 Rong Li1†
Rong Li1† Yong Feng2†
Yong Feng2† Haifeng Chen1Chanjuan Zhang1
Haifeng Chen1Chanjuan Zhang1 Yi Huang1
Yi Huang1 Limiao Chen1
Limiao Chen1 Qingnan Hao1Dong Cao1
Qingnan Hao1Dong Cao1 Songli Yuan1*
Songli Yuan1* Xinan Zhou1*
Xinan Zhou1*- 1Key Laboratory of Biology and Genetic Improvement of Oil Crops, Ministry of Agriculture and Rural Affairs of PRC, Oil Crops Research Institute of Chinese Academy of Agriculture Sciences, Wuhan, China
- 2State Key Laboratory of Agricultural Microbiology, Huazhong Agricultural University, Wuhan, China
In the present study, we sequenced the complete genome of Bradyrhizobium diazoefficiens 113-2. The genomic characteristics of six selected rhizobial strains (two fast-growing rhizobia, two medium-slow-growing rhizobia and two slow-growing rhizobia) with four different legume hosts were analyzed by comparative genomic analysis. Genomes of B. diazoefficiens 113-2 and B. diazoefficiens USDA110 were found to share a large synteny blocks and a high ANI value, supporting 113-2 as a strain of B. diazoefficiens. 5,455 singletons and 11,656 clusters were identified among the six rhizobia genomes, and most of the pair-wise comparisons clusters were shared by the two genomes of strains in the same genus. Similar genus-specific gene numbers in the assigned COG functional terms were present in the two strains of the same genus, while the numbers were decreased with the increase of growth rate in most of the COG terms. KEGG pathway analysis of B. diazoefficiens 113-2 suggested that the rhizobial genes in ABC transporters and Two-Component system were mainly species-specific. Besides, the candidate genes related to secretion system and surface polysaccharides biosynthesis in the genomes of the six strains were explored and compared. 39 nodulation gene families, 12 nif gene families and 10 fix gene families in the genomes of these six strains were identified, and gene classes in most of gene families and the types and total gene numbers of gene families were substantially different among these six genomes. We also performed synteny analyses for above-mentioned nod, nif, and fix gene groupings, and selected NodW, NolK, NoeJ, NifB, FixK, and FixJ gene families to perform phylogeny analyses. Our results provided valuable molecular insights into species specificity and host specificity. The genetic information responsible for host specificity will play important roles in expanding the host range of rhizobia among legumes, which might provide new clues for the understanding of the genetic determinants of non-legume-rhizobium symbiosis.
Introduction
Nitrogen-fixing symbioses between legumes and rhizobia provide the legume host with a large fraction of reduced atmospheric nitrogen in exchange for carbon source and shelter inside symbiosis-specific root nodules (Friesen, 2012). The efficiency of such cross-kingdom collaboration is mainly attributed to the symbiotic matching (symbiotic specificity), which is always associated with distinct nodulation phenotype (Jones et al., 2008; Hayashi et al., 2012; Yuan et al., 2016), leading to the existence of different legume-rhizobium associations. For example, Mesorhizobium loti MAFF303099 forms specific symbiosis with several host plants of Lotus (Estrella et al., 2009), Mesorhizobium huakuii 7653R can only form symbiosis with Astragalus sinicus (Wang et al., 2014), and Sinorhizobium meliloti can only nodulate Medicago, Melilotus, and Trigonella (Biondi et al., 2003; Radutoiu et al., 2007). The symbiotic specificity may be determined by a fine-tuned exchange of molecular signals between a host root and its inoculated rhizobial strains (Perret et al., 2000). These signals mainly include nodulation factors (NFs) (Lerouge et al., 1990; Schultze et al., 1992), surface polysaccharides (Skorupska et al., 2006; Jones et al., 2008) and secreted proteins/type III secretion system (T3SS) (Fauvart and Michiels, 2008; Okazaki et al., 2013; Nelson and Sadowsky, 2015). Lots of genes that affect the biological synthesis of these signaling molecules in the genomes of different strains have been explored by comparative genomics (Tian et al., 2012; Wang et al., 2014), and gene transfer between related taxa can alter the host range of symbionts (Temprano-Vera et al., 2018). Therefore, identifying the determinants responsible for host specificity plays important roles in expanding the host range of rhizobium.
According to the growth rate, rhizobia can be divided into fast-growing rhizobia (Rhizobium) (Keyser et al., 1982), slow-growing rhizobia (Bradyrhizobium) (Tampakaki et al., 2017) and medium-slow-growing rhizobia (Mesorhizobium) (Wang et al., 2014). Various genetic and environmental factors as well as the number of rRNA operons affect growth rates (Shrestha et al., 2007; Temprano-Vera et al., 2018; Cherni and Perret, 2019), yet carbon metabolism in cells with multiple carbon sources and high extent of carbon utilization in fast-growing rhizobia maybe tend to grow faster than others (Marsudi et al., 1999; Ansari and Rao, 2014). Compared with Bradyrhizobium, most of the Rhizobium have lower energy consumption as well as better environmental adaptability and nodulation competitiveness (Marsudi et al., 1999). Mesorhizobium, whose growth rate is intermediate between that of Rhizobium and Bradyrhizobium, is a genus of rhizobium with a narrow host range (Streit et al., 2004). An improved understanding of the genetic information differences among these rhizobia will provide molecular insights into understanding the characteristics of these three genera of rhizobia.
Bradyrhizobium diazoefficiens 113-2, a broad-host-range and highly efficient soybean rhizobium (isolated from soybean “monkey hair”), was collected from paddy fields in Hengyang area of Hunan Province, China in 1972 by Xuejiang Zhang, and it has been applied in sustainable agriculture in China, United States, and Canada. In our previous studies, B. diazoefficiens 113-2 had higher symbiotic matching abilities than B. diazoefficiens USDA110 and Sinorhizobium fredii USDA205 with soybean ‘Tianlong 1’ (Li et al., 2017b). The comparative analysis of symbiotic phenotypes of soybean ‘Tianlong 1’ with B. diazoefficiens 113-2 and S. fredii USDA205 (Li et al., 2017b) and the RNA-Seq analysis of differential gene expression responding to B. diazoefficiens 113-2 and S. fredii USDA205 in soybean roots (Yuan et al., 2016) have also been extensively studied. However, the genetic information of rhizobium responsible for the phenotypic differences among 113-2-soybean, B. diazoefficiens USDA110-soybean and USDA205-soybean associations, and different symbiotic matching abilities between 113-2-soybean and USDA205-soybean associations remains unclear, so comparative genomic analysis between B. diazoefficiens 113-2, B. diazoefficiens USDA110 and S. fredii USDA205 is an good ideal for discovering the genetic information of rhizobium related to the above-mentioned phenomenon.
In the present study, we investigated the entire genomic information of B. diazoefficiens 113-2 and provided useful insights into this strain’s symbiosis and its host-plant molecular interaction. Moreover, the comparative genomic investigation between B. diazoefficiens 113-2, B. diazoefficiens USDA110, M. huakuii 7653R, Mesorhizobium japonicum MAFF303099, S. fredii USDA205 and S. meliloti 2011 provided valuable insights into the species specificity and host specificity among different rhizobia.
Results
Complete Sequencing of the B. diazoefficiens 113-2 Genome
In the present study, a PacBio RS II platform and Illumina HiSeq 4000 platform were used to sequence the genome of B. diazoefficiens 113-2 in order to systematically investigate this strain’s symbiosis and its host-plant molecular interactions. The total sequence of the B. diazoefficiens 113-2 genome was 8,995,154 bp in length, consisting of only one chromosome (Figure 1). The GC content of the whole genome was 64.1% and shown on the circle map of the B. diazoefficiens 113-2 genome (Figure 1). Table 1 summarizes previously sequenced main genome characteristics of B. diazoefficiens 113-2 as well as the genomes of five other strains (B. diazoefficiens USDA110, M. huakuii 7653R, M. japonicum MAFF303099, S. fredii USDA205 and S. meliloti 2011). These six strains belonged to different genera and had different host plants. M. huakuii 7653R, M. japonicum MAFF303099 and S. meliloti 2011 have two plasmids each, B. diazoefficiens 113-2 and B. diazoefficiens USDA110 have none, and S. fredii USDA205 only have 255 contigs.
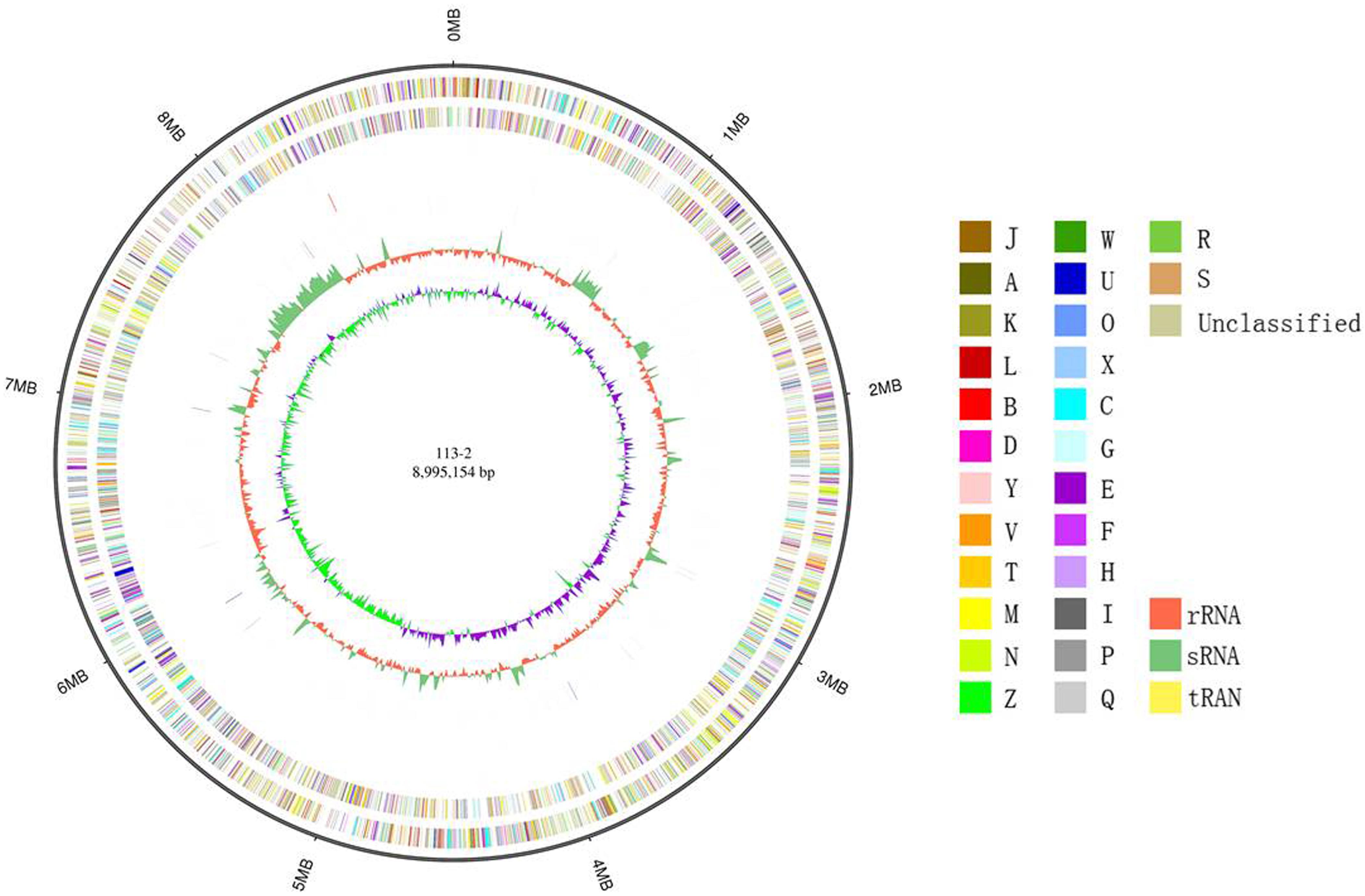
Figure 1. Circle map of the complete B. diazoefficiens 113-2 genome. Displayed circles from outer to inner: genome size (ring 1), forward strand gene, colored according to COG classification (ring 2), reverse strand gene, colored according to COG classification (ring 3), forward strand ncRNA (ring 4), reverse strand ncRNA (ring 5), repeat (ring 6), GC (ring 7), and GC-SKEW (ring 8).
