 Lina Liu
Lina Liu Yu Feng
Yu Feng Li Wei
Li Wei Fu Qiao
Fu Qiao Zhiyong Zong
Zhiyong Zong- 1Center of Infectious Diseases, West China Hospital, Sichuan University, Chengdu, China
- 2Center for Pathogen Research, West China Hospital, Sichuan University, Chengdu, China
- 3Division of Infectious Diseases, State Key Laboratory of Biotherapy, Chengdu, China
- 4Department of Infection Control, West China Hospital, Sichuan University, Chengdu, China
Kluyvera is a genus within the family Enterobacteriaceae and can cause various human infections but remains poorly studied. A carbapenem-resistant blaNDM–1-carrying Kluyvera strain 090646T was isolated from a hospital sink in Chengdu, Sichuan province, China. Whole genome sequencing of the strain revealed that it had 28.2 to 42.3% in silico DNA-DNA hybridization (isDDH) scores and 84.15 to 90.10% average nucleotide identity (ANI) values with other Kluyvera species. Both values are well below the ≥ 70.0% isDDH and ≥ 95–96% ANI cutoffs to define bacterial species, suggesting that the strain represents a novel species of the genus Kluyvera, for which the name Kluyvera sichuanensis. nov. is proposed. Type strain of K. sichuanensis is 090646T (=GDMCC 1.1872T =KCTC 82166T). Strain 090646T can be differentiated from other Kluyvera species by its ability to utilize D-sorbitol but not β-galactosidase (ONPG), D-mannose, glycerin, raffinose, nor sucrose. There were 47 genome sequences labeled as Kluyvera in GenBank, which were curated for precise species identification. Only 33 of the 47 genomes were indeed of Kluyvera and four novel Kluyvera genomospecies were identified, highlighting that the species assignation of bacterial genomes in GenBank need to be curated. Genome sequencing for more strains is required to understand the genus Kluyvera.
Introduction
Kluyvera is a genus of the family Enterobacteriaceae (Farmer et al., 1981) and can be found in soil, water, sewage, and healthcare environment (Farmer et al., 1981; Li et al., 2019; Mutoh et al., 2019). In human, Kluyvera strains appear to be largely colonized in the gastrointestinal tract but also cause a wide range of infections such as bacteremia, cholangitis, diarrhea, neonatal meningitis, peritonitis, and pneumonia with severity from mild to fatal diseases (Sarria et al., 2001; Lee et al., 2019). Although Kluyvera is of clinical significance, it remains poorly studied. At the time of writing, the Kluyvera genus consists of only four species: Kluyvera ascorbata, Kluyvera cryocrescens, Kluyvera georgiana, and Kluyvera intermedia (Farmer et al., 1981; Muller et al., 1996; Pavan et al., 2005). Of note, the previously known Kluyvera intestini (Tetz et al., 2017) has been moved to the genus Metakosakonia in 2017 (Alnajar and Gupta, 2017). In this study, we first reported a novel Kluyvera species by examining the taxonomy and characterizing phenotypes of a strain from a handwashing sink. Then, we curated the species assignation of all Kluyvera genomes (n = 47) in GenBank and found 4 novel Kluyvera genomospecies based on genome analysis. Last, we provided an updated taxonomy for the genus Kluyvera as a reference for future studies.
Materials and Methods
Strain Isolation and Identification
Strain 090646T (also called SCKS090646) was recovered from the residual water of a handwashing sink as part of an infection control surveillance program on sinks. The residual water was sampled using a sterile rayon swab (Copan; Brescia, Italy) moistened with tryptic soy broth (TSB) (Hopebio, Qingdao, China). The swab was placed into a 15 ml sterile tube containing 6 ml TSB and was incubated at 37°C overnight. Then the tube was centrifuged, and supernatant was discarded, and precipitants were resuspended with 1 ml TSB. A 50 μl suspension was streaked onto a CHROMagar Orientation colorimetric plate (Chromagar; Paris, France). Isolates from sinks were subjected to preliminary species identification using matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF; Bruker, Billerica, MA, United States) with a database version DB5989 according to the manufacturer’s instructions.
Analysis of the 16S rRNA Gene Sequence
A nearly complete sequence (1,411 bp) of the 16S rRNA gene was obtained from strain 090646T by PCR as described previously (Moreno et al., 2002). The corresponding 16S rRNA gene sequence of type strains of other Kluyvera species and other closely related genera sharing >97% nucleotide identity with that of strain 090646T were retrieved from GenBank. The 16S rRNA gene sequences were aligned using Clustal Omega (Madeira et al., 2019) and a maximum-likelihood phylogenetic tree (Figure 1) was inferred by RAxML v8.2.12 (Stamatakis, 2014) under GTRGAMMA model with a 1,000-boostrap test.
