 Xiaohan Yang1†
Xiaohan Yang1† Jingquan Dong
Jingquan Dong Panpan Zhao
Panpan Zhao- 1Jiangsu Key Laboratory of Marine Biological Resources and Environment, Jiangsu Key Laboratory of Marine Pharmaceutical Compound Screening, Co-Innovation Center of Jiangsu Marine Bio-industry Technology, School of Pharmacy, Jiangsu Ocean University, Lianyungang, China
- 2Jiangsu Institute of Oceanology and Marine Fisheries, Nantong, China
- 3Key Laboratory of Zoonosis Research by Ministry of Education, College of Veterinary Medicine, Jilin University, Changchun, China
As an important foodborne pathogen, Vibrio vulnificus gives a significant threat to food safety and public health. Rapid and accurate detection methods for V. vulnificus are required to control its spread. The conventional detection methods are time-consuming and labor-intensive, while the polymerase chain reaction (PCR)- and quantitative PCR (qPCR)-based methods are limited because of their dependence on laboratory equipment. Nucleic acid isothermal amplification technologies have been applied to develop simpler assays. In this study, a rapid detection method based on real-time recombinase polymerase amplification (RPA) targeting the extracellular metalloprotease (empV) gene of V. vulnificus has been established. The method finished the detection in 2–14 min at 39°C with good specificity. The limit of detection was 17 gene copies or 1 colony-forming unit (CFU) per reaction, or 1 CFU/10 g of spiked food with enrichment. In a clinical sample detection test, the results of real-time RPA were 100% consistent with bioassay and qPCR. Moreover, the method could resist the effect of food matrix and could tolerate crude templates. The real-time RPA method established in this study is rapid and simple and has the potential to be widely applied for V. vulnificus detection in food safety control.
Introduction
Foodborne infectious diseases are of significant public health concern. Researchers have focused on the development of rapid and reliable methods of pathogen detection for food safety. Vibrio vulnificus is a Gram-negative, halophilic bacterium found in coastal or estuarine environment worldwide and has been isolated from sediments, water, and a variety of seafood (Dalsgaard et al., 1996; Jones and Oliver, 2009). V. vulnificus is regarded as one of the major causes of seafood-associated diseases. Consumption of raw or undercooked seafood contaminated with V. vulnificus can result in severe infection that causes life-threatening septicemia and acute gastroenteritis (Jones and Oliver, 2009). Wound infections can also occur when one is exposed to contaminated sea water or seafood (Hlady and Klontz, 1996; Strom and Paranjpye, 2000). The mortality rate of V. vulnificus infection is up to 60%, making it a serious public health and food safety concern (Jones and Oliver, 2009). With the global warming, this bacterium inhabits a warm environment and causes outbreaks in more areas (Rippey, 1994; Huehn et al., 2014; Bonnin-Jusserand et al., 2019; Leng et al., 2019). Thus, rapid, specific, and reliable detection methods for V. vulnificus are particularly required to facilitate better control of its spread.
The conventional methods for the detection of V. vulnificus usually include pre-enrichment, isolation, and biochemical identifications, which are time-consuming and labor-intensive (Hartnell et al., 2019). Moreover, the culture-based methods are not sensitive to the viable but non-culturable (VBNC) status of the bacterium, and other bacteria with similar biochemical characteristics interfere (O’Hara et al., 2003; Li et al., 2014). Polymerase chain reaction (PCR)- and quantitative PCR (qPCR)-based methods have been developed for detecting V. vulnificus and have showed high sensitivity and specificity (Panicker and Bej, 2005; Kumar et al., 2006; D’Souza et al., 2019). However, these methods require sophisticated thermal-cycling equipment and other laboratory settings, and the usage is limited. Loop-mediated isothermal amplification (LAMP), a nucleic acid isothermal amplification technology, has been applied to develop simpler assays for the detection of V. vulnificus (Han and Ge, 2010; Surasilp et al., 2011).
Recombinase polymerase amplification (RPA), another isothermal amplification technology, has been widely applied for foodborne pathogen detections in recent years for its rapidness, simplicity, and convenience (Piepenburg et al., 2006; Du et al., 2018; Dong et al., 2020). The amplification products of RPA can be analyzed by gel electrophoresis, lateral flow chromatography, or fluorescence (Li et al., 2018). Among them, the fluorescence analysis makes real-time reading of the signal possible, and this “real-time RPA” assay has a good combination of speed, portability, and accessibility (Piepenburg et al., 2006). Briefly, the real-time RPA reaction contains an “exo probe” that anneals to one of the amplified strands and recruits an exonuclease to cleave off the tetrahydrofuran (THF) substitution on the probe to expose the 3’ end for strand extension. The cleavage also separates the fluorophore and the quenching group at the two sides of the THF on the probe, emitting a fluorescence signal (Piepenburg et al., 2006).
In this study, a real-time RPA method for rapid detection of V. vulnificus has been established. This method finishes the detection in 14 min with good specificity. The limit of detection was 17 gene copies or 1 colony-forming unit (CFU) per reaction, or 1 CFU/10 g of spiked food with enrichment. Detection results were 100% consistent with bioassay and qPCR for clinical food samples. Moreover, the method could resist the effect of food matrix and could tolerate crude templates, making it a well portable method to be widely applied for V. vulnificus detection in food safety control.
Materials and Methods
Bacteria Strains
A collection eight Vibrio vulnificus environmental strains and reference strains of Vibrio cholerae, Vibrio harveyi, Vibrio mediterranei, and Vibrio shilonii were obtained from Jiangsu Institute of Oceanology and Marine Fisheries (Nantong, China). Reference strains of V. vulnificus, Vibrio parahaemolyticus, Vibrio alginolyticus, Salmonella Typhimurium, Listeria monocytogenes, Bacillus cereus, and Staphylococcus aureus were kindly provided by the Wuhan Institute for Food and Cosmetic Control (Wuhan, China). Reference strains of Vibrio splendidus, Vibrio ichthyoenteri, Vibrio mimicus, Vibrio campbellii, Vibrio chagasii, Vibrio fluvialis, Vibrio natriegens, Vibrio sp., Vibrio ponticus, Vibrio rotiferianus, and Vibrio diabolicus were purchased from the Ministry of Natural Resources Third Institute of Oceanography (Xiamen, China). All the bacterial strains were confirmed by 16S rRNA sequencing (Hiergeist et al., 2016). The details of the strains are listed in Table 1.
