 Ning Zhang
Ning Zhang Zhenzhou Yang
Zhenzhou Yang Wenxing Liang
Wenxing Liang- Key Lab of Integrated Crop Pest Management of Shandong Province, College of Plant Health and Medicine, Qingdao Agricultural University, Qingdao, China
Lysine crotonylation (Kcr), a recently discovered post-translational modification, plays a key role in the regulation of diverse cellular processes. Botrytis cinerea is a destructive necrotrophic fungal pathogen distributed worldwide with broad ranging hosts. However, the functions of Kcr are unknown in B. cinerea or any other plant fungal pathogens. Here, we comprehensively evaluated the crotonylation proteome of B. cinerea and identified 3967 Kcr sites in 1041 proteins, which contained 9 types of modification motifs. Our results show that although the crotonylation was largely conserved, different organisms contained distinct crotonylated proteins with unique functions. Bioinformatics analysis demonstrated that the majority of crotonylated proteins were distributed in cytoplasm (35%), mitochondria (26%), and nucleus (22%). The identified proteins were found to be involved in various metabolic and cellular processes, such as cytoplasmic translation and structural constituent of ribosome. Particularly, 26 crotonylated proteins participated in the pathogenicity of B. cinerea, suggesting a significant role for Kcr in this process. Protein interaction network analysis demonstrated that many protein interactions are regulated by crotonylation. Furthermore, our results show that different nutritional conditions had a significant influence on the Kcr levels of B. cinerea. These data represent the first report of the crotonylome of B. cinerea and provide a good foundation for further explorations of the role of Kcr in plant fungal pathogens.
Introduction
Post-translational modifications (PTMs), in which functional groups are covalently introduced to amino acid residues, play important roles in regulating diverse biological processes (Khoury et al., 2011). With the application of liquid chromatography-mass spectrometry (LC-MS/MS)-based proteomics, an array of novel PTMs have been identified. Particularly, lysine residues are targeted by numerous PTMs, including acetylation, succinylation, butyrylation, propionylation, 2-hydroxyisobutyrylation, glutarylation, malonylation, lactylation, and crotonylation (Chen et al., 2007; Tan et al., 2011; Dai et al., 2014; Hirschey and Zhao, 2015; Zhang et al., 2019). Currently, there is a lack of research on the identification of substrates of these PTMs and elucidation of their functional impacts.
Lysine crotonylation (Kcr) was first identified on histones and its role in regulation of gene transcription has been well investigated (Tan et al., 2011). HDAC1, a member of the class I histone deacetylases (HDACs), was found to catalyze histone decrotonylation in mammalian cells (Wei et al., 2017a). Therefore, it is presumed that both protein crotonylation and acetylation are regulated by histone acetyltransferases (HATs) and HDACs. Other than histone proteins, crotonylation is newly discovered targeting non-histone proteins, regulating protein stability, enzymatic activity, protein localization, and many other cellular processes (Wei et al., 2017b; Xu et al., 2017). Advancements in novel enrichment strategies (LC-MS/MS) have made it possible to investigate Kcr on a proteomic level and large number of crotonylated proteins have been identified. Up to now, several organisms or cell lines have been reported with respect to Kcr, including Carica papaya (Liu K. et al., 2018), Nicotiana tabacum (Sun et al., 2017), Oryza sativa (Liu S. et al., 2018), Danio rerio (Kwon et al., 2018), and four human cell lines (Wei et al., 2017b; Wu et al., 2017; Xu et al., 2017; Huang et al., 2018). However, the understanding of the features of Kcr is still insufficient. Particularly in plant fungi, global identification of this kind of PTM has not been well reported.
Botrytis cinerea is an worldwide economically important fungal pathogen that can cause gray mold disease on a wide variety of hosts (Williamson et al., 2007). In recent years, extensive research has been conducted to interpret transcriptional regulation of virulence genes in B. cinerea (Weiberg et al., 2013; Brandhoff et al., 2017; Wang et al., 2017, 2018; Porquier et al., 2019). However, gaps in the protein level studies limited a deeper understanding of the molecular basis of B. cinerea pathogenesis. In this study, we conducted the first proteome-wide Kcr analysis in B. cinerea. In total, 3967 Kcr sites in 1041 proteins were identified. The crotonylated proteins were distributed in multiple compartments and associated with diversified biological processes. Particularly, 26 crotonylated proteins were found to be associated with virulence of B. cinerea. These proteins participates in diverse pathogenesis pathways including signal transduction, redox homeostasis, plant cell wall degrading, secretory of virulence factors, and secondary metabolites biosynthesis. This work provides an extensive dataset for further investigating the physiological role of Kcr in B. cinerea and other filamentous fungal pathogens.
Materials and Methods
Protein Extraction From B. cinerea
The B. cinerea strain B05.10 was cultured on PDA plates (2% dextrose, 20% potato, and 1.5% agar) at 25°C for 10 days, and then the conidia were collected and transferred into YEPD medium (2% glucose, 1% yeast extract, and 2% peptone) with shaking at 180 rpm for 14 h. The protein extraction was performed as previously described (Zhou et al., 2016). Briefly, the harvested mycelia were ground into powder in liquid nitrogen. Then the powder sample was suspended by lysis buffer (50 mM nicotinamide, 8 M urea, 65 mM dithiothreitol, 1% Triton-100, 0.1% protease inhibitor cocktail, 2 mM EDTA, and 3 μM Trichostatin A) and then sonicated. After centrifugation at 15,000 × g at 4°C for 15 min, 15% cold TCA was used to precipitate the proteins at −20°C for 2 h. The precipitates were washed three times with cold acetone after centrifugation at 4°C for 15 min. Finally, the target protein was re-dissolved in 8 M urea supplemented with 100 mM NH4CO3 (pH 8.0) and 2-D Quant kit (GE Healthcare) was used to determine protein concentration according to the manufacturer’s instructions.
Affinity Enrichment of Lysine Crotonylated Peptides
For affinity enrichment, the B. cinerea proteins were firstly digested into peptides by trypsin. The digestion reaction was performed with a previously described experimental approach (Zhou et al., 2016), briefly, the trypsin was added at 1:50 trypsin-to-protein mass ratio at the first time overnight and added again at 1:100 trypsin-to-protein mass ratio for another 4 h. The sample was separated into fractions by high PH reverse-phase HPLC using Agilent 300 Extend C18 column (5 μM particles, 4.6 mm ID, and 250 mm length) (Liu L. et al., 2016). The peptides were separated firstly into 80 fractions with a gradient of 2 to 60% acetonitrile in 10 mM (NH4)2CO3 (PH 10.0). Then, the peptides were combined into 8 fractions and dried by vacuum centrifuging. For Kcr peptides enrichment, the tryptic peptides were dissolved in NETN buffer (1 mM EDTA, 100 mM NaCl, 0.5% NP-40, and 50 mM Tris–HCl pH 8.0) and then separated into several fractions. Each fraction was incubated with pan anti-Kcr antibody (PTM-502, PTM Biolabs) conjugated agarose beads overnight at 4°C with gentle shaking. Then the peptides bound with beads were eluted with 0.1% trifluoroacetic acid after washing with NETN buffer and then the acquired peptides were cleaned with C18 Zip Tips (Millipore) (Sun et al., 2019).
LC-MS/MS Analysis
Liquid chromatography-mass spectrometry analysis of the crotonylated peptides was performed as described (Li et al., 2016; Liu L. et al., 2016; Lv et al., 2016; Zhou et al., 2016) by Micrometer Biotech Company (Hangzhou, China). Briefly, The Kcr peptides were separated using a reversed-phase analytical column (Acclaim PepMap RSLC C18 column, Thermo Scientific) on UPLC system. The gradient was composed of an increase from 2% formic acid (0.1%) to 10% formic acid (0.1% in 98% acetonitrile) for 6 min, 10 to 20% for 45 min, 20% climbing to 80% in 7 min and then holding at 80% at least for 4 min, all maintaining a flow rate of 250 nl/min. The peptides were subjected by to ESI/NSI sources followed by MS/MS in Q ExactiveTM Plus (Thermo Scientific) coupled online to UPLC. The Orbitrap was used to detect whole peptides and ion fragments at a resolution of 70,000 and 17,500, respectively, with NCE set at 30. The electrospray voltage was set at 2.0 kV. Automatic gain control (AGC) was used to avoid ion trap overfilling. The m/z range was from 350 to 1800 for MS scans. The MS fixed first mass was set at 100 m/z.
Generation of Bcpck1-GFP Strains
To generate Bcpck1-GFP overexpression construct, CDS (coding domain sequence) of Bcpck1 was cloned into pOPT-GFP vector that contains oliC promoter, niaD terminator, hygromycin phosphotransferase and optimized C-terminal eGFP sequence (Leroch et al., 2011). The constructed plasmid was then transformed into the B05.10 strain using protoplast transformation of B. cinerea (Gronover et al., 2001). The resulting transformants were selected by 100 μg/ml hygromycin B.
Immunoprecipitation and Western Blot Analysis
Total proteins were extracted using lysis buffer (0.5 mM EDTA, 150 mM NaCl, 0.5% NP-40, and 10 mM Tris–HCl pH 7.5) from mycelium of B. cinerea cultivated in liquid YEPD or MM (10 mM K2HPO4, 10 mM KH 2PO4, 4 mM (NH4)2SO4, 2.5 mM NaCl, 2 mM MgSO4, 0.45 mM CaCl2, 9 mM FeSO4, 50 mM glucose, and 1 L water, pH 6.5) with shaking at 180 rpm for 14 h. Anti-GFP agarose beads (KTSM1301, KT HEALTH) were subsequently added and the mixture was incubated for 2 h. The beads were washed 3 times with 500 μl of lysis buffer, and the bound proteins were eluted with SDS-PAGE loading buffer.
