 Yinhua Wu1
†
Yinhua Wu1
† Ang Li
Ang Li Xiaoyan Liu
Xiaoyan Liu- 1Department of Dermatology, The First Affiliated Hospital, School of Medicine of Zhejiang University, Hangzhou, China
- 2Department of the Second General Surgeon, The Yunhe People’s Hospital, Yunhe, China
- 3Physician Health Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 4Department of Henan Gene Hospital, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
Although the diversity and abundance of skin microbiome are mainly determined by intrinsic factors, including gender, age, anatomical site, and ethnicity, we question whether facial microbiome could be affected by long-term exposure to airborne pollution. Using 16S ribosomal RNA (rRNA) gene amplicon sequencing, we analyzed the facial bacterial microbiome of healthy and young Chinese women (25–35 years old) between two districts with different air quality indices (AQIs) in Zhejiang Province. The overall microbiome structure was obviously different between these two districts. It revealed an increase in both the abundance and diversity of facial bacterial microbiome in Hangzhou (HZ) with higher AQI compared with those in Yunhe (YH) with lower AQI. Linear discriminant analysis (LDA) and Lefse analysis identified a total of 45 genera showing significant overrepresentation in the HZ group. Furthermore, PICRUSt analysis showed that functional pathways associated with metabolism of saturated fatty acid were relatively more predominant in the HZ group, whereas those with DNA repair or mitochondrial DNA replication were more predominant in the YH group. Our present data can provide useful information for further researches on the composition and function of the skin microbiome related to air pollution factors as well as for the development of therapeutic agents targeting the microbes and their metabolites to resist damages of airborne pollutants.
Introduction
Human skin is the largest organ and acts as the first line of defense against environmental stressors. It harbors hundreds and thousands of resident microbes and maintains a delicate equilibrium, the microbiome, which is necessary for a healthy skin (Grice et al., 2009; Dréno et al., 2016). This balance is constantly influenced by both internal (host) and external (environmental) factors. These factors alter the composition of skin microbiome and further affect the barrier function of host skin.
With the rapid development of Chinese economy, environmental pollution and hazier weather have become a critical problem that cannot be overlooked. Air quality index (AQI) describes the extent of clean or polluted air and its impact on human health. The main air pollutants are mainly composed of particulate matter (PM2.5 or PM10), sulfur dioxide (SO2), nitrogen dioxide (NO2), ozone (O3), and carbon monoxide (CO; Araviiskaia et al., 2019), and they are also five major pollution standards to calculate the AQI (Woodall et al., 2017). It was reported that accumulation of SO2 and NOx could result in a lower pH value of rainwater (Li et al., 2019), and difference of pH would further lead to composition change in local microbiota (Vongsa et al., 2019). Available evidence indicate that long-term air pollution could accelerate skin aging (Vierktter et al., 2010; Ding et al., 2017), and some human skin diseases, such as acne (Liu et al., 2018), atopic dermatitis (AD; Ahn, 2014), and eczema (Pesce et al., 2015), were also reported to be associated with dysbiosis of skin microbiota (Fitz-Gibbon et al., 2013; Chng et al., 2016; Dybboe et al., 2017; Shibagaki et al., 2017; Kim et al., 2019; Reiger et al., 2020). On the one hand, it was uncovered that exposure to atmospheric-equivalent O3 levels for 2 h could halve the skin microbiome of women’s forearm (He et al., 2006). On the other hand, skin microbiome is thought to remain stable over months to years once intrinsic factors stabilize (Oh et al., 2016), and the stability of healthy skin microbiome established in early period might extend into adulthood (Capone et al., 2011). There was a very little impact of short-term exposure to coal combustion waste on the skin microbiome of adult (Hughey et al., 2016). However, a study conducted in four cities with different urbanization in China reported that skin microbiome in cities with better environmental standards and higher socioeconomic status had higher overall and phylogenetic diversities, which would be beneficial to human skin health (Kim et al., 2018). Therefore, whether airborne pollutants, especially long-term exposure to them, can affect the equilibrium of skin microbiome of healthy individuals has not directly been evaluated and needs to be further investigated.
One host factor, body mass index (BMI), was reported to be closely related to oral or vulva microbiota (Vongsa et al., 2019; de Andrade et al., 2020) besides gut microbiota (Ottosson et al., 2018). In addition, age also would impact the facial microbiome (Kim et al., 2019). In order to reduce the influence of these host factors on skin microbiome and mainly focus on discussing the poorly understood relationships between the facial microbiome composition of healthy women and air quality, we recruited 25–35-year-old healthy women with a BMI of 18.5–24.9 from Hangzhou (HZ, Zhejiang Province, China) or Yunhe (YH, Zhejiang Province, China) districts with different AQIs. As another host factor, skin sites vary dramatically in their levels of bacterial composition (Costello et al., 2009), and site-specific lipid and water levels were the most significant predictors of facial microbiome composition and diversity (Mukherjee et al., 2016). Thus, we collected skin samples from facial oily zone (forehead, nasal dorsum, and jaw) or dry zone (cheek) and characterized their bacterial microbiome using 16S ribosomal RNA (rRNA) sequencing. We aimed to explore the effect and probable mechanism of atmospheric pollution on healthy human skin.
Materials and Methods
Participants and Sampling
This study was approved by the Institutional Review Board of the First Affiliated Hospital, School of Medicine of Zhejiang University (reference number 2017-866). Written informed consent was obtained from all participants. For comparison of skin bacterial microbiome characteristics between two districts with different AQIs, we recruited 20 healthy women aged 25–35 years old for each group from HZ or YH, respectively. All of the selected subjects have been living in HZ or YH for more than 20 years. They had no history of chronic cutaneous diseases, such as acne, psoriasis, AD, and eczema. In addition, subjects would be excluded if they answered “yes” to any of the exclusion criteria (Supplementary Table S1) according to core microbiome sampling protocol for Human Microbiome Project (McInnes and Cutting, 2010). All subjects only used the provided cleanser and lotions for 2 weeks prior to sampling in order to minimize the impact of cosmetics. All microbial samples were collected in the same week in May 2019, in temperature‐ and humidity-controlled rooms (22°C, humidity 60%), 2 h after the last face wash. The skin area used for sampling was the oily zone (forehead, nasal dorsum, and jaw) and the dry zone (3 × 3 cm2 for the cheek). The samples were collected with sterile Catch-All Sample Collections Swabs (QEC091H, Epicentre, USA) and stored at −80°C within 15 min after collection until genomic DNA (gDNA) extraction. We also collected the clinical parameters of the participants, including BMI and age. BMI was calculated by the equation of weight (kg)/height (m2) and categorized in the World Health Organization guidelines: normal weight, BMI of 18.5–24.9 kg/m2. In order to explore whether different AQIs could lead to change in facial pH value and further impact the facial microbiome, the skin surface pH was measured using a skin pH-meter® pH905 (Courage + Khazaka Electronic GmbH, Germany).
Finally, 18 subjects from HZ and 11 subjects from YH were included due to discarding samples with low bacterial DNA. Among these 29 subjects, 10 subjects had both oily and dry-zone samples, so a total of 39 samples were used for further analysis. No significant differences in age were observed between these two groups, while the value of BMI in YH group tended to be a little higher (p = 0.035). The related clinical characteristics of tested subjects are shown in Table 1.
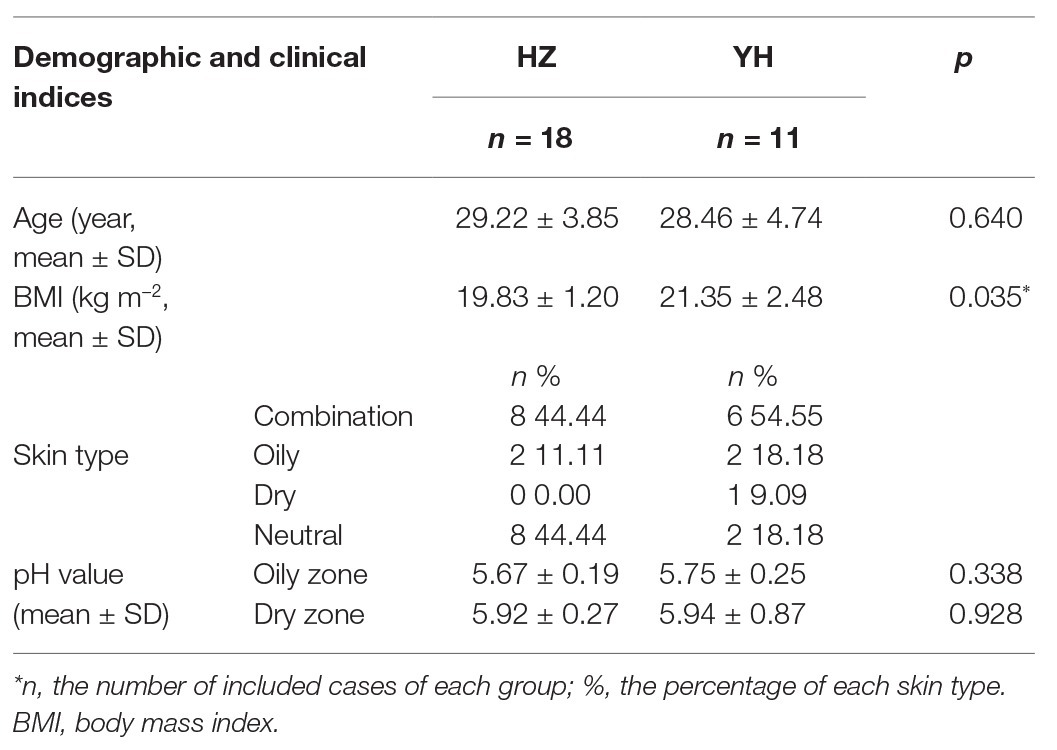
Table 1. Demographic and clinical details of recruited subjects.