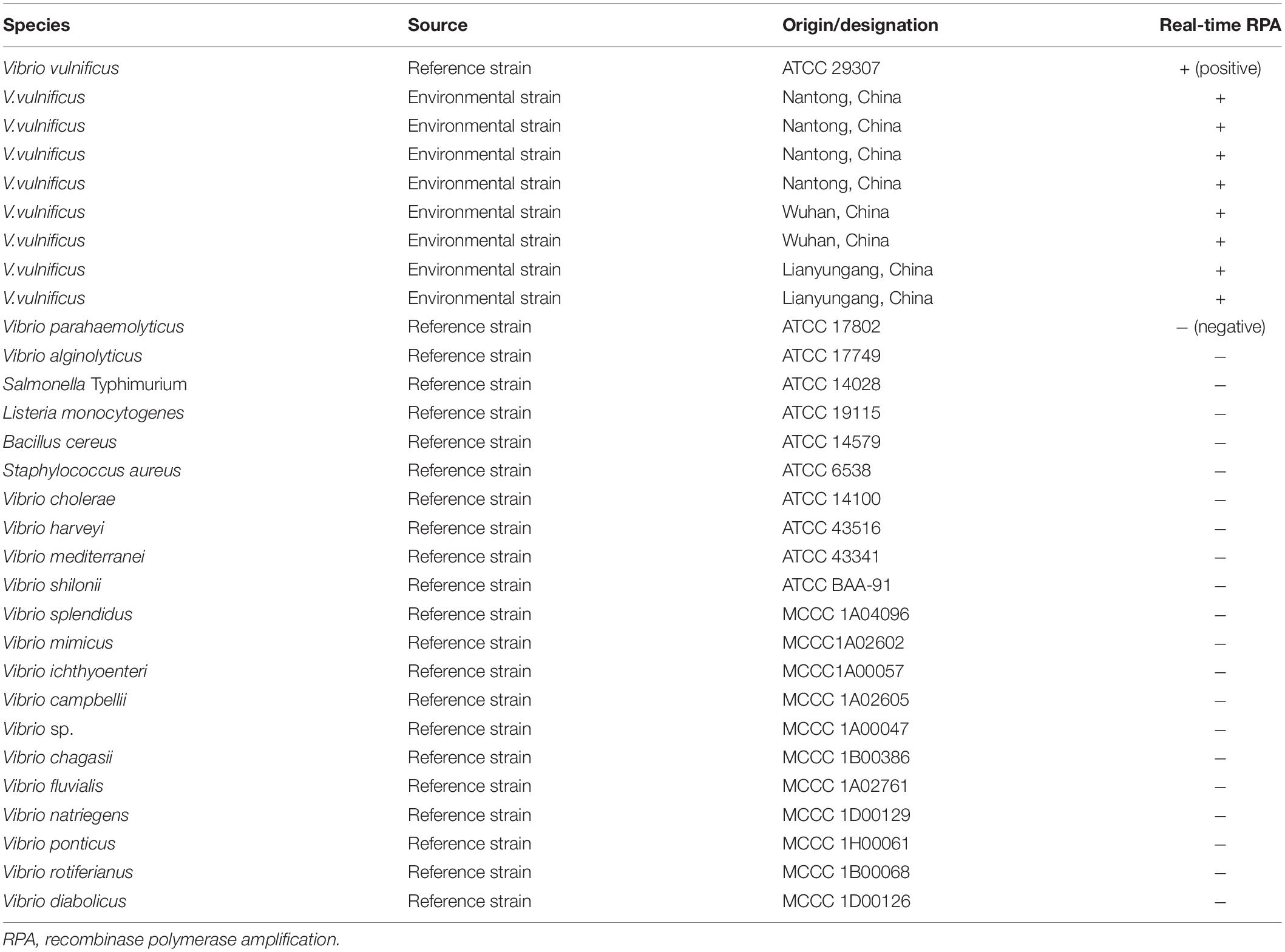
Table 1. Information of bacteria strains used in this study.
Sequence Analysis
Multiple sequence alignments were performed on empV and gyrB gene sequences of all V. vulnificus and other selected Vibrio species available in GenBank using Clustal1 with the default parameters; then the diverged sequences flanking the conservative core were ignored. A phylogenetic tree was constructed based on the alignments using MEGA42 with the neighbor-joining (NJ) method.
Primer/Probe Design
For design of primers, the FASTA sequence of empV gene (GenBank No. U50548.1) was input into the National Center for Biotechnology Information (NCBI) Primer-BLAST3. The primer BLAST was performed with the following criteria put into consideration: (1) the primer pair should only target the species of interest (V. vulnificus); (2) the primer pair should have less than four consecutive bases (and less than one if located at the 3’ end) pairing each other. The product size was set as minimum at 150 and maximum at 500. The database was set as Refseq representative genomes. The organism was set as V. vulnificus (taxid: 672). The primer size was set as minimum at 28 and maximum at 35. The primer guanine–cytosine (GC) content was set as minimum at 20 and maximum at 80. The maximal self-complementarity was set as any at 4 and 3’ at 1. The maximal pair complementarity was set as any at 4 and 3’ at 1. Other parameters were set as default. For the probe design, the sequence of region defined by the primer pair was input into the Primer Premier 5 software. The size of the probe was set as minimum at 46 and maximum at 52. The melting temperature (Tm) was set as minimum at 50 and maximum at 100. The GC content was set as minimum at 20 and maximum at 70. The maximum hairpin score was set as 9. The maximum primer-dimer score was set as 9. The maximum poly-X was set as 5. Other parameters were set as default. The probe had a C3 spacer (SpC3) at the 3’ end, which could block strand extension, and a THF group at the middle (position 32) to facilitate exonuclease III (exo) cutting. Only when the bases flanking the THF site of the probe had a good pairing with the template would the exo cutting occur to release the 3’ end from SpC3 blocking. Moreover, two T bases on the both sites of THF site were substituted by FAM (6-corboxy-fluorescein)-dT and BHQ1 (Black Hole Quencher 1)-dT. Primers and probe were synthesized by General Biosystems Co., Ltd., Anhui, China.
