 Dandan Song
Dandan Song Zhou Jiang
Zhou Jiang Teng Ma1,2
Teng Ma1,2 Yiran Dong
Yiran Dong Liang Shi
Liang Shi- 1School of Environmental Studies, China University of Geosciences, Wuhan, China
- 2State Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences, Wuhan, China
Clay layers are common in subsurface where microbial activities play an important role in impacting the biogeochemical properties of adjacent aquifers. In this study, we analyzed the community structure and abundance of bacteria and archaea in response to geochemical properties of six clay sediments at different depths in a borehole (112°34′0″E, 30°36′21″N) of Jianghan Plain (JHP), China. Our results suggested that the top two clay layers were oxic, while the remaining bottom four clay layers were anoxic. Both high-throughput sequencing and qPCR of 16S rRNA gene showed relatively high abundance of archaea (up to 60%) in three of the anoxic clay layers. Furthermore, microbial communities in these clay sediments showed distinct vertical stratification, which may be impacted by changes in concentrations of sulfate, HCl-extractable Fe2+ and total organic carbon (TOC) in the sediments. In the upper two oxic clay layers, identification of phyla Thaumarchaeota (11.2%) and Nitrosporales (1.2%) implied nitrification in these layers. In the two anoxic clay layers beneath the oxic zone, high abundances of Anaeromyxobacter, Chloroflexi bacterium RBG 16_58_14 and Deltaproteobacteria, suggested the reductions of nitrate, iron and sulfate. Remarkably, a significant portion of Bathyarchaeota (∼25%) inhabited in the bottom two anoxic clay layers, which may indicate archaeal anaerobic degradation of TOC by these organisms. The results of this study provide the first systematic understandings of microbial activities in subsurface clay layers at JHP, which may help develop microorganism-based solutions for mitigating subsurface contaminations.
Introduction
Clays and clay minerals are abundant in soils, sediments, and subsurface (Dong et al., 2009; Dong, 2012). In subsurface, clays and clay minerals are often found in the sediment layers with different depths, where they impact microbial activities substantially. For example, iron (Fe)-rich clays and clay minerals function as electron donors and/or acceptors to support microbial growth (for reviews, see Dong et al., 2009; Dong, 2012; Shi et al., 2016). The clays rich in aluminum oxides (Al2O3), however, inhibit the growth of sulfate-reducing bacteria (SRB) (Wong et al., 2004). This is consistent with the observations that microbial sulfate reduction activity in subsurface clay layers is lower compared to that in the adjacent sand layers (McMahon and Chapelle, 1991; Ulrich et al., 1998). It was also suggested that microorganisms in the subsurface clay layers converted complex organic matters into low-molecular-mass fatty acids and hydrogen gas (H2) via fermentation. These fermentation products diffused into the surrounding environments (e.g., aquifers) where they were used by SRB as electron donors to reduce sulfate (McMahon and Chapelle, 1991; McMahon et al., 1992; Krumholz et al., 1997; Fredrickson and Balkwill, 2006). Furthermore, recent results demonstrated that dissolved organic carbon diffused from the adjacent clay layer activated the microbial activity in aquifers, which increased the arsenic (As) level in the groundwater of aquifers (Mihajlov et al., 2020). Thus, microorganisms in the subsurface clay layers impact the geochemical properties of not only the clay layers, but also their surrounding environment, such as the groundwater of aquifers. Finally, microbial activity in subsurface clay layers also influences the diagenesis of subsurface sediments, such as aquitard (McMahon et al., 1992; Hesse and Schacht, 2011).
In addition to Fe(III)-reducing, Fe(II)-oxidizing and fermentation microorganisms, other microorganisms have been identified from subsurface clay layers. Bacteria related to Proteobacteria and Firmicutes were found in a clay layer that was 224 m deep (Boivin-Jahns et al., 1996). Analyses of different clay layers of coastal subseafloor sediments from Okhotsk sea identified a variety of bacteria and archaea, such as green non-sulfur bacteria and deep sea archaeal group (Inagaki et al., 2003). Different groups of bacteria were found in the Opalinus clay rock from Mont Terri, Switzerland and Kisameet Glacial clay from British Columbia, Canada, whose functions range from CO2 fixation to sulfate reduction (Bagnoud et al., 2016a, b; Svensson et al., 2017). Thus, a phylogenetically and physiologically diverse groups of bacteria and archaea are associated with subsurface clay layers.
Jianghan Plain (JHP) is a semi-closed Quaternary basin in Hubei province, China, which was formed by the alluvial sediments of the Yangtze River and Han River (Gan et al., 2014). Because of intensive human activities, the aquifer systems of JHP has been polluted with different contaminants, such as ammonium (Du et al., 2017), As (Gan et al., 2014), antibiotics (Tong et al., 2014), and phthalate esters (Liu et al., 2010), which pose substantial health risks to the local residents. Thus, the JHP subsurface has been subjected to rigorous investigation in order to find the science-based solutions for mitigating the risks. For example, microbiological investigation of the core samples from the shallow subsurface of JHP revealed that the subsurface bacterial communities and functional groups were influenced by a variety of environmental factors, such as pH, Fe content, and NH4+. Results also showed existence of unique dissimilatory arsenate-reducing bacterial communities in the JHP sediments (Lu et al., 2017). Further analyses suggested that Fe(III)-reducing and Fe(II)-oxidizing bacteria play crucial roles in controlling As dynamic in the shallow sediments of JHP (Deng et al., 2018; Zheng et al., 2019). Moreover, up to 192 bacteria species with pathogenic potential were identified from 156 samples collected from JHP groundwater (Wu et al., 2019). Geochemical analyses also revealed that the shallow aquifer system (<50 m below ground surface) in JHP consisted of multiple layers rich in clays (Deng et al., 2018; Gan et al., 2018). However, the bacterial and archaeal communities in these clay layers had never been systematically investigated.
In this study, we used high-throughput sequencing and q-PCR of 16S rRNA gene to investigate the bacterial and archaeal community structure and abundance of six clay sediments at different depths in a borehole from JHP shallow subsurface. We further analyzed vertical distribution characteristics of microbial community structure and functional potentials and determined environmental factors that significantly impact the microbial community structure.
Materials and Methods
Site Description and Sampling
The sampling site (112°34′0″E, 30°36′21″N) was near a paddy field at Guangmang village, Shayang county, Jingmen city, Hubei province, China (Figure 1). This sampling site is located in the northern part of JHP and lies on the western bank of Hang River. A borehole with a depth of 30 m was drilled in September, 2017 using direct-mud rotary drilling technique. Based on the lithological characters in this borehole, six representative clay sediments samples were collected from the depths of 3, 5, 7, 9, 12, and 26 m and were placed in sterile anaerobic bag (Lu et al., 2017). All the samples were stored and transported on dry ice in the field. The samples along the depth were herein designated A1, A2, A3, A4, A5, and A6, respectively.