Amplicon Generation, Sequencing, and Bioinformatics Analysis
DNA was extracted using PSP Spin DNA plus kit (Stratec, Berlin, Germany) according to manufacturer’s instructions. The V3–V4 region of 16S rRNA was PCR amplified with specific primers 341F 5'-CCTAYGGGRBGCASCAG-3' and 806R 5'-GGACTACNNGGGTATCTAAT-3' with the barcode. All PCR reactions were carried out in 30 μl reactions with 15 μl of Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM of forward and reverse primers, and about 10 ng template DNA. Thermal cycling consisted of initial denaturation at 98°C for 1 min, followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 50°C for 30 s, and elongation at 72°C for 30 s, and finally, 72°C for 5 min. PCR product was sequenced on Illumina Miseq sequencer. For bioinformatics analysis, refer to our previous study (Hu et al., 2019). Raw Illumina read data have been deposited in the EMBL database (study accession number PRJEB37433).
Statistics Analysis
Statistical analyses were performed on R version 3.6.1. R program package “phyloseq” was used to caculate unifrac distances. R program package “vegan” was used to perform permutational multivariate analysis of variance (PERMANOVA) and to calculate Shannon, Simpson, Inv-simpon, and observed species. R program package “fossil” was used to calculate Chao1 and ICE index.
Wilcoxon rank sum test was used to perform hypothesis test of alpha diversity and principle coordinates. Lefse version 1.0 (Segata et al., 2011) was used to find out whether taxons and Kyoto Encyclopedia of Genes and Genomes (KEGG) modules might be significantly associated destinations with default parameters. The Lefse analysis results were deposited in https://github.com/xiaoshubaba/Liuxiaoyan_SKIN_16Smetagenome.
The results of age, BMI, values of airborne pollutants, and AQI were expressed as the mean ± standard deviation. T-tests using SPSS software were used to assess significant differences between the HZ and TH groups. p < 0.05 was considered to be statistically significant.
Results
Permutational Multivariate Analysis of Variance
PERMANOVA showed that the effect of destination (cohort) was strongly associated with microbial composition in all of five distances, including JSD, Pearson, Spearman, and weighted UniFrac and unweighted UniFrac distances. PERMANOVA (Supplementary Table S2) revealed a weak association between skin type and microbial composition only in one distance (unweighted UniFrac distance, p = 0.020), which was much less significant than that in the destination. Furthermore, there was no obvious association between BMI and microbial composition.
Sequence Reads and Composition at the Whole Microbiome Level
A total of 1,041,112 qualified reads from 39 samples (mean, 26,695 ± 14,421) were sequenced, and 1,063 qualified operational taxonomy units (OTUs) were clustered for downstream analysis. For details, please refer to Supplementary Table S3.
Principal coordinate analysis (PCoA) was performed with five different distances at the OTU level. There was no significant difference on these principal coordinates between HZ and YH subjects (Supplementary Figure S1, p > 0.05).
Facial Microbial Diversity Changes Between the HZ and YH Groups
Shannon, Simpson, and inverse Simpson [invsimpson], which usually indicate diversity indexes, were used to assess facial microbial diversity. As shown in Figure 1, Shannon, Simpson, and Invsimpson index were greater in HZ oily zone compared with those in YH oily zone (p = 0.033, 0.033, and 0.033). However, no diversity changes in dry zone were observed between the two districts (p > 0.05). Furthermore, microbial richness in HZ subjects, as estimated by Obs, Chao1, and Ice, was higher than that in YH, not only in oily zone (p = 0.033, 0.048, and 0.028) but also in dry zone (p = 0.018, 0.023, and 0.018). It was revealed by these data that for the facial bacterial microbiome, the HZ group showed a higher alpha diversity and richness compared with the YH group.
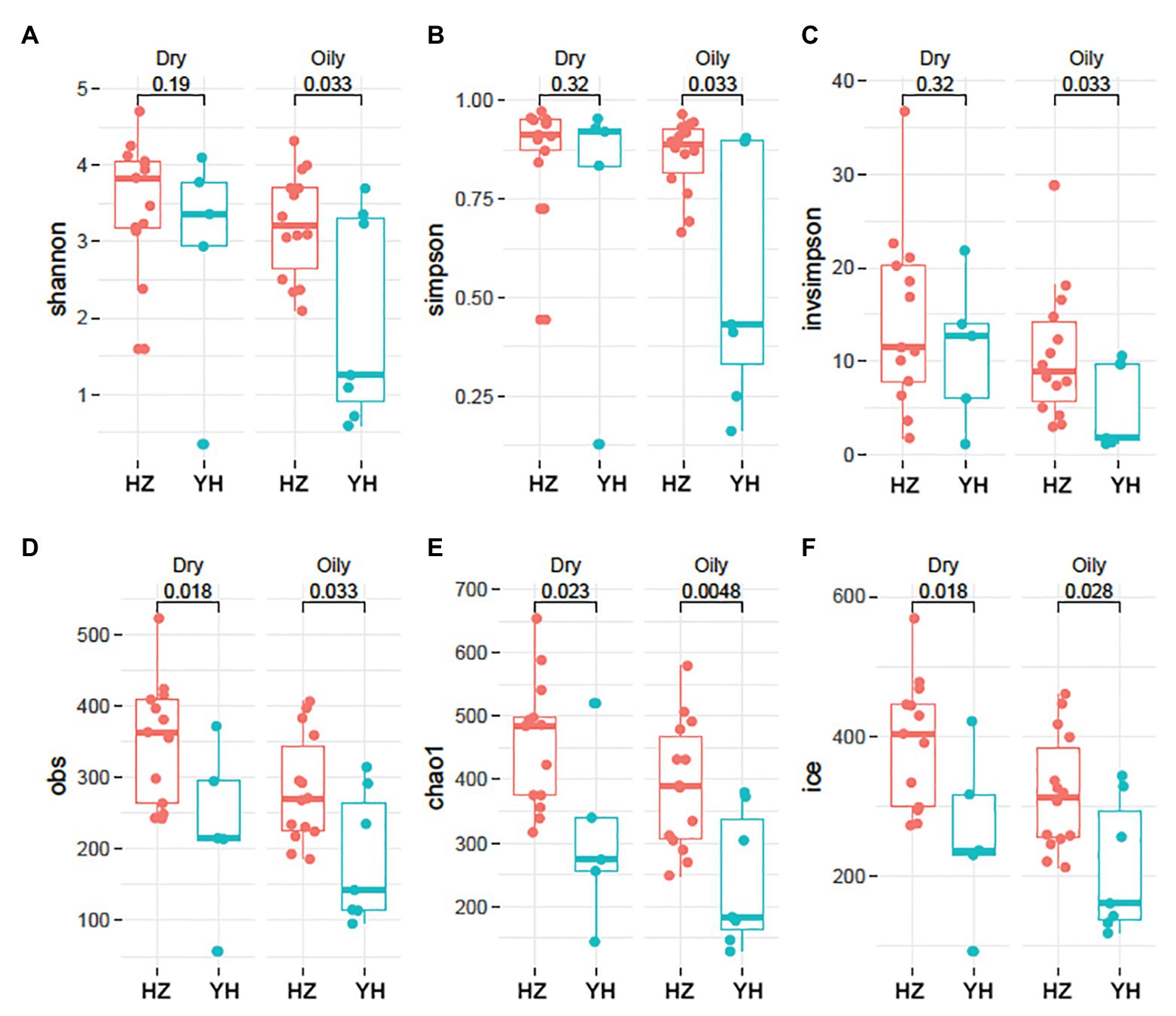
Figure 1. Phylogenetic diversity and richness of facial bacterial microbiome in HZ female compared to YH female individuals. (A–F) Alpha (α) diversity in Hangzhou (HZ) and Yunhe (YH) subjects. Oily represented samples from oily zone, and dry represented samples from dry zone. Each dot represented a sample. Shannon, Simpson, and inverse Simpson (invsimpson) indicate diversity indices, and Obs, Chao1, and Ice indicate microbial richness. Significance determined in Wilcoxon rank sum test with one-sided alternatives.
