 Monika Kopeckova
Monika Kopeckova Marek Link
Marek Link- Department of Molecular Pathology and Biology, Faculty of Military Health Sciences, University of Defence, Hradec Kralove, Czechia
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is well known for its involvement in numerous non-metabolic processes inside mammalian cells. Alternative functions of prokaryotic GAPDH are mainly deduced from its extracellular localization ability to bind to selected host proteins. Data on its participation in intracellular bacterial processes are scarce as there has been to date only one study dealing with this issue. We previously have reported several points of evidence that the GAPDH homolog of Francisella tularensis GapA might also exert additional non-enzymatic functions. Following on from our earlier observations we decided to identify GapA’s interacting partners within the bacterial proteome to explore its new roles at intracellular level. The quantitative proteomics approach based on stable isotope labeling of amino acids in cell culture (SILAC) in combination with affinity purification mass spectrometry enabled us to identify 18 proteins potentially interacting with GapA. Six of those interactions were further confirmed by alternative methods. Half of the identified proteins were involved in non-metabolic processes. Further analysis together with quantitative label-free comparative analysis of proteomes isolated from the wild-type strain strain with deleted gapA gene suggests that GapA is implicated in DNA repair processes. Absence of GapA promotes secretion of its most potent interaction partner the hypothetical protein with peptidase propeptide domain (PepSY) thereby indicating that it impacts on subcellular distribution of some proteins.
Introduction
The glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is known for its ability to exhibit multiple unrelated functions within a single polypeptide chain (Pancholi and Chhatwal, 2003; Jeffery, 2009). In mammalian cells, it is involved in diverse cellular non-metabolic processes, including gene regulation, DNA repair, cytoskeletal dynamics, cell death, vesicular transport, and many others. These so-called moonlighting roles are associated with GAPDH’s unusual, non-predictable localization. Such multifunctionality also has been reported for bacterial GAPDH in a number of independent studies (Pancholi and Fischetti, 1992; Modun and Williams, 1999; Boël et al., 2005; Butland et al., 2005; Arifuzzaman et al., 2006; Jin et al., 2011; Ferreira et al., 2015). In some human pathogens, the surface-localized and/or -secreted GAPDH is able to bind to several host proteins (Kopeckova et al., 2020), thus contributing to its virulence. The mechanisms by which this protein is transported out of the cell remain obscure, however, because it lacks any known recognition motif for membrane and/or extracytosolic trafficking (Pancholi and Chhatwal, 2003). Data on the intracellular non-enzymatic functions of GAPDH in bacteria are also scant. The results of a transcriptomic analysis on Streptococcus pyogenes mutant strain displaying higher intracellular levels of GAPDH suggest it has a role in transcriptional modulation of virulence and metabolic genes (Boël et al., 2005). Several large-scale protein–protein interaction studies performed on Escherichia coli have revealed that bacterial GAPDH, similar to its eukaryotic orthologs or analogs, is able to interact with other intracellular proteins, including metabolic enzymes as well as proteins involved in transcription, protein synthesis, and DNA repair (Butland et al., 2005; Arifuzzaman et al., 2006; Ferreira et al., 2013, 2015).
The pathogenic bacterium Francisella tularensis is a facultative intracellular, Gram-negative coccobacillus that causes the zoonotic disease tularemia (McCrumb et al., 1957). The subspecies tularensis (type A) strains exhibit extreme virulence and a mortality rate potentially as high as 60% (Steiner et al., 2014). Due to the very low infectious doses and easy transmission by aerosol, this bacterium has been classified as a potential biological weapon (Ellis et al., 2002; Meibom and Charbit, 2010; Barel and Charbit, 2013). F. tularensis thus constitutes a serious threat for humanity, and this is further heightened by the absence of a licensed vaccine. A persistent lack of knowledge regarding the molecular mechanisms of tularemia’s pathogenesis, however, hampers the development of effective and safe prophylactics.
We previously have shown that the GAPDH homolog GapA contributes to the full virulence manifestation of the F. tularensis subsp. holarctica FSC200 strain (Pavkova et al., 2017). Furthermore, its demonstrated extracytosolic localization and ability to bind to several host serum proteins have indicated that GapA performs other functions beyond its primary enzymatic role in glycolysis. In the presented study, we aimed to identify bacterial protein interaction partners to elucidate the potential non-enzymatic roles of F. tularensis GapA protein inside the bacterial cell. We employed for this purpose a stable isotope labeling of amino acids in cell culture (SILAC) strategy (Ong et al., 2002) in combination with affinity purification followed by quantitative mass spectrometry (SILAC-AP-MS/MS). This approach enabled us to identify 18 potential GapA interaction partners implicated in various intracellular processes. Several of them were selected for further confirmatory and/or functional characterization. We additionally analyzed the impact of gapA gene deletion on the F. tularensis proteome by quantitative label-free proteomic comparative analysis. Our data indicate that in F. tularensis the GapA is likely engaged in DNA reparation processes and in compartmentalization of proteins.
Materials and Methods
Bacterial Strain, Plasmids, and Culture Conditions
The F. tularensis and E. coli strains and plasmids used in this study are listed in Supplementary Table S1. Francisella strains were cultured on McLeod agar enriched with bovine hemoglobin (Becton Dickinson, Cockeysville, MD, United States) and IsoVitaleX (Becton Dickinson) and in liquid Chamberlain’s chemically defined medium (CDM) (Chamberlain, 1965) at 37°C. If not further specified, the E. coli strains were grown on Luria–Bertani (LB, Sigma–Aldrich, Schnelldorf, Germany) agar and in liquid LB broth or Terrific Broth medium at 37°C. When necessary, the following antibiotics were used: chloramphenicol 2.5 μg/ml (F. tularensis) or 25 μg/ml (E. coli), polymyxin B 75 μg/ml (F. tularensis), carbenicillin 50 μg/ml (F. tularensis), kanamycin 50 μg/ml (E. coli) or 20 μg/ml (F. tularensis).
Construction of F. tularensis Strain Expressing Strep-Tagged GapA Protein
The gapA gene (locus tag FTS_1117) was fused to the coding sequence of the Strep-tag (eight amino acids: WSHPQFEK) on its C-terminus using PCR amplification with specific primers (see Supplementary Table S2). The gapA gene (locus tag FTS_1117) was fused to the coding sequence of the Strep-tag (eight amino acids: WSHPQFEK) on its C-terminus using mutagenesis kit QuikChange II XL Site-Directed (Agilent, United States) with specific primers (see Supplementary Table S2). The final sequence (gapA-Strep) was introduced into the chromosome of F. tularensis FSC200 gapA deletion mutant strain (ΔgapA) prepared previously by Pavkova et al. (2017) by in-cis complementation strategy similar to in-frame deletion mutagenesis described below in section “Construction of In-Frame Deletion pepSY Mutant.” Briefly, the DNA construct encoding the gapA-Strep gene with introduced restriction sites XhoI and SpeI was generated by overlapping PCR amplification using the specific primers (Supplementary Table S2). The resulting DNA fragment was cloned into pCR4-TOPO vector (Supplementary Table S1) and verified by sequence analysis (ABI PRISM 3130xl, Applied Biosystems). Fragments from plasmids with verified inserts were cloned into suicide pDM4 plasmid (Supplementary Table S1), and then introduced into E. coli S17-1λpir (Supplementary Table S1). The plasmids were mobilized into the ΔgapA by conjugation followed by sucrose selection resulting finally to the allelic exchange on the FSC200 chromosome.
SILAC Metabolic Labeling, Cell Lysate Preparation, Affinity Purification
For metabolic labeling, the F. tularensis wild-type strain (wt) or strain expressing Strep-tagged GapA protein (FSC200/gapA_S) were cultivated in CDM containing the heavy labeled amino acids L-arginine hydrochloride [13C6 15N4] and L-lysine hydrochloride [13C6 15N2] (Sigma–Aldrich, Schnelldorf, Germany) in the same concentrations as in the light medium. After three generations, the incorporation levels were checked by mass spectrometry (MS) and the labeled bacteria were stored as frozen stocks at −150°C. For the experiment, the labeled bacterial stock was cultivated in heavy medium overnight at 37°C, 200 rpm. Non-labeled bacteria were grown in the light medium. The overnight cultures were diluted with fresh media to optical density at 600 nm (OD600nm) = 0.1 and cultivated to OD600nm = 0.8 (approximately 3.2 × 109 bacteria/ml, late exponential phase). Bacteria were then harvested by centrifugation and washed with phosphate-buffered saline (PBS, pH 7.4). At this point, the light and heavy bacteria were mixed in a 1:1 cell ratio. Three sets of biological replicates were prepared. Each set consisted of three samples. In two samples, the wt FSC200 was grown in light medium and the FSC200/gapA_S in heavy medium. For the third sample, the SILAC groups were swapped (FSC200/gapA_S was cultivated in light medium, wt FSC200 strain in heavy medium). After supplementation with protease inhibitor cocktail Complete EDTA-free (Roche Diagnostics, Mannheim, Germany) and benzonase (150 U/ml; Sigma–Aldrich), the bacterial cells were disrupted in a French press (Thermo IEC, Needham Heights, MA, United States) by two passages at 16,000 psi. Unbroken cells were removed by centrifugation at 6,000 × g for 40 min at 4°C. The clarified lysate supplemented with an appropriate amount of Avidin (11 U/mg; IBA Lifesciences, Göttingen, Germany) was applied on a column containing Strep-Tactin® affinity resin and the GapA/S protein was purified using the Strep-tag® purification system according to the manufacturer’s instructions (IBA Lifesciences).
Preparation of the Whole-Cell Lysate, Crude Membrane Fraction, and Culture Filtrate Proteins
Bacteria were grown in CDM at 37°C until the late logarithmic phase (0.7–0.8 OD600 nm). To obtain whole-cell lysate, the bacteria were lysed in a French pressure cell and unbroken cells and cell debris were removed by centrifugation. The pellet with membrane proteins was obtained by ultracentrifugation of the whole-cell lysate, and then suspended in PBS as described previously (Pavkova et al., 2017). The supernatant after the ultracentrifugation was used as cytosolic fraction in Western blot analysis. For the preparation of culture filtrate proteins, the bacteria were removed by centrifugation and the culture medium was vacuum-filtered through membranes (0.2 μm pore; Merck Millipore, Billerica, MA, United States). The filtrate was then concentrated using Stirred Ultrafiltration Cell (8200, Merck Millipore) with 5 kDa cutoff membrane from regenerated cellulose (Merck Millipore) followed by diafiltration using Amicon Ultra 3K devices (Merck Millipore) to further concentrate the protein sample and exchange the medium for 40 mM Tris-HCl (pH 7.3). The protein content was determined using a bicinchoninic acid assay.
