 Jorge G. Carvajal1
Jorge G. Carvajal1 Alberto J. Alaniz2
Alberto J. Alaniz2 Mario A. Carvajal1
Mario A. Carvajal1 Emily S. Acheson3
Emily S. Acheson3 Rodrigo Cruz4
Rodrigo Cruz4 Pablo M. Vergara1
Pablo M. Vergara1 Massimo Cogliati5*
Massimo Cogliati5*- 1Facultad Tecnológica, Universidad de Santiago de Chile, Santiago, Chile
- 2Facultad de Ingeniería, Universidad de Santiago de Chile, Santiago, Chile
- 3Department of Geography, The University of British Columbia, Vancouver, BC, Canada
- 4Laboratorio de Micología, Universidad de Valparaíso, Valparaíso, Chile
- 5Dipartimento di Scienze Biomediche per la Salute, Università degli Studi di Milano, Milan, Italy
In 2018 the fungal pathogen Cryptococcus bacillisporus (AFLP5/VGIII) was isolated for the first time in Chile, representing the only report in a temperate region in South America. We reconstructed the colonization process of C. bacillisporus in Chile, estimating the phylogenetic origin, the potential spread zone, and the population at risk. We performed a phylogenetic analysis of the strain and modeled the environmental niche of the pathogen projecting its potential spread zone into the new colonized region. Finally, we generated risk maps and quantified the people under potential risk. Phylogenetic analysis showed high similarity between the Chilean isolate and two clonal clusters from California, United States and Colombia in South America. The pathogen can expand into all the temperate Mediterranean zone in central Chile and western Argentina, exposing more than 12 million people to this pathogen in Chile. This study has epidemiological and public health implications for the response to a potential C. bacillisporus outbreak, optimizing budgets, routing for screening diagnosis, and treatment implementation.
Author Summary
Fungal diseases have been neglected over the last years, being cryptococcal disease a cause of high impact in global health. Here we determined the phylogenetic origin of the first C. bacillisporus strain found in a veterinary isolation in Chile which was identified as a new multi-locus sequence typing of a VGIII genotype closely related to two clonal clusters from California United States, and Colombia. Then, we found that the pathogen can expand into all the temperate Mediterranean zone of central Chile and western Argentina from 29.5° to 36.6° south latitude. Although no human case of cryptococcosis by C. bacillisporus have been reported in Chile, in last decades this pathogen has emerged in locations where veterinary cases were reported, affecting both immunocompromised and immunocompetent humans. For this reason, we generated risk maps and quantified the population under different exposure risk levels, finding that more than 12 million people are in risk in Chile, which 74% of them are in a very high risk level.
Introduction
The pathogenic Cryptococcus species complexes, Cryptococcus neoformans and Cryptococcus gattii, are the etiological agents of cryptococcosis, an invasive fungal disease with a high mortality rate and high public health costs worldwide (Mourad, 2018). These fungi generate different clinical and epidemiological consequences, which depend mainly on the immunity of the host and the pathogenicity of the agent (Rajasingham et al., 2017; George et al., 2018). The C. gattii species complex includes basidiomycetous yeasts that can be isolated from different environmental sources such as soil, decaying organic material and tree hollows, as well as from infected human and animal hosts where they grow as asexual budding yeasts (Cogliati, 2013). During sexual reproduction, this fungus presents a hyphal growth generating basidiospores which, together with asexual blastospores, play an important role as infectious propagules being dispersed by wind and then inhaled by hosts (Velagapudi et al., 2009; Voelz et al., 2013; You and Xu, 2018). Unlike cryptococcosis caused by the yeasts belonging to the C. neoformans species complex, which affects mostly immunocompromised hosts, C. gattii species complex strains also have the capacity to affect immunocompetent hosts as reported by recent studies (Mcmullan et al., 2013; Chen et al., 2014; Li et al., 2020). The C. gattii species complex has largely been historically restricted to tropical and subtropical regions, however records from 1990 in San Francisco California (United States), suggest their expansion into temperate zones in America (Pfeiffer and Ellis, 1991; Acheson et al., 2019). Since then, the most important outbreaks in the temperate zone were British Columbia Canada and the Northwest Pacific coast of the United States both in 1999, affecting immunocompromised and immunocompetent hosts and generating a public health alarm (Espinel-ingroff and Kidd, 2015). The species complex can be subdivided into two serotypes, B and C, based on the capsular antigenic pattern, and into five species named Cryptococcus gattii (VGI), Cryptococcus deuterogattii (VGII), Cryptococcus bacillisporus (VGIII), Cryptococcus tetragattii (VGIV), Cryptococcus decagattii (VGIV/VGIIIc) depending on molecular typing using different techniques (Boekhout et al., 2001; Meyer et al., 2003; Meyer et al., 2009; Feng et al., 2013; Cogliati et al., 2014; Firacative et al., 2016; Hagen et al., 2015). In addition, a further putative cryptic species denoted VGV has been recently identified (Farrer et al., 2019). However, this new taxonomy needs to be further validated and it is not yet accepted by the whole scientific community (Kwon-Chung et al., 2017). In North America, the preponderant species from clinical, veterinary, and environmental isolates are C. deuterogattii and C. bacillisporus. The former, C. deuterogattii, was identified in British Columbia, Canada and in Washington and Oregon in the United States (Kidd et al., 2004; Byrnes and Marr, 2011). By comparison, C. bacillisporus (VGIII) was identified in patients with HIV/AIDS in the United States, especially in Southern California, with fewer cases in Oregon, Washington, New Mexico, New Jersey, Michigan, and Alaska. Outside the United States, infections were reported in Latin America from Mexico, Colombia, Brazil, Paraguay, Argentina, Venezuela, and Guatemala (Byrnes et al., 2011; Walraven et al., 2011; Lockhart et al., 2013; Lizarazo et al., 2014; Singer et al., 2014; Springer et al., 2014; González et al., 2016; Vélez and Escandón, 2017; Berejnoi et al., 2019). However, a recent veterinary isolation in a domestic animal was reported for the first time in central Chile, in the locality of Limache, 15 kilometers south of the city of Valparaíso (30°59′S, 71°17′W), extending its southern geographical limit to a geographical area where it has never been recorded before (non-endemic species) (Vieille et al., 2018). This finding has generated concern in Chilean public health institutions and among researchers, mainly because this is the first time that a C. bacillisporus has been recorded in another American temperate zone. The similar climate conditions between Chile’s Mediterranean zone and California suggests a potential dispersal of the pathogen favored by similar climatic conditions and hence possible environmental suitability. Previous studies have identified the spatial distribution of C. neoformans and C. gattii species complex distributions in Europe through the ecological niche modeling of their bioclimatic conditions suitable for their survival (Cogliati et al., 2017; Alaniz et al., 2020), methodology that has also been succesfully applied to other recent global infectious diseases (Alaniz et al., 2017, 2018). Considering the latter, the aim of this study is to analyze: (i) the geographical and phylogenetic origin of the sample; (ii) the potential geographical spread in this new area; and (iii) the population exposed to the risk. The achievement of these objectives may contribute to generate focused surveillance and preventive actions that could help to understand the potential negative health consequences in the population.
Materials and Methods
Geographical Origin, Source, and Identification of the Chilean C. bacillisporus Isolate
The first autochthonous C. bacillisporus isolate (VGIII) was recently identified in Chile from a nasal injury in a domestic cat (Vieille et al., 2018). Mucocutaneous cryptococcosis was suspected in the animal, so nasal samples were taken to perform mycological studies. The nasal swab was analyzed in the Mycology Laboratory of the Universidad de Valparaíso, where the sample was cultured on Sabouraud dextrose agar under optimal conditions (37°C) obtaining encapsulated yeast cells after 48 h. The sample was sub-cultured on Staib agar, urea agar, and canavanine-glycine-bromothymol blue agar, and incubated at 30°C which, after 7 days, led to the classification of the yeast as C. gattii species complex. The isolate was then identified as C. bacillisporus, molecular type VGIII, by restriction fragment length polymorphism of URA5 and confirmed by duplex PCR (Meyer et al., 2003; Feng et al., 2013). Mating type allelic pattern αC was determined by multiplex PCR as described elsewhere (Cogliati et al., 2014).