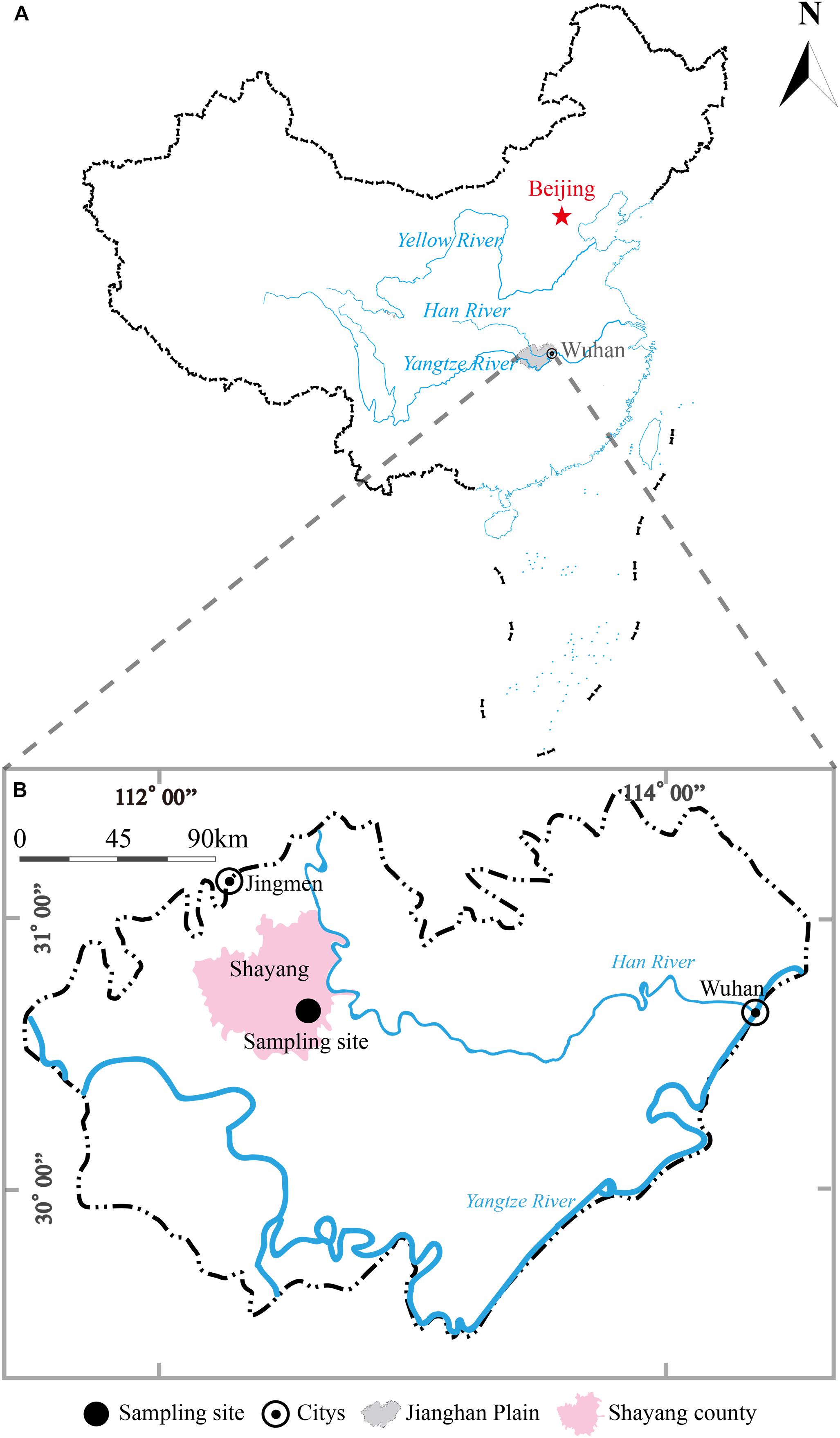
Figure 1. A geographic map showing locations of Jianghan Plain, China (A) and borehole site for clay sediment sampling in Jianghan Plain (B). The shadow area in map (A) refers to the boundary of Jianghan Plain.
Geochemical Analyses
In the laboratory, the external portions of the sediment cores that may be contaminated and oxidized during sampling were carefully removed in an anaerobic chamber filled with 97% N2 and 3% H2 (Coy Laboratory Products, MI, United States) (Lu et al., 2017). Only the internal parts were used for further analyses. Each sediment sample was homogenized and split into two portions to perform geochemical measurements and DNA extraction, respectively. Prior to chemical analysis, sediment samples were freeze-dried, homogenized and filtered through a 2.0 mm sieve. The pH value was analyzed using a Delta 320 pH Analyzer (Mettler Toledo, OH, United States) in a suspension of 5 g sediment and 25 mL 0.01 M calcium chloride. Concentrations of anion were measured by ion chromatograph (Dionex Aquion, Thermo Fisher Scientific, MA, United States) equipped with an ion exchange column (Dionex IonPac AS22 IC, Thermo Fisher Scientific, MA, United States) after extraction with ddH2O (a sediment-to-water ratio of 1:3). HCl-extractable Fe2+ was determined using the Ferrozine-based assay (Islam et al., 2004). Total Fe (FeTot), Mn (MnTot), and As (AsTot) concentrations in the sediments were extracted by aqua regia and measured by inductively coupled plasma optical emission spectrometer (ICP-OES, Avio 200, PerkinElmer, MA, United States) and atomic fluorescence spectrometry (AFS-9780, Haiguang, Beijing, China), respectively. Total organic carbon (TOC) was determined by a TOC analyzer (Vario TOC select, Elementar, Hesse, Germany). All the samples were run in triplicates.
DNA Extraction, 16S rRNA Gene Amplification and High-Throughput Sequencing
Genomic DNA in samples were extracted using DNeasy PowerMax Soil Kit (Qiagen, Shanghai, China) following the manufacturer’s instructions. DNA concentrations and quality were determined by a Nanodrop One UV-Vis Spectrophotometer (Thermo Fisher Scientific, MA, United States). The V4 region of bacteria and archaeal 16S rRNA gene was amplified using the primer 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) combined with Illumina adapter sequences, a pad and a linker, as well as barcodes on the reverse primers (Jiang et al., 2016). PCR amplification was carried out in a 50 μL reaction buffer containing 25 μL Premix Ex Taq (TaKaRa Biotechnology, Dalian, China), 0.5 μM of both forward and reverse primers, 60 ng DNA template using the following program: 5 min at 94°C for initialization; 30 cycles of 30 s denaturation at 94°C, 30 s annealing at 52°C, and 30 s extension at 72°C; 10 min final elongation at 72°C. Triplicate PCR reactions were performed per sample and pooled together. After confirmed by agarose gel electrophoresis, PCR products were mixed in equidensity ratios according to the GeneTools Analysis Software (Version 4.03.05.0, SynGene, Karnataka, India) and then purified with EZNA Gel Extraction Kit (Omega Bio-tek, GA, United States) (Chen et al., 2020). Sequencing libraries were prepared using NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, MA, United States) following the manufacturer’s recommendations. The library was sequenced on an Illumina Hiseq 2500 platform (Guangdong Magigene Biotechnology, Guangzhou, China) and 250 bp paired-end reads were generated. Raw sequencing read of 16S rRNA gene in the study were deposited in the NCBI Sequence Read Archive under the accession number SRP216165.
Sequencing Data Processing and Statistical Analyses
Raw sequence read of 16S rRNA with perfect matches to barcodes were split to sample libraries. Forward and reverse reads with at least 30 bp overlap and less than 25% mismatches were joined using Fast Length Adjustment of SHort reads (FLASH) (Magoc and Salzberg, 2011). The joined sequences were trimmed using Btrim with a QC threshold of greater than 30 over a 5 bp window size and a minimum length of 150 bp (Kong, 2011). After trimming the sequences containing N base and sequences with the lengths of <248 or >255 bp, the clean sequences were clustered to operational taxonomic units (OTUs) by Uparse at a similarity level of 97% (Edgar, 2013). For each representative sequence in OTUs, the silva1 database (SSU r132) was used to annotate taxonomic information at a minimal 50% confidence. Singletons in generated OTU tables were removed and then samples were rarefied at 42,887 sequences per sample based on the least number of sequences in the samples.
All statistical analyses in the study were performed with the Vegan and Stats packages in R2 (Jiang et al., 2016). Alpha diversity indices, such as Chao1, Shannon and equitability, and library coverage were calculated. Correlation analysis was used to evaluate the relationship between geochemical variables and diversity indices as well as microbial community compositions. The differences of microbial community composition among samples were determined using principal coordinates analysis (PCoA) (Wang et al., 2016). Three complimentary non-parametric multivariate statistical tests including adonis, ANOISM, and MRPP was performed to test the significance of microbial community differences among different groups (Jiang et al., 2016). Canonical correspondence analysis (CCA) was performed to elucidate the relationship between microbial community composition and geochemical variables. Geochemical variables were chosen for CCA analysis based on their significance calculated from individual CCA results and variance inflation factors (VIFs) calculated during CCA (Van Nostrand et al., 2009). Partial CCA (pCCA) was used to partition variation observed in microbial community composition to these selected geochemical variables (Lu et al., 2012b).