Real-Time Recombinase Polymerase Amplification Procedure
If bacterial cultures were used as the templates, they were diluted to the desired concentrations if necessary, treated at 100°C for 10 min and immediately used. For reactions using purified genomic DNA as the templates, the genomic DNA was extracted using TIANamp Genomic DNA Kit (Tiangen Biotech Co., Ltd., Beijing, China) and quantified using Qubit 4 (Thermo Fisher Scientific Inc., Wilmington, DE, United States). The copy number of targeted DNA fragment was calculated based on the genome size (4.97039 Mb). Real-time RPA reactions were set up according to the manufacturer’s instructions of TwistAmp DNA Amplification exo Kit (TwistDx Inc., Maidenhead, United Kingdom). The reaction contained 29.5 μl of rehydration buffer, 2.1 μl of each primer (10 μM), 0.6 μl of probe (10 μM), 12.2 μl of distilled water, 1 μl of the template, and a dried enzyme pellet. To initiate the reaction, 2.5 μl of magnesium acetate (280 mM) was added to the mixture. After a brief centrifugation, the reaction mixture was pre-incubated at 39°C for 4 min. Subsequently, the fluorescence signal was recorded in real-time on a Roche LightCycler 480 II qPCR machine at 39°C in the FAM channel, with signal reading at 13-s intervals for 25 min.
Preparation of Spiked Food Samples
Shrimp, oyster, fish flesh (pomfret), and crab were purchased from a local market and verified to be free of V. vulnificus by quantitative PCR (Kumar et al., 2006). Food samples were homogenized thoroughly using a handheld grinder (3rd Gen. TGrinder, Tiangen Biotech Co., Ltd.). Ten grams of the food homogenate was made to 100 ml with alkaline peptone broth (Sinopharm Chemical Reagent Co., Ltd., Beijing, China) and spiked with desired amounts of V. vulnificus. If no enrichment was needed, 1 ml of the solution was boiled at 100°C for 10 min and centrifuged at 5,000 g for 5 min. One microliter of the supernatant was used as the template for real-time RPA detection. If enrichment was needed, the spiked food samples were incubated at 30°C with 200-rpm shaking. A total of 1 ml of the enrichment solution was collected at different time points and centrifuged at 800 g for 10 min to remove food debris. Bacterial cells were pelleted with 5,000 g centrifugation for 5 min, resuspended with 200 μl of water, and boiled at 100°C for 10 min. Also, 1 μl of the boiled resuspension was used as the template for real-time RPA detection.
Clinical Samples
The clinical seafood samples (shrimp, fish, shellfish, and crab) were kindly provided by the Jiangsu Institute of Oceanology and Marine Fisheries (Nantong, China). After a disinfection treatment with ethanol, 9 ml of phosphate-buffered saline (PBS) was added to 1 g of each sample and homogenized thoroughly using the handheld grinder. Then 1 ml of the sample homogenate was boiled at 100°C for 10 min and centrifuged at 800 g for 10 min to remove food debris. A total of 1 μl of the supernatant was used as template for real-time RPA or quantitative PCR detection. For the bioassay, the procedure followed the National Standard of China DBS13/004-2016. Briefly, 10 g of the clinical seafood samples was added into 90 ml of alkaline saline peptone water and incubated at 37°C for 18–24 h. The cultures were line separated on mCPC medium plates at 37°C overnight. Suspected single colonies were inoculated onto Luria broth (LB) agar plates with 3% NaCl and incubated at 37°C for 18–24 h. Then oxidase test, gram stain, halophilic test, and ONPG test were performed.
Quantitative PCR
The qPCR detection of V. vulnificus followed an established method reported previously (Kumar et al., 2006). Specifically, the primer pair targeting the gyrB gene was used (Table 2). The qPCR mixture contained 25 μl of the 2 × SYBR Green qPCR Mix (Tiangen Biotech Co., Ltd.), 0.4 μl of each primer (10 μM), and 1 μl of the template. The cycling program was 95°C 10 min followed by 45 cycles of 95°C for 20 s, 55°C for 20 s, and 72°C for 30 s on a Roche LightCycler 480 II qPCR machine. The melting curve analysis was set as default. Cycle threshold (Ct) values less than 32 were considered as positive.

Table 2. Primer and probe sequences.
Statistical Analysis
For the limit of detection data of the real-time RPA assay, a probit regression analysis was performed using SPSS software (IBM, Armonk, NY, United States), and a semi-log regression analysis was carried out using GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA). For the clinical sample detection data, the correlation between threshold time (Tt) value of real-time RPA and cycle time (Ct) value of qPCR was analyzed using GraphPad Prism 8.0.
Results
Selection of the Target Gene and Primer/Probe Design
The conservative gene empV encoding the extracellular metalloproteinase of Vibrio vulnificus was selected as the detection target of the real-time RPA method (Chuang et al., 1997). The evolutionary conservation of the empV gene was confirmed by construction of an unrooted NJ phylogenetic tree based on multiple sequence alignments of empV gene sequences of 23 V. vulnificus strains and 10 other selected Vibrio species. All the 23 empV gene sequences of V. vulnificus strains were classified into one group that was a distant relative of the other Vibrio species (Figure 1). This result indicated that the empV gene had good specificity and evolutionary conservation. The NCBI Primer-BLAST search for primer candidates on the sequence of gene mepV returned with five potential primer pairs. These primers were tentatively screened by amplification of the target gene fragment with the no-template control. The amplification products were electrophorized on agarose gel to compare amplification performance of the target and primer-dimer formation in the no-template control. The primer pair showing the best amplification performance without a sign of primer-dimer formation was selected, and a probe was designed for this primer pair. The sequences of this primer-probe set are listed in Table 2.