Phylogenetic Analysis
To analyze the possible origin of the new Chilean C. bacillisporus isolate, we performed a phylogenetic analysis through the standardized genotyping technique called multi locus sequence typing (MLST) (Meyer et al., 2009), applied using the standard ISHAM MLST scheme by sequencing seven house-keeping loci (CAP59, GPD1, IGS1, LAC1, PLB1, SOD1, URA5) (Meyer et al., 2009). Allele types for each locus and the sequence type (ST), determined by the combination of the seven allele types, were assigned by matching the sequences with those present in the C. gattii MLST database1. A phylogenetic tree was reconstructed comparing the ST of the Chilean isolate with 69 different STs from VGIII global isolates previously reported (Byrnes et al., 2011; Lizarazo et al., 2014; Singer et al., 2014; Springer et al., 2014; González et al., 2016). Analysis was performed by MEGA software v6.062 using maximum likelihood algorithm, Tamura-Nei model, nearest-neighbor-interchange method starting from a neighbor-joining tree for tree inference, and bootstrap analysis on 1000 random repeats as phylogeny test. Allele type combinations were also input in Phyloviz software v2.03 to construct a minimum spanning tree in order to recognize clonal clusters among VGIII STs as well as the most probable founder genotypes.
Potential Geographical Spread in Chile
In order to predict the distribution range of C. bacillisporus, we used species distribution models (SDM) based on the maximum entropy algorithm with MaxEnt 3.4.1k software (Phillips et al., 2006). MaxEnt requires two types of input data: occurrence points of the organisms and environmental variables which constitute the predictors. The aim is to predict the environmental suitability for the species based on its ecological niche requirements. The spatial prediction of an SDM can be homologated to the potential abundance of organisms (Phillips et al., 2006), and it has proven to generate reliable results for the modeling of infectious disease vectors (Alaniz et al., 2017, 2018, 2020). We compiled and systematized an occurrence database consisting of 29 occurrences of C. bacillisporus environmental isolates according to the following criteria: (A) avoiding isolates from humans due to the uncertainty related to the movement of the patient, however we included four human occurrences from Lockhart et al., 2013 which denoted no travel history of patients, (B) excluding those without specific geographical coordinates (latitude and longitude) reducing the uncertainty on the location and (C) excluding the Chilean isolate to predict the distribution based on the source zone niche (Supplementary Appendix Figure S1 and Supplementary Appendix Table S1) (Pfeiffer and Ellis, 1991; Escandón et al., 2006; Mazza et al., 2013; Singer et al., 2014; Springer et al., 2014; Espinel-ingroff and Kidd, 2015). The environmental variables used as predictors of the spatial distribution model consisted of the bioclimatic layers of the WorldClim 2 project4 with 2.5 arc minutes spatial resolution (approximately 5 km × 5 km cells) (Fick, 2017), plus relative humidity (RH), enhanced vegetation index (EVI), mean annual solar radiation (SR), topographic diversity index (TDI), vegetation continuous fields (VCF), net primary productivity (NPP), mean annual wind speed, elevation, soil bulk density, soil organic carbon at 0–15 cm depth and mean soil pH at 0–15 cm depth (Supplementary Appendix Table S2). All the remote sensing variables such as VCF, TDI, NPP, and EVI were processed in Google Earth Engine Platform (Gorelick et al., 2017). To improve the quality of the data used for the modeling process, we carried out two measures. First, we reduced the spatial autocorrelation and geographical bias of the occurrences dataset by applying a spatial rarefy function in a GIS environment, retaining only the occurrences that were separated by at least 15 km. Second, we reduced the collinearity between predictor variables by generating a preliminary model with the complete set of variables. According to the percent contribution of each variable combined with a correlation matrix using the Shapiro–Wilk (normality distribution) test and Spearman (correlation) test, we excluded variables with a low percent of contribution and a high correlation coefficient (more than ± 0.7 in the correlogram). The final model was constructed using a bootstrap resampling method and retaining the values according to the 95% confidence interval. The accuracy of the model was assessed from the area under the curve (AUC) of the receiver operating characteristic, which estimates the sensitivity and specificity of the model by partitioning the occurrences dataset into a training and a test dataset, the latter used exclusively for model evaluation. Finally, we used the habitat suitability predicted by the model in Chile to estimate the potential spread of the C. bacillisporus. In the already occupied zones of the United States, we considered the suitability predicted by the model as the geographic distribution of the pathogen.
Estimation of the Population Under Exposure Risk
To estimate the risk, we included two components: (i) the potential abundance of the pathogen (threat); and (ii) the human population density (vulnerability). To assess the risk, we used human population density grid (version 4 of the year 2020) with a spatial resolution of 2.5 arc minutes from Socioeconomic Data and Application Centre of NASA5. We classified human population density in four levels: null (0–1 inhabitants/km2), low (>1–10 inhabitants/km2), medium (>10–100 inhabitants/km2), and high (>100 inhabitants/km2). Then, we assigned a numerical value to each of these categories (null = 0, low = 1, medium = 2, high = 3). On the other hand, we reclassified the suitability map of C. bacillisporus into four levels (null, low, medium, and high). The null level corresponded to the non-significant suitability value, considering as threshold the values under the 10th percentile. Then, the significant suitability values (>10th percentile) were divided into three categories considering equal intervals. We assigned a numeric value to each one of these suitability categories (null = 0, low = 1, medium = 2, and high = 3). Finally, we used a double entry matrix to multiply both reclassified rasters (Supplementary Appendix Figure S2) (population density and suitability) in a geographic information system. As product we obtained a map of potential exposure risk with five risk levels: very low, low, medium, high, and very high (Alaniz et al., 2017).
To quantify population at different risk levels we used the population count by square kilometer of NASA, which was overlapped with the generated exposure risk map. Population count product corresponds to an estimation of population per pixel based on national and subnational censuses and projected to the year 2020 (Doxsey-Whitfield et al., 2015). We quantified the population under risk exposure in both colonized (Chile) and the potential source (United States) zones, considering subnational administrative division per level of risk. For the product, we generated a series of maps and tables showing the population under risk per subnational administrative division and level of risk.
Results
Chilean Isolate Originated From One of the Two Major VGIII Clusters
Phylogenetic analysis of global C. bacillisporus isolates showed the presence of two major clusters reported as B1 and C1 (Figure 1), and further five minor clusters (B2, B3, C2, C3, and C4). Cluster B1 included 31 STs belonging to serotype B isolates mainly from United States and Mexico, cluster B2 included 3 STs from United States, and B3 grouped two phylogenetically distant STs (ST114 and ST64). Cluster C1 included 25 STs belonging to serotype C isolates from North and South America, cluster C2 was represented by two STs of isolates from Mexico and United States, and C4 grouped four STs from Mexico which are genetically very distant from the other VGIII STs. The Chilean C. bacillisporus (VGIII) isolate presented a new MLST profile (ST552: CAP59-20, GPD1-23, IGS1-112, LAC1-23, PLB1-23, SOD1-29, URA5-21), which did not match any of the profiles in the MLST database. Observing the phylogenetic tree, it was included in cluster C3, genetically related to cluster C1, together with an isolate from Paraguay belonging to genotype ST67, differing in IGS1 and PLB1 loci presenting one single nucleotide polymorphism each.
