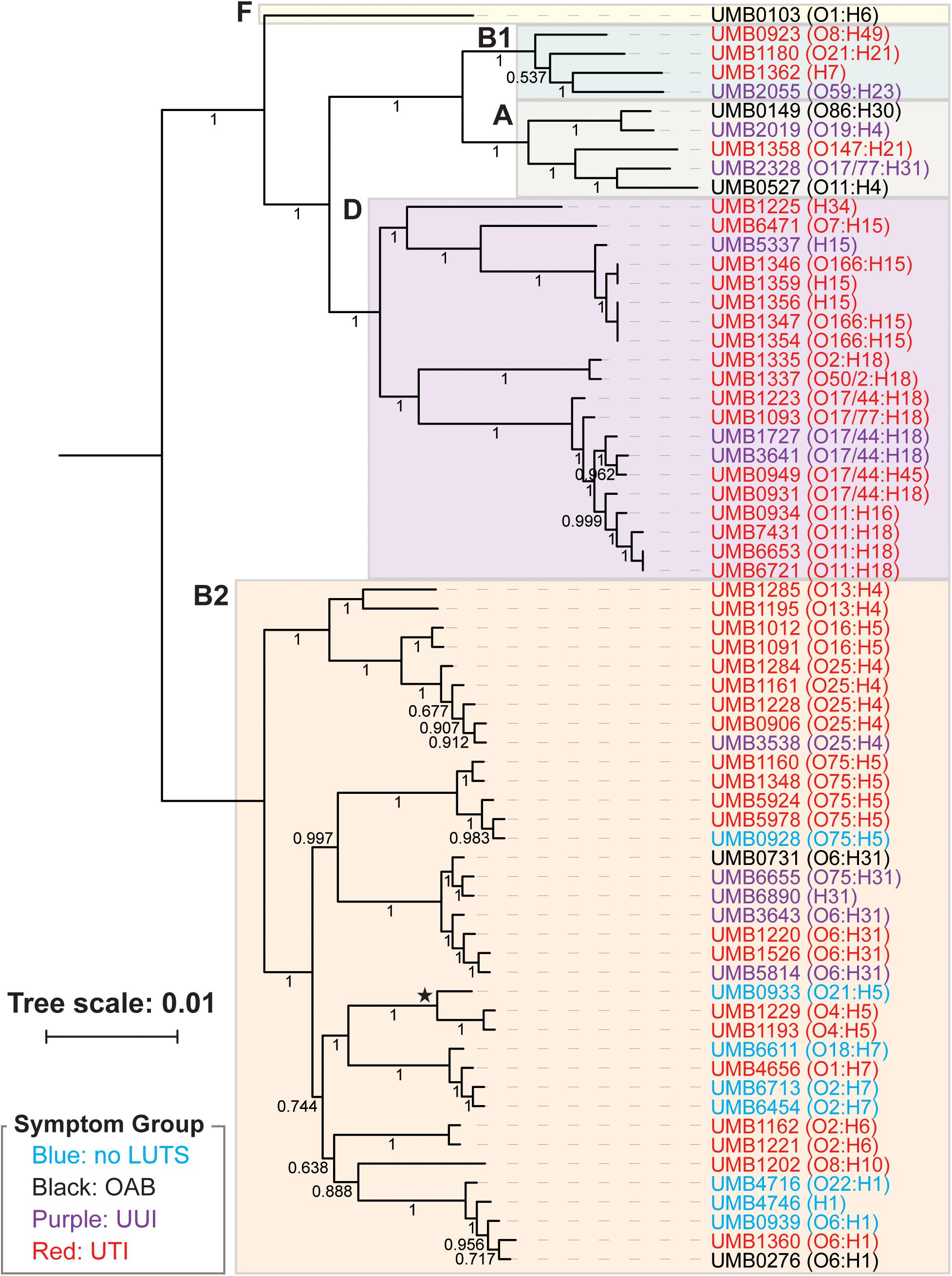
Figure 1. Phlyogenomic relationship of the 66 UTI (red) and non-UTI E. coli isolates from the bladder. Non-UTI symptom groups are indicated by color. Phylotypes are indicated by the colored boxes. The branch labeled with a star indicates the 3 isolates of the subphylotype B22. Serotypes for each isolate are listed next to the isolate name. Branch supports are shown.
Next we performed hierarchical clustering of the genes from the pangenome to ascertain if genome content was associated with symptom status (Figure 2). Here, genomes that share more genes in common cluster together. Again, we observed two major clades. Each isolate clustered within the same major clade based on gene content (Figure 2) as it did based on the core genome sequence (Figure 1). The clading structure, however, is not identical; for example, the single isolate in phylotype F, E. coli UMB0103, is no longer distinct from the isolates of phylotype D. Further comparison of these two trees reveals that some isolates are more similar with respect to their core gene sequences than they are with respect to the accessory genes that they contain. Using the gene presence matrix, the correlation between each gene’s presence/absence in the 66 E. coli genomes and the patient’s clinical diagnosis was calculated. Genes were found to have no or a weak correlation (r < 0.5) to clinical diagnosis.
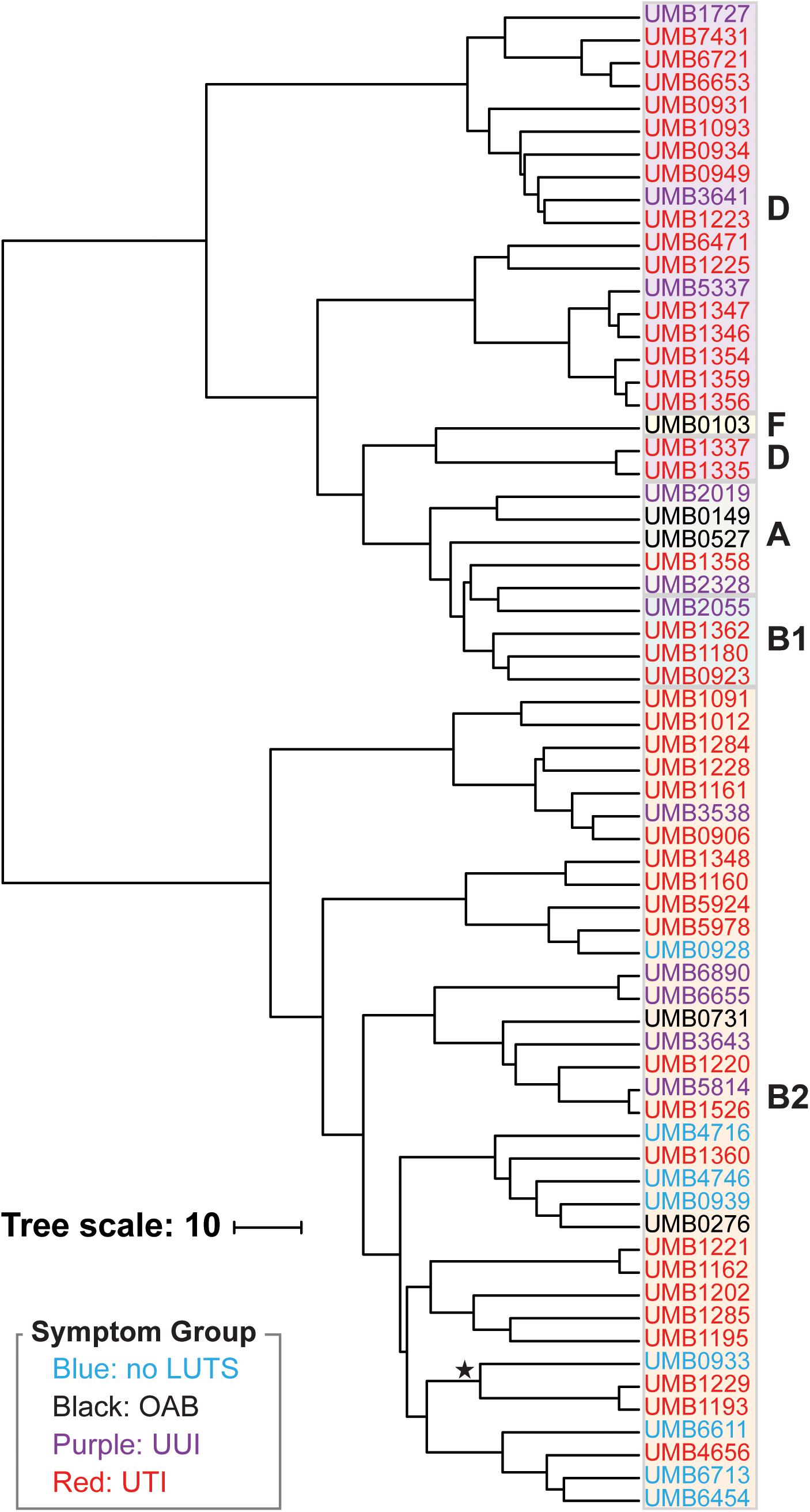
Figure 2. Hierarchical clustering of the 66 E. coli genomes based on gene content. Symptom groups are indicated by color. Phylotypes are indicated by the colored boxes. The branch labeled with a star indicates the 3 isolates of the subphylotype B22.