Quantitative Polymerase Chain Reaction
The universal primers bac331F (5′-TCCTACGGGAGGCAGC AGT-3′)-bac797R (5′-GGACTACCA GGGTCTAATCCT GTT-3′) and arch349F (5′-GYGCASCAGKCGMGAAW-3′)- arch 806R (5′-GGACTACVSGGGTATCTAAT-3′) were employed for quantification of bacterial and archaeal 16S rRNA gene, respectively. The bacterial 16S rRNA gene from Shewanella surugensis strain c959 and archaeal 16S rRNA gene from Methanospirillum sp. clone sagar113 were synthesized (Lin et al., 2012b) and cloned into pUC57-Kan as the quantitative polymerase chain reaction (qPCR) standards. PCR amplification was carried out in a 20 μL reaction buffer containing10 μL PowerUp SYBR Green Master Mix (Applied Biosystems, Thermo Fisher Scientific, MA, United States), 0.5 μM of both forward and reverse primers, 60 ng DNA template using the following program: 2 min at 95°C for initial denaturation; 40 cycles of 15 s denaturation at 95°C, 60 s annealing/extension at 60°C. All reactions were performed in triplicates. The melting curve analysis was performed to verify reaction specificity. The correlation coefficients (R2) of linear plots between the Ct value and log (copy numbers/reaction) for bacterial and archaeal 16S rRNA genes were 0.995 and 0.992, respectively. The qPCR amplification efficiencies were in the range of 92–103%.
Results and Discussion
Sediment Geochemistry
The grain size of sediments along the borehole generally ranged from clay to silt to fine-medium sand. The sediment color shifted dramatically from yellow-brown in upper layer (0–6 m) to gray in lower layer (6–30 m) (Figure 2). The physical properties of sediments in the borehole implied that an interface of vadose and saturated zone may occur near 6 m below the ground, which is very close to water level in JHP reported by Gan et al. (2018). Distinct geochemical characteristics occurred among upper layer samples (A1 and A2), middle layer samples (A3 and A4) and bottom layer samples (A5 and A6) (Figure 2). Sediment pH values were slightly acid, ranging from 6.55 at sample A4 to 6.92 at sample A2 (Figure 2). Nitrate and sulfate concentrations displayed a similar vertical distribution. The highest levels of nitrate and sulfate were found at sample A1 and A2, while the lowest were at sample A3 and A4 (Figure 2). TOC contents ranged from 1.54 mg/g (sample A4) to 16.31 mg/g (sample A5). High levels of Fe, Mn, and As were detected in all the samples, which is comparable to those reported before (Gan et al., 2014). The sample A3 contained the highest level of Fe, Mn, and As, while the A4 contained the lowest. Samples A1 and A2 contained few HCl-extractable Fe2+, which was detected in all other samples and their levels were much lower than that of total Fe detected. Similar to that for total Fe and Mn, the highest level of HCl-extractable Fe2+ was detected in sample A3 (Figure 2). Compared to that in samples A1 and A2, sample A3 contained much lower levels of nitrate and sulfate and higher level of Fe2+. These results suggest a change from microbial aerobic respiration of O2 in samples A1 and A2 to anaerobic respiration of nitrate, sulfate and Fe2+ at sample A3 as well as an oxic to anoxic transition zone between the depths of samples A2 and A3.
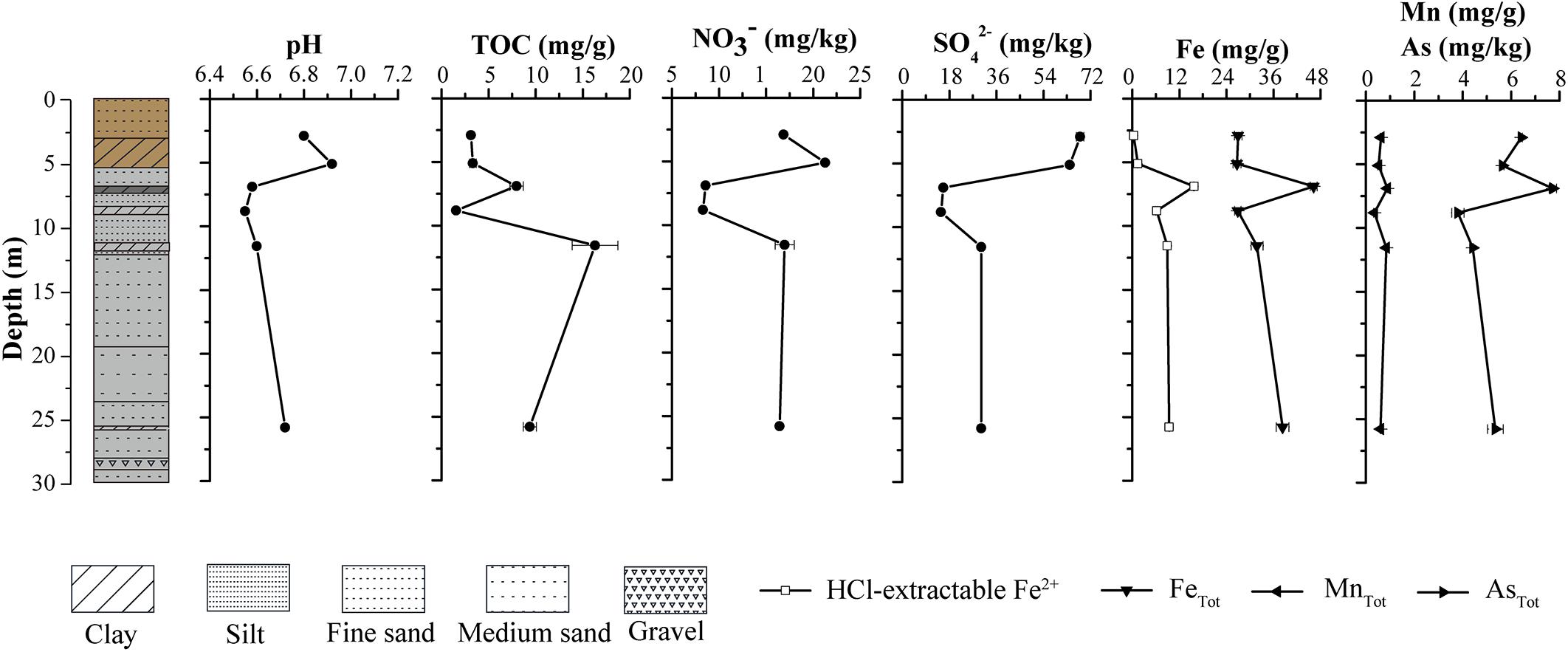
Figure 2. Vertical lithological profile of the borehole (left part) and physicochemical properties of sediment samples at different depths from the borehole (right part). The lithologies in the borehole were represented with the colors and textures defined for sediments at corresponding depths. The boxes represent core textures, which include clay, silt, fine sand, medium sand, and gravel. The colors depict the physical appearance of the core. The top portion of the core that includes A1 and A2 clay layer appears yellow-brown, while the remaining portion appears gray.