Sample Preparation for Mass Spectrometry Analysis
The protein samples were adjusted with 25 mM ammonium bicarbonate with 10% (w/v) sodium deoxycholate monohydrate (DOC) to final concentration 1%. Samples were next reduced with 4 mM dithiothreitol (DTT) at 60°C for 45 min, at 700 rpm, then alkylated with 16 mM iodoacetamide at room temperature for 30 min in darkness. The unreacted iodoacetamide was quenched with an additional 4 mM DTT at room temperature for 30 min. The proteins were digested with trypsin (Promega, Madison, WI, United States) overnight at 37°C. Thereafter 1 M hydrochloric acid was added to stop the digestion and precipitate DOC. The suspension was then mixed with an equal volume of ethyl acetate, vortexed vigorously for 1 min, centrifuged at 14,000 × g for 5 min, and the upper organic layer was removed. The extraction was repeated three times to completely extract DOC. The aqueous phases were desalted on EmporeTM C18-SD (4 mm/1 ml) extraction cartridges (Sigma–Aldrich) and dried in a vacuum.
Liquid Chromatography and Mass Spectrometry Analysis
The prepared bacterial peptide samples were dissolved in 20 μl 2% acetonitrile (ACN)/0.1% trifluoroacetic acid (TFA) and 1 μl was analyzed using the UltiMate 3000 high-performance liquid chromatography system (Dionex/Thermo Scientific, United States). This system with UV detection included a μ-Precolumn (300 μm × 5 mm, C18PepMap 5 μm 100 Å particles; Dionex) connected to the analytical NanoEase column (100 μm × 150 mm, Atlantis C18 3 μm 100 Å particles; Waters, Milford, MA, United States). Peptide separation was performed using the bilinear gradient of 3–44% ACN/0.1% TFA over 81 min under a flow rate of 360 nl/min and UV detection set to 215 nm. The data acquisition and chromatogram visualization were carried out in the Chromeleon software (v. 6.80, Dionex). The separation of peptides for nanoscale liquid chromatography-tandem mass spectrometry analysis was done using the UltiMate 3000 RSLC-nano high-performance liquid chromatography system (Dionex) with a trap column (75 μm × 20 mm) packed with 3 μm Acclaim PepMap100 C18 particles and a separation column (75 μm × 150 mm) packed with 2 μm Acclaim PepMap RSLC C18 particles. The samples were loaded onto the trap column with 2% ACN/0.05% TFA and peptide separation was performed with the dual linear gradient using 3–44% ACN in 0.1% formic acid over 89 min for all samples under the flow rate of 300 nl/min. Column temperature was set to 40°C. The separation was monitored with UV detection at 214 nm and directly coupled to mass spectrometry analysis with the Q Exactive system (Thermo Fisher Scientific, United States) in the positive mode with full MS scan (350–1650 m/z) at 70,000 full width at half maximum (FWHM), maximum filling time 100 ms and automatic gain control (AGC) target 1e6. The top 12 precursors in MS/MS at 17,500 FWHM with precursor charge 2+ or higher were selected for all samples with minimum threshold intensity of 5e4, maximum filling time 100 ms, and AGC target. Instrumental data acquisition was done using Xcalibur software v. 3.0.
Data Processing and Protein Identification
SILAC Quantification
Peptides and proteins were identified by searching the raw files against the F. tularensis subsp. holarctica (wt FSC200) protein sequences database (1,423 entries, downloaded from UniProt) appended with common protein contaminants (245 sequences, downloaded from MaxQuant homepage) using the Mascot v2.3 search engine (Matrix Science, Boston, MA, United States) within the Proteome Discoverer v2.2 software (Thermo Scientific). The parameters were as follow: trypsin was used as the enzyme, two missed cleavages were allowed, carbamidomethylation of cysteine was set as a fixed modification, oxidation of methionine was selected as a variable modification, and the mass tolerance of the precursor and fragment ions was set to 20 ppm and 0.02 Da, respectively. Arginine (13C6, 15N4) and lysine (13C6, 15N2) were set as labels in the heavy channel for peptide quantitation. A false discovery rate of 0.01, estimated by target-decoy approach, was used for accepting the identifications on the peptide-to-spectrum match and protein levels. The SILAC-based approach was used to reveal the candidate interacting proteins of GapA. These are expected to be detected in a single SILAC quantitation channel, contrary to background proteins, resulting in extreme abundance ratio values. Therefore, the low abundance resampling algorithm of the Proteome Discoverer was used to impute missing values of single-channel quantitative data on the peptide level. The GapA interacting proteins were found as significantly up-regulated proteins using a global mean rank test (Klammer et al., 2014) (R package MeanRankTest1) with parametric FDR level set to 0.05. Only the Francisella proteins quantified in all four replicates were allowed for testing. Data visualization was done in Perseus v1.6.2.3 software (Tyanova et al., 2016).
Label-Free Quantification
Proteome Discoverer software (Thermo Fisher Scientific, v. 2.4.0.305) was used for identification of MS/MS spectra and label-free quantification of the measured data. The raw files from the QExactive mass spectrometer were processed within the processing workflow containing spectrum files RC, spectrum selector, non-fragment filter, top N peaks filter, precursor detector, SequestHT search engine, and target decoy PSM validator. The parameters for SequestHT database searching were: protein database – UniProt Francisella tularensis subsp. holarctica FSC200 reference proteome UP000006302 (11 May 2018); enzyme – trypsin; maximum missed cleavage sites – 1; min. peptide length – 7; precursor mass tolerance – 15 ppm; fragment mass tolerance – 0.02 Da; weight of b- and y-ions – 1; static modifications – carbamidomethyl/ +57.021 (C); dynamic modifications – oxidation/ +15.995 Da (M); dynamic modifications (protein terminus) – acetyl/ +42.011 Da (N-terminus), Met-loss/-131.040 Da (M), Met-loss + acetyl/-89.030 Da (M); and dynamic modifications (peptide terminus) – Gln- > pyro-Glu/-17.027 Da (Q). The search results obtained in the msf file were further processed in the consensus workflow containing PSM Grouper, Peptide Validator, Protein and Peptide Filter, Protein Scorer, Protein FDR Validator, Protein Grouping, Protein in Peptide Annotation Nodes, Protein Marker, Modification Sites, Feature Mapper, and Precursor Ions Quantifier. Only proteins identified with at least two peptides at FDR level 0.01 with p-value less than 0.01 of the quantitation change (computed from peptide abundances in Proteome Discoverer software) greater than 1.5 were considered. The heat maps were visualized in the Proteome Discoverer graphic tools for Venn diagrams reports.
The raw MS data as well as the processed identification results have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD019739.
Expression and Purification of Recombinant Proteins Derived From F. tularensis
The genes encoding selected F. tularensis proteins were amplified by PCR with specific primers (Supplementary Table S2), and the gel-purified PCR products (Qiagen) were inserted into pET28b using either the NcoI and XhoI restriction sites (pepSY, rho, pepB, gene of YCII-related domain protein) or HindIII and XhoI restriction sites (trxA). The resulting plasmids encoded the aforementioned proteins with a C-terminal histidine tag. The final plasmid constructs were verified by direct DNA sequencing. For protein expression, the plasmids were transformed into E. coli NiCo21 expression strains (New England BioLabs, Ipswich, AM, United States). The cells were grown in Terrific Broth medium at 28°C overnight. Bacteria were lysed in a French pressure cell and the lysates were applied to a TALON® Metal Affinity Resin purification system (Clontech, Mountain View, CA, United States) for purification of His-tagged proteins. Finally, the elution buffer was changed to T100N150 (100 mM Tris-HCl, 150 mM NaCl, [pH 7.6]) using an Amicon® Ultra-15 centrifugal filter (Sigma–Aldrich). Eluted proteins were verified by SDS-PAGE followed by Coomassie staining (data not shown), and the protein concentrations were determined using Coomassie Plus Bradford assay reagent (Thermo Fisher).
Solid-Phase Ligand-Binding Assay
This assay was used to confirm the ability of GapA to bind to selected F. tularensis proteins and was conducted as described previously (Pavkova et al., 2017). Briefly, the purified recombinant proteins were coated onto a 96-well high binding microtiter plate overnight and then incubated with recombinant GapA (0.25–2.5 μg/ml). The amount of GapA bound to the proteins was determined spectrophotometrically (450 nm) in an enzyme-linked immunosorbent assay using anti-GapA antibody (Apronex, Vestec, Czechia).
Bacterial Two-Hybrid System
To confirm the interaction of GapA with UvrA, we performed bacterial two-hybrid assay using the BACTH (bacterial adenylate cyclase two-hybrid) System kit (Euromedex, Strasbourg, France). The uvrA and gapA genes were amplified from F. tularensis FSC200 genomic DNA, then inserted into the pKT25 and pUT18C using KpnI and BamHI restriction sites. The resulting plasmids were transformed into the BTH101 cells. Further protein–protein interaction analysis was performed according to the manufacturer’s protocols.
UV Tolerance Assay
The effect of UV on the survival of selected F. tularensis strains was evaluated as described by Karki and Ham (2016), albeit with minor modifications. The overnight bacterial cultures were resuspended in PBS to a density of OD600nm = 1.0 (approximately 4 × 109 bacteria/ml) and 10-fold serial dilutions of this bacterial suspension were plated onto McLeod agar. The inoculated plates (with open lids) were positioned directly below the UVLMS-38EL Series 3UVTM Lamp 8W (UVP, Cambridge, United Kingdom), at a distance of 37 cm, giving an irradiance of approximately 210 μW/cm2 at 254 nm. The bacteria were exposed to increasing applied doses of 254 nm UV light, using exposure times 0 s, 15 s, 30 s 60 s and 150 s, with dose calculated as irradiance (W/cm2) × exposure time (s). The plates were then incubated at 37°C in 5% CO2 for 5 days for CFU enumeration of the surviving bacteria.
H2O2 Susceptibility Assay
The bacteria were first cultured in CDM overnight at 37°C. The next morning, the bacterial culture was centrifuged and resuspended in fresh CDM to OD600 nm = 0.1 (approximately 4 × 108 bacteria/ml). The hydrogen peroxide was added at final concentration 1 mM and the bacteria were cultivated at 37°C, 200 rpm. After 0, 2.5, and 5 h of incubation, bacterial samples were collected and viable bacteria determined by plating 10-fold serial dilutions. The plates were incubated for 5 days at 37°C in 5% CO2 before enumeration of CFU.
