 Zhi Yuan Ma
Zhi Yuan Ma Xiu Min Zhang1
Xiu Min Zhang1 Min Wang
Min Wang Ting Liu
Ting Liu- 1CAS Key Laboratory for Agro-Ecological Processes in Subtropical Region, National Engineering Laboratory for Pollution Control and Waste Utilization in Livestock and Poultry Production, Institute of Subtropical Agriculture, Chinese Academy of Sciences, Changsha, China
- 2College of Resources and Environment, University of Chinese Academy of Sciences, Beijing, China
- 3College of Animal Science and Technology, Gansu Agricultural University, Lanzhou, China
QIAamp Fast DNA Stool Mini Kit (QIAGEN, Valencia, CA, United States) and RBB + C (Yu and Morrison, 2004) methodologies are widely employed to extract microbial DNA from rumen samples and can exhibit different efficiencies of obtaining DNA yield, quality, and downstream amplicon sequence analysis. No study has conducted to investigate the contributions of chemical and mechanical lysis on DNA extraction, which included chemical lysis from QIAamp Fast DNA Stool Mini Kit (QIA) and RBB + C (YM), bead (BB), and sand beating (SB). Effects of chemical lysis and bead beating (BB) were investigated by conducting a 2 × 2 factorial-designed experiment with four methodologies, including QIA without (QIA−) and with BB (QIA + BB), and YM without (YM−) and with BB (YM + BB). Comparisons between bead and sand were conducted by comparing methodologies of YM + BB and YM + SB. Comparing with QIA, YM had lower (P ≤ 0.10) OD260/280 and diversity of ZOTUs and length polymorphism of protozoal amplicons but harvested greater (P ≤ 0.086) DNA from fibrolytic bacteria such as Ruminococcaceae lineages. Including BB increased (P = 0.001) total DNA yield without affecting (P ≥ 0.55) OD260/280 and richness of bacterial ZOTUs but decreased (P ≤ 0.08) richness of both ZOTUs and length polymorphism of protozoal amplicon. Bead beating and SB showed no difference (P ≥ 0.19) in DNA yield and quality and bacterial and protozoal community. In summary, chemical lysis provided by RBB + C and QIAamp Fast DNA Stool Mini Kit should be better to extract DNA for analyzing bacterial and protozoal community, respectively. Sand can be an alternative beater for DNA extraction, and mechanical lysis is not recommended for protozoal community analysis.
Introduction
The quality (yield, purity, and integrity) of microbial DNA extracted from digesta samples is crucial for downstream analysis of amplicon sequencing (Henderson et al., 2013; Vaidya et al., 2018). Both QIAamp Fast DNA Stool Mini Kit (QIAGEN, Valencia, CA, United States) and RBB + C (Yu and Morrison, 2004) methodologies are widely used to extract microbial DNA from rumen samples. However, the RBB + C recovers greater microbial DNA yield from rumen fluid samples with 5-fold increase when compared to QIAamp Fast DNA Stool Mini Kit, implicating a more suitable methodology to extract microbial DNA from rumen fluid. Furthermore, downstream microbial community analysis showed distinctions between these two methodologies based on DGGE (Yu and Morrison, 2004) and amplicon sequencing (Henderson et al., 2013; Vaidya et al., 2018). Comparing with QIAamp Fast DNA Stool Mini Kit, RBB + C methodology not only has a different chemical lysis but also includes bead beating. Contributions of chemical lysis and bead beating have not been investigated in these two DNA extraction methodologies.
Mechanical lysis is another factor that influences efficiency of DNA extraction. Evidences show that including a beader beating step improves DNA yield (de Boer et al., 2010; Guo and Zhang, 2013; Pollock et al., 2018), bacterial diversity (Guo and Zhang, 2013), gram-positive bacteria (Salonen et al., 2010), spores (Salonen et al., 2010), and fungi (Fiedorova et al., 2019). Sand is raw material of bead and has a similar chemical composition of silicon dioxide as bead. Sand is irregularly shaped and may disrupt cell walls more efficiently than the evenly shaped round bead. Furthermore, sand is cheaper and more available than bead; replacing bead with sand may decrease the costs for DNA extraction. Including sand sufficiently extracts DNA from human feces for detecting pathogens (Karasartova et al., 2018). However, no study has been performed to evaluate the effect of sand beater on the efficiency of DNA extraction from rumen microorganisms.
The first objective of this study was to compare the effects of chemical lysis provided by RBB + C and QIAamp Fast DNA Stool Mini Kit and bead beating on microbial DNA quality and downstream amplicon analysis. The second objective is to compare bead and sand beating on microbial DNA quality and downstream amplicon analysis.
Materials and Methods
All animal procedures followed our institutional guidelines for the care and use of animals and were approved by the Animal Care Committee (Approval number ISA-W-201609), Institute of Subtropical Agriculture, Chinese Academy of Sciences, Changsha, China.
Experimental Design, Rumen Sampling, and DNA Extraction
Microbial DNA extraction briefly had three major steps from protocol of QIAamp Fast DNA Stool Mini Kit and RBB + C methodologies. Mechanical lysis (ML) was defined as pretreated processes with mechanical breakdown such as a violent vibration, while chemical lysis (CL) was defined as the chemical environment provided by processes assorted to their lysis buffers after ML to acquire raw DNA (Figure 1). The final purification was the processes to acquire good-quality DNA using silica matrix columns (Figure 1). QIAamp Fast DNA Stool Mini Kit methodology exhibited a different chemical lysis (QIA) in comparison with that of RBB + C methodology (YM) and did not contain a step of mechanical lysis. Mechanical lysis is a beating-based step and included bead (BB, 100–200 μm mesh, Omega Bio-Tek, Norcross, GA, United States) or sand (SB, 300–800 μm mesh, Sinopharm Chemical Reagent Co., Ltd., China) beating.