Alpha Diversity of Microbial Community and Abundance of Bacteria and Archaea
A total of 328,827 sequences were originally obtained from six sediment samples. After rarefaction at 42,887 sequences per sample, 257,322 sequences remained. A large number of taxa were detected in those samples, with 1,990–3,848 observed OTUs and 2,492–4,250 predicted OTUs (Chao1) (Supplementary Table 1). Library coverage of those samples ranged from 78.95 to 91.02%. Alpha diversity indices including richness, Shannon diversity and equitability generally declined along the borehole (Supplementary Figure 1). Moreover, Shannon diversity displayed a significantly negative relationship with depth (r = −0.829, p = 0.042). qPCR results showed the abundance of total 16S rRNA gene in the samples ranged from 9.26 × 105 to 5.96 × 107 copies/g sediment. The samples A3–A6 had approximately 10-fold less than the samples A1–A2 in 16S rRNA gene copies (Figure 3). The concurrent decrease of alpha diversity indices and 16S rRNA gene abundance along the borehole are most likely attributed to change of redox condition. This is consistent with geochemical data that suggest an oxic to anoxic transition between the depths of sample A2 and A3. Previous results also demonstrated that compared to that in the oxic zone of a terrestrial subsurface, microbial population was much lower in the anoxic zone (Lin et al., 2012a). Remarkably, archaea in these sediments constituted a far larger proportion of the whole microbial community (35.5% on average), as compared to other borehole sediments in aquifer system (Lin et al., 2012a). In contrast to that (5.4–9.4%) in the samples A1, A2, and A4, the relative abundances of archaea in samples A3, A5, and A6 were up to ∼60%, which suggests a dominant role of archaea in regulating biogeochemical reactions in these subsurface clay layers (Castelle et al., 2015; Anantharaman et al., 2016). A significantly positive correlation between archaeal relative abundance and TOC content (r = 0.846, p = 0.034) implied that extremely low TOC content in sample A4 (Figure 2) might lead to dramatically drop of archaeal relative abundance.
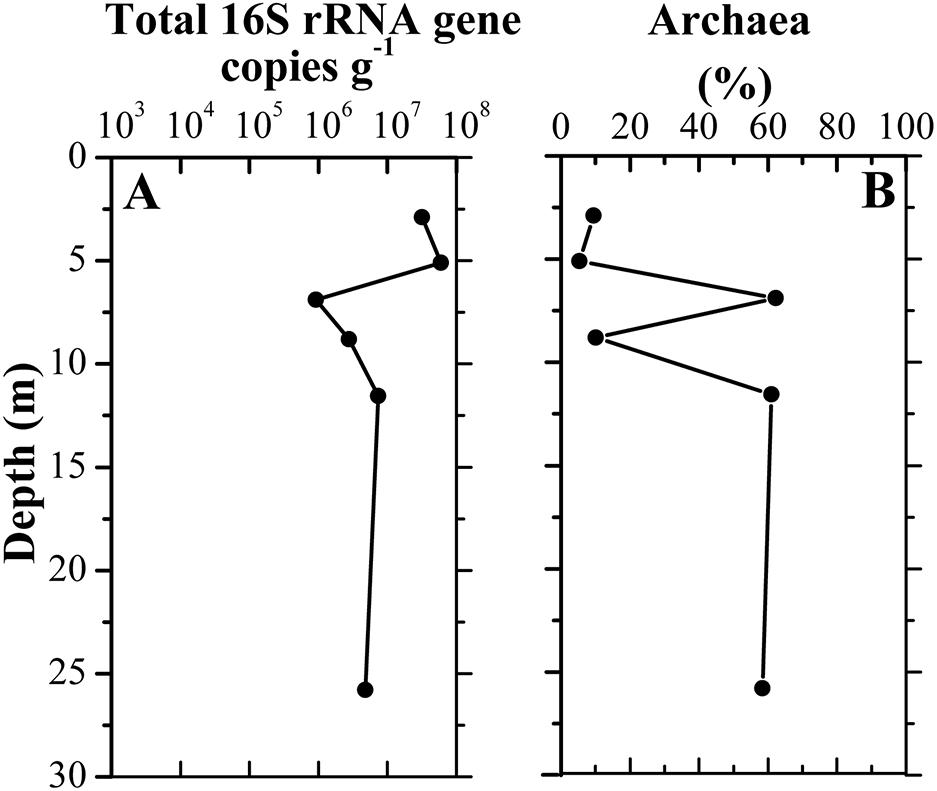
Figure 3. Vertical distribution of total 16S rRNA gene copy numbers (A) in clay sediments with different depths and the relative abundance of archaeal copy numbers to total 16S rRNA gene copy numbers (B).
Microbial Community Composition and Function
The microbial community in all six clay sediments generally comprised 67.5% bacterial phyla, 29.5% archaeal phyla, and 3% unclassified phyla (Figure 4A). The dominant phyla in bacteria mainly included Chloroflexi (17.5%), Proteobacteria (11.7%), Acidobacteria (7.1%), Planctomycetes (4.0%), Actinobacteria (3.7%), Patescibacteria (3.2%), Elusimicrobia (2.9%), NC10 (2.6%), and Firmicutes (2.3%), with respect to Bathyarchaeota (16.2%), Euryarchaeota (6.1%), Thaumarchaeota (3.9%), and Woesearchaeota (2.3%) being abundant in archaea (Figure 4A). The presence of 3% unclassified phyla in this study implied that subsurface clay layers in JHP maybe also nourish many novel bacterial or archaeal organisms as same as the aquifer adjacent to the Colorado River, near Rifle, CO, United States (Castelle et al., 2015; Anantharaman et al., 2016), which is deserved for the further investigation. The relative abundance of archaea revealed by 16S rRNA gene sequencing was comparable to that from q-PCR results, which highlights the importance of archaea in those samples. Microbial community composition in JHP subsurface clay layers is very similar to the aquifer adjacent to the Colorado River, both of which have dominant Chloroflexi, Proteobacteria and some archaeal phyla (Castelle et al., 2013; Hug et al., 2013). Vertically, NC10, Euryarchaeota, Thaumarchaeota, and Woesearchaeota were mainly found in samples A1 and A2, whereas Bathyarchaeota and Elusimicrobia dominate in samples A5 and A6. Besides, the abundances of Bathyarchaeota and Elusimicrobia in clay sediments had a significantly positive correlation with depth (Supplementary Table 2). Chloroflexi and Proteobacteria were predominant in the sample A3. At the class level, microbial community composition was more diverse and even, and about 12.2% sequences were not classified to known classes, suggesting the presence of novel lineages in these samples (Figure 4B). Compared to the relatively high proportions of Nitrososphaeria, Thermoplasmata, and Methylomirabilales in the upper samples A1 and A2, the bottom sediments from A5 and A6 were characterized by the presence of Dehalococcoidia of Chloroflexi, 4–29 of Elusimicrobia, Subgroup 6 and 12 of Bathyarchaeota. In addition, sample A3 was dominated by Anaerolineae and Deltaproteobacteria as well as subgroup 11 of Bathyarchaeota. Based on all OTUs detected, both PCoA and CCA ordination analyses showed that the microbial communities were similar between samples A1 and A2, between samples A3 and A4, and between samples A5 and A6, respectively, whereas microbial communities among the three groups (A1/A2, A3/A4, and A5/A6) were far apart (Figure 5A and Supplementary Figure 2), suggesting a distinct difference of microbial community among these three different zones of clay layers. Furthermore, the difference of microbial community among upper layer samples (A1 and A2), middle layer samples (A3 and A4) and bottom layer samples (A5 and A6) was tested to be significant, revealed by Adonis analysis (Supplementary Table 3). All of these results demonstrated the unique microbial communities resided in different subsurface clay layers of JHP.