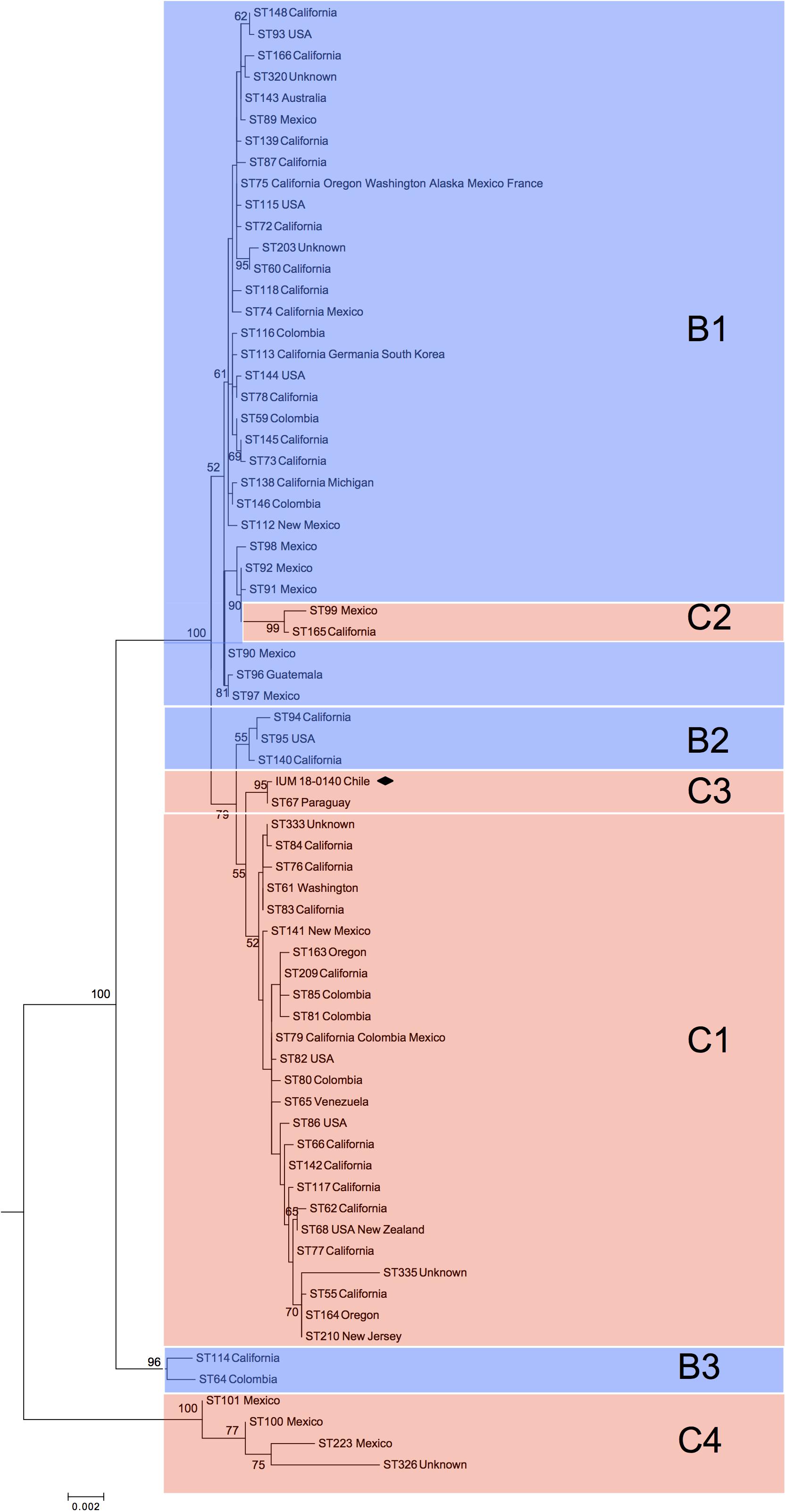
Figure 1. Phylogenetic reconstruction by maximum likelihood method resulting from the comparison of the Chilean isolate (black square) with 69 global Cryptococcus bacillisporus (VGIII) STs. C1, C2, C3, and C4 identify clusters including serotype C strains, while B1, B2, and B3 identify clusters including serotype B strains. Results of bootstrap analysis (>50%) are indicated beside each node.
The minimum spanning tree showed a similar picture with two main clonal clusters including most of the VGIII STs (Figure 2). Clonal cluster CC75 grouped 30 STs probably all originated from ST75 shared by a large number of VGIII isolates from the United States and Mexico. The probable founder for the second clonal cluster (CC79) was ST79, which was shared by both South and North American isolates and gave rise to the small cluster containing the Chilean and Paraguayan isolates.
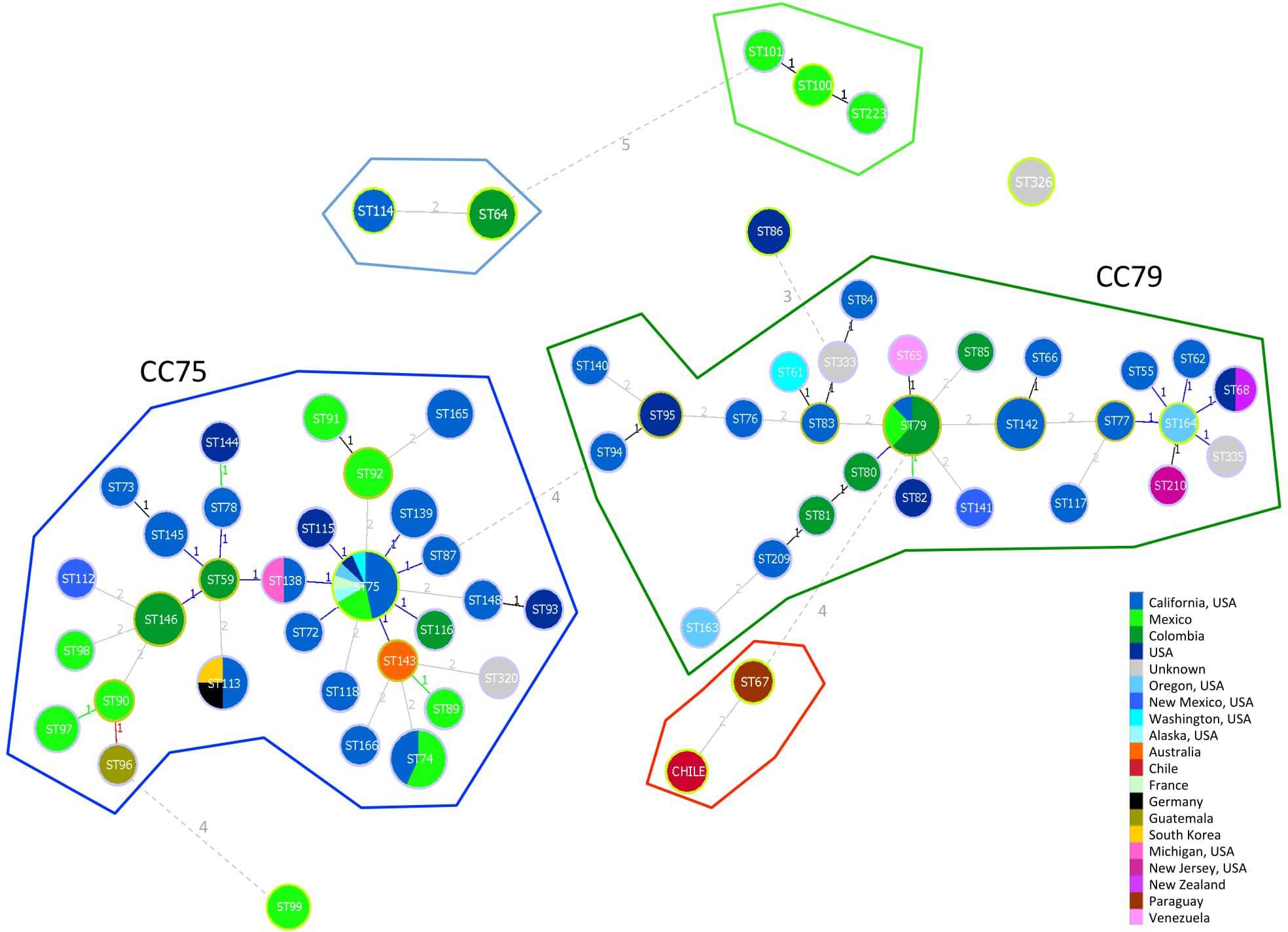
Figure 2. Minimum spanning tree reporting the main clonal clusters among C. bacillisporus VGIII sequence types. Circle sizes are proportional to the number of strains with the same sequence type. Different colors represent different geographic origins. Numbers beside the lines linking two circles indicate the number of different alleles. Yellow circles surrounding STs identify the founder of a clonal cluster. Clonal clusters, including STs differing in one or two alleles, are grouped inside closed lines with different colors.