The obtained proteins were then separated by 12% SDS-PAGE and immunoblotted using anti-GFP antibody (ab290, Abcam) and anti-Kcr antibody, respectively. Proteins were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore) according to the manufacturer’s protocol.
Database Search
MaxQuant and Andromeda search engine (v.1.5.1.8) were used to analyze the raw data of MS/MS (Cox and Mann, 2008; Cox et al., 2009). The tandem mass spectra collected were searched against B. cinerea B05.10 database from UniProt. Mass errors of fragment ions and precursor were set as 0.02 Da and 10 ppm, respectively. Trypsin/P was specified as cleavage enzyme allowing up to 4 missing cleavage, 5 charges and 5 modifications per peptide. Carbamidomethylation on Cysteine was specified as fixed modification and crotonylation on lysine was fixed as variable modification. The minimal peptide was set to seven, and the false discovery rate (FDR) threshold for modification sites and peptides were set as 1%. The Kcr site localization probability of <0.75 was exclude (Zhou et al., 2016; Yang et al., 2018).
Bioinformatics Analysis
Gene ontology (GO) of crotonylation proteome was performed from the UniProt-GOA database based on three categories: cellular component, molecular function, and biological process according to Zhou et al. (2016). The soft WoLF PSORT was used to predict the subcellular localization of the crotonylated protein (Horton et al., 2007). Proteins secondary structures (β-strand, α-helix, coil) were analyzed by the online tool NetSurfP (Chou and Schwartz, 2011). Soft MoMo (motif-x algorithm) was used to analyze the sequences model of crotonylated proteins composed of amino acids in distinct positions of modify-21-mers (10 amino acids up- and downstream of the Kcr site) in all protein sequences. To define the conservation of crotonylation, the BLASTP was used to compare the crotonylated protein sequences of B. cinerea with Carica papaya (Liu K. et al., 2018), Nicotiana tabacum (Sun et al., 2017), Oryza sativa (Liu S. et al., 2018), Danio rerio (Kwon et al., 2018), and four human cell lines including Human peripheral blood, Human Hela cells, Human H1299 cells, and Human A549 cells (Wei et al., 2017b; Wu et al., 2017; Xu et al., 2017; Huang et al., 2018). InterProScan was used to perform the protein domain functional annotation based on protein alignment method and the InterPro domain database. Kyoto Encyclopedia of Genes and Genomes (KEGG) database was employed to annotate protein pathway description (Kanehisa et al., 2004). Cytoscape software was used to analyze the protein-protein interactions which was obtained from the STRING database (Shannon et al., 2003; Szklarczyk et al., 2015). A two-tailed Fisher’s exact test was used to verify the enrichment of lysine crotonylated proteins against all database proteins. All projects with a corrected p-valve < 0.05 is considered significant.
Results
Identification and Analysis of Lysine-Crotonylated Sites and Proteins in B. cinerea
Lysine crotonylation is a newly discovered PTM which is poorly studied in B. cinerea, one of the most important plant fungi pathogen in the world. To extensively characterize protein crotonylation in B. cinerea, a proteome-wide analysis of Kcr was carried out. Proteins extracted from mycelium were digested by trypsin and the crotonylation peptides were enriched using anti-crotonyl lysine antibody, followed by high-resolution LC-MS/MS analysis. An overview of the experimental procedures was demonstrated in Supplementary Figure S1A. Mass errors of the identified peptides were checked first to confirm the dependability of the MS data. As shown in Supplementary Figure S1B, the distribution of mass errors was near zero, most were less than 10 PPM, indicating that the mass accuracy fits the requirement. Moreover, the length of most lysine-crotonylated peptides were between 7 and 20, which is consistent with tryptic peptides (Supplementary Figure S1C). The above results demonstrated that the sample preparation meets the standard.
After a global lysine crotonylome analysis using LC-MS/MS, 4913, 4839, and 4922 Kcr sites in 1242, 1230, and 1253 crotonylated proteins, respectively, were identified within three independent biological repeats. Moreover, 3967 Kcr sites and 1041 crotonylated proteins were overlapped in the individual triplicate experiments (Supplementary Figure S2 and Supplementary Table S1). MS/MS spectra of three crotonylated peptides were shown in Supplementary Figure S3. The number of crotonylated sites in the identified proteins was then calculated. As shown in Supplementary Figure S4, 371 (35%) proteins contained only one crotonylation site, whereas 670 (65%) proteins had multiple crotonylation sites.
Pattern Analysis of Crotonylated Sites
To evaluate the properties of Kcr sites in B. cinerea, we examined the sequence motif flanking the identified peptides. A total of nine conserved amino acid sequences from −10 to +10 around the crotonylated lysine were extracted from 3196 peptides (Figure 1A). Particularly, motifs YKcrE, FKcr, and KcrE were strikingly conserved. Among them, FKcr and KcrE have been identified as crotonylation motifs previously, while YKcrE was firstly found in our study, which may represent a characteristic feature of crotonylation in B. cinerea. To further analyze these motifs, heatmaps of the amino acid sequences surrounding the crotonylation sites were generated. The results showed that certain amino acid residues surrounding the Kcr were markedly enriched. K residues were observed to be enriched in the −10 to −5 and +5 to +9 positions, while residues A, D, E, F, Y were significantly enriched in −4 to −1, −3 to +3, −2 to +3, −3 to +1, and −3 to −1 positions, respectively (Figure 1B).
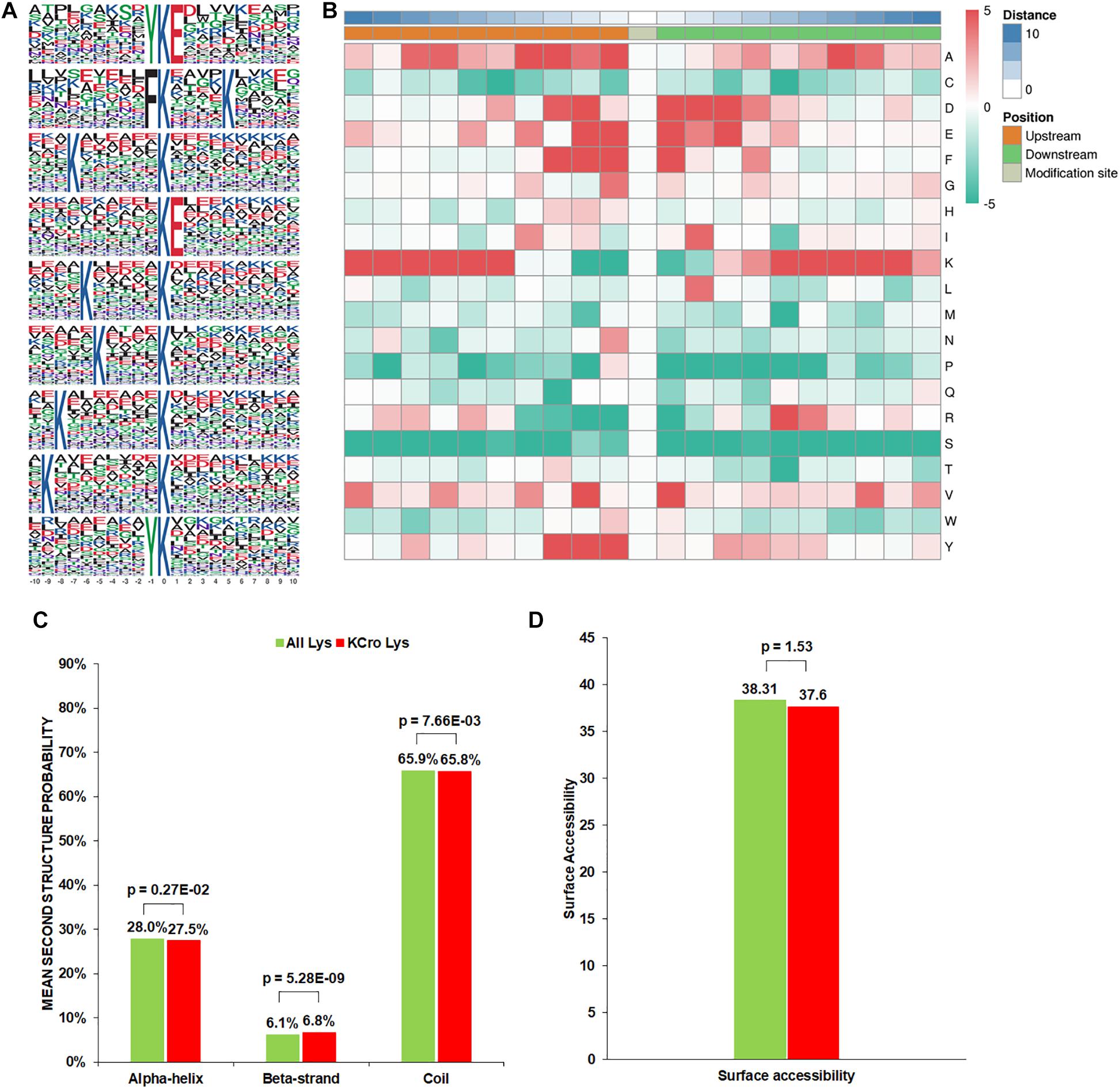
Figure 1. Properties of the Kcr peptides in B. cinerea. (A) Crotonylation sequence motifs for ±10 amino acids surrounding the Kcr sites. (B) Heatmap of the amino acid compositions of the Kcr sites demonstrating the frequency of certain amino acids around the modified lysine. Red indicates high frequency and green means low frequency. (C) Probabilities of Kcr in the structures of beta-strand, alpha-helix, and coil. (D) Predicted surface accessibility of Kcr sites.