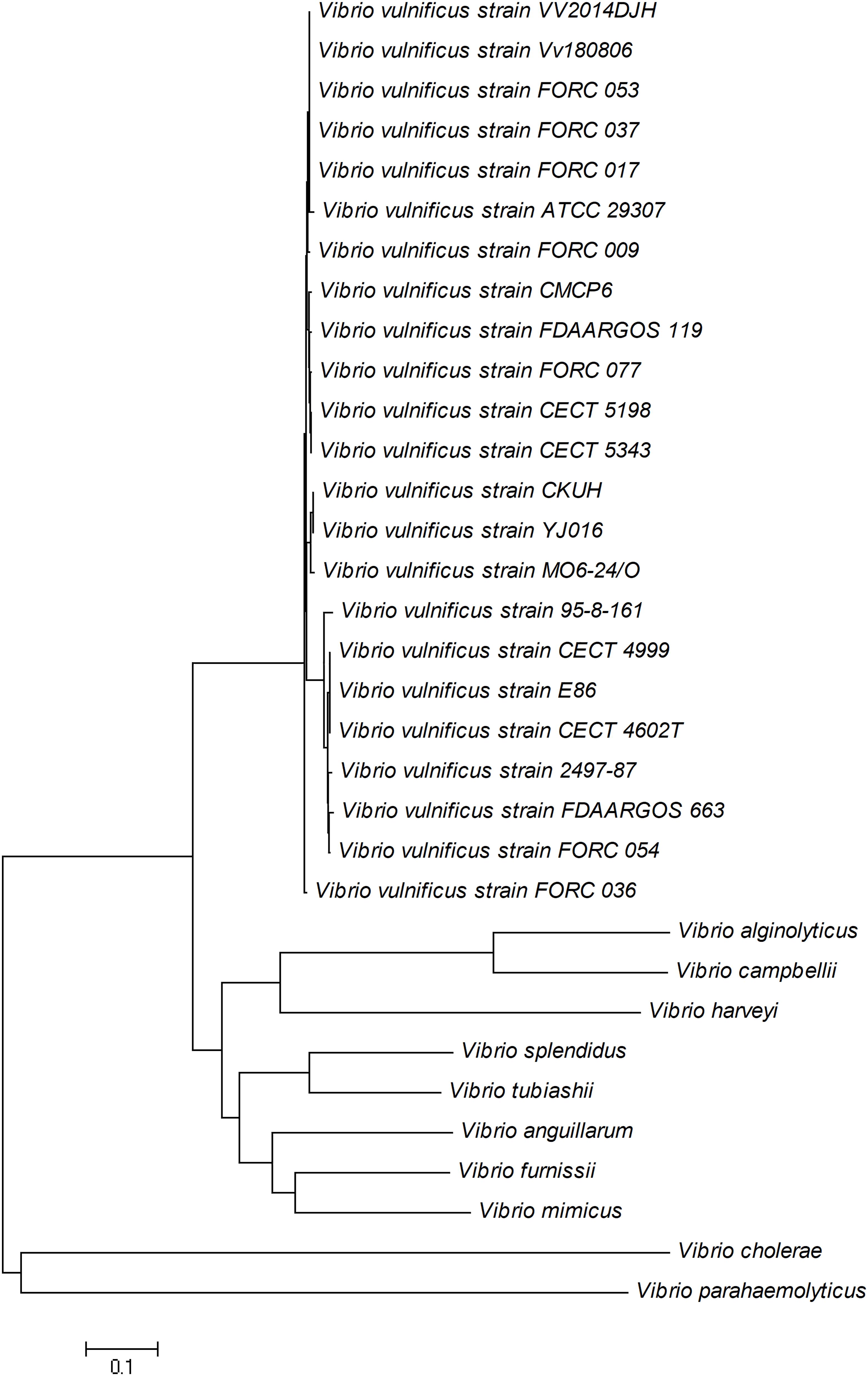
Figure 1. Neighbor-joining tree of empV gene of 23 Vibrio vulnificus strains and 10 other Vibrio species. GenBank accession numbers: V. vulnificus CMCP6 (CP037932.1), V. vulnificus 95-8-161 (AB540652.1), V. vulnificus 2497-87 (CP060048.1), V. vulnificus CECT 4602T (AM492792.1), V. vulnificus CECT 4999 (CP014637.1), V. vulnificus CECT 5198 (AB540651.1), V. vulnificus CECT 5343 (AB540651.1), V. vulnificus CKUH (AB540650.1), V. vulnificus E86 (DQ923325.1), V. vulnificus Env1 (CP017636.1), V. vulnificus FDAARGOS_119 (CP014048.2), V. vulnificus FDAARGOS_663 (CP044068.1), V. vulnificus FORC_009 (CP009985.1), V. vulnificus FORC_017 (CP012740.1), V. vulnificus FORC_036 (CP015513.1), V. vulnificus FORC_037 (CP016322.1), V. vulnificus FORC_053 (CP019291.1), V. vulnificus FORC_054 (CP019122.1), V. vulnificus FORC_077 (CP027031.1), V. vulnificus MO6-24/O (CP002470.1), V. vulnificus VV2014DJH (CP019321.1), V. vulnificus Vv180806 (CP044207.1), V. vulnificus YJ016 (BA000038.2), Vibrio alginolyticus (NZ_CP042449.1), Vibrio anguillarum (NZ_AEZA01000040.1), Vibrio campbellii (NZ_CP019634.1), Vibrio cholerae (NZ_AP014524.1), Vibrio furnissii (NZ_CP040990.1), Vibrio harveyi (NZ_CP009467.1), Vibrio mimicus (NZ_ADAF01000001.1), Vibrio splendidus (NZ_CP031055.1), Vibrio tubiashii (CP009354.1), and Vibrio parahaemolyticus (CP012950.1).
Specificity of the Method
To evaluate the specificity of the real-time RPA method, culture solutions of a number of bacterial strains, including environment isolates of V. vulnificus, other Vibrio species, and a number of other foodborne pathogenic bacteria, at a cell density of 106 CFU/ml, were tested (Table 1). The other Vibrio species and other foodborne pathogenic bacteria were all negative, and the V. vulnificus reference strain and environmental isolates were all detected. This result confirmed the specificity of the real-time RPA method.
Limit of Detection of the Method
The limit of detection of the real-time RPA method was evaluated under various conditions. Firstly, the purified genomic DNA of V. vulnificus with the concentration of 100–106 copies per microliter was tested (1 μl for each reaction) (Figure 2A). The real-time RPA assay was performed for eight independent repeats; 102 copies and above were detected in 8/8 runs, 101 copies were detected in 7/8 runs, and 100 copies were detected in 2/8 runs. A probit regression analysis was carried out for the data of the 8 repeats, and the result showed that the limit of detection was 17 copies/reaction in 95% of cases (Figure 2B). Semi-log regression analysis of the data of the 8 repeats showed that the reaction time lengths of the real-time RPA assay were 2–14 min for 106–101 copies (Figure 2C). Secondly, a 10-fold series dilution of V. vulnificus culture ranging from 106 to 100 CFU/ml (103–10–3 CFU per reaction) was tested. Signal of 100 CFU could be observed (Figure 3A). This limit of detection was not affected by the food matrix (shrimp homogenate) (Figure 3B). Thirdly, the limit of detection was tested in spiked food samples with different enrichment time. Ten grams of shrimp, oyster, fish flesh, and crab homogenate was spiked with 100–103 CFU of V. vulnificus and enriched for 0–24 h. The results showed that 1 CFU/10 g could be detected after 4 h of enrichment for all the four food matrix types (Table 3). Thus, the limit of detection of the real-time RPA method was 17 copies or 1 CFU per reaction, or 1 CFU/10 g in spiked food samples with 4 h of enrichment.