From the accessory genome alone, we first considered the 770 genes present in ≥80% of the UTI genomes and found that these genes were often present (≥50%) in the genomes from participants of the non-UTI symptom groups. There are no genes frequently present (≥80%) in the UTI genomes that are also absent from the non-UTI genomes. Only the gene that encodes the anti-adaptor protein IraD, which inhibits RssB-mediated degradation of RpoS (Battesti et al., 2011; Park et al., 2017), was frequently present in the UTI genomes (86%) and found in only 50% of the non-UTI genomes. All other genes meeting the ≥80% threshold of presence within the UTI genomes were also present in more than half of the non-UTI genomes. In contrast, three genes were underrepresented within the no LUTS group. The genes that encode the type II toxin-antitoxin system YhaV/PrlF and the type I toxin Hok were frequently present in the UTI genomes (≥85%). The genes that encode the YhaV/PrlF system were present in all of the genomes from OAB and UUI participants (n = 18) and Hok was present in most of the OAB and UUI participants (n = 15).
To further investigate the putative pathogenicity of the isolates, each genome was queried against the virulence factor database (VFDB), identifying 242 different virulence genes (associated with 99 virulence factors) amongst the 66 isolates (Supplementary Table 3). The only virulence factors found in all 42 UTI genomes were also found in all non-UTI (no LUTS, OAB, and UUI) genomes and included: Hemorrhagic E. coli pilus (related genes hcpA and hcpC), Type I fimbriae (related genes fimF, fimG, and fimH), enterohemorrhagic E. coli autotransporter B (related gene ehaB), and invasion of brain endothelial cells (related genes ibeB and ibeC). While there were no virulence factors found frequently and exclusively in the UTI isolates, the E. coli laminin-binding fimbriae (ELF) and non-LEE type III secretion system effector (TTSS) virulence factors were often present (30 and 40%, respectively) in UTI isolates and absent in all of the no LUTS isolates. However, genes related with these virulence factors were identified in OAB and UUI genomes. No association between virulence factors and symptom state be it UTI/non-UTI or UTI/OAB/UUI/no LUTS was identified.
To evaluate the hypothesis that UTIs can be the result of colonization of the urinary tract by E. coli strains from the gut, we examined the genome annotations for the presence of 52 genes associated with flagellar synthesis (flg, flh, and fli), flagellar rotation (mot), chemotactic signal transduction (che), and chemotactic membrane receptors (tar, tsr, tap, trg, and aer) (see section “Materials and Methods”). Twenty-four of the genomes contained all of the genes and at least one chemoreceptor, suggesting that these isolates are flagellated, motile, and chemotactic (Table 1). While all 66 genomes encode for the flh, mot, and che genes, the remaining 42 isolates are lacking one or more flg and/or fli genes. All 66 genomes include the tsr, tar, and trg chemoreceptors. UMB0276 is the only genome lacking the aer chemoreceptor and 36 lack the tap chemoreceptor (Supplementary Table 4).

Table 1. Number of genomes encoding for all flg, flh, fli, mot, and che genes, as well as at least one chemoreceptor.
Next, we considered pathogenicity relative to antibiotic resistance. Antibiotic resistance was predicted from the genome sequences (Supplementary Table 5). Genes associated with resistances to aminoglycosides, beta-lactams, macrolides, phenicol, quinolone, rifampicin, sulfonamides, tetracyclines, and trimethoprims were detected amongst the isolates. Most (85%) of these genes were chromosomal-mediated; only 21 strains carried plasmids encoding for an antibiotic resistance gene (Supplementary Table 5). While only UTI isolates encoded genes associated with phenicol (n = 2) and rifampicin (n = 1), all other resistances were observed in the genomes from both UTI and non-UTI isolates. Next, five antibiotics routinely prescribed for UTIs were tested: fosfomycin, ciprofloxacin, amoxicillin with clavulanic acid, sulfamethoxazole-trimethoprim, and cefpodoxime. Concurring with the genomic analysis, all of the isolates were sensitive to fosfomycin (Table 2). However, as indicated in Supplementary Table 6, computational predictions and experimental results were often discordant, e.g., resistance to ciprofloxacin was observed for 16 isolates even though quinolone resistance was only predicted for three isolates. Four of the isolates [UMB0731 (OAB) and UMB1193 (UTI), UMB1335 (UTI), and UMB1348 (UTI)] were sensitive to all five antibiotics tested. Statistical analysis found that sensitivity varied between non-UTI and UTI isolates by drug (p = 0.0086). Non-UTI isolates were more resistant to amoxicillin than UTI isolates and more sensitive to sulfamethoxazole-trimethoprim than UTI isolates. However, the variation in sensitivity between non-UTI and UTI isolates or amongst no LUTS, OAB, and UUI isolates was not statistically significant (p = 0.9682 and 0.8313, respectively).