Cryptococcus bacillisporus Potentially Colonizing Central Chile Region
The generated model reached an AUC = 0.979 ± 0.009 (Supplementary Appendix Figure S3), showing less than 20% uncertainty in the areas with the highest predicted suitability values (United States and Chile) (Supplementary Appendix Figure S4). The variables that mostly contributed to the final model were precipitation in the warmest quarter, precipitation in the coldest quarter, SR, and EVI, with PCs of 51.2, 22.1, 10.2, and 4.4%, respectively (Supplementary Appendix Figure S5 and Supplementary Appendix Table S3). The fungus has a suitability peak at 0 mm of precipitation in the warmest quarter, the same pattern observed for the precipitation of the coldest quarter variable, but with a peak at 350 mm. In the case of SR, we identified a suitability peak at 18,000 kJ m–2 day–1. Finally, in the case of EVI, the suitability presented a Gaussian pattern with a maximum value at 0.23.
In Chile, the predicted spread could occupy the central-north zones of Chile from 29.5°S to 36.6°S, mainly in the Coquimbo, Valparaíso, Santiago, O’Higgins, and El Maule regions, as well as the Andean boundary with Argentina until 40°S (Figure 3A).
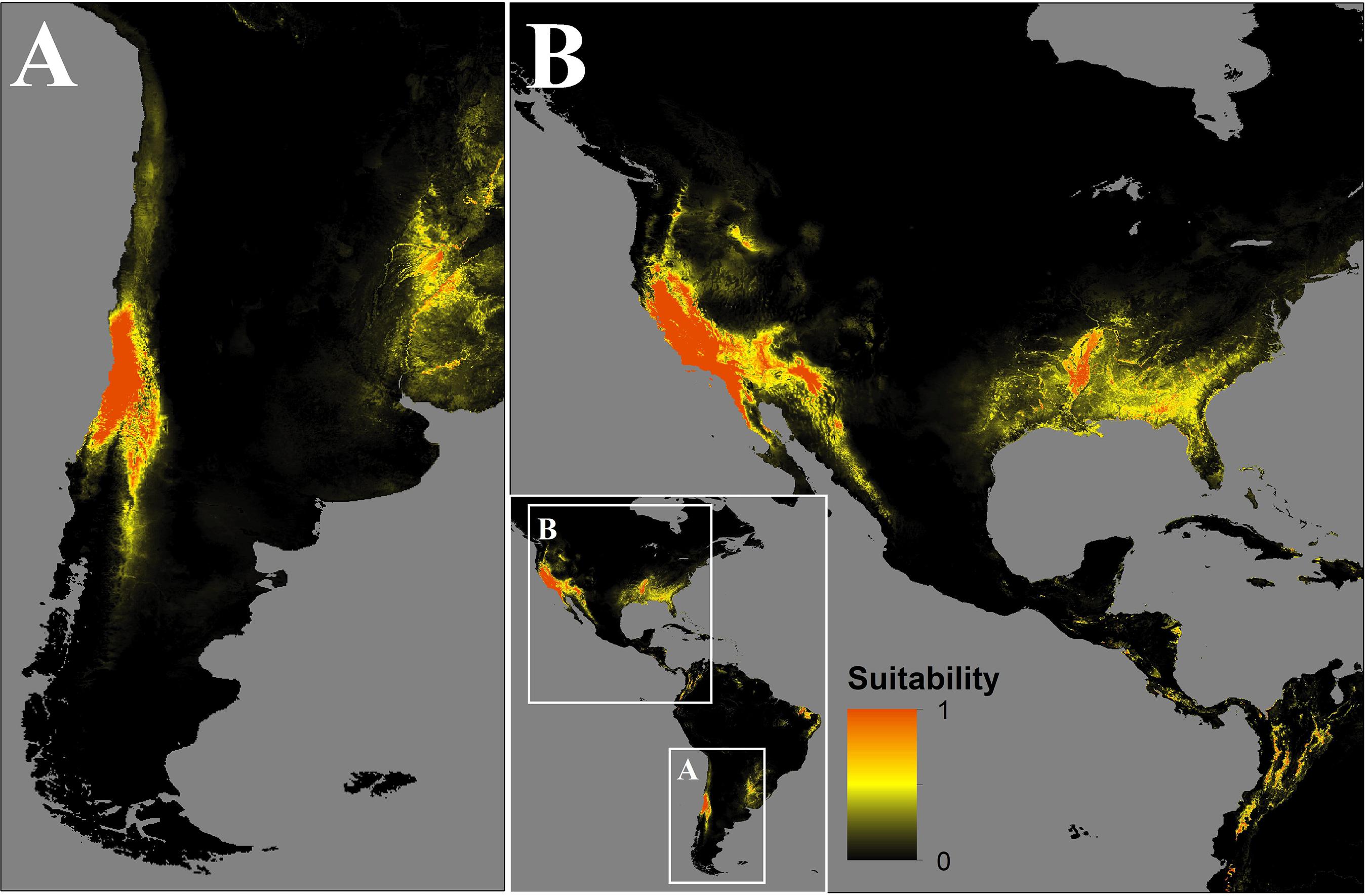
Figure 3. Map of the distribution of C. bacillisporus predicted by MaxEnt niche modeling in South (A) and North America (B).
In the source zone, the distribution of C. bacillisporus is mainly located in the west coast of the United States, mainly in California and Arizona, as well as in the northwest coastal zone of Mexico. On the other hand, in the east coast of the United States, Mississippi, Arkansas, Alabama, Georgia, South Carolina, Florida, and Louisiana showed high suitability for C. bacillisporus, but lower than the west coast (Figure 3B).
Potentially Exposed Population
In Chile the potentially exposed population reached 12,298,551 people, of which 12.8% and 74% correspond to high and very high exposure risk levels, respectively. The population exposed to a significant risk level (above medium), represents 56.7% of the total Chilean population by the year 2020. The most affected administrative regions are the Santiago Metropolitan, Valparaíso, and Libertador Bernardo O’Higgins regions, with 94, 83.3, and 81.3%, respectively, of their total population exposed to a significant risk level (Figure 4A and Table 1). Finally, the Maule, Coquimbo, and Biobío administrative regions have 42.4, 23.2, and 7.8% of their total population exposed to significant risk levels (Figure 4A and Table 1). The population per risk level and province is detailed in Supplementary Appendix Table S4.