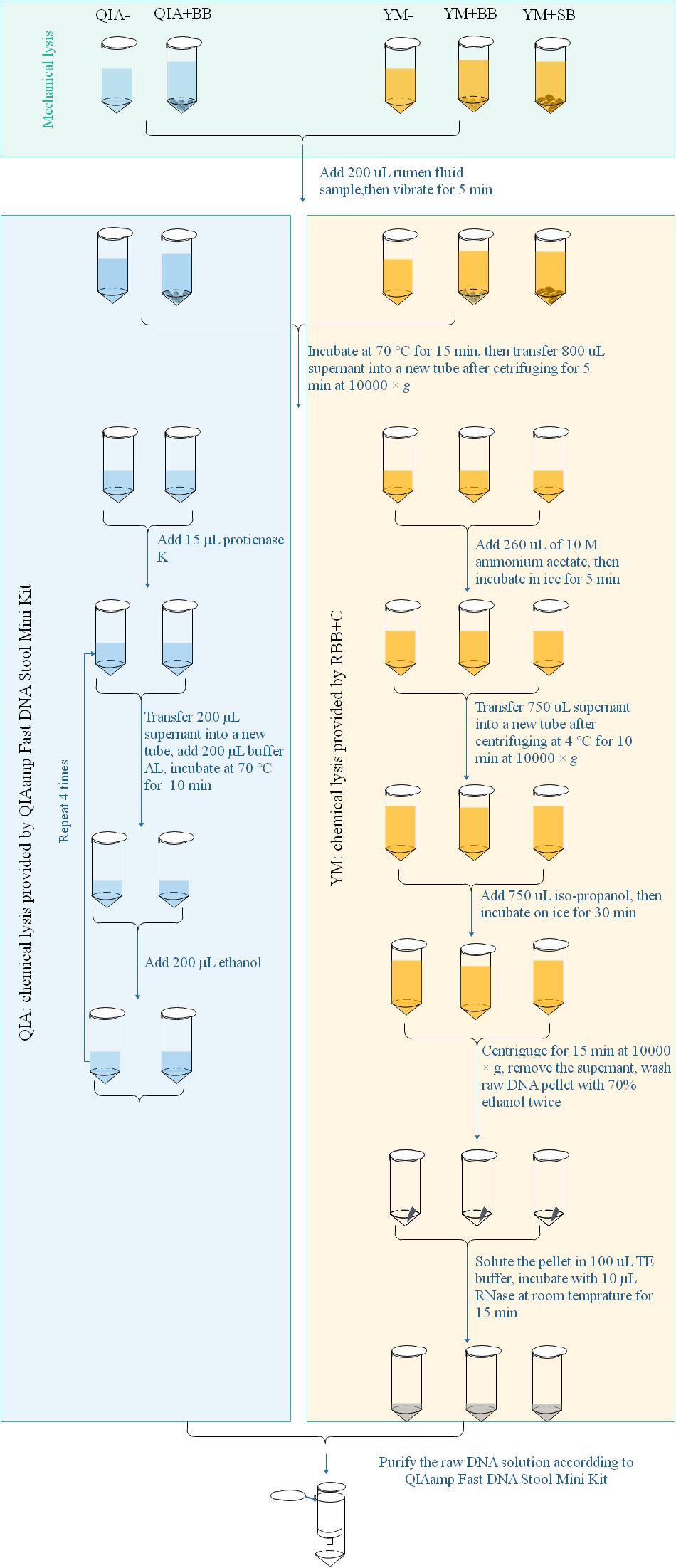
Figure 1. Scheme for DNA extraction procedures of five experimental methodologies. QIA, the chemical lysis provided by QIAamp Fast DNA Stool Mini Kit methodology; YM, the chemical lysis provided by RBB + C (Yu and Morrison, 2004); BB, bead beating; SB, sand beating; QIA–, QIA without mechanical lysis; QIA + BB, QIA with BB, YM–, YM without ML; YM + BB, QIA with BB; YM + SB, QIA with SB.
The procedure of five experimental methodologies included QIA without (QIA−) and with BB (QIA + BB), and YM without (YM−) and with BB (YM + BB) or SB (YM + SB), which were briefly described in Figure 1. Briefly speaking, for methodologies without beater beating including QIA− and YM−, samples were vibrated with 1 mL of designated lysis buffer. For methodologies with beater beating including QIA + BB, YM + BB, and YM + SB, 200 mg bead or sand was further added before vibrating. It needs to be noted that the original procedure of RBB + C methodology had a repeated lysis step to debris, which was discarded to ensure that the only difference between QIA− and YM− methodology came from two lysis buffers. Effects of CL and BB were investigated by conducting a 2 × 2 factorial-designed experiment with four methodologies, including QIA without (QIA−) and with BB (QIA + BB), and YM without (YM−) and with BB (YM + BB). Comparisons between bead and sand beading were conducted by comparing methodologies of YM + BB and YM + SB. Both two experiments share the YM + BB methodology.
Three rumen-cannulated goats aged 2, 5, and 6 years were used in this study. All goats were fed individually, and the diet was rice straw and concentrate (554 g corn grain, 198 g wheat bran, 185 g soybean meal, 30 g soybean oil, 12 g calcium carbonate, 11 g sodium chloride, and 10 g premix with vitamins and microelements per kg of DM) separately. The rumen contents were collected through the cannula 2 h after commencement of morning feeding. About 200 mL of rumen fluid was prepared for each animal by filtering the collected rumen contents through four layers of sterile cheesecloth into a prewarmed insulated bottle. Fresh samples were transferred to our laboratory within 10 min after detachment from rumen for microbial DNA extraction.
Microbial DNA extraction was conducted in triplicate by using 200 μL of rumen fluid sample of each goat according to the protocol of each methodology. All methodologies were vibrated by a vibrator (CEBO-48, Chebo BioTech Co., Shanghai, China) at a frequency of 50 Hz for 3 min, and raw DNA was purified through spin columns from QIAamp DNA Stool Mini Kit. The concentration and purity of DNA were measured using a spectrophotometer (ND-2000; NanoDrop Technologies, Wilmington, DE, United States). Two-microliter DNA samples were visualized by 1% agarose gel electrophoresis after pooling according to animals and methodologies.
Amplicon Sequencing and Bioinformatic Analysis
The V4 region amplicons of rrs genes were achieved by using primes of GTGYCAGCMGCCGCGGTAA (forward, 5′→3′) and GGACTACNVGGGTWTCTAAT (reverse, 5′→3′) for bacteria (Walters et al., 2015), and CGCGGTAATTCCAGCTCCA (forward, 5′→3′), TTGGYRAATGCTTTCGC (reverse, 5′→3′) for protozoa (Hugerth et al., 2014). Amplicon PCR procedure was performed by following previously described methods by Ma et al. (2018) and sequenced using the Illumina MiSeq platform by Allwegene Tech., Beijing, China. The sequences without adaptors were then demultiplexed according to their inserted barcodes for bioinformatic analysis.