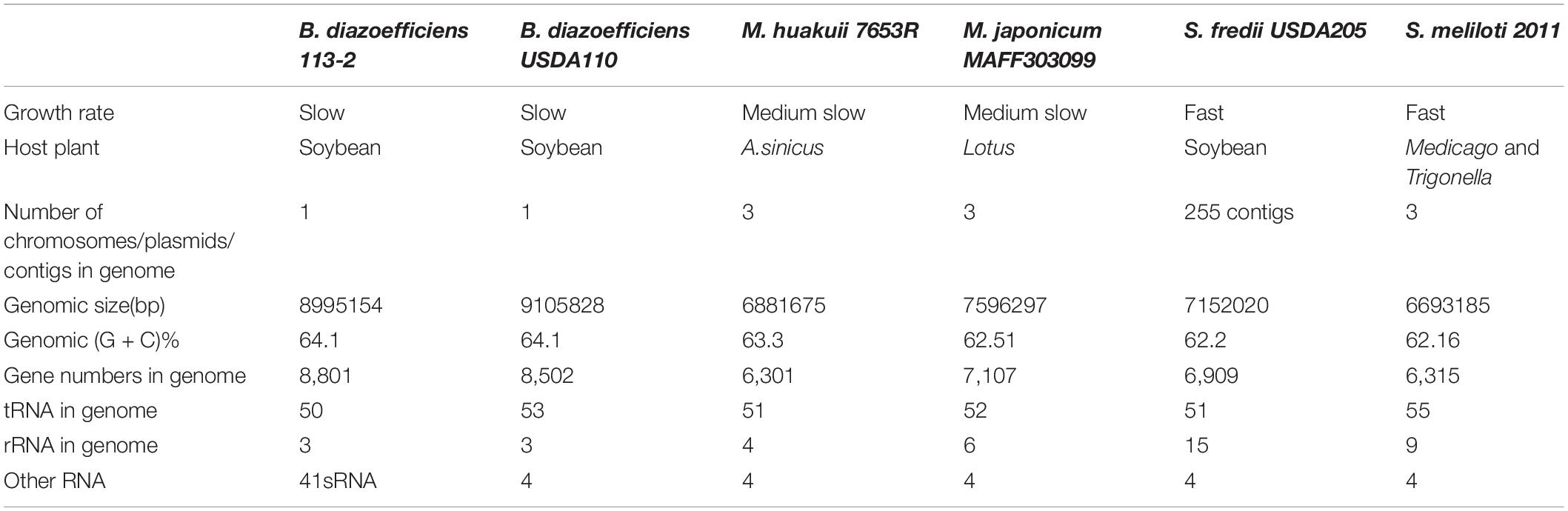
Table 1. General feature of B. diazoefficiens 113-2 and five other rhizobia genomes.
In order to study the characteristics and functions of B. diazoefficiens 113-2, we analyzed most of its genomic components, including gene, non-coding RNA, repeat sequence and prophage (Table 2). We predicted 8,801 genes in the B. diazoefficiens 113-2 genome (Supplementary Table S1), which was the highest among the six genomes (Table 1). The numbers of genes were basically consistent with the trend of genome size. However, in the Bradyrhizobium genus, the genome size of B. diazoefficiens 113-2 was smaller compared with B. diazoefficiens USDA110, while its gene number was greater compared with B. diazoefficiens USDA110 (Table 1). We predicted the numbers and types of rRNAs, tRNAs, and sRNAs of B. diazoefficiens 113-2 genome (Table 2 and Supplementary Table S2), and found that both the soybean Bradyrhizobium genomes and M. huakuii 7653R and M. japonicum MAFF303099 genomes had essentially identical numbers of rRNAs and tRNAs, while S. meliloti 2011 and S. fredii USDA205 genomes had dramatically different numbers of these RNAs (Table 1). Besides, we examined the species composition in terms of tandem repeat sequences (Supplementary Table S3) and environmental adaptability-related prophage of B. diazoefficiens 113-2 genome (Table 2).
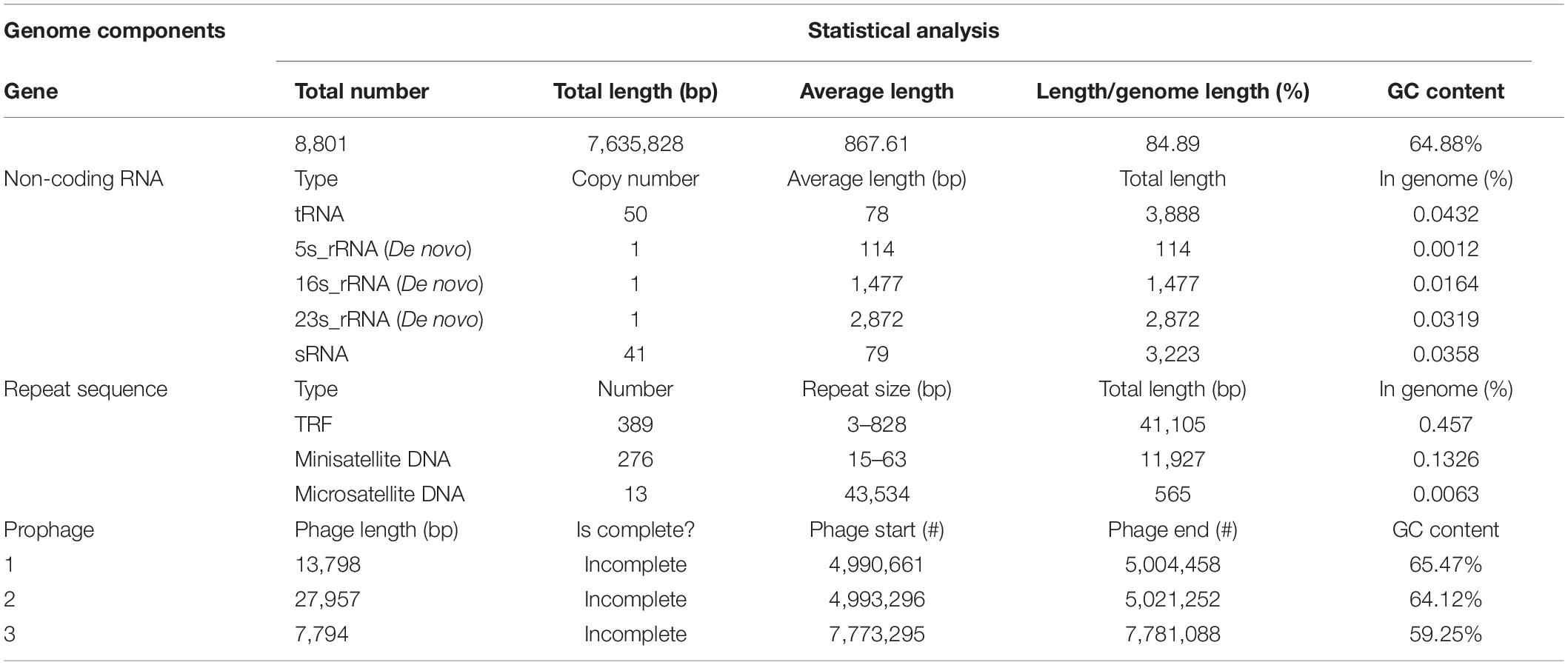
Table 2. Genome Component statistical analyses of B. diazoefficiens 113-2.
To evaluate the putative functions of B. diazoefficiens 113-2 gene set and provide clues for further research on finding target functional genes, we annotated the B. diazoefficiens 113-2 genome with 11 databases, including COG (Cluster of Orthologous Groups of proteins), GO (Gene Ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes), NR (No-Redundant Protein Database), Swiss-Prot (O’Donovan et al., 2002), IPR, T3SS (Type III secretion system Effector protein), PHI (Pathogen Host Interactions), VFDB (virulence factor database), ARDB (Antibiotic Resistance Genes Database), and CAZY (Carbohydrate-Active enZYmes Database), and Supplementary Table S4 lists the detailed information. Supplementary Table S5 illustrates the number and proportion of different B. diazoefficiens 113-2 gene sets annotated in each database. The results showed that 97% genes of B. diazoefficiens 113-2 had annotated functions, and the length of the most unannotated genes was less than 500 bp, suggesting that almost all meaningful predictive genes of B. diazoefficiens 113-2 had annotated functions.
Genome-Wide Synteny and ANI Analysis Among the Six Rhizobial Strains
To examine the phylogenetic relationships among the six strains, which belong to different genera and have different host plants, we firstly performed a synteny analysis based on the genome sequences of the above-mentioned five strains (except for S. fredii USDA205 with incompletely assembled genome) (Figure 2A). B. diazoefficiens 113-2 genome shared larger synteny blocks with B. diazoefficiens USDA110 compared with the other four strains. The gene consistency between the two strains in the same genus was higher than that in the different genera, and very few synteny blocks were shared between rhizobia of different genera.
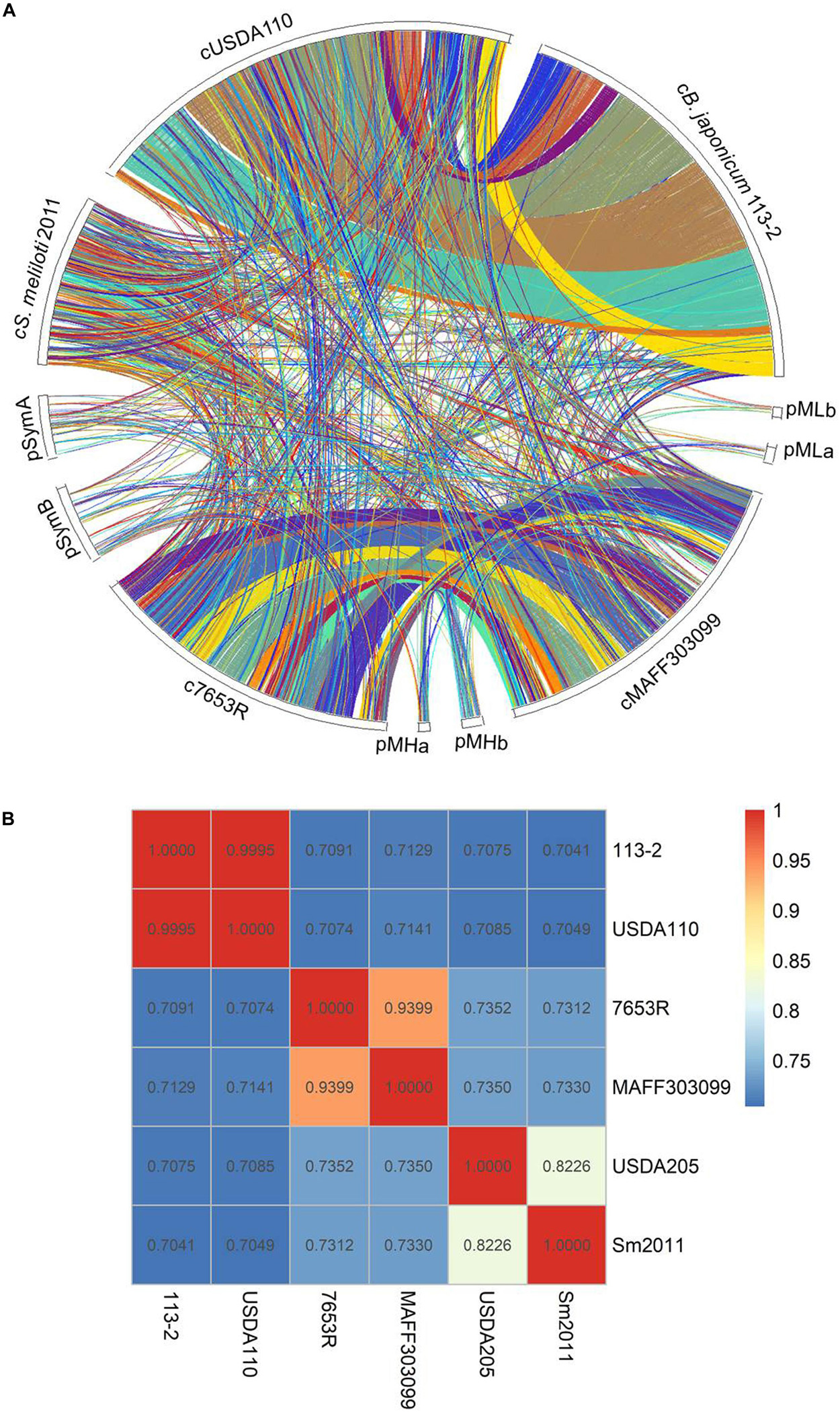
Figure 2. The circle map of genome synteny analysis and ANI analysis. (A) The circle map of genome synteny analysis among five rhizobial strains. Each colored block represents a synteny block and is internally independent from genomic rearrangement. (B) Summary of ANI calculations for the six rhizobial strains. The Average Nucleotide Identity (ANI) between the genome of B. diazoefficiens 113-2, M. huakuii 7653R, M. japonicum MAFF303099, S. meliloti 2011, B. diazoefficiens USDA110, and S. fredii USDA205 evaluated using the ANI Calculator.