Construction of In-Frame Deletion pepSY Mutant
The DNA construct encoding in-frame deletion for the pepSY gene with introduced restriction sites XhoI and SpeI (locus tag FTS_1731) was generated by overlapping PCR amplification using the specific primers (Supplementary Table S2). The resulting DNA fragment was cloned into pCR4-TOPO vector (Invitrogen, Carlsbad, CA, United States) and verified by sequence analysis (ABI PRISM 3130xl, Applied Biosystems, Foster City, CA, United States). Fragments from plasmids with verified inserts were cloned into the suicide pDM4 vector using the same restriction sites (Supplementary Table S1) and then introduced into E. coli S17-1γpir. The plasmids were mobilized into the F. tularensis FSC200 strain by conjugation followed by sucrose selection resulting finally to the allelic exchange on the FSC200 chromosome. Preparation of plasmid DNA, restriction enzyme digests, ligations, and transformations into E. coli all were performed essentially as previously described (Sambrook and Russell, 2001).
Intracellular Replication and Invasion Assays
To generate bone marrow macrophages (BMMs), bone marrow cells were collected from dissected femurs of female BALB/c mice 6–10 weeks old and differentiated into macrophages in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum and 20% L929-conditioned medium for 6–7 days. The differentiated BMMs were seeded at concentration 5 × 105 cells/well in 24-well plates and infected the next day with wt FSC200 at multiplicity of infection 50:1 (bacteria/cell). To synchronize infection, the infected cells were centrifuged at 400 × g for 5 min and incubated at 37°C for 30 min. The extracellular bacteria were then removed by gentamicin treatment (5 μg/ml) for 30 min. For the proliferation assay, the infected BMMs were lysed at selected time points with 0.1% DOC. To determine numbers of intracellular bacteria, the lysates were serially diluted and plated onto McLeod agar.
Animal Studies
For survival studies, groups of at least five female BALB/c mice 6–8 weeks’ old were infected subcutaneously with the ΔpepSY mutant strain (using doses of 102 CFU/mouse) or the wt FSC200 strain. Control groups of mice were inoculated with sterile saline only. Mice were housed in micro-isolator cages and provided sterilized water and food ad libitum.
Ethics Statement
This study was carried out in accordance with the recommendations of the guidelines of the Animal Care and Use Ethical Committee of the Faculty of Military Health Sciences, University of Defence, Czechia. The protocol was approved by the Ethical Committee of the Faculty of Military Health Sciences, University of Defence, Czechia.
Western Blotting
Protein samples (2 μg of whole-cell lysate or cytosol, 40 μg of crude membrane fraction, 5 μg of culture filtrate proteins for each strain) were separated on 12% acryl-amide gels and transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, United States). Membranes were blocked with 5% w/v skim milk powder, 0.1% v/v Tween in Tris-buffered saline and incubated in the same buffer with polyclonal rabbit anti-GapA antibody 1:1000 and swine anti-rabbit IgG/HRP (Dako, Glostrup, Denmark) 1:1000 as secondary antibody. Chemiluminescence was detected using a BM Chemiluminescence Blotting Substrate (POD) according to the manufacturer’s instructions (Roche Diagnostics). The blots obtained from biological duplicates were captured and analyzed using iBrightTM FL1000 Imaging System. Local background corrected density was used for band quantitation.
Statistical Analysis
The assays were performed in triplicate for each strain and time. Each experiment was independently repeated at least three times unless otherwise stated. Values are expressed as mean ± standard deviation (SD) and analyzed for significance using Student’s two-tailed t-test. Differences were considered statistically significant at p < 0.05.
Results
Identification of GapA Protein Interaction Partners Using SILAC-AP-MS/MS Strategy
We used a quantitative proteomic approach based on SILAC metabolic labeling in combination with affinity purification and mass spectrometry to screen for bacterial proteins that bind specifically to the GapA of F. tularensis (Figure 1). The interconnection of these techniques enables confident discrimination between specific and non-specific interactions (Gingras et al., 2007; Auweter et al., 2011). For this purpose, we first engineered F. tularensis FSC200 to express GapA-Streptag fusion protein. The wt FSC200 and the mutant strain FSC200/GapA/S were grown in CDM containing arginine and lysine labeled with regular abundance or heavy isotopes, respectively. The two bacterial cultures were mixed together (1:1 cell ratio) and lysed. The GapA together with its binding partners was purified using StrepTactin. The effector complexes were treated with trypsin followed by LC-MS/MS analysis. Proteins bonding specifically to the Gap/S were significantly enriched in heavy labeled amino acids, resulting in high heavy:light SILAC ratios, whereas proteins that interacted non-specifically with the Strep-Tactin showed equal peak intensities for heavy and light forms.
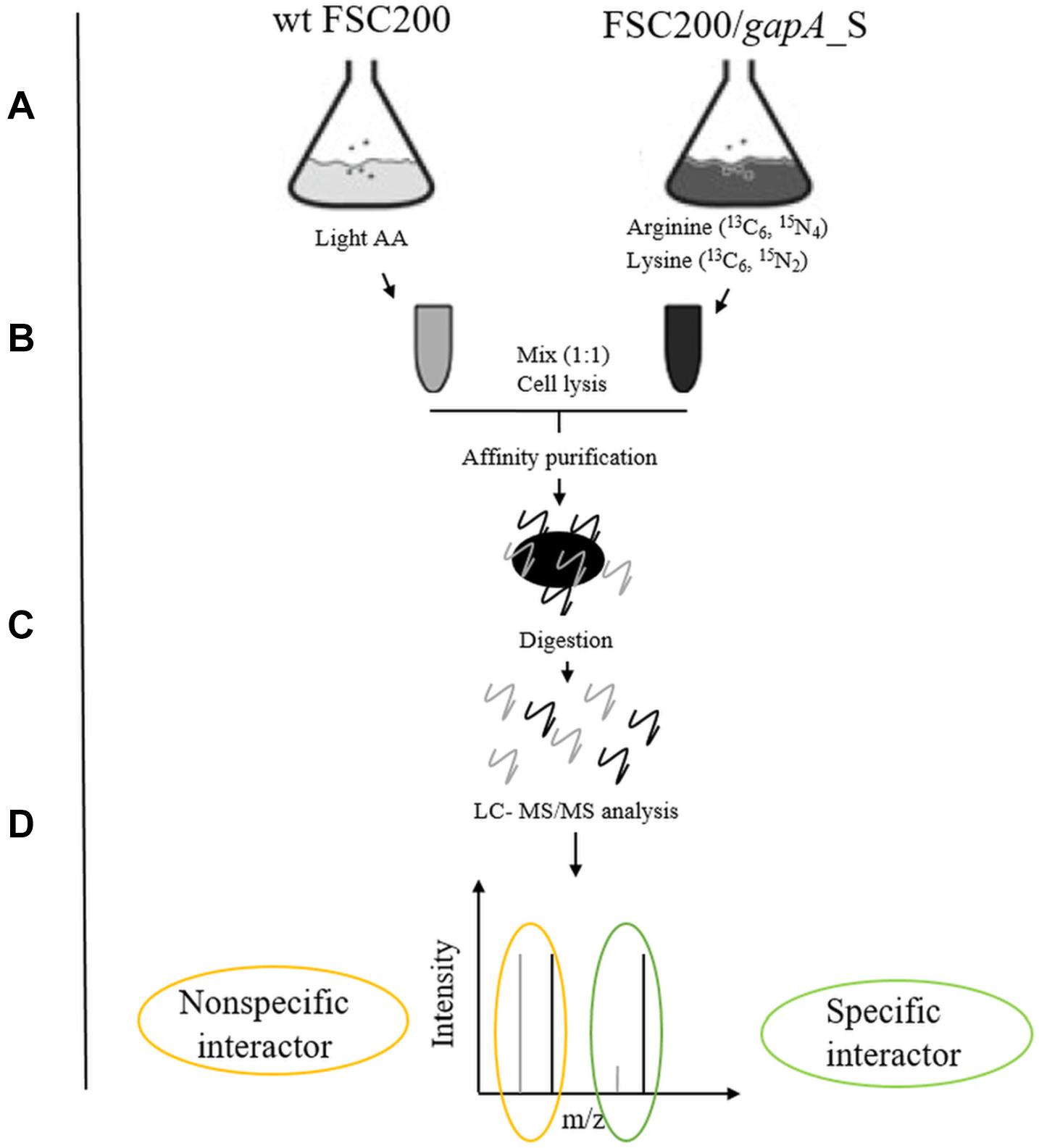
Figure 1. Schematic overview of SILAC-AP-MS/MS strategy. (A) The bacterial strains wt FSC200 and FSC200/gapA_S were grown in CDM with light or heavy amino acids (arginine 13C6, 15N4; lysine 13C6, 15N2). (B) Bacterial cultures (OD600 nm = 0.8) were mixed in 1:1 cell ratio and lysed by French press. (C) GapA/S together with interacting partners was purified using Strep-tag® purification system and digested with trypsin. (D) Tryptic peptides were analyzed by LC-MS/MS.
Based on the criteria described in the section “Materials and Methods,” we were able to detect 18 potential binding partners (Table 1). The functional classification revealed that the largest group of the identified proteins (nine proteins) were involved in metabolic processes and might be related to the enzymatic function of GapA. The identified phosphoglycerate kinase forms in complex with GAPDH a functional ATP-generating unit that provides ATP production for glycolysis and might thus be considered as a method validation indicator. Another five proteins are engaged in genetic information processing, like transcription, translation, DNA repair, or protein folding. The remaining four proteins are only poorly characterized, but the presence of a functional domain in three of them indicates their putative role also in transcription processes.
