 Brittni R. Kelley1†
Brittni R. Kelley1† J. Christopher Ellis2†
J. Christopher Ellis2† Annabel Large2
Annabel Large2 Liesel G. Schneider3
Liesel G. Schneider3 Daniel Jacobson2,4
Daniel Jacobson2,4 Jeremiah G. Johnson1*
Jeremiah G. Johnson1*- 1Department of Microbiology, The University of Tennessee, Knoxville, Knoxville, TN, United States
- 2Biosciences, Oak Ridge National Laboratory, Oak Ridge, TN, United States
- 3Department of Animal Science, The University of Tennessee, Knoxville, Knoxville, TN, United States
- 4Bredesen Center, The University of Tennessee, Knoxville, Knoxville, TN, United States
As a leading cause of bacterial-derived gastroenteritis worldwide, Campylobacter jejuni has a significant impact on human health in both the developed and developing worlds. Despite its prevalence as a human pathogen, the source of these infections remains poorly understood due to the mutation frequency of the organism and past limitations of whole genome analysis. Recent advances in both whole genome sequencing and computational methods have allowed for the high-resolution analysis of intraspecies diversity, leading multiple groups to postulate that these approaches may be used to identify the sources of Campylobacter jejuni infection. To address this hypothesis, our group conducted a regionally and temporally restricted sampling of agricultural and environmental Campylobacter sources and compared isolated C. jejuni genomes to those that caused human infections in the same region during the same time period. Through a network analysis comparing genomes from various sources, we found that human C. jejuni isolates clustered with those isolated from cattle and chickens, indicating these as potential sources of human infection in the region.
Introduction
As a leading cause of bacterial-derived gastroenteritis worldwide, Campylobacter species have a significant impact on human health (Kaakoush et al., 2015; Crofts et al., 2018) with approximately 96 million global cases (Kirk et al., 2015) and 1.3 million cases in the United States, annually (Fitzgerald et al., 2016). Symptoms of acute campylobacteriosis in the developed world typically include bloody and/or watery diarrhea, lethargy, and abdominal cramps (Connerton and Connerton, 2017). While the majority of cases are self-limiting and subside after several days, several post-infectious disorders have been associated with Campylobacter infections, including the development of Guillain-Barré Syndrome, post-infectious reactive arthritis, and irritable bowel syndrome (Backert et al., 2017; Halpin et al., 2018). In the developed world, consumption of undercooked or improperly prepared poultry has historically been implicated as the predominant source of human infection (Young et al., 2007; Kaakoush et al., 2015); however, direct contact with live cattle, pigs, sheep, and contaminated drinking water may also serve as sources of infection (Ashbolt, 2004; Mou et al., 2015; Kempf et al., 2017; Sacher et al., 2018). Because Campylobacter infections significantly impact human health and there are several potential sources of human infection, it is important to public health that these sources and the proportion of human infections attributed to each be thoroughly understood in both the developed and developing worlds.
Relative to foodborne pathogens like E. coli and Salmonella, successful source-tracking of Campylobacter has proven challenging. For example, pulse-field gel electrophoresis (PFGE) was highly impactful in identifying foodborne outbreaks and performing bacterial source-tracking of E. coli and Salmonella serotypes (Johnson et al., 1995; Fugett et al., 2007). Unfortunately, since this method relies on gel-based resolution of genomic restriction fragments, it can fail to discriminate between strains of bacterial species that experience even minor genomic variation, like Campylobacter, since the restriction fragment pattern can be altered (Fitzgerald et al., 2005). To circumvent these limitations, multi-locus sequence typing (MLST) was used by state and federal public health agencies to discriminate between strains and identify sources of infections for pathogens that were not amenable to PFGE. MLST differs from PFGE in that seven conserved housekeeping genes are amplified and sequenced, allowing for a SNP-based comparison of sequences to those previously deposited in online databases. This analysis allowed a queried strain to be designated as a sequence type (ST), which allowed for the assignment of particular STs to specific sources (Kittl et al., 2013; Kovanen et al., 2014). Similar to PFGE, this method works well to discriminate between strains of genomically stable pathogens, but has limited efficacy when distinguishing between strains of Campylobacter due to inherent hypervariability of the genome (Parkhill et al., 2000). Such variability has led to an overabundance of ST assignments within Campylobacter, making outbreak detection and source attribution challenging (Thépault et al., 2018b). Recently, U.S. public health agencies have begun shifting from PFGE- and MLST-based analyses to whole genome sequencing (WGS) approaches. These changes were instituted due to several perceived advantages, including the ability to analyze bacterial genomes with single nucleotide-level resolution, regular access of more laboratories to sequencing technology, rapidly decreasing costs of sequencing a genome, and increased speed of sequencing (Salipante et al., 2015).
Using these WGS technologies, groups have begun investigating the genetic relatedness of Campylobacter isolates from various sources (Dearlove et al., 2016; Kovanen et al., 2019), including whole genome MLST (wgMLST) analysis (Cody et al., 2013). For example, wgMLST has been used to examine C. jejuni and C. coli clonal complexes (Sheppard et al., 2012), the presence of Campylobacter antibiotic resistant genotypes (Zhao et al., 2016), and the comparison of isolates recovered in processing plants and on chicken meat (Ma et al., 2014; Guyard-Nicodème et al., 2015). Additionally, wgMLST studies have analyzed isolates from agricultural sources, including cattle and chickens, to examine for links to human infections based on genomic similarity and ST assignments (Sheppard et al., 2009; Oporto et al., 2011; Sheppard et al., 2012; Rosner et al., 2017). Due to the challenges of identifying Campylobacter outbreaks in real time, clinical cases were considered to occur sporadically. However, retrospective studies using WGS in conjunction with epidemiological data have been employed to investigate the genetic relatedness of clinical isolates and identify potential sources indicated by epidemiological data (Revez et al., 2014a, b; Clark et al., 2016; Moffatt et al., 2016; Joensen et al., 2018; Montgomery et al., 2018; Oakeson et al., 2018).
Despite the success of these studies and the increasing availability of sequencing technologies in health department laboratories, the ability to process and analyze the resulting sequences in a timely and accurate manner remains a limiting factor in public health investigations (Fricke and Rasko, 2014). Reliable protocols to perform source-tracking have not been verified for most pathogens, including Campylobacter (Dearlove et al., 2016), and the lack of continued systemic surveillance and reliance on sequence repositories can make the real-time detection of outbreaks difficult (Llarena et al., 2017). Taken together, these observations indicate that it is becoming increasingly urgent that streamlined surveillance and WGS-based approaches be developed and vetted for public health investigations, especially for regions with limited resources.
The objective of this work was to collect agricultural, environmental, and clinical C. jejuni isolates from East Tennessee during a defined time period and with collaborators at Oak Ridge National Laboratory (ORNL) conduct a novel network-based analysis to identify potential sources of human infections in the region. A total of 630 samples were collected between October 2016 and October 2018, resulting in 144 PCR-confirmed C. jejuni isolates. Subsequent whole-genome sequencing and assembly resulted in 80 high quality genomes collected during this study, while an additional 87 high quality genome assemblies were identified from the GenomeTrakr database. Together, these 167 genomes were incorporated into a robust reference-independent network analysis. Using this bioinformatic approach, we found that the human isolates clustered with those from cattle and chickens, which are known to be common sources of human Campylobacter infections. Working within the temporal and geographical constraints of this study, we were able to isolate C. jejuni from a variety of sources highlighting the importance of broad surveillance, while supporting the potential of whole-genome sequencing for source-tracking by utilizing a novel network analysis approach for comparison of isolates from different sources.
Materials and Methods
This study was an observational survey that utilized samples from confirmed human cases and convenience sampling from water, foods, and fresh excreted feces from domestic animals over the course of 2 years.
Regional Sampling of Water and Food
Samples from local fresh water sources (rivers, streams, and tributaries) in East Tennessee were aseptically collected in sterile 100 mL screw top glass bottles. A minimum of two samples were taken from each sampling site. Samples were stored on ice until filtration with a 0.2 μm vacuum filter. Filters were aseptically removed and placed in a 15 mL tube with 5 mL sterile 1x PBS and vortexed vigorously for 20 s before serial dilution and plating on Campylobacter-selective media consisting of Mueller-Hinton (MH) agar supplemented with 10% defibrinated sheep blood, cefoperazone (40 μg/ml), cycloheximide (100 μg/ml), trimethoprim (10 μg/ml), and vancomycin (100 μg/ml). Plates were incubated for 48 h under microaerobic conditions (85% N2, 10% CO2, 5% O2) at 37°C.
Meat (raw chicken, pork, and beef), fruit (unwashed berries, citrus fruit, apples, bananas, peaches, and plums), and vegetable (unwashed potatoes, kale leaves, and carrots) samples were obtained from local grocery chains and farmers markets in Knox County, TN and the surrounding region of eastern Tennessee. Food samples were cut into approximately 3 cm3 pieces using a disinfected cutting board and razor blade. The resulting cubes were placed in sterile 15 ml conical tubes with enough MH broth to cover the samples (approximately 3 mls) and allowed to shake overnight under microaerobic conditions at 37°C. Following incubation, samples were serially diluted and plated on Campylobacter-selective media as previously described. Only city and sample type were collected as metadata.
Regional Sampling of Animal Feces
Convenience fecal samples obtained from animals under observation for routine veterinary care from the University of Tennessee College of Veterinary Medicine were collected from the outside of examination gloves and weighed out in 200 mg aliquots and placed in sterile 15 ml conical tubes containing 2 mls sterile 1x PBS. Samples were then vortexed vigorously for 20 s before serial dilution and plating as previously described for other samples. Organically raised chicken fecal isolates were obtained from fecal samples on the ground using sterilized tongue depressors and sterile 15 ml conical tubes, before processing and plating for isolation as described above. Only city and host species were collected as metadata.