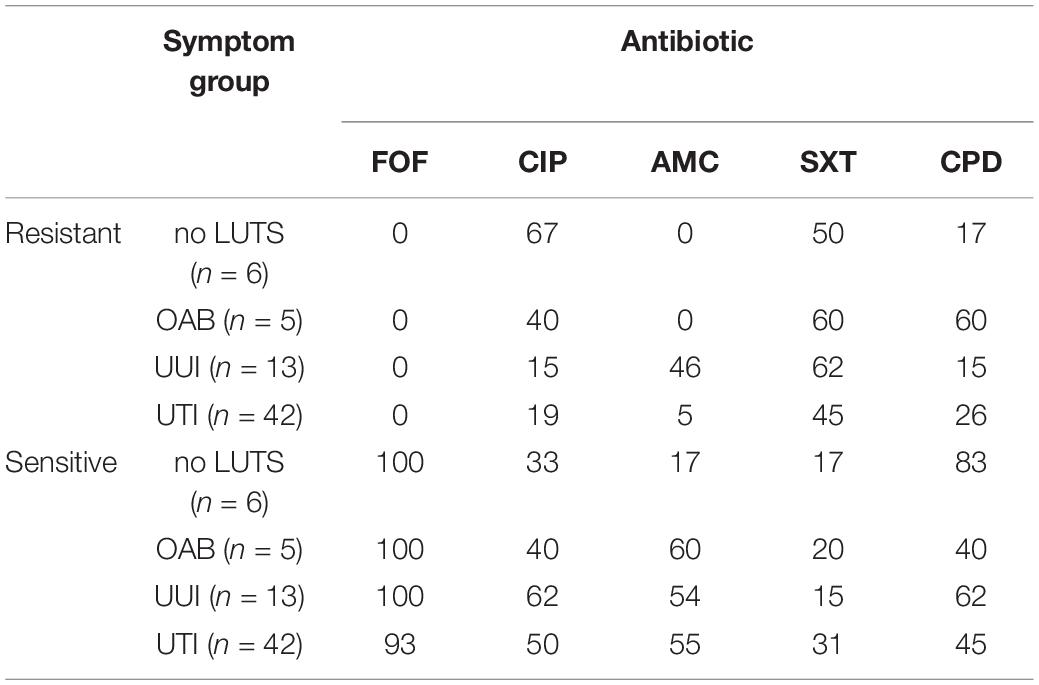
TABLE 2. Percentage of E. coli isolates resistant or sensitive to the antibiotics fosfomycin (FOF), ciprofloxacin (CIP), amoxicillin with clavulanic acid (AMC), sulfamethoxazole-trimethoprim (SXT), and cefpodoxime (CPD).
As prior studies have attributed the gastrointestinal (GI) tract as the source of UPEC strains, we next sought to compare our 66 bladder isolates with all publicly available E. coli genomes. It is important to note that many of the genomes deposited in GenBank were isolated either from the GI tract or from urine collected by an unspecified method during a UTI. The bladder isolates aligned to E. coli genomes in NCBI’s nr/nt database with 83–98% query coverage (Supplementary Table 7). These hits included isolates from urine (n = 24) and stool (n = 17) of humans, canines, cattle, mice, birds, and pigs (Table 3). Two isolates, UMB0149 (OAB) and UMB2019 (UUI), exhibited greatest similarity to isolates from forest soil and hospital wastewater, respectively (Supplementary Table 7).
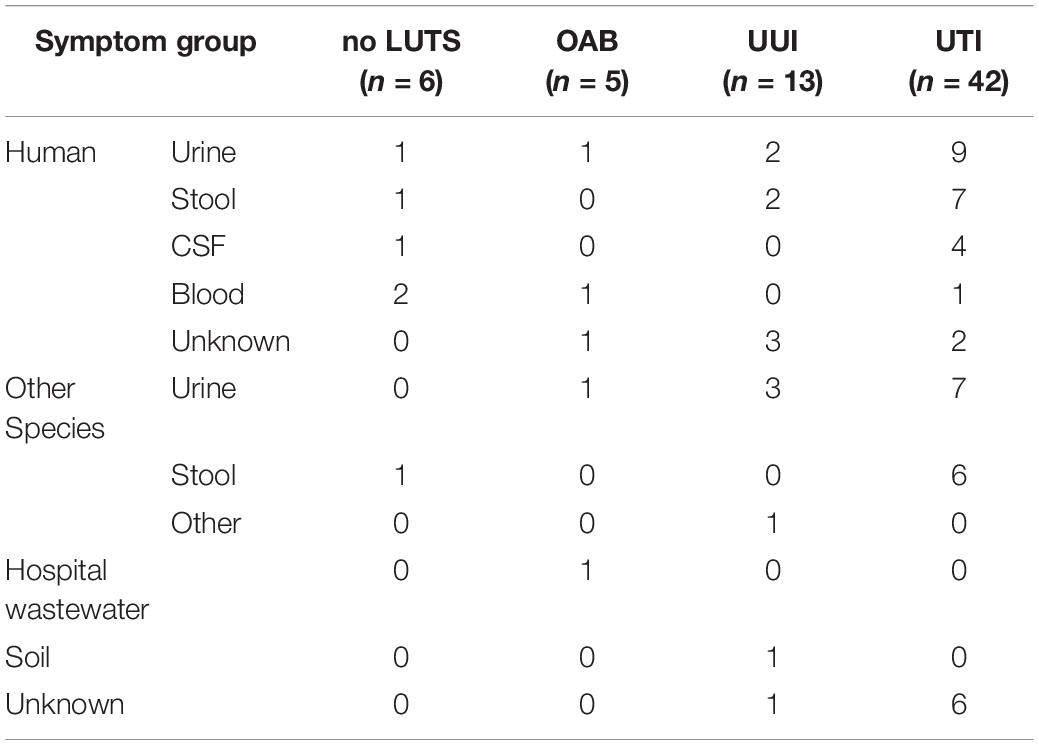
Table 3. Isolation source of genomes most similar to the bladder E. coli isolates.
Because our results suggest that the E. coli genome itself does not correspond with UTI symptoms, we next considered the E. coli isolates as a member of the bladder community. During the isolation of each these E. coli isolates, other bacterial taxa of the urobiome were also identified (Supplementary Table 8). As Figure 3 shows, several different species were found within the urobiomes of these women. Fifteen womens’ urobiomes included only E. coli; only 9 of these were from UTI patients. E. coli was the predominant member or was present in high abundance within 18 (75%) and 9 (37.5%), respectively, of the urobiomes of women without UTI symptoms. Within the UTI cohort, the urobiomes of 6 women included species from the genera Staphylococcus, Lactobacillus, Klebsiella, and Gardnerella that were in greater abundance than E. coli. To improve the statistical robustness of our analyses, we supplemented our urobiome community data, including CFU/mL counts for all constituents, for the 64 participants from which the 66 E. coli genomes were collected with an additional 135 participant data sets for a total of 109 UTI participants and 90 non-UTI no LUTS participants. While we found that E. coli presence was highly significant (p-value = 1.39E-09), the effect was very small. Our model predicted that, for a unit increase in E. coli, the expected odds of UTI increases by 0.003%. Thus, the presence of E. coli alone is not sufficient to distinguish between the urobiomes of individuals with UTI and those with no LUTS.
