 Ran Mei
Ran Mei Masaru K. Nobu
Masaru K. Nobu Takashi Narihiro2
Takashi Narihiro2 Wen-Tso Liu
Wen-Tso Liu- 1Department of Civil and Environmental Engineering, University of Illinois at Urbana-Champaign, Urbana, IL, United States
- 2Bioproduction Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Japan
Current understanding of amino acid (AA) degraders in anaerobic digesters is mainly based on cultured species, whereas microorganisms that play important roles in a complex microbial community remain poorly characterized. This study investigated short-term enrichments degrading single AAs using metagenomics and metatranscriptomics. Metagenomic analysis revealed that populations related to cultured AA degraders had an abundance <2.5% of the sequences. In contrast, metagenomic-assembled bins related to uncultured Bacteroidales collectively accounted for >35% of the sequences. Phylogenetic analyses suggested that these Bacteroidales populations represented a yet-to-be characterized family lineage, i.e., Bacteroidetes vadinHA17. The bins possessed the genetic capacity related to protein degradation, including surface adhesion (3–7 genes), secreted peptidase (52–77 genes), and polypeptide-specific transporters (2–5 genes). Furthermore, metatranscriptomics revealed that these Bacteroidales populations expressed the complete metabolic pathways for degrading 16 to 17 types of AAs in enrichments fed with respective substrates. These characteristics were distinct from cultured AA degraders including Acidaminobacter and Peptoclostridium, suggesting the uncultured Bacteroidales were the major protein-hydrolyzing and AA-degrading populations. These uncultured Bacteroidales were further found to be dominant and active in full-scale anaerobic digesters, indicating their important ecological roles in the native habitats. “Candidatus Aminobacteroidaceae” was proposed to represent the previously uncharted family Bacteroidetes vadinHA17.
Introduction
Anaerobic digestion (AD) is a biological process where complex polymers are converted to small molecules and mineralized to methane and carbon dioxide. Protein is one of the polymers and accounts for a significant portion (25.4%–37.9% of the volatile solids content) of organic feed to anaerobic digesters (McInerney, 1988). It is also abundant in wastewater produced from food industries that process whey, cheese, and fish (Tang et al., 2005). Protein degradation can be the rate-limiting step in AD, and has been shown as a slower process than carbohydrates degradation (Gujer and Zehnder, 1983; Yang et al., 2015). Protein is subsequently hydrolyzed to amino acids (AAs), which are the key groups of intermediates in AD metabolism. Despite the importance of AAs in anaerobic environments, few studies explicitly identify microorganisms that can degrade AAs especially in a complex microbial community like AD (Tang et al., 2005). Furthermore, AA-degrading microorganisms were primarily studied in the 1980s based on clostridia pure cultures (McInerney, 1988). Considering most anaerobic microorganisms have not been cultured, important AA degraders in AD remain to be uncovered.
Our recent study characterized microbial communities that were inoculated with AD sludge and enriched with individual AAs (Mei et al., 2020). During continuous transfer spanning 18 months, analyses of 16S rRNA and rRNA gene revealed a shift in predominant populations from uncultured members of the order Bacteroidales in the early enrichment to cultured AA fermenters including Acidaminobacter and Peptoclostridium at the end of enrichment. This observation implied that although continuous enrichment is the classic strategy to characterize novel microorganisms, it may fail to capture species that are functionally important in the native habitats due to the bias associated with artificial cultivation conditions (Kamagata, 2015). The predominance of uncultured Bacteroidales populations (>20% 16S rRNA abundance) in the short-term enrichment communities further indicated their important roles involved in AA degradation.
The order Bacteroidales is one of the most prevalent in AD, and its abundance can reach as high as 15% in a community (Mei et al., 2016, 2018; Narihiro et al., 2018; Zealand et al., 2018; Nobu et al., 2020). It is also widely distributed in different non-AD habitats (Gupta, 2004) such as animal intestinal microflora (Dick et al., 2005) and marine bacterioplankton (Fernández-Gomez et al., 2013). Many studies (Narihiro et al., 2015; Kim et al., 2016; Ormerod et al., 2016; Ramayo-Caldas et al., 2016; Vanwonterghem et al., 2016; Reichardt et al., 2017; Lagkouvardos et al., 2019) have suggested populations related to Bacteroidales as saccharolytic because carbohydrate degradation is a common feature among most non-marine isolated species of this order (Rosenberg et al., 2014). However, there are still more than 20 uncultured family-level lineages in this order based on the most recent SILVA database (release 138) (Yilmaz et al., 2013), including Bacteroidetes vadinHA17, Bacteroidetes BD2-2, and Bacteroidales RF16, suggesting the metabolic diversity of this order is not fully uncovered. Some of those uncharacterized Bacteroidales are likely capable of protein and amino acids degradation given their high abundances observed in anaerobic reactors treating protein-rich substrates such as bovine serum albumin (Tang et al., 2005) and casein (Ley et al., 2006). In addition, a few Bacteroidales isolates have been reported to be capable of non-saccharolytic metabolisms [e.g., Proteiniphilum saccharofermentans (Tomazetto et al., 2018), Williamwhitmania taraxaci (Pikuta et al., 2017), and Salinivirga cyanobacteriivorans (Ben Hania et al., 2017)]. Therefore, further investigation is clearly warranted to expand our understanding about the diverse roles that uncultured Bacteroidales could play, including protein and amino acids degradation.
The present study conducted metagenomic and metatranscriptomic analyses to decipher the roles of the uncultured Bacteroidales that were abundant in the short-term enrichment communities we previously established (Mei et al., 2020). The combination of short-term enrichment and meta-omics analyses can exert sufficient selective pressure while avoiding enrichment bias. Functions related to protein and AA degradation were assessed in detail based on the recovered genomes and gene expression profile of the Bacteroidales populations. Their ecological prevalence in full-scale ADs and sediment environments was also evaluated.