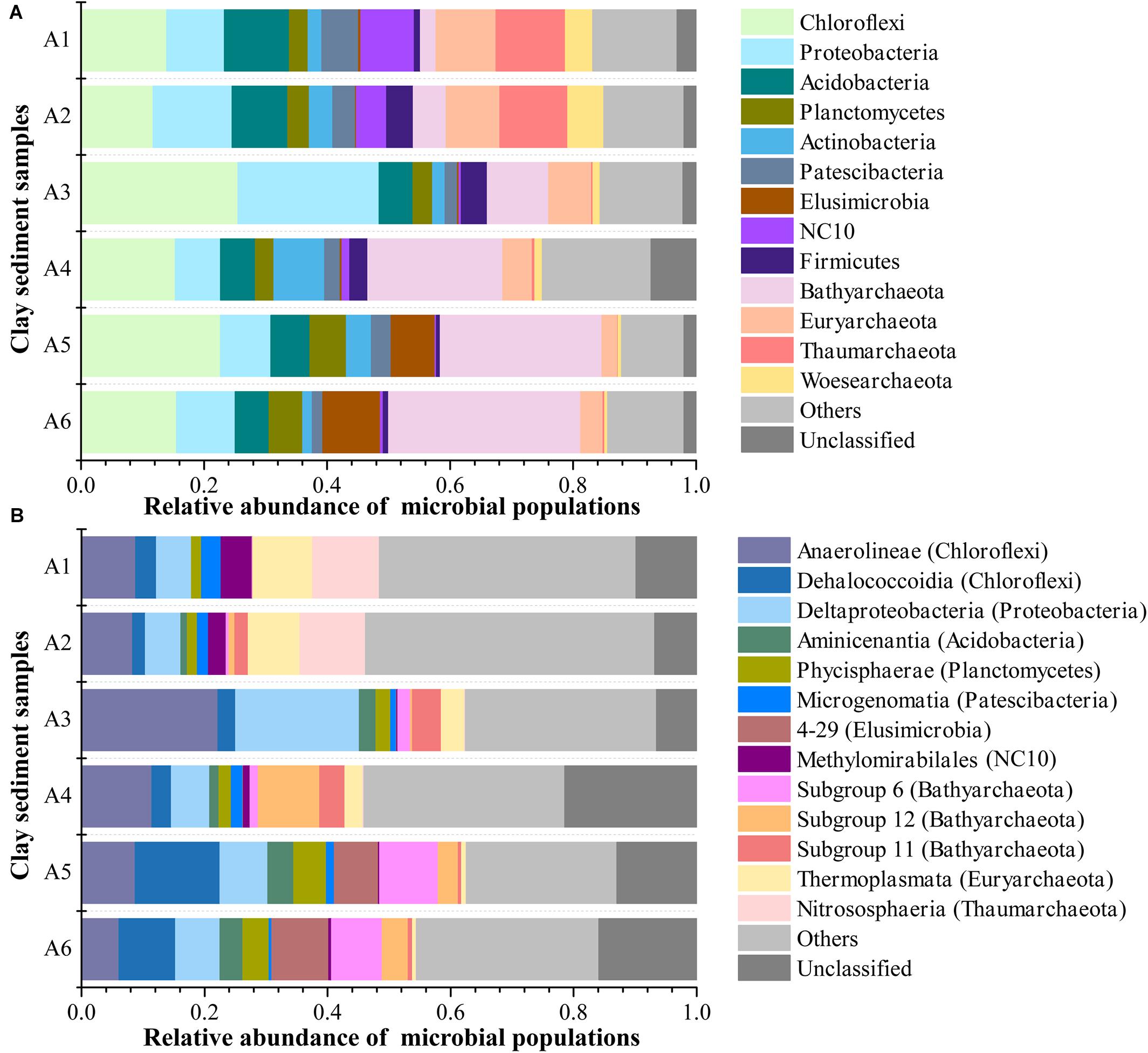
Figure 4. Microbial community structure of sediment samples at the (A) phylum- and (B) class-level. Only top 13 phyla and classes in abundance were displayed, the rest phyla and classes were summed and assigned to others.
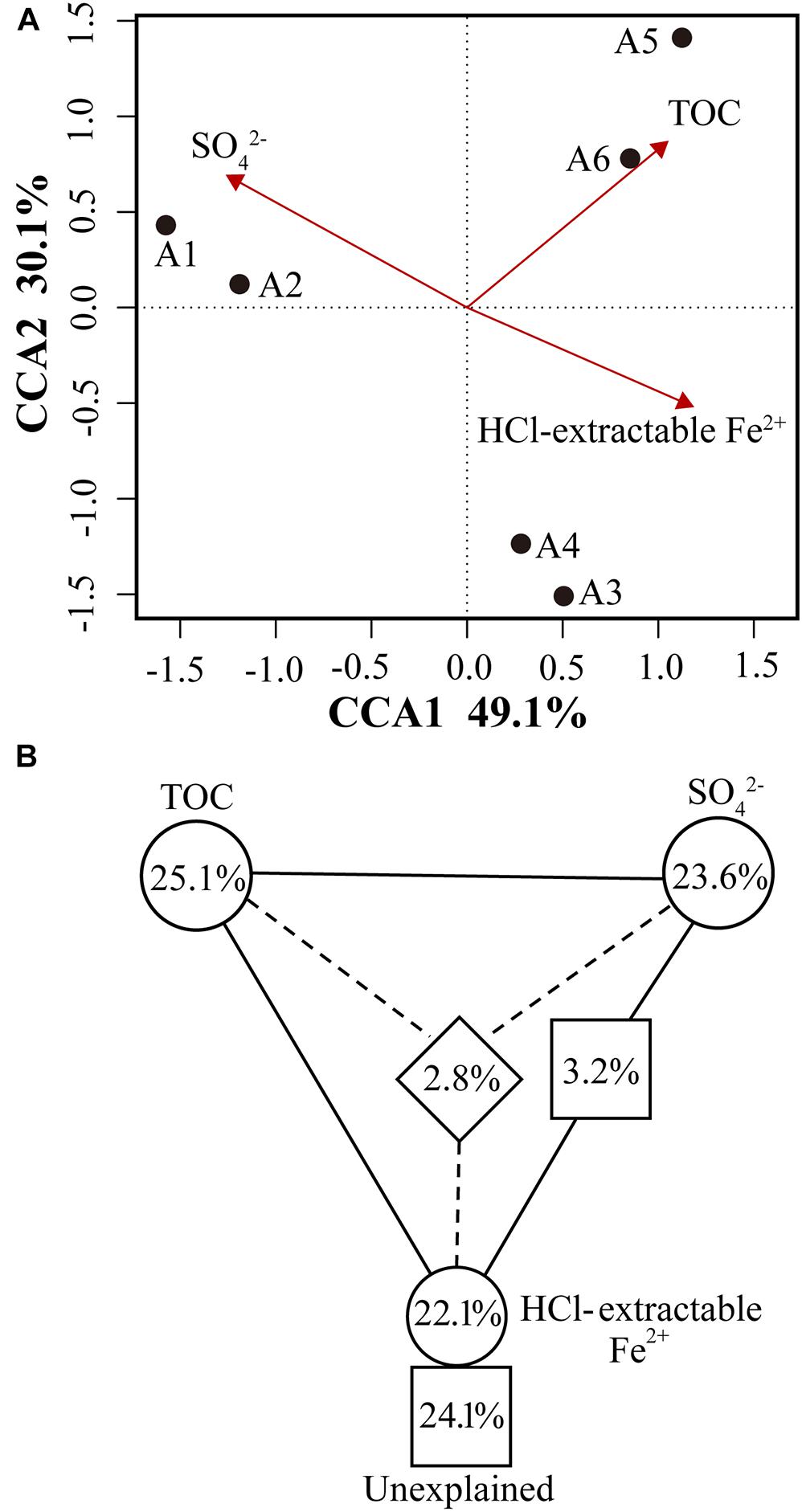
Figure 5. (A) Canonical correspondence analysis (CCA) of microbial community composition and geochemical variables (TOC, sulfate, HCl-extractable Fe2+). Geochemical variables were chosen based on significance calculated from individual CCA results and VIFs calculated during CCA. The percentage of variation explained by each axis is shown, and the relationship is significant (p = 0.014). (B) Variation partitioning of microbial community composition respond to significant geochemical variables.