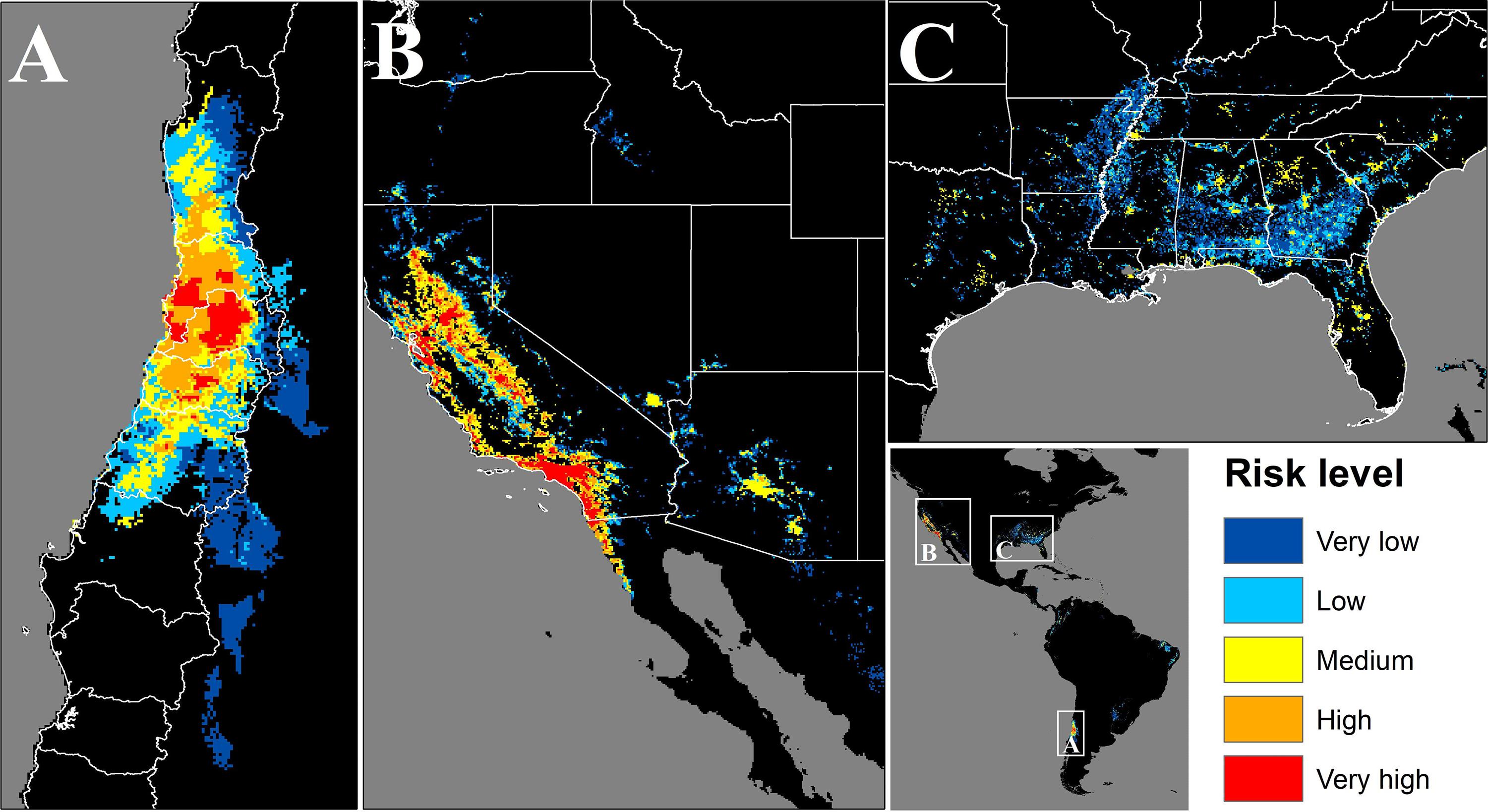
Figure 4. Map of the areas where population could be potentially exposed to a significant risk of contact with C. bacillisporus in Chile (A), Western United States (B), and Eastern United States (C). CQ = Coquimbo, VP = Valparaiso, ST = Santiago Metropolitan region, OH = Libertador Bernardo O’Higgins, MA = Maule, BB = Biobío, CA = California, NV = Nevada, OR = Oregon, WS = Washington, CO = Colorado, ID = Idaho, AZ = Arizona, TX = Texas, LA = Louisiana, AR = Arkansas, MO = Missouri, MS = Mississippi, AL = Alabama, GA = Georgia, FL = Florida, TN = Tennessee, NC = North Carolina, SC = South Carolina.
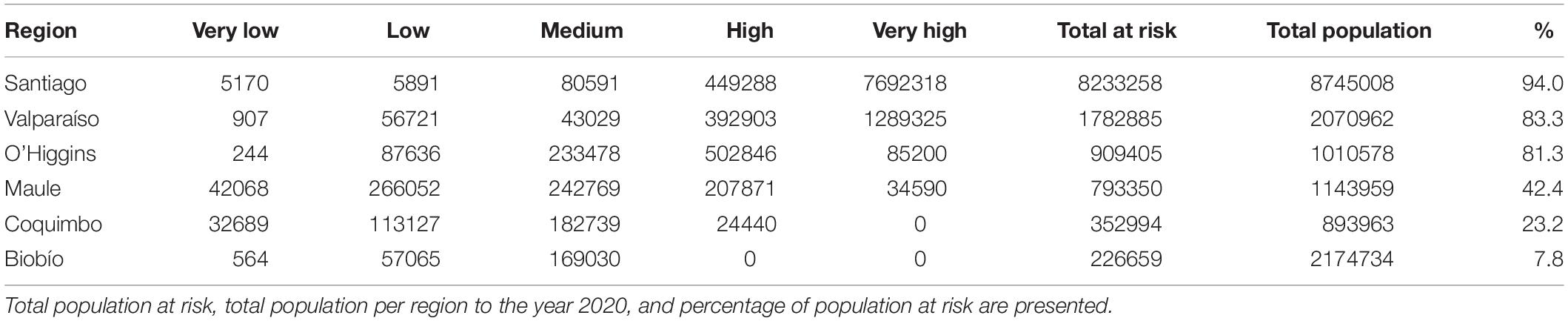
Table 1. Population per risk level and region in Chile.
In the source zone, the population under exposure risk reached 63,521,158 people, of which 5.9 and 47.5% corresponds to high and very high exposure risk levels. On the other hand, 41.3% of the population is exposed to medium risk level, and only 4.3 and 0.9% are under low and very low levels, respectively. In the United States, California, Nevada, and Arizona are the most affected states, with 86, 76.2, and 64.6%, respectively of their total population exposed to significant risk levels. On the other hand, Alabama, Georgia, Mississippi, and South Carolina have more than 20% of their total population exposed to significant risk level, while Tennessee, Florida, Louisiana, and Arkansas have more than 12%. Finally, thirteen states have less than 8% of their total population exposed to risk level above medium (Figures 4B,C and Table 2). The population per risk level and district is detailed in Supplementary Appendix Table S5.