The barcodes and primers were stripped by using vsearch (Rognes et al., 2016). The stripped fastq pairs were merged by usearch v11 (Edgar, 2010). Quality controls of merged sequences were performed according to a vsearch pipeline1. Length filtered sequences were achieved by setting as -fastq_minlen 248 -fastq_maxlen 255 for bacteria, and -fastq_minlen 50 -fastq_maxlen 500 for protozoa. The chimera, singletons, and doubletons were removed after dereplicating of sequences. The ZOTUs were clustered by using usearch v11 (Edgar, 2010). The ZOTU table with amplicon counts of each ZOTU was created by mapping length-filtered sequences to representative ZOTU sequences.
The downstream analysis of taxonomical annotation and beta diversity based on Bray–Curtis dissimilar matrix (Bray and Curtis, 1957) were performed with Mothur v 1.41.1 (Schloss et al., 2009) according to the instruction of MiSeq SOP2. The reference of Silva.nr.132 (Quast et al., 2013) was used for bacteria and protozoa putative taxonomy annotation. The alpha diversity of ZOTUs was estimated by using ACE and Pielou as richness and evenness, respectively. The principle coordination analysis plots and genera relative abundance was visualized by ggplots23.
Statistical Analyses
All data was analyzed using the lm model by R v3.6.34 except the relative abundance of microbial genera. The analysis model of first experiment was expressed as follows:
where Yikmj is the response; μ is the general mean; Ai is the effect of animals (i = 3); Ck is the effect of chemical lysis (k = 2); Bm is the fixed effect of bead beater (m = 2); and eikmj is the random error term. When significant interactions occurred, a pair-wise comparison was conducted to determine differences among the four methodologies. Differences of P ≤ 0.05 were considered significant and 0.05 < P ≤ 0.1 were accepted as tendencies.
The analysis model of second experiment was expressed as follows:
where Yikmj is the response; μ is the general mean; Ai is the effect of animals (i = 3); Ck is the effect of mechanical lysis (k = 2); and eikj is the random error term.
As relative abundance of microbial taxa commonly does not fit a normal distribution, we used a permutational multivariate analysis of variance (PMAV) method (Fisher, 1925) for linear models described above. The PMAV was performed by lmperm v 2.1.05, and permutation time was determined by the method of Anscombe (1953) by setting perm = “Prob.” The Bray–Curtis dissimilarity matrix among sources of variation was parted according to the algorithm proposed by Anderson (2001) and was performed by vegan v2.5 (Dixon, 2003) with 9999 permutation. All P-values were adjusted according to the method of Benjamini and Hochberg (1995) by p.adjust() function of R v3.6.3 (see text footnote 4).
Results and Discussion
DNA Yield
It has been widely reported that RBB + C extracts more microbial DNA from rumen fluid samples than QIAamp Fast DNA Stool Mini Kit (Yu and Morrison, 2004; Henderson et al., 2013). Such result can be caused by BB in RBB + C methodology, as the original QIAamp Fast DNA Stool Mini Kit methodology does not contain BB. Our results indicated that CL did not exhibit different DNA yields (P = 0.14, Table 1), indicating a similar efficiency to harvest DNA by QIA and YM. However, both agarose-gel electrophoresis image (Figure 2) and OD260 measurement indicated that BB greatly enhanced (P = 0.001; Table 1) DNA yield. Previous studies also indicate that BB effectively increases DNA yield in fecal samples (de Boer et al., 2010; Karasartova et al., 2018). Furthermore, the lack of interaction between CL and BB was another important finding of this study (P ≥ 0.48, Table 1), which indicates that the improved DNA yield caused by BB is independently on CL.
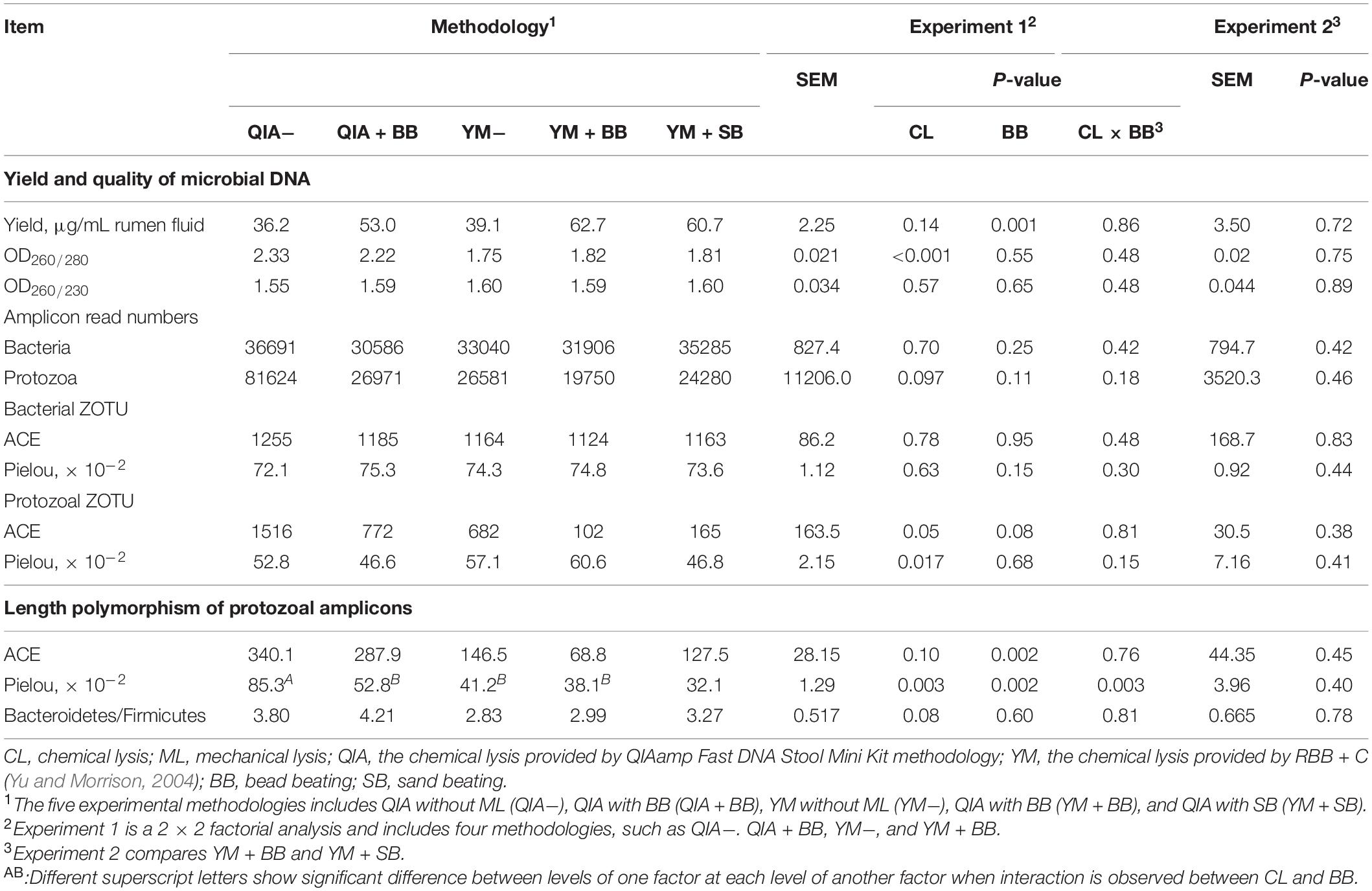
Table 1. Effect of chemical and mechanical lysis on DNA yield, quality, alpha diversity of bacterial and protozoal ZOTU, and length polymorphism of protozoal amplicons (n = 3).