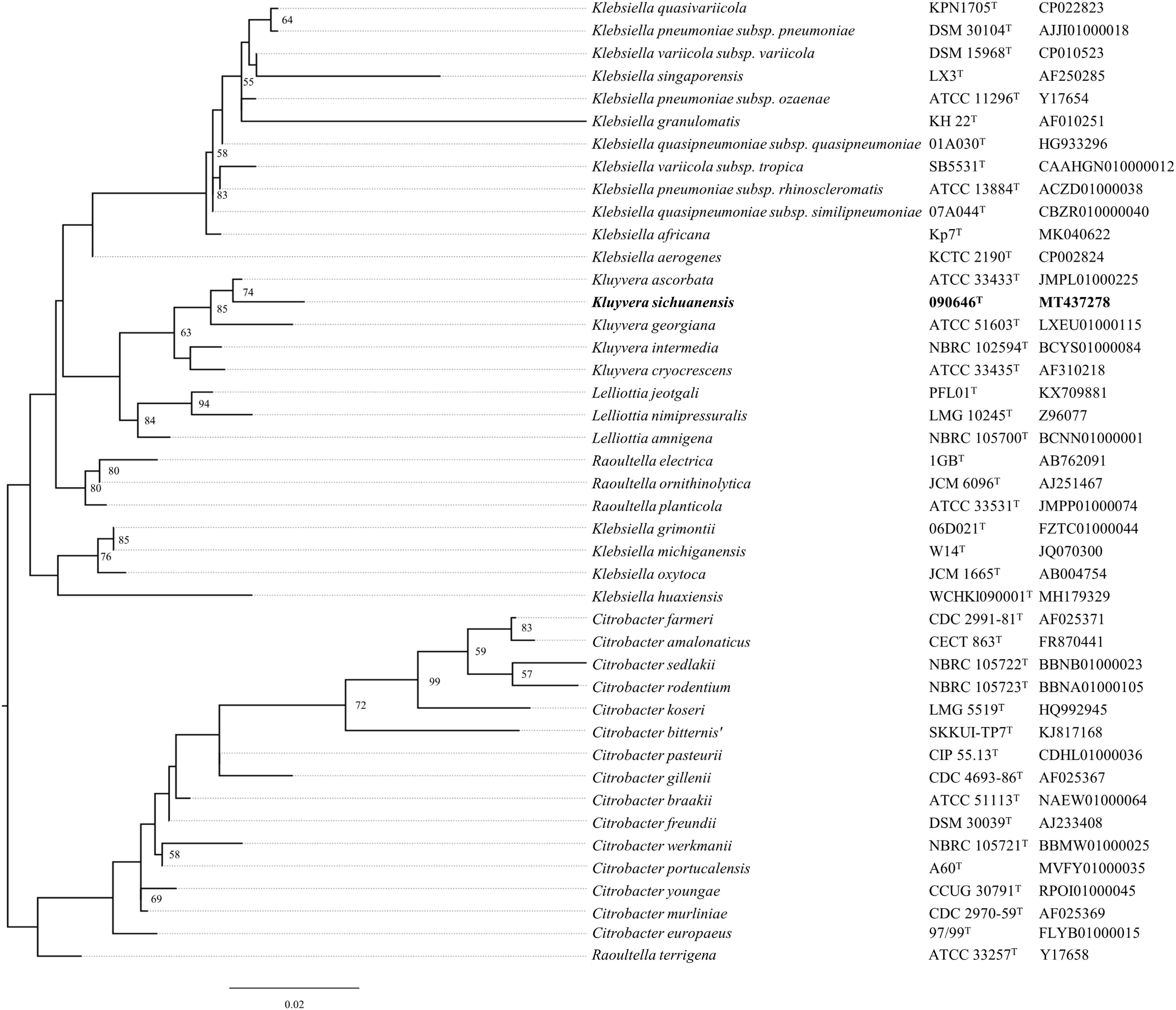
Figure 1. Phylogenetic tree of Kluyvera sichuanensis 090646T, other Kluyvera species and other closely related genera based on 16S rRNA gene sequences. The tree was inferred using the maximum-likelihood method. Bootstrap values >50% (based on 1,000 resamplings) are shown. Bar, 0.02 substitutions per nucleotide position.
Phenotypic Characterization
Gram-staining reaction was performed as described previously (Smibert and Krieg, 1994). Cell motility was examined by observing the bacterial growth and diffusion on the deep semi-solid nutrient agar medium of 0.3% (w/v) agar (Hopebio, Qingdao, China). Anaerobic growth was examined by streaking the bacterial cultures on Brain Heart Infusion agar plates and placed in the GasPakTM EZ Anaerobic Bag (BD; Franklin Lakes, NJ, United States) at 35°C for 3 days. After incubation in nutrient broth at 37°C for 3 days, flagella of strain 090646T was observed with a H-7650 transmission electron microscope (Hitachi; Tokyo, Japan) as described previously (Hanada et al., 2002). Briefly, the centrifuged bacterial precipitation was fixed with 5% (v/v) glutaraldehyde and 1% (v/v) osmium tetroxide. Ultrathin sections of the sample were embedded in epoxy resin using a Reichert ultrathin microtome (AMETEK; Berwyn, PA, United States). The samples were stained with uranyl acetate and lead citrate and examined using the microscope.
The growth of strain 090646T was examined in 5 ml aliquots of nutrient broth dispensed into tubes (16 mm, inner diameter) at temperatures of 4, 8, 18, 28, 32, 37, 42, 45, 48, and 50°C. Salt and pH tolerances were measured using nutrient broth at 37°C for 2 days at different NaCl concentrations (0.5, 1, 2, 3, 4, 5, 7.5, 10, and 15%, w/v) and at various pH values (with a pH unit of 4.0–12.0, in increments of 1.0 unit), respectively as described previously (Hu et al., 2017). Catalase activity test was conducted by examining the production of bubbles after addition of 3% (v/v) hydrogen peroxide solution, while oxidase activity was tested by using 1% tetramethyl-p-phenylenediamine dihydrochloride solution. DNase activity was detected with 1M HCl using DNase agar (Solarbio; Beijing, China) after 3 days of incubation at 30°C. Malonate, phenylalanine deaminase and potassium cyanide (KCN) experiments were performed using biochemical identification tubes (Huankai; Guangzhou, Guangdong, China). Commercially available API 20E, API 50CH, and API ZYM kits (bioMérieux; Marcy l’Etoile, France) were used for testing biochemical features and enzyme activities according to the manufacturer’s instructions with Escherichia coli strain ATCC 25922 and Pseudomonas aeruginosa strain ATCC 27853 as controls. All of the above experiments were performed in triplicate.
Fatty Acid Analysis
The analysis of cellular fatty acids was performed by the Guangdong Institute of Microbiology (Guangzhou, Guangdong, China). Briefly, fatty acid methyl esters were extracted and analyzed by gas chromatography according to the instructions of the Sherlock Microbial Identification System (MIDI Inc.; Newark, DE, United States) as described previously (Sasser, 1990; Pandey et al., 2002). Peaks were automatically integrated, and fatty acid proportions were calculated using the MIDI identification database RTSBA6 (version 6.00; MIDI Inc.).
Antimicrobial Susceptibility and Antimicrobial Resistance Genes of Strain 090646T
Minimum inhibitory concentrations (MICs) of amikacin, ampicillin, ampicillin-sulbactam, aztreonam, ceftriaxone, ceftazidime, cefepime, cefotaxime, cefuroxime, chloramphenicol, ciprofloxacin, colistin, imipenem, meropenem, piperacillin-tazobactam, sulfamethoxazole-trimethoprim, and tigecycline were determined using the microdilution broth method of the Clinical and Laboratory Standards Institute (CLSI) (CLSI, 2020). Breakpoints defined by CLSI (2020) for the Enterobacteriaceae were applied except for tigecycline, for which breakpoints defined by the European Committee on Antimicrobial Susceptibility Testing (EUCAST)1 were used. Antimicrobial resistance genes of clinical strain 090646T were identified from genome sequences using the ABRicate program2 to query the ResFinder database3.