Except for distribution, the value of p is also greatly impacted by sample size, so we used a bootstrap sampling (randomly chose five HZ dry-zone and seven HZ oily zone samples), then repeated this process for 100 times. We found that the majority of value distribution of p (Supplementary Figure S2) is still significant (median p < 0.05), although YH had only five dry‐ and seven oily zone samples.
Differences in Taxonomic Composition of Facial Bacterial Microbiome Between the HZ and YH Groups
We further investigated the taxonomic profiles in the destination-related diversification of facial microbiome. The taxonomy resolution of all samples is shown in Figure 2A, and the ratio at genus level was about 77.21%. As shown in Figures 2B,C, Firmicutes, Proteobacteria, Actinobacteria, and Bacteroidetes were the four phyla that composed the major facial bacterial microbiome. Further comparison of the phyla abundance in both groups uncovered that Firmicutes and Proteobacteria were the two most predominant in the HZ group, accounting for the average abundance of 35.63 and 30.37% in the oily zone and 35.97 and 29.27% in the dry zone, respectively. However, in the YH group, Actinobacteria was the most predominant (43.59%) in the oily zone, and Firmicutes accounted for 52.18% in the dry zone. Overall, these data indicated that the HZ group had a tendency to have more Proteobacteria and less Actinobacteria in the facial bacterial microbiome than the YH group.
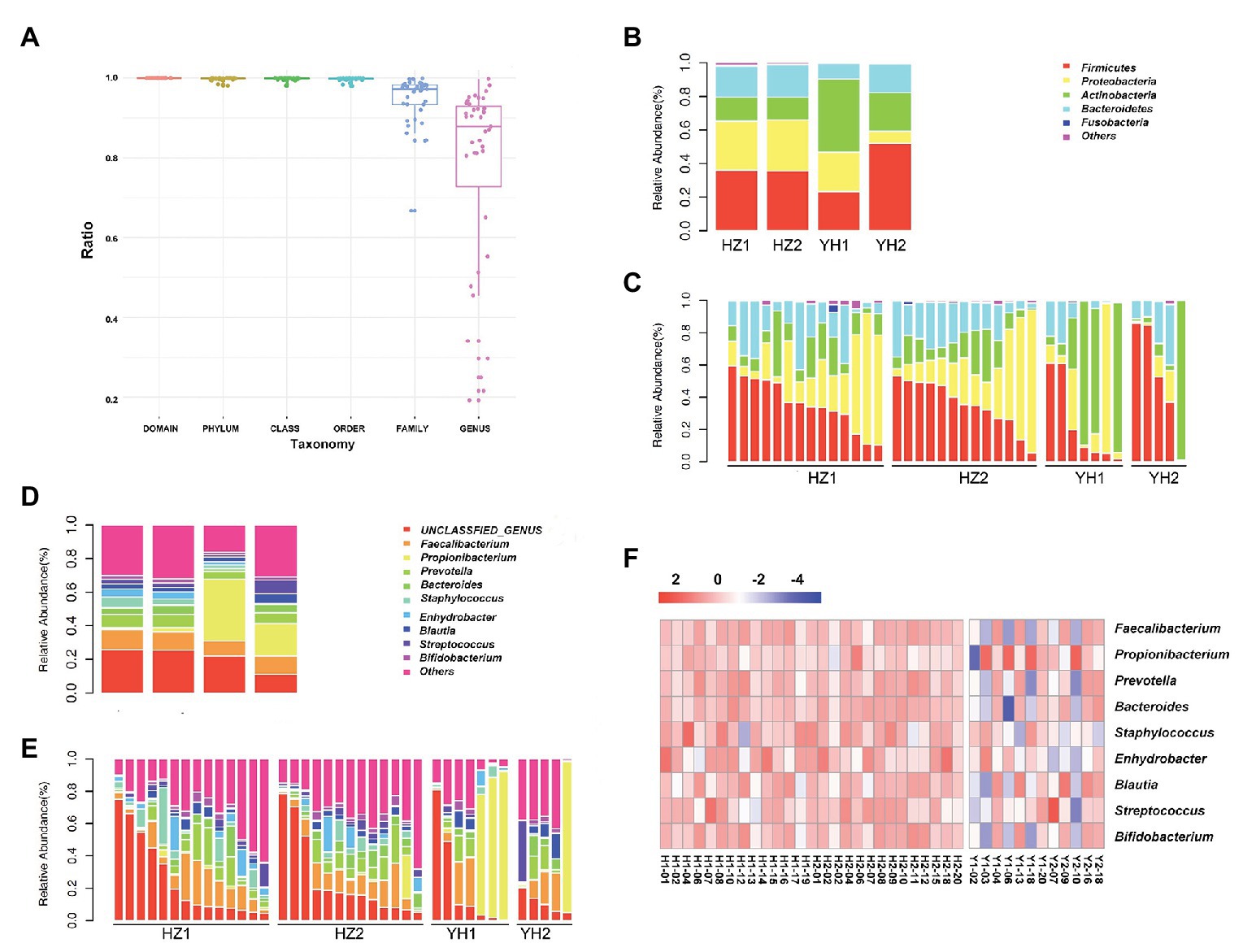
Figure 2. Taxonomy resolution of facial bacterial microbiome in healthy female individuals between the HZ and YH groups. (A) Taxonomy resolution in all samples; each bar represented ratio of reads annotated at that level. (B,C) Relative abundance levels of the phyla presented in samples from the HZ and YH healthy female individuals. (D,E) Relative abundance levels of genera presented in samples from the HZ and YH female individuals. (F) The heatmap of the most dominated genera of the facial microbiome. HZ1 and H1 indicated data of oily zone in the HZ district, HZ2 and H2 indicated data of dry zone in the HZ district, YH1 and Y1 indicated data of oily zone in the YH district, and YH2 and H2 indicated data of dry zone in the YH district.
In addition, Faecalibacterium, Propionibacterium, Prevotellaceae, Bacteroides, Staphylococcus, Enhydrobacter, Blautia, Streptococcus, and Bigidobacterium were found to be the top nine genera in all microbiome. Notably, a comparison at the genus level between the two groups showed that although value of p > 0.05, the abundance of Propionibacterium was much lower in the HZ group (1.78%) compared to the that in the YH group (29.43%), in both oily and dry zones (Figures 2D–F, Supplementary Table S4). It might partly interpret the marked reduction in Actinobacteria in the facial bacterial microbiomes of the HZ subjects since Propionibacterium is the major genus belonging to this phylum. In contrast, the abundance of Bacteroides, Staphylococcus, and Enhydrobacter were significantly increased in the HZ group compared to that in the YH group (p < 0.05, Figures 2D–F, Supplementary Table S4).
Linear discriminant analysis (LDA) effect size (Lefse) method was further used to clarify the species that contributed to the destination-related difference in the facial bacterial microbiome between the HZ and YH groups. A total of 45 genera were chosen by LDA score > 2, all of which were overrepresented in the HZ group (p < 0.05, Figure 3A). Among them, Enhydrobacter, Staphylococcus, Bachybacterum, Micrococcus, Paracoccus, Neisseria, Lysobacter, Streptophyta, Deinococcus, and Anoxybacillus were the top 10 increased genera. In addition, in Proteobacteria phylum, Enhydrobacter, Paracoccus, Neisseria, Lysobacter, Brevundimonas, Haemophilus, Sandaracinobacter, and Xanthomonas genera were greatly enriched in HZ subjects, accounting for eight from the top 20.