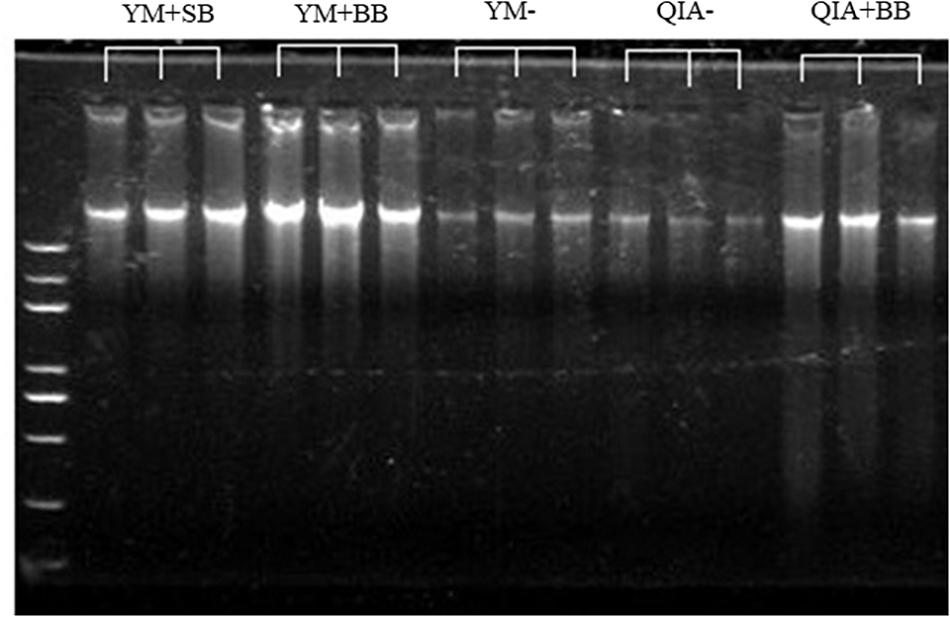
Figure 2. The agarose-gel electrophoresis graph of extracted DNA of five experimental methodologies. QIA, the chemical lysis provided by QIAamp Fast DNA Stool Mini Kit methodology; YM, the chemical lysis provided by RBB + C (Yu and Morrison, 2004); BB, bead beating; SB, sand beating; QIA–, QIA without mechanical lysis; QIA + BB, QIA with BB; YM–, YM without ML; YM + BB, QIA with BB; YM + SB, QIA with SB.
Bead size can be another factor that influences efficiency of DNA extraction. Rantakokko-Jalava and Jalava (2002) reported that the smaller bead beating has greater DNA extracted from mock bacterial samples, as smaller beads provide greater relative surface and hence interact with large numbers of bacteria. In our study, although the mesh size of sand (300–800 μm) was three times greater than that of bead (100–200 μm), the DNA yield was not statistically different between BB and SB (P = 0.72, Table 1). It seems that both BB and SB have similar efficiency to harvest DNA yield. We speculate that the irregular shape of sand may compensate its disadvantage in mesh size.
DNA Quality
The OD260/280 value of microbial DNA solution is an important indicator of DNA quality and can be increased by RNA remains or the hyperchromic effect of degraded microbial DNA (Felsenfeld and Sandeen, 1962; Desjardins and Conklin, 2010). Fiedorova et al. (2019) reported that RBB + C methodology produces a lower OD260/280 value than QIAamp Fast DNA Stool Mini Kit methodology (2.06 vs. 2.20). We suspect that such better DNA quality extracted by RBB + C methodology can be caused by YM, which has not been reported before. In our study, YM had lower OD260/280 closer to the ideal value of 1.8 than QIA (P < 0.001, Table 1). Such observation was further upheld by agarose gel electrophoresis image, especially that YM + BB methodology exhibited a lower shearing effect than QIA + BB methodology (Figure 2). It seems that chemical lysis of RBB + C methodology tends to obtain a better DNA quality of rumen samples. Although beater beating can increase the efficiency of DNA extraction, it may cause server shearing of DNA (Yu and Morrison, 2004), which can be revealed by a poor DNA integrity with decreased OD260/280 values (Desjardins and Conklin, 2010) and a diffused DNA band in agarose gel electrophoresis images (Yu and Morrison, 2004). However, our study showed that BB and SB had similar DNA integrity (Figure 2) and OD260/280 value (Table 1), indicating unchanged fragmenting DNA proportion by ML. Both QIAamp Fast DNA Stool Mini Kit and RBB + C methodologies have an identical final purification step, which is supposed to have similar ability to remove contaminants in the final DNA solution. In consistency with this theory, the OD260/230 value was not affected (P ≥ 0.48) by CL, BB, or SB in our study (Table 1). We speculate that YM helps to obtain a better quality of DNA extracted from rumen fluid samples, while proper use of ML may not affect the DNA quality.