Secondly, we carried out ANI analysis among these six rhizobial strains, and calculated the ANI values of each two rhizobial strains based on the nucleotide sequences (Figure 2B). M. huakuii 7653R and M. japonicum MAFF303099 were two strains in the same species (Wang et al., 2014), and the ANI value between these two strains was 0.9399. The ANI value between B. diazoefficiens 113-2 and B. diazoefficiens USDA110 was 0.9995, which even higher than that of M. huakuii 7653R and M. japonicum MAFF303099, indicating that these two strains were also in the same species. S. fredii USDA205 and S. meliloti 2011 were two strains in the fast-growing rhizobia genus, and the ANI value between these two strains was 0.8226. Besides, all of the ANI values between the two strains in different genera were less than 0.75, suggesting that relative lower correlation between the genomes of strains in different genera.
Genome-Wide Ortholog Analysis Among the Six Rhizobial Strains
We compared the six genomes and identified the singletons of each strain and the numbers of shared clusters of each strain (Table 3). 1839 core-clusters were identified and similar numbers of shared variable-clusters predicted in the two strains in the same genus. There were significant differences in the proportions of singletons, which were mainly increased as the growth rate of bacteria was increased (Table 3). In the group of fast-growing rhizobia, about 19.3% (1,211) of the proteins in S. meliloti 2011 and 18.3% (1,094) in S. fredii USDA205 were singletons. In the medium-slow rhizobia group, about 13.9% (973) of the proteins in M. japonicum MAFF303099 and 10.3% (680) in M. huakuii 7653R were singletons. In the Bradyrhizobium genus, about 12.3% (1081) of the proteins in B. diazoefficiens 113-2 were singletons, while only 5.2% (416) in B. diazoefficiens USDA110 (Table 3), suggesting that B. diazoefficiens 113-2 had more unique functions compared with B. diazoefficiens USDA110. The detailed protein ID information of these singletons was shown in Supplementary Table S6.
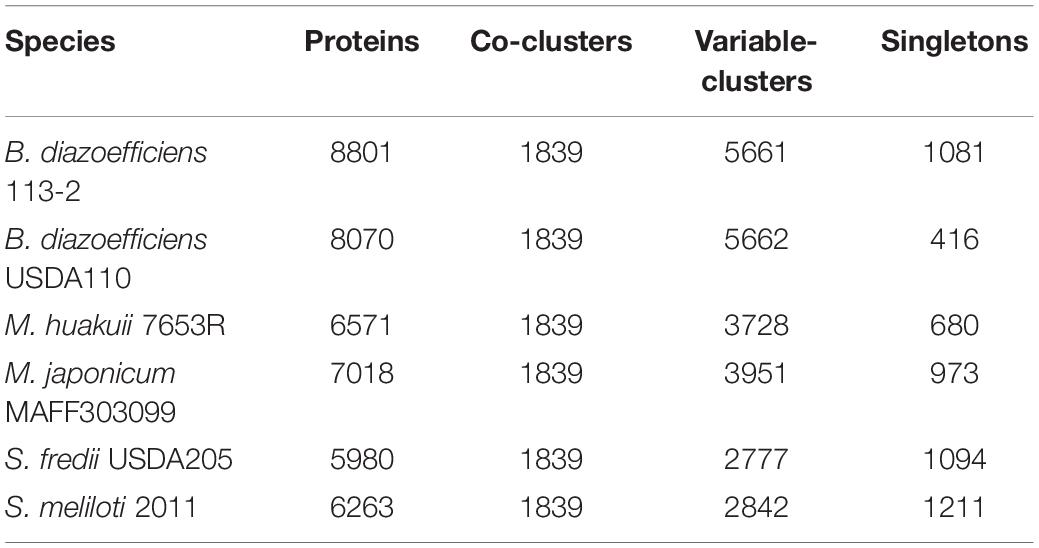
Table 3. The cluster-singleton analysis of the six rhizobial strains genomes.
We identified 11,656 clusters among the six rhizobia genomes (Figure 3), and the proteins in each cluster were shown in Supplementary Table S7. Of these clusters, 1839 (15.8%) clusters (including 1,615 single-copy gene clusters) were found to be shared by all of the six strains genomes and 266 clusters were existed in only one strain genome. An additional 383, 1,570, and 964 clusters were shared by five, four, and three of the six genomes, respectively. The remaining 6,634 clusters were observed to be present in two of the six genomes, among all these pair-wise comparisons, the 113-2-USDA110 pair was found to share the most abundant clusters (3978, 60.0%), followed by the 7653R-MAFF303099 (1,631, 24.6%) and S. meliloti 2011-S. fredii USDA205 (806, 12.1%), and there were very few clusters shared by the two genomes of strains in different genera.
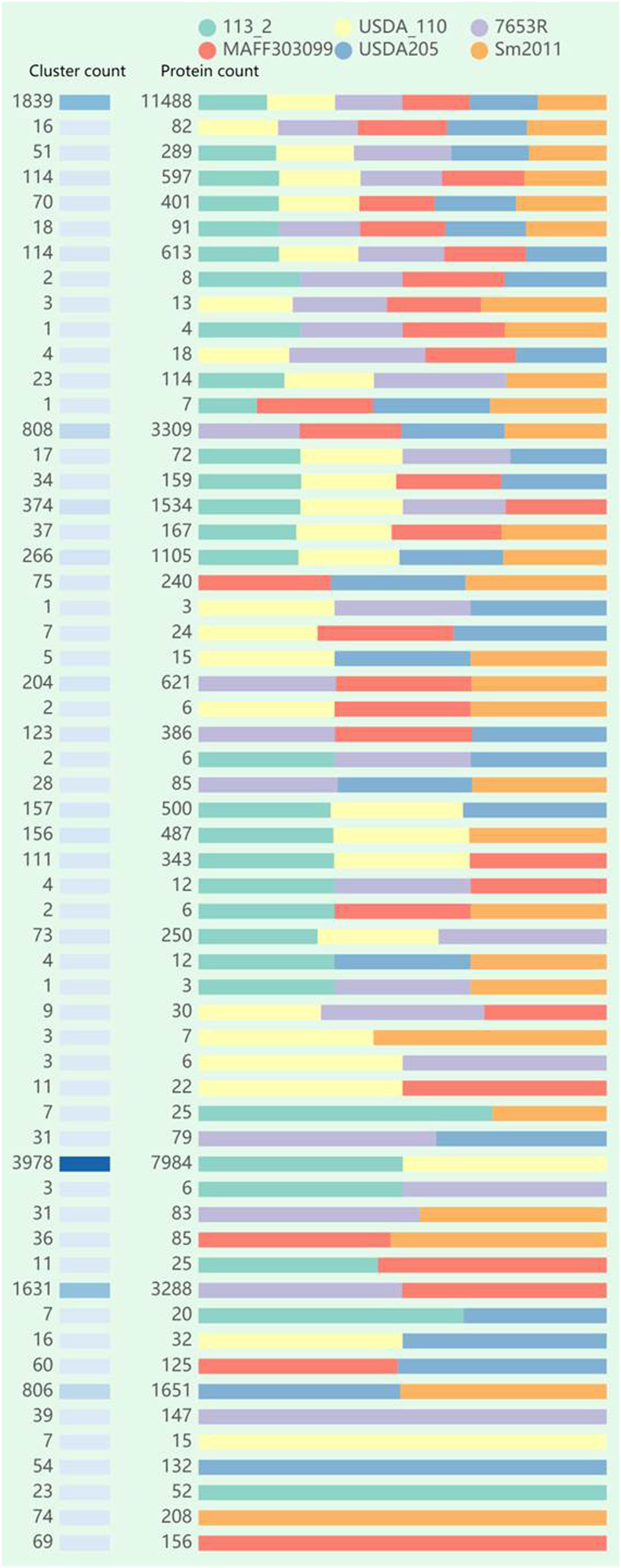
Figure 3. Summary of the distribution of orthologous clusters and protein data among the six rhizobial strains using OrthoVenn2.
COG Function Classification of Singletons and Clusters Genes in the Six Rhizobial Strains
To investigate whether the strain specificity and species specificity were related to the difference of protein numbers involved in various biological processes in rhizobia, we analyzed the COG assignments of the core-clusters genes, strain unique-clusters genes, species specificity-clusters genes and singletons (Figure 4), and Supplementary Table S8 lists the detailed gene ID information and annotation information. Similar numbers of core-clusters genes predicted in these COG functional terms were present in these six genomes, and the functions mainly focused on amino acid transport and metabolism (E), energy production and conversion (C), translation, ribosomal structure and biogenesis (J), Inorganic ion transport and metabolism (P), and Transcription (K) (Figure 4A), whereas there were significant differences in the numbers of singletons and strain unique-clusters genes (Figures 4B,C). The B. diazoefficiens 113-2 was found to have the highest proportion (about 82.1%) of the not annotated or Function unknown (S) singletons, and in most COG terms (15 out of 20), the numbers of singletons in S. meliloti 2011 or S. fredii USDA205 were more compared with the other strains, especially for energy production and conversion (C), amino acid transport and metabolism (E), carbohydrate transport and metabolism (G), transcription (K), and inorganic ion transport and metabolism (P) (Figure 4B). Compared with singletons, the strain unique-clusters genes did not assigned in the three COG functional terms (D, cell cycle control/cell division/chromosome partitioning; N, Cell motility; U, Intracellular trafficking/secretion/vesicular transport). All of the unique-clusters genes in B. diazoefficiens USDA110 were not annotated or Function unknown (S), half of the unique-clusters genes (26 out 52) in B. diazoefficiens 113-2 were annotated and predicted to have the function of Replication, recombination and repair (L), and more unique-clusters genes and more assigned COG functional terms of the rest four strains (Figure 4C). Similar gene numbers in these COG functional terms were present in the two strains of the same genus, while the numbers were decreased with the increase of growth rate (except for D, cell cycle control/cell division/chromosome partitioning; F, Nucleotide transport and metabolism; L, Replication, recombination and repair; N, Cell motility and U, intracellular trafficking/secretion/vesicular transport) (Figure 4D).