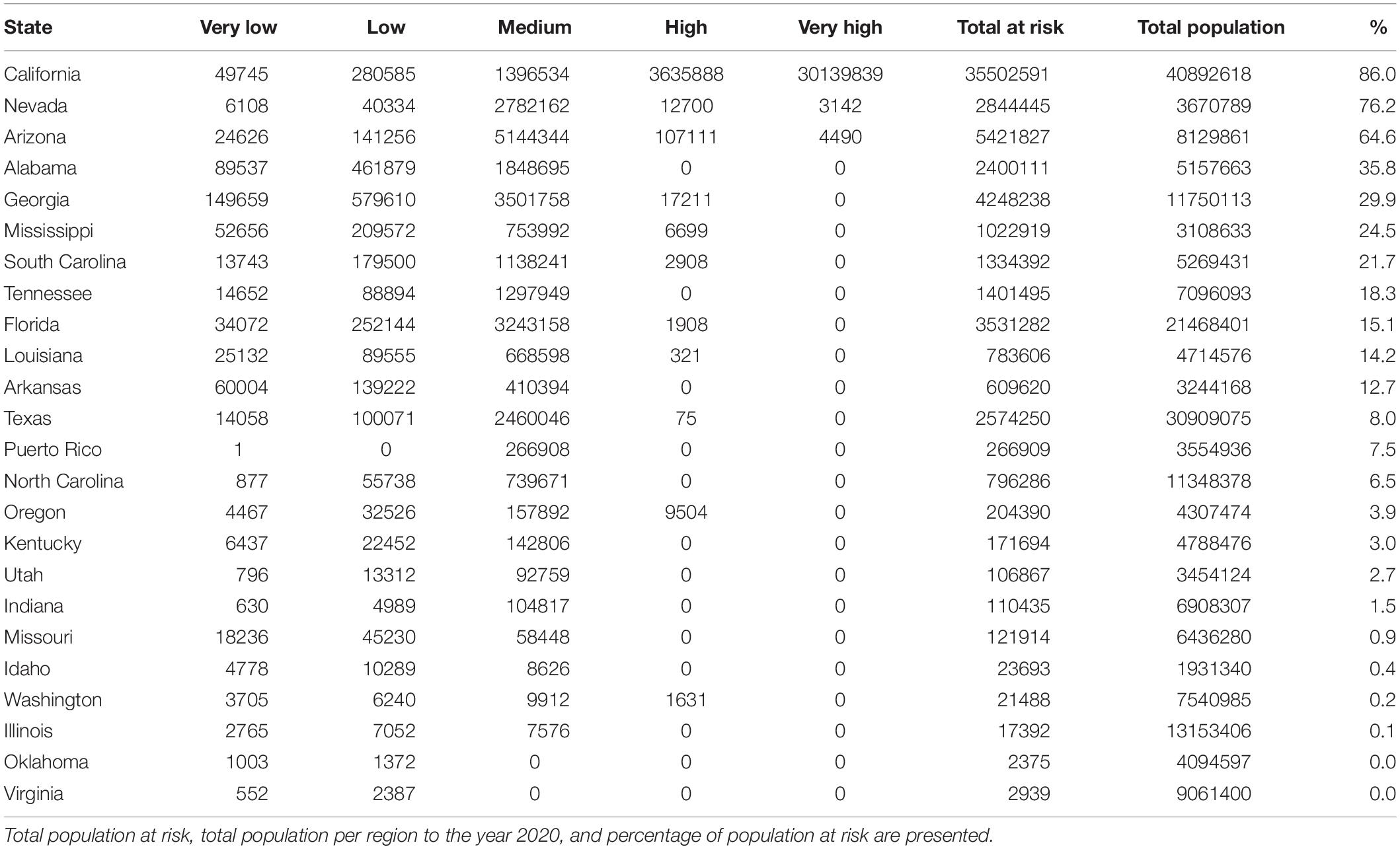
Table 2. Population per risk level and region in the United States.
Discussion
Assumptions and Limitations
This study incorporates some main assumptions and uncertainties which are necessary to discuss. First, the methodology used to predict the potential distribution of C. bacillisporus was based on 29 isolations from the genetically identified potential source zone (California and Colombia), not including the occurrence recorded from Chile. We decided to not include the Chilean occurrence to test if the high-risk zone predicted by the model overlaps with this record. We identified that the occurrence recorded in Chile (Limache at 30°59′S, 71°17′W) coincides with the high risk zone predicted by the model, confirming its reliability. However, since there is only one occurrence record for Chile, it is needed to further verify in the time and space by developing random environmental surveys if the fungus is actually present in the complete predicted high risk range. In this context our study represents a baseline to analyze the potential arrival, spread and establishment of C. bacillisporus in Chile, presenting a possible scenario that needs to be monitored and tracked in the light of this first isolation that could represent the actual arrival of the pathogen. Second, our prediction is based on the Niche Conservatism Principle which assumes that the species maintains they environmental niche requirements in the colonized zone (Gallien et al., 2012). However, non-native alien species can increase their distributional ranges in the colonized zone (non-stability hypothesis), thus our results could underestimate the potential distribution range (Broennimann et al., 2007). Third, it is also important to identify at species and molecular type level all isolates from cases of cryptococcosis to verify if they correspond to infections due to C. bacillisporus. Fourth, a potential source of uncertainty for our study could be related with the four clinical occurrences included from Lockhart et al. (2013), in which it is possible that some of those patients got infected in states other than the ones in which they were recorded. However, these occurrences represent only the 13.8% of the complete dataset.
Insights and Implications
In the present study, we examined the potential role of C. bacillisporus as emerging pathogen in Chile. Even when this pathogen has never been documented to infect a human in this country, and our study is based on the single isolation of the fungus in a veterinary case, a possible outbreak cannot be ruled out. During the last decades C. bacillisporus has emerged as a cause of disease in immunocompromised humans in southern California, United States, areas where according epidemiological studies cats are most often infected with C. bacillisporus compared with other animals (Trivedi et al., 2011; Singer et al., 2014).
Two main genotypes, ST75 and ST79, were identified as founders of two large phylogenetically distinct clonal clusters containing the majority of the globally identified STs. The presence of two VGIII phylogenetic clusters, one including serotype B and one serotype C isolates, were previously reported by other authors (Meyer et al., 2003; Byrnes et al., 2011; Walraven et al., 2011; Lockhart et al., 2013; Singer et al., 2014; Springer et al., 2014). The majority of STs included in the CC75 were from North America particularly from California United States, while the ones grouped in the CC79 included several STs identified in South America, particularly from Colombia. This is in agreement with a recent phylogenetic study carried out using whole genome sequencing of a set of VGIII global isolates, which describes Mexico and the United States as the origin of the VGIII serotype B population, and Colombia as the origin of serotype C population (Firacative et al., 2016). The phylogenetic analysis grouped the Chilean isolate (C3) with a strain from Paraguay, which suggests a close link to the genotype ST79 related to Colombia (Figure 2) in the CC79 cluster. There is also possible that the populations from CC79 cluster could have migrated north, stablishing in North America and further developing the CC75 cluster, as it happened before with the spread of VGII molecular type from South American Amazonian Rainforests to North America and Columbia, Canada (Chen et al., 2014; Souto et al., 2016; Engelthaler and Casadevall, 2019).
The spatial methodological framework presented here was successfully used in a recent study to determine the risk zones in Europe to different cryptococcal pathogens (Alaniz et al., 2020). The suitable geographic areas for C. bacillisporus survival are characterized by scarce or null rainfall during the dry season and moderate rainfall during the cold season, high SR and scarce vegetation corresponding to the warm summer Mediterranean climate present in California and northwestern Mexico (Acheson et al., 2019). The same climatic conditions are present in Central Chile, where the C. bacillisporus isolate was recovered, suggesting that VGIII is spreading outside the endemic areas and is potentially able to colonize a wide area in Chile covering about 150,000 km2. The same pattern of colonization has been present in other organisms that colonized central Chile, reinforcing the results presented here (Carvajal et al., 2019). This area corresponds to the most populated area of the country, including the capital city of Santiago. The colonization of the whole area by C. bacillisporus would expose more than 12 million people to contact with this pathogen, posing a serious risk for sensitive categories such as patients living with HIV, which in Chile has a prevalence of 0.34%, of whom only one half has access to antiretroviral therapy6.
On the other hand, the information generated here may provide guidance for decision making about this pathogen in the United States, which can be highly valuable considering the large amount of people under exposure risk. The generated maps can represent a tool for the design of contingency plans, public health strategies, budget optimization and routing for screening, diagnosis and treatments in the high-risk zones.