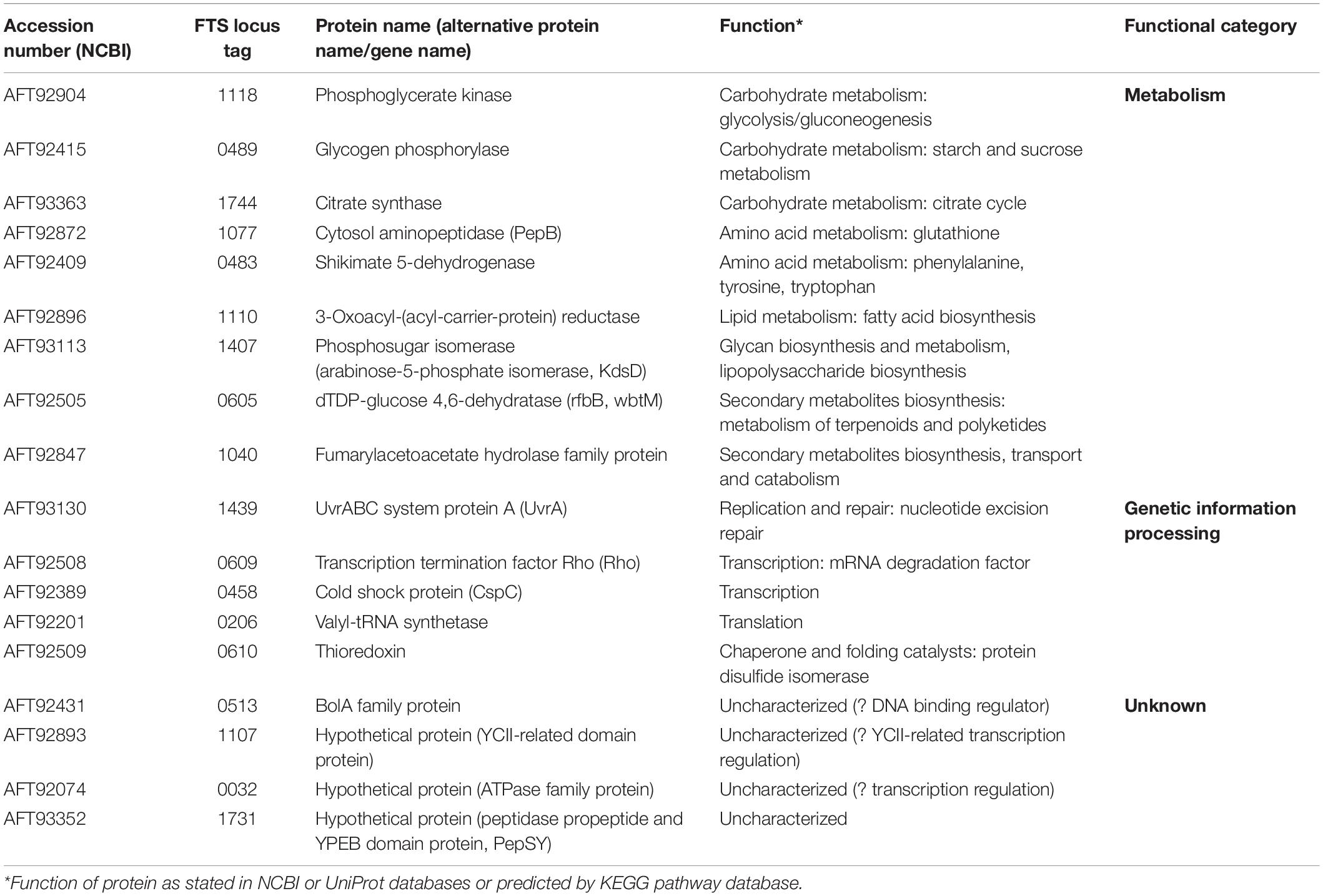
Table 1. Candidate GapA interaction partners identified by SILAC-AP-MS/MS strategy.
Confirmation of Selected Interactions
To validate our proteomic screen, we further aimed to confirm the GapA interaction with the seven most potent hits based on significance in the global mean rank test. Using the solid-phase ligand-binding assay, we were able to confirm the binding ability of purified recombinant GapA with the protein candidates PepSY, Rho, thioredoxin, PepB, and YCII-related domain protein in vitro (Figure 2A). Due to problems with purification of recombinant UvrA, this interaction was successfully confirmed using the bacterial two-hybrid system (Figure 2B).
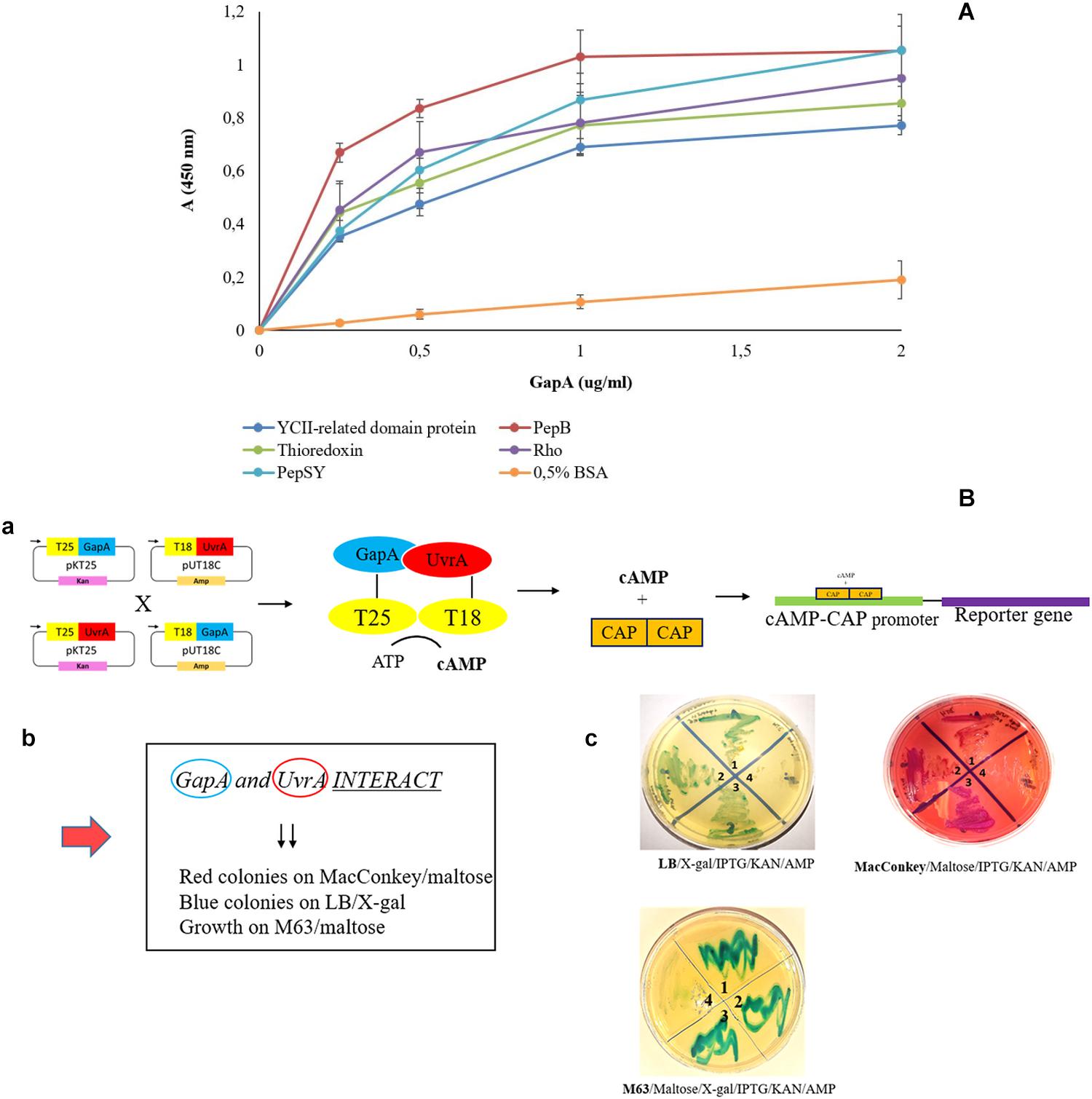
Figure 2. Validation of F. tularensis GapA binding ability to selected bacterial proteins. (A) Solid-phase binding assay of PepSY, Rho, thioredoxin, PepB, YCII-related domain protein, and BSA (negative control) coated on 96-well microtiter plate and reacted with different concentrations of purified GapA. Data are presented as means ± SD from three independent experiments. (B) Validation of GapA and UvrA interaction using the BACTH system. (a) Principle of the method: GapA and uvrA genes were cloned into the plasmids pKT25 and pUT18C and fused with T25 and T18 fragments. During the interaction of GapA with UvrA, complementation occurs between T25 and T18 fragments with subsequent cAMP synthesis. The complex of cAMP/CAP is a pleiotropic regulator of gene expression (E. coli) that initiates expression of reporter genes (lac, mal operon). (b) Manifestation of positive interaction. (c) BTH101 cells were co-transformed with the following plasmid combinations: (1) pKT25-gapA × pUT18C-uvrA, (2) pUT18C-gapA × pKT25-uvrA, (3) positive control plasmids pKT25 zip × pUT18C zip, and (4) negative control plasmids pkT25 empty + pUT18C empty. Co-transformed BTH101 cells with a plasmid combination were grown on selective agar plates (LB/X-gal/IPTG/KAN/AMP or MacConkey/Maltose/IPTG/KAN/AMP or M63/Maltose/X-gal/IPTG/KAN/AMP). The bacteria grown on LB agar plates were passaged on MacConkey agar plates and vice versa. We observed the results to be the same (data not shown).
Sensitivity of GapA-Deficient Cells to DNA Damaging Conditions
The UvrA protein identified in our study as a potential GapA interacting partner forms together with UvrB and UvrC proteins a multienzyme complex responsible for nucleotide excision repair (NER) in DNA damaged primarily by ultraviolet (UV) radiation (Sancar and Reardon, 2004). We thus decided to examine the sensitivity of bacteria with deleted gapA gene to UV irradiation. As seen in Figure 3A, however, the survival rates of the ΔgapA mutant were almost comparable to those of wt FSC200. In contrast, the mutant strain with disrupted uvrA gene (uvrA insertion mutant – uvrAin), published previously in use as a positive control (Pavkova et al., 2017), was far more sensitive to UV killing. None of the three strains showed any difference in their susceptibility to alkylating agent methyl methanesulfonate (data not shown). As DNA damage caused by alkylating agents is predominantly repaired by the base excision repair pathway, this indicates that neither the UvrA nor GapA are essential for this DNA repair process.