Obtaining Regional Human Isolates
Campylobacter jejuni strains used in this study were isolated previously from human clinical samples by the Tennessee Department of Health using commercially available Campylobacter blood free selective media (CCDA) plates. After a 48 h incubation at 42°C in a GasPak container with a Campy sachet, isolates were used to conduct a Gram stain, hippurate hydrolysis assay, catalase, indole, and oxidase test, in addition to MALDI-TOF confirmation (data not shown) as described previously (Kelley et al., 2018). Personal identifiable information was removed before isolates were shipped to researchers at the University of Tennessee in accordance with the IRB protocol: UTK IRB-17-03683-XP. Only city and gender were collected as metadata. Once received, isolates were passaged on Campylobacter-selective media and grown for 48 h under microaerobic conditions at 37°C. This growth was harvested and used for genomic DNA extraction (below) and stocked in MH broth with 20% glycerol at -80C.
Isolation of Campylobacter From Regional Samples
After incubation on selective media under the conditions described above, plates from each sample type were enumerated and 2-5 individual colonies were passaged onto the Campylobacter-selective media as described above and incubated for another 48 h under microaerobic conditions at 37°C. Resulting growth was harvested and used for both genomic DNA extraction (below) and stocked in MH broth with 20% glycerol at −80°C.
Genomic DNA Preparation and PCR-Based Identification
Genomic DNA was obtained from growth of isolated colonies following the protocol described previously (Kelley et al., 2018). Briefly, growth was resuspended in sterile genomic lysis buffer (50 mM Tris Base - pH7.5, 50 mM EDTA, 1% SDS, 10 mM NaCl) before adding protein precipitation solution (Promega – A795A). Following DNA precipitation, the pellet was dried before resuspension in 100 μl ultra-pure water. Genomic DNA was stored at −20°C. Designation of samples as either C. jejuni or C. coli was conducted via PCR with primers that specifically amplify either mapA (Forward-TCAATGCAGTTCTTGTGAAA; Reverse-TTCAGAGATTAAACTAGCTGC) or ceuE (Forward–ATGAAAAAATATTTAGTTTTTGCA; Reverse-ATTTTATTATTTGTAGCAGCG), respectively under the following conditions: 95°C-5min; 50°C - 30 sec, 45°C - 30 sec, 72°C - 1min for 30 cycles; 72°C - 7 min (Gonzalez et al., 1997; Dekker, 2016). Resulting amplicons were imaged using a 1.0% agarose gel stained with ethidium bromide to check for a band of the corresponding sizes: mapA-550 bp or ceuE-893 bp.
Preparation for Whole-Genome Sequencing
Isolates confirmed as C. jejuni were utilized for WGS. Each sample was RNase-treated by incubating 44 μl genomic DNA with 5 μl buffer and 1 μl RNase (Invitrogen – AM2294) for 1 h at 37°C before heat inactivating at 70°C for 20 min. The resulting RNase-treated DNA was cleaned using a Zymo Genomic DNA Clean and Concentrate Kit (D4011) following the manufacturer’s instructions. Genomic DNA sample concentrations were quantified on a NanoDrop 2000 spectrophotometer and visualized on a 1.0% agarose gel to confirm the presence of intact genomic DNA. Samples were aliquoted in nuclease-free 96-well plates and shipped to the Center for Genomics and Bioinformatics at Indiana University for WGS1.
Whole-Genome Sequencing
Library preparation, multiplexing, and barcoding was conducted utilizing NEXTflex kits (PerkinElmer) following the manufacturer’s protocol (Perkinelmer-Appliedgenomics.com, 2020). DNA concentrations were obtained on a Qubit3 fluorometer before running on a 2200 TapeStation bioanalyzer. Sequencing was performed utilizing the Illumina NextSeq 500 platform with 150 × 150 paired end reads. The resulting paired-end reads were demultiplexed using bcl2fastq software (Support.Illumina.com, 2020). Resulting reads were accessed by researchers at Oak Ridge National Laboratory for genome assembly, annotation, and further bioinformatic analyses.
Obtaining Reads From GenomeTrakr Database
After identifying the sampling period and region of interest, raw reads for 87 isolates deposited in the GenomeTrakr database were accessed and downloaded to incorporate into the network analysis. The accession numbers for reads used in the study can be found in Table 2.
Bioinformatic Analysis of C. jejuni Genomes
The initial analysis of the resulting sequence reads and reads obtained from the GenomeTrakr database was performed as previously described (Kelley et al., 2018). Briefly, read quality for each sample was analyzed and adapter sequences were trimmed using Atropos (Didion et al., 2017). Genomes were de novo assembled from the remaining paired-end sequences using the up-to-date version (3.12.0) of SPAdes (Bankevich et al., 2012). For all genomes, quality was assessed using CheckM and low quality genome assemblies were removed (Parks et al., 2015). The Prokka genome annotation software package was utilized to predict protein-coding genes (Prodigal) and non-coding RNA genes (RNAmmer, tRNAscan-SE) (Lowe and Eddy, 1997; Lagesen et al., 2007; Hyatt et al., 2010; Seemann, 2014). Predicted proteins were annotated using Prokka and hmmscan (Finn et al., 2011) and were clustered into orthologous groups and designated as either belonging to the pan or core genomes using PIRATE (Bayliss et al., 2019). For each genome, a quantitative matrix was constructed and imported into Cytoscape for network analysis with a threshold of 0.93 (Shannon et al., 2003). Community clustering was then performed at 0.93 threshold to visualize distinct community groups (Newman, 2004; Su et al., 2010).
To compare the network-based analysis to previously established pipelines, we performed SNP calling using a CFSAN-based workflow with a minimal alternative allele frequency threshold of 90%, (Peerj.com, 2020). The following parameters were applied to assembled genomes – COV: 10, Rel. COV: 10%, SNP quality 30, MQ: 25, Z-score: 1.96, and distance between SNPs: 10. The C. jejuni 81-176_G1_B7 genome was used as a reference for the SNP analysis and tree assembly. The SNP tree was then visualized in iTOL (Letunic and Bork, 2016). In addition, assembled genomes were also assigned to ST clonal complexes for comparison. This was done by querying each assembled genome against the Campylobacter PubMLST database (Pubmlst.org, 2020).
Antibiotic Susceptibility Testing of C. jejuni Isolates
Isolates used for susceptibility testing were cultured on MH plates containing trimethoprim (TMP) at 10 μg/ml and incubated under microaerobic conditions for 48 h at 37°C. Growth was harvested into 500 μl MH broth and a sterile cotton swab was used to spread each suspension on a large, 14 cm MH agar plate. Using the standard Kirby-Bauer method, Oxoid brand antibiotic disks of the following antibiotics and concentrations: Amoxycillin/Clavulanic Acid (30 μg), Ampicillin (10 μg), Azithromycin (15 μg), Ceftriaxone (30 μg), Cephazolin (30 μg), Ciprofloxacin (5 μg), Doxycycline (30 μg), Erythromycin (15 μg), Gentamicin (10 μg), Levofloxacin (5 μg), Meropenem (10 μg), and Tetracycline (30 μg), were dispensed onto each plate and incubated for 48–72 h under microaerobic conditions before zones of inhibition were measured. Measurements for individual antibiotics across all isolates were averaged and measurements for each isolate were subtracted from the average to determine strains that were more sensitive and more resistant in relation to the calculated average for each antibiotic.
Results
Sample Collection and Campylobacter Isolation
Through the combined efforts of our group, veterinarians at the University of Tennessee College of Veterinary Medicine, and the Tennessee Department of Health, a total of 630 samples were collected (Figure 1). Of these, 293 fecal samples were collected from various animal sources, including alpaca, camel, cat, chicken, cow, dog, falcon, goat, goose, horse, pig, raccoon, sheep, snake, and zebra throughout the sampling period (Table 1). During the same period, 65 food samples were collected from local farmer’s markets and grocery stores, including vegetables (unwashed potatoes, kale leaves, and carrots), fruits (unwashed berries, citrus fruit, apples, bananas, peaches, and plums), and raw meats (chicken, pork, and beef). Sampling of local commercial meat processing plants resulted in fecal samples from cattle and pigs, all collected post-slaughter, included in the total listed above. Local surface water was also collected throughout the sampling period, with 165 samples collected from the banks of local running rivers and small streams, including the Tennessee River, the French Broad River, the Holston River, and lower tributaries.