In order to clarify the relationship between crotonylation and the presence of protein structures in B. cinerea, a structure analysis of the crotonylated proteins was performed. As shown in Figure 1C, 34.3% of the crotonylated sites were located in regions with ordered secondary structures. Among them, 27.5% were located in alpha-helix, and 6.8% were in a beta-strands. The residual 65.8% of the crotonylated sites were distributed in disordered protein regions. However, considering the distribution patterns of crotonylated lysines and all lysines are similar, there was no tendency of Kcr in B. cinerea. The surface accessibility of Kcr sites was further evaluated. The results showed that the exposure of crotonylation sites on the protein surface is close to that of all lysine residues (Figure 1D). Therefore, Kcr may not be affected by the surface properties of proteins in B. cinerea.
Conservation Analysis of Lysine Crotonylated Proteins
Increasing studies in recent years have revealed improved number of crotonylated proteins in various species or cell lines (Sun et al., 2017; Wei et al., 2017b; Wu et al., 2017; Xu et al., 2017; Huang et al., 2018; Kwon et al., 2018; Liu K. et al., 2018; Liu S. et al., 2018). However, the conservation of Kcr in these organisms is unknown. As such, we compared the crotonylated proteins in B. cinerea with those in eight organisms or cell lines that have determined crotonylomes. Totally, 3019 orthologs of the crotonylproteins in B. cinerea were identified in these organisms (Supplementary Table S2). As shown in Figure 2A, 790 crotonylated proteins have orthologs in Oryza sativa (359 proteins), Nicotiana tabacum (301 proteins), Human peripheral blood (225 proteins), Human Hela cells (182 proteins), Human H1299 cells (522 proteins), Human A549 cells (682 proteins), Danio rerio (129 proteins), and Carica papaya (619 proteins), which account for 75.9% (790/1041 proteins) of the total crotonylproteins in B. cinerea. We further classified the crotonylated proteins of B. cinerea depending on the number of the orthologous proteins in these organisms. The data demonstrated that the percentage of completely conserved proteins (have 8 orthologs), well-conserved proteins (have 6 to 7 orthologs), conserved proteins (have 3 to 5 orthologs) and poorly conserved proteins (have 1 to 2 orthologs) were 3.9% (41/1041 proteins), 12.1% (126/1041 proteins), 36.9% (384/1041 proteins), and 22.9% (239/1041 proteins) (Figure 2B), respectively. Furthermore, 24.1% (251/1041 proteins) of the crotonylated proteins in B. cinerea was characterized as novel proteins since no orthologs were found in these organisms (Figure 2B). These results suggest that Kcr plays both common and specific roles in different species.
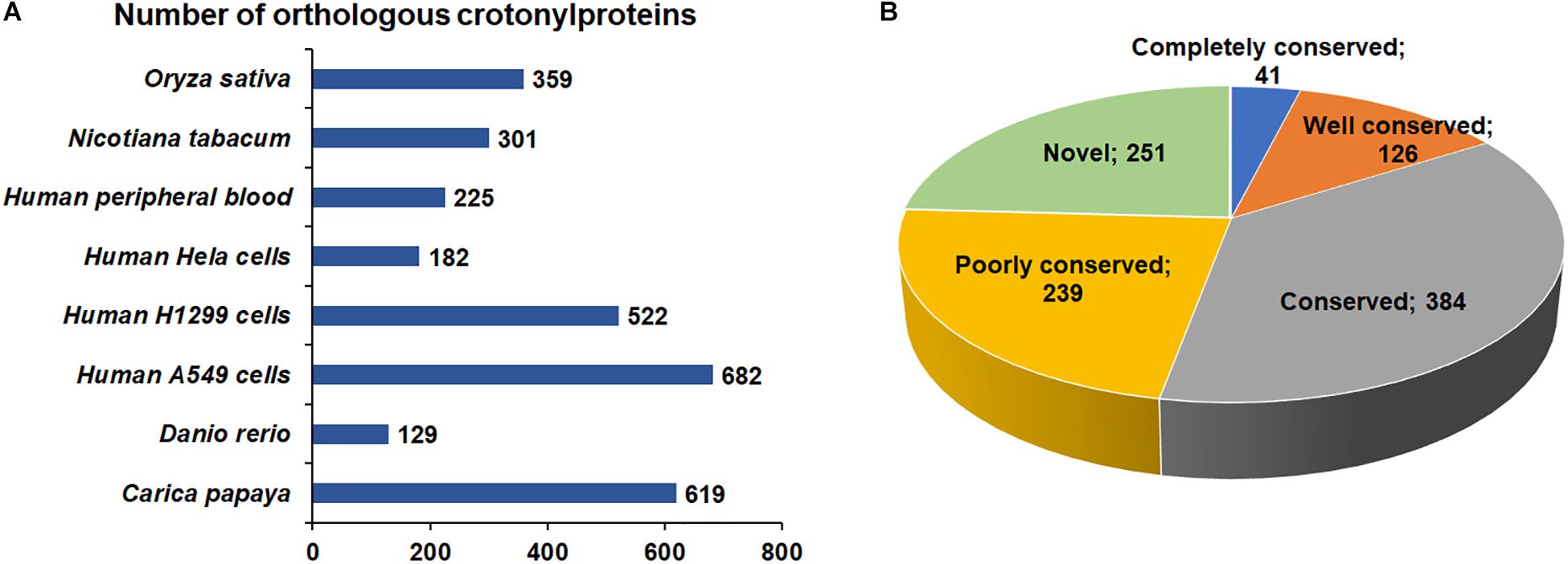
Figure 2. Conservation analysis of lysine crotonylated proteins. (A) Number of orthologous crotonylproteins in eight organisms or cell lines with reported crotonylomes. (B) A pie chart of conservation of crotonylproteins in Oryza sativa, Nicotiana tabacum, Human peripheral blood, Human Hela cells, Human H1299 cells, Human A549 cells, Danio rerio, and Carica papaya. Classification was performed as follows: Completely conserved group, 8 orthologs; Well conserved group, 6 to 7 orthologs; Conserved group, 3 to 5 orthologs; Poorly conserved group, 1 to 2 orthologs; Novel group, 0 orthologs.
Functional Annotation and Cellular Localization of Crotonylated Proteins in B. cinerea
To obtain a comprehensive overview of potential roles of crotonylated proteins in B. cinerea, we conducted the Gene Ontology (GO) functional classification analysis of all crotonylated proteins based on their biological process, cellular component, and molecular function (Figure 3 and Supplementary Table S3). The result of biological process analysis showed that crotonylated proteins were widely distributed within different kinds of metabolic and cellular processes (Figure 3A). For cellular component analysis, crotonylated proteins were distributed among various cellular compartments, mostly in intracellular (21%), intracellular organelle (19%), and membrane-bounded organelle (16%) (Figure 3B). Within the molecular function category, the majority of the modified proteins were composed of those related to heterocyclic compound binding (15%), organic cyclic compound binding (15%), protein binding (8%), ion binding (8%), and hydrolase activity (8%) (Figure 3C).