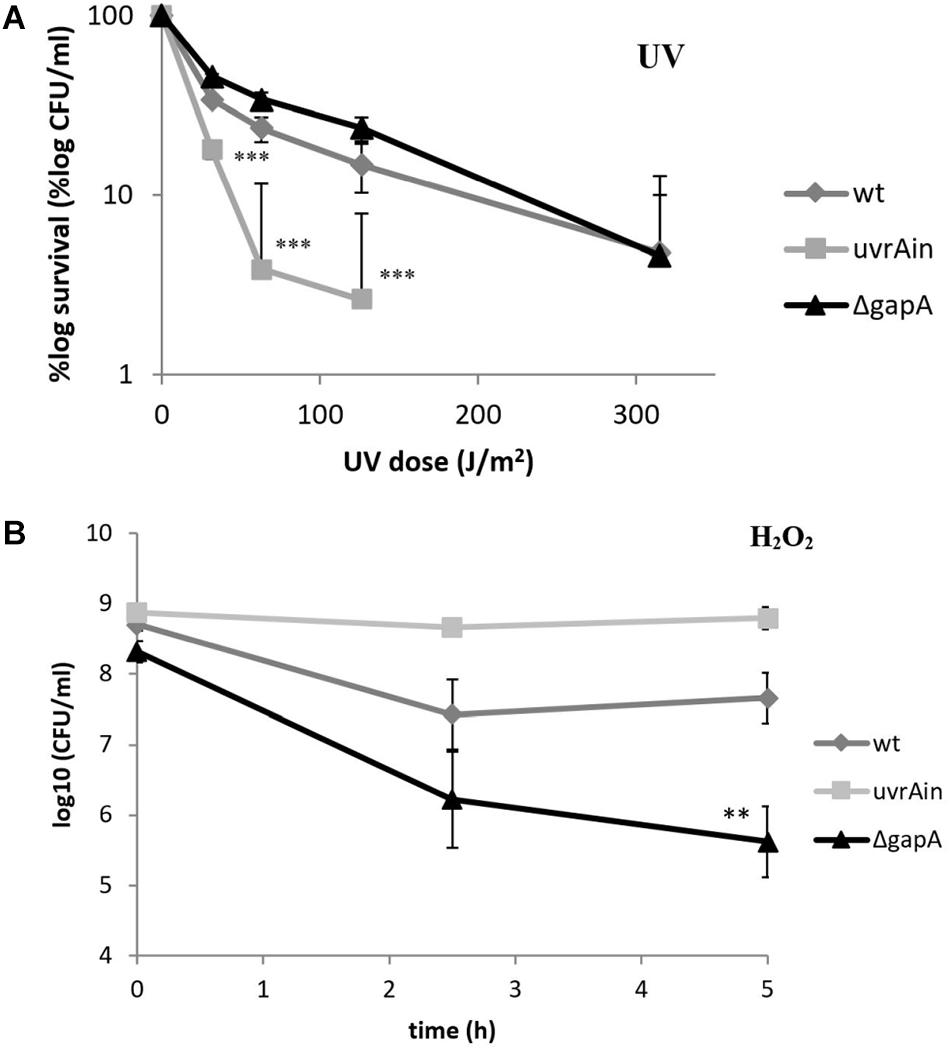
Figure 3. Sensitivity of F. tularensis ΔgapA, wtFSC200, and uvrAin to UV irradiation (A) and to H2O2 (B). For UV tolerance assay (A), 10-fold serially diluted bacteria plated on McLeod agar were irradiated for various lengths of time under a 254 nm germicidal lamp. After UV irradiation the bacteria were cultivated at 37°C and the number of survivors was calculated by plate counts. For the uvrAin, no viable colonies could be detected when irradiated for 150 s. For the H2O2 susceptibility assay (B), bacteria in CDM were exposed to 1 mM H2O2 for 2.5 and 5 h. The viable bacteria were determined by plating 10-fold serial dilutions. The values shown in both graphs represent means of triplicates ± SD. Three independent experiments were performed. Asterisks indicate statistically significant differences according to the Student’s two-tailed t-test: **P < 0.01; ***P < 0.001 (comparing mutant strain with the wt strain).
In contrast, the ΔgapA was observed to be significantly more sensitive upon exposure to H2O2, a highly reactive molecule inducing oxidative DNA lesions. The viability of ΔgapA treated with 1 mM H2O2 gradually decreased by about 2.7 log10 during the 5 h of cultivation. Meanwhile, the viability of the wt strain dropped only during the first 2.5 h to by about 1.3 log10 and then ceased to decline (Figure 3B). Notably, the viability of uvrAin remained nearly undisturbed. The concentration used had previously been shown not to affect the viability of virulent strains wt FSC200 and SchuS4 (Lindgren et al., 2007).
The PepSY Protein Is Not Essential for F. tularensis Virulence and Does Not Affect the GapA Extracellular Localization
The protein displaying the highest significance score in the global mean rank test of the SILAC-AP-MS/MS analysis was the PepSY domain-containing protein (hypothetical protein FTS_1731, PepSY). According to UniProtKB, the protein contains a signal sequence and two PepSY domains2. Although this domain is believed to have a protease inhibitory function, the role of PepSY-containing proteins is not clear to date. We wanted to test whether this protein might play a role in F. tularensis virulence, and therefore we introduced the pepSY in-frame deletion mutant in strain wt FSC200 (ΔpepSY). The ΔpepSY mutant was shown to enter and replicate in BMMs to the same extent as did the wt strain (Figure 4A). In addition, all mice infected subcutaneously with the mutant strain at doses 102 CFU died within 7–8 days, as did mice infected with the equal dose of wt strain (Figure 4B). Based on our observations, deletion of the gene encoding the PepSY protein did not result in any obvious changes in F. tularensis phenotype and thus that protein is not essential for the virulence of this pathogen.
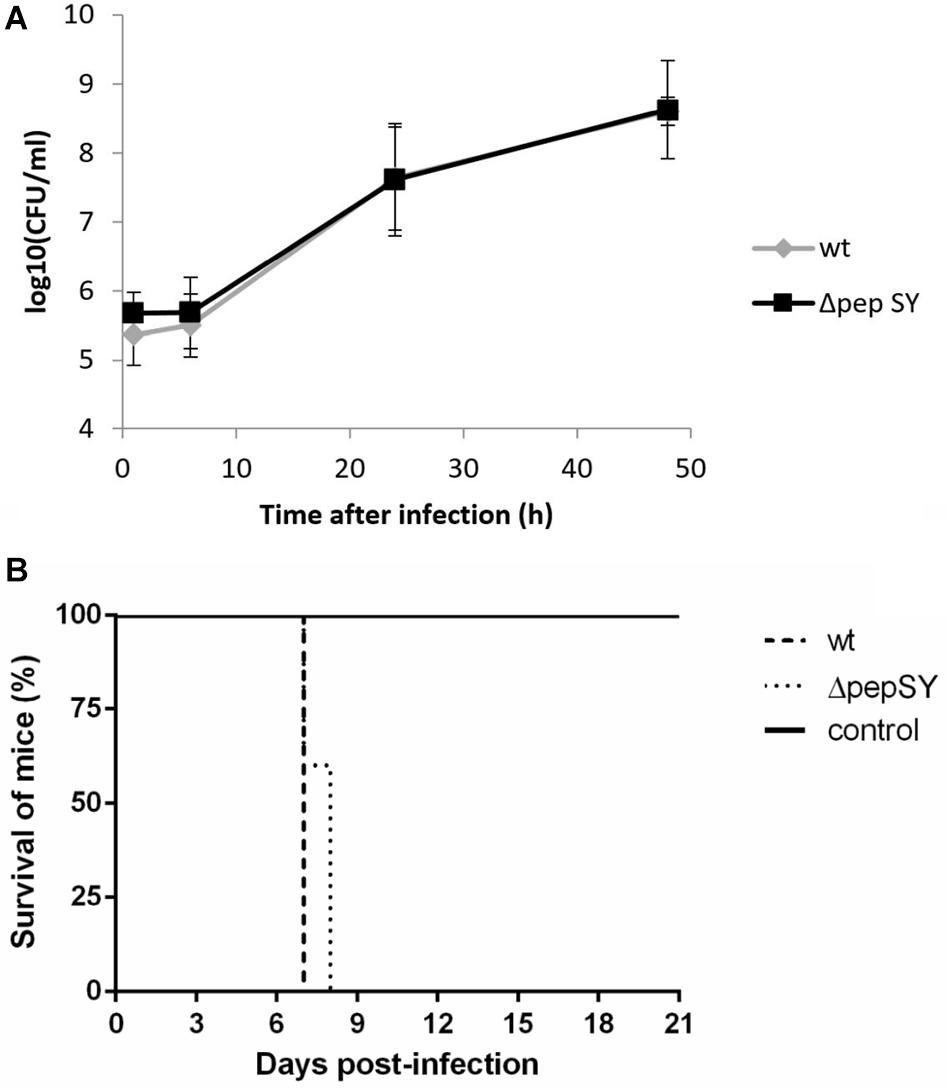
Figure 4. In vitro proliferation of ΔpepSY and wt FSC200 in BMMs (A). The cells were infected at multiplicity of infection 50:1 with the indicated strains. BMMs were harvested at 1, 6, 24, and 48 h. The data shown are means ± SD of triplicate samples and the results shown are representatives of three independent experiments. Survival of mice infected with ΔpepSY mutant (B). Groups of five BALB/c mice were infected subcutaneously with the wt and ΔpepSY mutant using a dose of 102 CFU/mouse, the control group was inoculated with sterile saline.
In a previously published study, we were able to demonstrate extracellular localization of F. tularensis GapA. The GapA sequence, however, lacks a signal sequence or other known sorting motif for extracytosolic trafficking, so the mechanism of its export to the exterior of the cell is unclear. As a signal sequence was predicted in the PepSY, we hypothesized that this protein could assist GapA through the bacterial membrane. To follow this hypothesis, we compared the GapA abundance in the ΔpepSY and wt strains. The whole-cell lysates, cytosolic proteins, crude membrane fraction, and culture filtrate proteins were separated on SDS-PAGE, which was followed by Western blot and immunodetection of GapA with polyclonal antibody. The Western blot analysis did not reveal any significant changes in GapA distribution across all the analyzed fractions between the wt and ΔpepSY strain (Figure 5). It might thus be suggested that the PepSY protein has no impact on GapA localization.
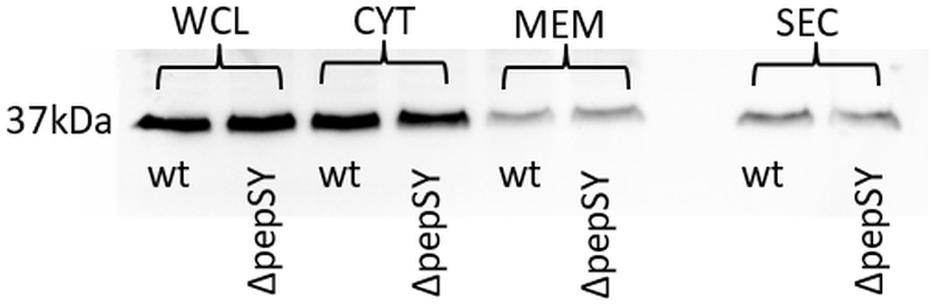
Figure 5. Representative immunodetection of GapA protein (Mw 37 kDa) in whole-cell lysate (WCL), cytosol (CYT), crude membrane fraction (MEM), and culture filtrate protein (SEC) isolated from F. tularensis FSC200 wt or ΔpepSY mutant strain. Protein samples (2 μg of WCL and CYT, 40 μg of MEM, 5 μg of SEC for each strain) were separated on 12% SDS-PAGE. Western blots were probed with polyclonal rabbit anti-GapA antibody (Apronex), imaged and analyzed using the iBright Imaging System (Invitrogen).