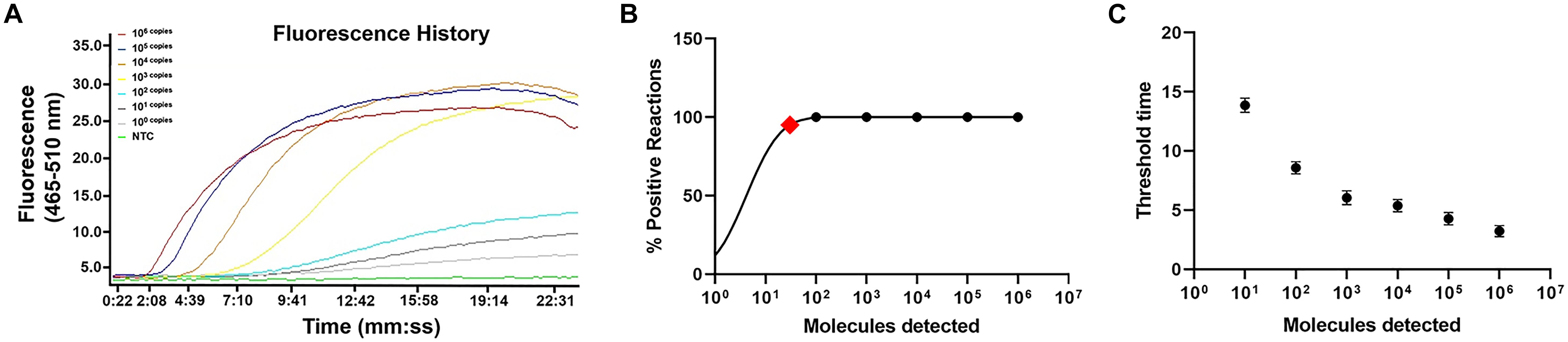
Figure 2. Limit of detection of real-time recombinase polymerase amplification (RPA) method. (A) The fluorescence history diagram of the results of real-time RPA with different amounts (in copies) of Vibrio vulnificus. The amounts tested are indicated with different colors. NTC, no-template control. The diagram was one typical outcome of eight independent experiments. (B) Probit regression analysis of the data collected from the eight real-time RPA repeats using SPSS software. The limit of detection at 95% probability (17 copies/reaction) is depicted by a red rhomboid. (C) Semi-logarithmic regression of the data collected from the eight real-time RPA repeats using GraphPad Prism 8.0. The run time of the real-time RPA was ∼2–14 min for the templates at 106–100 copies/reaction.
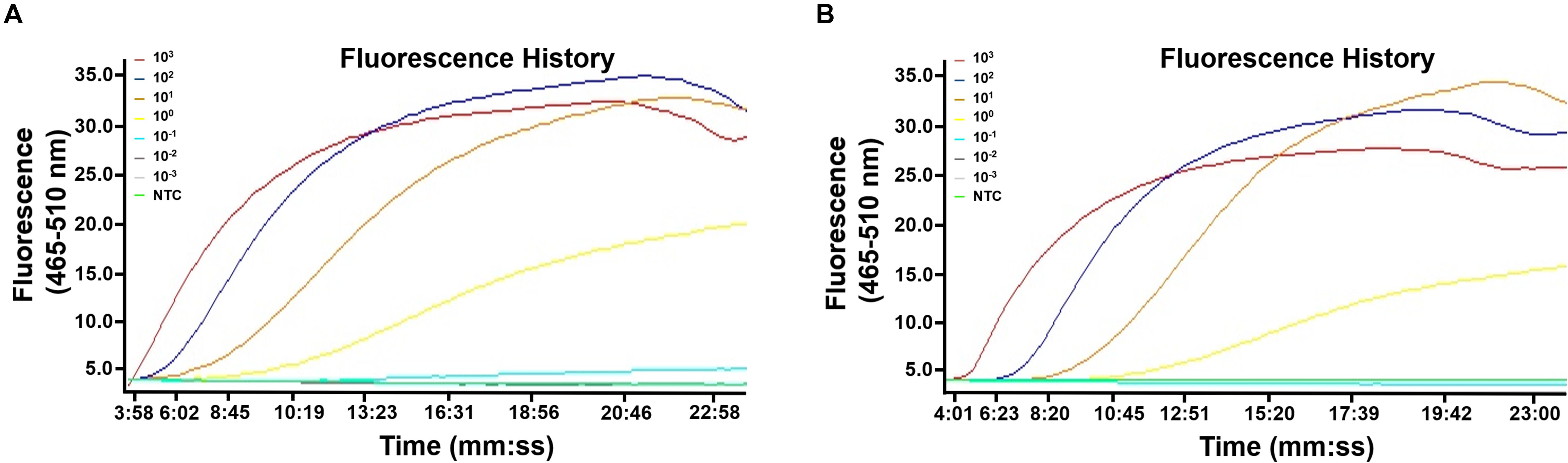
Figure 3. Limit of detection of the real-time recombinase polymerase amplification (RPA) method. The fluorescence history diagrams show the results of real-time RPA with different amounts (in CFU) of Vibrio vulnificus. The amounts tested are indicated with different colors. Templates from culture solution (A) or spiked shrimp samples (B) were tested. NTC, no-template control. The diagrams are one typical outcome of three independent experiments.