Materials and Methods
Enrichment Cultures and Sequencing
Enrichment cultures were described in our previous publication (Mei et al., 2020). In brief, anaerobic digester sludge taken from a municipal wastewater treatment plant (Urbana, IL, United States) was used as inoculum (2 mL sludge into 80 mL media). Twenty individual amino acids were used individually as single substrates (5 mM final concentration). The cultures were incubated under 35°C in dark without strong mixing to mimic the anaerobic digester environment. When the substrates were used up, as determined by ceased methane production, and prior to further culture transfer, 14 of the 20 enrichments were selected for metagenomic sequencing and 19 for metatranscriptomic sequencing (with duplicates, Supplementary Table 1). Sequencing was performed at the Joint Genomic Institute (JGI) in US Department of Energy using the Illumina HiSeq-2500 1TB platform. The raw sequences are available at the JGI Genome Portal (Supplementary Table 1). JGI also performed sequencing filtering and assembling. For metagenomic sequences, BBDuk v38.08 was used to remove contaminants and trim reads that contained adapter sequence and had low quality1. Reads mapped to masked human, cat, dog and mouse references and other common microbial contaminants were also removed. Filtered reads were further corrected using BFC vr181 (Li, 2015) and assembled using SPAdes assembler v3.11.1 (Nurk et al., 2017). For metatranscriptomic sequences, quality filtering was performed in the same way as metagenomic sequences with additional removal rRNA reads.
Metagenomic Binning
Two binning softwares, MetaBAT v2.12.1 (Kang et al., 2015) and MaxBin v2.2.5 (Wu et al., 2015), were applied to the metagenomic assembly and generated two independent sets of bins from each sample. Binning_refiner v1.2 (Song and Thomas, 2017) was used to compare and merge the results of MetaBAT and MaxBin. The resulting bins were further improved using RefineM v0.0.24 (Parks et al., 2017). The quality, i.e., completeness and contamination, of the final bins was assessed using CheckM v1.0.12 (Parks et al., 2015). Bins with >80% completeness and <5% contamination, were selected for downstream analysis. Phylophlan v0.99 (Segata et al., 2013) was used to identify and align marker proteins from bins. The alignment was further used to construct a phylogenomic tree using maximum likelihood method with bootstrap value of 100 in MEGA X (Kumar et al., 2018). Average amino acids identity (AAI) between bins was calculated using CompareM v0.0.23 (Parks et al., 2017). Based on the results of phylogenomics and AAI, closely related bins from different samples were grouped using Spine (Ozer et al., 2014) with only core genome content being kept. In total, 56 final bins were obtained including pangenomes. The taxonomy of the final bins was further assessed using GTDB-TK v1.0.2 (Chaumeil et al., 2019). Quality-filtered metagenomic and metatranscriptomic reads were mapped to the 56 final bins using BBmap v38.46 to estimate the representativeness of the genomes. The metagenomic abundance of the genomes in the community was calculated as described previously (Woodcroft et al., 2018). Briefly, quality-filtered metagenomic reads from each sample were mapped to the genomes using BamM v1.7.3 (Parks et al., 2017). Low-quality mappings were removed, and the length-weighted coverage of each contig was averaged to calculate the coverage of the genomes in the community. The relative abundance of each genome in each sample was calculated as its coverage divided by the total coverage of all the genomes. The draft genomes were deposited in NCBI GenBank under the accession WXFB00000000-WXFH00000000. 16S rRNA genes found in the genomes were deposited under the accession MK990229–MK990231.
Comparative Genomics
Five Bacteroidales bins and two bins related to Peptoclostridium and Acidaminobacter were analyzed in detail. Initial annotation was performed with Prokka v1.13.3 (Seemann, 2014). The 16S rRNA gene sequences obtained from Bacteroidales bin 2, 3, and 4 were analyzed in ARB (Ludwig et al., 2004) using maximum likelihood method RAxML with 100 time bootstrapping. Phylogenomic analysis was performed using Phylophlan v0.99 and MEGA X. The annotated genomes were then checked and corrected manually by searching for similar proteins (>50% similarity and >50% coverage) in the UniProt Knowledgebase2 using BLASTP v2.10.0. Peptidase, lipase, transporter, hydrogenase were identified by BLASTP v2.10.0 search using the MEROPS database release 12.1 (Rawlings et al., 2017), Lipase Engineering Database (Widmann et al., 2010), Transporter Classification Database (Saier et al., 2015), HydDB (Søndergaard et al., 2016), with >50% amino acids similarity and >50% coverage. Carbohydrate-active enzyme and signal peptide were annotated using dbCAN2 (Zhang et al., 2018) and SignalP 5.0 (Armenteros et al., 2019). Genes related to surface adhesion were found using the Pfam search as described previously (Fernández-Gomez et al., 2013). SusC/RagA-like protein sequences described previously (Ben Hania et al., 2017) were obtained from GenBank or Uniprot. Alignment of the protein sequences was performed using ClustalW, and phylogenetic analysis was performed using maximum likelihood algorithm with 100 bootstraps implemented in MEGA X. BBmap v38.46 was used to calculate the gene expression levels as reads per kilobase transcript per million reads (RPKM). Public metagenomic and metatranscriptomic sequences obtained from full-scale anaerobic digesters (Nobu et al., 2020) or sediments (Urich et al., 2014; Dyksma et al., 2016; Voorhies et al., 2016; Chu et al., 2018; Orsi et al., 2018) were mapped to the bins to estimate prevalence of the studied populations.