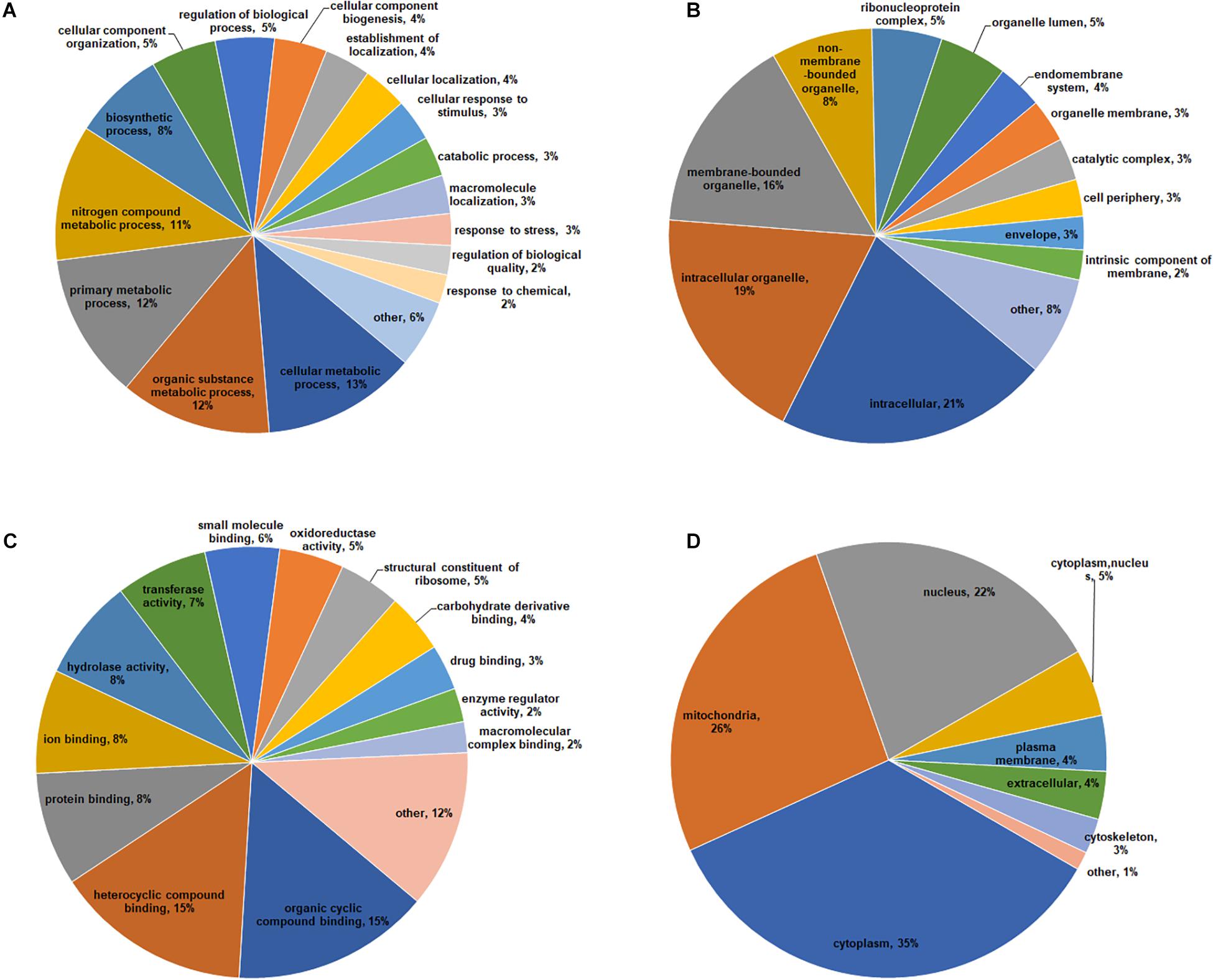
Figure 3. Functional classification of lysine crotonylated proteins based on GO and subcellular location in B. cinerea. GO classification of crotonylated proteins based on biological process (A), cellular component (B), and molecular function (C). (D) Subcellular localization classification of the crotonylated proteins.
Subcellular localization analysis of the crotonylated proteins was also performed. As shown in Figure 3D, a large number of the crotonylated proteins in B. cinerea were distributed in the cytoplasm (35%), mitochondria (26%), and nucleus (22%). These data demonstrated that the crotonylated proteins, with diversified cellular distribution, are involved in various biological processes in B. cinerea.
Functional Enrichment Analysis of Crotonylated Proteins
To better elucidate the characteristics of crotonylaed proteins in B. cinerea, enrichment analyses of GO (Gene Ontology), protein domain and KEGG pathway were conducted. The enrichment analysis of biological process revealed that the majority of crotonylaed proteins were related to cytoplasmic translation (Figure 4A and Supplementary Table S4). In support of these observations, large numbers of the modified proteins were involved in structural constituent of ribosome in molecular function enrichment analysis (Figure 4B and Supplementary Table S4). Based on cellular component enrichment analysis, proteins located to cytosolic part were more likely to be crotonylated (Figure 4C and Supplementary Table S4).
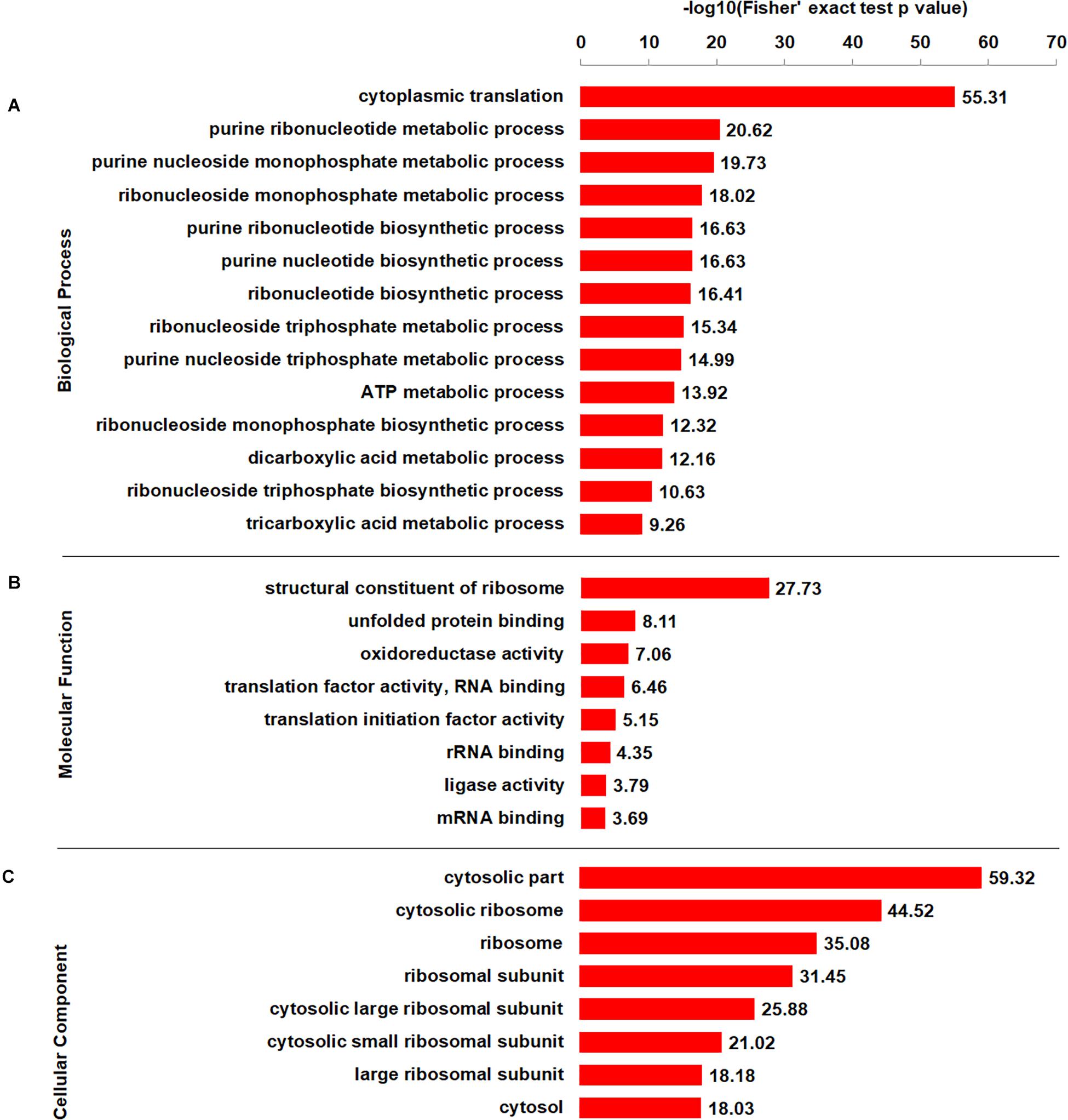
Figure 4. GO-based enrichment analysis of crotonylated proteins according to biological process (A), molecular function (B), and cellular component (C).
Further enrichment analyses of protein domain and KEGG pathway obtained similar results. The protein domain enrichment analysis demonstrated that the proteins with domains of proteasome subunit were more likely to have crotonylation (Supplementary Figure S5A and Supplementary Table S5). Consistent with these findings, enrichment analysis of KEGG pathway indicated that a large proportion of crotonylated proteins were associated with ribosome (Supplementary Figure S5B and Supplementary Table S6). Overall, our data indicate that lysine crotonylated proteins are widely distributed and involved in various pathways, suggesting important roles of crotonylation in cell metabolism.
Analysis of Crotonylated Proteins Related to Fungal Pathogenicity
To further understand the pathogenesis regulated by crotonylation in B. cinerea, we summarized the crotonylated proteins reported to be involved in fungal pathogenicity and listed in Table 1 and Supplementary Table S7. A total of 26 proteins were identified, with diversified numbers of Kcr sites varied from one to eight. These proteins participate in diverse pathogenesis pathways including signal transduction (6 proteins), redox homeostasis (6 proteins), plant cell wall degrading (4 proteins), secretory of virulence factors (3 proteins), and secondary metabolites biosynthesis (7 proteins), indicating that Kcr has a broad range of effects on pathogenicity of B. cinerea. Importantly, most of the crotonylated sites were located or near to the functional domains of these proteins. For example, cytochrome P450 reductase (Bccpr1) was found to be crotonylated at K290, which is next to R292, one of the conserved key sites of flavin adenine dinucleotide (FAD) domain of Cytochrome P450 reductase (Ebrecht et al., 2019; Figure 5A). Glutathione reductase (Bcglr1) carried the crotonylated K379 which is spatially close to one of the key sites of this enzyme, R371 (Yu and Zhou, 2007; Figure 5B). These data suggests that Kcr may have effect on the adjacent key domain sites, and thus regulate the function of these proteins involved in pathogenicity.

Table 1. Crotonylated proteins associated with pathogenicity in B. cinerea.