Impact of gapA Gene Deletion on F. tularensis Proteome
In order to reveal the broader spectrum of GapA functions, we also decided to analyze the impact of gapA gene deletion on F. tularensis proteome. For this purpose, we performed a comparative nanoscale liquid chromatography-tandem mass spectrometry analysis of whole-cell lysates, crude membrane fractions, and culture filtrate proteins obtained from the FSC200 wt and ΔgapA mutant strains. We were able to identify in total 1,092 proteins in whole-cell lysates, 985 proteins in crude membrane fractions, and 748 proteins in culture filtrates. The label-free quantification approach enabled us to select 38 proteins whose amounts significantly differed between the two tested strains (Table 2). In whole-cell lysates, 13 proteins were significantly more and seven proteins significantly less abundant in ΔgapA compared to wt. To increase the depth of our proteome analysis we have also analyzed the crude membrane fraction. This sample enrichment enabled in the identification of additional 16 differently abundant proteins – 10 of them were significantly increased and 6 decreased in the mutant strain. Otherwise, it might also be assumed that the distribution of some proteins had changed as a consequence of gapA deletion. The biclustering analysis and heat maps obtained by comparing the differentially expressed proteins in these two fractions revealed that the protein profile of the ΔgapA was different from that of the wt FSC200 strain (Figure 6). The analysis of culture filtrate proteins revealed an extremely increased abundance of PepSY in ΔgapA, indicating that PepSY secretion might be related to the absence of GapA in ΔgapA. These findings taken together suggest that GapA really is able to affect the compartmentalization of other proteins.
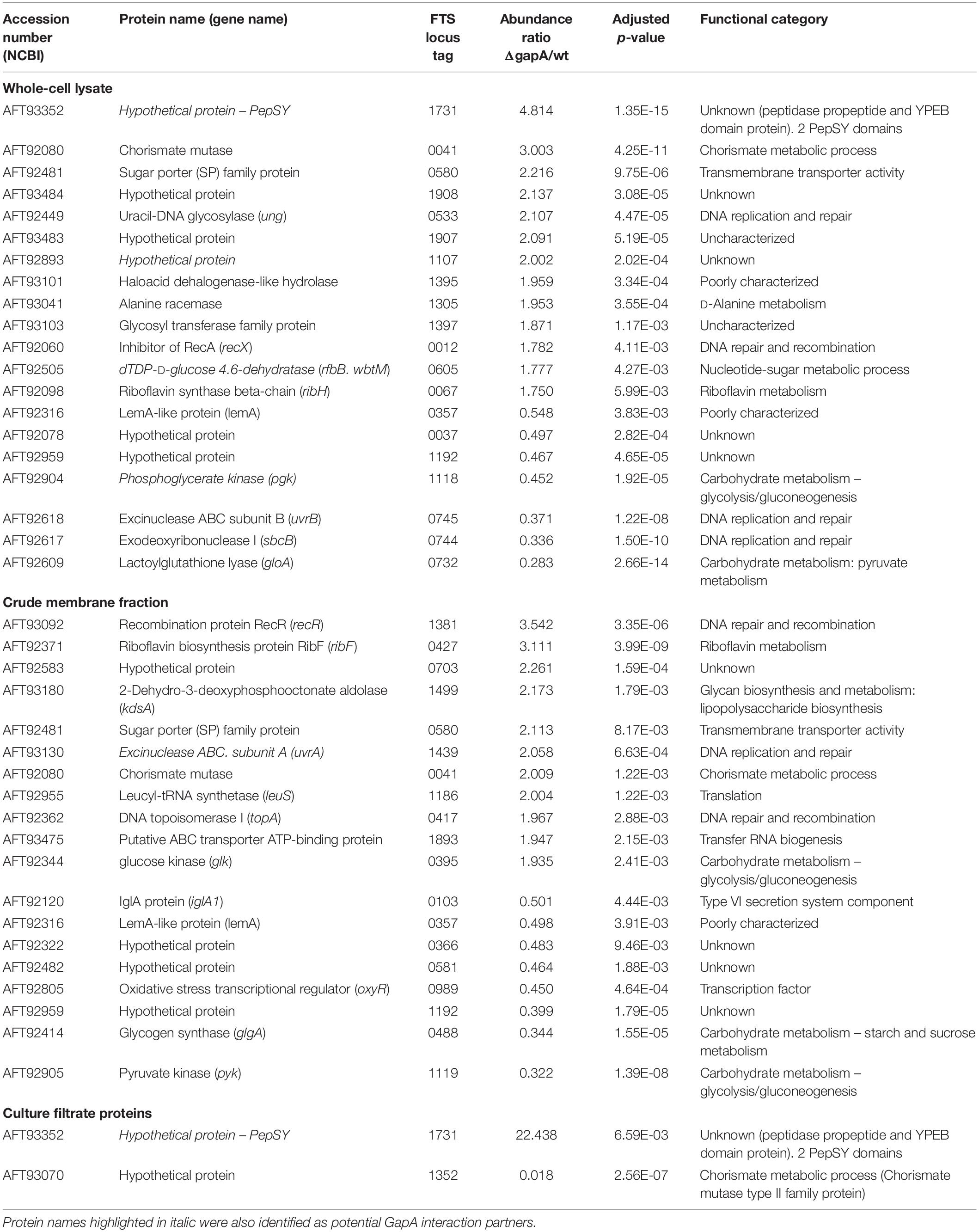
Table 2. Proteins with significantly altered expression in ΔgapA compared to the FSC200 wt strain detected by label-free quantitative shotgun approach.
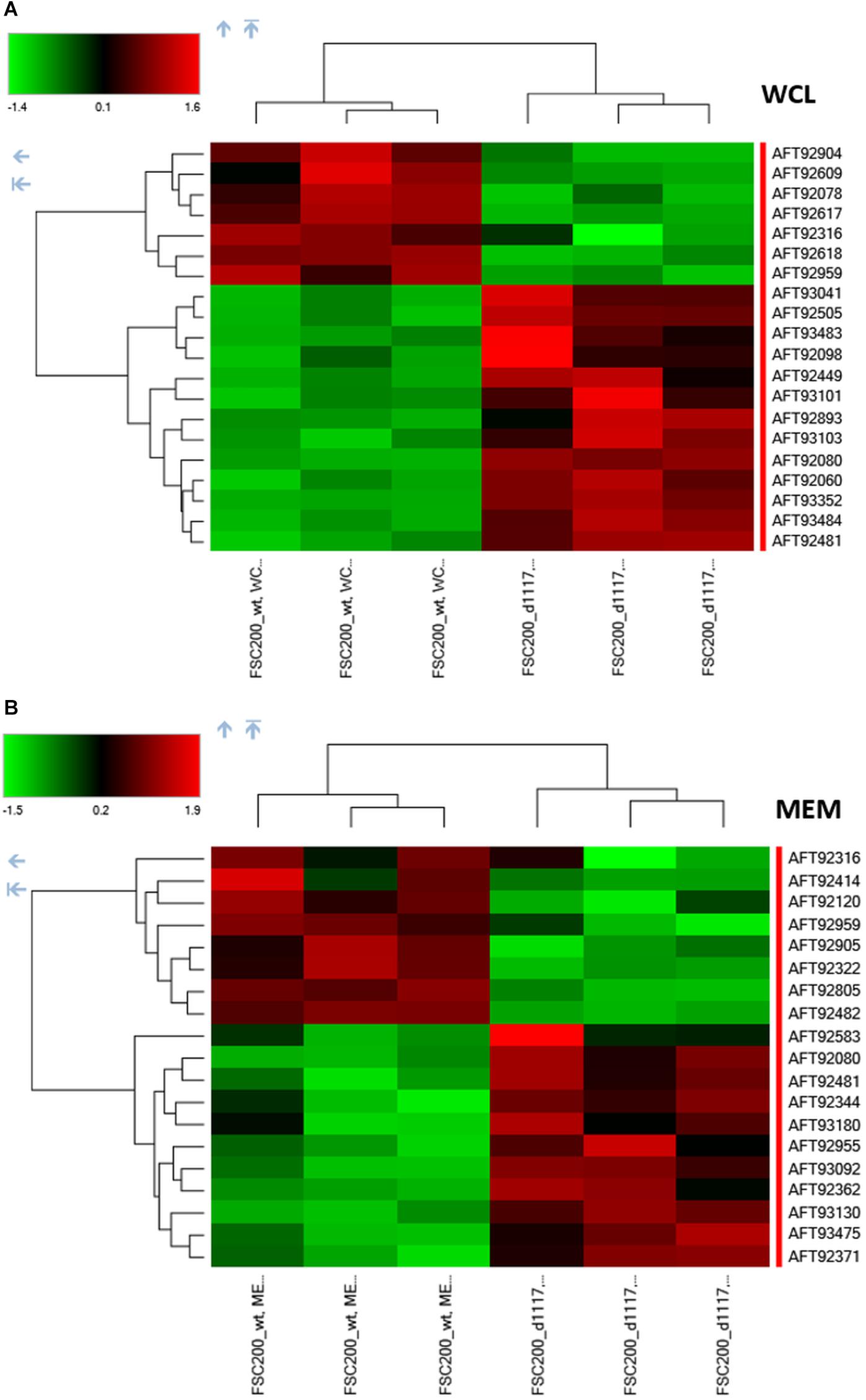
Figure 6. Biclustering and heat maps of proteins differentially expressed and identified from label-free MS analysis of whole-cell lysates (A) and crude membrane fractions (B) of wt FSC200 and ΔgapA strains.
Based on the functional classification, deletion of the gapA gene resulted in differentially expressed proteins involved in metabolic (12 proteins) and genetic information processing (11 proteins). Another 13 proteins were only poorly characterized and their function remains so far unknown. As expected, proteins of the carbohydrate metabolic pathway were the most under-represented in the proteome of the ΔgapA strain. The changed levels of several enzymes engaged in other metabolic processes might indicate the effort of F. tularensis to compensate the disturbed glycolytic pathway. Interestingly, most of the proteins from the functional category “genetic information processing” are engaged in distinct DNA reparation processes (UvrA, UvrB, uracil-DNA glycosylase, exodeoxyribonuclease I, RecR, RecX). Four proteins significantly increased in response to gapA deletion were also identified as potential interaction partners: UvrA, hypothetical YCII-related protein, PepSY, and dTDP-glucose 4,6-dehydratase. This finding supports the assumption that these proteins interact with GapA.
Discussion
Although moonlighting of the eukaryotic GAPDH is a well-known phenomenon reported in a number of published studies, current knowledge about the non-enzymatic roles of GAPDH in prokaryotes remains limited. Several studies have been able to demonstrate the involvement of this protein in host cell manipulation (Terao et al., 2006; Alvarez-Dominguez et al., 2008; Boradia et al., 2014), but its potential role in other non-metabolic processes at the intracellular level remains nearly unexplored. Because GAPDH has no functional domain or motif other than the enzymatic one, prediction of such additional functions is almost impossible. Identification of protein interaction partners thus provides valuable clues to identify new functions for such a protein (Rao et al., 2014). The interaction of eukaryotic GAPDH with various proteins has been studied for many decades (Kumagai and Sakai, 1983; Glaser et al., 2002; Andrade et al., 2004; Azam et al., 2008; Qvit et al., 2016). On the other hand, similar knowledge on bacterial GAPDH is still very limited. To date, only a single reported study has focused on the identification of intracellular protein interactors of GAPDH derived from E. coli. Using immunopurification followed by MS, the authors of that study identified several potential GAPDH interactions, some of which indicated its involvement in DNA repair processes (Ferreira et al., 2013).