Results and Discussion
AA-Degrading Community Structure Based on Metagenomics
Among 20 short-term enrichment cultures degrading individual AAs established in our previous study (Mei et al., 2020), 14 were further selected for metagenomic sequencing (Supplementary Table 1). The remaining cultures were not selected because they had very similar 16S rRNA gene-based community structures (i.e., cultures fed with cysteine, serine, aspartic acid, glutamine, and proline) or had no growth (i.e., cultures fed with tyrosine). From the 14 metagenomic datasets, a total of 796 metagenomics-assembled bins were obtained and 281 bins with >80% completeness and <5% contamination were selected for downstream analysis (Supplementary Figure 1). After merging bins closely clustered with each other based on phylogenomic analysis, 56 final bins were obtained. These 56 final bins showed good representativeness of the populations in the enrichment cultures as majority of the metagenomic (67.4% on average) and metatranscriptomic reads (86.2% on average) in each culture could be mapped to them (Supplementary Figure 2).
Figure 1 showed the metagenomic abundances of those 56 bins. Among them, five bins were related to known AA fermenters, such as Peptoclostridium acidaminophilum and Acidaminobacter hydrogenoformans. They had low abundance (total abundance of 1.2%) and low occurrence (detected in < 3 cultures with > 1% abundance), suggesting the marginal roles of these populations in the short-term enrichment. Seven bins were related to syntrophic acids degraders (total abundance of 20%), including Syntrophaceae (7.7% on average), Syntrophorhabdus (3.5%), and Syntrophobacter (2.4%). These syntrophs could degrade diverse intermediates that were produced from AA fermentation and biomass degradation (Nobu et al., 2015). Specifically, the bins related to Syntrophaceae and Syntrophorhabdus had a high abundance (maximum abundance > 15%) in cultures fed with leucine, isoleucine, valine, tryptophan, or phenylalanine (Supplementary Figure 3), suggesting they played important roles in degrading intermediates generated from specific AA fermentation, such as branch-chain fatty acids, heterocyclic compounds, and phenolic compounds. For methanogens, five bins were detected (total abundance of 7%), including those related to Methanosaeta (0.5% on average), Methanoculleus (1.3%), and Ca. Methanofastidiosa (2.7%). They represented different types methanogenic pathways, i.e., aceticlastic, hydrogenotrophic, and methyl-reducing. Specifically, bins related to Ca. Methanofastidiosa (26.6%) and Methanomassiliicoccaceae (3.1%) were abundant in the culture fed with methionine, consistent with previous reports that they could reduce methylated compounds and produce methane (Dridi et al., 2012; Nobu et al., 2016). Last, populations with unknown functions were consistently observed with high abundance (total abundance of 72%) in all the 14 enrichments, including five bins related to the order Bacteroidales. Among the five Bacteroidales bins, bin1, bin2, and bin4 were more abundant (9.1%, 2.6%, and 20.2%, respectively) than bin3 (0.8%) and bin5 (0.2%).
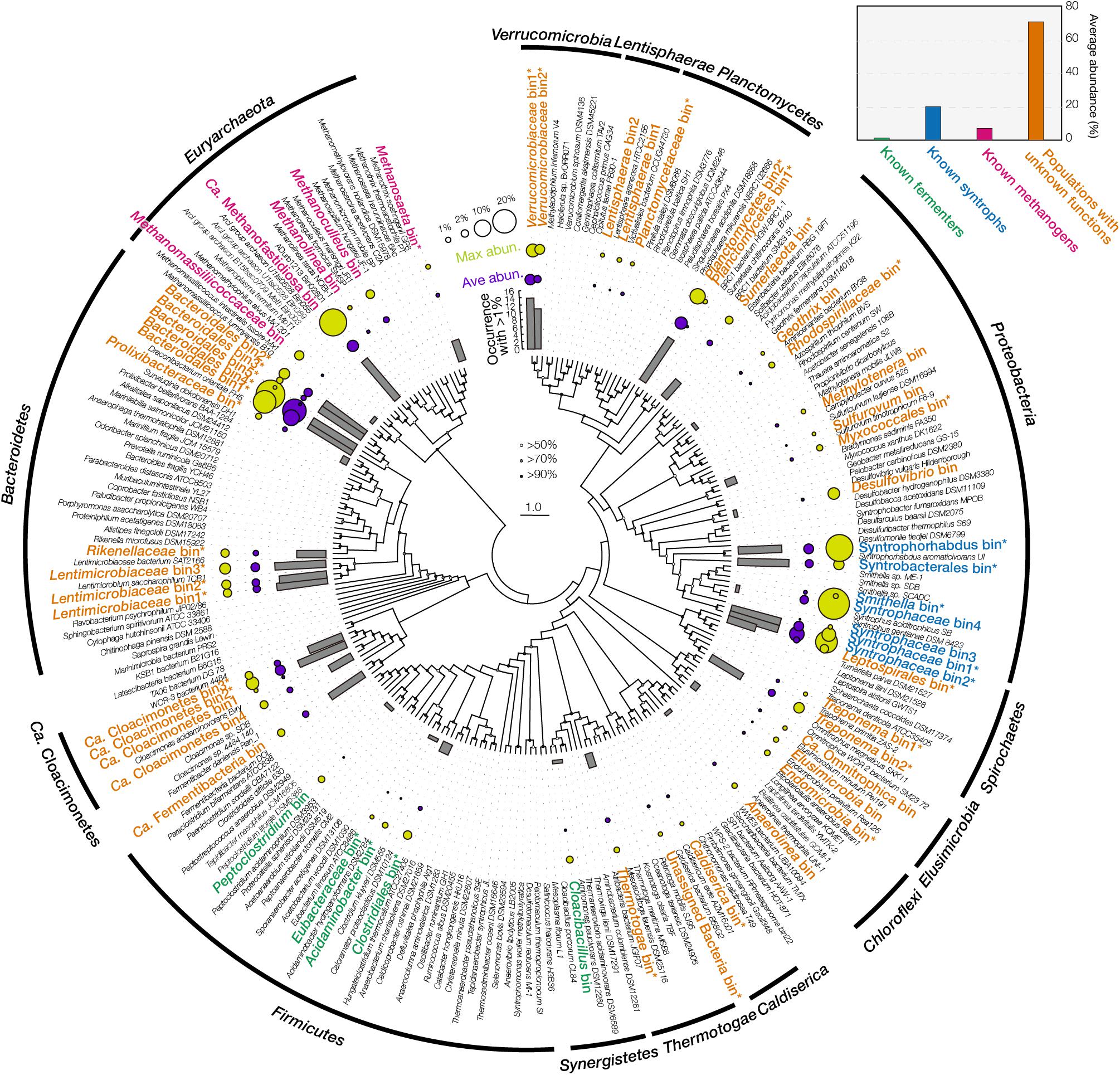
Figure 1. Fifty-six final bins assembled from metagenome. The phylogenetic tree was built using maximum likelihood method with 100 bootstraps. Bootstrap values are denoted using bubbles on the node. Bars in the inner circle denote the occurrence of each bin with an abundance >1% among the 14 metagenomic datasets. Bubbles in the middle circle denote the average metagenomic abundance. Bubbles in the outer circle denote the maximum metagenomic abundance. Bins’ name is colored according to their potential functions. Asterisk after the bins’ name indicates this final bin was merged from multiple bins from different samples clustered together in this tree. Small panel at the top-right corner shows the average abundance of four functional groups.