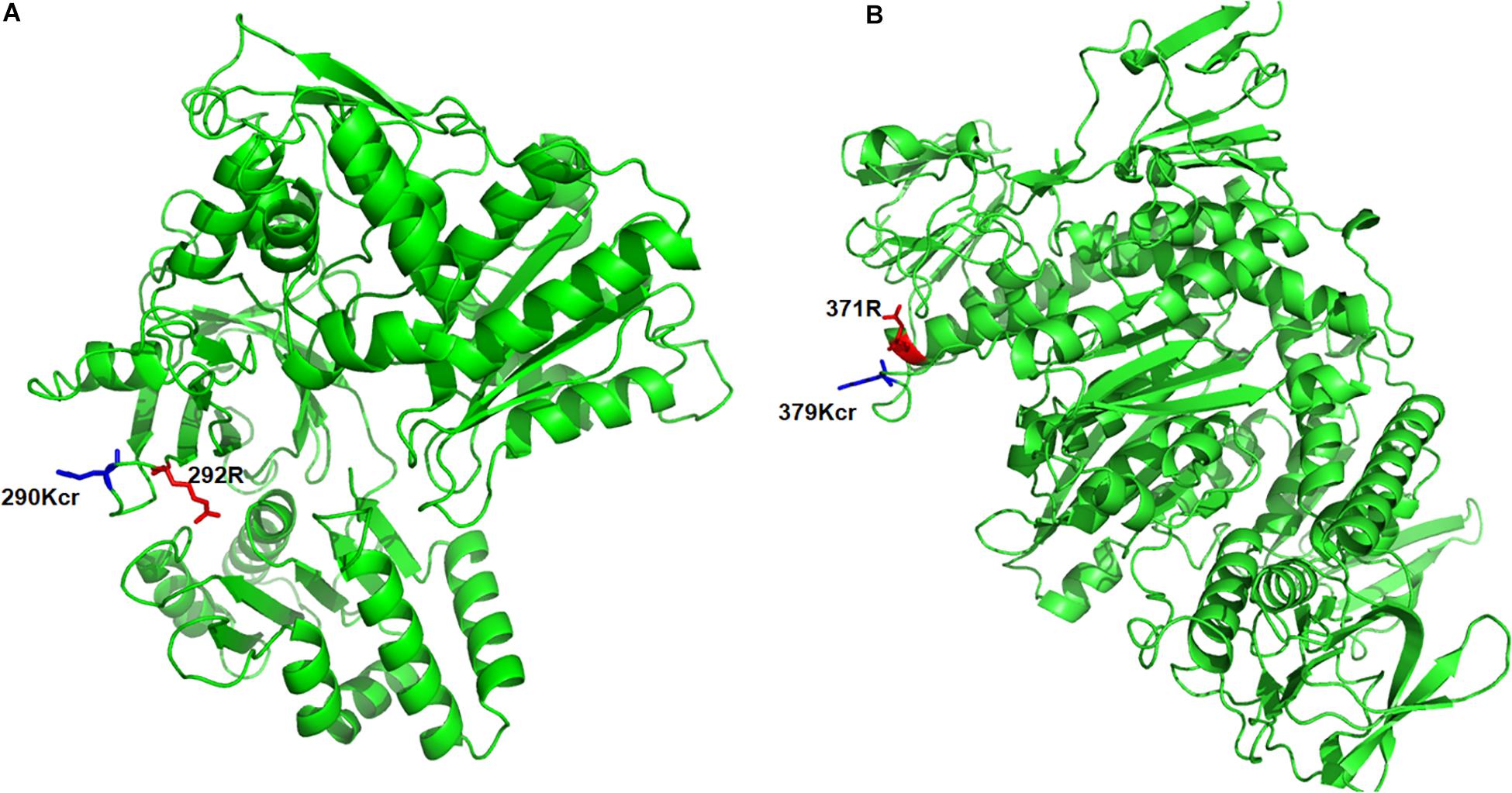
Figure 5. Three-dimensional structure of Bccpr1 (A) and Bcglr1 (B) with identified crotonylation site. The structure was derived from PDB database. The crotonylated lysine residues and adjacent arginine residues were indicated by blue and red colored sticks, respectively.
PPI Network of Crotonylated Proteins in B. cinerea
To better investigate the cellular processes regulated by crotonylation in B. cinerea, a PPI network of all identified crotonylated proteins was assembled using Cytoscape software. As shown in Figure 6 and Supplementary Table S8, 783 crotonylated proteins were mapped to the PPI network, exhibiting an overview of diverse pathways modulated by these proteins in B. cinerea. Five greatly interconnected clusters of crotonylated proteins were retrieved, including ribosome, carbon metabolism, proteasome, oxidative phosphorylation, and aminoacyl-tRNA biosynthesis (Figure 6 and Supplementary Table S8). These findings indicate that physiological interactions among these complexes may conduce to their harmonization and cooperation in B. cinerea.
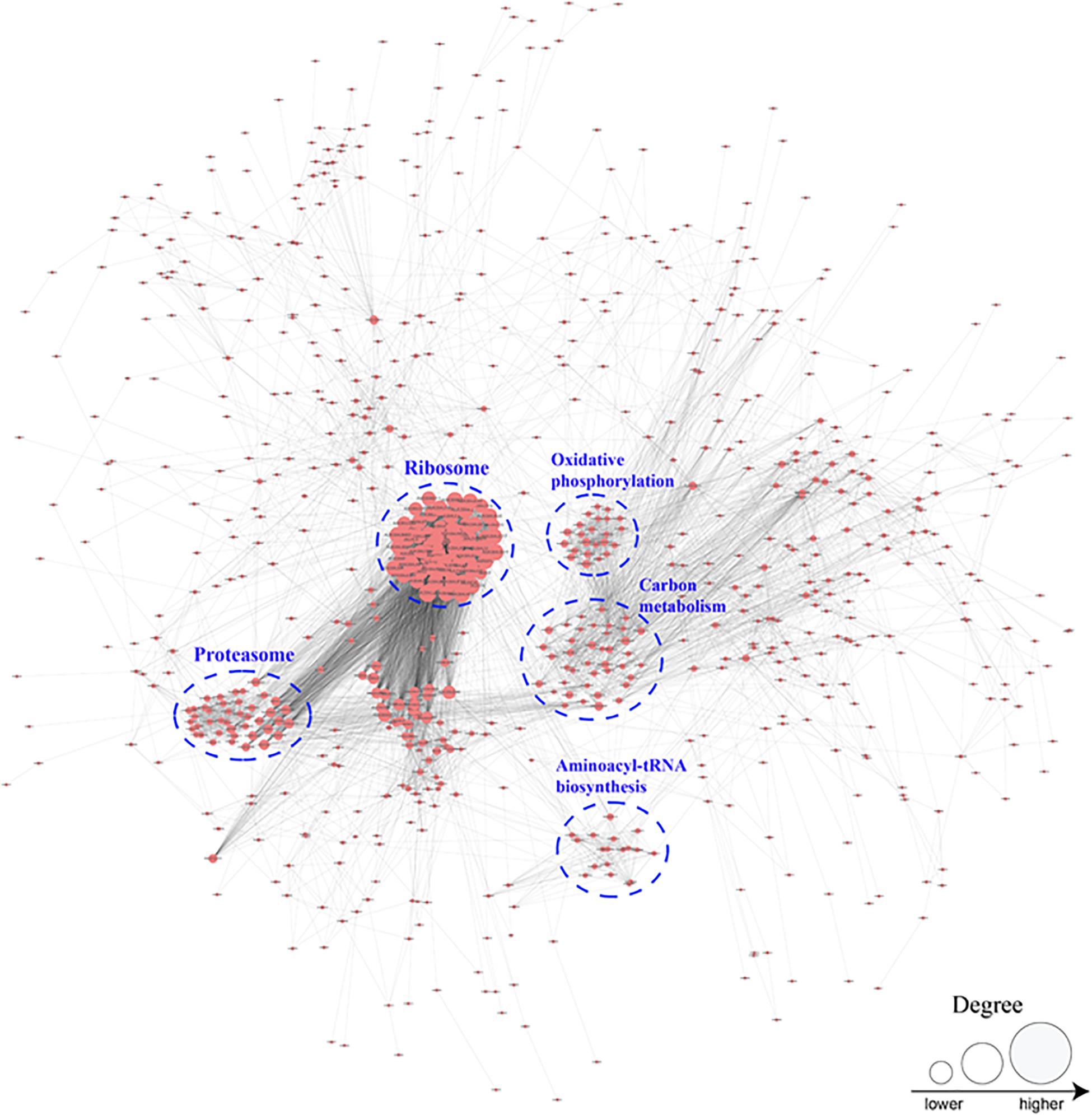
Figure 6. Protein interaction networks of the crotonylation proteins in B. cinerea.
Kcr Levels of B. cinerea Under Different Metabolic Conditions
To test whether the crotonylome differs under different physiological conditions, we carried out immunoblotting of the crotonylated proteins of B. cinerea cultivated in YEPD or MM (minimal medium), representing rich or limited nutrient condition. As shown in Figure 7A, the whole protein Kcr levels were increased in YEPD than in MM. Moreover, the Kcr level of Bcpck1, a critical enzyme in the nutrient-starvation adaptation before host invasion (Liu J.K. et al., 2018), was also elevated in YEPD (Figure 7B). Thus the above results suggested that Kcr may participate in nutrient metabolic regulation and it will be interesting to characterize the function of crotonylated substrates under different metabolic conditions.