Whole Genome Sequencing and Species Identification Based on Genome Sequences
Genomic DNA of strain 090646T was prepared using the QIAamp DNA mini kit (Qiagen; Hilden, Germany) and DNA sequencing libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB; Ipswich, MA, United States). Whole genome sequencing was performed using the HiSeq X10 Sequencer (Illumina; San Diego, CA, United States) with a layout in 150 bp paired-end and aiming for at least 200× depth of coverage. Adaptor sequences and bases of low quality at the end were trimmed from raw reads using Trimmomatic v0.39 (Bolger et al., 2014) invoked in the pipeline of Shovill v1.1.04 with default settings. Reads that passed the quality control were downsampled to approximately 150× in depth before being assembled into a draft genome of 090646T using SPAdes v3.14.0 (Bankevich et al., 2012) under “–isolate” model. Contigs shorter than 200 bp or with coverage lower than 2× were discarded in the final assembly prior to being assessed in QUAST v5.0.2 (Gurevich et al., 2013). Genome completeness and contamination were examined using CheckM (Parks et al., 2015) with the marker set of Enterobacteriaceae. The genome sequences were reported following recommendations of standards for describing a new taxonomy (Chun et al., 2018). Plasmid replicon type was determined using the PlasmidFinder tool available from the Center for Genomic Epidemiology5.
Genome sequences of type strains of all Enterobacteriaceae species (Supplementary Table S1) were retrieved from GenBank and were annotated using Prokka v1.14.6 (Seemann, 2014). Single-copy genes shared by all genomes were identified using PIRATE v1.0.4 (Bayliss et al., 2019) with default settings and were defined as core genes (n = 684). Nucleotide sequences of core genes were aligned and concatenated using MAFFT v7.313 (Katoh et al., 2002) and AMAS v0.98 (Borowiec, 2016) prior to being fed into RAxML v8.2.12 (Stamatakis, 2014) for inferring a maximum-likelihood phylogenomic tree (Figure 2) under GTRGAMMA model with a 1,000-bootstrap test.
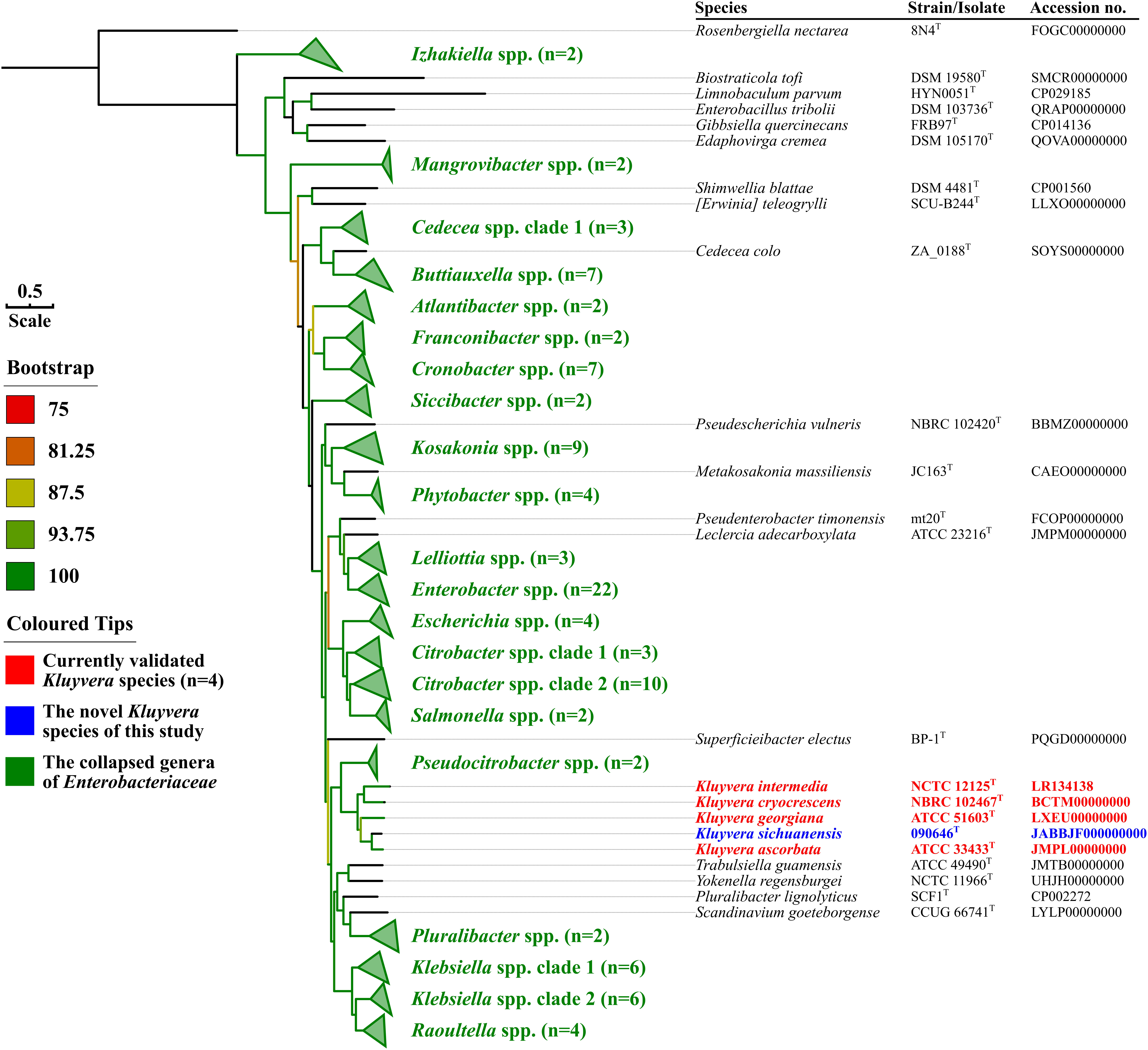
Figure 2. Maximum-likelihood phylogenomic tree of Kluyvera sichuanensis 090646T and other species in the family Enterobacteriaceae based on the core genome. The four Kluyvera species known before this study are highlighted in red, while K. sichuanensis is shown in blue. Genera comprising multiple species are depicted in green with the number of species shown in parentheses. There are two clades for the genera Cedecea, Citrobacter, and Klebsiella. Cedecea colo belongs to a separate clade, while the remaining three Cedecea species (Supplementary Table S1) form Cedecea spp. clade 1. Citrobacter spp. clade 1 comprises Citrobacter amalonaticus, Citrobacter rodentium, and Citrobacter sedlakii, while the remaining 10 Citrobacter species (Supplementary Table S1) belong to Citrobacter spp. clade 2. Klebsiella spp. clade 1 comprises Klebsiella aerogenes, Klebsiella africana, Klebsiella pneumoniae, Klebsiella quasipneumoniae, Klebsiella quasivariicola, and Klebsiella variicola, while the remaining six Klebsiella species belongs to Klebsiella spp. clade 2. For genera or clade comprising only one species and Kluyvera species, the type strains and their nucleotide accession numbers are listed alongside the names of species. The tree was inferred using the maximum likelihood method under GTRGAMMA model with a 1,000-bootstrap test and branches with support over 75% are indicated by different colors. Bar, value indicates the nucleotide substitution per site.