Phylogenetic Analysis of Bacteroidales Bins
Phylogenetic affiliations of the Bacteroidales bins were examined by first analyzing the nearly full-length 16S rRNA gene sequences found in three of the five Bacteroidales bins (bins 2, 3, and 4). We did not find 16S rRNA genes in bins 1 and 5 after several trials. These Bacteroidales bins were distinct from all known Bacteroidales families containing cultured representatives (Figure 2A), but were affiliated with an uncultured family-level lineage Bacteroidetes vadinHA17. The sequence similarity between the three 16S rRNA gene sequences and representative sequences of other Bacteroidales families ranged from 79.2% to 88.6% (Supplementary Table 2), suggesting that vadinHA17 represented a novel family based on the sequence similarity threshold of 86.5% for family and 94.5% for genus (Yarza et al., 2014). Analysis based on universally conserved protein sequences further supported that the five Bacteroidales bins formed a cluster that was distinct from all cultured Bacteroidales families (Figure 2B). The average amino acid identity (AAI) of the five Bacteroidales bins against representative genomes of cultured families ranged from 49.7% to 57.6% (Supplementary Table 2), indicating that they were not associated with existing cultured families based on a cutoff of ∼60% (Konstantinidis and Tiedje, 2005).

Figure 2. Phylogenetic analysis of Bacteroidales bins based on (A) 16S rRNA gene sequences and (B) universally conserved protein sequences.
Protein Degradation by the Bacteroidales Species
Figure 3 shows the general lifestyle of protein degradation, AA metabolism and energy conservation based on the annotation of Bacteroidales bins 1, 2, and 4 that were abundant in the cultures (abundance >2%). In addition, two bins that were closely related to cultured AA degraders, i.e., Acidaminobacter (Stams and Hansen, 1984) and Peptoclostridium (Zindel et al., 1988), were analyzed and used us references. Domains involved in surface adhesion to biomass were first examined (Fernández-Gomez et al., 2013). The Bacteroidales bins were observed to encode more adhesion genes than the cultured AA degraders (Supplementary Table 3). Bins 1, 2, and 4 encoded 3, 6, and 7 adhesion genes, including the Bac_surface_Ag family (PF01103) related to outer membrane surface antigen, the F5_F8_Type_C family (PF00754) related to cell adhesion protein discoidin and the HYR family (PF02494) related to cell adhesion (Baumgartner et al., 1998; Callebaut et al., 2000; Feng et al., 2012). Particularly, genes related to the HYR family had the highest expression in the three bins (2.3-4.4 times of the bins’ median activity), suggesting the critical role of the cell adhesion function. In contrast, the Acidaminobacter bin and Peptoclostridium bin did not encode genes related to those families. The Acidaminobacter bin only encoded one gene related to the fasciclin family (PF02496), an ancient cell adhesion domain (Huber and Sumper, 1994). These observations suggested that the Bacteroidales populations had distinct surface adhesion properties from cultured AA degraders.