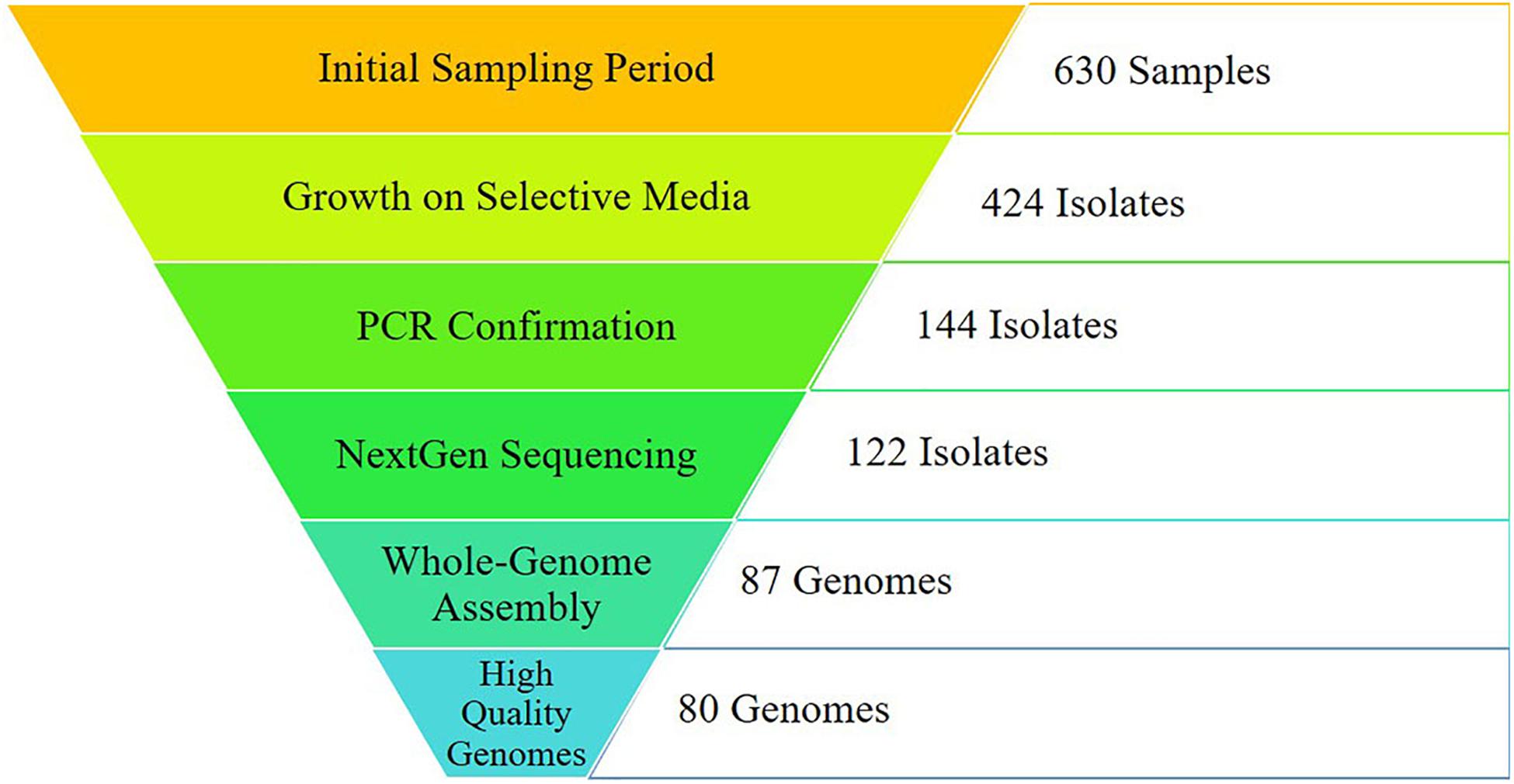
Figure 1. Sampling results from initial sampling to final analyses. Initial sampling was conducted across the region of East Tennessee from October 2016- October 2018, including environmental and human samples. Campylobacter-specific (CS) media was used for selective plating. Isolates were confirmed as either C. jejuni or C. coli utilizing primers specific for each. Quality control was conducted on raw sequencing reads before de novo assembly. Genomes assembling with 95% completeness or higher were utilized for downstream analyses. A total of 167 genomes (80 from this study and 87 from GenomeTrakr) were used for analysis.
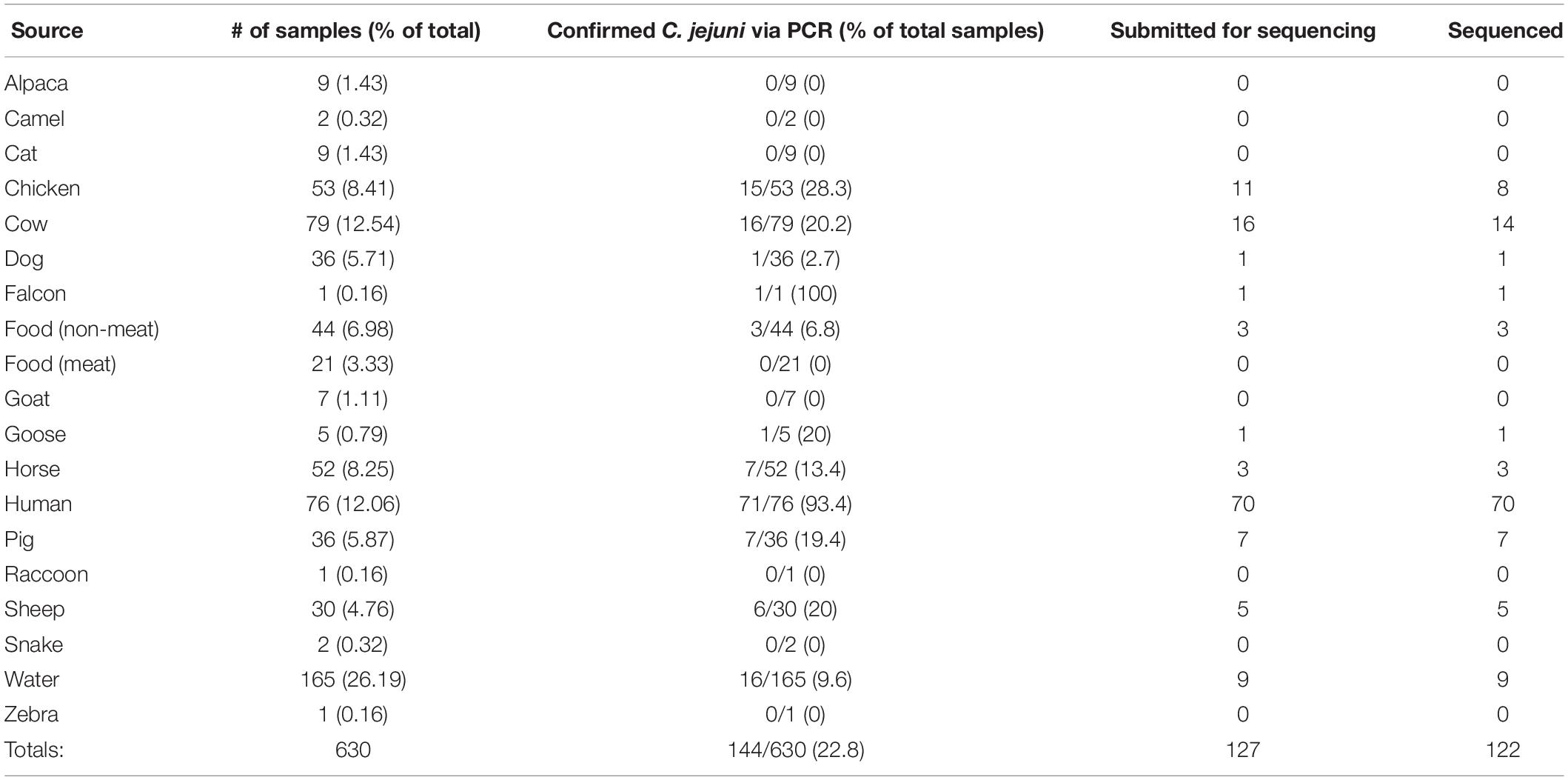
Table 1. Breakdown of sampling numbers by source type, percent of total for each source type, PCR-confirmed C. jejuni samples, isolates submitted for sequencing, and total sequenced.
Successful isolation and PCR-confirmation of C. jejuni varied greatly by source. Samples yielded PCR-positive isolates as follows: chickens-15, cattle-16, sheep-6, horse-7, pig-7, dog-1, falcon-1, goose-1, non-meat food-3, and water-16. We did not obtain PCR-confirmed C. jejuni isolates from alpaca, camel, cat, goat, raccoon, snake, and zebra samples. Human isolates were collected by the Tennessee Department of Health during the previously described sampling period. Of the 76 human isolates received by our group, 71 were C. jejuni, 4 were C. coli, and a single isolate was C. hyointestinalis. Overall, a single PCR-confirmed C. jejuni isolate from each sample was prepared and submitted for whole-genome sequencing and downstream analyses. Of the 127 genomes submitted for sequencing, 122 samples (Chicken-8, Cow-14, Dog-1, Falcon-1, Non-meat food-3, Goose-1, Horse-3, Human-70, Pig-7, Sheep-5, Water-9) passed quality control standards employed by the sequencing facility (DNA quality/quantity) and resulted in high quality reads. At least 50 million reads were generated for each isolate, representing 100x coverage of each C. jejuni genome (∼1.7 Mb).
Whole-Genome Assembly and Quality Filtering
All environmental genomes utilized for the whole genome analyses are between 98.0% and 99.9% complete as determined by analysis with CheckM (Figures 2A,B) (Parks et al., 2015). The range of completeness for the environmental genomes varied between isolate source, with human isolates demonstrating a range in completeness of 99.0% - > 99.8%. The range of completeness for chicken isolates was 98.0% – 99.8% across all genomes analyzed, while cattle isolates presented a range in completeness (99.3% – 99.875%) similar to that of humans. Only genomes mapping to a reference genome (C. jejuni 81-176_G1_B7) with 90% identity or above to ensure species identity were utilized for the study. Overall, the genomes from various sources mapped to the reference genome at an average of 92% identity, with the exception of the water samples. The genomes from water isolates that clustered with an identity close to 98% of the reference genome. Interestingly, there was some variability in percent identity of human isolates to the reference, with several close to 70% identity to the reference genome (Figure 2A) and therefore excluded from further analyses. The number of genomes covering each position in the reference sequence was also relatively high, although some positions in the reference were covered by only a few genomes suggesting more novel regions in the reference genome (Figure 2C).