The average nucleotide identity (ANI), in silico DNA-DNA hybridization (isDDH), and the percentages of conserved proteins (POCP) values between strain 090646T and type strains of all Kluyvera species were determined. ANI was determined using the JSpecies with a ≥95–96% cutoff for defining species (Richter and Rossello-Mora, 2009), while isDDH was performed using Genome-to-Genome Distance Calculator (formula 2) with a the ≥70.0% cutoff for defining species (Meier-Kolthoff et al., 2013). POCP was calculated as described previously (Qin et al., 2014).
Curation of Species Identification for Kluyvera Genome Species in GenBank
We searched the NCBI GenBank database and found 47 genome sequences labeled as Kluyvera including 20 assemblies and another 27 short-read data (Dataset S1 in the Supplementary, accessed by 10-06-2020). All of the 47 genome sequences were retrieved. Strains from NCBI collection with short-read data available were fed into the same pipeline as assembling strain 090646T as described above. A maximum-likelihood phylogenomic tree based on core genes (Figure 3) was inferred for all Kluyvera genomes using RAxML v8.2.12 (Stamatakis, 2014) under GTRGAMMA model with a 1,000-bootstrap test as described above. All Kluyvera genomes were also subjected to precise species identification using ANI and isDDH as described above. Strains that have a <70% isDDH value and a <96% ANI value with any known Kluyvera species belong to a novel species, which is temporarily assigned genomospecies (genomosp.) here as the establishment of a novel species also requires phenotypic characterizations in addition to genome analysis.

Figure 3. Maximum-likelihood phylogenomic tree of Kluyvera sichuanensis 090646T and the 47 genomes labeled as Kluyvera based on the core genome. Strains and their nucleotide accession numbers are listed alongside the names of species. Type strains are depicted in blue. For genomes that are labeled as Kluyvera in GenBank but need to be revised as suggested in this study (depicted in red), the revised species names are shown first, while the current labels are shown after the slash. Genomosp. 1–4 are tentative Kluyvera species (Supplementary Table 3). The tree was inferred using the maximum likelihood method under GTRGAMMA model with a 1,000-bootstrap test and branches with support over 75% are indicated by different colors. Bar, value indicates the nucleotide substitution per site.
Comparative Genomic Analysis
Genomes labeled as Kluyvera and those of type strains of other species in Enterobacteriaceae (Supplementary Table S1) were annotated using Prokka v1.14.6 (Seemann, 2014) prior to being fed into PIRATE v1.0.4 (Bayliss et al., 2019) with 85% amino acid identity as the cutoff for comparative genomic analysis. Universal primers to amplify the complete sequence of the ferric reductase-encoding gene fes from all Kluyvera genomes were designed manually by examining the upstream and downstream sequences. Primers were checked for melting temperature (Tm), secondary structure, and primer dimer using oligoevaluator6. Virulence factors present in all Kluyvera genomes were determined using genome sequences to query the virulence determinants of the Enterobacteriaceae including E. coli, Klebsiella pneumoniae, Salmonella enterica, and Shigella flexneri in the VFDB database (Liu et al., 2019).
Results
Strain Identification
Strain 090646T was recovered from the residual water of a handwashing sink at an ICU in Chengdu, China, on April 2019 as part of an infection control surveillance program on sinks. The strain was preliminarily identified as Kluyvera ascorbata by MALDI-TOF. A nearly complete sequence (1,411 bp) of the 16S rRNA gene of strain 090646T has the highest identity with that of Kluyvera ascorbata ATCC 33433T (99.22%) and is also highly similar with those of K. intermedia NCTC 12125T (98.88%), K. cryocrescens NBRC 102467T (98.58%), K. georgiana ATCC 51603T (98.30%), Klebsiella aerogenes KCTC 2190T (98.23%), Raoultella terrigena ATCC 33257T (98.09%), Citrobacter braakii ATCC 51113T (97.95%), and Lelliottia amnigena NBRC 105700T (97.94%). In a maximum-likelihood phylogenetic tree (Figure 1) based on the 16S rRNA gene sequence alignment, strain 090646T was allocated within the Kluyvera clade. However, strain 090646T forms a relatively long branch separating from other Kluyvera species (Figure 1) and it is well known that analysis on 16S rRNA gene sequence is insufficient for accurate bacterial species assignation (Mulet et al., 2020). We therefore performed whole genome sequencing by HiSeq X 10 (Illumina; San Diego, CA, United States) for strain 090646T. A total of 1.42 gigabyte bases were generated, which were then assembled into 117 contigs (N50, 144,979 bp). The draft genome of strain 090646T is 5,476,810 bp with a 54.51 mol% G + C content, which has been deposited at DDBJ/EMBL/GenBank under the accession JABBJF000000000.
A total of 684 core genes (Supplementary Dataset S2 in the Supplementary file) were identified in genome sequences of type strains of all Enterobacteriaceae species (Supplementary Table S1). In the maximum-likelihood phylogenomic tree based on core genes (Figure 2), strain 090646T was also allocated within the Kluyvera clade. The POCP values between strain 090646T and type strains of all Kluyvera species ranged from 90.81 to 93.18% (Table 1), suggesting that strain 090646T indeed belonged to the genus Kluyvera. The ANI values between strain 090646T and type strains of all Kluyvera species ranged from 84.15 to 90.10% (Table 1), lower than the ≥95–95% cutoff for defining species (Richter and Rossello-Mora, 2009). Consistently, isDDH values between strain 090646T and type strains of all Kluyvera species ranged from 28.2 to 42.3% (Table 1), which are well below the ≥70.0% cutoff to define species (Richter and Rossello-Mora, 2009; Meier-Kolthoff et al., 2013). Both ANI and isDDH analyses clearly suggest that strain 090646T represents a novel species of the genus Kluyvera. We proposed the species name as Kluyvera sichuanensis (si.chuan.en’sis. N.L. adj. sichuanensis, referring to Sichuan Province, China, where the type strain was recovered) after phenotypic characterization (see below). The type strain 090646T has been deposited into Guangdong Microbial Culture Collection Center as GDMCC 1.1872T and into the Korean Collection for Type Cultures as KCTC82166T.