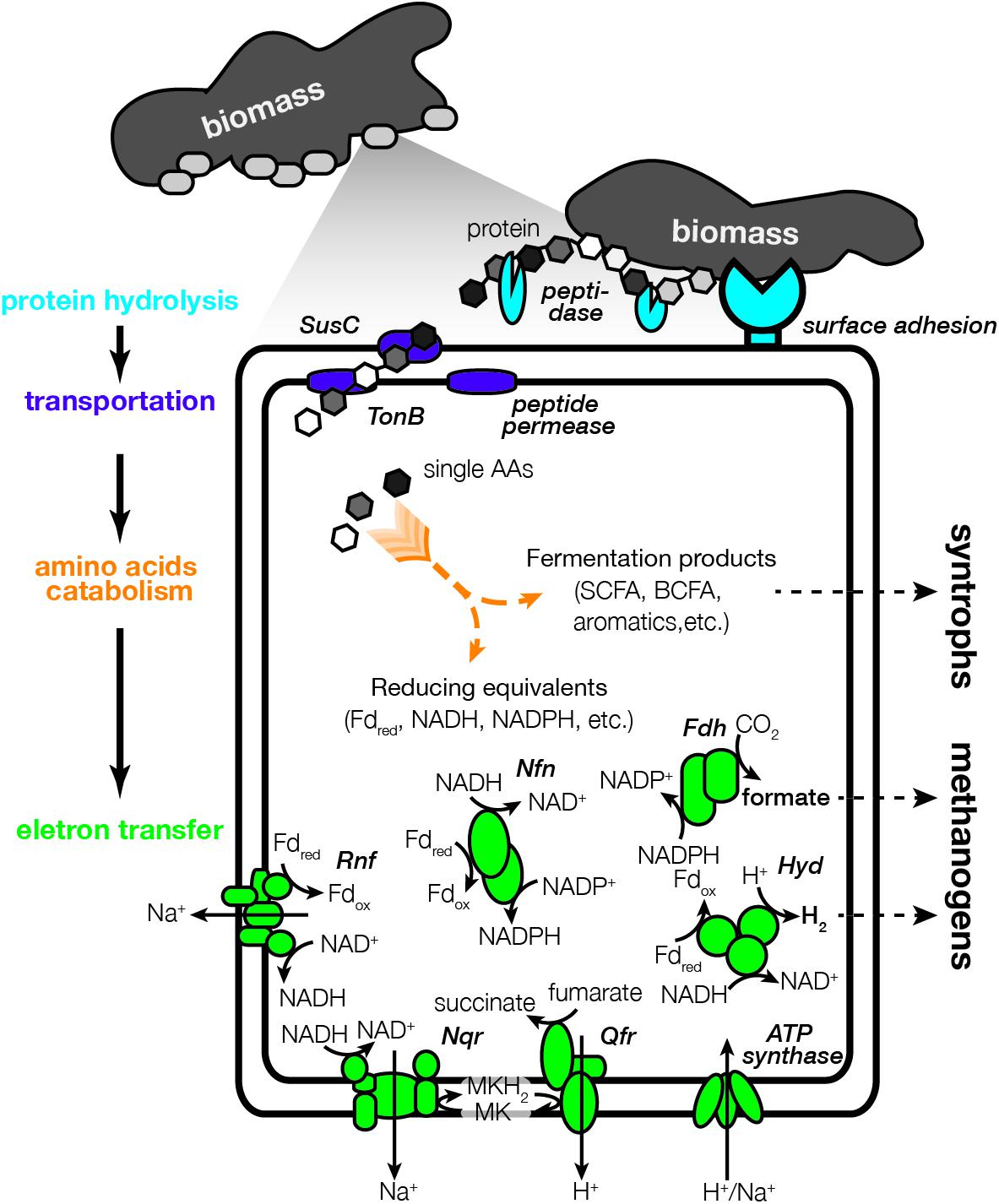
Figure 3. Illustration of the proteolytic amino acids degrading lifestyle of the Bacteroidales populations.
After biomass adhesion, we analyzed extracellular peptidases that can hydrolyze the protein fractions in the biomass (Figure 3). Bins 1, 2, and 4 encoded 172, 109, and 100 peptidase genes, respectively (Supplementary Table 4). Metallo (M) and serine (S) peptidases were the most abundant types (>40 genes), followed by cysteine (C) peptidases (>15 genes) and small numbers of other types (<5 genes). These genes included known extracellular peptidases such as papain (C01A), thermitase (S08A), and tricorn core protease (S41B). To specifically identify peptidases that were secreted out of the membrane and involved in protein hydrolysis, putative signal sequence motifs that translocate peptidases toward the secretory system were examined (Nguyen et al., 2019). Bins 1, 2, and 4 encoded 72, 55, and 68 secreted peptidase genes, respectively, higher than the Acidaminobacter bin (12 genes) and Peptoclostridium bin (8 genes). Among secreted peptidases that were encoded, bins 1, 2, and 4 highly expressed 38, 16, and 30 genes, respectively (> the bins median activity) (Supplementary Figure 4), significantly higher than the number expressed by the Acidaminobacter bin (2.4 genes on average) and Peptoclostridium bin (0.3 genes on average). The Bacteroidales bins also encoded genes for secreted carbohydrates and lipids hydrolysis. But these genes had lower expression level compared with secreted peptidases, suggesting the Bacteroidales were specialized in hydrolyzing protein but not polysaccharides or lipids in the short-term enrichments. Their activities in degrading other macromolecule substrates in different environments remain further investigation.
After protein hydrolysis, the Bacteroidales species were observed with the capacity of transporting protein hydrolysis products using the TonB-dependent receptor SusC protein (Figure 3). While SusC protein is usually considered to be related to polysaccharide uptake and transport (Reeves et al., 1997), recent evidences also revealed its role in taking up non-polysaccharide substrates such as peptides (Ben Hania et al., 2017) and other degradation products of protein (Nagano et al., 2007). The types of substrates that SusC protein utilized can be also inferred from its sequence similarity (Ben Hania et al., 2017). Bins 1, 2, and 4 encoded 3 to 8 susC genes, and more than half were located in a cluster specialized for polypeptide transport based on the phylogenetic analysis of SusC protein sequences found in diverse Bacteroidetes genomes (Supplementary Figure 5). This polypeptide-specific cluster also included most Bacteroidales species known to utilize proteinaceous substrates, including Williamwhitmania taraxaci (Pikuta et al., 2017), Porphyromonas gingivalis (Nagano et al., 2007), Acetobacteroides hydrogenigenes (Su et al., 2014), and Proteiniphilum saccharofermentans (Tomazetto et al., 2018). We also found TonB proteins and oligopeptide permeases in Bacteroidales bins, which could facilitate the cross-membrane transport of protein hydrolysis products. In contrast, no susC gene was identified in the Acidaminobacter bin and Peptoclostridium bin. Overall, the characterization of biomass adhesion, secreted peptidase, and cross-membrane transporter strongly suggested that the Bacteroidales populations had a proteolytic lifestyle, which was not observed with Acidaminobacter and Peptoclostridium that degrade soluble AA monomers. They were dominant in the short-term enrichment because there were plenty of insoluble macromolecules in the inoculum biomass (2.5 ml anaerobic digester sludge). These observations were consistent with our previous results that the Bacteroidales populations were competed out after long-term enrichment where biomass was significantly diluted after a series of transfer (Mei et al., 2020).