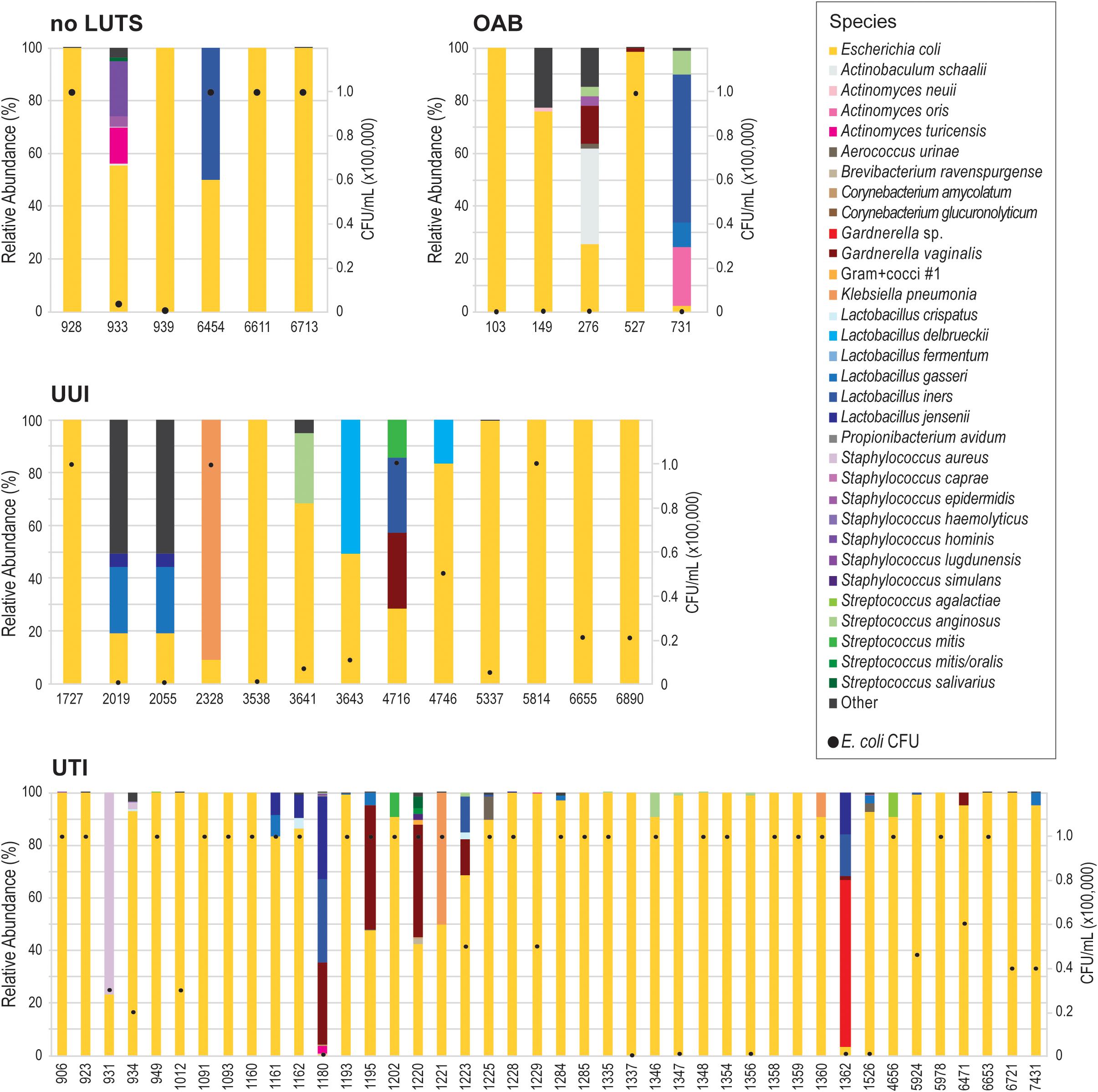
Figure 3. Urobiomes from which E. coli was isolated. Each bar represents the relative abundance of microorganisms within the urobiome, color coded by species, for the individual patient. The black circles on each stacked bar show the abundance of E. coli in the urobiome. Urobiomes are presented according to the symptom group.
Discussion
As prior studies have shown, E. coli can be found in the bladders of women with or without UTI symptoms (Price et al., 2016, 2020; Thomas-White et al., 2018b). Furthermore, it can be found in abundance in women without UTI symptoms [(Price et al., 2016); see also review (Ferroni and Taylor, 2015), shown also in this study (Supplementary Table 1)]. The 66 E. coli genomes sequenced and analyzed here include isolates obtained from women with or without UTI symptoms, as well as women with other LUTS. To our knowledge, this is the first study that has investigated E. coli genomes from the urobiome of women with either OAB or UUI. Prior comparative genomic studies have focused on E. coli isolates from individuals with or without UTI symptoms only (Rasko et al., 2008; Moriel et al., 2016, see also review; Lo et al., 2017). We also investigated the genomes of E. coli isolates from women without LUTS, a scenario previously called asymptomatic bacteriuria (Rubin et al., 1992; Nicolle, 2003; Nicolle et al., 2019). A recent study of 224 adult women without LUTS showed that E. coli is relatively uncommon in this population, but it can be present and can be the predominant taxon especially in older women (Price et al., 2020). As Figures 1, 2 show, the phylogenetic history and genomic content of E. coli isolates from women with or without UTI does not correspond with the participant’s UTI status. While the isolates from women with no LUTS are only found within the B2 phylotype clade, our sample size is small. We expect with an increased sample size that isolates would represent other phylotypes as isolates from the non-UTI group, which includes no LUTS, OAB, and UUI, were found in both clades of the trees.
Of the 66 isolates sequenced here, 3 were from the same individual, a woman with recurrent UTIs. The three isolates from this woman, UMB6653, UMB6721, and UMB7431, were most similar to each other in the core genome phylogeny and gene content. UMB6653 and UMB6721 are identical, according to their core gene sequences, suggesting that the same strain is responsible for the woman’s reinfection. This reinfection could be the result of persistence within the bladder over time, potentially through intracellular reservoirs, as has previously been observed within recurrent UTI patients (Mysorekar and Hultgren, 2006; Thänert et al., 2019). Our gene content analysis (Figure 2) provides evidence that this strain has been evolving over time through gene acquisition/loss; 69 genes were present in only one isolate, whereas 86 genes were present in only two of the three isolates.
Focusing our investigation of these genomes on characteristics advantageous to infection and persistence, our analysis of the accessory genome and virulence factors once again showed no distinction between those traits and symptom status. There was no gene frequently present (≥80%) in the UTI genomes that was not present in the non-UTI genomes. Prior investigations of the E. coli accessory genome have likewise found that symptom status and even source (urinary versus fecal) cannot reliably be distinguished by the accessory genome (Nielsen et al., 2017). Isolates from so-called asymptomatic bacteriuric individuals contain virulence factors despite the lack of symptoms (Yun et al., 2014). Here, we also have examined the genomes of E. coli isolates that represent a small population and/or fraction of the entire urobiome community. No distinction in virulence factors was observed between E. coli isolates from urobiomes in which E. coli was prevalent (≥100,000 CFU/ml) or dominant and those for which it was not. Together, this suggests that E. coli strains within the bladder have the potential for virulence, although they may not presently cause UTI symptoms.
While the E. coli genomes suggested that many of our isolates were resistant to antibiotics, these predictions were not significantly different between symptom states for the 9 classes of antibiotics considered (Supplementary Table 5). From our subsequent testing of 5 antibiotics, distinction between antibiotic resistance of isolates from women with or without UTI symptoms was not statistically significant. Bioinformatic predictions showed their limitations here. For example, more strains were resistant to ciprofloxacin and sulfamethoxazole-trimethoprim (16 and 32, respectively) than predicted (3 and 26, respectively). Neither the frequency of resistance predicted or observed within the no LUTS, OAB, and UUI cohorts suggests that E. coli strains may persist as a minor constituent of the normal bladder flora after antibiotic treatment because of acquired resistance.