DNA Amplicon Sequence Analysis of Bacteria
It has been reported that RBB + C and QIAamp Fast DNA Stool Mini Kit methodologies have similar overall bacteria community (Fiedorova et al., 2019). In this study, we collected the rumen fluid samples from three goats with ages being 2, 5, and 6 years old, which were supposed to have different microbial communities. The five employed DNA extraction methodologies generated a total of 1785 bacteria ZOTUs and shared a total of 1200 bacterial ZOTUs (Figure 3A). The PCoA cluster analysis clearly indicated that animals (P < 0.001), other than CL and BB, were clearly separated (Figure 4A), indicating that the bacterial community was different among three goats and can be compared by using four emploryed DNA extraction methodologies in the first experiment.
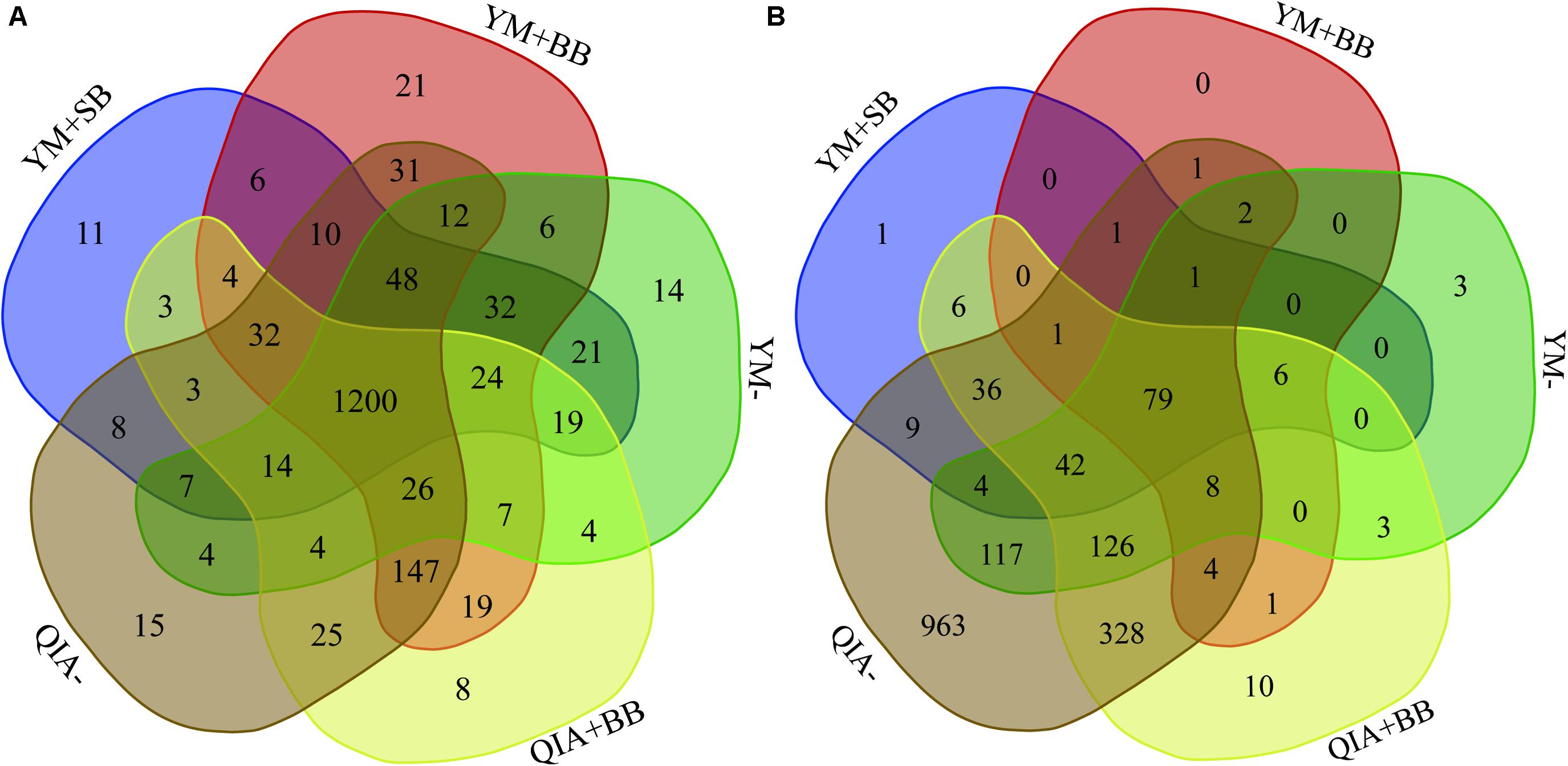
Figure 3. Venn diagram of bacterial (A) and protozoal (B) ZOTUs of five experimental methodologies. QIA, the chemical lysis provided by QIAamp Fast DNA Stool Mini Kit methodology; YM, the chemical lysis provided by RBB + C (Yu and Morrison, 2004); BB, bead beating; SB, sand beating; QIA–, QIA without mechanical lysis; QIA + BB, QIA with BB; YM–, YM without ML; YM + BB, QIA with BB; YM + SB, QIA with SB.