Table 1. ANI, isDDH and POCP values between 090646T and the type strains of other Kluyvera species.
Phenotypic Characterization
We performed physiological and biochemical characterization for strain 090646T (Table 2). The utilization of D-sorbitol combined with negative reactions for β-galactosidase (ONPG), D-mannose, glycerin, raffinose, nor sucrose is able to differentiate strain 090646T from other Kluyvera species. Detailed results of the above tests are given in the species descriptions. The major cellular fatty acids of strain 090646T are C16:0 (31.28%), C17:0 cyclo (17.56%), and summed in feature 8 (C18:1ω7c) (15.74%). By contrast, the composition of fatty acids in other Kluyvera species has not been reported.
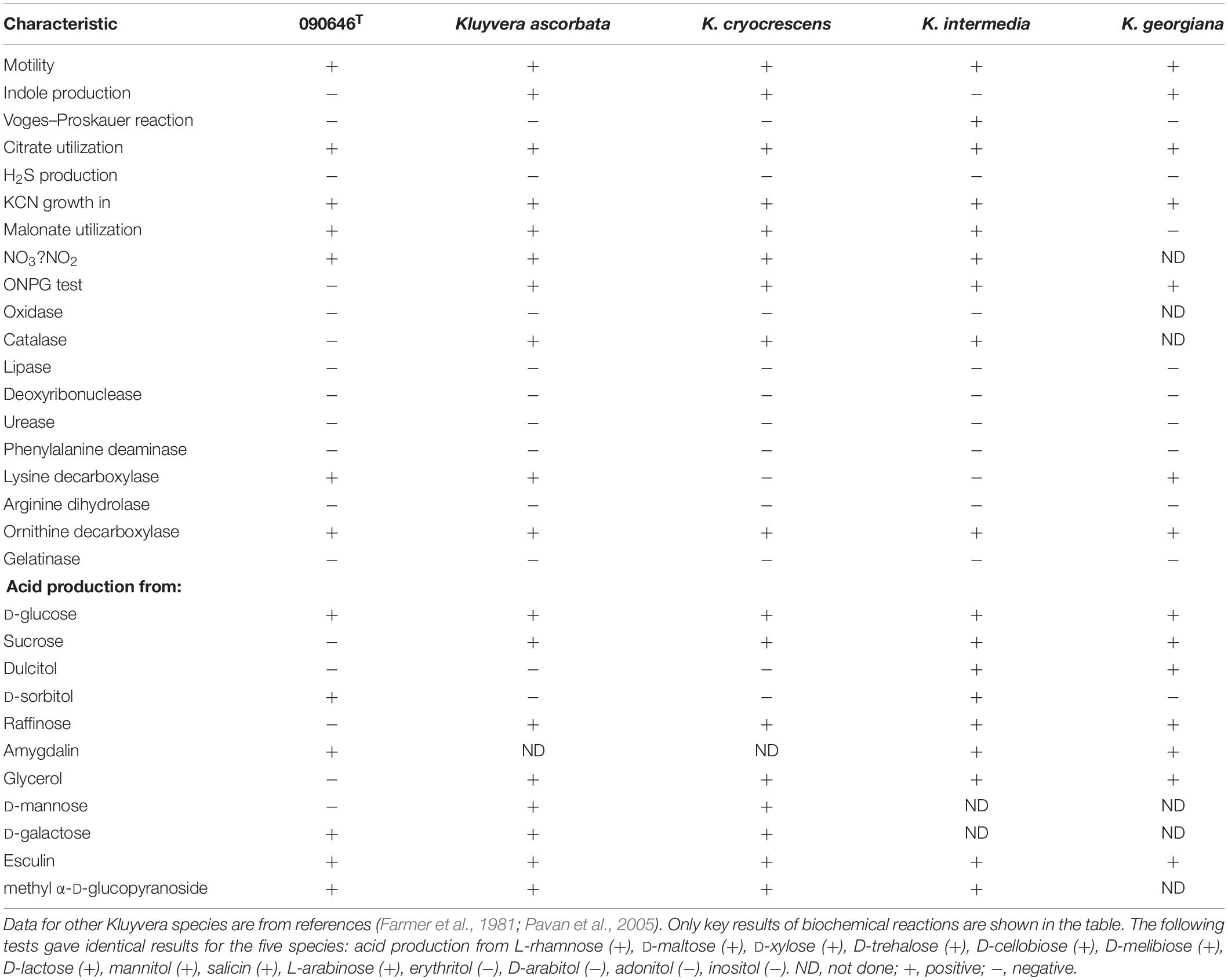
Table 2. Biochemical characteristics of strain 090646T and other Kluyvera species.
Antimicrobial Susceptibility and Antimicrobial Resistance Genes
Strain 090646T was resistant to ampicillin, ampicillin-sulbactam, aztreonam, ceftriaxone, ceftazidime, cefepime, cefotaxime, cefuroxime, ciprofloxacin, imipenem, meropenem, piperacillin-tazobactam, but was susceptible to amikacin, chloramphenicol, colistin, sulfamethoxazole-trimethoprim, and tigecycline (Supplementary Table S2). Strain 090646T had blaNDM–1, encoding a metallo-β -lactamase to confer resistance to most β -lactams including carbapenems. Like strains of all other Kluyvera species, strain 090646T harbors an intrinsic chromosomal blaCTX–M gene, which encodes a CTX-M enzyme sharing the highest amino acid identity (90.72%, 264/291) with CTX-M-95 from Kluyvera ascorbata. Strain 090646T also had blaCTX–M–14, blaOXA–1, blaTEM–1, and blaSFO–1 to mediate resistance to aztreonam, cephalosporins and penicillins, aminoglycoside-resistance genes aac(6′)-Ib-cr, aac(3)-IId, aph(6)-Id, and aph(3″)-Ib, fosfomycin-resistance gene fosA3, and quinolone-resistance gene qnrS1. Strain 090646T contains seven plasmid replicons including IncFIB(pHCM2), RepA_pKPC-CAV1321, ColRNAI, Col(MGD2), ColpVC, Col440I, and Col440II. The association between antimicrobial resistance genes and plasmids warrants further investigations, which is, however, out of the scope of this study focusing on taxonomy.