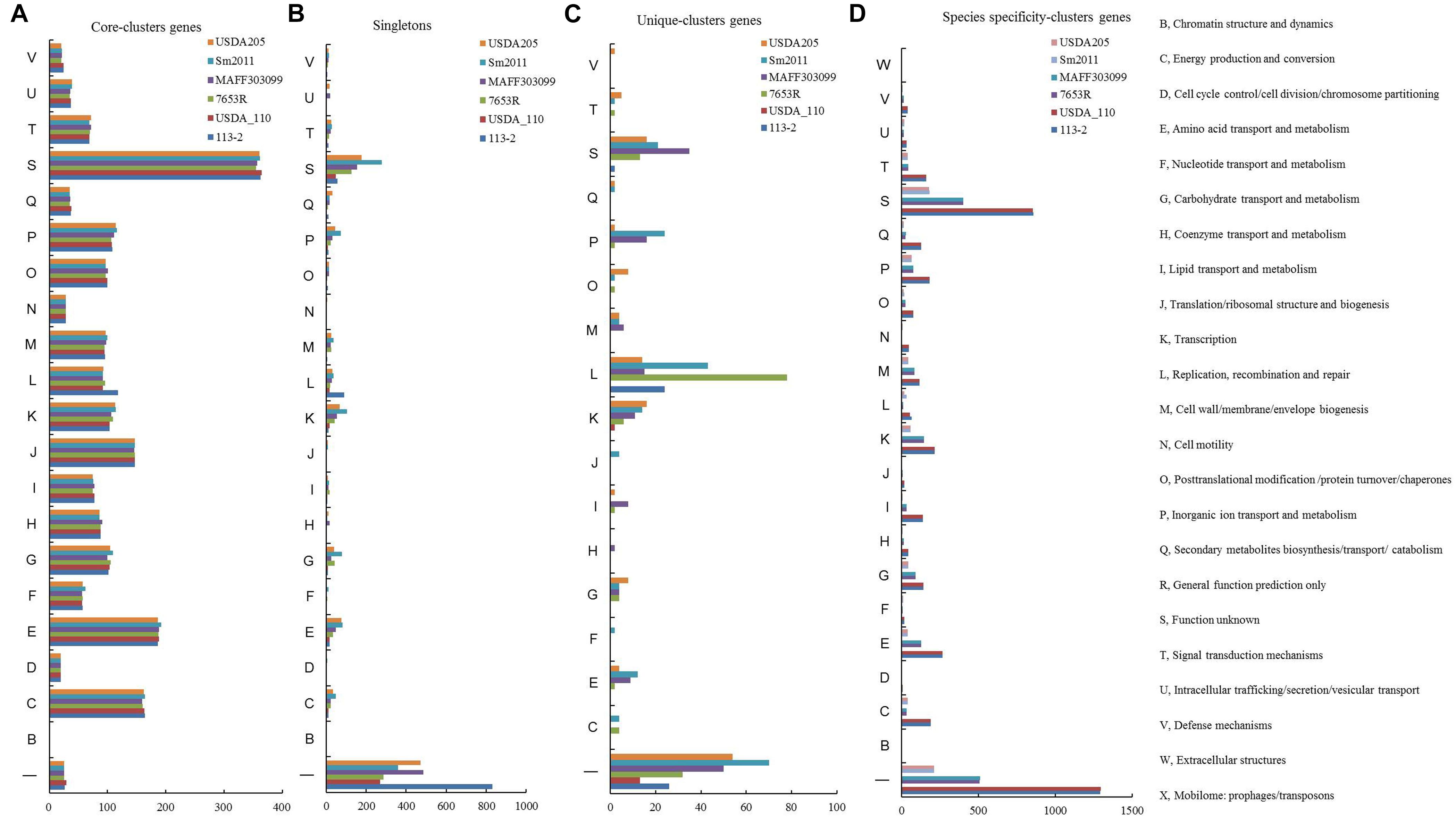
Figure 4. Cluster of Orthologous Groups of proteins (COG) functional classification of singletons and clusters genes in the six rhizobial strains. (A) COG functional classification of the core-clusters genes in the six rhizobial strains. (B) COG functional classification of singletons in the six rhizobial strains. (C) COG functional classification of the Unique-clusters genes in the six rhizobial strains. (D) COG functional classification of the species specificity-clusters genes in the six rhizobial strains.
KEGG Pathways Analysis of B. diazoefficiens 113-2
Kyoto Encyclopedia of Genes and Genomes is the major public pathway-related database, and a total of 26 KEGG pathways were listed in Figure 5A and divided into five categories as follows: cellular processes, environmental information processing, genetic information processing, metabolism and organismal systems. Most of the annotated genes were attributed to metabolism pathways, and the associated pathways primarily contained amino acid metabolism, carbohydrate metabolism, energy metabolism and global and overview maps. These results confirmed a preference for metabolism of amino acid, carbohydrates and energy.
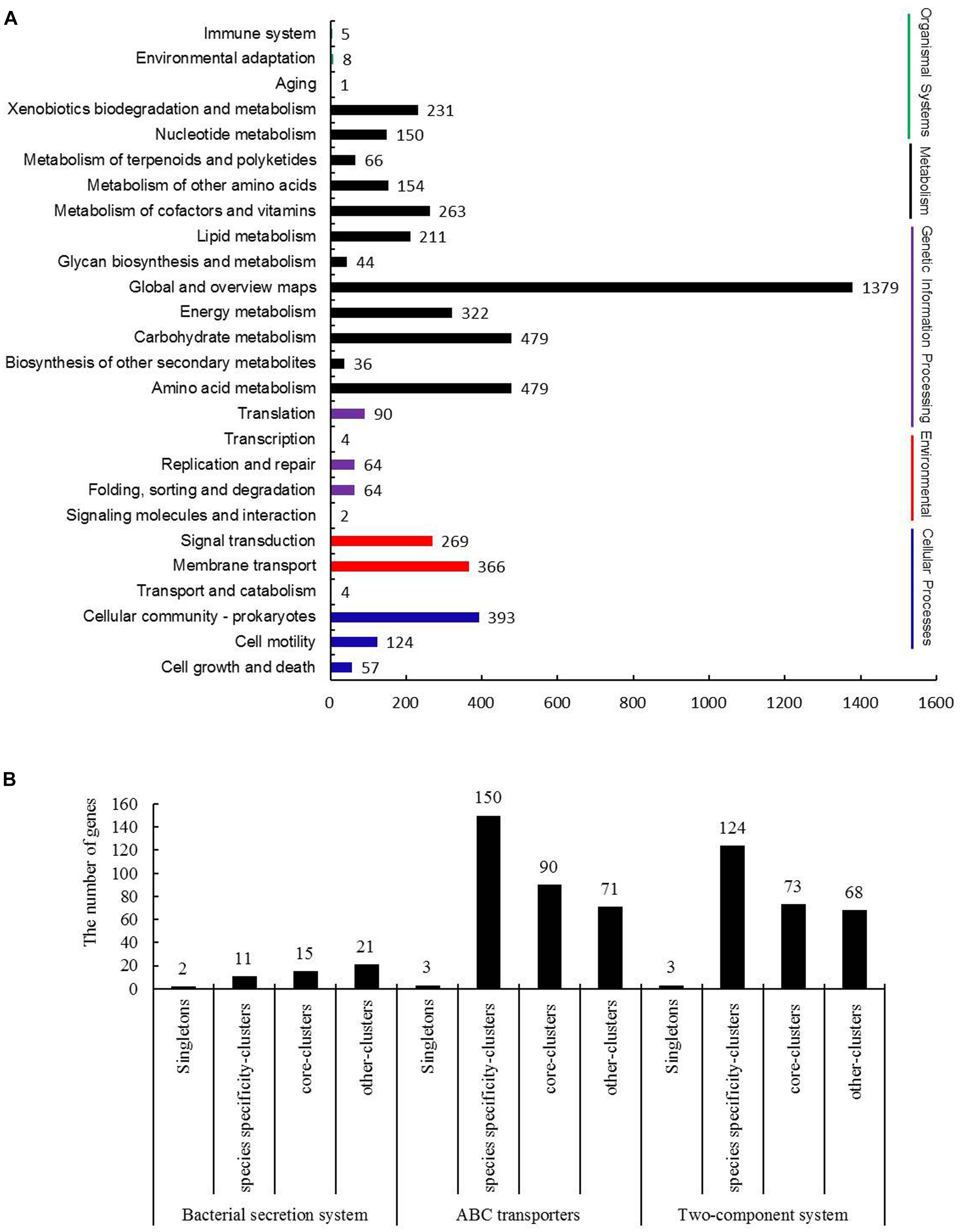
Figure 5. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of B. diazoefficiens 113-2. (A) KEGG annotation of B. diazoefficiens 113-2. (B) Three KEGG pathways analysis of singletons and clusters genes of B. diazoefficiens 113-2.
To investigate whether the strain specificity and species specificity were related to the difference of protein numbers involved in various KEGG pathways in rhizobia, we mainly analyzed Bacterial secretion system (k03060), ABC transporters (k02010), and Two-Component system (k02020) of the core-clusters genes, strain unique-clusters genes, species specificity-clusters genes and singletons of B. diazoefficiens 113-2 (Figure 5B). The detailed gene ID information of the pathway genes was shown in Supplementary Table S9. The numbers of singletons for bacterial secretion system, ABC transporters and two-component system pathways were two, three and three, respectively, no strain unique-clusters genes for these three pathways, and about 27.3 ∼ 28.8% genes were core-clusters genes in these three pathways. The numbers of species specificity-clusters genes for bacterial secretion system was 11 (22.4%), however, the numbers for ABC transporters and two-component system pathways were 150 (47.8%) and 124 (46.3%), respectively (Figure 5B), indicating that the rhizobial genes in these two pathways were mainly species-specific.
Host Specificity Analysis
The above-mentioned six genomes displayed drastically different host (Table 1). Because NFs, surface polysaccharides and secreted proteins are important determinants of host specificity of a rhizobium (Fauvart and Michiels, 2008), we explored genes that affect the biological synthesis of these signaling molecules in the genomes of these six strains.
Secretion System
Proteins secreted by rhizobial strains are necessary for beneficial symbiosis establishment (Wang et al., 2014). By means of gene families searches using secretion proteins of B. diazoefficiens 113-2 identified in the Bacterial secretion system (Figure 5B) as queries, we identified the genes related to secretory processes in the six strain genomes. We mainly analyzed two separate type-I systems, three type-II systems, type-III system, three type-VI systems, a twin-arginine (TAT) secretion system, a OmpA/MotB domain protein system, a flagellar-related protein system and a TraG family system (Figure 6). For the three strains that nodulate soybean, similar numbers of proteins in these analyzed secretion systems are present in B. diazoefficiens 113-2 and B. diazoefficiens USDA110, while S. fredii USDA205 had different types and numbers of secretion proteins with the other two strains. Different numbers of secreted proteins of most of the analyzed secretion systems (except for TolC, HlyD, TAT, and flagellar-related protein systems) were present in M. huakuii 7653R, M. japonicum MAFF303099, S. fredii USDA205, and S. meliloti 2011. Besides, M. huakuii 7653R had the same number of secreted proteins as M. japonicum MAFF303099 in type-III system, and S. fredii USDA205 had the same number of secreted proteins as S. meliloti 2011 in Sec pathway system. The detailed information of these secreted proteins was shown in Supplementary Table S10.