This study supplies basilar information for monitoring the evolution of C. bacillisporus epidemiology in Chile. The potential distribution map of the pathogen as well as the risk map reported here represent an important tool to plan future environmental surveys and clinical screening of patients exposed to the pathogen in the high risk areas. Identification of limited geographical areas at high risk allows for punctual intervention of public health, avoiding dispersion and waste of scarce and highly valuable health resources. Finally, the framework used in this study can be applied to other environmentally driven pathogens, helping to foresee the impacts and repercussions in public health before the actual potential spread. The linkage of phylogenetic analysis with ecological niche modeling allows to generate a new level of precision and accuracy in the prediction of pathogen distributions.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The present study analyzed data already reported and published by other authors and thus it does not require an approval from an ethical committee.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer KF-P declared a past collaboration with one of the authors MC to the handling editor.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.02117/full#supplementary-material
Footnotes
- ^ www.mycologylab.org
- ^ www.megasoftware.net
- ^ www.phyloviz.net
- ^ www.worldclim.org
- ^ https://sedac.ciesin.columbia.edu
- ^ https://www.unaids.org/en/regionscountries/countries/chile
References
Acheson, E. S., Galanis, E., Bartlett, K., and Klinkenberg, B. (2019). Climate classification system – based detection of Cryptococcus gattii sensu lato. Emerg. Infect. Dis. 25, 1723–1726. doi: 10.3201/eid2509.181884
Alaniz, A. J., Bacigalupo, A., and Cattan, P. E. (2017). Original article Spatial quantification of the world population potentially exposed to Zika virus. Int. J. Epidemiol. 46, 966–975. doi: 10.1093/ije/dyw366
Alaniz, A. J., Carvajal, J. G., Carvajal, M. A., Cogliati, M., and Vergara, P. M. (2020). Spatial quantification of the population exposed to Cryptococcus neoformans and Cryptococcus gattii species complexes in Europe: estimating the immunocompetent and HIV/AIDS patients under risk. Risk Anal. 40, 524–533. doi: 10.1111/risa.13410
Alaniz, A. J., Carvajal, M. A., Bacigalupo, A., and Cattan, P. E. (2018). Global spatial assessment of Aedes aegypti and Culex quinquefasciatus: a scenario of Zika virus exposure. Epidemiol. Infect. 147, 1–11.
Berejnoi, A., Taverna, C. G., Mazza, M., Vivot, M., Isla, G., Córdoba, S., et al. (2019). Case report first case report of cryptococcosis due to Cryptococcus decagattii in a pediatric patient in Argentina. Rev. Soc. Bras. Med. Trop. 52:e20180419.
Boekhout, T., Theelen, B., Diaz, M., Fell, J. W., Hop, W. C. J., Abeln, E. C. A., et al. (2001). Hybrid genotypes in the pathogenic yeast Cryptococcus neoformans. Microbiology 147, 891–907. doi: 10.1099/00221287-147-4-891
Broennimann, O., Treier, U. A., Müller-Schärer, H., Thuiller, W., Peterson, A. T., and Guisan, A. (2007). Evidence of climatic niche shift during biological invasion. Ecol. Lett. 10, 701–709. doi: 10.1111/j.1461-0248.2007.01060.x
Byrnes, E. J. III, Li, W., Ren, P., Lewit, Y., Voelz, K., Fraser, J. A., et al. (2011). A diverse population of Cryptococcus gattii molecular type VGIII in southern Californian HIV/AIDS patients. PLoS Pathog. 7:e1002205. doi: 10.1371/journal.ppat.1002205
Byrnes, E. J., and Marr, K. A. (2011). The outbreak of Cryptococcus gattii in western north America: epidemiology and clinical issues. Curr. Infect. Dis. Rep. 13, 256–261. doi: 10.1007/s11908-011-0181-0
Carvajal, M. A., Alaniz, A. J., Núñez-Hidalgo, I., and González-Césped, C. (2019). Spatial global assessment of the pest Bagrada hilaris (Burmeister) (Heteroptera: Pentatomidae): current and future scenarios. Pest. Manag. Sci. 75, 809–820. doi: 10.1002/ps.5183
Chen, S. C., Meyer, W., and Sorrell, C. (2014). Cryptococcus gattii infections. Clin. Microbiol. Rev. 27, 980–1024. doi: 10.1128/cmr.00126-13
Cogliati, M. (2013). Global molecular epidemiology of Cryptococcus neoformans and Cryptococcus gattii: an atlas of the molecular types. Scientifica (Cairo) 2013, 675213.
Cogliati, M., Amicis, R. D., and Tortorano, A. M. (2014). Cryptococcus gattii sero-mating type allelic pattern determined by multiplex PCR. Clin. Microbiol. Infect. 21, 190.e1–190.e4.
Cogliati, M., Puccianti, E., Montagna, M. T., De Donno, A., Susever, S., Ergin, C., et al. (2017). Fundamental niche prediction of the pathogenic yeasts Cryptococcus neoformans and Cryptococcus gattii in Europe. Environ. Microbiol. 19, 4318–4325.
Doxsey-Whitfield, E., MacManus, K., Adamo, S. B., Pistolesi, L., Squires, J., Borkovska, O., et al. (2015). Taking advantage of the improved availability of census data: a first look at the gridded population of the world: Version 4. Pap. Appl. Geogr. 1, 226–234. doi: 10.1080/23754931.2015.1014272
Engelthaler, D. M., and Casadevall, A. (2019). On the emergence of Cryptococcus gattii in the Pacific Northwest: ballast tanks, tsunamis, and black swans. mBio 10:e02193-19.
Escandón, P., Sanchez, A., Martínez, M., Meyer, W., and Casta, E. (2006). Molecular epidemiology of clinical and environmental isolates of the Cryptococcus neoformans species complex reveals a high genetic diversity and the presence of the molecular type VGII mating type a in Colombia. Med. Mycol. 48, 675–678.
Espinel-ingroff, A., and Kidd, S. E. (2015). Current trends in the prevalence of Cryptococcus gattii in the United States and Canada. Infect. Drug Resist. 8, 89–97. doi: 10.2147/idr.s57686
Farrer, R. A., Chang, M., Davis, M. J., van Dorp, L., Yang, D.-H., Shea, T., et al. (2019). A n ew lineage of Cryptococcus gattii (VGV) discovered in the Central Zambezian Miombo Woodlands. mBio 10:e02306-19.
Feng, X., Fu, X., Ling, B., Wang, L., Liao, W., and Pan, W. (2013). Rapid differentiation of cryptic species within Cryptococcus gattii by a duplex PCR Assay. J. Clin. Microbiol. 51, 3110–3112. doi: 10.1128/jcm.01455-13
Fick, S. E. (2017). WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 37, 4302–4315. doi: 10.1002/joc.5086
Firacative, C., Roe, C. C., Malik, R., Ferreira-Paim, K., Escandón, P., Sykes, J. E., et al. (2016). MLST and whole-genome-based population analysis of Cryptococcus gattii VGIII links clinical, veterinary and environmental strains, and reveals divergent serotype specific sub-populations and distant ancestors. PLoS Negl. Trop. Dis. 10:e0004861. doi: 10.1371/journal.pntd.0004861
Gallien, L., Douzet, R., Pratte, S., Zimmermann, N. E., and Thuiller, W. (2012). Invasive species distribution models - how violating the equilibrium assumption can create new insights. Glob. Ecol. Biogeogr. 21, 1126–1136. doi: 10.1111/j.1466-8238.2012.00768.x
George, I. A., Spec, A., Powderly, W. G., and Santos, C. A. Q. (2018). Comparative epidemiology and outcomes of HIV, non-HIV non-transplant comparative epidemiology and outcomes of human immunodeficiency virus (HIV), solid organ transplant associated cryptococcosis: a population-based study. Clin. Infect. Dis. 66, 608–611. doi: 10.1093/cid/cix867
González, G. M., Casillas-vega, N., Garza-gonzález, E., Hernández-bello, R., Rivera, G., and Rodríguez, J. A. (2016). Molecular typing of clinical isolates of Cryptococcus neoformans / Cryptococcus gattii species complex from Northeast Mexico. Folia Microbiol. 61, 51–56. doi: 10.1007/s12223-015-0409-8
Gorelick, N., Hancher, M., Dixon, M., Ilyushchenko, S., Thau, D., and Moore, R. (2017). Google Earth engine: planetary-scale geospatial analysis for everyone. Rem. Sens. Environ. 202, 18–27. doi: 10.1016/j.rse.2017.06.031
Hagen, F., Khayhan, K., Theelen, B., Kolecka, A., Polacheck, I., Sionov, E., et al. (2015). Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet. Biol. 78, 16–48. doi: 10.1016/j.fgb.2015.02.009
Kidd, S. E., Hagen, F., Tscharke, R. L., Huynh, M., Bartlett, K. H., Fyfe, M., et al. (2004). A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc. Natl. Acad. Sci. U.S.A. 101, 17258–17263.