Species Curation for Kluyvera Genomes in GenBank
Except strain 090464T, there are 47 strains labeled as Kluyvera with genomes available in GenBank including 32 SRA and 20 assemblies (5 genomes with both SRA and assemblies) (Dataset S1, accessed by 10-06-2020). Among the 47 strains labeled as Kluyvera, 33 (70.2%, 33/47) were indeed of Kluyvera, while 13 strains actually belonged to Phytobacter ursingii, a non-Kluyvera species of the family Enterobacteriaceae (Pillonetto et al., 2018), and one strain was in fact of Klebsiella aerogenes. Twenty-five of the 33 Kluyvera strains could be correctly assigned to known Kluyvera species including Kluyvera ascorbata (n = 7), K. cryocrescens (n = 8), K. georgiana (n = 3), and K. intermedia (n = 7) including one strain labeled as Kluyvera ascorbata belonging to K. georgiana instead. No strain belongs to K. sichuanensis is found. The remaining eight strains were labeled as one of the four known Kluyvera species but actually do not belong to any known Kluyvera species. Instead, the 8 strains can be assigned to four novel Kluyvera genomospecies, genomosp. 1, 2, 3, and 4, based on ANI and isDDH values (Table 3) as well as illustrated in the phylogenomic tree (Figure 3). Species names are not assigned to the novel genomospecies here as they need to be characterized by phenotype methods. Currently, the Kluyvera genus therefore comprises nine species (Table 3). However, as there were only 34 Kluyvera genomes available, genome sequencing for more Kluyvera strains is required to further untangle the taxonomic composition of the genus. Collectively, nearly a half (n = 23) of the 47 Kluyvera genomes need to be curated for species assignations including species misidentification of 14 non-Kluyvera genomes and one Kluyvera genomes and update of 8 genomes to novel genomospecies.
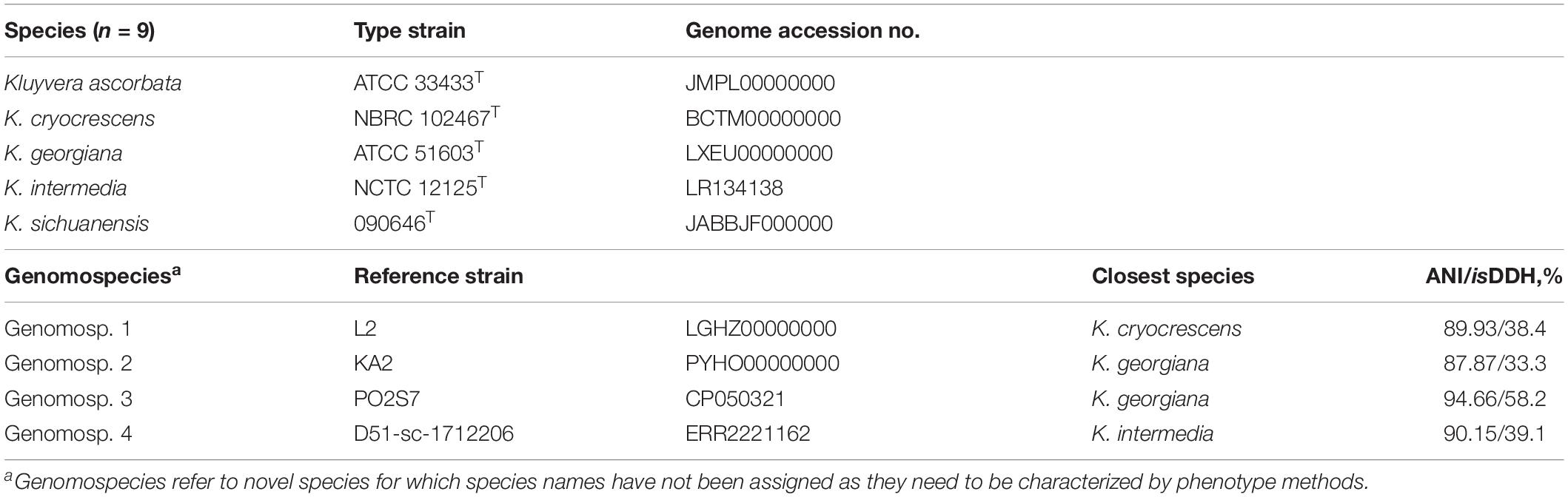
Table 3. Updated Classification and nomenclature of the genus Kluyvera.
Virulence Factors
Strain 090646T had multiple known virulence genes, i.e., the chuS-U-W-X-Y, entA-B-C-E-F-S, and fepA-B-C-D-G clusters, fes, iutA, and shuV for iron acquisition, csgG (encoding curli production assembly/transport protein), the fimC-D-F-G-H cluster (encoding type 1 fimbriae), the kpsD-M-T cluster (encoding K1 capsule), and ompA (encoding outer membrane protein A) (Supplementary Dataset S3 in the Supplementary). All of the virulence factors in strain 090646T were also present in other Kluyvera genomes (Supplemenatry Dataset S3).
Designing a Universal Pair of Primers for Differentiating Kluyvera Species by Comparative Genomics
As there are only few genomes for each Kluyvera species, we did not design a panel of primers targeting individual species but instead used a universal PCR approach. By comparative genomics, we identified that a ferric reductase-encoding gene fes was present in all Kluyvera genomes and had significant nucleotide variations for each Kluyvera species. We therefore designed a pair of primers (fes-up, 5′-TACGCTATTGCAAATGCAAA; fes-dw, 5′-TTGCATACCAAACTCCTGTC) able to in silico amplify the complete sequence (1,206 to 1,257 bp, Supplementary Table S3) of fes in all available Kluyvera genomes with <94% inter-species and >97% intra-species identities (Supplementary Dataset S4 in the Supplementary). Tm of the primers is 61.7 and 60.2°C, respectively, and therefore PCR with this pair of primers could be performed with a 55°C annealing temperature. Subsequent Sanger sequencing of the amplicons has the ability to differentiate all Kluyvera species with a >97% nucleotide identity cutoff. This approach warrants verification by experiments in the future.
Discussion
In this study, we firstly described a novel Kluyvera species, K. sichuanensis, from a sink, using both genome and phenotypic analyses. The type strain 090646T was recovered from a handwashing sink in an ICU, exhibited multi-drug resistance including resistance to carbapenems, and carried the blaNDM–1 carbapenemase gene and therefore was of clinical significance.