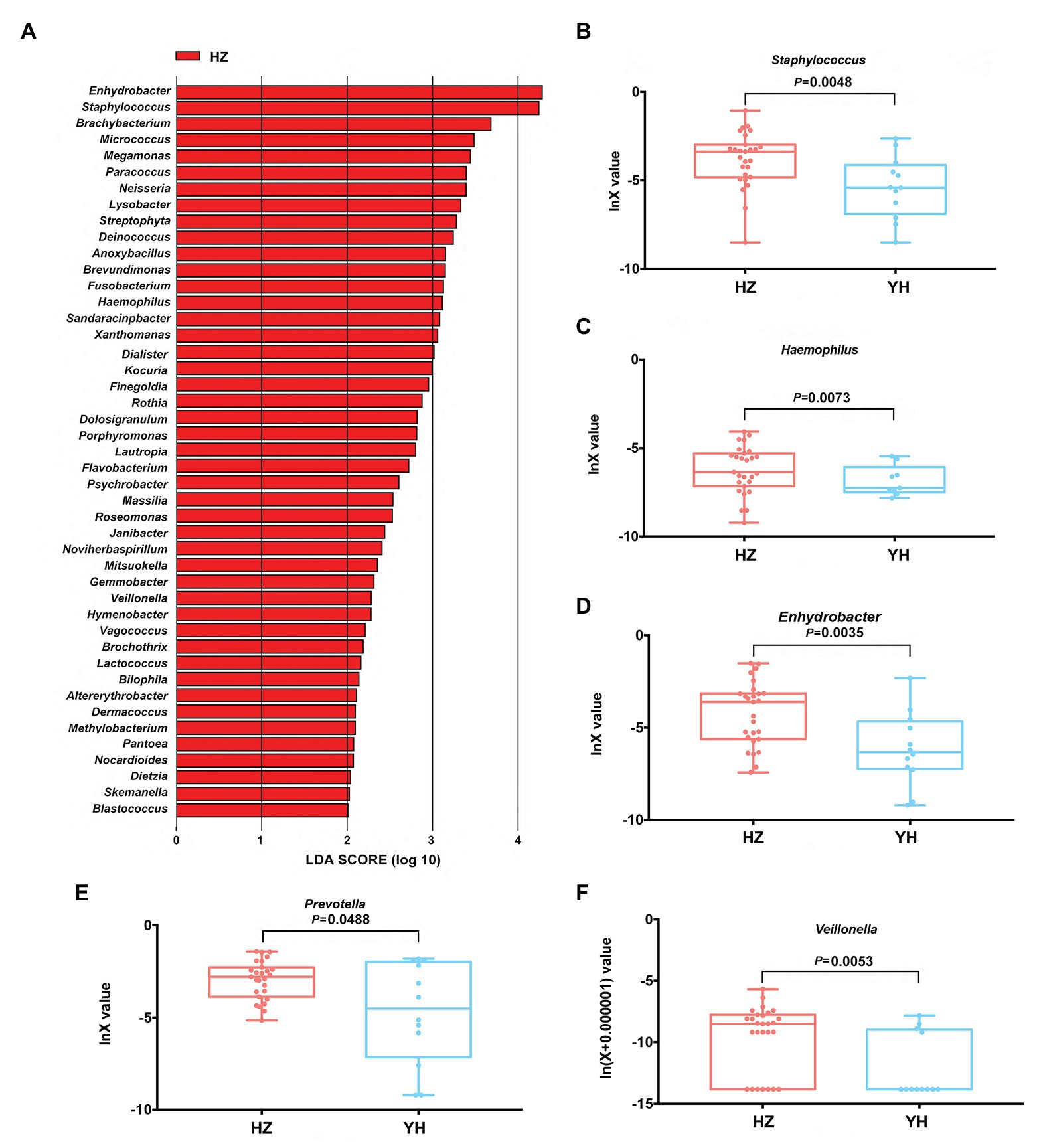
Figure 3. Differences in taxonomic composition of healthy women’s face between the HZ and YH groups using Lefse analysis and linear discriminant analysis (LDA). (A) Overexpressed bacterial genera in the HZ group chosen by LDA score > 2 (p < 0.05). (B–F) The relative expression of Staphylococcus, Haemophilus, Enhydrobacter, Prevotella, and Veillonella in the HZ and YH groups (p < 0.05). Significance determined in Wilcoxon rank sum test with one-sided alternatives.
Based on the previous report (Mukherjee et al., 2016), Haemophilus, Enhydrobacter, Granulicatella, Streptococcus, Veillonella, Bacillus, and Prevotella might be a hint of skin aging; we thus focused on the differential expressions of these genera between the HZ and YH groups in addition to Staphylococcus, one of the most predominant genera. It was shown that the expressions of Staphylococcus, Haemophilus, Enhydrobacter, Prevotella, and Veillonella were much higher in the HZ group than those in the YH group (p < 0.05, Figures 3B–F).
Functional Changes Associated With Microbiome Changes Between HZ and YH Groups
Using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) built-in reference database, a total of 31 Kyoto Encyclopedia of Genes and Genomes (KEGG) orthodoxies were significantly different between HZ and YH female individuals (p < 0.05, LDA score > 2; showing the top 10 in Figure 4). Notably, three pathways from the top 10 in the HZ group involved saturated fatty acid elongation, fatty acid β-oxidation I (generic), and fatty acid salvage, while pathways in the YH group were related to adenosine deoxyribonucleotides de novo biosynthesis II, guanosine deoxyribonucleotides de novo biosynthesis II, and pyrimidine deoxyribonucleosides salvage.
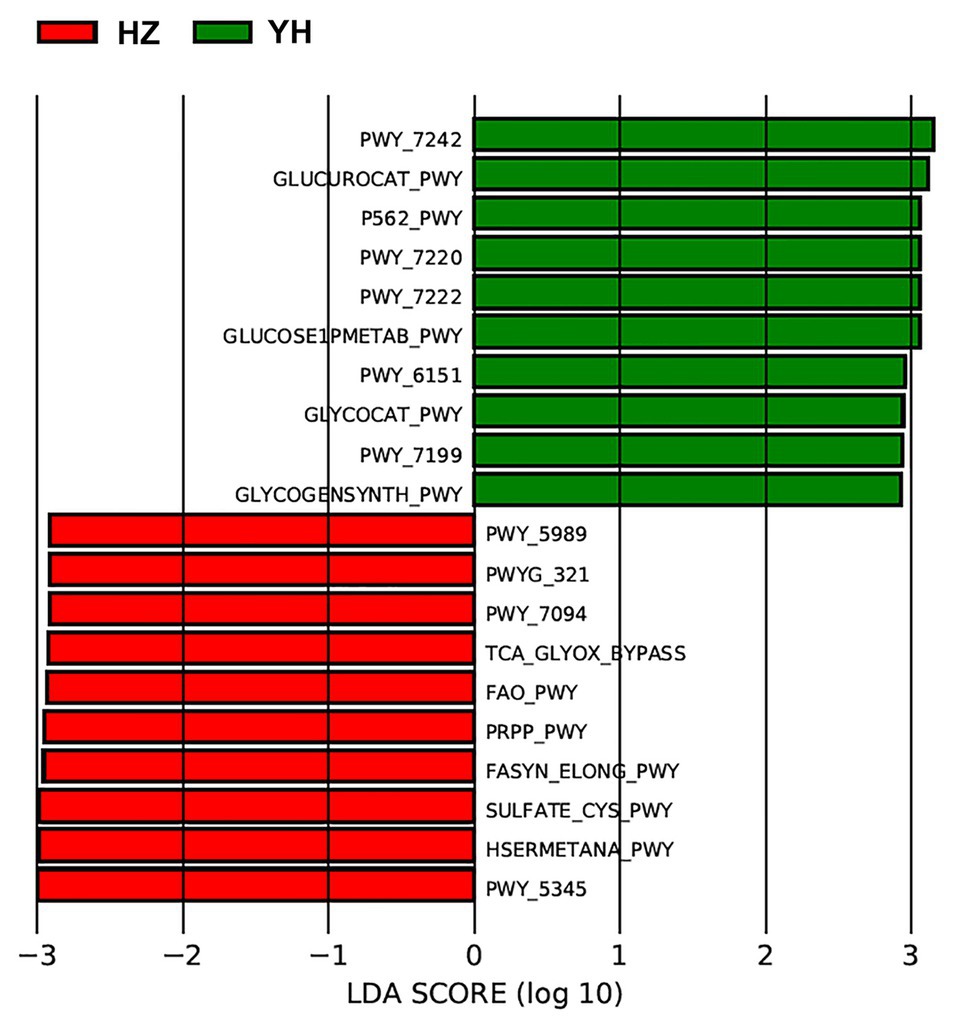
Figure 4. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) revealed different profiles of gene function in HZ female individuals compared with YH female individuals. The red bars indicated the Kyoto Encyclopedia of Genes and Genomes (KEGG) modules dominated in the HZ group, and the green ones indicated those in the YH group (p < 0.05, LDA score > 2).