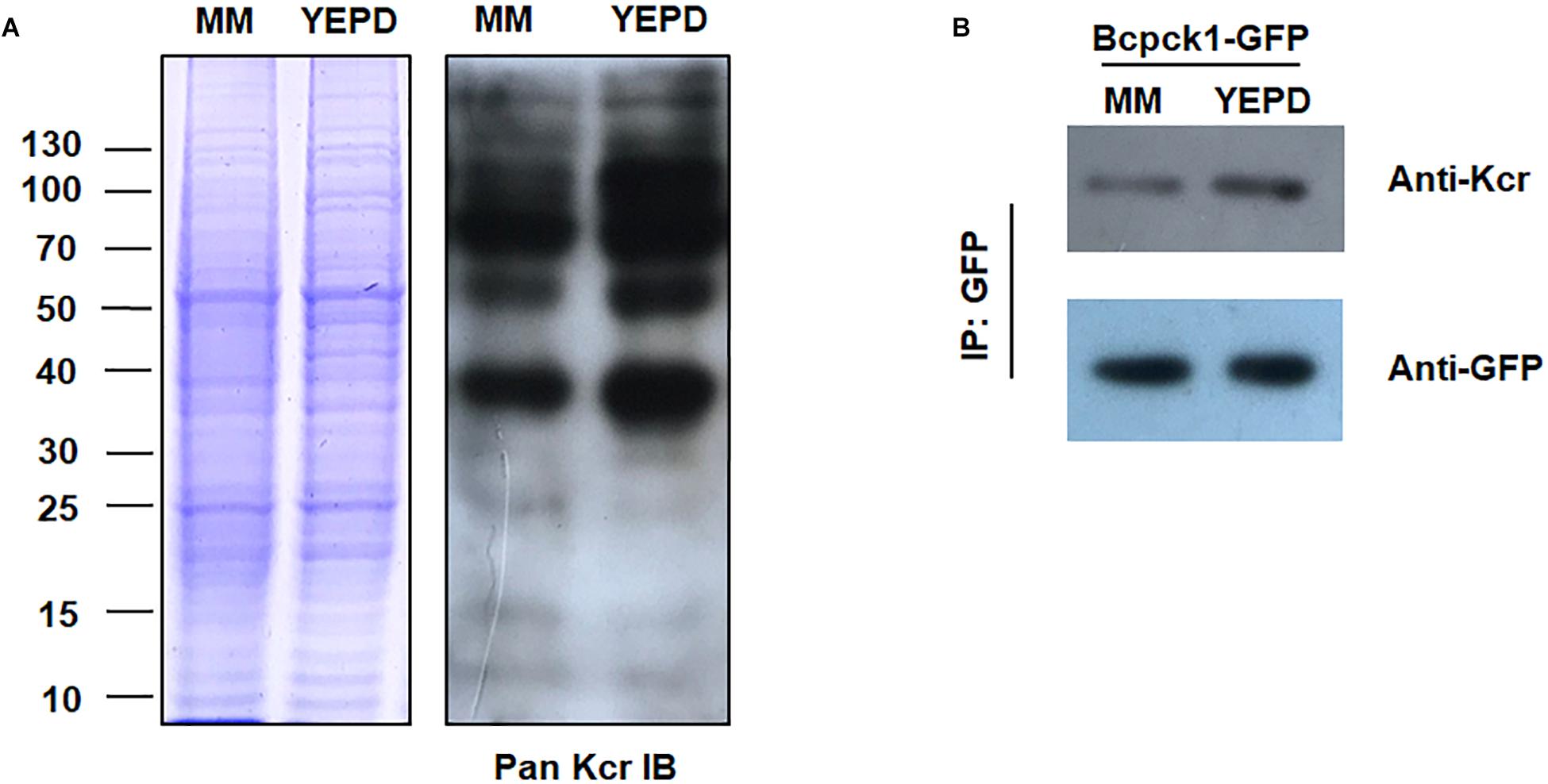
Figure 7. Kcr levels of B. cinerea under different physiological conditions. (A) Immunoblot analysis of crotonylated proteins with pan anti-Kcr antibody of B. cinerea cultivated in liquid YEPD or MM. The loading control by Coomassie blue staining was used to ensure that equal amounts of protein were loaded in each lane. (B) Protein extracts containing Bcpck1-GFP were immunoprecipitated (IP) using anti-GFP beads. The isolated proteins were resolved by SDS-PAGE. Anti-GFP and anti-Kcr antibodies were used to detect Bcpck1-GFP and its crotonylated isoform, respectively.
Discussion
Histone crotonylation is a newly discovered PTM which plays critically important roles for gene transcription in mammalian cells (Tan et al., 2011; Wei et al., 2017a). Other than histones, recent findings on global profiling of crotonylated proteins in various species have expanded the repertoire of modified proteins (Sun et al., 2017, 2019; Wei et al., 2017b; Wu et al., 2017; Xu et al., 2017; Liu J.K. et al., 2018; Liu K. et al., 2018; Liu S. et al., 2018; Yang et al., 2018), which exhibited widely distributed cellular localization and diversified biological function. However, study of Kcr is surprisingly lacking and has not yet been studied in plant fungi. In this study, we performed a global Kcr analysis in B. cinerea to bridge this knowledge gap in fungal pathogens.
A total of 1041 crotonylated proteins with 3967 Kcr sites were identified, which exceeds the Kcr levels of most reported organisms or cell lines including Danio rerio, Oryza sativa, Nicotiana tabacum, Human Hela cells, Human peripheral blood, Human H1299 cells, and less than Human A549 cells and Carica papaya. These data indicate that Kcr is a widespread PTM among organisms. Further functional annotation and enrichment analysis demonstrated that the crotonylated proteins were widely distributed and involved in diverse cellular processes. The identification of specific lysine motifs indicates substrate preference of Kcr in B. cinerea. Furthermore, PPI network analysis showed that a mass of protein interactions are regulated by crotonylation. These data represents the first proteome-wide view of the crotonylome in B. cinerea and fungal pathogens.
Recently, proteomic studies into crotonylation have indicated that abundant proteins are crotonylated in various organisms, suggesting that crotonylation is a common mechanism of metabolic regulation. However, it remains unclear whether such biochemical changes play crucial roles in physiological processes. In this study, we found that the crotonylome of B. cinerea differs under different nutrient conditions (Figure 7). Moreover, the crotonylation of Bcpck1, a PEP-carboxykinase which is important in gluconeogenesis, changed significantly under different metabolic conditions. Future studies are needed to uncover the effects of crotonylation on regulating protein functions and to interpret the underlying mechanisms behind protein crotonylation’s ability to modulate diverse physiological processes.
In our study, 85 ribosomal proteins were crotonylated, accounting for 8% of the total crotonylated proteins, suggesting that Kcr of ribosomal proteins may have effect on protein translational control and ribosome assembly. Afterward, 26 crotonylated proteins were reported to be associated with pathogenicity in B. cinerea, participating in various pathogenesis processes (Table 1 and Supplementary Table S7). The infection process of B. cinerea is regulated by a complicated signal transduction network, such as Mitogen-activated protein kinases (MAPKs), the cAMP-dependent signal cascade, and small GTPases. In our study, several components involved in these signaling pathways were identified to be crotonylated including MAPKs bmp1 (K298), bmp3 (K72), cAMP-dependent protein kinase Bcpka1 (K205), G-protein alpha subunit Bcg1 (K51,141), and Ras-related small GTPase Bcras1 (K153). Mutants in genes encoding MAPK bmp1 and bmp3 showed defects in germination, vegetative growth, and pathogenicity (Zheng et al., 2000; Rui and Hahn, 2007). Bcpka1 and Bcg1 play important roles in growth and virulence in B. cinerea, regulating invasive process of plant tissue via a cAMP-independent pathway (Schumacher et al., 2008a,b). Bcras1, which influences both MAPK and cAMP-dependent signal transduction, is necessary for development and virulence in B. cinerea (Minz Dub et al., 2013). These data indicates that Kcr might play a role in signal transduction and thus modulate multiple steps of fungal growth and pathogenicity.
The rapid production of reactive oxygen species (ROS) and maintaining a balanced redox homeostasis are crucial for colonization process of necrotrophic pathogen B. cinerea. Cu-Zn superoxide dismutase Bcsod1, an important virulence factor through stimulating ROS generation in B. cinerea (López-Cruz et al., 2017), was found to be crotonylated at K89, K106, and K129 sites. Thioredoxin and glutathione are two major cellular redox systems to keep redox homoeostasis during stage of infection for B. cinerea. Thioredoxins Bctrx1 (K74, K114), and Bctrx2 (K57), thioredoxin reductase Bctrr1 (K331), and glutathione reductase Bcglr1 (K72, K275, K379) were found to be crotonylated in this study. Knock out mutants of these enzymes showed retarded growth and impaired virulence (Viefhues et al., 2014). In addition, protein disulfide-isomerase Bcpdi1, a new interaction partner of NADPH oxidase complexes which is essential for the conservation of the redox homeostasis in B. cinerea (Marschall and Tudzynski, 2017), was identified to be crotonylated at multiple sites in our study.
D-galacturonic acid, one of the major components of plant cell wall, is an important carbon source for filamentous fungi. The mutants in each step of the catabolic pathway of converting D-galacturonic acid of plant to pyruvate and l-glyceraldehyde showed reduced virulence (Zhang et al., 2011). In our study, a group of D-galacturonate catabolic enzymes were identified to be crotonylated including galacturonate reductase Bcgar1 (K276, K286), galactonate dehydratase Bclgd1 (K173, K186, K410), and 2-keto-3-deoxy-l-galactonate aldolase Bclga1 (K247, K307). In addition, endopolygalacturonase Bcpg1, which function as a cell-wall-degrading enzyme and is required for full virulence of B. cinerea (ten Have et al., 1998), was found to be crotonylated at multiple sites in this study.