We then curated all other Kluyvera genomes (n = 47) in GenBank and found the misidentification of 13 P. ursingii and one Klebsiella aerogenes of other genera as Kluyvera. This highlights that species identification for bacterial genomes in GenBank need to be carefully curated. Researchers who use bacterial genomes should be fully aware of the fact that the species labeled may be incorrect and therefore are recommended to check the precise species assignation by using genome-based analysis methods such as ANI and isDDH. In this study, we identified four novel genomospecies from the genome sequences in GenBank and therefore at present the Kluyvera genus comprises nine species (Table 3). However, as there were only 34 Kluyvera genomes available, genome sequencing for more Kluyvera strains is required to further untangle the taxonomic composition of the genus.
Conclusion
In conclusion, a novel Kluyvera species, K. sichuanensis sp. nov., is identified and is characterized by both genome analysis and phenotypic methods. Four novel genomospecies are also identified, which warrant further phenotypic characterization. Genome sequencing for more Kluyvera strains is required to understand the genus of clinical relevance.
Descriptions
Description of Kluyvera sichuanensis sp. nov.
Kluyvera sichuanensis (si.chuan.en’sis. N.L. fem. adj. sichuanensis, referring to Sichuan Province, China, where the type strain was recovered). The species status is determined using ANI, isDDH (Table 1 and Supplementary Dataset S1), core gene-based phylogenomic analysis (Figure 2) and phenotypic assays (see below).
Cells are Gram-stain-negative, facultatively anaerobic, motile, non-spore-forming and short-rod shaped (0.5–0.8 μm wide and 1.0–2.0 μm long, Supplementary Figure S1). Colonies growing on the nutrient agar after 12 h are round, smooth, convex, white. Growth occurs between 8 and 42°C, in the 0 to 5% NaCl (w/v), and at pH 5.0 to 9.0. Nitrate reduction, citrate utilization, and activities of lysine decarboxylase and ornithine decarboxylase are positive. It is able to assimilate esculin, amygdalin, arbulin, D-cellobiose, D-fructose, D-glucose, D-galactose, D-lactose, D-maltose, D-melibiose, D-sorbitol, D-trehalose, D-xylose, 5-keto-gluconate, L-arabinose, L-rhamnose, mannitol, malonate, methyl-α-D-glucopyranoside, N-acetylglucosamine, potassium gluconate, ribose, and salicin. It is negative for acetoin production (Voges–Proskauer), catalase, DNase, H2S production, indole production, oxidase and phenylalanine deaminase, and activities of arginine dihydrolase, β-galactosidase (ONPG), gelatinase, tryptophan deaminase and urease. It does not utilize adonitol, D-mannose, D-saccharose, dulcitol, erythritol, glycerol, inositol, raffinose and sucrose. The major cellular fatty acids are C16:0 (31.28%), C17:0 cyclo (17.56%) and summed in feature 8 (C18:1ω7c) (15.74%).
The type strain is 090646T (also called SCKS090646T), recovered from a hospital sink in Chengdu, Sichuan province, China. Strain 090646T has been deposited into Guangdong Microbiology Culture Center as GDMCC 1.1872T and into Korean Collection for Type Cultures as KCTC 82166T. The draft genome of the type strain is 5,476,810 bp with a 54.51 mol% G + C content (DDBJ/EMBL/GenBank accession no. JABBJF000000000).
Description of Kluyvera Genomospecies 1
As delineation of the strains could be determined using ANI and isDDH (Table 3 and Supplementary Dataset S1) as well as core gene-based phylogenomic analysis (Figure 3), it is proposed to designate a novel genomospecies, Kluyvera genomospecies 1, represented by strain L2 (GenBank accession no. LGHZ00000000 for the genome sequence).
Description of Kluyvera Genomospecies 2
As delineation of the strains could be determined using ANI and isDDH (Table 3 and Supplementary Dataset S1) as well as core gene-based phylogenomic analysis (Figure 3), it is proposed to designate a novel genomospecies, Kluyvera genomospecies 2, represented by strain KA2 (GenBank accession no. PYHO00000000 for the genome sequence).
Description of Kluyvera Genomospecies 3
As delineation of the strains could be determined using ANI and isDDH (Table 3 and Supplementary Dataset S1) as well as core gene-based phylogenomic analysis (Figure 3), it is proposed to designate a novel genomospecies, Kluyvera genomospecies 3, represented by strain PO2S7 (GenBank accession no. CP050321 for the genome sequence).
Description of Kluyvera Genomospecies 4
As delineation of the strains could be determined using ANI and isDDH (Table 3 and Supplementary Dataset S1) as well as core gene-based phylogenomic analysis (Figure 3), it is proposed to designate a novel genomospecies, Kluyvera genomospecies 4, represented by strain D51-sc-1712206 (GenBank accession no. ERR2221162 for the genome sequence).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
ZZ designed the study. LL, YF, LW, and FQ performed the study. LL, YF, and ZZ analyzed the data. LL and ZZ drafted the manuscript. All authors contributed to approve the manuscript.
Funding
The work was supported by grants from the National Natural Science Foundation of China (project nos. 81772233, 81661130159, and 81861138055), West China Hospital of Sichuan University (1.3.5 project for disciplines of excellence, project no. ZYYC08006 and grant no. 312190022), and the Newton Advanced Fellowships, Royal Society, United Kingdom (NA150363).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.579306/full#supplementary-material
Footnotes
- ^ http://www.eucast.org/
- ^ https://github.com/tseemann/abricate
- ^ http://genomicepidemiology.org/
- ^ https://github.com/tseemann/shovill
- ^ http://genomicepidemiology.org/
- ^ http://www.oligoevaluator.com
References
Alnajar, S., and Gupta, R. S. (2017). Phylogenomics and comparative genomic studies delineate six main clades within the family Enterobacteriaceae and support the reclassification of several polyphyletic members of the family. Infect. Genet. Evol. 54, 108–127. doi: 10.1016/j.meegid.2017.06.024
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bayliss, S. C., Thorpe, H. A., Coyle, N. M., Sheppard, S. K., and Feil, E. J. (2019). PIRATE: a fast and scalable pangenomics toolbox for clustering diverged orthologues in bacteria. Gigascience 8:giz119. doi: 10.1093/gigascience/giz119
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Borowiec, M. L. (2016). AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4:e1660. doi: 10.7717/peerj.1660
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., Da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
CLSI (2020). Performance Standards for Antimicrobial Susceptibility Testing; Thirty-first Informational Supplement. M100-S31. Wayne, PA: Clinical and Laboratory Standards Institute.