Correlation of Facial Bacterial Microbiome Abundance With Clinical Characteristics
In order to identify the impact of clinical parameters on microbiome abundance, we also evaluated the correlations of the facial bacterial microbiome with clinical characteristics. As shown in Figure 5, we found that BMI was negatively correlated with Methylobacterium, Serratia, Holdemania, Microbacterium, Sandaracinobacter, Caulobacter, Klebsiella, and Rheinheimera amounts but positively correlated with Gluconobacter and Atopobium abundance (p < 0.05). Furthermore, age was negatively correlated with the levels of Arcobacter, Bhargavaea, Eubacterium, Eggerthella, Nevskia, Pseudacidovorax, and Megasphaera but positively correlated with Senegalimassilia, Azospirillum, and Rheinherimera (p < 0.05). The pH value in both oily and dry zones was positively correlated with Leptotrichia, Aggregatibacter, Gemella, Neisseria, Oribacterium, Actinomyces, and Peptostreptococcaceae_incertae _sedis abundance, while it was negatively correlated with Aerococcus and Janibacter abundance (p < 0.05).
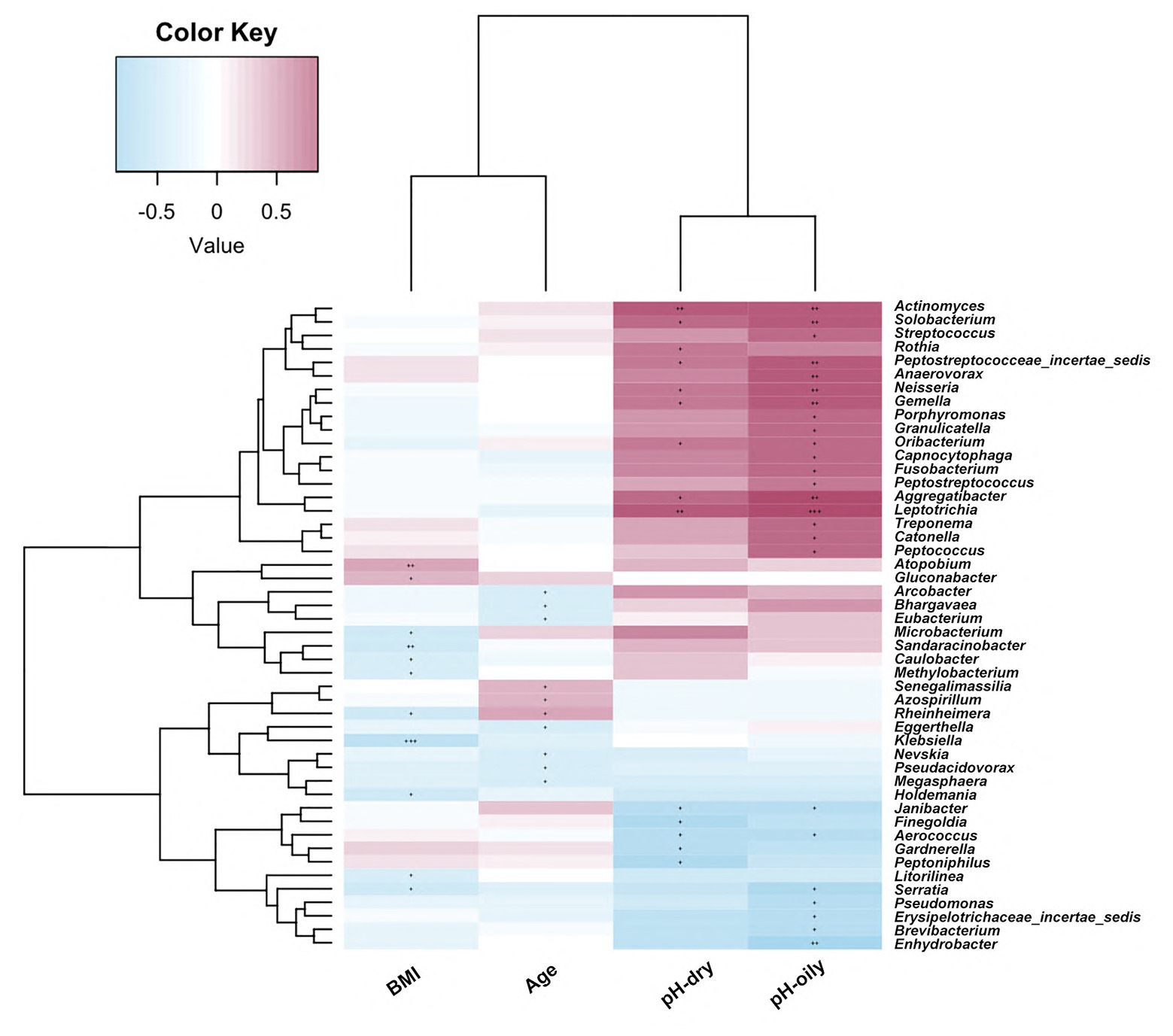
Figure 5. Correlations of facial bacterial microbiome abundance with clinical characteristics. The heat map of Spearman’s rank correlation coefficients between the facial bacterial microbiome abundance and clinical parameters. +p < 0.05; ++p < 0.01; +++p < 0.001; BMI, body mass index.
Discussion
In recent years, it had been reported that the diversity and abundance of skin microbiota vary with intrinsic factors, including gender, age, anatomical site, and ethnicity (Costello et al., 2009; Findley et al., 2013). External factors, such as cosmetics, soaps, and hygienic products (Lopes et al., 2015; Martin et al., 2015; Gonzalez et al., 2016; Shi et al., 2016; Urban et al., 2016), also contribute to the variation in skin microbiome. With the rapid development of national economy, environmental pollution is becoming a pressing issue. Skin, as the first line to interface with the air, is the target of various environmental stressors (Valacchi et al., 2012; Dong et al., 2019). Basic and clinical studies have provided growing evidence that airborne pollutants exacerbate skin aging (Vierktter et al., 2010; Ding et al., 2017) and even some chronic inflammatory skin diseases (Ahn, 2014; Pesce et al., 2015; Liu et al., 2018), which also exists when there is microbiome imbalance (He et al., 2006; Nielsen and Jiang, 2019). However, whether airborne pollutants have an impact on microbial communities of healthy skin, especially facial microbiome, still needs to be further explored.
Independent researches (Costello et al., 2009; Grice et al., 2009) from different regions characterized four main phyla in American population: Actinobacteria (36.6–51.8%), Firmicutes (24.4–34.3%), Proteobacteria (11.9–16.5%), and Bacteroides (6.3–9.5%). While for Chinese population, it was revealed by xi’an data (Kim et al., 2019) that Bacteroidetes and Firmicutes phyla were significantly higher in 20–30-year-old women’s face, while Proteobacteria and Actinobacteria were the top two abundant phyla in the 50–60s group. In addition, they also found that the alpha diversity of the younger group was higher than that of the older group in both species richness and phylogenetic diversity, which is in contrast to the results of a cohort of Japanese volunteers (Shibagaki et al., 2017). These discrepancies suggested that the formation of skin microbial community might be significantly affected by the individual’s residential environment and lifestyle.
Herein, focused on facial microbiome, we choose healthy and young women from two districts of Zhejiang Province in China. They have the same ethnicity, similar lifestyle, and dietary habits but with obviously different AQIs between these two districts because of different industrial development. Data of air quality collected from www.aqistudy.cn during 2014–2019 revealed a higher AQI together with higher levels of PM2.5, PM10, SO2, CO, and NO2 in HZ compared with those in YH where is there is well-developed agriculture (Supplementary Figure S3). Differed from previous report that skin’s microbial communities including bacteria, fungi, and virus were largely stable over months and years (1 month–2 years; Mukherjee et al., 2016; Oh et al., 2016), our microbiome data revealed an obvious difference in facial bacterial microbiome structure for healthy female individuals between these two districts, indicating that it might be affected by long-term exposure (>10 years) to airborne pollutes.
We further found an increase in both the taxonomic composition and the diversity of healthy female individuals in HZ. We hypothesized that similar to being inhaled into the human bodies to trigger immune response (Vawda et al., 2014) or respiratory tract infection (Miller and Peden, 2014), bacterial microorganisms, virus particles, and fungal spores (mostly sizing at the range of 0.2–2 μm) easily attached on PM particles and transferred with airflow, adhered to skin surfaces, and further caused microbiome changes. A latest study (Qin et al., 2019) reported that after exposure to high levels of PM2.5 and PM10, the abundance of the genera of Streptococcus, Haemophilus, Moraxella, and Staphylococcus in the human pharyngeal microbiome increased significantly, which is consistent with our results.