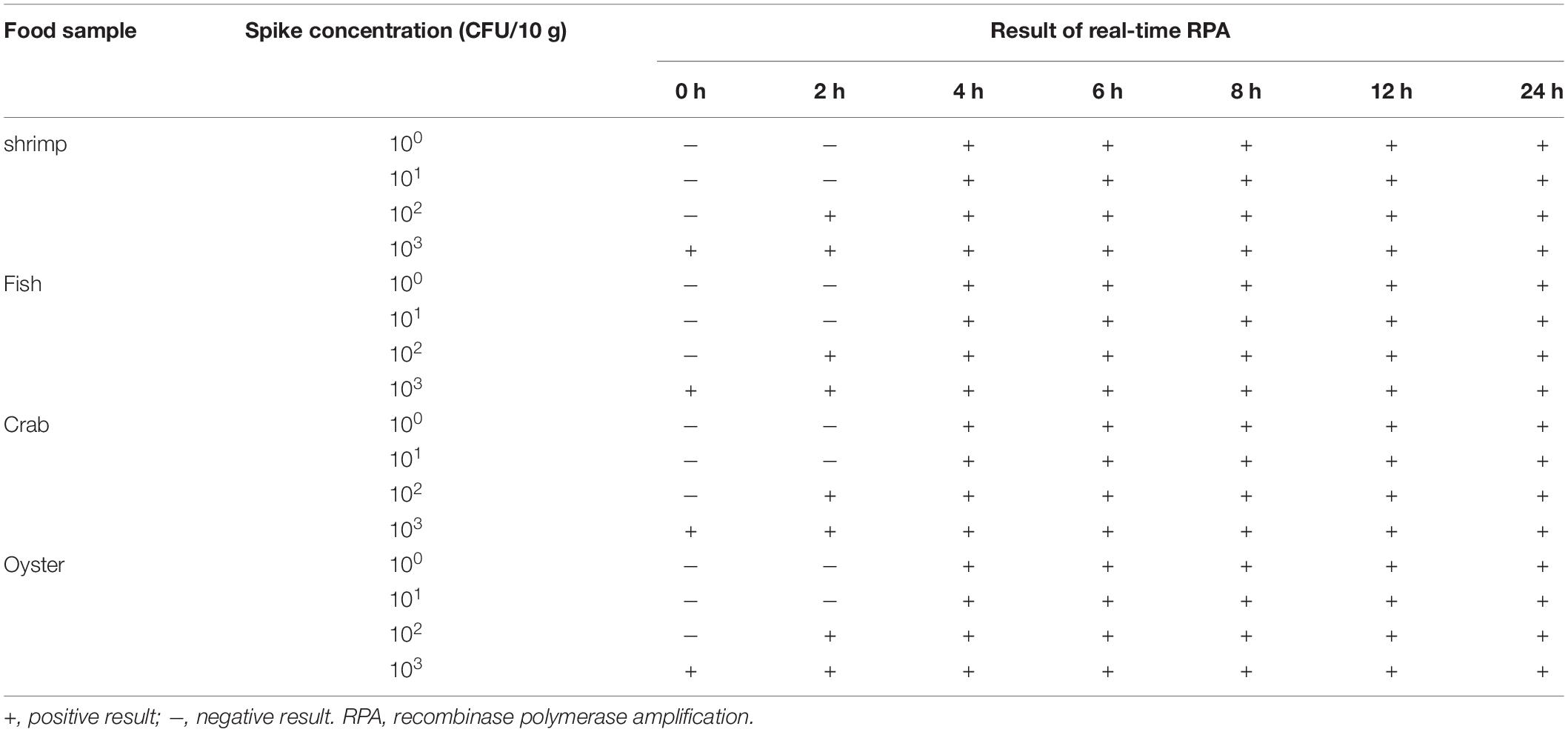
Table 3. Detection of Vibrio vulnificus in spiked food samples.
Clinical Sample Detection
A total of 65 clinical seafood samples including 44 shrimp, 12 fish, 5 shellfish, and 4 crab samples were tested for V. vulnificus with real-time RPA, culturing-based bioassay (National Standard of China DBS13/004-2016), and qPCR. The detection results of real-time RPA were consistent with the bioassay and qPCR results (Figure 4A and Supplementary Table S1). Moreover, the threshold time (Tt value) of real-time RPA ranged from 2.05 to 14.23 min, while the cycle threshold (Ct value) of qPCR ranged from 13.58 to 35 cycles. The correlation of the Ct value and Tt value of the 24 positive samples was analyzed. The result showed that a well correlation was observed with an R2 value of 0.8936 (Figure 4B).
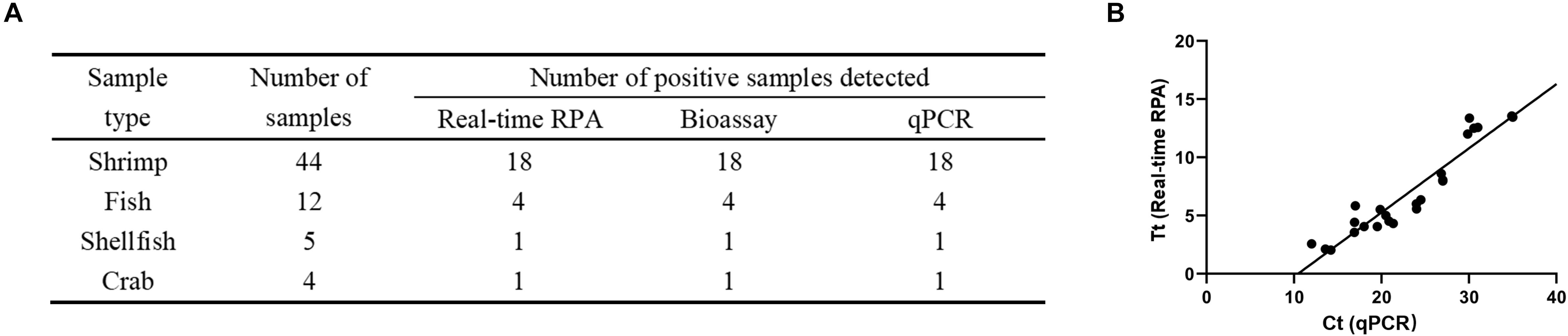
Figure 4. Detection of Vibrio vulnificus in clinical samples. (A) The detection of V. vulnificus for each clinical sample was performed by real-time recombinase polymerase amplification (RPA), bioassay, and qPCR. (B) Linear regression analysis of real-time RPA threshold time (Tt) and qPCR cycle threshold values (Ct) using GraphPad Prism software. R2 = 0.8936.
Discussion
Vibrio vulnificus is a prevalent foodborne pathogen that leads to significant economic losses and public health concern (Jones and Oliver, 2009). Rapid and accurate detection method of V. vulnificus in food can help control the related epidemics and diseases. This study described the development and evaluation of a real-time RPA method for detecting V. vulnificus. The real-time RPA method finishes in 14 min with good specificity and sensitivity and has satisfactory tolerance for crude samples. Being rapid and simple, the method is potentially applied in a wide range of conditions.