Farmer, J. J., Fanning, G. R., Huntley-Carter, G. P., Holmes, B., and Brenner, D. J. (1981). Kluyvera, a new (redefined) genus in the family Enterobacteriaceae: identification of Kluyvera ascorbata sp. nov. and Kluyvera cryocrescens sp. nov. in clinical specimens. J. Clin. Microbiol. 13, 919–933. doi: 10.1128/JCM.13.5.919-933.1981
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hanada, S., Takaichi, S., Matsuura, K., and Nakamura, K. (2002). Roseiflexus castenholzii gen. nov., sp. nov., a thermophilic, filamentous, photosynthetic bacterium that lacks chlorosomes. Int. J. Syst. Evol. Microbiol. 52, 187–193. doi: 10.1099/00207713-52-1-187
Hu, Y., Feng, Y., Zhang, X., and Zong, Z. (2017). Acinetobacter defluvii sp. nov., recovered from hospital sewage. Int. J. Syst. Evol. Microbiol. 67, 1709–1713. doi: 10.1099/ijsem.0.001847
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Lee, J., Hwang, J. H., Jo, D. S., Lee, H. S., and Hwang, J. H. (2019). Kluyvera ascorbata as a pathogen in adults and children: clinical features and antibiotic susceptibilities in a single center study. Jpn. J. Infect. Dis. 72, 142–148. doi: 10.7883/yoken.JJID.2018.375
Li, Y., Luo, L., Xiao, Z., Wang, G., Li, C., Zhang, Z., et al. (2019). Characterization of a carbapenem-resistant Kluyvera cryocrescens isolate carrying blaNDM-1 from hospital sewage. Antibiotics 8:149. doi: 10.3390/antibiotics8030149
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Madeira, F., Park, Y. M., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Goker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Moreno, C., Romero, J., and Espejo, R. T. (2002). Polymorphism in repeated 16S rRNA genes is a common property of type strains and environmental isolates of the genus Vibrio. Microbiology 148, 1233–1239. doi: 10.1099/00221287-148-4-1233
Mulet, M., Lalucat, J., and García-Valdés, E. (2020). DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 12, 1513–1530. doi: 10.1111/j.1462-2920.2010.02181.x
Muller, H. E., Brenner, D. J., Fanning, G. R., Grimont, P. A., and Kampfer, P. (1996). Emended description of Buttiauxella agrestis with recognition of six new species of Buttiauxella and two new species of Kluyvera: Buttiauxella ferragutiae sp. nov., Buttiauxella gaviniae sp. nov., Buttiauxella brennerae sp. nov., Buttiauxella izardii sp. nov., Buttiauxella noackiae sp. nov., Buttiauxella warmboldiae sp. nov., Kluyvera cochleae sp. nov., and Kluyvera georgiana sp. nov. Int. J. Syst. Bacteriol. 46, 50–63. doi: 10.1099/00207713-46-1-50
Mutoh, Y., Kobe, T., Hirano, T., Ichihara, T., Takenaka, H., Niinomi, T., et al. (2019). The first case of third-generation cephalosporins resistant Kluyvera ascorbata biliary tract infection in Japan: a case report and review of the literature. IDCases 15:e00498. doi: 10.1016/j.idcr.2019.e00498
Pandey, K. K., Mayilraj, S., and Chakrabarti, T. (2002). Pseudomonas indica sp. nov., a novel butane-utilizing species. Int. J. Syst. Evol. Microbiol. 52, 1559–1567. doi: 10.1099/00207713-52-5-1559
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pavan, M. E., Franco, R. J., Rodriguez, J. M., Gadaleta, P., Abbott, S. L., Janda, J. M., et al. (2005). Phylogenetic relationships of the genus Kluyvera: transfer of Enterobacter intermedius Izard et al. 1980 to the genus Kluyvera as Kluyvera intermedia comb. nov. and reclassification of Kluyvera cochleae as a later synonym of K. intermedia. Int. J. Syst. Evol. Microbiol. 55, 437–442. doi: 10.1099/ijs.0.63071-0
Pillonetto, M., Arend, L. N., Faoro, H., D’espindula, H. R. S., Blom, J., Smits, T. H. M., et al. (2018). Emended description of the genus Phytobacter, its type species Phytobacter diazotrophicus (Zhang 2008) and description of Phytobacter ursingii sp. nov. Int. J. Syst. Evol. Microbiol. 68, 176–184. doi: 10.1099/ijsem.0.002477
Qin, Q. L., Xie, B. B., Zhang, X. Y., Chen, X. L., Zhou, B. C., Zhou, J., et al. (2014). A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215. doi: 10.1128/JB.01688-14
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Sarria, J. C., Vidal, A. M., and Kimbrough, R. C. III (2001). Infections caused by Kluyvera species in humans. Clin. Infect. Dis. 33, E69–E74. doi: 10.1086/322686
Sasser, M. (1990). Identification of bacteria by gas chromatography of cellular fatty acids. MIDI Tech. Note 101, 1–7.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Smibert, R. M., and Krieg, N. R. (1994). “Phenotypic characterization,” in Methods for General and Molecular Bacteriology, eds P. Gerhardt, R. G. E. Murray, W. A. Wood, and N. R. Krieg (Washington, DC: American Society for Microbiology), 607–654.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Keywords: Kluyvera sichuanensis, Kluyvera, genome sequences, sinks, taxonomy
Citation: Liu L, Feng Y, Wei L, Qiao F and Zong Z (2020) Precise Species Identification and Taxonomy Update for the Genus Kluyvera With Reporting Kluyvera sichuanensis sp. nov.. Front. Microbiol. 11:579306. doi: 10.3389/fmicb.2020.579306
Received: 02 July 2020; Accepted: 25 August 2020;
Published: 16 September 2020.
Edited by:
Denis Grouzdev, Federal Center Research Fundamentals of Biotechnology (RAS), RussiaReviewed by:
Alba Pérez-Cataluña, Institute of Agrochemistry and Food Technology (IATA), SpainSwapnil Prakash Doijad, University Hospital of Giessen and Marburg, Germany
Copyright © 2020 Liu, Feng, Wei, Qiao and Zong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiyong Zong, em9uZ3poaXlAc2N1LmVkdS5jbg==
†These authors have contributed equally to this work