There is evidence that the gut of individuals can be a reservoir for E. coli and thus a source of UTI (Yamamoto et al., 1997; Larsen et al., 2014). Without paired stool samples for the isolates investigated here, we cannot directly determine the source of the E. coli isolates. As a proxy, we compared the genomes of these isolates to publicly available genome sequences for which there are currently >19,000 from a wide variety of sources. While the most hits (36%) were to isolates from urine (Table 3), these only constitute ∼6.5% of the publicly available genomes. A substantial number of hits to isolates from stool were also observed, 10 to human stool samples and 7 to stool samples from other animals. Colonization of the urinary tract by a gut bacterium necessitates motility. However, only 24 of our isolates encode for all of the flagellar and motility genes (Table 1). For example, nine isolates lack fliC, which is required for the production of flagella (Lane et al., 2005). Two other isolates do not encode for flgM, a situation that hinders motility (Lane et al., 2005). Our investigation into these flagella-related genes suggest limited if any motility. However, as paired studies have shown, there is more than one way a UPEC strain can cause UTI recurrence. In addition to an external source, UPEC strains may persist within the urinary tract (Thänert et al., 2019).
Here, we found that neither E. coli presence nor E. coli genomic variation can predict UTI symptoms. Our analysis of urobiome compositions found that E. coli was a weak predictor of UTI status. Prior comparisons of urobiomes from women with or without UTI symptoms identified several differences in bacteria abundances including Aerococcus urinae, Klebsiella pneumoniae, Enterococcus faecalis, E. coli, Staphylococcus aureus, Streptococcus agalactiae, and Streptococcus anginosus (Price et al., 2016). These taxa include several emerging uropathogens that have increasingly been associated with UTIs (see review; Kline and Lewis, 2016). Furthermore, UTI diagnosis based upon E. coli abundance alone ignores recent evidence that UTI symptoms can be the result of more than one bacterial species, also known as a polymicrobial infection (see review; Kline and Lewis, 2016).
Because E. coli presence, abundance, and genomic content are weak predictors of UTI status, we hypothesize that UTI symptoms are more likely the result of multiple factors, including the urobiome composition. The urobiome appears to protect against post-treatment UTI (Brubaker et al., 2014; Thomas-White et al., 2018b). For instance, we previously observed that Lactobacillus crispatus is frequently in the urobiomes of women with no LUTS (Pearce et al., 2014). Prior studies have found that Lactobacilli can inhibit the growth of E. coli (Atassi et al., 2006, 2019; Cadieux et al., 2009; Hudson et al., 2020). In contrast, E. faecalis can promote E. coli growth under conditions for which it would otherwise not be able to thrive (Keogh et al., 2016). It is likely that other interactions between members of the urobiome could inhibit or contribute to symptoms. Metagenomic sequencing of “clean catch” urine specimens from individuals with or without UTI symptoms has identified urobiome compositions associated with UTIs, often dominated by Proteobacteria (Moustafa et al., 2018). These compositions require further investigation to identify constituents contributing to symptoms. In addition to microbes, other factors must also be considered, e.g., urine components (Forsyth et al., 2018; Reitzer and Zimmern, 2019) and the human host’s genetics (Ragnarsdóttir et al., 2011; Adebayo et al., 2020). Transcriptomic and proteomic studies have already begun to identify transcript and protein level changes associated with infection (Bielecki et al., 2014; Subashchandrabose et al., 2014; Sintsova et al., 2019; see also review; Lo et al., 2017). With the aid of these other ‘omic technologies, the likely multiple causes of UTI symptoms can be determined, be it a single bacterial taxon or a particular microbial community.
The E. coli genomes, including the presence of virulence factors and antibiotic resistance genes, from the urobiomes of women with or without UTIs cannot be distinguished. Furthermore, the E. coli genomes found within the urobiomes of women with OAB and UUI, which have not been previously studied, do not differ from E. coli genomes of isolates from women with UTI symptoms. As our analysis shows, E. coli’s ability to dominate the community and cause symptoms cannot be associated with a gene or set of genes. While UTI can be the result of colonization of E. coli, it is likely that UTI symptoms can be caused by multiple different bacteria. Recent evidence supports this hypothesis. The growing evidence of E. coli in the urobiomes of women without UTI symptoms suggests that E. coli may be a persistent member of the urinary community. This suggests that other constituents of the urobiome may directly or indirectly contribute to the development of UTI symptoms. Future investigations of the etiology of UTI symptoms should consider the entire urobiome.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, GCF_003885035, GCF_003885055, GCF_003885095, GCF_003885125, GCF_003885145, GCF_003885155, GCF_003 885195, GCF_003885215, GCF_003885225, GCF_003885245, GCF_003885255, GCF_003885295, GCF_003885305, GCF_0038 85875, GCF_003885915, GCF_003885965, GCF_003885995, GC F_003886005, GCF_003886015, GCF_003886035, GCF_003886 045, GCF_003886095, GCF_003886105, GCF_003886115, GC F_003886135, GCF_003886175, GCF_003886185, GCF_00388 6195, GCF_003886225, GCF_003886245, GCF_003886275, GC F_003886285, GCF_003886295, GCF_003886325, GCF_00388 6345, GCF_003886375, GCF_003886385, GCF_003886395, GC F_003886435, GCF_003886445, GCF_003886455, GCF_003886 495, GCF_003886515, GCF_003886535, GCF_003886545, GC F_003886565, GCF_003886615, GCF_003886635, GCF_00388 6655, GCF_003886675, GCF_003886695, GCF_003886735, GC F_003892355, GCF_003892375, GCF_003892435, GCF_00389 2445, GCF_003892455, GCF_003892475, GCF_003892485, GC F_003892535, GCF_003892545, GCF_003892555, GCF_00389 2595, GCF_003892605, GCF_003892635, and GCF_003892645.