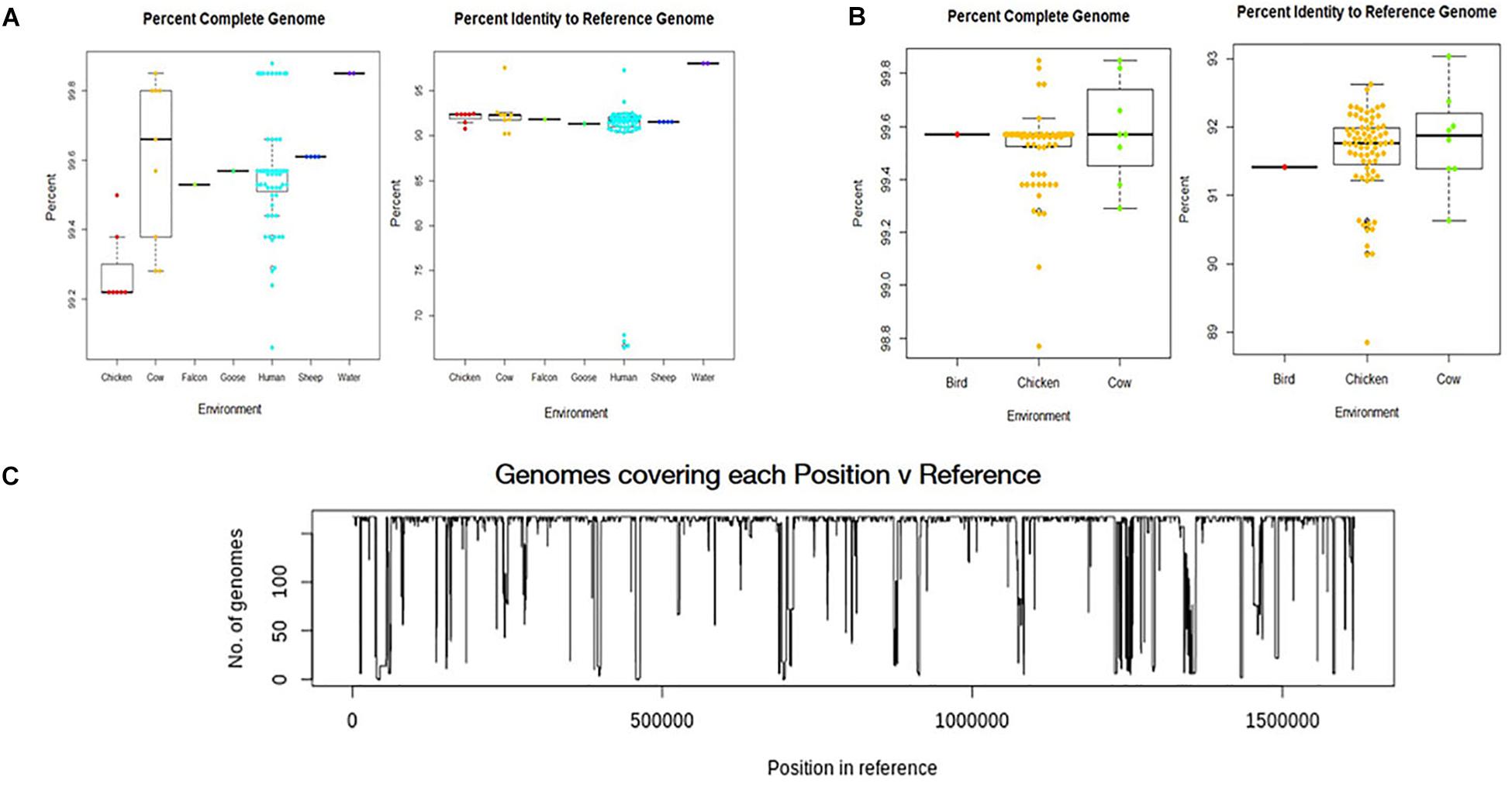
Figure 2. Analysis of genome assemblies for completeness, identity to reference, and coverage. (A) Box and whisker plot of genome completeness and percent identity to the reference were compiled for each source type for samples collected in this study. (B) Box and whisker plots were also compiled for GenomeTrakr genomes. Completeness across all assembled genomes ranged from 98.0% to above 99.8% and assembled genomes had an average alignment of 92% to the reference genome (C. jejuni 81-176_G1_B7). used for this study. (C) Coverage of each position in the reference genome by the genomes analyzed.
Core and Pan Genome Assembly and Analysis
Following quality filtering and genome assembly, 167 high quality genomes were used to construct an analytical pipeline that was used to define both the core and pan genome, which were based on the presence of predicted gene families (Figure 3). In all, there were 4710 gene families identified in the 167 C. jejuni genomes analyzed by PIRATE with 225 gene families containing greater than one allele at the 90–95% threshold. The pangenome of C. jejuni comprised 4710 gene families of which 1384 were classified as core (defined as genes found in >95% of the genomes) and 3326 accessory genes. The large number of accessory genes identified in the pan genome analysis indicates Campylobacter isolates have remarkable genetic variability and likely contribute to their broad host distribution
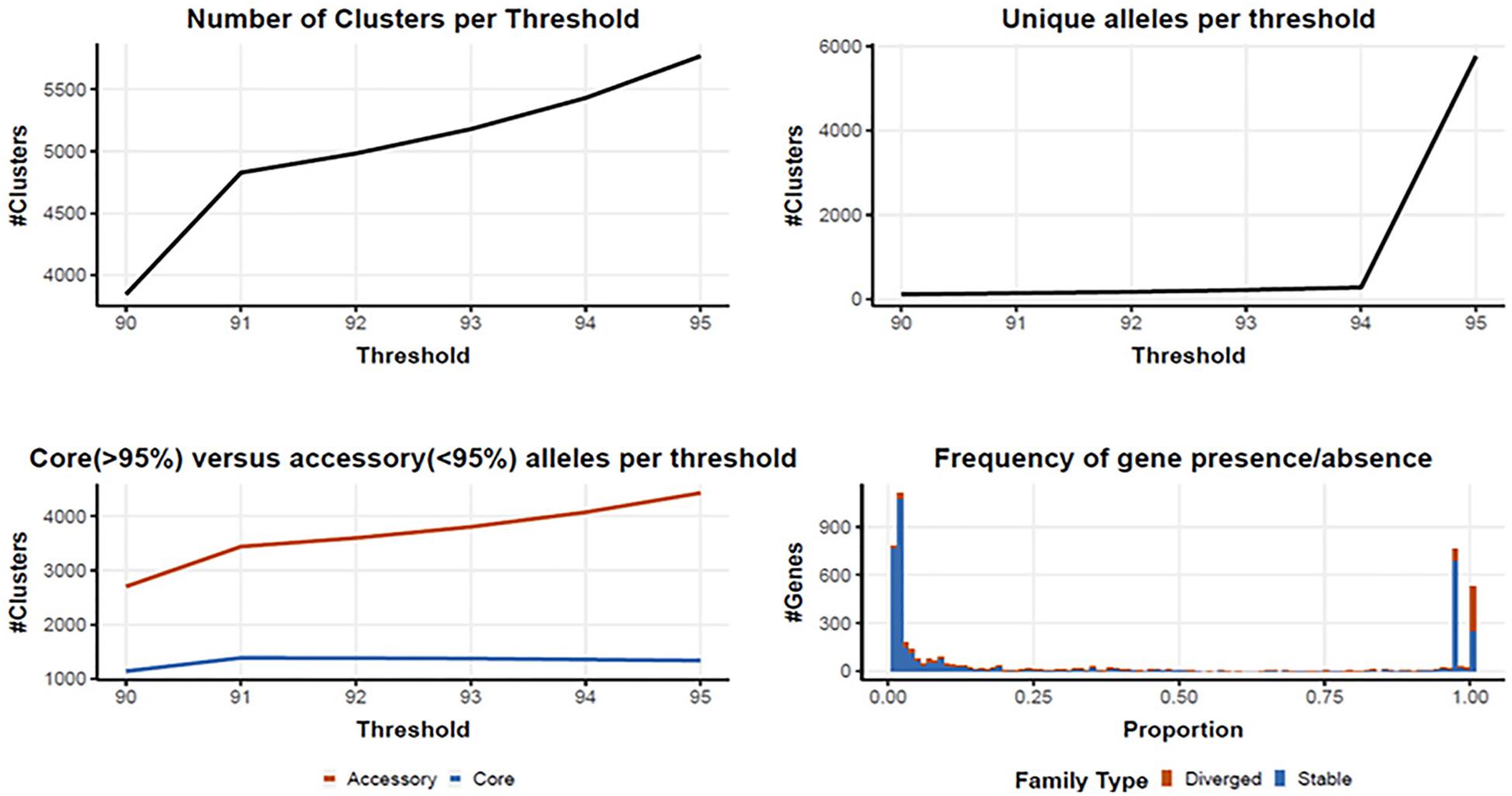
Figure 3. Core and pan genome assembly and gene family distribution and identification of new genes. One hundred and sixty seven genomes were used to identify a core and pan genome. There were 4710 gene families identified in the pangenome, with 225 gene families containing greater than one allele at the 90–95% threshold. The core genome is comprised of 1,384 gene families, with 3326 accessory gene families.
SNP and Network Analysis for Comparison of Assembled Genomes
To investigate the potential of whole genome comparison as a means for Campylobacter source-tracking, genomes underwent a SNP analysis to determine the potential relatedness of various isolates. A tree of relatedness was produced of all isolates using C. jejuni 81-176_G1_B7 as a reference genome (Figure 4). Isolate source is denoted by color. We also employed a non-reference-based approach for whole genome comparisons using network analysis. The genomes from this study and GenomeTrakr were analyzed by comparing coding regions to produce a network for visualization of relatedness (Figure 5). While the distance between nodes is arbitrary, lines connecting individual nodes indicate they meet or exceed a threshold of 0.93 across the entire coding region of the two genomes. The network was further characterized by clustering genomes with community clustering (Figure 5B). Community clustered genomes reveals human isolates cluster with other human isolates, but also cluster with those from other sources including cows/chickens (Cluster 1 and 3), cows/water (Cluster 5), and chickens/other birds (Clusters 2, 4, and 6).