Consistent with geochemical characteristics in the sediment samples, the function potentials of microbial community also displayed a distinct distribution along the depth. In the clay sediments of A1 and A2, nitrification was identified. Ammonia-oxidizing archaea (AOA) might outcompete ammonia-oxidizing bacteria (AOB) and contribute to ammonia oxidation in upper clay sediments, as indicated by high abundance of Thaumarchaeota (Nitrosopumilales and Nitrososphaerales) relative to these representative AOB (Supplementary Figure 3; Herber et al., 2020). Nitrite produced by AOA could serve as substrate for nitrite-oxidizing bacteria (NOB), such as Nitrosporales, leading to nitrate accumulation in upper samples (Figure 2; Daims et al., 2016). AOA and NOB abundance significantly positively correlated to sulfate concentration and negatively correlated to HCl-extractable Fe2+ concentration, which further suggested the oxic condition in samples A1 and A2 (Supplementary Figure 2 and Supplementary Table 2). In addition, Woesearchaeota were mainly found in upper sediment samples from the borehole (Supplementary Figure 3), which coincide with its wide ecological niches ranging from oxic to anoxic biotopes and might provide a series of intermediates for other aerobes and anaerobes due to reported heterotrophic and syntrophic lifestyle (Castelle et al., 2015; Liu et al., 2018). The sediments of A3 and A4 were characterized by the significant portion of microbial populations associated with reductions of nitrate (Anaeromyxobacter and nitrate-dependent methanotrophic Methanoperedens-like archaea), Fe (Anaeromyxobacter and Chloroflexi bacterium RBG_16_58_14), and sulfate (Deltaproteobacteria) (Supplementary Figure 3; He and Sanford, 2003; Muyzer and Stams, 2008; Anantharaman et al., 2016; Welte et al., 2016; Bell et al., 2020). This is consistent with observations of low levels of nitrate and sulfate, but high level of HCl-extractable Fe2+ in the samples A3 and A4, which further support the idea of an oxic to anoxic transition between the sediments of A2 and A3 (Figure 2). These microbial groups may use TOC as energy and electron sources to reduce nitrate, Fe, and sulfate. More importantly, reduction of iron and sulfate causes As mobilization in JHP groundwater system (Deng et al., 2018; Zheng et al., 2019). In aquifers, As is mainly adsorbed to solid-phase iron minerals (Fendorf et al., 2010). Indeed, our results also showed that amounts of AsTot detected correlated with those of FeTot and HCl-extractable Fe2+, which are all detected in the highest amounts in sample A3 (Figure 2). These iron-reducing bacteria and sulfide produced by SRB in sample A3 most likely reductively dissolve the iron minerals adsorbed with As, which could release adsorbed As into the pore water of clay sediments. It has documented that the pore water containing dissolved As and As-mobilizing solutes, such as dissolved organic carbon and competing ions, are expelled to the underlying aquifer with extensive groundwater pumping, thus making As contamination in aquifers even worse (Erban et al., 2013; Mihajlov et al., 2020; Xiao et al., 2020). Methanogens, such as Methanobacteriales, Methanocellales, and Methanosarcinales, were also relatively abundant in A3 and A4 sediments, where methane might be oxidized by methanotrophic microorganisms by coupling with nitrate reduction (Supplementary Figure 3). Significantly different with A1–A4 sediments, a substantial portion of Bathyarchaeota (∼25%) consisting of mainly subgroup 6 and subgroup 12 were identified in the bottom A5 and A6 clay layers. Given the common acetyl-CoA-centralized heterotrophic lifestyle shared by all Bathyarchaeota subgroups (Zhou et al., 2018; Feng et al., 2019), the Bathyarchaeota in these sediments could use organic substrates to grow. This was supported by the significantly positive correlation between Bathyarchaeota subgroup 6 abundance and TOC content (Supplementary Table 2). The metabolites produced by Bathyarchaeota might be used by other methanogens, such as members from Methanofastidiosales and Crenarchaeote enrichment culture clone_61-15f detected in the same layer sediments, to produce methane (Supplementary Figure 3; Pavlova et al., 2014; Evans et al., 2019). Furthermore, Bathyarchaeota itself in the bottom clay layers might also have a potential of methanogenesis, which was supported by methane metabolism found in Bathyarchaeota genomes (BA1 and BA2) recovered from a deep aquifer (Evans et al., 2015; Lloyd, 2015). Elusimicrobia, another microbial population appearing in bottom samples A5 and A6 (Figure 4), might contribute to various substrates for methanogens by sugar fermentation to acetate, malate and butyrate, suggested by recent reconstructed Elusimicrobia genomes analysis from groundwater and other natural environments (Meheust et al., 2020). Beyond methanogenesis, these fermentation products could also to be used by the members in class Dehalococcoidia residing in bottom samples A5 and A6 (Figure 4B) to respire the organohalide, a widespread recalcitrant pollutant in groundwater systems (Yang et al., 2020).
Relationship Between Microbial Community Structure and Sediment Geochemistry
Canonical correspondence analysis was used to determine the most significant geochemical variables to shape the microbial community structure of samples. On the basis of significance (p < 0.05) calculated from individual CCA results and VIFs calculated during CCA (Van Nostrand et al., 2009), TOC, HCl-extractable Fe2+ and sulfate were chosen from all geochemical variables to perform CCA. The specified CCA model was significant (p = 0.014) and described 79.2% of the total variation (the first axis explained 47.3% and the second axis explained 14.3%) (Figure 5A). The microbial community structure in sediments from A1–A2, A3–A4, and A5–A6 appeared to be strongly affected by the concentrations of sulfate, HCl-extractable Fe2+ and TOC, respectively, as indicated by their proximity to those arrows in Figure 5A. To further assess the contribution of above three variables to microbial community structure, variation partitioning analysis was performed (Lu et al., 2012a). Three variables explained a large portion of the variation observed, leaving 24.1% of the variation unexplained by these factors. HCl-extractable Fe2+ alone accounted for 22.1% (p = 0.019), 23.5% of the variation was attributed to sulfate (p = 0.026) and TOC explained the largest amount of variation, 25.1% (p = 0.013) (Figure 5B). Interactions between and among variables only explained <3.5% of variation. These results suggest that sulfate, HCl-extractable Fe2+ and TOC significantly influence the microbial community structures of subsurface clay layers in JHP, which was also reflected by functional potentials of microbial populations.