Author Contributions
AGa and CP conceived of the experiment and drafted the manuscript. AGa, TM-E, AE, ZM, AS, AGe, JWS, and SB performed the data analyses. AGa, TM-E, AE, BB, GJ, RM, LM, MB, and NS assisted with experimental assays. CM assisted with writing and editing of the manuscript. AJW provided the samples and assisted with editing of the manuscript. All other authors assisted with the DNA extractions and genome assemblies as part of Loyola University Chicago’s Bacterial Genomics course. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Science Foundation (award # 1661357) to CP and the National Institutes of Health (award # R01DK104718) to AJW. AGa was supported by Loyola University’s Carbon Research Fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
Acknowledgments
Genome sequencing was conducted as part of Loyola University Chicago’s Department of Biology Bacterial Genomics course. For prior patient recruitment, we acknowledge the Loyola Urinary Education and Research Collaborative (LUEREC) and the patients who provided the samples for this study. We thank Gina Kuffel at Loyola’s Genomics Facility for her assistance in sequencing these isolates.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.02094/full#supplementary-material
Footnotes
- ^ https://github.com/najoshi/sickle
- ^ https://sourceforge.net/projects/bbmap/
- ^ www.scipy.org
- ^ https://www.R-project.org/
References
Adebayo, A., Ackermann, G., Bowyer, R., Wells, P., Humphreys, G., Knight, R., et al. (2020). The urinary tract microbiome in older women exhibits host genetics and environmental influences. Cell Host Microbe 28, 298–305. doi: 10.1016/j.chom.2020.06.022
Aho, K., Derryberry, D., and Peterson, T. (2014). Model selection for ecologists: the worldviews of AIC and BIC. Ecology 95, 631–636. doi: 10.1890/13-1452.1
Akaike, H. (1974). A new look at the statistical model identification. IEEE Trans. Autom. Control 19, 716–723. doi: 10.1109/TAC.1974.1100705
Atassi, F., Brassart, D., Grob, P., Graf, F., and Servin, A. L. (2006). Vaginal Lactobacillus isolates inhibit uropathogenic Escherichia coli. FEMS Microbiol. Lett. 257, 132–138. doi: 10.1111/j.1574-6968.2006.00163.x
Atassi, F., Pho Viet Ahn, D. L., and Lievin-Le Moal, V. (2019). Diverse expression of antimicrobial activities against bacterial vaginosis and urinary tract infection pathogens by cervicovaginal microbiota strains of Lactobacillus gasseri and Lactobacillus crispatus. Front. Microbiol. 10:2900. doi: 10.3389/fmicb.2019.02900
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barber, M. D., Kuchibhatla, M. N., Pieper, C. F., and Bump, R. C. (2001). Psychometric evaluation of 2 comprehensive condition-specific quality of life instruments for women with pelvic floor disorders. Am. J. Obstet. Gynecol. 185, 1388–1395. doi: 10.1067/mob.2001.118659
Battesti, A., Majdalani, N., and Gottesman, S. (2011). The RpoS-mediated general stress response in Escherichia coli. Annu. Rev. Microbiol. 65, 189–213. doi: 10.1146/annurev-micro-090110-102946
Bielecki, P., Muthukumarasamy, U., Eckweiler, D., Bielecka, A., Pohl, S., Schanz, A., et al. (2014). In vivo mRNA profiling of uropathogenic Escherichia coli from diverse phylogroups reveals common and group-specific gene expression profiles. mBio 5:e01075-14. doi: 10.1128/mBio.01075-14
Brubaker, L., Nager, C. W., Richter, H. E., Visco, A., Nygaard, I., Barber, M. D., et al. (2014). Urinary bacteria in adult women with urgency urinary incontinence. Int. Urogynecol. J. 25, 1179–1184. doi: 10.1007/s00192-013-2325-2
Brubaker, L., and Wolfe, A. J. (2017). The female urinary microbiota, urinary health and common urinary disorders. Ann. Transl. Med. 5:34. doi: 10.21037/atm.2016.11.62
Cadieux, P. A., Burton, J., Devillard, E., and Reid, G. (2009). Lactobacillus by-products inhibit the growth and virulence of uropathogenic Escherichia coli. J. Physiol. Pharmacol. 60(Suppl. 6), 13–18.
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Chen, S. L., Wu, M., Henderson, J. P., Hooton, T. M., Hibbing, M. E., Hultgren, S. J., et al. (2013). Genomic diversity and fitness of E. coli strains recovered from the intestinal and urinary tracts of women with recurrent urinary tract infection. Sci. Transl. Med. 5:184ra60. doi: 10.1126/scitranslmed.3005497
Chu, C. M., and Lowder, J. L. (2018). Diagnosis and treatment of urinary tract infections across age groups. Am. J. Obstetr. Gynecol. 219, 40–51. doi: 10.1016/j.ajog.2017.12.231
Clayson, D., Wild, D., Doll, H., Keating, K., and Gondek, K. (2005). Validation of a patient-administered questionnaire to measure the severity and bothersomeness of lower urinary tract symptoms in uncomplicated urinary tract infection (UTI): the UTI Symptom Assessment questionnaire. BJU Int. 96, 350–359. doi: 10.1111/j.1464-410X.2005.05630.x
Coorevits, L., Heytens, S., Boelens, J., and Claeys, G. (2017). The resident microflora of voided midstream urine of healthy controls: standard versus expanded urine culture protocols. Eur. J. Clin. Microbiol. Infect. Dis. 36, 635–639. doi: 10.1007/s10096-016-2839-x
Coyne, K., Revicki, D., Hunt, T., Corey, R., Stewart, W., Bentkover, J., et al. (2002). Psychometric validation of an overactive bladder symptom and health-related quality of life questionnaire: the OAB-q. Qual. Life Res. 11, 563–574. doi: 10.1023/a:1016370925601
Cramer, J. S. (2002). The Origins of Logistic Regression. Tinbergen Institute Working Paper No. 2002-119/4. doi: 10.2139/ssrn.360300
Dune, T. J., Price, T. K., Hilt, E. E., Thomas-White, K. J., Kliethermes, S., Brincat, C., et al. (2017). Urinary symptoms and their associations with urinary tract infections in urogynecologic patients. Obstet. Gynecol. 130, 718–725. doi: 10.1097/AOG.0000000000002239
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Eto, D. S., Jones, T. A., Sundsbak, J. L., and Mulvey, M. A. (2007). Integrin-mediated host cell invasion by type 1–piliated uropathogenic Escherichia coli. PLoS Pathog. 3:e100. doi: 10.1371/journal.ppat.0030100
Ferroni, M., and Taylor, A. K. (2015). Asymptomatic bacteriuria in noncatheterized adults. Urol. Clin. North Am. 42, 537–545. doi: 10.1016/j.ucl.2015.07.003
Flores-Mireles, A. L., Walker, J. N., Caparon, M., and Hultgren, S. J. (2015). Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 13, 269–284. doi: 10.1038/nrmicro3432
Forsyth, V. S., Armbruster, C. E., Smith, S. N., Pirani, A., Springman, A. C., Walters, M. S., et al. (2018). Rapid growth of uropathogenic Escherichia coli during human urinary tract infection. mBio 9:e00186-18. doi: 10.1128/mBio.00186-18
Fouts, D. E., Pieper, R., Szpakowski, S., Pohl, H., Knoblach, S., Suh, M.-J., et al. (2012). Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med. 10:174. doi: 10.1186/1479-5876-10-174
Foxman, B. (2014). Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect. Dis. Clin. North Am. 28, 1–13. doi: 10.1016/j.idc.2013.09.003