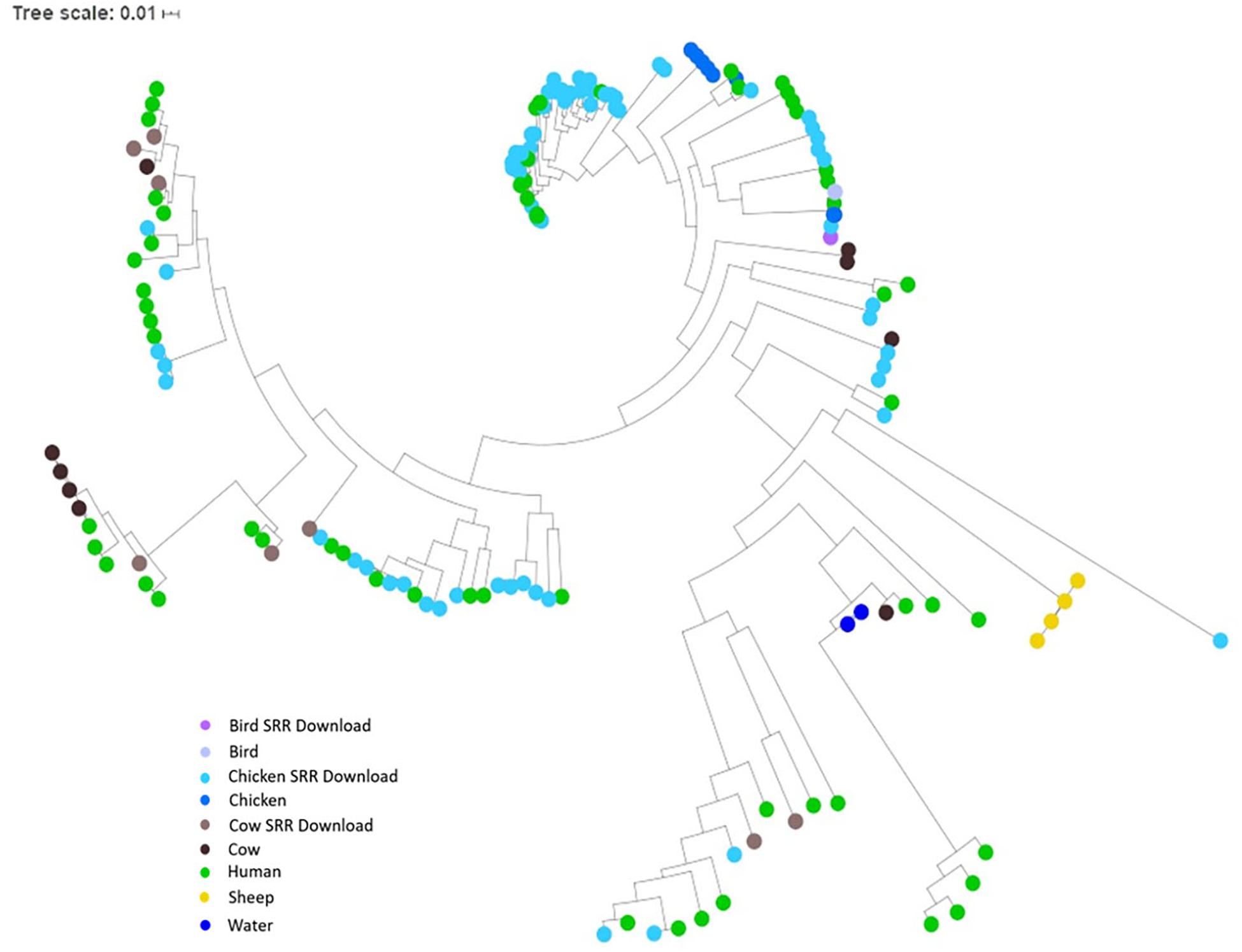
Figure 4. SNP tree of environmental isolates demonstrating relatedness. A tree of relatedness was created using 167 assembled genomes, organized by source, utilizing C. jejuni 81-176_G1_B7 as a reference. Colors denote sample source, with GenomeTrakr genomes identified as SRR Download.
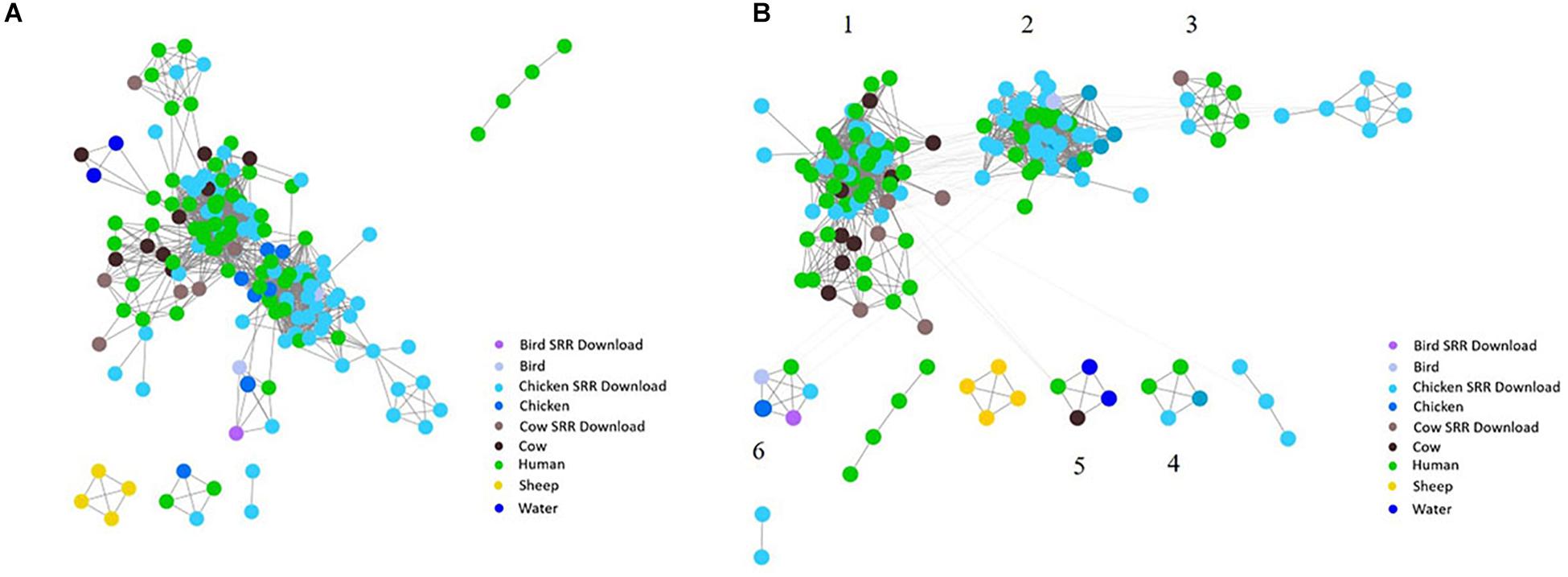
Figure 5. Network analysis with assembled genomes. A network analysis of 167 genomes and the coding regions across all genomes. Distance between nodes is arbitrary, but edges connecting individual nodes indicate these meet or surpass a threshold of 0.93 similarity across the entire coding portion of the two genomes. (A) The initial unclustered genomes falling above the threshold of 0.93 similarity. (B) The same network with community clustering applied. GenomeTrakr downloads are labeled SRR Download.
ST Assignments of Isolates Based on Whole-Genomes
The whole genome of each isolate identified within clusters of the network analysis were queried against the PubMLST online database of Campylobacter strains. Isolates Human-40, Cow-2, Chicken(meat)-6, Chicken(live)-7, Chicken (meat)-12, Chicken(meat)-14, Chicken(meat)-38, Cow-16, Cow-18, Chicken(meat)-58, Chicken(meat)-59, Chicken(meat)-61, and Cow-22 were not assigned to a clonal complex, but were assigned to ST groups, as shown in Table 2. Additionally, five genomes did not match to any previously identified ST group or clonal complex. ST-353 was the most common assignment across all the genomes, especially the GenomeTrakr isolates (Table 2).
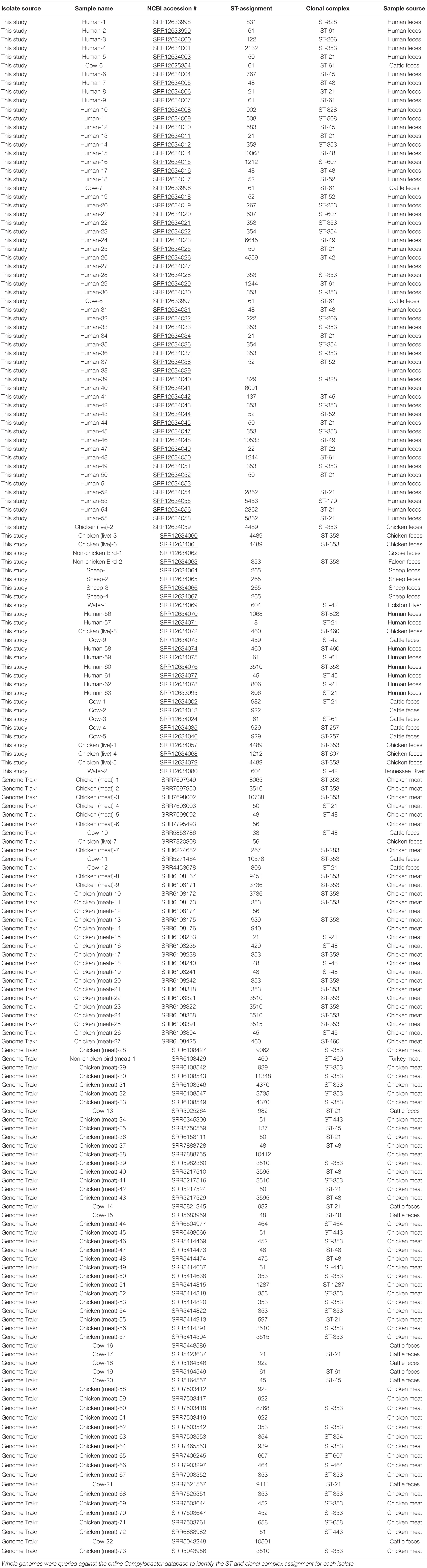
Table 2. ST assignments for each isolate identified in the cluster analysis.