Conclusion
Microbial community composition residing in different subsurface clay layers of JHP generally consisted of 67.5% bacterial phyla, 29.5% archaeal phyla, and 3% unclassified phyla. Geochemical and microbial characterizations suggest that the top two clay layers are oxic and the bottom four clay layers are anoxic. High abundance of archaea (up to 60%) were observed in three of the anoxic clay layers. The concentrations of sulfate, HCl-extractable Fe2+ and total organic carbon significantly shaped the microbial community structure in different clay layers of subsurface. A distinctly vertical stratification of microbial communities in clay sediments was evident along the borehole. In the upper two oxic clay layers, nitrification, nitrite-dependent methane oxidation, and ammonium oxidation may occur. Reduction of nitrate, iron and sulfate were mainly found in the two anoxic clay layers beneath the oxic zone. The bottom two anoxic clay layers were dominated by archaeal anaerobic degradation of TOC and potential methanogenesis. Additionally, large amounts of unclassified sequences suggested that microorganisms with novel lineages might inhabit in the subsurface clay layers of JHP. These results for the first provide comprehensive insights into bacterial and archaeal community structure and functional potentials in shallow subsurface clay layers of JHP, which will help develop the science-based solutions for mitigating the health risks associated with JHP subsurface.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
LS conceived and designed the experiments. DS conducted the field sampling and laboratory experiments. DS and ZJ analyzed the data and wrote the draft manuscript. All authors contributed to the revisions of the manuscript and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC41630318, 91851211, 41772363), One Hundred Talents Program of Hubei Province, and the Fundamental Research Funds for the Universities of Chinese Central Government, China University of Geosciences, Wuhan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.572560/full#supplementary-material
Footnotes
References
Anantharaman, K., Brown, C. T., Hug, L. A., Sharon, I., Castelle, C. J., Probst, A. J., et al. (2016). Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 7:13219. doi: 10.1038/ncomms13219
Bagnoud, A., Chourey, K., Hettich, R. L., de Bruijn, I., Andersson, A. F., Leupin, O. X., et al. (2016a). Reconstructing a hydrogen-driven microbial metabolic network in Opalinus Clay rock. Nat. Commun. 7:12770. doi: 10.1038/ncomms12770
Bagnoud, A., de Bruijn, I., Andersson, A. F., Diomidis, N., Leupin, O. X., Schwyn, B., et al. (2016b). A minimalistic microbial food web in an excavated deep subsurface clay rock. FEMS Microbiol. Ecol. 92:fiv138. doi: 10.1093/femsec/fiv138
Bell, E., Lamminmaki, T., Alneberg, J., Andersson, A. F., Qian, C., Xiong, W., et al. (2020). Active sulfur cycling in the terrestrial deep subsurface. ISME J. 14, 1260–1272. doi: 10.1038/s41396-020-0602-x
Boivin-Jahns, V., Ruimy, R., Bianchi, A., Daumas, S., and Christen, R. (1996). Bacterial diversity in a deep-subsurface clay environment. Appl. Environ. Microbiol. 62, 3405–3412. doi: 10.1128/aem.62.9.3405-3412.1996
Castelle, C. J., Hug, L. A., Wrighton, K. C., Thomas, B. C., Williams, K. H., Wu, D., et al. (2013). Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat. Commun. 4:2120. doi: 10.1038/ncomms3120
Castelle, C. J., Wrighton, K. C., Thomas, B. C., Hug, L. A., Brown, C. T., Wilkins, M. J., et al. (2015). Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr. Biol. 25, 690–701. doi: 10.1016/j.cub.2015.01.014
Chen, X., Sun, H., Jiang, F., Shen, Y., Li, X., Hu, X., et al. (2020). Alteration of the gut microbiota associated with childhood obesity by 16S rRNA gene sequencing. PeerJ 8:e8317. doi: 10.7717/peerj.8317
Daims, H., Lucker, S., and Wagner, M. (2016). A new perspective on microbes formerly known as nitrite-oxidizing bacteria. Trends Microbiol. 24, 699–712. doi: 10.1016/j.tim.2016.05.004
Deng, Y., Zheng, T., Wang, Y., Liu, L., Jiang, H., and Ma, T. (2018). Effect of microbially mediated iron mineral transformation on temporal variation of arsenic in the Pleistocene aquifers of the central Yangtze River basin. Sci. Total Environ. 619-620, 1247–1258. doi: 10.1016/j.scitotenv.2017.11.166
Dong, H. (2012). Clay-microbe interactions and implications for environmental mitigation. Elements 8, 95–100. doi: 10.2113/gselements.8.2.113
Dong, H., Jaisi, D. P., Kim, J., and Zhang, G. (2009). Microbe-clay mineral interactions. Am. Mineral. 94, 1505–1519. doi: 10.2138/am.2009.3246
Du, Y., Ma, T., Deng, Y., Shen, S., and Lu, Z. (2017). Sources and fate of high levels of ammonium in surface water and shallow groundwater of the Jianghan Plain, central China. Environ. Sci. Process. Impacts 19, 161–172. doi: 10.1039/c6em00531d
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Erban, L. E., Gorelick, S. M., Zebker, H. A., and Fendorf, S. (2013). Release of arsenic to deep groundwater in the Mekong Delta, Vietnam, linked to pumping-induced land subsidence. Proc. Natl. Acad. Sci. U.S.A 110, 13751–13756. doi: 10.1073/pnas.1300503110
Evans, P. N., Boyd, J. A., Leu, A. O., Woodcroft, B. J., Parks, D. H., Hugenholtz, P., et al. (2019). An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol. 17, 219–232. doi: 10.1038/s41579-018-0136-7
Evans, P. N., Parks, D. H., Chadwick, G. L., Robbins, S. J., Orphan, V. J., Golding, S. D., et al. (2015). Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350, 434–438. doi: 10.1126/science.aac7745
Fendorf, S., Michael, H. A., and van Geen, A. (2010). Spatial and temporal variations of groundwater arsenic in South and Southeast Asia. Science 328, 1123–1127. doi: 10.1126/science.1172974
Feng, X., Wang, Y., Zubin, R., and Wang, F. (2019). Core metabolic features and hot origin of Bathyarchaeota. Engineering 5, 498–504. doi: 10.1016/j.eng.2019.01.011
Fredrickson, J. K., and Balkwill, D. L. (2006). Geomicrobial processes and biodiversity in the deep terrstrial subsurface. Geomicrobiol. J. 23, 345–356. doi: 10.1080/01490450600875571
Gan, Y., Wang, Y., Duan, Y., Deng, Y., Guo, X., and Ding, X. (2014). Hydrogeochemistry and arsenic contamination of groundwater in the Jianghan Plain, central China. J. Geochem. Explor. 138, 81–93. doi: 10.1016/j.gexplo.2013.12.013
Gan, Y., Zhao, K., Deng, Y., Liang, X., Ma, T., and Wang, Y. (2018). Groundwater flow and hydrogeochemical evolution in the Jianghan Plain, central China. Hydrogeol. J. 26, 1609–1623. doi: 10.1007/s10040-018-1778-2
He, Q., and Sanford, R. A. (2003). Characterization of Fe(III) reduction by chlororespiring Anaeromyxobacter dehalogenans. Appl. Environ. Microbiol. 69, 2712–2718. doi: 10.1128/aem.69.5.2712-2718.2003
Herber, J., Klotz, F., Frommeyer, B., Weis, S., Straile, D., Kolar, A., et al. (2020). A single Thaumarchaeon drives nitrification in deep oligotrophic lake constance. Environ. Microbiol. 22, 212–228. doi: 10.1111/1462-2920.14840
Hesse, R., and Schacht, U. (2011). “Early diagenesis of deep-sea sediments,” in Deep-Sea Sediments, eds H. HüNeke and T. Mulder (Amsterdam: Elsevier), 557–713. doi: 10.1016/b978-0-444-53000-4.00009-3
Hug, L. A., Castelle, C. J., Wrighton, K. C., Thomas, B. C., and Banfield, J. F. (2013). Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1, 22–22. doi: 10.1186/2049-2618-1-22
Inagaki, F., Suzuki, M., Takai, K., Oida, H., Sakamoto, T., Aoki, K., et al. (2003). Microbial communities associated with geological horizons in coastal subseafloor sediments from the sea of okhotsk. Appl. Environ. Microbiol. 69, 7224–7235. doi: 10.1128/aem.69.12.7224-7235.2003