The method targets the empV gene encoding the extracellular metalloprotease. This gene has a highly conservative property (Chuang et al., 1997), and the phylogenetic analysis showed that all the empV gene sequences of 23 V. vulnificus strains were classified into one group while other Vibrio species were classified into other groups, predicting good detection coverage and specificity. The gyrB gene has also been utilized as a target for detection of V. vulnificus (Kumar et al., 2006; D’Souza et al., 2019). Just as in the case of empV, the phylogenetic tree of gyrB gene sequences also predicted good detection coverage and specificity, indicating equivalent evolutionary conservation of the two genes (Supplementary Figure S1). The results in this study showed excellent specificity to V. vulnificus and good coverage for the isolated environment strains. Moreover, in the clinical sample tests, results of real-time RPA were consistent with those of qPCR (targeting gyrB gene) and culture-based bioassay. Therefore, the empV gene is an effective detection target.
The real-time RPA method showed good detection sensitivity that was comparable with that of qPCR and LAMP. The limit of detection was 17 copies/reaction in 95% of the cases or 1 CFU per reaction, or 1 CFU/10 g in spiked food samples with enrichment. In other reports, the limit of detection of V. vulnificus was 0.1–1 CFU/reaction with qPCR-based methods or 10–100 CFU/reaction with LAMP (Panicker and Bej, 2005; Wang et al., 2016; D’Souza et al., 2019). The limit of detection of the real-time RPA method was also comparable with that of other RPA-based methods for detection of other pathogens, such as ∼10 CFU per reaction (Vibrio harveyi and Listeria monocytogenes) or 50 copies per reaction (Vibrio parahaemolyticus) (Garrido-Maestu et al., 2019; Pang et al., 2019; Yang et al., 2020).
As reported, the RPA reaction has a good tolerance for crude samples (Moore and Jaykus, 2017). This is also true for the real-time RPA method of this study. In our culture, spiked food, and clinical samples, DNA was released by simple boiling and directly used for the detection, and the limit of detection and accuracy were not affected. This makes the overall procedure of detection even simpler. Although we used a qPCR machine to read the fluorescence signal in this study, a portable tube scanner (such as Genie III from Beijing Suntrap Science & Technology Co., Ltd.) could also give satisfactory results (Geng et al., 2019). Thus, the real-time RPA method is composed of two rapid and simple steps: sample boiling and isothermal amplification with real-time signal reading, which are very portable and can be applied widely.
In conclusion, a real-time RPA method was developed for rapid detection of V. vulnificus. It is an efficient and reliable detection tool for V. vulnificus in the food safety control.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
JD directed the program. SG, JD, and XY designed the research. XY and XZ performed the experiments. HS, GJ, and YW processed the data. XY and PZ wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31470275), the Open Foundation of Jiangsu Key Laboratory of Marine Pharmaceutical Compound Screening (HY201805), the Fishery Science and Technology Innovation Program (Y2018-14), the Nantong Municipal Science and Technology Plan (GJZ17077), the Lianyungang Science and Technology Project of China (SF2003), the Science and Technology Project of Lianyungang High-tech Zone of China (HZ201901), and the Priority Academic Program Development of Jiangsu Higher Education Institutions of China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.586981/full#supplementary-material
Footnotes
- ^ http://www.clustal.org/
- ^ http://www.megasoftware.net/mega4/mega.html
- ^ https://www.ncbi.nlm.nih.gov/tools/primer-blast
References
Bonnin-Jusserand, M., Copin, S., Bris, C. L., Brauge, T., Gay, M., Brisabios, A., et al. (2019). Vibrio species involved in seafood-borne outbreaks (Vibrio cholerae, V. parahaemolyticus and V. vulnificus): review of microbiological versus recent molecular detection methods in seafood products. Crit. Rev. Food Sci. Nutr. 59, 597–610. doi: 10.1080/10408398.2017.1384715
Chuang, Y. C., Chang, T. M., and Chang, M. C. (1997). Cloning and characterization of the gene (empV) encoding extracellular metalloprotease from Vibrio vulnificus. Gene 189, 163–168. doi: 10.1016/s0378-1119(96)00786-x
Dalsgaard, A., Frimodt-Moller, N., Bruun, B., Hoi, L., and Larsen, J. L. (1996). Clinical manifestations and molecular epidemiology of Vibrio vulnificus infections in Denmark. Eur. J. Clin. Microbiol. Infect. Dis. 15, 227–232. doi: 10.1007/BF01591359
Dong, Y., Zhao, P., Chen, L., Wu, H., Si, X., Shen, X., et al. (2020). Fast, simple and highly specific molecular detection of Vibrio alginolyticus pathogenic strains using a visualized isothermal amplification method. BMC Vet Res. 16:76. doi: 10.1186/s12917-020-02297-4
D’Souza, C., Kumar, B. K., Rai, P., Deekshit, V. K., and Karunasagar, I. (2019). Application of gyrB targeted SYBR green based qPCR assay for the specific and rapid detection of Vibrio vulnificus in seafood. J. Microbiol. Methods 166:105747. doi: 10.1016/j.mimet.2019.105747
Du, X. J., Zang, Y. X., Liu, H. B., Li, P., and Wang, S. (2018). Recombinase polymerase amplification combined with lateral flow strip for Listeria monocytogenes detection in Food. J. Food Sci. 83, 1041–1047. doi: 10.1111/1750-3841.14078
Garrido-Maestu, A., Azinheiro, S., Carvalho, J., and Prado, M. (2019). Combination of immunomagnetic separation and real-time recombinase polymerase amplification (IMS-qRPA) for specific detection of Listeria monocytogenes in Smoked salmon samples. J. Food Sci. 84, 1881–1887. doi: 10.1111/1750-3841.14662