Amino Acids Metabolism by the Bacteroidales Species
The Bacteroidales species were further observed to possess the capacity to metabolize individual AAs that were available in the cell (Figure 3). Catabolic pathways of each AA were manually identified, and one pathway was defined as complete only when all the genes of this pathway were found in the bins. For example, bins 1, 2, and 4 were identified to be able to degrade alanine because they encoded the gene for alanine dehydrogenase, a key enzyme that converts alanine to pyruvate (Supplementary Table 5 and Supplementary Figure 6). For AAs that require multi-step pathways such as glycine, bins 1, 2, and 4 could degrade glycine to pyruvate because all the six genes of the glycine decarboxylating pathway were identified in each bin. In total, bins 1, 2, and 4 were observed to encode the complete pathways for degrading 16, 17, and 17 types of AAs, respectively (Supplementary Table 6). Such genetic capacity was comparable to the Acidaminobacter bin and Peptoclostridium bin, which possessed pathways to degrade 13 and 16 types of AAs, respectively.
The expression of these pathways was further examined in the enrichments fed with respective substrate based on metatranscriptomics (Figure 4). For example, in the enrichment fed with alanine, all three Bacteroidales bins expressed alanine dehydrogenase whereas the Acidaminobacter bin and the Peptoclostridium bin did not express the gene. In the enrichment fed with glycine, bin1 and bin4 expressed all the six genes of the glycine decarboxylating pathway. In contrast, there were at least one gene not expressed by bin2, the Acidaminobacter bin, and the Peptoclostridium bin. In summary, bins 1, 2, and 4 expressed the complete degradation pathways of 16, 11, and 17 types of AAs (Supplementary Table 6). These gene expression results were supported by the abundance profile that bin1 and bin4 were more abundant than bin2 (9.1% and 20.2% vs. 2.6%). As a comparison, the bins related to Acidaminobacter and Peptoclostridium only expressed the pathways of five and two types of AAs, respectively, which was in line with their low abundances (<0.5%). These results indicated that although Acidaminobacter and Peptoclostridium were well-known AA degraders, the Bacteroidales populations were the actual active degraders in the short-term enrichment culture.
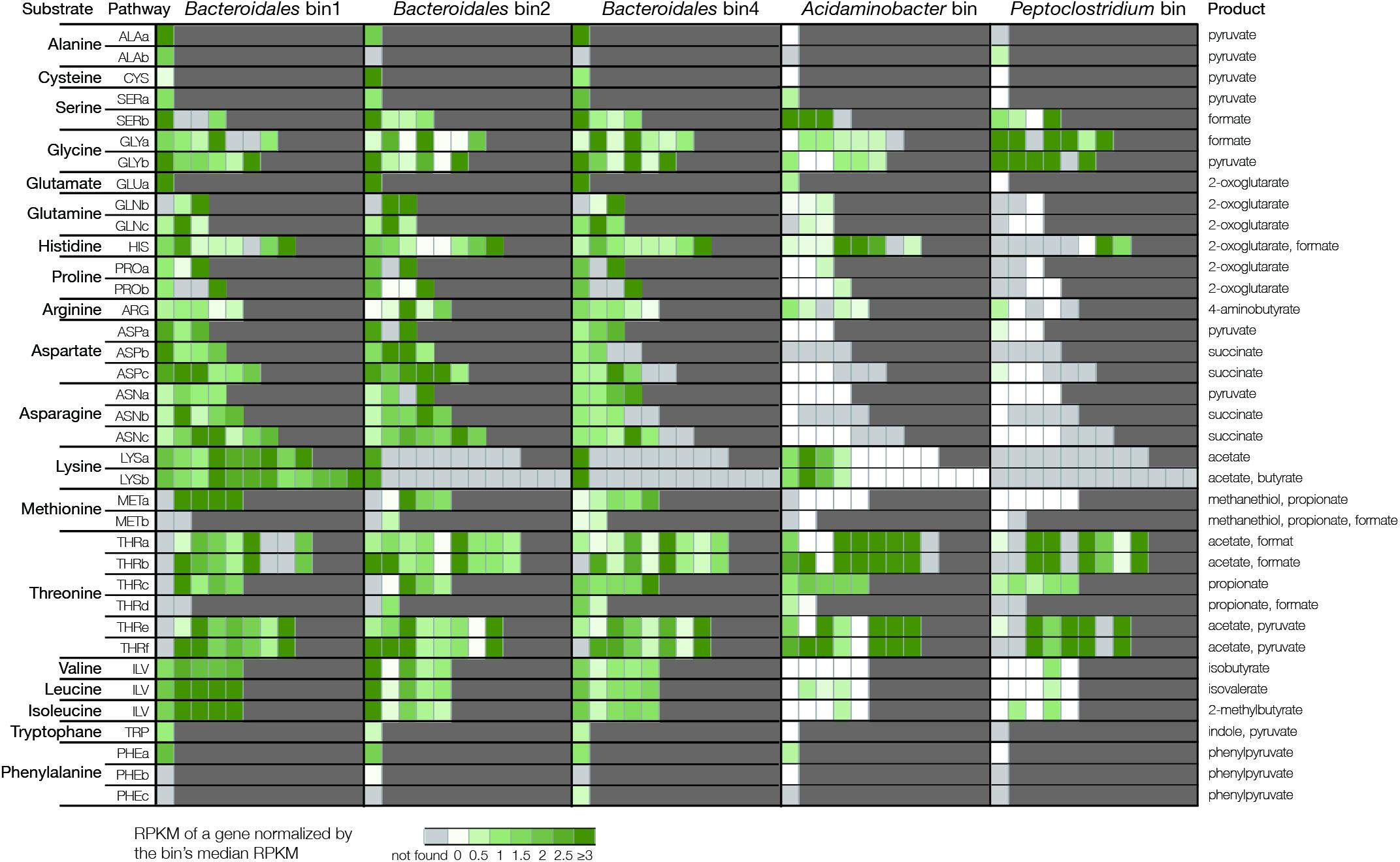
Figure 4. Gene expression of amino acids degradation pathways in the respective cultures. Each substrate can have multiple pathways (details of the pathways are listed in Supplementary Table 5). Genes in each pathway are represented by individual cells. Light gray color indicates the gene is not found in the bins. Color scale from white to green indicates the level of expression based on normalized RPKM value.