Antibiotic Susceptibility Testing
We assayed isolates identified in this study against two antibiotics each from six different antibiotic classes to investigate phenotypic differences (Figure 6). The greatest amount of variability for a single antibiotic class appears for tetracycline and doxycycline which display a broader spectrum of sensitivity across all the isolates, regardless of cluster, when compared to results for the other antibiotics. Human isolates displayed the greatest variability in antibiotic sensitivity across the antibiotics assayed. With a few exceptions, cattle isolates demonstrated a pattern of lower overall susceptibility across the spectrum of antibiotics assayed when compared to other isolates, while isolates from chickens displayed increased susceptibility to the majority of antibiotics when compared to other isolates (Figure 6A). When separated based on ST assignment, the overall broader sensitivity to tetracycline and doxycycline can be observed as well (Figure 6B).
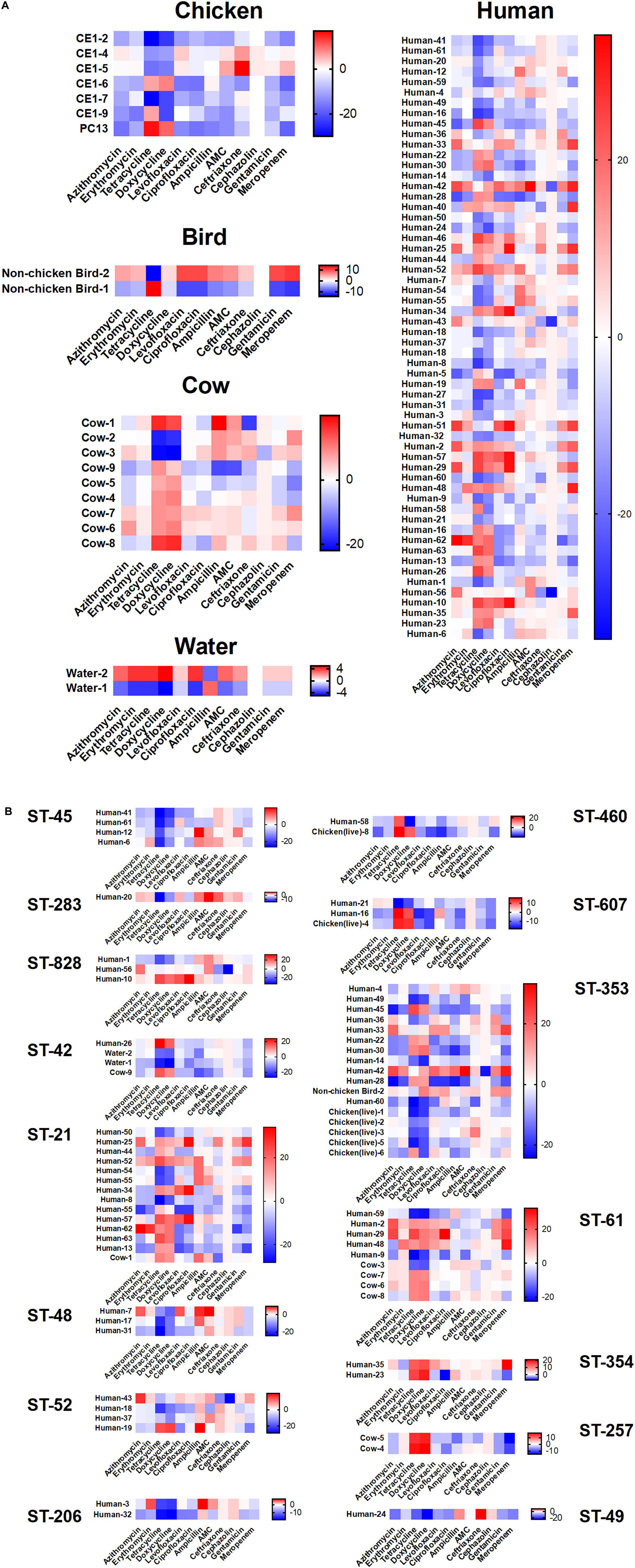
Figure 6. Antibiotic susceptibility testing results according to cluster. Antibiotic susceptibility to several classes of antibiotics were compared across source type (A) and ST grouping (B) by subtracting measurements for each isolate from the overall average for each antibiotic. Positive numbers (red) indicate isolates less susceptible to the corresponding antibiotic, while negative numbers (blue) indicate isolates more susceptible. No obvious patterns were distinguishable across the groupings despite extensive heterogeneity.
Discussion
To investigate whether a whole genome-based approach is a viable method for source-tracking human Campylobacter infections in a geographically and temporally restricted manner, we conducted active surveillance of agricultural and environmental sources in eastern Tennessee between October 2016 and October 2018. Confirmed C. jejuni isolates obtained during the sampling period were subjected to whole-genome sequencing and bioinformatically compared to clinical isolates deposited with the Tennessee Department of Health and GenomeTrakr sequences from the same geographical area during the indicated sampling period. Historically, East Tennessee has a higher incidence of C. jejuni infections (8–11 per 100,000 persons) when compared to the rest of the state (5–7 per 100,000 persons) (Weisent et al., 2011; Tn.gov, 2020). Despite this incidence, East Tennessee is home to relatively few poultry farms and other agricultural operations, although there is some variability between counties in the region (Nass.Usda.Gov,, 2020). This observation is particularly pronounced in Knox County, where numerous human isolates were collected, but where agricultural production is limited. In addition to agricultural operations, the region also contains numerous rivers, streams, and tributaries that experience flooding throughout the year, and may lead to contamination of groundwater or recreational waters, increasing the risk of human exposure. During the study period, East Tennessee was also impacted by a C. jejuni outbreak that was attributed to puppies sold by a large pet store chain (Montgomery et al., 2018). Taken together, these factors suggested that C. jejuni infections in the region may not be due solely to the consumption of undercooked or contaminated poultry meat, but may also be the result of interactions with other infected animals or contaminated water sources.
Our study is unique in that the analyzed strains were isolated from the same region during the same period, which provided a “real world” scenario of sampling for source-tracking using whole-genome sequencing in a region that had not been previously investigated. Of over 600 samples collected, we were able to generate 80 complete, high quality genomes for our analyses. With less than one quarter (∼12.7%) of the collected samples resulting in a high quality genome, successful surveillance of a region like the one described in this study would likely be time consuming and costly. This was a considerable obstacle in creating an adequate set of reference isolates within our geographical area, but was addressed by supplementing with genomes submitted to an online database from the same region and time period, which underscores the importance of efforts like the GenomeTrakr program. In addition to the relatively low rate of high quality genome generation, several samples yielded growth on Campylobacter-specific media, but could not be identified as either C. jejuni or C. coli by PCR. Such a result indicates that other Campylobacter species may be present in the environmental sources, which may be worth further investigation in the future.
Assembly of the C. jejuni genomes from raw sequence reads was successful and utilized a high threshold for completeness, ensuring that only the most informative genomes were used. It is possible to lower the threshold of completeness in order to include more genomes that are less complete, but doing so may negatively affect the results and skew downstream analyses. Although we were able to identify a core and pan genome using the data provided from the combined 167 genomes, utilizing a larger set of genomes would ensure all genes in the core genome are identified, as well as all potential genes in the pan genome. In subsequent studies, the resulting pan genome could then be used to identify host-specific determinants, virulence factors that impact human campylobacteriosis severity, and bacterial factors that promote survival within different environments. Since C. jejuni isolates were the most commonly isolated species from human clinical samples, the Campylobacter jejuni 81-176_G1_B7 served as a reference strain to determine genome assembly quality. While the potential for identity bias should be kept in mind when selecting a reference genome for other types of analyses, the analyses described in this study avoid the introduction of these biases by only using the reference genome to ensure genomes are from C. jejuni before direct comparison of the genomes to each other.
The unique network analysis described in this study produced clusters of human and environmental isolates, suggesting whole genome comparisons may be a viable method for linking human infections to potential source types, although tracking to a specific site may not be possible. Interestingly, human isolates most frequently clustered with cattle and chicken isolates as indicated by the number of edges linking human isolates to those from cattle, chickens, or both. We believe the network analysis provides a visual representation of the similarities between isolates by denoting clusters that may not be indicated or obvious in the SNP dendrogram. Portraying the data in a way such as the network analysis also potentially increases the ability to identify clusters or isolates that should be examined more closely. This method could also prove useful since the threshold of similarity can be adjusted as necessary based on the organism of interest and the level of similarity desired for comparison.
The genetic variability between isolates from different sources led us to question whether phenotypic variability also exists between isolates and if patterns can be detected between sampling sources. This study utilized the Kirby-Bauer disk diffusion method to obtain preliminary phenotypic data by comparing the zones of inhibition for individual isolates to the overall average zone of inhibition for each antibiotic, allowing for intraspecies comparison across an array of antibiotics. Overall, a large amount of variability was easily detected between the isolates making the identification of phenotypic patterns challenging. Cattle isolates collected at random during routine checks of healthy animals on farms demonstrated lower overall susceptibility across the spectrum of antibiotics assayed. While no data was collected regarding antibiotic use on these farms, further epidemiological work may provide insights into potential links to antibiotic usage and susceptibility in the coordinating isolates. As a preliminary analysis focused on direct phenotypic comparisons, this analysis did not take into account minimum inhibitory concentration (MIC) data or clinical breakpoints for the antibiotics tested. However, the observed variability between isolates from the same source type indicate the potential for future work to investigate antibiotic susceptibility of environmental isolates in a more clinically relevant manner.
This work demonstrates the potential of whole-genome sequencing as a means of microbial source tracking for C. jejuni, but also identifies issues that must be addressed before this technique can be adequately utilized in an effective manner. WGS can provide a breadth of genomic information for comparison, but the sequencing quality and computing power necessary to conduct such in-depth analyses can be limiting factors. While the analyses described in this study would be useful for investigating the genomic relatedness of Campylobacter in the environment and the clinic, the ability to perform these in-depth analyses may not be realistic for groups without access to supercomputing facilities. Additionally, the level of surveillance necessary to maintain a current profile of C. jejuni genomic information from environmental sources in an accessible database would require continual sampling from numerous sources, which may also prove to be a limiting factor for state and federal health departments. Again, this need underscores the importance and continued support for resources like the FDA GenomeTrakr program.