To colonize the host tissue, B. cinerea usually secretes a battery of virulence factors to the extracellular environment. The Rab GTPase Bcsas1, reported to play a critical role in the secretion of polygalacturonases, polysaccharide hydrolases, xylanases and proteases (Zhang et al., 2014), was identified to be crotonylated at K172. Deletion of gene encoding vesicle coat complex protein Bcsec31 resulted in a reduction in the protein secretion and virulence of B. cinerea (Zhang et al., 2016). This protein was found to be crotonylated at K444 and K571 sites in our study. In addition, actin protein BcactA was found crotonylated at multiple sites, which was recently discovered to influence the secretome of B. cinerea and is crucial for extracellular virulence factors regulation (Li et al., 2020).
The secondary metabolites play critical roles in fungal survival and host killing. In our study, several key enzymes responsible for the committed biosynthetic steps were found to be crotonylated. DHN melanin has different effects during the life cycles of the fungus, particularly, contributing to the invading process of the penetration structures and the longevity of the reproduction structures (Zhang et al., 2015; Schumacher, 2016). In this study, three key enzymes of melanogenesis pathway, tetrahydroxynaphthalene reductases Bcbrn1, Bcbrn2, and scytalone dehydratase Bcscd1, were found to be crotonylated at multiple sites. Besides, the phosphoenolpyruvate carboxykinase Bcpck1, which was identified to be crotonylated at multiple sites in our study, plays an important role in gluconeogenesis and is critical in the nutrient-starvation adaptation before host invasion (Liu J.K. et al., 2018). Furthermore, Bccpr1, which function as a cytochrome P450 oxidoreductase regulating the fungal ABA production (Siewers et al., 2004), was identified to be a crotonylated protein with multiple modified sites. Further studies are needed to explore the functions of crotonylation of these substrate proteins to deeply understand mechanisms of B. cinerea pathogenesis at protein level.
Conclusion
We have conducted the first crotonyl-proteome of B. cinerea, an important plant fungal pathogen, showing abundant Kcr among a large number of proteins. The lysine crotonylome in this study provides a good resource for in-depth exploration of the functions of Kcr in the growth, development and pathogenicity of B. cinerea and other filamentous fungi. Investigating the functions of the crotonylation of the target proteins may help to design drugs with high specificity and low toxicity to prevent and control gray mold disease.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD019358.
Author Contributions
NZ, WL, and ML generated the hypothesis and planned the experiments. NZ and ZY performed the experiments. NZ and WL wrote the manuscript. All other authors provided comments on the manuscript.
Funding
This research was supported by the Ministry of Agriculture of China (2016ZX08009003-001), the National Natural Science Foundation of China (31722044 and 31972213), the Shandong Provincial Natural Science Foundation (ZR2019BC070), Shandong Postdoctoral Innovation Program (201903081), Taishan Scholar Construction Foundation of Shandong Province (tshw20130963) and “First class grassland science discipline” program in Shandong Province, China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.564350/full#supplementary-material
Supplementary Figure 1 | (A) Systematic analysis of Kcr in B. cinerea. (B) Mass error distribution of the Kcr peptides. (C) Distribution of Kcr peptides based on their length.
Supplementary Figure 2 | Overlaps of three individual MS/MS spectra of the crotonylated peptides.
Supplementary Figure 3 | MS/MS spectra of three crotonylated peptides: A0A384J6Z0 (Pyruvate kinase), A0A384K3A6 (Bcrpl5), and A0A384K3C6 (Ribosomal protein L15).
Supplementary Figure 4 | Number of crotonylation sites per protein in B. cinerea.
Supplementary Figure 5 | Enrichment analyses of protein domain (A) and KEGG pathway (B).
Supplementary Table 1 | The identified Kcr sites in B. cinerea.
Supplementary Table 2 | Conservation analysis of Kcr proteins.
Supplementary Table 3 | Protein GO terms distribution.
Supplementary Table 4 | GO enrichment based on molecular function, cellular component, and biological process.
Supplementary Table 5 | Enrichment analysis of KEGG protein pathway.
Supplementary Table 6 | Enrichment analysis of protein domain.
Supplementary Table 7 | Crotonylated proteins associated with pathogenicity in B. cinerea.
Supplementary Table 8 | Protein interaction network of the crotonylated proteins.
References
Brandhoff, B., Simon, A., Dornieden, A., and Schumacher, J. (2017). Regulation of conidiation in Botrytis cinerea involves the light-responsive transcriptional regulators BcLTF3 and BcREG1. Curr. Genet. 63, 931–949. doi: 10.1007/s00294-017-0692-9
Chen, Y., Sprung, R., Tang, Y., Ball, H., Sangras, B., Kim, S. C., et al. (2007). Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell Proteomics 6, 812–819. doi: 10.1074/mcp.M700021-MCP200
Chou, M. F., and Schwartz, D. (2011). Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinformatics Chapter 13:Unit1315–24. doi: 10.1002/0471250953.bi1315s35
Cox, J., and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. doi: 10.1038/nbt.1511
Cox, J., Matic, I., Hilger, M., Nagaraj, N., Selbach, M., Olsen, J. V., et al. (2009). A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4, 698–705. doi: 10.1038/nprot.2009.36
Dai, L., Peng, C., Montellier, E., Lu, Z., Chen, Y., Ishii, H., et al. (2014). Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 10, 365–370. doi: 10.1038/nchembio.1497
Ebrecht, A. C., Van Der Bergh, N., Harrison, S. T. L., Smit, M. S., Sewell, B. T., and Opperman, D. J. (2019). Biochemical and structural insights into the cytochrome P450 reductase from Candida tropicalis. Sci. Rep. 9:20088. doi: 10.1038/s41598-019-56516-6
Gronover, C. S., Kasulke, D., Tudzynski, P., and Tudzynski, B. (2001). The role of G protein alpha subunits in the infection process of the gray mold fungus Botrytis cinerea. Mol. Plant Microbe Interact. 14, 1293–1302. doi: 10.1094/mpmi.2001.14.11.1293
Hirschey, M. D., and Zhao, Y. (2015). Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol. Cell Proteomics 14, 2308–2315. doi: 10.1074/mcp.R114.046664
Horton, P., Park, K. J., Obayashi, T., Fujita, N., Harada, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Huang, H., Wang, D. L., and Zhao, Y. (2018). Quantitative crotonylome analysis expands the roles of p300 in the regulation of lysine crotonylation pathway. Proteomics 18:e1700230. doi: 10.1002/pmic.201700230
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280. doi: 10.1093/nar/gkh063
Khoury, G. A., Baliban, R. C., and Floudas, C. A. (2011). Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep. 1:90. doi: 10.1038/srep00090
Kwon, O. K., Kim, S. J., and Lee, S. (2018). First profiling of Kcr of myofilament proteins and ribosomal proteins in zebrafish embryos. Sci. Rep. 8:3652. doi: 10.1038/s41598-018-22069-3
Leroch, M., Mernke, D., Koppenhoefer, D., Schneider, P., Mosbach, A., and Doehlemann, G. (2011). Living colors in the gray mold pathogen Botrytis cinerea: codon-optimized genes encoding green fluorescent protein and mCherry, which exhibit bright fluorescence. Appl. Environ. Microbiol. 77, 2887–2897. doi: 10.1128/aem.02644-10
Li, D., Lv, B., Tan, L., Yang, Q., and Liang, W. (2016). Acetylome analysis reveals the involvement of lysine acetylation in diverse biological processes in Phytophthora sojae. Sci. Rep. 6:29897. doi: 10.1038/srep29897
Li, H., Zhang, Z., Qin, G., He, C., Li, B., and Tian, S. (2020). actin is required for cellular development and virulence of Botrytis cinerea via the mediation of secretory proteins. mSystems 5:e00732-19. doi: 10.1128/mSystems.00732-19
Liu, J. K., Chang, H. W., Liu, Y., Qin, Y. H., Ding, Y. H., Wang, L., et al. (2018). The key gluconeogenic gene PCK1 is crucial for virulence of Botrytis cinerea via initiating its conidial germination and host penetration. Environ. Microbiol. 20, 1794–1814. doi: 10.1111/1462-2920.14112
Liu, K., Yuan, C., Li, H., Chen, K., Lu, L., Shen, C., et al. (2018). A qualitative proteome-wide Kcr profiling of papaya (Carica papaya L.). Sci. Rep. 8:8230. doi: 10.1038/s41598-018-26676-y