As one of the most predominant genera, Staphylococcus was obviously abundant in the HZ group than that in the YH group. On the one hand, Staphylococcus epidermidis, as a skin commensal, can activate Toll-like receptor 2 (TLR2; Lai et al., 2010) or produce phenol-soluble modulins (Cogen et al., 2010) to increase antimicrobial defense and butyric acid to downregulate the production of proinflammatory cytokines (Keshari et al., 2019). On the other hand, Staphylococcus aureus, usually as a potential pathogen, was found to be overgrown in atopic dermatitis (AD; Chng et al., 2016; Geoghegan et al., 2018) and could bind to TLR2 heterodimers to initiate long-lasting cutaneous inflammation (Biedermann et al., 2015). AD patients with greater S. aureus predominance showed more severe symptoms, while the ones with greater S. epidermidis predominance showed less severity (Bydr et al., 2017). Furthermore, the link between AD or eczema and pollution has been discussed before. A cohort in Southwest China (Li et al., 2018) revealed a positive association between the visits for eczema outpatients and airborne pollutants of NO2, SO2, and PM10, and another study from the Seoul Metropolitan Area (Kim et al., 2017) reported that a 10-U increase in PM10, NO2, and O3 could aggravate same-day symptoms of AD. Alpine climate treatment led to a reduction in the Staphylococcus genus, further resulting in significant changes in the skin microbiome composition in AD children (van Mierlo et al., 2019). However, which strain of Staphylococcus was the dominant one in our present study and how much of the ratio between S. aureus and S. epidermidis could indicate the risk of inflammatory skin diseases, especially atopic eczema, need to be further investigated.
As another important commensal organism of the skin, Propionibacterium was found abundant in both healthy individuals and acne patients (Fitz-Gibbon et al., 2013; Barnard et al., 2016). Higher relative abundances and less diversities of Propionibacteria and Propionibacterium acnes phage were found in healthy skin (Barnard et al., 2016). One study from India (Mukherjee et al., 2016) was focused on the cheek and forehead areas of young and healthy female individuals and revealed that Propionibacterium was positively, while Haemophilus, Enhydrobacter, Granulicatella, Streptococcus, Veillonella, Bacillus, and Prevotella were negatively associated with the cheek sebum that is significantly related to skin aging, which is characterized by reduced sweat and sebum secretion (Bolognia, 1995; Farage et al., 2008). Another study from Japan (Shibagaki et al., 2017) found that sebum levels were positively correlated with the Propionibacterium abundance, and the relative abundance of Propionibacterium decreased significantly in the elderly group. Both studies from Mexico (Lefebvre et al., 2015) and Shanghai (Lefebvre et al., 2016) reported that exposure to pollution could increase the facial skin dryness of individuals. More interestingly, Propionibacterium usually benefits the host by secreting short-chain fatty acid, antimicrobial substances, and its immunomodulation (Christensen and Bruggemann, 2014), and growth of pathogens such as S. aureus and Streptococcus pyogenes could be inhibited due to free fatty acids released by P. acnes (Grice and Segre, 2011). A decrease in Staphylococcus abundance and an increase in Propionibacterium abundance after coal tar treatment for AD suggested that the microbial communities of AD shifted toward those of healthy controls (Smits et al., 2020). In our study, an obvious decrease in Propionibacterium abundance, albeit not significant due to the small size of YH samples, together with a significant increase in Haemophilus, Enhydrobacter, Veillonella, and Prevotella was observed in the HZ group, although subjects in these two groups were age matched. Therefore, it may suggest that, to a certain extent, long-term exposure to airborne pollution would affect the physiological state of skin further to change the skin’s microbiome expression independent of chronological age.
We further identified functional variation based on the differential microbial communities between the HZ and YH group and predicted KEGG pathway on the basis of MetaCyc metabolic pathway data. The most prevalent processes of fasyn-elong-pwy, fao-pwy, and pwy-7094 in the HZ group are associated with metabolism of saturated fatty acid. Unlike unsaturated fatty acids as protective factors, saturated fatty acids exacerbate skin inflammation of keratinocytes (Herbert et al., 2018), affect the innate immune response of endothelial cells (Zhang et al., 2017), and induce cellular apoptosis (Lee et al., 2017). While in the YH group, the most dominated pathways involved the pathways for deoxyribonucleotide metabolism, such as pwy-7220, pwy-7222, and pwy-7199, which are very important for DNA repair or mitochondrial DNA replication in many types of organisms and cells (Kamiya, 2003; Setzer et al., 2008). These functional differences might provide meaningful information on the microbial role in the relationship between airborne pollution and skin microbiome communities. However, they needed to be confirmed experimentally in the future.
We also investigated the association between relevant clinical parameters with facial bacterial microbiome. As one important parameter, BMI was related with alternation in oral and vulva microbiome (Vongsa et al., 2019; de Andrade et al., 2020). A positive correlation between oral bacterial community with increased BMI was found by fingerprint analysis (de Andrade et al., 2020). High BMI (>30 kg/m2) was associated with higher pH in vulva and further modulated vulva microbiota, characterized by predomination of Finegoldia and Corynebacterium. However, the BMI-associated microbiota shift was not observed in abdominal skin samples (Vongsa et al., 2019). In our results, BMI was found to be negatively correlated with the abundance of several genera and positively with other two genera amounts, as shown in Figure 5. However, none of these genera differed between HZ and YH groups. It was indicated that although there was a statistical difference in BMI between the two groups, changes in facial microbiome were due to airborne pollutants rather than BMI discrepancy. In addition, similar results were also observed in the association between age and facial microbiome. A previous study (Kim et al., 2019) revealed a pattern similarity between the age type in young group of 20–30 years old and the group of 50–60 years old in the metabolism category, suggesting that an individual skin microbiome rather than chronological age might be a major determinant for skin age. As we discussed above, a decrease in Propionibacterium together with increases in Haemophilus, Enhydrobacter, Veillonella, and Prevotella might be a hint of skin aging. Then, in view of pH, the abundance of Janibacter was found to be negatively correlated with pH value and was significantly greater in the HZ group (Supplementary Table S4). However, there was no obvious difference in skin pH value in both oily and dry zones between the HZ and YH groups, which might be due to mild or moderate air pollution in HZ that is not sufficient to cause change in facial pH value or the mixed influence of free fatty acids released by Propionibacterium (Fluhr et al., 2001). Whether or not the increase in Janibacter was caused by the reduction in facial pH value as a consequence of air pollutants needs to be further verified by larger size samples.
There are some limitations to this study. The number of enrolled subjects in each group was relatively small. A total of respective 20 subjects were enrolled for each group in the beginning, while only seven oily-zone samples and five dry-zone samples from YH were used for further analysis because of the low bacterial DNA quantity obtained by swab-sampling method. Second, some skin physical parameters, such as sebum content, moisture content, and transepidermal water loss (TWEL), were not measured. The association of skin physical state and facial microbiome was not analyzed as well. Moreover, we did not perform a prospective cohort research, and whether or not air factors would have a dynamic effect on facial microbiome still remains unclear. Moreover, we noticed a higher variance of bacterial diversity and richness in YH samples. It may partly due to a little higher variance in age and BMI of YH participants and also because of a very low Shannon diversity of some samples (YH1-3, YH1-6, and YH2-10) in the YH group. An increase in sample size in our future study may reduce the impact of variance in results and conclusion.
In conclusion, this study indicated that long-term exposure to airborne pollutants could lead to significant alterations with an increase in both the abundance and diversity of facial bacterial microbiome, including metabolic pathways. Our data can provide useful information for further researches on the composition and function of the skin microbiome related to air quality as well as for the development of therapeutic agents targeting the microbes and their metabolites to resist damages of airborne pollutants.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of the First Affiliated Hospital, School of Medicine of Zhejiang University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
XL conceived the study. YW, YZ, and XL performed the literature search and wrote the protocols. ZW, YW, YZ, and LR recruited the subjects and collected the specimens. AL performed the laboratory analyses and analyzed the data. XL and AL interpreted the data. All authors contributed to the writing and editing of the report and approved the final version.