As demonstrated by the network analyses, Campylobacter can be clustered by potential source, but the inherent genomic variability between individual isolates may make linking cases to a specific source challenging. Previous studies have demonstrated the benefits of whole-genome sequencing for comparing environmental and human isolates through the incorporation of sequences deposited in online databases (Wilson et al., 2008; Dearlove et al., 2016; Buchanan et al., 2017). By utilizing the GenomeTrakr database, we were able to increase the number of genomes used for our analyses and enhance the quality of the network. The resulting clusters support the conclusion that both chickens and cattle may serve as sources for human infections in East Tennessee, which has been observed in similar studies conducted in France and Germany (Rosner et al., 2017; Thépault et al., 2018a, b). Using this form of network analysis with a thorough examination of the corresponding epidemiological data could provide insight into risk factors leading to C. jejuni infections in East Tennessee.
Our study is the first to utilize WGS technology to preliminarily analyze isolates collected from a variety of sources in East Tennessee during a set sampling period and incorporate sequence information deposited in an online repository to compare to human isolates from the same region. Based on our results, we believe whole-genome sequencing is a beneficial technique that can provide an abundance of genomic data and source-tracking information. Additionally, the in-depth curation and analyses of epidemiological data by state and federal health agencies are necessary for source-tracking of human Campylobacter infections, along with concurrent surveillance of potential reservoirs throughout the region. While this may prove a major hurdle, we believe the implementation of WGS technology and our network analysis method can provide valuable information about the presence of environmental C. jejuni in regions like East Tennessee, and when compiled with epidemiological data, can aid in the identification of potential sources of human infection.
Data Availability Statement
The datasets presented in this study can be found in GenBank under the BioProject number PRJNA644378 for samples collected in the study. NCBI accession numbers for all samples used in this study can be found in Table 2.
Ethics Statement
The studies involving human participants were reviewed and approved by University of Tennessee Institutional Review Board. The ethics committee waived the requirement of written informed consent for participation. Ethical review and approval was not required for the animal study because the researchers were never in contact with animals from which samples were acquired. Samples from livestock were obtained from licensed DVMs during routine health exams. Written informed consent for participation was not obtained from the owners because Written informed consent was not required as these were indirect samples obtained during routine health exams. The researchers never had contact with the animals and no identifiable information was provided by the veterinarians.
Author Contributions
BK, JE, DJ, and JJ conceived and designed the experiments. BK and JE performed the experiments and analyzed the data. AL aided in analysis of bioinformatic analyses. LS performed the epidemiological analysis. BK, JE, LS, and JJ wrote and revised the manuscript. All the authors have read and approved the final manuscript.
Funding
Funding was provided by the Joint Directed Research and Development (JDRD) initiative between the University of Tennessee and Oak Ridge National Labs (ORNL) JDRD# R013318090 awarded to JJ. Funding also came from University of Tennessee startup funds awarded to JJ. Additional funding was provided by the Plant Microbe Interface (PMI) Scientific Focus Area by the Office of Biological and Environmental Research in the DOE Office of Science and the Oak Ridge National Laboratory’s, Laboratory Directed Research and Development (LDRD) program, project 8321. AL acknowledges that this project was supported in part by appointments to the Higher Education Research Experiences (HERE) Program at Oak Ridge National Laboratory, administered by the Oak Ridge Institute for Science and Education (ORISE). ORISE is managed by Oak Ridge Associated Universities (ORAU) for the U.S. Department of Energy (DOE). This manuscript has been co-authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Tennessee Department of Health, especially Steffany Cavallo and Dr. John Dunn, for graciously supplying human isolates for the time period described. The authors also would like to thank Dr. Andrea Lear, Dr. Meggan Graves, and Heidi Wyrosdick from the University of Tennessee College of Veterinary Medicine for generously providing animal fecal samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.571064/full#supplementary-material
Supplementary Figure 1 | Percentage identity of gene family at lowest threshold. Breakdown of percent identity of gene families at the lowest threshold. Identified gene families were classified as either fission/fusion, fission/fusion + multicopy, multicopy, or single copy.
Supplementary Table 1 | Sampling location and date. Sampling source is listed for individual samples collected in this study. Sampling locations are listed by county in Tennessee, along with the collection date for each sample.
Footnotes
References
Ashbolt, N. J. (2004). Microbial contamination of drinking water and disease outcomes in developing regions. Toxicology 198, 229–238. doi: 10.1016/j.tox.2004.01.030
Backert, S., Tegtmeyer, N., Cróinín, T. Ó, Boehm, M., and Heimesaat, M. M. (2017). “Human campylobacteriosis,” in Campylobacter Features, Detection, and Prevention of Foodborne Disease, ed. G. Klein, (Cambridge, MA: Academic Press), 1–25. doi: 10.1016/b978-0-12-803623-5.00001-0
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bayliss, S. C., Thorpe, H. A., Coyle, N. M., Sheppard, S. K., and Feil, E. J. (2019). PIRATE: a fast and scalable pangenomics toolbox for clustering diverged orthologues in bacteria. Gigascience 8:giz119.
Buchanan, C. J., Webb, A. L., Mutschall, S. K., Kruczkiewicz, P., Barker, D. O., Hetman, B. M., et al. (2017). A genome-wide association study to identify diagnostic markers for human pathogenic Campylobacter jejuni strains. Front. Microbiol. 8:1224. doi: 10.3389/fmicb.2017.01224
Clark, C. G., Berry, C., Walker, M., Petkau, A., Barker, D. O., Guan, C., et al. (2016). Genomic insights from whole genome sequencing of four clonal outbreak Campylobacter jejuni assessed within the global C. jejuni population. BMC Genomics 17:990. doi: 10.1186/s12864-016-3340-8
Cody, A. J., Mccarthy, N. D., Jansen Van Rensburg, M., Isinkaye, T., Bentley, S. D., Parkhill, J., et al. (2013). Real-time genomic epidemiological evaluation of human Campylobacter isolates by use of whole-genome multilocus sequence typing. J. Clin. Microbiol. 51, 2526–2534. doi: 10.1128/jcm.00066-13
Connerton, I., and Connerton, P. (2017). “Campylobacter foodborne disease,” in Foodborne Diseases, eds C. E. R. Dodd, T. Aldsworth, R. A. Stein, D. O. Cliver, and H. P. Riemann, (Amsterdam: Elsevier), 209–221. doi: 10.1016/b978-0-12-385007-2.00008-5
Crofts, A. A., Poly, F. M., Ewing, C. P., Kuroiwa, J. M., Rimmer, J. E., Harro, C., et al. (2018). Campylobacter jejuni transcriptional and genetic adaptation during human infection. Nat. Microbiol. 3, 494–502. doi: 10.1038/s41564-018-0133-7
Dearlove, B. L., Cody, A. J., Pascoe, B., Méric, G., Wilson, D. J., and Sheppard, S. K. (2016). Rapid host switching in generalist Campylobacter strains erodes the signal for tracing human infections. ISME J. 10, 721–729. doi: 10.1038/ismej.2015.149
Dekker, J. P. (2016). Commentary: molecular assay validation using genomic sequence databases. J. Clin. Microbiol. 54, 2854–2856. doi: 10.1128/jcm.01797-16
Didion, J. P., Martin, M., and Collins, F. S. (2017). Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ 5:e3720. doi: 10.7717/peerj.3720
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37.
Fitzgerald, C., Patrick, M., Gonzalez, A., Akin, J., Polage, C. R., Wymore, K., et al. (2016). Multicenter evaluation of clinical diagnostic methods for detection and isolation of Campylobacter spp. from stool. J. Clin. Microbiol. 54, 1209–1215. doi: 10.1128/jcm.01925-15
Fitzgerald, C., Sails, A. D., and Fields, P. I. (2005). “Campylobacter jejuni strain variation,” in Campylobacter-Molecular and Cellular Biology, Horizon Bioscience, eds J. M. Ketley, and M. E. Konkel, (Wymondham: Horizon Bioscience), 59–77.
Fricke, W. F., and Rasko, D. A. (2014). Bacterial genome sequencing in the clinic: bioinformatic challenges and solutions. Nat. Rev. Genet. 15, 49–55. doi: 10.1038/nrg3624
Fugett, E. B., Schoonmaker-Bopp, D., Dumas, N. B., Corby, J., and Wiedmann, M. (2007). Pulsed-field gel electrophoresis (PFGE) analysis of temporally matched Listeria monocytogenes isolates from human clinical cases, foods, ruminant farms, and urban and natural environments reveals source-associated as well as widely distributed PFGE types. J. Clin. Microbiol. 45, 865–873. doi: 10.1128/jcm.01285-06
Gonzalez, I., Grant, K. A., Richardson, P. T., Park, S. F., and Collins, M. D. (1997). Specific identification of the enteropathogens Campylobacter jejuni and Campylobacter coli by using a PCR test based on the ceuE gene encoding a putative virulence determinant. J. Clin. Microbiol. 35, 759–763. doi: 10.1128/jcm.35.3.759-763.1997
Guyard-Nicodème, M., Rivoal, K., Houard, E., Rose, V., Quesne, S., Mourand, G., et al. (2015). Prevalence and characterization of Campylobacter jejuni from chicken meat sold in French retail outlets. Int. J. Food Microbiol. 203, 8–14. doi: 10.1016/j.ijfoodmicro.2015.02.013
Halpin, A. L., Gu, W., Wise, M., Sejvar, J., Hoekstra, R., and Mahon, B. (2018). Post-Campylobacter Guillain Barré syndrome in the USA: secondary analysis of surveillance data collected during the 2009–2010 novel Influenza A (H1N1) vaccination campaign. Epidemiol. Infect. 146, 1740–1745. doi: 10.1017/s0950268818001802
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Joensen, K., Kuhn, K., Müller, L., Björkman, J., Torpdahl, M., Engberg, J., et al. (2018). Whole-genome sequencing of Campylobacter jejuni isolated from Danish routine human stool samples reveals surprising degree of clustering. Clin. Microbiol. Infect. 24, 201.e5–201.e8.