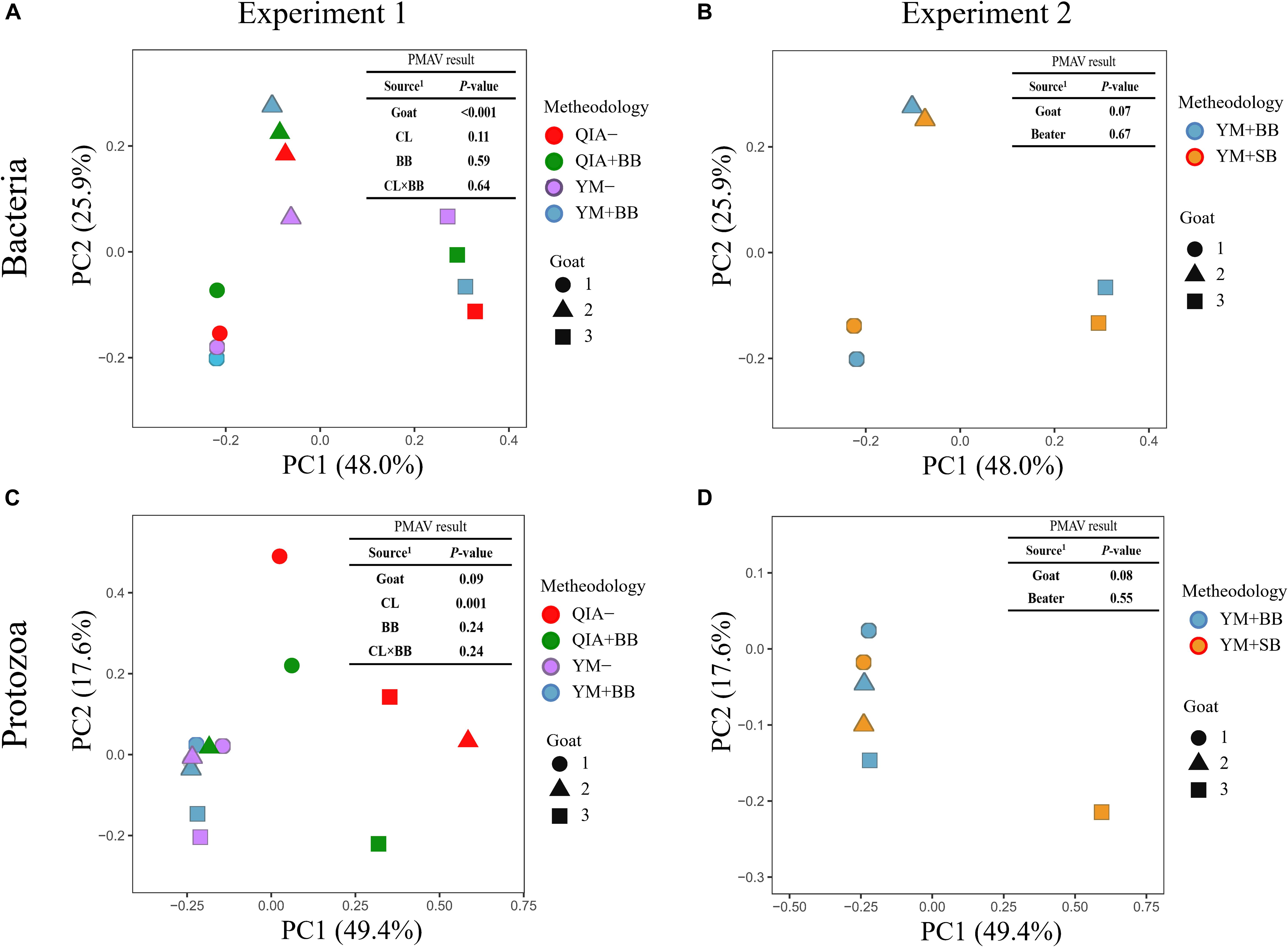
Figure 4. Principal coordinate analysis (PCoA) plots for the community of bacteria (A,B) and protozoa (C,D) based on the Bray–Curtis dissimilarity matrix. QIA, the chemical lysis provided by QIAamp Fast DNA Stool Mini Kit methodology; YM, the chemical lysis provided by RBB + C (Yu and Morrison, 2004); BB, bead beating; SB, sand beating; QIA–, QIA without mechanical lysis; QIA + BB, QIA with BB; YM–, YM without ML; YM + BB, QIA with BB; YM + SB, QIA with SB; PMAV, permutational multivariate analysis of variance with 999 permutations. Experiment 1 is a 2 × 2 factorial analysis and includes four methodologies, such as QIA–, QIA + BB, YM–, and YM + BB (A,C). Experiment 2 compares YM + BB and YM + SB (B,D).
Fibrolytic bacteria have the ability to infiltrate the fiber surface layer, therefore tightly attaching to it (McAllister et al., 1994) and making it more difficult to be lysed by microbial DNA extraction methodologies (Whitehouse et al., 1994). We speculate that sodium dodecyl sulfate (SDS), an ionic surfactant which can dissolve non-fiber contents in measuring NDF, may also help to release fibrolytic bacteria. In consistency with this speculation, YM tended to have greater (P ≤ 0.086) relative abundance of Ruminococcaceae lineage of Ruminococcus_1, Ruminococcaceae_NK4A214, Ruminococcaceae_UCG-005, and Ruminococcaceae_ge, in comparison with QIA (Figure 5 and Supplementary Table S1). Although CL did not affect (P = 0.11, Figure 4A) the overall bacterial community, YM increased (P ≤ 0.086, Figure 5 and Supplementary Table S1) some fibrolytic bacterial genera with relative abundance being ranged from 0.33 to 0.78%.
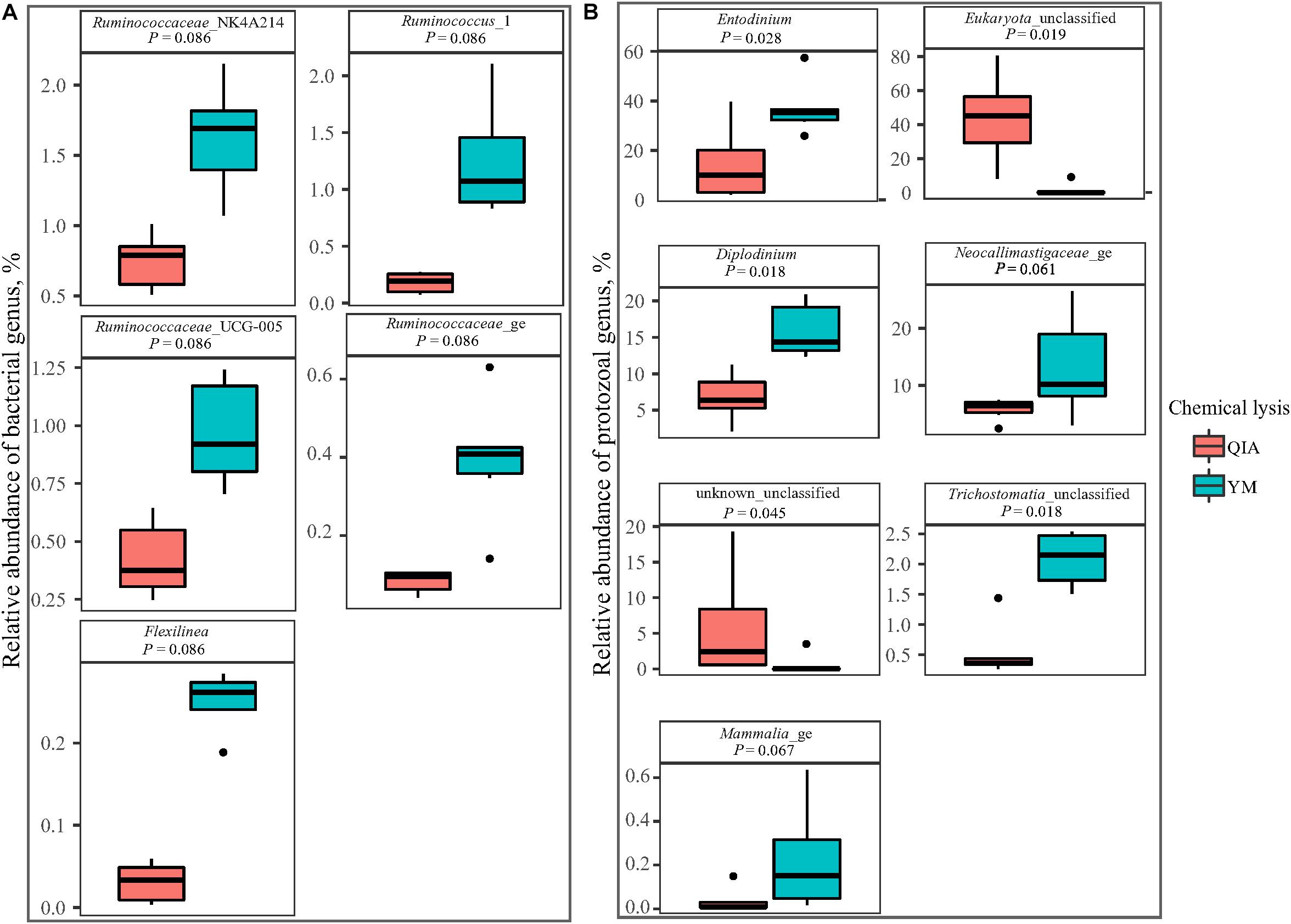
Figure 5. Boxplot of influenced (P ≤ 0.10) relative abundance of bacterial (A) and protozoal (B) genera by chemical lysis. QIA, the chemical lysis provided by QIAamp Fast DNA Stool Mini Kit methodology; YM, the chemical lysis provided by RBB + C (Yu and Morrison, 2004).
The cell wall of gram-positive bacteria is more difficult to be lysed than that of gram-negative bacteria (Pollock et al., 2018). Including mechanical lysis, such as beater beating, has been reported to enhance hard lysis cells such as Firmicutes in fecal samples (Salonen et al., 2010). However, in our study, BB did not alter the overall bacteria community (Figure 4A) and relative abundances of major bacterial genera (>0.1%, Supplementary Table S1), as well as the ratio of major gram-negative bacteria to major gram-positive bacteria (i.e., Bacteroidetes/Firmicutes) (P = 0.60, Table 1). Furthermore, both BB and SB also exhibited a similar overall bacteria community (Figure 4B). It seems that proper use of ML has similar efficiency in lysis of different bacterial groups, leading to an unchanged relative abundance of hard lysis bacteria in rumen fluid samples.
DNA Amplicon Sequence Analysis of Protozoa