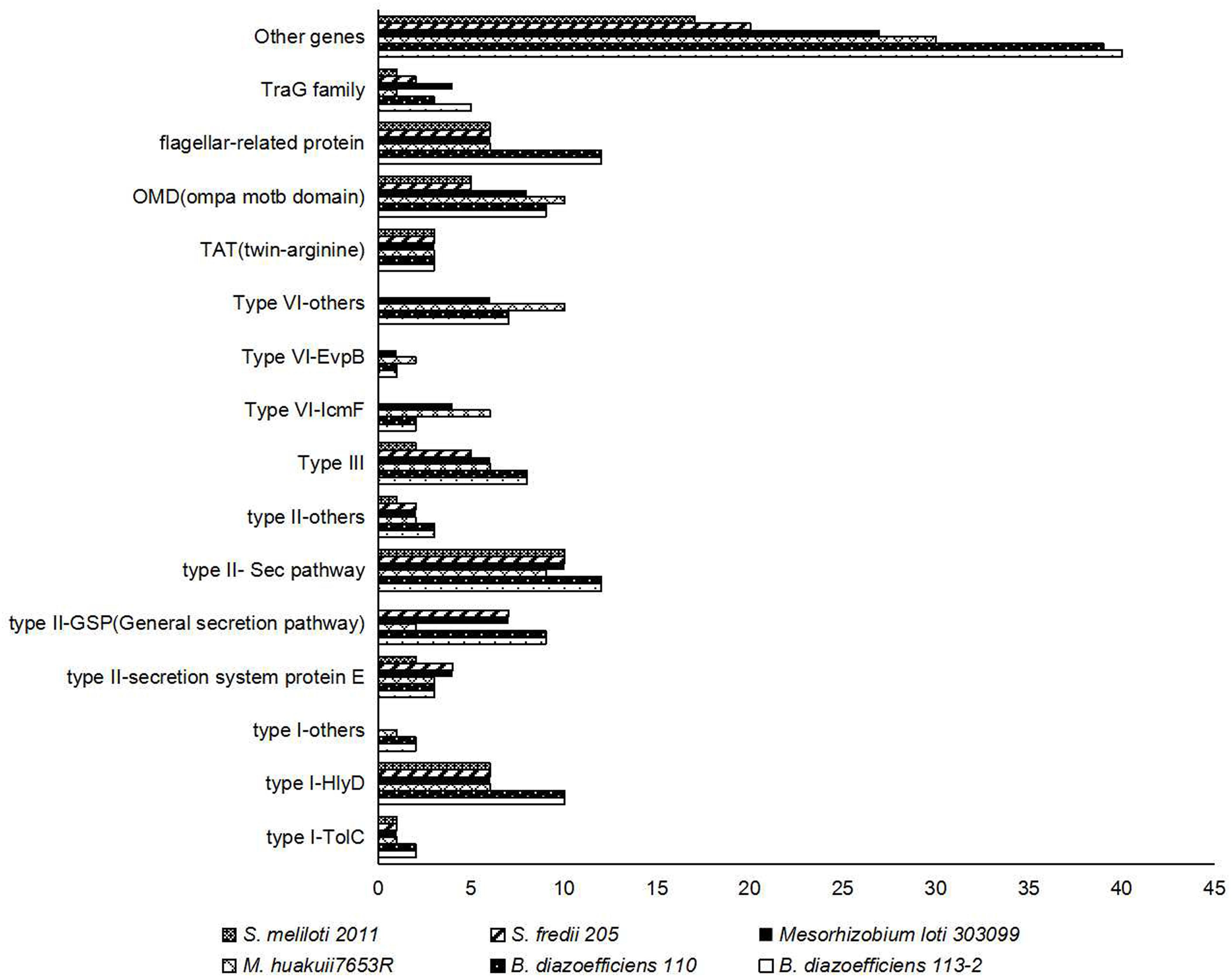
Figure 6. Numbers and distributions of genes related to different types of secretion systems in the six strain genomes.
Surface Polysaccharides Biosynthesis
Rhizobial cell-surface polysaccharidess, especially for exopolysaccharides (EPSs) and lipo- polysaccharides (LPSs), play important roles in establishing effective RNS with their hosts (Janczarek et al., 2010). We used the genes related to the biosynthesis of EPSs and LPSs identified in M. huakuii 7653R (Wang et al., 2014) as queries to identify the families of these genes. We explored and compared 19 EPS biosynthesis gene families (Figure 7A and Supplementary Table S11) and 17 LPS biosynthesis gene families in the genomes of the six strains (Figure 7B and Supplementary Table 11). Among them, four LPS biosynthesis gene (LpxB, LpxC, LpxXL, and AcpXL) families had the same numbers in all of the six strains. For the three strains that nodulate soybean, S. fredii USDA205 had different numbers of most of the genes related to surface polysaccharides biosynthesis with the other two strains. Besides, most of the genes had the similar numbers in the two strains of the same genus, while vary different between rhizobia of different genera. The detailed ID information of these genes related to surface polysaccharides biosynthesis was shown in Supplementary Table S11.
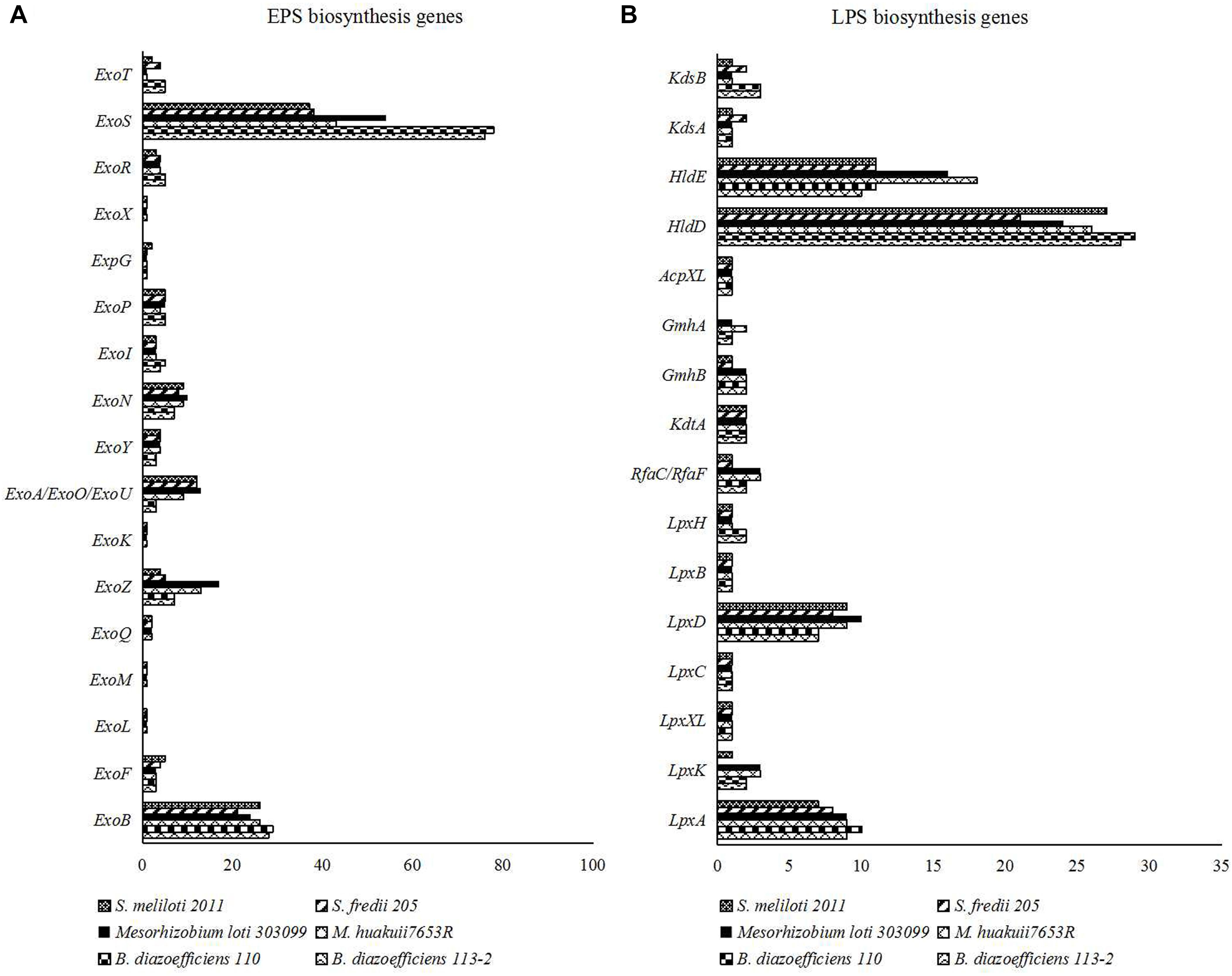
Figure 7. (A) Numbers of the genes related to the biosynthesis of EPSs in the six strain genomes. (B) Numbers of the genes related to the biosynthesis of LPSs in the six strain genomes.
Nodulation, nif and fix Gene Family Analysis
Nodulation factors, which are produced by rhizobial strains in response to flavonoids secreted by legume root hairs, play key roles in the determinants of host specificity of a rhizobium. We applied the nod, nif and fix genes in M. huakuii 7653R and/or M. japonicum MAFF303099 (Wang et al., 2014) as queries to search the nodulation, nif and fix genes in the genomes of these six strains. We firstly identified and analyzed 39 NF families (22 nod gene families, 11 nol gene families, five noe gene families and one nfe gene family). Among them, nine gene families had core genes, and only NodA was a single-copy-core-ortholog among these genomes. Moreover, 25 gene families had unique genes, 26 gene families had species specificity genes, and 31 gene families had other types of genes (Table 4). Six genes (NodF, NodH, NodQ, NolL, NolX, and NoeB) did not exist in B. diazoefficiens 113-2 and B. diazoefficiens USDA110, two genes (NodY and NoeB) did not exist in M. huakuii 7653R and M. japonicum MAFF303099, and 11 genes (NodB, NodY, NodZ, NolB, NolL, NolT, NolU, NolV, NolX, NolW, and NoeL) were not found in S. fredii USDA205 and S. meliloti 2011. Besides, NodY did not exist in B. diazoefficiens 113-2, four genes (NodU, NodZ, NolL, and NoeL) were not found in M. huakuii 7653R, and two genes (NodF and NoeB) did not exist in S. fredii USDA205 (Table 4). Supplementary Table S12 lists the detailed gene information.
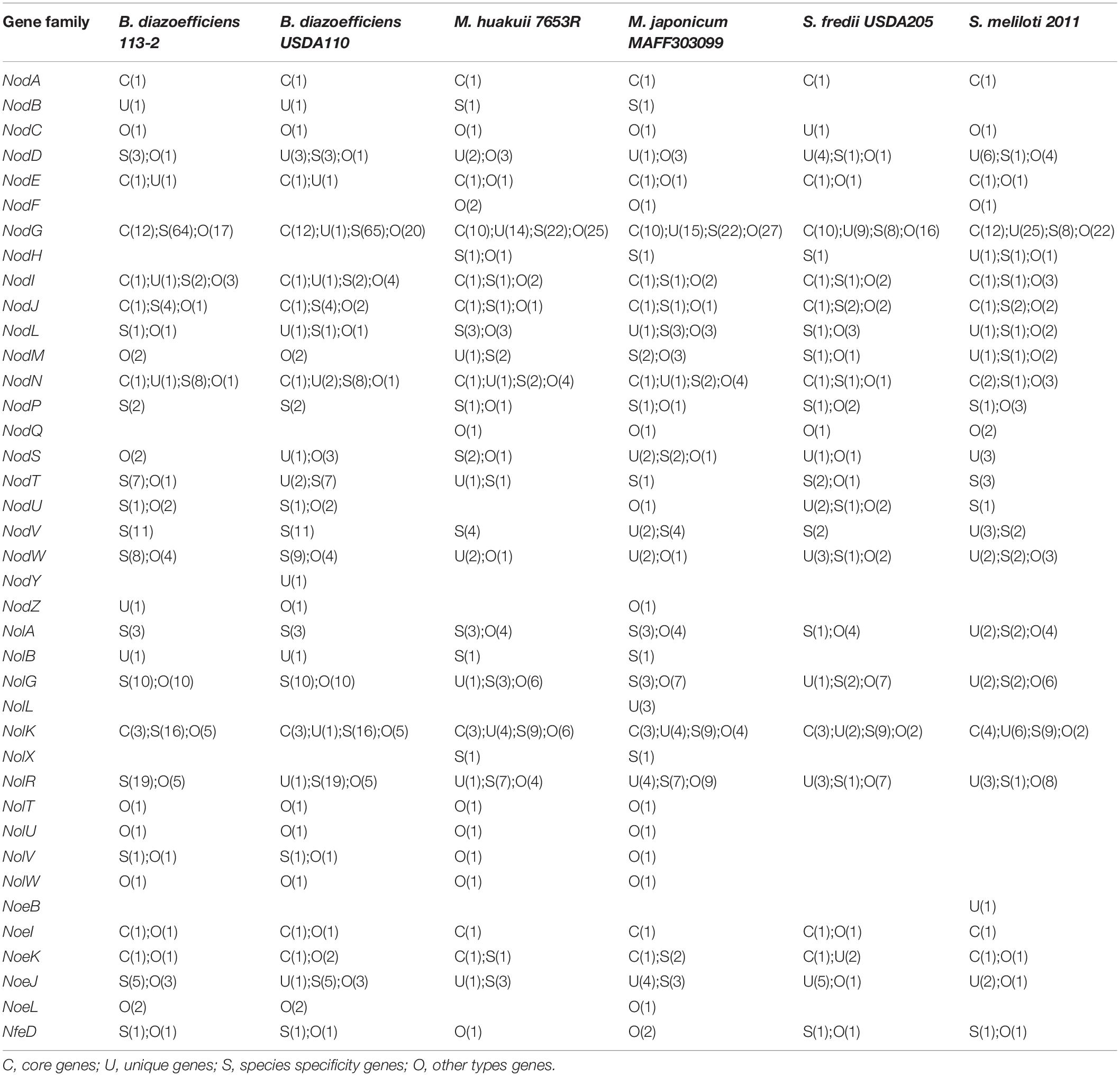
Table 4. List of the gene numbers of nodulation gene families among the six genomes.