Electron Transfer by the Bacteroidales Species
Figure 3 further shows the mechanisms that the Bacteroidales populations used to transfer electrons and conserve energy. During AA metabolism, electrons are generated and carried by reducing equivalents such as reduced ferredoxin, NADH, and NADPH. The Bacteroidales bins possessed diverse approaches to reoxidize these electron carriers for sustainable fermentation and simultaneous energy conservation (Supplementary Table 7). In the cytoplasm, genes related to hydrogen production (Hyd) and formate production (Fdh) were consistently expressed, which allowed the Bacteroidales bins to use proton or CO2 as the sink for electron disposal. The production of hydrogen and formate was further supported by the consistent detection of hydrogen- and formate-utilizing methanogens including Methanolinea, Ca. Methanofastidiosa, and Methanoculleus in the enrichments. In terms of membrane-bound enzymes, the Bacteroidales bins contained Rnf-type ion-translocating NADH:ferredoxin oxidoreductase complex that can leverage the low-potential reduced ferredoxin to generate a sodium gradient for ATP synthesis. Similarly, the quinol:fumarate oxidoreductase can couple with the NADH:quinone oxidoreductase to translocate proton and sodium ion across the membrane, which can be further used for energy conservation. These electron transfer and energy conservation mechanisms suggested that the Bacteroidales populations had an AA-fermentation lifestyle in the enrichment cultures.
Micro-Heterogeneity Among Bacteroidales Species
Differences in AA metabolism were further observed between individual Bacteroidales bins. Bin2 and bin4 were more similar compared to bin1. Among the tested AAs, bin2 and bin4 shared most degradation pathways. Also, they both missed alanine transaminase in alanine degradation, glutamate synthase large chain in glutamine degradation, 1-pyrroline-5-carboxylate dehydrogenase in proline degradation, and eight genes in lysine degradation (Supplementary Table 5). In contrast, bin1 encoded the genes that were missing in bin2 and bin4, including alanine transaminase, 1-pyrroline-5-carboxylate dehydrogenase, and the entire lysine degradation pathway. On the other hand, bin1 missed genes that were detected in both bin2 and bin4, including methenyltetrahydrofolate cyclohydrolase for histidine degradation, formate C-acetyltransferase for methionine degradation, and the entire threonine degradation capacity due to the lack of L-threonine aldolase, threonine 3-dehydrogenase, and L-threonine ammonia-lyase. In addition, bin1 was observed in a sister cluster with bin2 and bin4 in the genome tree (Figure 2), implying heterogeneity at sub-family level within the clade vadinHA17. However, due to the lack of 16S rRNA gene in bin1, we were not able to accurately determine whether bin1 represent a different species or genus. The difference in phylogeny was consistent with other physiological differences. For example, bin1 expressed less glycoside hydrolases (21 genes) and lipases (3 genes) than bin2 (57 and 12 genes) and bin4 (41 and 12 genes) (Supplementary Figure 4). Bin1 also encoded one susC gene for α-glucan transportation, whereas bin2 and bin4 encoded susC genes specific for β-glucan transportation (Supplementary Figure 5). Overall, based on the phylogenetic affiliation and metabolic features of the Bacteroidales bins, we propose the provisional name “Candidatus Aminobacteroidaceae” fam. nov. for the lineage previous known as Bacteroidetes vadinHA17 and “Candidatus Aminobacteroides proteolyticus” gen. nov. sp. nov. for Bacteroidales bin4, the most abundant bin found in the enrichment culture.
Ecological Prevalence of Uncultured Bacteroidales
We further evaluated the environmental prevalence of the family Ca. Aminobacteroidaceae (i.e., Bacteroidetes vadinHA17) represented by the Bacteroidales bins recovered in this study. This family contained 333 full-length 16S rRNA gene sequences in the most recent release of SILVA NR database. The majority of sequences were obtained from sediments (150 sequences) and anaerobic bioreactors (140 sequences), where proteinaceous substrates are abundant. The abundance of Ca. Aminobacteroidaceae populations was further reported to reach over 15% of the microbial community in up-flow anaerobic sludge bed reactors (Chen et al., 2017; Wang et al., 2018), suggesting their important roles in anaerobic environments. Furthermore, we mapped metagenomic and metatranscriptomic sequences obtained from full-scale ADs and sediments to the Ca. Aminobacteroidaceae bins. Among 55 metagenomic datasets retrieved from full-scale ADs, the bins were consistently detected (Figure 5). Bin1 and bin4 were abundant, accounting for up to 3.5% of metagenomics sequences. Among 24 metatranscriptomic datasets retrieved from full-scale ADs, the maximum abundances of bin1 and bin4 were >25%. In comparison, the bins related to Acidaminobacter and Peptoclostridium only accounted for <0.01% of metagenomic or metatranscriptomic sequences. Overall, the abundance of Ca. Aminobacteroidaceae bins based on metagenomic and metatranscriptomic sequences suggested they were the dominant populations that performed proteolytic AA degradation in full-scale AD ecosystems. In contrast, among 26 metagenomic and 13 metatranscriptomic datasets retrieved from marine and lake sediments, the five Bacteroidales bins were not detected in most samples and the maximum abundance was < 0.03%. The results suggested that these Ca. Aminobacteroidaceae populations were unique to AD environments whereas the niche of protein and AA degradation in sediments could be occupied by other microorganisms.