Kwon-Chung, K. J., Bennett, J. E., Wickes, B. L., Meyer, W., Cuomo, C. A., Wollenburg, K. R., et al. (2017). The case for adopting the “Species Complex” nomenclature for the etiologic agents of cryptococcosis. mSphere 2:e00357-16.
Li, Y., Zou, M., Yin, J., Liu, Z., and Lu, B. (2020). Microbiological, epidemiological, and clinical c characteristics of patients with cryptococcal meningitis at a tertiary hospital in China: a 6- year retrospective analysis. Front. Microbiol. 11:1837. doi: 10.3389/fmicb.2020.01837
Lizarazo, J., Escandon, P., Agudelo, C., Firacative, C., Meyer, W., and Castan, E. (2014). Retrospective study of the epidemiology and clinical manifestations of Cryptococcus gattii infections in Colombia from 1997 – 2011. PLoS Negl. Trop. Dis. 8:e3272. doi: 10.1371/journal.pntd.0003272
Lockhart, S. R., Iqbal, N., Harris, J. R., Grossman, N. T., Debess, E., Wohrle, R., et al. (2013). Cryptococcus gattii in the United States: genotypic diversity of human and veterinary isolates. PLoS One 8:e74737. doi: 10.1371/journal.pone.0074737
Mazza, M., Bosco-borgeat, E., Taverna, C. G., Trovero, A. C., and Rog, A. (2013). Cryptococcus gattii in urban trees from cities in North-eastern Argentina. Mycoses 56, 646–650. doi: 10.1111/myc.12084
Mcmullan, B. J., Sorrell, T. C., and Chih-ann, S. (2013). Cryptococcus gattii infections: contemporary aspects of epidemiology, clinical manifestations and management of infection. Future Microbiol. 8, 1613–1631. doi: 10.2217/fmb.13.123
Meyer, W., Aanensen, D. M., Boekhout, T., Cogliati, M., Diaz, M. R., Esposto, M. C., et al. (2009). Consensus multi-locus sequence typing scheme for Cryptococcus neoformans and Cryptococcus gattii. Med. Mycol. 47, 561–570.
Meyer, W., Castañeda, A., Jackson, S., Huynh, M., and Castañeda, E. (2003). Molecular typing of IberoAmerican Cryptococcus neoformans Isolates. Emerg. Infect. Dis. 9, 189–195.
Mourad, A. (2018). Present and future therapy of Cryptococcus infections. J. Fungi (Basel) 4:79. doi: 10.3390/jof4030079
Pfeiffer, T., and Ellis, D. (1991). Environmental isolation of Cryptococcus neoformans gattii from California Hickman catheter-related protothecal algaemia in an immunocompromised child. J. Infect. Dis. 163, 929–930. doi: 10.1093/infdis/163.4.929
Phillips, S. J., Anderson, R. P., Dudík, M., Schapire, R. E., and Blair, M. E. (2006). Maximum entropy modeling of species geographic distributions. Ecol. Model. 190, 231–259. doi: 10.1016/j.ecolmodel.2005.03.026
Rajasingham, R., Smith, R. M., Park, B. J., Jarvis, J. N., Govender, N. P., Chiller, T. M., et al. (2017). Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect. Dis. 17, 873–881. doi: 10.1016/s1473-3099(17)30243-8
Singer, L. M., Meyer, W., Firacative, C., Iii, R. T., Samitz, E., and Sykes, E. (2014). Antifungal drug susceptibility and phylogenetic diversity among Cryptococcus isolates from dogs and c ats in North America. J. Clin. Microbiol. 52, 2061–2070. doi: 10.1128/jcm.03392-13
Souto, A. C., Bonfietti, L. X., Ferreira-Paim, K., Trilles, L., Martins, M., Ribeiro-Alves, M., et al. (2016). Population genetic analysis reveals a high genetic diversity in the Brazilian Cryptococcus gattii VGII population and shifts the global origin from the Amazon rainforest to the semi-arid desert in the Northeast of Brazil. PLoS Negl. Trop. Dis. 10:e0004885. doi: 10.1371/journal.pntd.0004885
Springer, D. J., Billmyre, R. B., Filler, E. E., Voelz, K., Pursall, R., Mieczkowski, P. A., et al. (2014). Cryptococcus gattii VGIII isolates causing infections in HIV/AIDS patients in Southern California: identification of the local environmental source as arboreal. PLoS Pathog. 10:e1004285. doi: 10.1371/journal.ppat.1004285
Trivedi, S. R., Sykes, J. E., Cannon, M. S., Wisner, E. R., Meyer, W., Sturges, B. K., et al. (2011). Clinical features and epidemiology of cryptococcosis in cats and dogs in California: 93 cases (1988-2010). J. Am. Vet. Med. Assoc. 239, 357–369. doi: 10.2460/javma.239.3.357
Velagapudi, R., Hsueh, Y. P., Geunes-Boyer, S., Wright, J. R., and Heitman, J. (2009). Spores as infectious propagules of Cryptococcus neoformans. Infect. Immun. 77, 4345–4355. doi: 10.1128/iai.00542-09
Vélez, N., and Escandón, P. (2017). Report on novel environmental niches for Cryptococcus neoformans and Cryptococcus gattii in Colombia: Tabebuia guayacan and Roystonea regia. Med. Mycol. 55, 794–797.
Vieille, P., Cruz, R., León, P., Cáceres, N., and Giusiano, G. (2018). Medical mycology case reports isolation of Cryptococcus gattii VGIII from feline nasal injury. Med. Mycol. Case Rep. 22, 55–57. doi: 10.1016/j.mmcr.2018.09.003
Voelz, K., Ma, H., Phadke, S., Byrnes, E. J., Zhu, P., Mueller, O., et al. (2013). Transmission of hypervirulence traits via sexual reproduction within and between lineages of the human fungal pathogen Cryptococcus gattii. PLoS Genet. 9:e1003771. doi: 10.1371/journal.pgen.1003771
Walraven, C. J., Gerstein, W., Hardison, S. E., Wormley, F., Lockhart, S. R., Julie, R., et al. (2011). Fatal disseminated Cryptococcus gattii infection in New Mexico. PLoS One 6:e28625. doi: 10.1371/journal.pone.0028625
Keywords: Cryptococcus bacillisporus, molecular type VGIII, Chile, niche modeling, MLST
Citation: Carvajal JG, Alaniz AJ, Carvajal MA, Acheson ES, Cruz R, Vergara PM and Cogliati M (2020) Expansion of the Emerging Fungal Pathogen Cryptococcus bacillisporus Into America: Linking Phylogenetic Origin, Geographical Spread and Population Under Exposure Risk. Front. Microbiol. 11:2117. doi: 10.3389/fmicb.2020.02117
Received: 04 June 2020; Accepted: 11 August 2020;
Published: 28 August 2020.
Edited by:
László Galgóczy, University of Szeged, HungaryReviewed by:
Patricia Escandon, Instituto Nacional de Salud, ColombiaKennio Ferreira-Paim, Universidade Federal do Triângulo Mineiro, Brazil
Copyright © 2020 Carvajal, Alaniz, Carvajal, Acheson, Cruz, Vergara and Cogliati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimo Cogliati, bWFzc2ltby5jb2dsaWF0aUB1bmltaS5pdA==