Secondly, the numbers of nif and fix genes were found to be different among these six genomes. Two nif gene families (NifS and NifU) and two fix gene families (FixA and FixS) had core genes, 13 gene families had unique genes, 14 gene families had species specificity genes, and 18 gene families had other types of genes (Table 5). NifQ had no ortholog in S. fredii USDA205 and S. meliloti 2011, Nif11 was not found in B. diazoefficiens 113-2 and S. meliloti 2011, NifW had no ortholog in M. huakuii 7653R and S. meliloti 2011 genomes, and eight genes (NifA, NifD/E/N/K, NifH, NifQ, NifT, NifX, NifZ, and FixU) were not found in the S. fredii USDA205 genome. Two genes (FixJ and FixK) had larger numbers in B. diazoefficiens 113-2 and B. diazoefficiens USDA110 genomes compared with the other strains (Table 5). Table S13 lists the detailed gene information.

Table 5. List of the gene numbers of nif, fix gene families among the six genomes.
Thirdly, a synteny analysis based on the gene sequences of above-mentioned nod (Figure 8A), nif (Figure 8B), and fix (Figure 8C) genes (except for the genes in S. fredii USDA205) was performed to estimate the phylogenetic relationships of these genes among the five strains. In the three gene groupings, very few synteny blocks were shared by all of the five strains. The consistencies of the genes in the three groupings (especially for nod gene grouping) between the two strains in the same genus was higher than that in the different genera. Besides, we selected NodW, NolK, NoeJ, NifB, FixK, and FixJ gene families to perform phylogeny analyses (Supplementary Figures S1–S6), and the results revealed closer phylogenetic relationships between the two strains in the same genus, and only a small branch of NodW gene family especially for the three strains that nodulate soybean (Supplementary Figure S1).
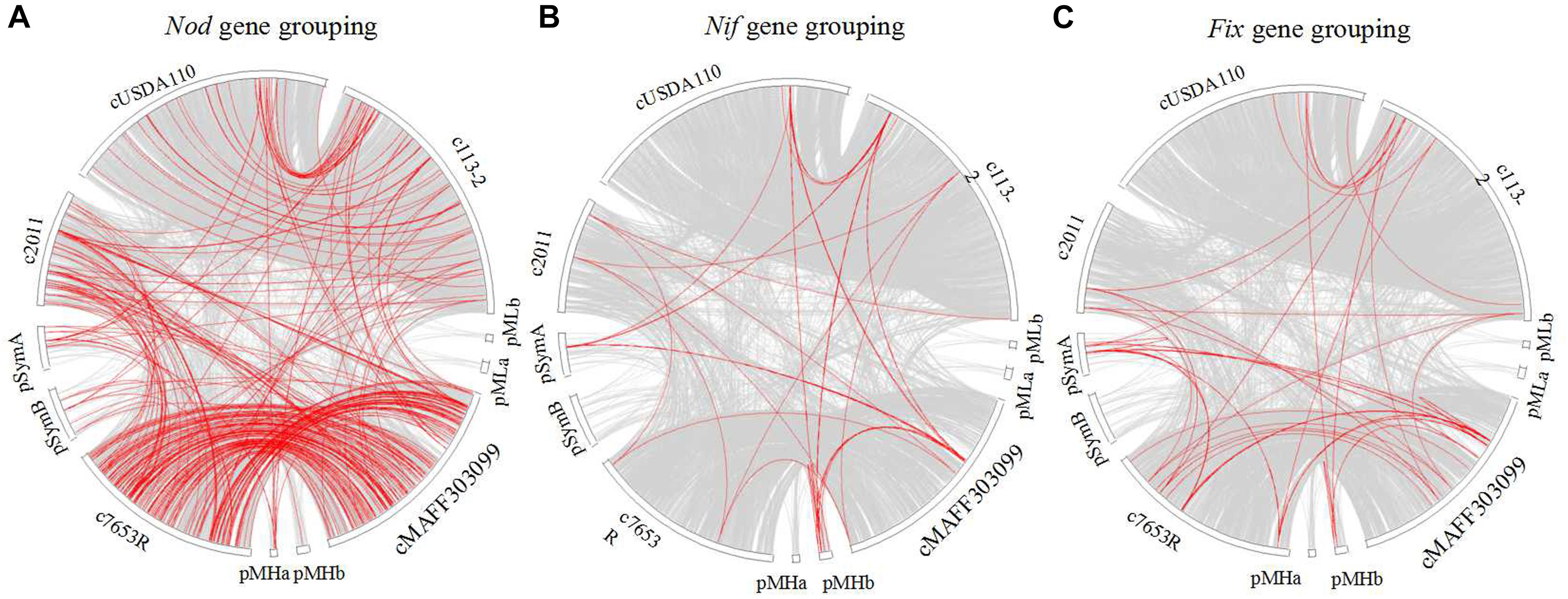
Figure 8. Synteny analysis of the nod, nif, and fix gene groupings among five rhizobial strains. Each gray block represents a synteny block and is internally independent from genomic rearrangement, red block represent Nod (A), Nif (B), and Fix (C) genes.
Discussion
The symbiotic nitrogen fixation system of leguminous plants and rhizobia is of great significance in the development of sustainable green agriculture. Although whole-genome sequencing of a series of rhizobial strains and comparative genomics among different rhizobial strains have provided valuable genetic information for symbiotic rhizobia (Tian et al., 2012; Sugawara et al., 2013; Wang et al., 2014), the genomic features responsible for species specificity among different rhizobial species with different growth rates still remain largely unexplored. Lots of genes (especially for nod genes) responsible for host specificity in the genomes of different strains have been explored by comparative genomics (Tian et al., 2012; Wang et al., 2014), while few studies on the homology classification analysis of the genes in these key gene families. In the present report, we sequenced and annotated the B. diazoefficiens 113-2 genome. The genomic characteristics of six rhizobia from different species and hosts were analyzed by comparative genomic analysis. Besides, the candidate genes related to secretion system, surface polysaccharides biosynthesis and RNS in the genomes of the six strains were explored and compared. Our results enriched the genomic library of rhizobia, and provided new insights and basic gene materials for species - specificity and strain - specificity of rhizobia.
Genomic Evidence Supporting 113-2 as a Strain of B. diazoefficiens
Bradyrhizobium diazoefficiens 113-2 is a broad-host-range and highly efficient soybean rhizobium, and had higher symbiotic matching abilities than B. diazoefficiens USDA110 with soybean ‘Tianlong 1’ (Li et al., 2017a). The general feature and structure of B. diazoefficiens 113-2 genome were similar to B. diazoefficiens USDA110 (Table 1). B. diazoefficiens 113-2 genome shared a large proportion of synteny blocks and high ANI value (0.9995) with B. diazoefficiens USDA110 (Figure 2). Typically, the ANI values between genomes of the same species are above 95% (Goris et al., 2007). About 53% clusters in B. diazoefficiens 113-2 and B. diazoefficiens USDA110 genome were species specificity-clusters (113-2-USDA110 pair, Figure 3), and similar species specificity-clusters gene numbers in the assigned COG functional terms were present in these two genomes (Figure 4D). For nodulation, nif and fix genes, similar gene numbers of species specificity existed in B. diazoefficiens 113-2 and B. diazoefficiens USDA110 genomes (Tables 4, 5). These results supported a closer phylogenetic relationship between B. diazoefficiens 113-2 and B. diazoefficiens USDA110 compared with the other strains, and our current findings provided molecular evidence that B. diazoefficiens 113-2 and B. diazoefficiens USDA110 were two strains in the same species. Compared with B. diazoefficiens 113-2, B. diazoefficiens USDA110 contained more nodulation, nif and fix genes, including the unique genes in the nine nodulation gene families (NodD, NodG, NodL, NodS, NodT, NodY, NolK, NolR, and NoeJ) and NifS gene family (Tables 4, 5 and Supplementary Tables S12, S13), and these particular genes might be play key roles in the difference of the symbiotic matching abilities between B. diazoefficiens 113-2 and B. diazoefficiens USDA110 strains.
Compared with the other five genomes, B. diazoefficiens 113-2 had more small RNAs (Tables 1, 2 and Supplementary Table S2), which act as signal molecules modulating the host nodulation (Ren et al., 2019). Moreover, 1,081 (about 12.3%) singletons, which are unique genes of a species (Grose et al., 2014), were found in B. diazoefficiens 113-2 genome (Table 3), and most of them (831 out of 1,081) had no assigned COG functional terms (Figure 4B and Supplementary Table S8), suggesting that B. diazoefficiens 113-2 had more or unique functions compared with the other five strains. Besides, B. diazoefficiens 113-2 also had singletons in the selected KEGG pathways (Figure 5B) and had unique nodulation, nif and fix genes (Tables 4, 5), which are important for host specificity (Andrews and Andrews, 2017). These results suggested that B. diazoefficiens 113-2 had unique characteristics of genomic and symbiotic functions.
Contrasting Genomic Features of Three Species of Rhizobia With Different Growth Rates
The development and maintenance process of legume-rhizobium symbiosis is a high resource-consuming process (Ferguson et al., 2019). Therefore, the equilibrium between the nitrogen fixation efficiency and energy consumption in legume-rhizobium symbiosis is particularly important in legume cultivation. To screen rhizobia with both high symbiotic efficiency and low energy consumption (fast growth rate and/or short cycle), we compared the genomic characteristics of three rhizobial species with different growth rates. Firstly, the genomic size, genomic (G + C)% and gene numbers were relatively consistent in the same genus, which were increased as the growth rate of bacteria was slowed down (Table 1). This finding was consistent with an earlier report (Tian et al., 2012). Secondly, similar genome structures and high ANI values were existed between the two strains in the same genus, while greatly varied genome structures and low ANI values were found among the strains in different genera with different growth rates (Figure 2). The difference in genomes might be the reason for the different symbiotic characters of different rhizobia (Siqueira et al., 2014) or rhizobia in different genera (Tian et al., 2012; Sugawara et al., 2013; Alaswad et al., 2019). Thirdly, the numbers of genus-specific cluster genes were decreased with the increase of growth rate of the strains in most of these COG functional terms (Figure 4C), indicating that there were more genes involved in various processes in slow-growing rhizobia. These genes would improve the ecological success of slow-growing rhizobia growing under more diverse soil conditions with limit but various resources (Konstantinidis and Tiedje, 2004; Tian et al., 2012), which might be the reason for that the adaptation of slow-growing rhizobia is wider compared with the other rhizobia (Tian et al., 2012). Fourthly, for candidate genes related to secretion system, surface polysaccharides biosynthesis and RNS, the numbers of genus-specific genes were relatively consistent in the strains of the same genus, while there were great differences among strains in different species of rhizobia (Figures 6–8 and Tables 4, 5). These differences might be the key factors to distinguish the host ranges as well as the nodulation and nitrogen fixation characteristics between rhizobia of different species (Tian et al., 2012; Wang et al., 2014; Zhang et al., 2014; Alaswad et al., 2019).