Our previously published results clearly indicate that the GAPDH homolog in pathogenic F. tularensis – GapA – might fulfill functions other than the enzymatic one. To gain deeper insights into the new intracellular functions of GapA, we performed a quantitative proteomic screen for the potential intracellular interaction partners of F. tularensis GapA. We took advantage of SILAC technology and designed an experimental workflow based on the affinity purification of tagged protein bait from the SILAC-labeled bacterial cultures followed by LC-MS/MS analysis. Such approach allows for eliminating the non-specific background that presents the major challenge to traditional AP-MS (Gingras et al., 2007). In this way, we were able to identify several bacterial proteins that seem to bind to the F. tularensis GapA. Interestingly, none of the proteins we found here had been identified in the similar study performed with E. coli GAPDH (Ferreira et al., 2013). This might be due to the different experimental approach and/or to differences in cellular processes across the various bacterial species. The interaction with six selected proteins was successfully validated by solid-phase ligand binding assay or bacterial two-hybrid assay. To further corroborate our findings, we additionally analyzed changes in the F. tularensis proteome induced by the gapA gene deletion.
A number of proteins identified in both our proteomic analyses are related to various metabolic processes. This finding is not surprising inasmuch as GAPDH is at the intersection of multiple metabolic pathways and its interaction with other enzymes may point to its regulatory role in these processes. In this context, the levels of some metabolic enzymes changed significantly in response to gapA deletion. The identification of proteins involved in amino acid metabolism points to the dispensability of glycolysis for F. tularensis demonstrated by several published studies, including ours (Raghunathan et al., 2010; Brissac et al., 2015; Pavkova et al., 2017; Radlinski et al., 2018). Francisella is able to utilize amino acids for energy production, especially during its intracellular lifestyle. As the main goal of our study was to obtain information about the GapA functions not related to metabolism, we paid much more attention to those proteins with no direct impact on metabolism.
The cysteine of GAPDH’s catalytic site is very sensitive to overoxidation by reactive H2O2, resulting in irreversible inactivation of its enzymatic activity. The active site is efficiently protected by S-thiolation mediated by glutathione in eukaryotes, Gram-negative and some Gram-positive bacteria, by bacillithiols in most Gram-positives (Kosova et al., 2017) or by mycothiols in Actinomycetes (Newton et al., 2008). This modification results in reversible decrease of catalytic activity, and reduction by glutaredoxins or thioredoxins is required for the regeneration of protein function. The S-thiolation protects GAPDH also against hypochlorite or sulfide stress (Imber et al., 2016; Peng et al., 2018). Here we demonstrate that thioredoxin is able to form a complex with F. tularensis GapA, thus being responsible for maintaining the reduced active protein form. The interaction of thioredoxin with GapA has also been reported in E. coli by an interactome analysis in vivo (Arts et al., 2016). We further observed that the ΔgapA mutant strain is more susceptible to H2O2 compared to the wild-type strain. GapA seems thus to contribute to cell protection against oxidative stress induced by H2O2. F. tularensis has at its disposal several functional antioxidative systems, and the fully virulent strains of type A and B are highly resistant against the destructive effect of H2O2 (Lindgren et al., 2007). The mammalian GAPDH is known to act as a redox sensor. The oxidative modification by hydrogen peroxide induces alterations in GAPDH oligomerization that result in its interaction with other proteins and/or translocation into other cellular compartments to fulfill its moonlighting activities. Whether the same is true for bacterial GAPDH homologs remains thus far unknown (Hancock et al., 2006; Russell et al., 2020). The enhanced deleterious effect of H2O2 on the ΔgapA viability may also be related to the decreased levels of OxyR, a transcriptional regulator of genes encoding primary antioxidant proteins, like glutathione, catalase, peroxidase, and others. In F. tularensis it regulates the oxidative stress response and provides resistance against reactive oxygen species (Binesse et al., 2015; Ma et al., 2016).
In bacteria, the hydrogen peroxide is able to induce oxidative lesions in the DNA molecule leading to cell death. The damaged DNA is repaired by several DNA repair mechanisms. The potential role of mammalian GAPDH in DNA repair was further indicated in several studies summarized by Kosova et al. (2017), and Ferreira et al. (2013, 2015) described this also for bacterial GAPDH. In E. coli, the GAPDH was shown to interact with proteins implicated in SOS response, the single-stranded DNA binding protein, as well as uracil-DNA-glycosylase and endonuclease IV from the base excision repair pathway. Moreover, the gapA-silenced E. coli cells were more susceptible compared to the wild-type cells to alkylating agents like methyl methanesulfonate but not to H2O2. In contrast, we were able to identify the UvrA protein from the NER pathway as a putative interaction partner of F. tularensis GapA. An absence of GapA, however, had no effect on F. tularensis susceptibility to UV irradiation or the alkylating agent methyl methanesulfonate that is the causative agent of DNA lesions repaired by base excision repair (data not shown). Notably, the GapA depletion affected the abundance of proteins of other reparation pathways inclusive of the aforementioned uracil-DNA. Taken together, we observe that GapA seemingly participates in DNA reparation processes in F. tularensis. Its involvement is not essential, however, and differs from that in E. coli.
The most potent GapA interaction partner detected in our protein interaction studies was the PepSY protein. The presented PepSY domain is widespread across all bacterial species. It occurs mainly in the N-terminal propeptide of the M4 peptidase family, where it seems to inhibit protease activity. Furthermore, this domain is present in a diverse family of secreted and cell wall-associated proteins, where its role has not yet been established. These proteins are hypothesized to contribute to the regulation of protease activity in the cell’s local environment, thus being important for pathogenesis and formation of microbial communities (Yeats et al., 2004). Because proteins with this domain are widely found in both pathogenic and non-pathogenic bacteria, they are not presumed to act as virulence factors. Accordingly, the PepSY-containing protein is not essential for virulence in the F. tularensis pathogenic strain. We further undertook to elucidate the role of its interaction with GapA. Because the role of PepSY in GapA secretion was not established in our experiments, we wondered whether, on the contrary, the GapA could assist in PepSY secretion. Similarly, the GAPDH homolog of Streptococcus pyogenes has been found to facilitate the export of Streptococcal pyogenic exotoxin B, a virulence factor with cysteine protease activity (Jin et al., 2011). Unexpectedly, the results of our comparative proteomics indicated that PepSY seems to be secreted only in the strain not expressing GapA. Thus it might be assumed that the GapA retains this protein inside the cell. The functional importance of this observation remains to be further examined.
This is the first study demonstrating the ability of F. tularensis GapA to interact with several bacterial proteins. The identified non-metabolic proteins point to several processes wherein the GapA might assist as a moonlighting protein. Such information is crucial for follow-up studies aimed at identifying further non-enzymatic functions of F. tularensis GapA at the intracellular level.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/pride/archive/, PXD019739.
Ethics Statement
The animal study was reviewed and approved by Ethical Committee of the Faculty of Military Health Sciences, University of Defence, Czechia.
Author Contributions
MK and IP designed and performed the research, analyzed the data, and wrote the manuscript. ML and PR performed the research and analyzed the data. JS reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Ministry of Defence of the Czechia – Long-term organization development plan Medical Aspects of Weapons of Mass Destruction of the Faculty of Military Health Sciences, University of Defence.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are very thankful to Prof. Ales Macela for critical reading of the manuscript and valuable advice and Lenka Luksikova for excellent technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.576618/full#supplementary-material
Footnotes
- ^ https://www.evotec.com/en/execute/science-pool/resources
- ^ https://www.uniprot.org/uniprot/A0A0B3VXZ9
References
Alvarez-Dominguez, C., Madrazo-Toca, F., Fernandez-Prieto, L., Vandekerckhove, J., Pareja, E., Tobes, R., et al. (2008). Characterization of a Listeria monocytogenes protein interfering with Rab5a. Traffic Cph. Den. 9, 325–337. doi: 10.1111/j.1600-0854.2007.00683.x
Andrade, J., Pearce, S. T., Zhao, H., and Barroso, M. (2004). Interactions among p22, glyceraldehyde-3-phosphate dehydrogenase and microtubules. Biochem. J. 384, 327–336. doi: 10.1042/BJ20040622
Arifuzzaman, M., Maeda, M., Itoh, A., Nishikata, K., Takita, C., Saito, R., et al. (2006). Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 16, 686–691. doi: 10.1101/gr.4527806
Arts, I. S., Vertommen, D., Baldin, F., Laloux, G., and Collet, J.-F. (2016). Comprehensively Characterizing the Thioredoxin Interactome In Vivo Highlights the Central Role Played by This Ubiquitous Oxidoreductase in Redox Control. Mol. Cell. Proteomics MCP 15, 2125–2140. doi: 10.1074/mcp.M115.056440
Auweter, S. D., Bhavsar, A. P., de Hoog, C. L., Li, Y., Chan, Y. A., van der Heijden, J., et al. (2011). Quantitative mass spectrometry catalogues Salmonella pathogenicity island-2 effectors and identifies their cognate host binding partners. J. Biol. Chem. 286, 24023–24035. doi: 10.1074/jbc.M111.224600