Hilt, E. E., McKinley, K., Pearce, M. M., Rosenfeld, A. B., Zilliox, M. J., Mueller, E. R., et al. (2014). Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J. Clin. Microbiol. 52, 871–876. doi: 10.1128/JCM.02876-13
Hooton, T. M. (2001). Recurrent urinary tract infection in women. Int. J. Antimicrob. Agents 17, 259–268. doi: 10.1016/s0924-8579(00)00350-2
Hudson, P. L., Hung, K. J., Bergerat, A., and Mitchell, C. (2020). Effect of vaginal Lactobacillus species on Escherichia coli growth. Female Pelvic Med. Reconstr. Surg. 26, 146–151. doi: 10.1097/SPV.0000000000000827
Joensen, K. G., Tetzschner, A. M. M., Iguchi, A., Aarestrup, F. M., and Scheutz, F. (2015). Rapid and easy In Silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 53, 2410–2426. doi: 10.1128/JCM.00008-15
Johnson, J. R., O’Bryan, T. T., Delavari, P., Kuskowski, M., Stapleton, A., Carlino, U., et al. (2001). Clonal relationships and extended virulence genotypes among Escherichia coli isolates from women with a first or recurrent episode of cystitis. J. Infect. Dis. 183, 1508–1517. doi: 10.1086/320198
Johnson, J. R., Porter, S., Johnston, B., Kuskowski, M. A., Spurbeck, R. R., Mobley, H. L. T., et al. (2015). Host characteristics and bacterial traits predict experimental virulence for Escherichia coli bloodstream isolates from patients with urosepsis. Open Forum Infect. Dis. 2:ofv083. doi: 10.1093/ofid/ofv083
Karstens, L., Asquith, M., Davin, S., Stauffer, P., Fair, D., Gregory, W. T., et al. (2016). Does the urinary microbiome play a role in urgency urinary incontinence and its severity? Front. Cell. Infect. Microbiol. 6:78. doi: 10.3389/fcimb.2016.00078
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keogh, D., Tay, W. H., Ho, Y. Y., Dale, J. L., Chen, S., Umashankar, S., et al. (2016). Enterococcal metabolite cues facilitate interspecies niche modulation and polymicrobial infection. Cell Host Microbe 20, 493–503. doi: 10.1016/j.chom.2016.09.004
Khasriya, R., Sathiananthamoorthy, S., Ismail, S., Kelsey, M., Wilson, M., Rohn, J. L., et al. (2013). Spectrum of bacterial colonization associated with urothelial cells from patients with chronic lower urinary tract symptoms. J. Clin. Microbiol. 51, 2054–2062. doi: 10.1128/JCM.03314-12
Kline, K. A., and Lewis, A. L. (2016). Gram-positive uropathogens, polymicrobial urinary tract infection, and the emerging microbiota of the urinary tract. Microbiol. Spectr. 4. doi: 10.1128/microbiolspec.UTI-0012-2012
Lane, M. C., Alteri, C. J., Smith, S. N., and Mobley, H. L. T. (2007). Expression of flagella is coincident with uropathogenic Escherichia coli ascension to the upper urinary tract. Proc. Natl. Acad. Sci. U.S.A. 104, 16669–16674. doi: 10.1073/pnas.0607898104
Lane, M. C., Lockatell, V., Monterosso, G., Lamphier, D., Weinert, J., Hebel, J. R., et al. (2005). Role of motility in the colonization of uropathogenic Escherichia coli in the urinary tract. Infect. Immun. 73, 7644–7656. doi: 10.1128/IAI.73.11.7644-7656.2005
Larsen, P., Dynesen, P., Nielsen, K. L., and Frimodt-Møller, N. (2014). Faecal Escherichia coli from patients with E. coli urinary tract infection and healthy controls who have never had a urinary tract infection. J. Med. Microbiol. 63, 582–589. doi: 10.1099/jmm.0.068783-0
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Lo, A. W., Moriel, D. G., Phan, M.-D., Schulz, B. L., Kidd, T. J., Beatson, S. A., et al. (2017). “Omic” approaches to study uropathogenic Escherichia coli virulence. Trends Microbiol. 25, 729–740. doi: 10.1016/j.tim.2017.04.006
Luo, Y., Ma, Y., Zhao, Q., Wang, L., Guo, L., Ye, L., et al. (2012). Similarity and divergence of phylogenies, antimicrobial susceptibilities, and virulence factor profiles of Escherichia coli isolates causing recurrent urinary tract infections that persist or result from reinfection. J. Clin. Microbiol. 50, 4002–4007. doi: 10.1128/JCM.02086-12
Marchesi, J. R., Sato, T., Weightman, A. J., Martin, T. A., Fry, J. C., Hiom, S. J., et al. (1998). Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl. Environ. Microbiol. 64, 795–799.
Moriel, D. G., Tan, L., Goh, K. G. K., Phan, M.-D., Ipe, D. S., Lo, A. W., et al. (2016). A novel protective vaccine antigen from the core Escherichia coli genome. mSphere 1:e00326-16. doi: 10.1128/mSphere.00326-16
Moustafa, A., Li, W., Singh, H., Moncera, K. J., Torralba, M. G., Yu, Y., et al. (2018). Microbial metagenome of urinary tract infection. Sci. Rep. 8:4333. doi: 10.1038/s41598-018-22660-8
Mysorekar, I. U., and Hultgren, S. J. (2006). Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. U.S.A. 103, 14170–14175. doi: 10.1073/pnas.0602136103
Nicolle, L. E. (2003). Asymptomatic bacteriuria: when to screen and when to treat. Infect. Dis. Clin. N. Am. 17, 367–394.
Nicolle, L. E., Gupta, K., Bradley, S. F., Colgan, R., DeMuri, G. P., Drekonja, D., et al. (2019). Clinical practice guideline for the management of asymptomatic bacteriuria: 2019 update by the infectious diseases society of America. Clin. Infect. Dis. 68, e83–e110. doi: 10.1093/cid/ciy1121
Nielsen, K. L., Dynesen, P., Larsen, P., and Frimodt-Møller, N. (2014). Faecal Escherichia coli from patients with E. coli urinary tract infection and healthy controls who have never had a urinary tract infection. J. Med. Microbiol. 63, 582–589. doi: 10.1099/jmm.0.068783-0
Nielsen, K. L., Stegger, M., Kiil, K., Godfrey, P. A., Feldgarden, M., Lilje, B., et al. (2017). Whole-genome comparison of urinary pathogenic Escherichia coli and faecal isolates of UTI patients and healthy controls. Int. J. Med. Microbiol. 307, 497–507. doi: 10.1016/j.ijmm.2017.09.007
Nienhouse, V., Gao, X., Dong, Q., Nelson, D. E., Toh, E., McKinley, K., et al. (2014). Interplay between bladder microbiota and urinary antimicrobial peptides: mechanisms for human urinary tract infection risk and symptom severity. PLoS One 9:e114185. doi: 10.1371/journal.pone.0114185
Park, H., McGibbon, L. C., Potts, A. H., Yakhnin, H., Romeo, T., and Babitzke, P. (2017). Translational repression of the RpoS antiadapter IraD by CsrA is mediated via translational coupling to a short upstream open reading frame. mBio 8:e01355-17. doi: 10.1128/mBio.01355-17
Pearce, M. M., Hilt, E. E., Rosenfeld, A. B., Zilliox, M. J., Thomas-White, K., Fok, C., et al. (2014). The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. mBio 5:e01283-14. doi: 10.1128/mBio.01283-14
Pearce, M. M., Zilliox, M. J., Rosenfeld, A. B., Thomas-White, K. J., Richter, H. E., Nager, C. W., et al. (2015). The female urinary microbiome in urgency urinary incontinence. Am. J. Obstet. Gynecol. 213, 347.e1–347.e11. doi: 10.1016/j.ajog.2015.07.009
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Price, T. K., Dune, T., Hilt, E. E., Thomas-White, K. J., Kliethermes, S., Brincat, C., et al. (2016). The clinical urine culture: enhanced techniques improve detection of clinically relevant microorganisms. J. Clin. Microbiol. 54, 1216–1222. doi: 10.1128/JCM.00044-16