Host Specificity
In most rhizobia, expression of genes related to secretion system, surface polysaccharides biosynthesis and RNS is needed for inducing nodule organogenesis and nodule development (Putnoky et al., 1988; Lorkiewicz, 1997; Fauvart and Michiels, 2008; Li et al., 2014), and the type and/or number of these nodule-related genes are often play important roles in host specificity (Horvath et al., 1986; Philip-Hollingsworth et al., 1989; Wang et al., 2014). Among our six tested strains, S. fredii USDA205 nodulated the same legume host (soybean) with B. diazoefficiens 113-2 and B. diazoefficiens USDA110 (Table 1), while there were no genes related to secretion system, surface polysaccharides biosynthesis and RNS that were both specific and common to these three strains (Figures 6, 7 and Tables 4, 5), suggesting that there was no gene specifically shared by rhizobia of different species to establish symbiosis with soybean, which was consistent with a previous study (Tian et al., 2012). M. huakuii 7653R and S. meliloti 2011 form indeterminate nodules (Cheng et al., 2007; Sallet et al., 2013), and the other four strains form determinate nodules (Kaneko et al., 2000; Yuan et al., 2016, 2017; Shah and Subramaniam, 2018). However, this phenomenon was consistent with the above-mentioned findings, and no gene was specifically shared by M. huakuii 7653R and S. meliloti 2011 or the rest four strains, indicating that the formation of determinate nodules or indeterminate nodules was mainly determined by host legume plants. The two strains nodulate different legume hosts in the group of medium-slow-growing rhizobia or fast-growing rhizobia (Table 1). In these four rhizobial strains, the types and total numbers of genes related to secretion system, surface polysaccharides biosynthesis and RNS were substantially different (Figures 6, 7 and Tables 4, 5). Among the RNS-related gene families, only 11 gene families (Nod A, Nod E, Nod G, Nod I, Nod J, Nod P, Nod Q, Nol K, Nol R, Fix G, and Fix H) had same gene types in these four strains, and three of them (Nod G, Nol K, and Nol R) had unique genes (Tables 4, 5). These differences might contribute to the establishment of differential legume-rhizobium symbiosis.
Collectively, the B. diazoefficiens 113-2 genome was sequenced, assembled and annotated in the present study. The synteny, ANI and ortholog analysis firmly establish 113-2 as a strain of B. diazoefficiens. The genomic characteristics of the six rhizobial strains from different species and different hosts were analyzed by comparative genomic analysis. The candidate genes related to secretion system, surface polysaccharides biosynthesis and RNS in the genomes of the six strains were explored and compared. Our results enriched the genomic library of rhizobia and provided valuable insights into the species-specificity and host specificity among different rhizobial strains.
Materials and Methods
Bacterial Strains and DNA Preparation
Bradyrhizobium diazoefficiens 113-2 (Stored in our lab) was cultured in YMA plate for 4 days at 28°C. Cells of B. diazoefficiens 113-2 were harvested by centrifugation at 1,3000 rpm for 30 min. Genomic DNA was extracted by Beijing Genomics Institute (BGI, Shenzhen, China) using a Genomic DNA Mini Preparation Kit.
Genome Sequencing, Assembly and Component Prediction
De novo sequencing of B. diazoefficiens 113-2 genome was performed by BGI using PacBio RS II platform and Illumina HiSeq 4000 platform. The proportion of clean data (1,144 Mb) was 87.95% among the total acquired reads (1,301 Mb) in the Illumina platform. The proportion of Subreads Post Filter data (544,740,462 bp) was about 99.7% among the Polymerase Read Post Filter (546,403,010 bp) in the PacBio platform. The analysis results of 15-kmer (Supplementary Figure S7) and GC-depth (Supplementary Figure S8) indicated that the sequencing was of good quality. Sequence assembly was done with SOAP de novo (Luo et al., 2012). Glimmer 3.021 with Hidden Markov models was used to perform the gene prediction of B. diazoefficiens 113-2 genome assembly. RNAmmer 1.2 (Lagesen et al., 2007), Rfam 9.1 (Gardner et al., 2009) and tRNA scan-SE (Lowe and Eddy, 1997) were used to identify tRNA, rRNA, and sRNAs in B. diazoefficiens 113-2 genome. The tandem repeats annotation was obtained using the Tandem Repeat Finder2. The prophages were predicted using the PHAST (PHAge Search Tool) (Grissa et al., 2007).
Genome Annotation
Gene function annotation of B. diazoefficiens 113-2 was performed by using Basic Local Alignment Search Tool (BLAST) against 11 different databases. These databases are COG (Clusters of Orthologous Groups), GO (Gene Ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes), NR (Non-Redundant Protein Database databases), Swiss-Prot (O’Donovan et al., 2002), IPR, Type III secretion system (T3SS), PHI (Pathogen Host Interactions), VFDB (Virulence Factors of Pathogenic Bacteria), ARDB (Antibiotic Resistance Genes Database), and CAZy (Carbohydrate-Active enZYmes Database).
Synteny Analysis and ANI Analysis
The complete nucleotide sequences and genomic features of strains B. diazoefficiens USDA110, M. huakuii 7653R, M. japonicum MAFF303099, S. fredii USDA205 and S. meliloti 2011 were obtained from GenBank (accession numbers: USDA 110, NC_004463; 7653R, NC_002678, NC_002679, and NC_002682; MAFF 303099, NC_002678, NC_002679 and NC_002682; USDA205, GCA_009601405; S. meliloti 2011, NC_020528, NC_020527, and NC_020560). The sequences were organized according to their chromosomal origins of replication for intuitive comparison. Genome sequence alignments were created using NCBI BLAST + and visualized using MCScanX (Wang et al., 2012) and Mauve software. The Average Nucleotide Identity (ANI) between the six genomes was performed using the ANI Calculator, available at https://www.ezbiocloud.net/tools/ani (Yoon et al., 2017).
Singletons-Clusters Analysis
Orthologous clustering analysis were performed with the web server OrthoVenn2 with bacteria group parameters and an E-value cutoff of 1e−5 (Xu et al., 2019). The protein FASTA file containing predicted protein sequences for strains B. diazoefficiens USDA110, M. huakuii 7653R, M. japonicum MAFF303099, S. fredii USDA205 and S. meliloti 2011 were used to predict the orthologous gene clusters.
Core-Pan Genes Analysis
Core/Pan genes of above-mentioned six strains were clustered by the CD-HIT 4.663 rapid clustering of similar proteins software (Edgar, 2004) with a threshold of 50% pairwise identity and 0.7 length difference cutoff in amino acid, and the final gene pool after clustered analysis is called the pan gene pool. Proteins existed in all of the six genomes in the clustering results act as the core gene pool. Proteins only existed in one genome are classified as the specific gene pool. The rest of the Pan proteins after removing core proteins are called the Dispensable gene pool.
Phylogenetic Analysis
The different NolKs or FixKs were applied for multi-species phylogenetic analysis. Multiple alignments of the full-length deduced amino acid sequences of these genes were conducted with Clustal W program. And the multi-species phylogenetic tree was performed using MEGAX software (Kumar et al., 2018) with Neighbor-Joining (NJ) method, and bootstrap analysis was conduct educing 1,000 replicates with the p-distance model.
Nucleotide Sequence Accession Numbers
Complete genome sequences of B. diazoefficiens 113-2 have been submitted to GenBank under the assigned accession number (CP055233).
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
SY and XZ designed this work. SY wrote the manuscript. SY, RL, and YF performed most of the experiments and analysis. HC, CZ, YH, LC, QH, and DC contributed substantially to the completion of this work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funds from the National Natural Science Foundation of China (grant no. 31701346), Fundamental Research Funds for Central Non-profit Scientific Institution (grant no. 1610172018001), the National Transgenic Project of China (2016ZX08004-005), and the Agricultural Science and Technology Innovation Program of CAAS (CAAS-ASTIP-2016-OCRI). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.576800/full#supplementary-material
Supplementary Figure 1 | Phylogenetic relationships among the NodWs from the six strains.
Supplementary Figure 2 | Phylogenetic relationships among the NoeJs from the six strains.
Supplementary Figure 3 | Phylogenetic relationships among the NolKs from the six strains.
Supplementary Figure 4 | Phylogenetic relationships among the NifBs from the six strains.
Supplementary Figure 5 | Phylogenetic relationships among the FixJs from the six strains.
Supplementary Figure 6 | Phylogenetic relationships among the FixKs from the six strains.
Supplementary Figure 7 | 15-mer analysis on sample.
Supplementary Figure 8 | GC content and depth correlative analysis.
Supplementary Table 1 | The sequence information of genes in B. diazoefficiens 113-2 genome.
Supplementary Table 2 | The detail information of different types of RNAs in B. diazoefficiens 113-2 genome.
Supplementary Table 3 | The detail information of Tandem Repeats in B. diazoefficiens 113-2 genome.
Supplementary Table 4 | The annotation information of the B. diazoefficiens 113-2 genome in 11 different databases.
Supplementary Table 5 | Function annotation assignment from different databases.
Supplementary Table 6 | The detail protein ID information of the singletons of the six rhizobial strains.
Supplementary Table 7 | The detail protein ID information in the 11656 clusters.
Supplementary Table 8 | The detail gene ID information in COG classification and annotation information of the genes in the six rhizobia.
Supplementary Table 9 | The detailed gene ID information of the three pathway genes in B. diazoefficiens 113-2 genome.
Supplementary Table 10 | The detailed information of the genes related to different secretion system in the six genomes.
Supplementary Table 11 | The detail ID information of the genes related to the biosynthesis of EPSs and LPSs in the six genomes.
Supplementary Table 12 | The detail information of the genes of nodulation gene families in the six genomes.
Supplementary Table 13 | The detail information of the genes of nif, fix gene families in the six genomes.
Footnotes
- ^ http://ccb.jhu.edu/software/glimmer/index.shtml
- ^ http://tandem.bu.edu/trf/trf.html
- ^ http://weizhongli-lab.Org/cd-hit/
References
Alaswad, A. A., Oehrle, N. W., and Krishnan, H. B. (2019). Classical Soybean (Glycine max (L.) Merr) Symbionts, Sinorhizobium fredii USDA191 and Bradyrhizobium diazoefficiens USDA110, reveal contrasting symbiotic phenotype on Pigeon Pea (Cajanus cajan (L.) Millsp). Int. J. Mol. Sci. 20:1091. doi: 10.3390/ijms20051091
Andrews, M., and Andrews, M. E. (2017). Specificity in legume-rhizobia symbioses. Int. J. Mol. Sci. 18, 705. doi: 10.3390/ijms18040705
Ansari, P. G., and Rao, D. L. (2014). Differentiating Indigenous Soybean Bradyrhizobium and Rhizobium spp. of Indian Soils. Indian J. Microbiol. 54, 190–195. doi: 10.1007/s12088-013-0430-z
Biondi, E. G., Pilli, E., Giuntini, E., Roumiantseva, M. L., Andronov, E. E., Onichtchouk, O. P., et al. (2003). Genetic relationship of Sinorhizobium meliloti and Sinorhizobium medicae strains isolated from Caucasian region. FEMS Microbiol. Lett. 220, 207–213. doi: 10.1016/s0378-1097(03)00098-3
Cheng, G., Li, Y., Xie, B., Yang, C., and Zhou, J. (2007). Cloning and identification of lpsH, a novel gene playing a fundamental role in symbiotic nitrogen fixation of Mesorhizobium huakuii. Curr. Microbiol. 54, 371–375. doi: 10.1007/s00284-006-0471-1
Cherni, A. E., and Perret, X. (2019). Deletion of rRNA operons of Sinorhizobium fredii strain NGR234 and impact on symbiosis with legumes. Front. Microbiol. 10:154. doi: 10.3389/fmicb.2019.00154
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 5:113. doi: 10.1186/1471-2105-5-113
Estrella, M. J., Munoz, S., Soto, M. J., Ruiz, O., and Sanjuan, J. (2009). Genetic diversity and host range of rhizobia nodulating Lotus tenuis in typical soils of the salado river basin (Argentina). Appl. Environ. Microbiol. 75, 1088–1098. doi: 10.1128/aem.02405-08
Fauvart, M., and Michiels, J. (2008). Rhizobial secreted proteins as determinants of host specificity in the rhizobium-legume symbiosis. FEMS Microbiol. Lett. 285, 1–9. doi: 10.1111/j.1574-6968.2008.01254.x
Ferguson, B. J., Mens, C., Hastwell, A. H., Zhang, M., Su, H., Jones, C. H., et al. (2019). Legume nodulation: the host controls the party. Plant Cell Environ. 42, 41–51. doi: 10.1111/pce.13348
Friesen, M. L. (2012). Widespread fitness alignment in the legume-rhizobium symbiosis. New Phytol. 194, 1096–1111. doi: 10.1111/j.1469-8137.2012.04099.x
Gardner, P. P., Daub, J., Tate, J. G., Nawrocki, E. P., Kolbe, D. L., Lindgreen, S., et al. (2009). Rfam: updates to the RNA families database. Nucl. Acids Res. 37, D136–D140. doi: 10.1093/nar/gkn766
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57(Pt. 1), 81–91. doi: 10.1099/ijs.0.64483-0
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucl. Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Grose, J. H., Jensen, G. L., Burnett, S. H., and Breakwell, D. P. (2014). Genomic comparison of 93 Bacillus phages reveals 12 clusters, 14 singletons and remarkable diversity. BMC Genomics 15:855. doi: 10.1186/1471-2164-15-855
Hayashi, M., Saeki, Y., Haga, M., Harada, K., Kouchi, H., and Umehara, Y. (2012). Rj (rj) genes involved in nitrogen-fixing root nodule formation in soybean. Breed Sci. 61, 544–553. doi: 10.1270/jsbbs.61.544
Horvath, B., Kondorosi, E., John, M., Schmidt, J., Török, I., Györgypal, Z., et al. (1986). Organization, structure and symbiotic function of Rhizobium meliloti nodulation genes determining host specificity for alfalfa. Cell 46, 335–343. doi: 10.1016/0092-8674(86)90654-9
Janczarek, M., Kutkowska, J., Piersiak, T., and Skorupska, A. (2010). Rhizobium leguminosarum bv. trifolii rosR is required for interaction with clover, biofilm formation and adaptation to the environment. BMC Microbiol. 10:284. doi: 10.1186/1471-2180-10-284
Jones, K. M., Sharopova, N., Lohar, D. P., Zhang, J. Q., VandenBosch, K. A., and Walker, G. C. (2008). Differential response of the plant Medicago truncatula to its symbiont Sinorhizobium meliloti or an exopolysaccharide-deficient mutant. Proc. Natl. Acad. Sci. U.S.A. 105, 704–709. doi: 10.1073/pnas.0709338105
Kaneko, T., Nakamura, Y., Sato, S., Asamizu, E., Kato, T., Sasamoto, S., et al. (2000). Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 7, 331–338. doi: 10.1093/dnares/7.6.331
Keyser, H. H., Bohlool, B. B., Hu, T. S., and Weber, D. F. (1982). Fast-growing rhizobia isolated from root nodules of soybean. Science 215, 1631–1632. doi: 10.1126/science.215.4540.1631
Konstantinidis, K. T., and Tiedje, J. M. (2004). Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. U.S.A. 101, 3160–3165. doi: 10.1073/pnas.0308653100
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucl. Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lerouge, P., Roche, P., Faucher, C., Maillet, F., Truchet, G., Prome, J. C., et al. (1990). Symbiotic host-specificity of Rhizobium meliloti is determined by a sulphated and acylated glucosamine oligosaccharide signal. Nature 344, 781–784. doi: 10.1038/344781a0
Li, R., Yuan, S., Chen, H., Zhang, C., Chen, L., Hao, Q., et al. (2017a). Comparative analysis of symbiotic phenotypes of soybean “Tianlong 1” with two different rhizobia and co-inoculation at different developmental stages. Oil Crop Sci. 2, 160–168.