Azam, S., Jouvet, N., Jilani, A., Vongsamphanh, R., Yang, X., Yang, S., et al. (2008). Human Glyceraldehyde-3-phosphate Dehydrogenase Plays a Direct Role in Reactivating Oxidized Forms of the DNA Repair Enzyme APE1. J. Biol. Chem. 283, 30632–30641. doi: 10.1074/jbc.M801401200
Barel, M., and Charbit, A. (2013). Francisella tularensis intracellular survival: to eat or to die. Microbes Infect. 15, 989–997. doi: 10.1016/j.micinf.2013.09.009
Binesse, J., Lindgren, H., Lindgren, L., Conlan, W., and Sjöstedt, A. (2015). Roles of Reactive Oxygen Species-Degrading Enzymes of Francisella tularensis SCHU S4. Infect. Immun. 83, 2255–2263. doi: 10.1128/IAI.02488-14
Boël, G., Jin, H., and Pancholi, V. (2005). Inhibition of cell surface export of group A streptococcal anchorless surface dehydrogenase affects bacterial adherence and antiphagocytic properties. Infect. Immun. 73, 6237–6248. doi: 10.1128/IAI.73.10.6237-6248.2005
Boradia, V. M., Raje, M., and Raje, C. I. (2014). Protein moonlighting in iron metabolism: glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Biochem. Soc. Trans. 42, 1796–1801. doi: 10.1042/BST20140220
Brissac, T., Ziveri, J., Ramond, E., Tros, F., Kock, S., Dupuis, M., et al. (2015). Gluconeogenesis, an essential metabolic pathway for pathogenic Francisella. Mol. Microbiol. 98, 518–534. doi: 10.1111/mmi.13139
Butland, G., Peregrín-Alvarez, J. M., Li, J., Yang, W., Yang, X., Canadien, V., et al. (2005). Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433, 531–537. doi: 10.1038/nature03239
Chamberlain, R. E. (1965). Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl. Microbiol. 13, 232–235. doi: 10.1128/aem.13.2.232-235.1965
Ellis, J., Oyston, P. C. F., Green, M., and Titball, R. W. (2002). Tularemia. Clin. Microbiol. Rev. 15, 631–646. doi: 10.1128/cmr.15.4.631-646.2002
Ferreira, E., Giménez, R., Aguilera, L., Guzmán, K., Aguilar, J., Badia, J., et al. (2013). Protein interaction studies point to new functions for Escherichia coli glyceraldehyde-3-phosphate dehydrogenase. Res. Microbiol. 164, 145–154. doi: 10.1016/j.resmic.2012.11.002
Ferreira, E., Giménez, R., Cañas, M. A., Aguilera, L., Aguilar, J., Badia, J., et al. (2015). Glyceraldehyde-3-phosphate dehydrogenase is required for efficient repair of cytotoxic DNA lesions in Escherichia coli. Int. J. Biochem. Cell Biol. 60, 202–212. doi: 10.1016/j.biocel.2015.01.008
Gingras, A.-C., Gstaiger, M., Raught, B., and Aebersold, R. (2007). Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8, 645–654. doi: 10.1038/nrm2208
Glaser, P. E., Han, X., and Gross, R. W. (2002). Tubulin is the endogenous inhibitor of the glyceraldehyde 3-phosphate dehydrogenase isoform that catalyzes membrane fusion: implications for the coordinated regulation of glycolysis and membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 99, 14104–14109. doi: 10.1073/pnas.222542999
Hancock, J., Desikan, R., Harrison, J., Bright, J., Hooley, R., and Neill, S. (2006). Doing the unexpected: proteins involved in hydrogen peroxide perception. J. Exp. Bot. 57, 1711–1718. doi: 10.1093/jxb/erj180
Imber, M., Huyen, N. T. T., Pietrzyk-Brzezinska, A. J., Loi, V. V., Hillion, M., Bernhardt, J., et al. (2016). Protein S-Bacillithiolation Functions in Thiol Protection and Redox Regulation of the Glyceraldehyde-3-Phosphate Dehydrogenase Gap in Staphylococcus aureus Under Hypochlorite Stress. Antioxid. Redox Signal. 28, 410–430. doi: 10.1089/ars.2016.6897
Jeffery, C. J. (2009). Moonlighting proteins–an update. Mol. Biosyst. 5, 345–350. doi: 10.1039/b900658n
Jin, H., Agarwal, S., Agarwal, S., and Pancholi, V. (2011). Surface Export of GAPDH/SDH, a Glycolytic Enzyme, Is Essential for Streptococcus pyogenes Virulence. mBio 2:e00068-11. doi: 10.1128/mBio.00068-11
Karki, H., and Ham, J. H. (2016). Testing the Effect of UV Radiation on the Survival of Burkholderia glumae. BIO-Protoc. 6:e1755. doi: 10.21769/BioProtoc.1755
Klammer, M., Dybowski, J. N., Hoffmann, D., and Schaab, C. (2014). Identification of significant features by the Global Mean Rank test. PLoS One 9:e104504. doi: 10.1371/journal.pone.0104504
Kopeckova, M., Pavkova, I., and Stulik, J. (2020). Diverse Localization and Protein Binding Abilities of Glyceraldehyde-3-Phosphate Dehydrogenase in pathogenic bacteria: the key to its multifunctionality? Front. Cell. Infect. Microbiol. 10:89. doi: 10.3389/fcimb.2020.00089
Kosova, A. A., Khodyreva, S. N., and Lavrik, O. I. (2017). Role of Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) in DNA Repair. Biochem. Biokhimiia 82, 643–654. doi: 10.1134/S0006297917060013
Kumagai, H., and Sakai, H. (1983). A porcine brain protein (35 K protein) which bundles microtubules and its identification as glyceraldehyde 3-phosphate dehydrogenase. J. Biochem. 93, 1259–1269. doi: 10.1093/oxfordjournals.jbchem.a134260
Lindgren, H., Shen, H., Zingmark, C., Golovliov, I., Conlan, W., and Sjöstedt, A. (2007). Resistance of Francisella tularensis strains against reactive nitrogen and oxygen species with special reference to the role of KatG. Infect. Immun. 75, 1303–1309. doi: 10.1128/IAI.01717-06
Ma, Z., Russo, V. C., Rabadi, S. M., Jen, Y., Catlett, S. V., Bakshi, C. S., et al. (2016). Elucidation of a mechanism of oxidative stress regulation in Francisella tularensis live vaccine strain: regulation of oxidative stress resistance in Francisella tularensis. Mol. Microbiol. 101, 856–878. doi: 10.1111/mmi.13426
McCrumb, F. R., Snyder, M. J., and Woodward, T. E. (1957). Studies on human infection with Pasteurella tularensis; comparison of streptomycin and chloramphenicol in the prophylaxis of clinical disease. Trans. Assoc. Am. Physicians 70, 74–79.
Meibom, K. L., and Charbit, A. (2010). The unraveling panoply of Francisella tularensis virulence attributes. Curr. Opin. Microbiol. 13, 11–17. doi: 10.1016/j.mib.2009.11.007
Modun, B., and Williams, P. (1999). The Staphylococcal Transferrin-Binding Protein Is a Cell Wall Glyceraldehyde-3-Phosphate Dehydrogenase. Infect. Immun. 67, 1086–1092. doi: 10.1128/iai.67.3.1086-1092.1999
Newton, G. L., Buchmeier, N., and Fahey, R. C. (2008). Biosynthesis and Functions of Mycothiol, the Unique Protective Thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. MMBR 72, 471–494. doi: 10.1128/MMBR.00008-08
Ong, S.-E., Blagoev, B., Kratchmarova, I., Kristensen, D. B., Steen, H., Pandey, A., et al. (2002). Stable isotope labeling by amino acids in cell culture. SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics MCP 1, 376–386. doi: 10.1074/mcp.m200025-mcp200
Pancholi, V., and Chhatwal, G. S. (2003). Housekeeping enzymes as virulence factors for pathogens. Int. J. Med. Microbiol. IJMM 293, 391–401. doi: 10.1078/1438-4221-00283
Pancholi, V., and Fischetti, V. A. (1992). A major surface protein on group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J. Exp. Med. 176, 415–426. doi: 10.1084/jem.176.2.415
Pavkova, I., Kopeckova, M., Klimentova, J., Schmidt, M., Sheshko, V., Sobol, M., et al. (2017). The Multiple Localized Glyceraldehyde-3-Phosphate Dehydrogenase Contributes to the Attenuation of the Francisella tularensis dsbA Deletion Mutant. Front. Cell. Infect. Microbiol. 7:503. doi: 10.3389/fcimb.2017.00503
Peng, H., Zhang, Y., Trinidad, J. C., and Giedroc, D. P. (2018). Thioredoxin Profiling of Multiple Thioredoxin-Like Proteins in Staphylococcus aureus. Front. Microbiol. 9:2385. doi: 10.3389/fmicb.2018.02385
Qvit, N., Joshi, A. U., Cunningham, A. D., Ferreira, J. C. B., and Mochly-Rosen, D. (2016). Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) Protein-Protein Interaction Inhibitor Reveals a Non-catalytic Role for GAPDH Oligomerization in Cell Death. J. Biol. Chem. 291, 13608–13621. doi: 10.1074/jbc.M115.711630
Radlinski, L. C., Brunton, J., Steele, S., Taft-Benz, S., and Kawula, T. H. (2018). Defining the Metabolic Pathways and Host-Derived Carbon Substrates Required for Francisella tularensis Intracellular Growth. mBio 9:e01471-18. doi: 10.1128/mBio.01471-18
Raghunathan, A., Shin, S., and Daefler, S. (2010). Systems approach to investigating host-pathogen interactions in infections with the biothreat agent Francisella. Constraints-based model of Francisella tularensis. BMC Syst. Biol. 4:118. doi: 10.1186/1752-0509-4-118
Rao, V. S., Srinivas, K., Sujini, G. N., and Kumar, G. N. S. (2014). Protein-protein interaction detection: methods and analysis. Int. J. Proteomics 2014:147648. doi: 10.1155/2014/147648
Russell, G., Veal, D., and Hancock, J. T. (2020). Is Glyceraldehyde-3-Phosphate Dehydrogenase a Central Redox Mediator? React. Oxyg. Species 9, 48–69.
Sambrook, J. F., and Russell, D. (2001). Molecular Cloning: A Laboratory Manual (3-Volume Set). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Sancar, A., and Reardon, J. T. (2004). Nucleotide excision repair in E. coli and man. Adv. Protein Chem. 69, 43–71. doi: 10.1016/S0065-3233(04)69002-4
Steiner, D. J., Furuya, Y., and Metzger, D. W. (2014). Host–pathogen interactions and immune evasion strategies in Francisella tularensis pathogenicity. Infect. Drug Resist. 7, 239–251. doi: 10.2147/IDR.S53700
Terao, Y., Yamaguchi, M., Hamada, S., and Kawabata, S. (2006). Multifunctional glyceraldehyde-3-phosphate dehydrogenase of Streptococcus pyogenes is essential for evasion from neutrophils. J. Biol. Chem. 281, 14215–14223. doi: 10.1074/jbc.M513408200
Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M. Y., Geiger, T., et al. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. doi: 10.1038/nmeth.3901
Keywords: protein–protein interaction, multifunctional enzyme, Francisella tularensis, glyceraldehyde-3-phosphate dehydrogenase, SILAC
Citation: Kopeckova M, Pavkova I, Link M, Rehulka P and Stulik J (2020) Identification of Bacterial Protein Interaction Partners Points to New Intracellular Functions of Francisella tularensis Glyceraldehyde-3-Phosphate Dehydrogenase. Front. Microbiol. 11:576618. doi: 10.3389/fmicb.2020.576618
Received: 26 June 2020; Accepted: 19 August 2020;
Published: 10 September 2020.
Edited by:
Haike Antelmann, Freie Universität Berlin, GermanyReviewed by:
Xiangmin Lin, Fujian Agriculture and Forestry University, ChinaVladimir Muronetz, Lomonosov Moscow State University, Russia
Copyright © 2020 Kopeckova, Pavkova, Link, Rehulka and Stulik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivona Pavkova, aXZvbmEucGF2a292YUB1bm9iLmN6