Johnson, J. M., Weagant, S. D., Jinneman, K. C., and Bryant, J. L. (1995). Use of pulsed-field gel electrophoresis for epidemiological study of Escherichia coli O157: H7 during a food-borne outbreak. Appl. Environ. Microbiol. 61, 2806–2808. doi: 10.1128/aem.61.7.2806-2808.1995
Kaakoush, N. O., Castaño-Rodríguez, N., Mitchell, H. M., and Man, S. M. (2015). Global epidemiology of Campylobacter infection. Clin. Microbiol. Rev. 28, 687–720. doi: 10.1128/cmr.00006-15
Kelley, B. R., Ellis, J. C., Hyatt, D., Jacobson, D., and Johnson, J. (2018). Isolation and whole-genome sequencing of environmental Campylobacter. Curr. Protoc. Microbiol. 51:e64. doi: 10.1002/cpmc.64
Kempf, I., Kerouanton, A., Bougeard, S., Nagard, B., Rose, V., Mourand, G., et al. (2017). Campylobacter coli in organic and conventional pig production in France and Sweden: prevalence and antimicrobial resistance. Front. Microbiol. 8:955. doi: 10.3389/fmicb.2017.00955
Kirk, M. D., Pires, S. M., Black, R. E., Caipo, M., Crump, J. A., Devleesschauwer, B., et al. (2015). World Health Organization estimates of the global and regional disease burden of 22 foodborne bacterial, protozoal, and viral diseases, 2010: a data synthesis. PLoS Med. 12:e1001921. doi: 10.1371/journal.pmed.1001921
Kittl, S., Heckel, G., Korczak, B. M., and Kuhnert, P. (2013). Source attribution of human Campylobacter isolates by MLST and fla-typing and association of genotypes with quinolone resistance. PLoS One 8:e81796. doi: 10.1371/journal.pone.0081796
Kovanen, S., Rossi, M., Pohja-Mykra, M., Nieminen, T., Raunio-Saarnisto, M., Sauvala, M., et al. (2019). Population genetics and characterization of Campylobacter jejuni isolates from western jackdaws and game birds in Finland. Appl. Environ. Microbiol. 85:e02365-18.
Kovanen, S. M., Kivistö, R. I., Rossi, M., Schott, T., Kärkkäinen, U.-M., Tuuminen, T., et al. (2014). MLST and whole—genome MLST of human Campylobacter jejuni isolates from three districts during a seasonal peak in Finland. J. Clin. Microbiol. 52, 4147–4154.
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245.
Llarena, A.-K., Taboada, E., and Rossi, M. (2017). Whole-genome sequencing in epidemiology of Campylobacter jejuni infections. J. Clin. Microbiol. 55, 1269–1275. doi: 10.1128/jcm.00017-17
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Ma, L., Wang, Y., Shen, J., Zhang, Q., and Wu, C. (2014). Tracking Campylobacter contamination along a broiler chicken production chain from the farm level to retail in China. Int. J. Food Microbiol. 181, 77–84.
Moffatt, C., Greig, A., Valcanis, M., Gao, W., Seemann, T., Howden, B., et al. (2016). A large outbreak of Campylobacter jejuni infection in a university college caused by chicken liver pâté, Australia, 2013. Epidemiol. Infect. 144, 2971–2978. doi: 10.1017/s0950268816001187
Montgomery, M. P., Robertson, S., Koski, L., Salehi, E., Stevenson, L. M., Silver, R., et al. (2018). Multidrug-resistant Campylobacter jejuni outbreak linked to puppy exposure—United States, 2016–2018. Morb. Mortal. Wkly. Rep. 67, 1032–1035.
Mou, K. T., Muppirala, U. K., Severin, A. J., Clark, T. A., Boitano, M., and Plummer, P. J. (2015). A comparative analysis of methylome profiles of Campylobacter jejuni sheep abortion isolate and gastroenteric strains using PacBio data. Front. Microbiol. 5:782. doi: 10.3389/fmicb.2014.00782
Nass.Usda.Gov, (2020). Available at: https://www.nass.usda.gov/publications/agcensus/.2017/online_resources/county_profiles/tennessee/index.php (accessed July 2020).
Oakeson, K. F., Wagner, J. M., Rohrwasser, A., and Atkinson-Dunn, R. (2018). Whole-genome sequencing and bioinformatic analysis of isolates from foodborne illness outbreaks of Campylobacter jejuni and Salmonella enterica. J. Clin. Microbiol. 56:e00161-18.
Oporto, B., Juste, R., López-Portolés, J., and Hurtado, A. (2011). Genetic diversity among Campylobacter jejuni isolates from healthy livestock and their links to human isolates in Spain. Zoonoses Public Health 58, 365–375.
Parkhill, J., Wren, B., Mungall, K., Ketley, J., Churcher, C., Basham, D., et al. (2000). The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403, 665–668.
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055.
Peerj.com, (2020). Available at: https://peerj. com/articles/cs-20/ (accessed July 2020).
Perkinelmer-Appliedgenomics.com, (2020). Available at: https://perkinelmer-appliedgenomics.com/home/library-preparation-kits/dna-library-prep-kits/nextflex-rapid-dna-seq-kit/ (accessed July 2020).
Pubmlst.org, (2020). Available at: https://pubmlst. org/campylobacter/ (accessed July 2020).
Revez, J., Llarena, A.-K., Schott, T., Kuusi, M., Hakkinen, M., Kivistö, R., et al. (2014a). Genome analysis of Campylobacter jejuni strains isolated from a waterborne outbreak. BMC Genomics 15:768. doi: 10.1186/1471-2164-15-768
Revez, J., Zhang, J., Schott, T., Kivistö, R., Rossi, M., and Hänninen, M.-L. (2014b). Genomic variation between Campylobacter jejuni isolates associated with milk-borne-disease outbreaks. J. Clin. Microbiol. 52, 2782–2786.
Rosner, B. M., Schielke, A., Didelot, X., Kops, F., Breidenbach, J., Willrich, N., et al. (2017). A combined case-control and molecular source attribution study of human Campylobacter infections in Germany, 2011–2014. Sci. Rep. 7:5139.
Sacher, J., Yee, E., Szymanski, C., and Miller, W. (2018). Complete genome sequence of Campylobacter jejuni strain 12567, a livestock-associated clade representative. Genome Announc. 6:e00513-18.
Salipante, S. J., Sengupta, D. J., Cummings, L. A., Land, T. A., Hoogestraat, D. R., and Cookson, B. T. (2015). Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J. Clin. Microbiol. 53, 1072–1079.
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498– 2504.
Sheppard, S. K., Dallas, J. F., Strachan, N. J., Macrae, M., Mccarthy, N. D., Wilson, D. J., et al. (2009). Campylobacter genotyping to determine the source of human infection. Clin. Infect. Dis. 48, 1072–1078.
Sheppard, S. K., Jolley, K. A., and Maiden, M. C. (2012). A gene-by-gene approach to bacterial population genomics: whole genome MLST of Campylobacter. Genes 3, 261–277.
Su, G., Kuchinsky, A., Morris, J. H., States, D. J., and Meng, F. (2010). GLay: community structure analysis of biological networks. Bioinformatics 26, 3135–3137.
Support.Illumina.com, (2020). Available at: https://support.illumina.com/content/dam/illumina-support/documents/documentation/software_documentation/bcl2fastq2-v2-20-software-guide-15051736-03.pdf (accessed July 2020).
Thépault, A., Poezevara, T., Quesne, S., Rose, V., Chemaly, M., and Rivoal, K. (2018a). Prevalence of thermophilic Campylobacter in cattle production at slaughterhouse level in France and link between C. jejuni bovine strains and campylobacteriosis. Front. Microbiol. 9:471. doi: 10.3389/fmicb.2018.00471
Thépault, A., Rose, V., Quesne, S., Poezevara, T., Béven, V., Hirchaud, E., et al. (2018b). Ruminant and chicken: important sources of campylobacteriosis in France despite a variation of source attribution in 2009 and 2015. Sci. Rep. 8:9305.
Tn.gov, (2020). Available at: https://www.tn.gov/health/cedep/reportable-diseases/campylobacteriosis-campylobacter-species.html (accessed July 2020).
Weisent, J., Rohrbach, B., and Dunn, J. R. (2011). Detection of high risk campylobacteriosis clusters at three geographic levels. Geospat. Health 6, 65–76.
Wilson, D. J., Gabriel, E., Leatherbarrow, A. J., Cheesbrough, J., Gee, S., Bolton, E., et al. (2008). Tracing the source of campylobacteriosis. PLoS Genet. 4:e1000203. doi: 10.1371/journal.pgen.1000203
Young, K. T., Davis, L. M., and Dirita, V. J. (2007). Campylobacter jejuni: molecular biology and pathogenesis. Nat. Rev. Microbiol. 5, 665–679.
Keywords: Campylobacter (C. jejuni), whole-genome sequencing (WGS), environmental isolation, agricultural isolates, human campylobacteriosis
Citation: Kelley BR, Ellis JC, Large A, Schneider LG, Jacobson D and Johnson JG (2020) Whole-Genome Sequencing and Bioinformatic Analysis of Environmental, Agricultural, and Human Campylobacter jejuni Isolates From East Tennessee. Front. Microbiol. 11:571064. doi: 10.3389/fmicb.2020.571064
Received: 09 June 2020; Accepted: 08 October 2020;
Published: 05 November 2020.
Edited by:
Greta Gölz, Freie Universität Berlin, GermanyReviewed by:
Ben Pascoe, University of Bath, United KingdomYosra A. Helmy, The Ohio State University, United States
Copyright © 2020 Kelley, Ellis, Large, Schneider, Jacobson and Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremiah G. Johnson, ampvaG4zNThAdXRrLmVkdQ==
†These authors have contributed equally to this work