The employed five DNA extraction methodologies generated a total of 1785 protozoal ZOTUs (Figure 3B), indicating that protozoa had almost similar ZOTUs with bacteria. Such result is contrary to the theory that diversity of protozoa is much lower than bacteria (Mcsweeney and Mackie, 2012). A total of 79 protozoal ZOTUs was shared by five DNA extraction methodologies and contributed to 89.4% of total reads of protozoa. Although the number of protozoal ZOTUs was unexpected large, protozoal ZOTUs representing the majority reads were reasonable.
Unlike the bacterial community, the PCoA cluster analysis indicated that both CL and animals separated the protozoal community (P ≤ 0.09, Figure 4A). The QIA increased the unclassified protozoa than YM (about 24-fold; P ≤ 0.045, Figure 5 and Supplementary Table S2). As only 29 ciliates and seven Flagellate genomes are known, higher unclassified protozoal genera generated by QIA may be caused by lacking protozoal gene contents in the Silva.nr.132 (Quast et al., 2013) database. Length polymorphisms of marker genes of eukaryotes such as protozoa are diverse (Hugerth et al., 2014). The QIA tended to generate more protozoal amplicons and had higher richness in amplicon length polymorphism than YM (P ≤ 0.1, Table 1). We speculate that the QIA can harvest DNA from greater diverse protozoa cells than YM.
Mechanical lysis is not recommended in extracting high-molecular-weight DNA (Rosewarne et al., 2011). The genome of protozoa is large, which can be vulnerated to mechanical lysis such as beater beating. For example, the genome of Entodinium caudatum, the most abundant protozoa in rumen, is 107 Mb6, which is about 30 times as rumen bacteria. In our study, neither the BB nor the SB influenced the overall protozoal community (P ≥ 0.24, Figures 4C,D) or annotation results (P ≥ 0.17, Supplementary Table S2). However, BB decreased (P ≤ 0.08) the richness of protozoal ZOTUs and length polymorphism of protozoal amplicons (Table 1). As limited protozoal genomes are included in the reference, only 6.9% protozoal ZOTUs were annotated at the genus level. The diversity of protozoal ZOTUs and length polymorphism can better indicate protozoal diversity than annotation results. Although ML did not influence overall protozoal community, it greatly decreased the richness of protozoa.
In summary, the QIA harvests DNA from higher diverse protozoa cells in comparison with YM. Although the YM has a similar bacterial community as QIA, it can help to harvest more DNA from fibrolytic bacteria such as Ruminococcaceae lineages. Proper use of ML can increase total DNA yield without affecting DNA quality and bacterial community but decreases protozoal amplicon diversity. Sand can be an alternative beater for DNA extraction, as both BB and SB have similar total DNA yield and quality and bacterial and protozoal community. Our study highlights that DNA extraction methodology should be slightly different for analyzing the bacterial and protozoal community.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below https://www.ncbi.nlm.nih.gov/, PRJNA644440.
Ethics Statement
The animal study was reviewed and approved by the Animal Care Committee of Institute of Subtropical Agriculture, CAS (Approval number ISA-W-201609).