Li, R., Yuan, S., Chen, H., Zhang, C., Chen, L., Hao, Q., et al. (2017b). Symbiotic matching between soybean ‘Tianlong 1’ and rhizobia. Oil Crop Sci. 1, 64–70.
Li, X., Deng, Z., Liu, Z., Yan, Y., Wang, T., Xie, J., et al. (2014). The genome of Paenibacillus sabinae T27 provides insight into evolution, organization and functional elucidation of nif and nif-like genes. BMC Genomics 15:723. doi: 10.1186/1471-2164-15-723
Lorkiewicz, Z. (1997). Nodulation genes in the Rhizobium–plant signal exchange. Acta Biochim. Pol. 44, 1–12. doi: 10.18388/abp.1997_4434
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl. Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217x-1-18
Marsudi, N. D. S., Glenn, A. R., and Dilworth, M. J. (1999). Id∗entification and characterization of fast- and slow-growing root nodule bacteria from South-Western Australian soils able to nodulate Acacia saligna. Soil Biol. Biochem. 31,
Nelson, M. S., and Sadowsky, M. J. (2015). Secretion systems and signal exchange between nitrogen-fixing rhizobia and legumes. Front. Plant Sci. 6:491. doi: 10.3389/fpls.2015.00491
O’Donovan, C., Martin, M. J., Gattiker, A., Gasteiger, E., Bairoch, A., and Apweiler, R. (2002). High-quality protein knowledge resource: SWISS-PROT and TrEMBL. Brief Bioinform. 3, 275–284. doi: 10.1093/bib/3.3.275
Okazaki, S., Kaneko, T., Sato, S., and Saeki, K. (2013). Hijacking of leguminous nodulation signaling by the rhizobial type III secretion system. Proc. Natl. Acad. Sci. U.S.A. 110, 17131–17136. doi: 10.1073/pnas.1302360110
Perret, X., Staehelin, C., and Broughton, W. J. (2000). Molecular basis of symbiotic promiscuity. Microbiol. Mol. Biol. Rev. 64, 180–201. doi: 10.1128/mmbr.64.1.180-201.2000
Philip-Hollingsworth, S., Hollingsworth, R. I., Dazzo, F. B., Djordjevic, M. A., and Rolfe, B. G. (1989). The effect of interspecies transfer of Rhizobium host-specific nodulation genes on acidic polysaccharide structure and in situ binding by host lectin. J. Biol. Chem. 264, 5710–5714.
Putnoky, P., Grosskopf, E., Ha, D. T., Kiss, G. B., and Kondorosi, A. (1988). Rhizobium fix genes mediate at least two communication steps in symbiotic nodule development. J. Cell Biol. 106, 597–607. doi: 10.1083/jcb.106.3.597
Radutoiu, S., Madsen, L. H., Madsen, E. B., Jurkiewicz, A., Fukai, E., Quistgaard, E. M., et al. (2007). LysM domains mediate lipochitin-oligosaccharide recognition and Nfr genes extend the symbiotic host range. Embo J. 26, 3923–3935. doi: 10.1038/sj.emboj.7601826
Ren, B., Wang, X., Duan, J., and Ma, J. (2019). Rhizobial tRNA-derived small RNAs are signal molecules regulating plant nodulation. Science 365, 919–922. doi: 10.1126/science.aav8907
Sallet, E., Roux, B., Sauviac, L., Jardinaud, M. F., Carrère, S., Faraut, T., et al. (2013). Next-generation annotation of prokaryotic genomes with EuGene-P: application to Sinorhizobium meliloti 2011. DNA Res. 20, 339–354. doi: 10.1093/dnares/dst014
Schultze, M., Quiclet-Sire, B., Kondorosi, E., Virelizer, H., Glushka, J. N., Endre, G., et al. (1992). Rhizobium meliloti produces a family of sulfated lipooligosaccharides exhibiting different degrees of plant host specificity. Proc. Natl. Acad. Sci. U.S.A. 89, 192–196. doi: 10.1073/pnas.89.1.192
Shah, V., and Subramaniam, S. (2018). Bradyrhizobium japonicum USDA110: a representative model organism for studying the impact of pollutants on soil microbiota. Sci. Total Environ. 624, 963–967. doi: 10.1016/j.scitotenv.2017.12.185
Shrestha, P. M., Noll, M., and Liesack, W. (2007). Phylogenetic identity, growth-response time and rRNA operon copy number of soil bacteria indicate different stages of community succession. Environ. Microbiol. 9, 2464–2474. doi: 10.1111/j.1462-2920.2007.01364.x
Siqueira, A. F., Ormeño-Orrillo, E., Souza, R. C., Rodrigues, E. P., Almeida, L. G., Barcellos, F. G., et al. (2014). Comparative genomics of Bradyrhizobium japonicum CPAC 15 and Bradyrhizobium diazoefficiens CPAC 7: elite model strains for understanding symbiotic performance with soybean. BMC Genomics 15:420. doi: 10.1186/1471-2164-15-420
Skorupska, A., Janczarek, M., Marczak, M., Mazur, A., and Krol, J. (2006). Rhizobial exopolysaccharides: genetic control and symbiotic functions. Microb. Cell Fact. 5:7. doi: 10.1186/1475-2859-5-7
Streit, W. R., Schmitz, R. A., Perret, X., Staehelin, C., Deakin, W. J., Raasch, C., et al. (2004). An evolutionary hot spot: the pNGR234b replicon of Rhizobium sp. strain NGR234. J. Bacteriol. 186, 535–542. doi: 10.1128/jb.186.2.535-542.2004
Sugawara, M., Epstein, B., Badgley, B. D., Unno, T., Xu, L., Reese, J., et al. (2013). Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol 14:R17. doi: 10.1186/gb-2013-14-2-r17
Tampakaki, A. P., Fotiadis, C. T., Ntatsi, G., and Savvas, D. (2017). Phylogenetic multilocus sequence analysis of indigenous slow-growing rhizobia nodulating cowpea (Vigna unguiculata L.) in Greece. Syst. Appl. Microbiol. 40, 179–189. doi: 10.1016/j.syapm.2017.01.001
Temprano-Vera, F., Rodriguez-Navarro, D. N., Acosta-Jurado, S., Perret, X., Fossou, R. K., Navarro-Gomez, P., et al. (2018). Sinorhizobium fredii strains HH103 and NGR234 form nitrogen fixing nodules with diverse wild soybeans (Glycine soja) from central china but are ineffective on northern china accessions. Front. Microbiol. 9:2843. doi: 10.3389/fmicb.2018.02843
Tian, C. F., Zhou, Y. J., Zhang, Y. M., Li, Q. Q., Zhang, Y. Z., Li, D. F., et al. (2012). Comparative genomics of rhizobia nodulating soybean suggests extensive recruitment of lineage-specific genes in adaptations. Proc. Natl. Acad. Sci. U.S.A. 109, 8629–8634. doi: 10.1073/pnas.1120436109
Wang, S., Hao, B., Li, J., Gu, H., Peng, J., Xie, F., et al. (2014). Whole-genome sequencing of Mesorhizobium huakuii 7653R provides molecular insights into host specificity and symbiosis island dynamics. BMC Genomics 15:440. doi: 10.1186/1471-2164-15-440
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucl. Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucl. Acids Res. 47, W52–W58. doi: 10.1093/nar/gkz333
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Yuan, S., Li, R., Chen, S., Chen, H., Zhang, C., Chen, L., et al. (2016). RNA-seq analysis of differential gene expression responding to different rhizobium strains in soybean (Glycine max) Roots. Front. Plant Sci. 7:721. doi: 10.3389/fpls.2016.00721
Yuan, S. L., Li, R., Chen, H. F., Zhang, C. J., Chen, L. M., Hao, Q. N., et al. (2017). RNA-Seq analysis of nodule development at five different developmental stages of soybean (Glycine max) inoculated with Bradyrhizobium japonicum strain 113-2. Sci. Rep. 7:42248. doi: 10.1038/srep42248
Zhang, X. X., Guo, H. J., Wang, R., Sui, X. H., Zhang, Y. M., Wang, E. T., et al. (2014). Genetic divergence of bradyrhizobium strains nodulating soybeans as revealed by multilocus sequence analysis of genes inside and outside the symbiosis island. Appl. Environ. Microbiol. 80, 3181–3190. doi: 10.1128/aem.00044-14
Keywords: Bradyrhizobium diazoefficiens 113-2, whole-genome sequencing, comparative analysis, species specificity, host specificity
Citation: Li R, Feng Y, Chen H, Zhang C, Huang Y, Chen L, Hao Q, Cao D, Yuan S and Zhou X (2020) Whole-Genome Sequencing of Bradyrhizobium diazoefficiens 113-2 and Comparative Genomic Analysis Provide Molecular Insights Into Species Specificity and Host Specificity. Front. Microbiol. 11:576800. doi: 10.3389/fmicb.2020.576800
Received: 27 June 2020; Accepted: 20 October 2020;
Published: 16 November 2020.
Edited by:
Gustavo Santoyo, Universidad Michoacana de San Nicolás de Hidalgo, MexicoReviewed by:
Elena Erika Fedorova, Timiryazev Institute of Plant Physiology RAS, RussiaLuis Rey, Polytechnic University of Madrid, Spain
Copyright © 2020 Li, Feng, Chen, Zhang, Huang, Chen, Hao, Cao, Yuan and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Songli Yuan, eXl5eXktMDkwOUAxNjMuY29t; Xinan Zhou, emhvdW9jcmlAc2luYS5jb20=
†These authors have contributed equally to this work