Islam, F. S., Gault, A. G., Boothman, C., Polya, D. A., Charnock, J. M., Chatterjee, D., et al. (2004). Role of metal-reducing bacteria in arsenic release from Bengal delta sediments. Nature 430, 68–71. doi: 10.1038/nature02638
Jiang, Z., Li, P., Van Nostrand, J. D., Zhang, P., Zhou, J., Wang, Y., et al. (2016). Microbial communities and arsenic biogeochemistry at the outflow of an alkaline sulfide-rich hot spring. Sci. Rep. 6:25262. doi: 10.1038/srep25262
Kong, Y. (2011). Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98, 152–153. doi: 10.1016/j.ygeno.2011.05.009
Krumholz, L. R., McKinley, J. P., Ulrich, G. A., and Sulfita, J. W. (1997). Confined subsurface microbial communities in Cretaceous rock. Nature 386, 64–66. doi: 10.1038/386064a0
Lin, X., Kennedy, D., Fredrickson, J., Bjornstad, B., and Konopka, A. (2012a). Vertical stratification of subsurface microbial community composition across geological formations at the Hanford Site. Environ. Microbiol. 14, 414–425. doi: 10.1111/j.1462-2920.2011.02659.x
Lin, X., Kennedy, D., Peacock, A., McKinley, J., Resch, C. T., Fredrickson, J., et al. (2012b). Distribution of microbial biomass and potential for anaerobic respiration in Hanford Site 300 Area subsurface sediment. Appl. Environ. Microbiol. 78, 759–767. doi: 10.1128/AEM.07404-11
Liu, H., Liang, H., Liang, Y., Zhang, D., Wang, C., Cai, H., et al. (2010). Distribution of phthalate esters in alluvial sediment: a case study at JiangHan Plain, Central China. Chemosphere 78, 382–388. doi: 10.1016/j.chemosphere.2009.11.009
Liu, X., Li, M., Castelle, C. J., Probst, A. J., Zhou, Z., Pan, J., et al. (2018). Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome 6:102. doi: 10.1186/s40168-018-0488-2
Lu, X., Wang, N., Wang, H., Deng, Y., Ma, T., Wu, M., et al. (2017). Molecular characterization of the total bacteria and dissimilatory arsenate-Reducing bacteria in core sediments of the Jianghan Plain, Central China. Geomicrobiol. J. 34, 467–479. doi: 10.1080/01490451.2016.1222468
Lu, Z., Deng, Y., Van Nostrand, J. D., He, Z., Voordeckers, J., Zhou, A., et al. (2012a). Microbial gene functions enriched in the deepwater horizon deep-sea oil plume. ISME J. 6, 451–460. doi: 10.1038/ismej.2011.91
Lu, Z., He, Z., Parisi, V. A., Kang, S., Deng, Y., Van Nostrand, J. D., et al. (2012b). GeoChip-based analysis of microbial functional gene diversity in a landfill leachate-contaminated aquifer. Environ. Sci. Technol. 46, 5824–5833. doi: 10.1021/es300478j
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
McMahon, P. B., and Chapelle, F. H. (1991). Microbial productioin of organic acids in aquitard sediments and its role in aquifer geochemistry. Nature 349, 233–235. doi: 10.1038/349233a0
McMahon, P. B., Chapelle, F. H., Falls, W. F., and Bradley, P. M. (1992). Role of microbial processes in linking sandstone diagenesis with organic-rich clays. J. Sediment. Petrol. 62, 1–10. doi: 10.1306/d4267870-2b26-11d7-8648000102c1865d
Meheust, R., Castelle, C. J., Matheus Carnevali, P. B., Farag, I. F., He, C., and Chen, L. X. (2020). Groundwater Elusimicrobia are metabolically diverse compared to gut microbiome Elusimicrobia and some have a novel nitrogenase paralog. ISME J. [Epub ahead of print]. doi: 10.1038/s41396-020-0716-1
Mihajlov, I., Mozumder, M. R. H., Bostick, B. C., Stute, M., Mailloux, B. J., Knappett, P. S. K., et al. (2020). Arsenic contamination of Bangladesh aquifers exacerbated by clay layers. Nat. Commun. 11:2244. doi: 10.1038/s41467-020-16104-z
Muyzer, G., and Stams, A. J. (2008). The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 6, 441–454. doi: 10.1038/nrmicro1892
Pavlova, O. N., Bukin, S. V., Lomakina, A. V., Kalmychkov, G. V., Ivanov, V. G., Morozov, I. V., et al. (2014). Production of gaseous hydrocarbons by microbial communities of Lake Baikal bottom sediments. Microbiology 83, 798–804. doi: 10.1134/s0026261714060137
Shi, L., Dong, H., Reguera, G., Beyenal, H., Lu, A., Liu, J., et al. (2016). Extracellular electron transfer mechanisms between microorganisms and minerals. Nat. Rev. Microbiol. 14, 651–662. doi: 10.1038/nrmicro.2016.93
Svensson, S. L., Behroozian, S., Xu, W., Surette, M. G., Li, L., and Davies, J. (2017). Kisameet glacial clay: an unexpected source of bacterial diversity. mBio 8:e00590-17. doi: 10.1128/mBio.00590-17
Tong, L., Huang, S., Wang, Y., Liu, H., and Li, M. (2014). Occurrence of antibiotics in the aquatic environment of Jianghan Plain, central China. Sci. Total Environ. 497-498, 180–187. doi: 10.1016/j.scitotenv.2014.07.068
Ulrich, G. A., Martino, D., Burger, K., Routh, J., Grossman, E. L., Ammerman, J. W., et al. (1998). Sulfur cycling in the terrestrial subsurface: commensal interactions, spatial scales, and microbial heterogeneity. Microb. Ecol. 36, 141–151. doi: 10.1007/s002489900101
Van Nostrand, J. D., Wu, W. M., Wu, L., Deng, Y., Carley, J., Carroll, S., et al. (2009). GeoChip-based analysis of functional microbial communities during the reoxidation of a bioreduced uranium-contaminated aquifer. Environ. Microbiol. 11, 2611–2626. doi: 10.1111/j.1462-2920.2009.01986.x
Wang, Y., Li, P., Jiang, Z., Sinkkonen, A., Wang, S., Tu, J., et al. (2016). Microbial community of high arsenic groundwater in agricultural irrigation area of Hetao Plain, Inner Mongolia. Front. Microbiol. 7:1917. doi: 10.3389/fmicb.2016.01917
Welte, C. U., Rasigraf, O., Vaksmaa, A., Versantvoort, W., Arshad, A., Op, et al. (2016). Nitrate- and nitrite-dependent anaerobic oxidation of methane. Environ. Microbiol. Rep. 8, 941–955. doi: 10.1111/1758-2229.12487
Wong, D., Suflita, J. M., McKinley, J. P., and Krumholz, L. R. (2004). Impact of clay minerals on sulfate-reducing activity in aquifers. Microb. Ecol. 47, 80–86. doi: 10.1007/s00248-003-1021-z
Wu, G., Yang, J., Jiang, H., Deng, Y., and Lear, G. (2019). Distribution of potentially pathogenic bacteria in the groundwater of the Jianghan Plain, central China. Int. Biodeterior. Biodegrad. 143:104711. doi: 10.1016/j.ibiod.2019.05.028
Xiao, C., Ma, T., Du, Y., Liu, Y., Liu, R., and Zhang, D. (2020). Impact process of the aquitard to regional arsenic accumulation of the underlying aquifer in Central Yangtze River Basin. Environ. Geochem. Health [Epub ahead of print]. doi: 10.1007/s10653-020-00541-2
Yang, Y., Sanford, R., Yan, J., Chen, G., Capiro, N. L., Li, X., et al. (2020). Roles of organohalide-respiring Dehalococcoidia in carbon cycling. mSystems 5:e00757-19.
Zheng, T., Deng, Y., Wang, Y., Jiang, H., O’Loughlin, E. J., Flynn, T. M., et al. (2019). Seasonal microbial variation accounts for arsenic dynamics in shallow alluvial aquifer systems. J. Hazard. Mater. 367, 109–119. doi: 10.1016/j.jhazmat.2018.12.087
Keywords: bacteria, archaea, diversity and abundance, clay sediment, Jianghan Plain
Citation: Song D, Jiang Z, Ma T, Dong Y and Shi L (2020) Bacterial and Archaeal Diversity and Abundance in Shallow Subsurface Clay Sediments at Jianghan Plain, China. Front. Microbiol. 11:572560. doi: 10.3389/fmicb.2020.572560
Received: 14 June 2020; Accepted: 01 September 2020;
Published: 22 October 2020.
Edited by:
Fengping Wang, Shanghai Jiao Tong University, ChinaReviewed by:
Changyi Zhang, University of Illinois at Urbana–Champaign, United StatesWenyuan Han, Huazhong Agricultural University, China
Copyright © 2020 Song, Jiang, Ma, Dong and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhou Jiang, amlhbmd6aG91QGN1Zy5lZHUuY24=; Liang Shi, bGlhbmcuc2hpQGN1Zy5lZHUuY24=