Liu, S., Xue, C., Fang, Y., Chen, G., Peng, X., Zhou, Y., et al. (2018). Global involvement of lysine crotonylation in protein modification and transcription regulation in rice. Mol. Cell Proteomics 17, 1922–1936. doi: 10.1074/mcp.RA118.000640
Liu, L., Wang, G., Song, L., Lv, B., and Liang, W. (2016). Acetylome analysis reveals the involvement of lysine acetylation in biosynthesis of antibiotics in Bacillus amyloliquefaciens. Sci. Rep. 6:20108. doi: 10.1038/srep20108
López-Cruz, J., Óscar, C. S., Emma, F. C., Pilar, G. A., and Carmen, G. B. (2017). Absence of Cu-Zn superoxide dismutase BCSOD1 reduces Botrytis cinerea virulence in Arabidopsis and tomato plants, revealing interplay among reactive oxygen species, callose and signalling pathways. Mol. Plant Pathol. 18, 16–31. doi: 10.1111/mpp.12370
Lv, B., Yang, Q., Li, D., Liang, W., and Song, L. (2016). Proteome-wide analysis of lysine acetylation in the plant pathogen Botrytis cinerea. Sci. Rep. 6:29313. doi: 10.1038/srep29313
Marschall, R., and Tudzynski, P. (2017). The protein disulfide isomerase of Botrytis cinerea: an ER protein involved in protein folding and redox homeostasis influences NADPH oxidase signaling processes. Front. Microbiol. 8:960. doi: 10.3389/fmicb.2017.00960
Minz Dub, A., Kokkelink, L., Tudzynski, B., Tudzynski, P., and Sharon, A. (2013). Involvement of Botrytis cinerea small GTPases BcRAS1 and BcRAC in differentiation, virulence, and the cell cycle. Eukaryot Cell 12, 1609–1618. doi: 10.1128/ec.00160-13
Porquier, A., Moraga, J., Morgant, G., Dalmais, B., Simon, A., Sghyer, H., et al. (2019). Botcinic acid biosynthesis in Botrytis cinerea relies on a subtelomeric gene cluster surrounded by relics of transposons and is regulated by the Zn(2)Cys(6) transcription factor BcBoa13. Curr. Genet. 65, 965–980. doi: 10.1007/s00294-019-00952-4
Rui, O., and Hahn, M. (2007). The Slt2-type MAP kinase Bmp3 of Botrytis cinerea is required for normal saprotrophic growth, conidiation, plant surface sensing and host tissue colonization. Mol. Plant Pathol. 8, 173–184. doi: 10.1111/j.1364-3703.2007.00383.x
Schumacher, J. (2016). DHN melanin biosynthesis in the plant pathogenic fungus Botrytis cinerea is based on two developmentally regulated key enzyme (PKS)-encoding genes. Mol. Microbiol. 99, 729–748. doi: 10.1111/mmi.13262
Schumacher, J., Kokkelink, L., Huesmann, C., Jimenez-Teja, D., Collado, I. G., Barakat, R., et al. (2008a). The cAMP-dependent signaling pathway and its role in conidial germination, growth, and virulence of the gray mold Botrytis cinerea. Mol. Plant Microbe Interact. 21, 1443–1459. doi: 10.1094/mpmi-21-11-1443
Schumacher, J., Viaud, M., Simon, A., and Tudzynski, B. (2008b). The Galpha subunit BCG1, the phospholipase C (BcPLC1) and the calcineurin phosphatase co-ordinately regulate gene expression in the grey mould fungus Botrytis cinerea. Mol. Microbiol. 67, 1027–1050. doi: 10.1111/j.1365-2958.2008.06105.x
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Siewers, V., Smedsgaard, J., and Tudzynski, P. (2004). The P450 monooxygenase BcABA1 is essential for abscisic acid biosynthesis in Botrytis cinerea. Appl. Environ. Microbiol. 70, 3868–3876. doi: 10.1128/aem.70.7.3868-3876.2004
Sun, H., Liu, X., Li, F., Li, W., Zhang, J., Xia, Z., et al. (2017). First comprehensive proteome analysis of Kcr in seedling leaves of Nicotiana tabacum. Sci. Rep. 7:3013. doi: 10.1038/s41598-017-03369-6
Sun, J., Qiu, C., Qian, W., Wang, Y., Sun, L., Li, Y., et al. (2019). Ammonium triggered the response mechanism of lysine crotonylome in tea plants. BMC Genomics 20:340. doi: 10.1186/s12864-019-5716-z
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. doi: 10.1093/nar/gku1003
Tan, M., Luo, H., Lee, S., Jin, F., Yang, J. S., Montellier, E., et al. (2011). Identification of 67 histone marks and histone Kcr as a new type of histone modification. Cell 146, 1016–1028. doi: 10.1016/j.cell.2011.08.008
ten Have, A., Mulder, W., Visser, J., and Van Kan, J. A. (1998). The endopolygalacturonase gene Bcpg1 is required for full virulence of Botrytis cinerea. Mol. Plant Microbe Interact. 11, 1009–1016. doi: 10.1094/mpmi.1998.11.10.1009
Viefhues, A., Heller, J., Temme, N., and Tudzynski, P. (2014). Redox systems in Botrytis cinerea: impact on development and virulence. Mol. Plant Microbe Interact. 27, 858–874. doi: 10.1094/mpmi-01-14-0012-r
Wang, M., Weiberg, A., Dellota, E. Jr., Yamane, D., and Jin, H. (2017). Botrytis small RNA Bc-siR37 suppresses plant defense genes by cross-kingdom RNAi. RNA Biol. 14, 421–428. doi: 10.1080/15476286.2017.1291112
Wang, Y., Zhou, J., Zhong, J., Luo, D., Li, Z., Yang, J., et al. (2018). Cys(2)His(2) zinc finger transcription factor BcabaR1 positively regulates abscisic acid production in Botrytis cinerea. Appl. Environ. Microbiol. 84:AEM.00920-18. doi: 10.1128/aem.00920-18
Wei, W., Liu, X., Chen, J., Gao, S., Lu, L., Zhang, H., et al. (2017a). Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 27, 898–915. doi: 10.1038/cr.2017.68
Wei, W., Mao, A., Tang, B., Zeng, Q., Gao, S., Liu, X., et al. (2017b). Large-scale identification of protein crotonylation reveals its role in multiple cellular functions. J. Proteome Res. 16, 1743–1752. doi: 10.1021/acs.jproteome.7b00012
Weiberg, A., Wang, M., Lin, F. M., Zhao, H., Zhang, Z., Kaloshian, I., et al. (2013). Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 342, 118–123. doi: 10.1126/science.1239705
Williamson, B., Tudzynski, B., Tudzynski, P., and Van Kan, J. A. (2007). Botrytis cinerea: the cause of grey mould disease. Mol. Plant Pathol. 8, 561–580. doi: 10.1111/j.1364-3703.2007.00417.x
Wu, Q., Li, W., Wang, C., Fan, P., Cao, L., Yang, J., et al. (2017). Ultradeep lysine crotonylome reveals the crotonylation enhancement on both histones and nonhistone proteins by SAHA treatment. J. Proteome Res. 16, 3664–3671. doi: 10.1021/acs.jproteome.7b00380
Xu, W., Wan, J., Zhan, J., Li, X., He, H., Shi, Z., et al. (2017). Global profiling of crotonylation on non-histone proteins. Cell Res. 27, 946–949. doi: 10.1038/cr.2017.60
Yang, Q., Li, Y., Apaliya, M. T., Zheng, X., Serwah, B. N. A., Zhang, X., et al. (2018). The response of rhodotorula mucilaginosa to patulin based on lysine crotonylation. Front. Microbiol. 9:2025. doi: 10.3389/fmicb.2018.02025
Yu, J., and Zhou, C. Z. (2007). Crystal structure of glutathione reductase Glr1 from the yeast Saccharomyces cerevisiae. Proteins 68, 972–979. doi: 10.1002/prot.21354
Zhang, C., He, Y., Zhu, P., Chen, L., Wang, Y., Ni, B., et al. (2015). Loss of bcbrn1 and bcpks13 in Botrytis cinerea not only blocks melanization but also increases vegetative growth and virulence. Mol. Plant Microbe Interact. 28, 1091–1101. doi: 10.1094/mpmi-04-15-0085-r
Zhang, D., Tang, Z., Huang, H., Zhou, G., Cui, C., Weng, Y., et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. doi: 10.1038/s41586-019-1678-1
Zhang, L., Thiewes, H., and Van Kan, J. A. (2011). The D-galacturonic acid catabolic pathway in Botrytis cinerea. Fungal Genet. Biol. 48, 990–997. doi: 10.1016/j.fgb.2011.06.002
Zhang, Z., Li, H., Qin, G., He, C., Li, B., and Tian, S. (2016). The MADS-Box transcription factor Bcmads1 is required for growth, sclerotia production and pathogenicity of Botrytis cinerea. Sci. Rep. 6:33901. doi: 10.1038/srep33901
Zhang, Z., Qin, G., Li, B., and Tian, S. (2014). Knocking out Bcsas1 in Botrytis cinerea impacts growth, development, and secretion of extracellular proteins, which decreases virulence. Mol. Plant Microbe Interact. 27, 590–600. doi: 10.1094/mpmi-10-13-0314-r
Zheng, L., Campbell, M., Murphy, J., Lam, S., and Xu, J. R. (2000). The BMP1 gene is essential for pathogenicity in the gray mold fungus Botrytis cinerea. Mol. Plant Microbe Interact. 13, 724–732. doi: 10.1094/mpmi.2000.13.7.724
Keywords: crotonylome, Botrytis cinerea, LC-MS/MS, fungal pathogenicity, lysine crotonylation
Citation: Zhang N, Yang Z, Liang W and Liu M (2020) Global Proteomic Analysis of Lysine Crotonylation in the Plant Pathogen Botrytis cinerea. Front. Microbiol. 11:564350. doi: 10.3389/fmicb.2020.564350
Received: 21 May 2020; Accepted: 08 October 2020;
Published: 23 October 2020.
Edited by:
Chengshu Wang, Chinese Academy of Sciences, ChinaReviewed by:
Wolfgang Buckel, University of Marburg, GermanyWende Liu, Chinese Academy of Agricultural Sciences, China
Copyright © 2020 Zhang, Yang, Liang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenxing Liang, d2xpYW5nMUBxYXUuZWR1LmNu; Mengjie Liu, bWpsbGl1QHFhdS5lZHUuY24=