Geng, Y., Liu, G., Liu, L., Deng, Q., Zhao, L., Sun, X. X., et al. (2019). Real-time recombinase polymerase amplification assay for the rapid and sensitive detection of Campylobacter jejuni in food samples. J. Microbiol. Methods 157, 31–36. doi: 10.1016/j.mimet.2018.12.017
Han, F., and Ge, B. (2010). Quantitative detection of Vibrio vulnificus in raw oysters by real-time loop-mediated isothermal amplification. Int. J. Food Microbiol. 142, 60–66. doi: 10.1016/j.ijfoodmicro.2010.05.029
Hartnell, R. E., Stockley, L., Keay, W., Rosec, J. P., Hervio-Heath, D., Van den Berg, H., et al. (2019). A pan-European ring trial to validate an international standard for detection of Vibrio cholerae, Vibrio parahaemolyticus and Vibrio vulnificus in seafoods. Int. J. Food Microbiol. 288, 58–65. doi: 10.1016/j.ijfoodmicro.2018.02.008
Hiergeist, A., and Reischl, U. Priority Program Intestinal Microbiota Consortium/Quality Assessment Participants Gessner, A. (2016). Multicenter quality assessment of 16S ribosomal DNA-sequencing for microbiome analyses reveals high inter-center variability. Int. J. Med. Microbiol. 306, 334–342. doi: 10.1016/j.ijmm.2016.03.005
Hlady, W. G., and Klontz, K. C. (1996). The epidemiology of Vibrio infections in Florida, 1981-1993. J. Infect. Dis. 173, 1176–1183. doi: 10.1093/infdis/173.5.1176
Huehn, S., Eichhorn, C., Urmersbach, S., Breidenbach, J., Bechlars, S., Bier, N., et al. (2014). Pathogenic Vibrios in environmental, seafood and clinical sources in Germany. Int. J. Med. Microbiol. 304, 843–850. doi: 10.1016/j.ijmm.2014.07.010
Jones, M. K., and Oliver, J. D. (2009). Vibrio vulnificus: disease and pathogenesis. Infect. Immun. 77, 1723–1733. doi: 10.1128/IAI.01046-08
Kumar, H. S., Parvathi, A., Karunasagar, I., and Karunasagar, I. (2006). A gyrB-based PCR for the detection of Vibrio vulnificus and its application for direct detection of this pathogen in oyster enrichment broths. Int. J. Food Microbiol. 111, 216–220. doi: 10.1016/j.ijfoodmicro.2006.05.007
Leng, F., Lin, S., Wu, W., Zhang, J., Song, J., and Zhong, M. (2019). Epidemiology, pathogenetic mechanism, clinical characteristics, and treatment of Vibrio vulnificus infection: a case report and literature review. Eur. J. Clin. Microbiol. Infect. Dis. 38, 1999–2004. doi: 10.1007/s10096-019-03629-5
Li, J., Macdonald, J., and von Stetten, F. (2018). Review: a comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 144, 31–67. doi: 10.1039/c8an01621f
Li, L., Mendis, N., Trigui, H., Oliver, J. D., and Faucher, S. P. (2014). The importance of the viable but non-culturable state in human bacterial pathogens. Front. Microbiol. 5:258. doi: 10.3389/fmicb.2014.00258
Moore, M. D., and Jaykus, L. A. (2017). Development of a recombinase polymerase amplification assay for detection of epidemic human noroviruses. Sci. Rep. 7:40244. doi: 10.1038/srep40244
O’Hara, C. M., Sowers, E. G., Bopp, C. A., Duda, S. B., and Strockbine, N. A. (2003). Accuracy of six commercially available systems for identification of members of the family Vibrionaceae. J. Clin. Microbiol. 41, 5654–5659. doi: 10.1128/jcm.41.12.5654-5659.2003
Pang, J., Wang, Q., Fei, Y., Zhu, P., Qiao, L., Huang, H., et al. (2019). A real-time recombinase polymerase amplification assay for the rapid detection of Vibrio harveyi. Mol. Cell. Prob. 44, 8–13. doi: 10.1016/j.mcp.2019.01.001
Panicker, G., and Bej, A. K. (2005). Real-time PCR detection of Vibrio vulnificus in oysters: comparison of oligonucleotide primers and probes targeting vvhA. Appl. Environ. Microbiol. 71, 5702–5709. doi: 10.1128/AEM.71.10.5702-5709.2005
Piepenburg, O., Williams, C. H., Stemple, D. L., and Armes, N. A. (2006). DNA detection using recombination proteins. PLoS Biol. 4:e204. doi: 10.1371/journal.pbio.0040204
Rippey, S. R. (1994). Infectious diseases associated with molluscan shellfish consumption. Clin. Microbiol. Rev. 7, 419–425. doi: 10.1128/cmr.7.4.419
Strom, M. S., and Paranjpye, R. N. (2000). Epidemiology and pathogenesis of Vibrio vulnificus. Microb. Infect. 2, 177–188. doi: 10.1016/s1286-4579(00)00270-7
Surasilp, T., Longyant, S., Rukpratanporn, S., Sridulyakul, P., Sithigorngul, P., and Chaivisuthangkura, P. (2011). Rapid and sensitive detection of Vibrio vulnificus by loop-mediated isothermal amplification combined with lateral flow dipstick targeted to rpoS gene. Mol. Cell. Prob. 25, 158–163. doi: 10.1016/j.mcp.2011.04.001
Wang, Y., Li, D., Wang, Y., Li, K., and Ye, C. (2016). Rapid and sensitive detection of Vibrio parahaemolyticus and Vibrio vulnificus by multiple endonuclease restriction real-time loop-mediated isothermal amplification technique. Molecules 21:E111. doi: 10.3390/molecules21010111
Keywords: Vibrio vulnificus, real-time recombinase polymerase amplification, rapid detection, recombinase polymerase amplification, extracellular metalloprotease
Citation: Yang X, Zhang X, Wang Y, Shen H, Jiang G, Dong J, Zhao P and Gao S (2020) A Real-Time Recombinase Polymerase Amplification Method for Rapid Detection of Vibrio vulnificus in Seafood. Front. Microbiol. 11:586981. doi: 10.3389/fmicb.2020.586981
Received: 24 July 2020; Accepted: 12 October 2020;
Published: 06 November 2020.
Edited by:
Dario De Medici, National Institute of Health (ISS), ItalyReviewed by:
Jessica L. Jones, United States Food and Drug Administration, United StatesEva Sanjuan, University of Valencia, Spain
Copyright © 2020 Yang, Zhang, Wang, Shen, Jiang, Dong, Zhao and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingquan Dong, MjAxODAwMDAyOUBqb3UuZWR1LmNu; Panpan Zhao, emhhb3BwMTlAbWFpbHMuamx1LmVkdS5jbg==; Song Gao, Z2Fvc0Bqb3UuZWR1LmNu; MjAxMzAwMDAwMkBqb3UuZWR1LmNu
†These authors have contributed equally to this work