Funding
This study was supported by 2017 CMA-L’OREAL China Skin/Hair Grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.548618/full#supplementary-material
References
Ahn, K. (2014). The role of air pollutants in atopic dermatitis. J. Allergy Clin. Immunol. 134, 993–999. doi: 10.1016/j.jaci.2014.09.023
Araviiskaia, E., Berardesca, E., Bieber, T., Gontijo, G., Sanchez-Viera, M., Marrot, L., et al. (2019). The impact of airborne pollution on skin. J. Eur. Acad. Dermatol. Venereol. 33, 1496–1505. doi: 10.1111/jdv.15583
Barnard, E., Shi, B., Kang, D., Craft, N., and Li, H. (2016). The balance of metagenomic elements shapes the skin microbiome in acne and health. Sci. Rep. 6:39491. doi: 10.1038/srep39491
Biedermann, T., Skabytska, Y., Kaesler, S., and Volz, T. (2015). Regulation of T cell immunity in atopic dermatitis by microbes: the Yin and Yang of cutaneous inflammation. Front. Immunol. 6:353. doi: 10.3389/fimmu.2015.00353
Bydr, A. L., Deming, C., Cassidy, S. K. B., Harrison, O. J., Ng, W. I., Conlan, S., et al. (2017). Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci. Transl. Med. 9:eaa14651. doi: 10.1126/scitranslmed.aal4651
Capone, K. A., Dowd, S. E., Stamatas, G. N., and Nikolovski, J. (2011). Diversity of the human skin microbiome early in life. J. Invest. Dermatol. 131, 2026–2032. doi: 10.1038/jid.2011.168
Chng, K. R., Tay, A. S., Li, C., Ng, A. H., Wang, J., Suri, B. K., et al. (2016). Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare. Nat. Microbiol. 1:16106. doi: 10.1038/nmicrobiol.2016.106
Christensen, G. J., and Bruggemann, H. (2014). Bacterial skin commensals and their role as host guardians. Benefic. Microbes 5, 201–215. doi: 10.3920/BM2012.0062
Cogen, A. L., Yamasaki, K., Sanchez, K. M., Dorschner, R. A., Lai, Y., MacLeod, D. T., et al. (2010). Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J. Invest. Dermatol. 130, 192–200. doi: 10.1038/jid.2009.243
Costello, E. K., Lauber, C. L., Hamady, M., Fierer, N., Gordon, J. I., and Knight, R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486
de Andrade, P. A. M., Giovani, P. A., Araujo, D. S., de Souza, A. J., Pedroni-Pereira, A., Kantovitz, K. R., et al. (2020). Shifts in the bacterial community of saliva give insights on the relationship between obesity and oral microbiota in adolescents. Arch. Microbiol. 202, 1085–1095. doi: 10.1007/s00203-020-01817-y
Ding, A., Yang, Y., Zhao, Z., Hüls, A., Vierkötter, A., Yuan, Z., et al. (2017). Indoor PM2.5 exposure affects skin aging manifestation in a Chinese population. Sci. Rep. 7:15329. doi: 10.1038/s41598-017-15295-8
Dong, Y. M., Liao, L. Y., Li, L., Yi, F., Meng, H., He, Y. F., et al. (2019). Skin inflammation induced by ambient particulate matter in China. Sci. Total Environ. 682, 364–373. doi: 10.1016/j.scitotenv.2019.05.155
Dréno, B., Araviiskaia, E., Berardesca, E., Gontijo, G., Sanchez-Viera, M., Xiang, L. F., et al. (2016). Microbiome in healthy skin, update for dermatologists. J. Eur. Acad. Dermatol. Venereol. 30, 2038–2047. doi: 10.1111/jdv.13965
Dybboe, R., Bandier, J., Skov, L., Engstrand, L., and Johansen, J. D. (2017). The role of the skin microbiome in atopic dermatitis: a systematic review. Br. J. Dermatol. 177, 1272–1278. doi: 10.1111/bjd.15390
Farage, A., Miller, W., Elsner, P., and Maibach, I. (2008). Functional and physiological characteristics of the aging skin. Aging Clin. Exp. Res. 20, 195–200. doi: 10.1007/bf03324769
Findley, K., Oh, J., Yang, J., Conlan, S., Deming, C., Meyer, J. A., et al. (2013). Topographic diversity of fungal and bacterial communities in human skin. Nature 498, 367–370. doi: 10.1038/nature12171
Fitz-Gibbon, S., Tomida, S., Chiu, B. H., Nguyen, L., Du, C., Liu, M., et al. (2013). Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J. Invest. Dermatol. 133, 2152–2160. doi: 10.1038/jid.2013.21
Fluhr, J. W., Kao, J., Jain, M., Ahn, S. K., Feingold, K. R., and Elias, P. M. (2001). Generation of free fatty acids from phospholipids regulates stratum corneum acidification and integrity. J. Invest. Dermatol. 117, 44–51. doi: 10.1046/j.0022-202x.2001.01399.x
Geoghegan, J. A., Irvine, A. D., and Foster, T. J. (2018). Staphylococcus aureus and atopic dermatitis: a complex and evolving relationship. Trends Microbiol. 26, 484–497. doi: 10.1016/j.tim.2017.11.008
Gonzalez, M. E., Schaffer, J. V., Orlow, S. J., Gao, Z., and Li, H. (2016). Cutaneous microbiome effects of fluticasone propionate cream and adjunctive bleach baths in childhood atopic dermatitis. J. Am. Acad. Dermatol. 75, 481.e8–493.e8. doi: 10.1016/j.jaad.2016.04.066
Grice, E. A., Kong, H. H., Conlan, S., Deming, C. B., Davis, J., Young, A. C., et al. (2009). Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192. doi: 10.1126/science.1171700
Grice, E. A., and Segre, J. A. (2011). The skin microbiome. Nat. Rev. Microbiol. 9, 244–253. doi: 10.1038/nrmicro2537
He, Q. C., Tavakkol, A., Wietecha, K., Begum-Gafur, R., Ansari, S. A., and Polefka, T. (2006). Effects of environmentally realistic levels of ozone on stratum corneum function. Int. J. Cosmet. Sci. 28, 349–357. doi: 10.1111/j.1467-2494.2006.00347.x
Herbert, D., Franz, S., Popkova, Y., Anderegg, U., Schiller, J., and Schwede, K. (2018). High-fat diet exacerbates early psoriatic skin inflammation independent of obesity: saturated fatty acids as key players. J. Invest. Dermatol. 138, 1999–2009. doi: 10.1016/j.jid.2018.03.1522
Hu, S., Li, A., Huang, T., Lai, J., Li, J., Sublette, M. E., et al. (2019). Gut microbiota changes in patients with bipolar depression. Adv. Sci. 6:1900752. doi: 10.1002/advs.201900752
Hughey, M. C., Walke, J. B., Becker, M. H., Umile, T. P., Burzynski, E. A., Minbiole, K. P. C., et al. (2016). Short-term exposure to coal combustion waste has little impact on the skin microbiome of adult spring peepers (Pseudacris crucifer). Appl. Environ. Microbiol. 82, 3493–3502. doi: 10.1128/AEM.00045-16
Kamiya, H. (2003). Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: approaches using synthetic oligonucleotides and nucleotides: survey and summary. Nucleic Acids Res. 31, 517–531. doi: 10.1093/nar/gkg137
Keshari, S., Balasubramaniam, A., Myagmardoloonjin, B., Herr, D. R., Negari, I. P., and Huang, C. M. (2019). Butyric acid from probiotic Staphylococcus epidermidis in the skin microbiome down-regulates the ultraviolet-induced pro-inflammatory IL-6 cytokine via short-chain fatty acid receptor. Int. J. Mol. Sci. 20:E4477. doi: 10.3390/ijms20184477
Kim, Y. M., Kim, J., Han, Y., Joen, B. H., Cheong, H. K., and Ahn, K. (2017). Short-term effects of weather and air pollution on atopic dermatitis symptoms in children: a panel study in Korea. PLoS One 12:e0175229. doi: 10.1371/journal.pone.0175229
Kim, H. J., Kim, H., Kim, J. J., Myeong, N. R., Kim, T., Park, T., et al. (2018). Fragile skin microbiomes in megacities are assembled by a predominantly nich-based process. Sci. Adv. 4:e1701581. doi: 10.1126/sciadv.1701581
Kim, H. J., Kim, J. J., Myeong, N. R., Kim, T., Kim, D., An, S., et al. (2019). Segregation of age-related skin microbiome characteristics by functionality. Sci. Rep. 9:16748. doi: 10.1038/s41598-019-53266-3
Lai, Y., Cogen, A. L., Radek, K. A., Park, H. J., Macleod, D. T., Leichtle, A., et al. (2010). Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J. Invest. Dermatol. 130, 2211–2221. doi: 10.1038/jid.2010.123
Lee, S. H., Cunha, D., Piermarocchi, C., Paternostro, G., Pinkerton, A., and Ladriere, L. (2017). High-throughput screening and bioinformatic analysis to ascertain compounds that prevent saturated fatty acid-induced β-cell apoptosis. Biochem. Pharmacol. 138, 140–149. doi: 10.1016/j.bcp.2017.05.007
Lefebvre, M. A., Pham, D. M., Boussouira, B., Bernard, D., Camus, C., and Nguyen, Q. L. (2015). Evaluation of the impact of urban pollution on the quality of skin: a multicentre study in Mexico. Int. J. Cosmet. Sci. 37, 329–338. doi: 10.1111/ics.12203
Lefebvre, M. A., Pham, D. M., Boussouira, B., Qiu, H., Ye, C., Long, X., et al. (2016). Consequences of urban pollution upon skin status. A controlled study in Shanghai area. Int. J. Cosmet. Sci. 38, 217–223. doi: 10.1111/ics.12270
Li, A., Fan, L., Xie, L., Ren, Y., and Li, L. (2018). Associations between air pollution, climate factors and outpatient visits for eczema in West China Hospital, Chengdu, South-Western China: a time series analysis. J. Eur. Acad. Dermatol. Venerol. 32, 486–494. doi: 10.1111/jdv.14730
Li, J., Li, R., Cui, R., Meng, Y., and Fu, H. (2019). Spacial and temporal variation of inorganic irons in rainwater in Sichuan province from 2011 to 2016. Environ. Pollut. 254:112941. doi: 10.1016/j.envpol.2019.07.109
Liu, W., Pan, X., Vierkotter, A., Guo, Q., Wang, X., Wang, Q., et al. (2018). A time-series study of the effect of air pollution on outpatient visits for acne vulgaris in Beijing. Skin Pharmacol. Physiol. 31, 107–113. doi: 10.1159/000484482
Lopes, C., Soares, J., Tavaria, F., Duarte, A., Correia, O., Sokhatska, O., et al. (2015). Chitosan coated textiles may improve atopic dermatitis severity by modulating skin staphylococcal profile: a randomized controlled trial. PLoS One 10:e0142844. doi: 10.1371/journal.pone.0142844
Martin, R., Henley, J. B., Sarrazin, P., Sarrazin, P., and Seité, S. (2015). Skin microbiome in patients with psoriasis before and after balneotherapy at the thermal care center of La Roche-Posay. J. Drugs Dermatol. 14, 1400–1405.