Price, T. K., Hilt, E. E., Thomas-White, K., Mueller, E. R., Wolfe, A. J., and Brubaker, L. (2020). The urobiome of continent adult women: a cross-sectional study. BJOG 127, 193–201. doi: 10.1111/1471-0528.15920
Ragnarsdóttir, B., Lutay, N., Grönberg-Hernandez, J., Köves, B., and Svanborg, C. (2011). Genetics of innate immunity and UTI susceptibility. Nat. Rev. Urol. 8, 449–468. doi: 10.1038/nrurol.2011.100
Rasko, D. A., Rosovitz, M. J., Myers, G. S. A., Mongodin, E. F., Fricke, W. F., Gajer, P., et al. (2008). The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 190, 6881–6893. doi: 10.1128/JB.00619-08
Reitzer, L., and Zimmern, P. (2019). Rapid growth and metabolism of uropathogenic Escherichia coli in relation to urine composition. Clin. Microbiol. Rev. 33:e00101-19. doi: 10.1128/CMR.00101-19
Rubin, R. H., Shapiro, E. D., Andriole, V. T., Davis, R. J., and Stamm, W. E. (1992). Evaluation of new anti-infective drugs for the treatment of urinary tract infection. Infectious Diseases Society of America and the Food and Drug Administration. Clin. Infect. Dis. 15(Suppl. 1), S216–S227. doi: 10.1093/clind/15.supplement_1.s216
Schreiber, H. L., Conover, M. S., Chou, W.-C., Hibbing, M. E., Manson, A. L., Dodson, K. W., et al. (2017). Bacterial virulence phenotypes of Escherichia coli and host susceptibility determine risk for urinary tract infections. Sci. Transl. Med. 9:eaaf1283. doi: 10.1126/scitranslmed.aaf1283
Sintsova, A., Frick-Cheng, A. E., Smith, S., Pirani, A., Subashchandrabose, S., Snitkin, E. S., et al. (2019). Genetically diverse uropathogenic Escherichia coli adopt a common transcriptional program in patients with UTIs. eLife 8:e49748. doi: 10.7554/eLife.49748
Subashchandrabose, S., Hazen, T. H., Brumbaugh, A. R., Himpsl, S. D., Smith, S. N., Ernst, R. D., et al. (2014). Host-specific induction of Escherichia coli fitness genes during human urinary tract infection. Proc. Natl. Acad. Sci. U.S.A. 111, 18327–18332. doi: 10.1073/pnas.1415959112
Subashchandrabose, S., and Mobley, H. L. T. (2015). Virulence and fitness determinants of uropathogenic Escherichia coli. Microbiol. Spectr. 3. doi: 10.1128/microbiolspec.UTI-0015-2012
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Terlizzi, M. E., Gribaudo, G., and Maffei, M. E. (2017). UroPathogenic Escherichia coli (UPEC) infections: virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front. Microbiol. 8:1566. doi: 10.3389/fmicb.2017.01566
Thänert, R., Reske, K. A., Hink, T., Wallace, M. A., Wang, B., Schwartz, D. J., et al. (2019). Comparative genomics of antibiotic-resistant uropathogens implicates three routes for recurrence of urinary tract infections. mBio 10:e01977-19. doi: 10.1128/mBio.01977-19
Thomas-White, K. J., Forster, S. C., Kumar, N., Van Kuiken, M., Putonti, C., Stares, M. D., et al. (2018a). Culturing of female bladder bacteria reveals an interconnected urogenital microbiota. Nat. Commun. 9:1557. doi: 10.1038/s41467-018-03968-5
Thomas-White, K. J., Gao, X., Lin, H., Fok, C. S., Ghanayem, K., Mueller, E. R., et al. (2018b). Urinary microbes and postoperative urinary tract infection risk in urogynecologic surgical patients. Int. Urogynecol. J. 29, 1797–1805. doi: 10.1007/s00192-018-3767-3
Thomas-White, K. J., Hilt, E. E., Fok, C., Pearce, M. M., Mueller, E. R., Kliethermes, S., et al. (2016). Incontinence medication response relates to the female urinary microbiota. Int. Urogynecol. J. 27, 723–733. doi: 10.1007/s00192-015-2847-x
Thomas-White, K. J., Kliethermes, S., Rickey, L., Lukacz, E. S., Richter, H. E., Moalli, P., et al. (2017). Evaluation of the urinary microbiota of women with uncomplicated stress urinary incontinence. Am. J. Obstet. Gynecol. 216, 55.e1–55.e16. doi: 10.1016/j.ajog.2016.07.049
Uebersax, J. S., Wyman, J. F., Shumaker, S. A., McClish, D. K., and Fantl, J. A. (1995). Short forms to assess life quality and symptom distress for urinary incontinence in women: the incontinence impact questionnaire and the urogenital distress inventory. Continence Program for Women Research Group. Neurourol. Urodyn. 14, 131–139. doi: 10.1002/nau.1930140206
Waters, N. R., Abram, F., Brennan, F., Holmes, A., and Pritchard, L. (2020). Easy phylotyping of Escherichia coli via the EzClermont web app and command-line tool. Access Microbiol. doi: 10.1099/acmi.0.000143
Wolfe, A. J., Toh, E., Shibata, N., Rong, R., Kenton, K., Fitzgerald, M., et al. (2012). Evidence of uncultivated bacteria in the adult female bladder. J. Clin. Microbiol. 50, 1376–1383. doi: 10.1128/JCM.05852-11
Yamamoto, S., Tsukamoto, T., Terai, A., Kurazono, H., Takeda, Y., and Yoshida, O. (1997). Genetic evidence supporting the fecal-perineal-urethral hypothesis in cystitis caused by Escherichia coli. J. Urol. 157, 1127–1129.
Yun, K. W., Kim, H. Y., Park, H. K., Kim, W., and Lim, I. S. (2014). Virulence factors of uropathogenic Escherichia coli of urinary tract infections and asymptomatic bacteriuria in children. J. Microbiol. Immunol. Infect. 47, 455–461. doi: 10.1016/j.jmii.2013.07.010
Keywords: E. coli, UPEC, UTI, urinary microbiome, urobiome
Citation: Garretto A, Miller-Ensminger T, Ene A, Merchant Z, Shah A, Gerodias A, Biancofiori A, Canchola S, Canchola S, Castillo E, Chowdhury T, Gandhi N, Hamilton S, Hatton K, Hyder S, Krull K, Lagios D, Lam T, Mitchell K, Mortensen C, Murphy A, Richburg J, Rokas M, Ryclik S, Sulit P, Szwajnos T, Widuch M, Willis J, Woloszyn M, Brassil B, Johnson G, Mormando R, Maskeri L, Batrich M, Stark N, Shapiro JW, Montelongo Hernandez C, Banerjee S, Wolfe AJ and Putonti C (2020) Genomic Survey of E. coli From the Bladders of Women With and Without Lower Urinary Tract Symptoms. Front. Microbiol. 11:2094. doi: 10.3389/fmicb.2020.02094
Received: 29 May 2020; Accepted: 10 August 2020;
Published: 04 September 2020.
Edited by:
François J. M. A. Meurens, UMR INRAE-Oniris 1300 Oniris – Nantes Atlantic National College of Veterinary Medicine, FranceReviewed by:
Bert Devriendt, Ghent University, BelgiumJo-Anne Renaud Dillon, University of Saskatchewan, Canada
Pierre Germon, Institut National de la Recherche Agronomique (INRA), France
Copyright © 2020 Garretto, Miller-Ensminger, Ene, Merchant, Shah, Gerodias, Biancofiori, Canchola, Canchola, Castillo, Chowdhury, Gandhi, Hamilton, Hatton, Hyder, Krull, Lagios, Lam, Mitchell, Mortensen, Murphy, Richburg, Rokas, Ryclik, Sulit, Szwajnos, Widuch, Willis, Woloszyn, Brassil, Johnson, Mormando, Maskeri, Batrich, Stark, Shapiro, Montelongo Hernandez, Banerjee, Wolfe and Putonti.
*Correspondence: Catherine Putonti, Y3B1dG9udGlAbHVjLmVkdQ==
†Present address: Andrea Garretto, Department of Microbiology & Immunology, University of Michigan, Ann Arbor, MI, United States; Nicole Stark, Department of Biology, Indiana University, Bloomington, IN, United States