Author Contributions
ZM, XZ, and MW conceived this study. ZM, XZ, and RW conducted this experiment. ZM and XZ carried out the statistical analysis. ZM and MW wrote the manuscript. TL and ZT revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors thank the National Natural Science Foundation of China (Grant Nos. 31561143009 and 31922080), Major Project for Science and Technology of Hunan Province (Grant No. 2017NK1020), and Research Foundation for Public Enrolled Doctor of Gansu Agricultural University (Grant No. GSAU-RCZX201710) for the joint financial support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.581227/full#supplementary-material
Footnotes
- ^ https://github.com/torognes/vsearch/wiki/VSEARCH-pipeline
- ^ https://mothur.org/wiki/MiSeq_SOP
- ^ https://github.com/tidyverse/ggplot2
- ^ https://www.r-project.org/
- ^ https://github.com/mtorchiano/lmPerm
- ^ https://www.ncbi.nlm.nih.gov/genome/?term=Entodinium
References
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austr. Ecol. 26, 32–46. doi: 10.1111/j.2517-6161.1953.tb00121.x
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
de Boer, R., Peters, R., Gierveld, S., Schuurman, T., Kooistra-Smid, M., and Savelkoul, P. (2010). Improved detection of microbial DNA after bead-beating before DNA isolation. J. Microbiol. Methods 80, 209–211. doi: 10.1016/j.mimet.2009.11.009
Desjardins, P., and Conklin, D. (2010). NanoDrop microvolume quantitation of nucleic acids. J. Vis. Exp. 2010:2565. doi: 10.3791/2565
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Felsenfeld, G., and Sandeen, G. (1962). The dispersion of the hyperchromic effect in thermally induced transitions of nucleic acids. J. Mol. Biol. 5, 587–610. doi: 10.1016/s0022-2836(62)80088-6
Fiedorova, K., Radvansky, M., Nemcova, E., Grombirikova, H., Bosak, J., Cernochova, M., et al. (2019). The impact of DNA extraction methods on stool bacterial and fungal microbiota community recovery. Front. Microbiol. 10:821. doi: 10.3389/fmicb.2019.00821
Guo, F., and Zhang, T. (2013). Biases during DNA extraction of activated sludge samples revealed by high throughput sequencing. Appl. Microbiol. Biotechnol. 97, 4607–4616. doi: 10.1007/s00253-012-4244-4
Henderson, G., Cox, F., Kittelmann, S., Miri, V. H., Zethof, M., Noel, S. J., et al. (2013). Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities. PLoS One 8:e74787. doi: 10.1371/journal.pone.0074787
Hugerth, L. W., Muller, E. E., Hu, Y. O., Lebrun, L. A., Roume, H., Lundin, D., et al. (2014). Systematic design of 18S rRNA gene primers for determining eukaryotic diversity in microbial consortia. PLoS One 9:e95567. doi: 10.1371/journal.pone.0095567
Karasartova, D., Gureser, A. S., Ruh, E., Turegun-Atasoy, B., Calgin, M. K., Tasci, L., et al. (2018). An alternative DNA extraction method for detection of Blastocystis spp. in human fecal samples. Exp. Parasitol. 186, 36–41. doi: 10.1016/j.exppara.2018.01.019
Ma, Z., Wang, R., Wang, M., Zhang, X., Mao, H., and Tan, Z. (2018). Short communication: variability in fermentation end-products and methanogen communities in different rumen sites of dairy cows. J. Dairy Sci. 101, 5153–5158. doi: 10.3168/jds.2017-14096
McAllister, T. A., Bae, H. D., Jones, G. A., and Cheng, K. J. (1994). Microbial attachment and feed digestion in the rumen. J. Anim. Sci. 72, 3004–3018. doi: 10.2527/1994.72113004x
Mcsweeney, C., and Mackie, R. (2012). Commission on Genetic Resources for Food and Agriculture. Micro-organisms and Ruminant Digestion: State of Knowledge, Trends and Future Prospects. Rome: FAO.
Pollock, J., Glendinning, L., Wisedchanwet, T., and Watson, M. (2018). The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 84:e02627-17. doi: 10.1128/AEM.02627-17
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rantakokko-Jalava, K., and Jalava, J. (2002). Optimal DNA isolation method for detection of bacteria in clinical specimens by broad-range PCR. J. Clin. Microbiol. 40, 4211–4217. doi: 10.1128/jcm.40.11.4211-4217.2002
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahe, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Rosewarne, C. P., Pope, P. B., Denman, S. E., McSweeney, C. S., O’Cuiv, P., and Morrison, M. (2011). High-yield and phylogenetically robust methods of DNA recovery for analysis of microbial biofilms adherent to plant biomass in the herbivore gut. Microb. Ecol. 61, 448–454. doi: 10.1007/s00248-010-9745-z
Salonen, A., Nikkila, J., Jalanka-Tuovinen, J., Immonen, O., Rajilic-Stojanovic, M., Kekkonen, R. A., et al. (2010). Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 81, 127–134. doi: 10.1016/j.mimet.2010.02.007
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Vaidya, J. D., van den Bogert, B., Edwards, J. E., Boekhorst, J., van Gastelen, S., and Saccenti, E. (2018). The effect of DNA extraction methods on observed microbial communities from fibrous and liquid rumen fractions of dairy cows. Front. Microbiol. 9:92. doi: 10.3389/fmicb.2018.00092
Walters, W., Hyde, E. R., Berg-Lyons, D., Ackermann, G., and Humphrey, G. (2015). Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1, e9–e15. doi: 10.1128/mSystems.00009-15
Whitehouse, N. L., Olson, V. M., Schwab, C. G., Chesbro, W. R., Cunningham, K. D., and Lykos, T. (1994). Improved techniques for dissociating particle-associated mixed ruminal microorganisms from ruminal digesta solids. J. Anim. Sci. 72, 1335–1343. doi: 10.2527/1994.7251335x
Keywords: chemical lysis, mechanical lysis, DNA extraction, rumen fluid, bacteria, protozoa
Citation: Ma ZY, Zhang XM, Wang R, Wang M, Liu T and Tan ZL (2020) Effects of Chemical and Mechanical Lysis on Microbial DNA Yield, Integrity, and Downstream Amplicon Sequencing of Rumen Bacteria and Protozoa. Front. Microbiol. 11:581227. doi: 10.3389/fmicb.2020.581227
Received: 09 July 2020; Accepted: 19 October 2020;
Published: 16 November 2020.
Edited by:
Anil Kumar Puniya, National Dairy Research Institute (ICAR), IndiaReviewed by:
Ravinder Nagpal, Wake Forest School of Medicine, United StatesAbhishek Baghela, Agharkar Research Institute, India
Sanjay Kumar, University of Georgia, United States
Gunjan Goel, Central University of Haryana, India
Copyright © 2020 Ma, Zhang, Wang, Wang, Liu and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Wang, bXdhbmdAaXNhLmFjLmNu