McInnes, P., and Cutting, M. A. (2010). Core microbiome sampling protocol A HMP Protocol # 07-001. Manual of Procedures—Human Microbiome Project 11, 101-109.
Miller, R. L., and Peden, D. B. (2014). Environmental effects on immune responses in patients with atopy and asthma. J. Allergt Clin. Immunol. 134, 1001–1008. doi: 10.1016/j.jaci.2014.07.064
Mukherjee, S., Mitra, R., Maitra, A., Gupta, S., Kumaran, S., Chakrabortty, A., et al. (2016). Sebum and hydration levels in specific regions of human face significantly predict the nature and diversity of facial skin microbiome. Sci. Rep. 6:36062. doi: 10.1038/srep36062
Nielsen, M. C., and Jiang, S. C. (2019). Alterations of the human skin microbiome after ocean water exposure. Mar. Pollut. Bull. 145, 595–603. doi: 10.1016/j.marpolbul.2019.06.047
Oh, J., Byrd, A. L., Park, M., NISC Comparative Sequencing ProgramKong, H. H., and Segre, J. A. (2016). Temporal stability of the human skin microbiome. Cell 165, 854–866. doi: 10.1016/j.cell.2016.04.008
Ottosson, F., Brunkwall, L., Ericson, U., Nilsson, P. M., Almgren, P., Fernandez, C., et al. (2018). Connection between BMI-related plasma metabolite profile and gut microbiota. J. Clin. Endocrinol. Metab. 103, 1491–1501. doi: 10.1210/jc.2017-02114
Pesce, G., Marcon, A., Carosso, A., Antonicelli, L., Cazzoletti, L., Ferrari, M., et al. (2015). Adult eczema in Italy: prevalence and associations with environmental factors. J. Eur. Acad. Dermatol. Venereol. 29, 1180–1187. doi: 10.1111/jdv.12784
Qin, T., Zhang, F., Zhou, H., Ren, H., Du, Y., Liang, S., et al. (2019). High-level PM2.5/PM10 exposure is associated with alterations in the human pharyngeal microbiota composition. Front. Microbiol. 10:54. doi: 10.3389/fmicb.2019.00054
Reiger, M., Traidl-Hoffmann, C., and Neumann, A. U. (2020). The skin microbiome as a clinical biomarker in atopic eczema: promises, navigation, and pitfalls. J. Allergy Clin. Immunol. 145, 93–96. doi: 10.1016/j.jaci.2019.11.004
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Setzer, B., Lebrecht, D., and Walker, U. A. (2008). Pyrimidine nucleoside depletion sensitizes to the mitochondrial hepatotoxicity of the reverse transcriptase inhibitor stavudine. Am. J. Pathol. 172, 681–690. doi: 10.2353/ajpath.2008.070613
Shi, B., Bangayan, N. J., Curd, E., Taylor, P. A., Gallo, R. L., Leung, D. Y. M., et al. (2016). The skin microbiome is different in pediatric versus adult atopic dermatitis. J. Allergy Clin. Immunol. 138, 1233–1236. doi: 10.1016/j.jaci.2016.04.053
Shibagaki, N., Suda, W., Clavaud, C., Bastien, P., Takayasu, L., Iioka, E., et al. (2017). Aging-related changes in the diversity of women’s skin microbiomes associated with oral bacteria. Sci. Rep. 7:10567. doi: 10.1038/s41598-017-10834-9
Smits, J. P. H., Ederveen, T. H. A., Rikken, G., van den Brink, N. J. M., van Vlijmen-Willems, I. M. J. J., and Boekhorst, J. (2020). Targeting the cutaneous microbiota in atopic dermatitis by coal tar via AHR-dependent induction of antimicrobial peptides. J. Invest. Dermatol. 140, 415.e10–424.e10. doi: 10.1016/j.jid.2019.06.142
Urban, J., Fergus, D. J., Savage, A. M., Ehlers, M., Menninger, H. L., Dunn, R. R., et al. (2016). The effect of habitual and experimental antiperspirant and deodorant product use on the armpit microbiome. PeerJ 4:e1605. doi: 10.7717/peerj.1605
Valacchi, G., Sticozzi, C., Pecorelli, A., Cervellati, F., Cervellati, C., and Maioli, E. (2012). Cutaneous responses to environmental stressors. Ann. N. Y. Acad. Sci. 1271, 75–81. doi: 10.1111/j.1749-6632.2012.06724.x
van Mierlo, M. M. F., Totté, J. E. E., Fieten, K. B., van den Broek, T. J., Schuren, F. H. J., and Pardo, L. M. (2019). The influence of treatment in alpine and moderate maritime climate on the composition of the skin microbiome in patients with difficult to treat atopic dermatitis. Clin. Exp. Allergy 49, 1437–1445. doi: 10.1111/cea.13492
Vawda, S., Mansour, R., Takeda, A., Funnell, P., Kerry, S., Mudway, I., et al. (2014). Associations between inflammatory and immune response genes and adverse respiratory outcomes following exposure to outdoor air pollution: a HuGE systematic review. Am. J. Epidemiol. 179, 432–442. doi: 10.1093/aje/kwt269
Vierktter, A.Schikowski, T., Ranft, U., Sugiri, D., Matsui, M., Krämer, U., et al. (2010). Airborne particle exposure and extrinsic skin aging. J. Invest. Dermatol. 130, 2719–2726. doi: 10.1038/jid.2010.204
Vongsa, R., Hoffman, D., Shepard, K., and Koenig, D. (2019). Comparative study of vulva and abdominal skin microbiota of healthy females with high and average BMI. BMC Microbiol. 19:16. doi: 10.1186/s12866-019-1391-0
Woodall, G. M., Hoover, M. D., Williams, R., Benedict, K., Harper, M., Soo, J. C., et al. (2017). Interpreting mobile and handheld air sensor readings in relation to air quality standards and health effect reference values: tackling the challenges. Atmosphere 8:182. doi: 10.3390/atmos8100182
Keywords: airborne pollution, air quality index, 16S rRNA gene, microbiome, healthy women
Citation: Wu Y, Wang Z, Zhang Y, Ruan L, Li A and Liu X (2020) Microbiome in Healthy Women Between Two Districts With Different Air Quality Index. Front. Microbiol. 11:548618. doi: 10.3389/fmicb.2020.548618
Edited by:
M. Pilar Francino, Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunitat Valenciana (FISABIO), SpainReviewed by:
Osiris Gaona, National Autonomous University of Mexico, MexicoIsaac G. Santoyo, National Autonomous University of Mexico, Mexico
Luisa I. Falcon, National Autonomous University of Mexico, Mexico
Copyright © 2020 Wu, Wang, Zhang, Ruan, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Liu, aHpsaXV4aWFveWFuQHpqdS5lZHUuY24=; Ang Li, bGlhQHpqdS5lZHUuY24=
†These authors have contributed equally to this work