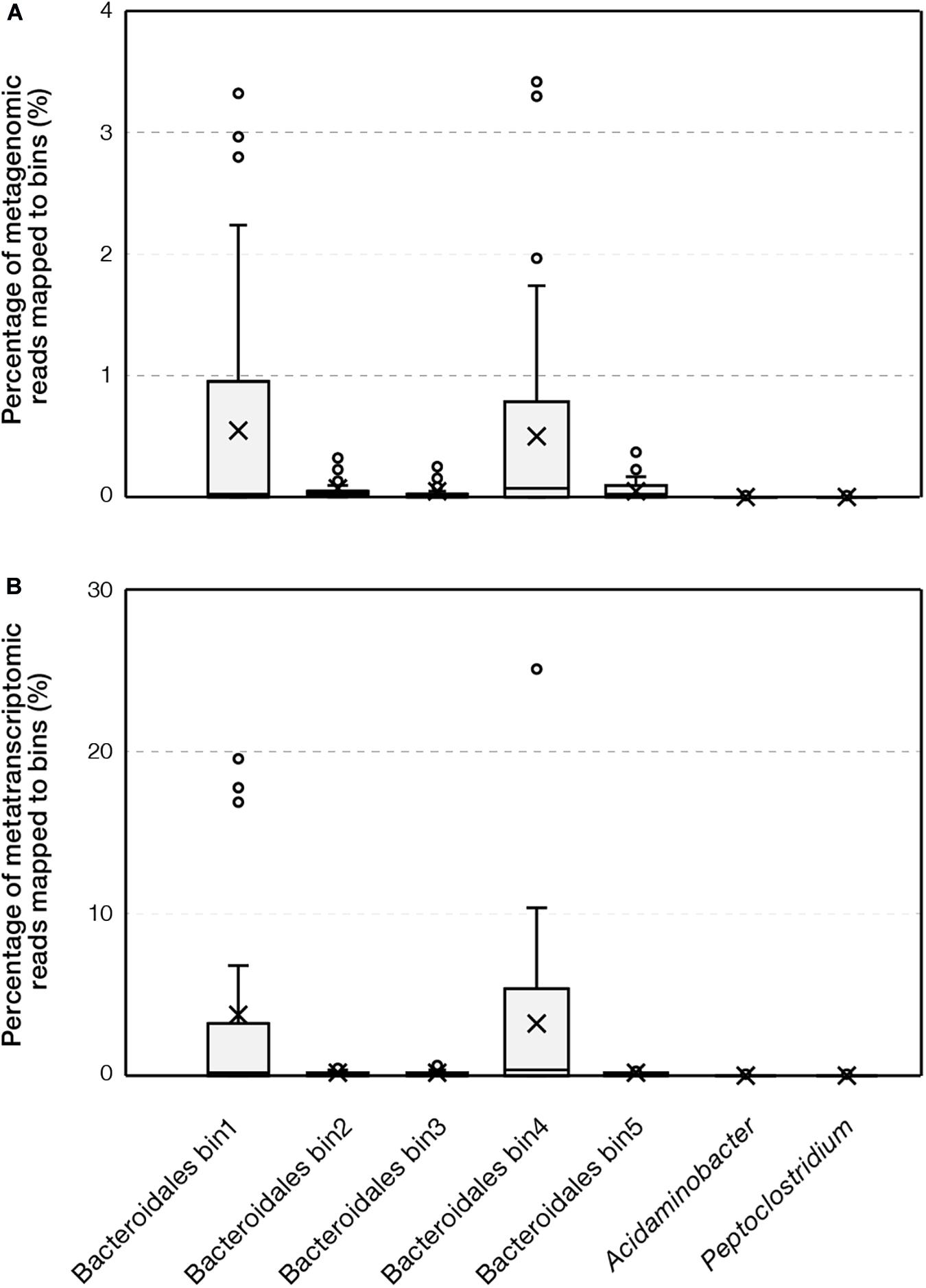
Figure 5. Percentage of reads that were mapped to bins based on (A) metagenomic and (B) metatranscriptomic datasets obtained from full-scale anaerobic digesters.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material.
Author Contributions
RM designed the study, conducted the experiments, analyzed the data, and wrote the manuscript. MN, TN, and W-TL designed the study, collaboratively interpreted the results, and revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The metagenomic and metatranscriptomic sequencing was supported by the Office of Science of the DOE under Contract No. DE-AC02-05CH1123. The staff at the U.S. Department of Energy (DOE) Joint Genome Institute are gratefully acknowledged for the high-throughput sequencing assists.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.593006/full#supplementary-material
Footnotes
References
Armenteros, J. J. A., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Baumgartner, S., Hofmann, K., Bucher, P., and Chiquet-Ehrismann, R. (1998). The discoidin domain family revisited: new members from prokaryotes and a homology-based fold prediction. Protein Sci. 7, 1626–1631. doi: 10.1002/pro.5560070717
Ben Hania, W., Joseph, M., Bunk, B., Spröer, C., Klenk, H.-P., Fardeau, M.-L., et al. (2017). Characterization of the first cultured representative of a Bacteroidetes clade specialized on the scavenging of cyanobacteria. Environ. Microbiol. 19, 1134–1148. doi: 10.1111/1462-2920.13639
Callebaut, I., Mornon, J.-P., Gilgès, D., and Vigon, I. (2000). HYR, an extracellular module involved in cellular adhesion and related to the immunoglobulin-like fold. Protein Sci. 9, 1382–1390. doi: 10.1110/ps.9.7.1382
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927. doi: 10.1093/bioinformatics/btz848
Chen, C., Liang, J., Yoza, B. A., Li, Q. X., Zhan, Y., and Wang, Q. (2017). Evaluation of an up-flow anaerobic sludge bed (UASB) reactor containing diatomite and maifanite for the improved treatment of petroleum wastewater. Bioresource Technol. 243, 620–627. doi: 10.1016/j.biortech.2017.06.171
Chu, B. T. T., Petrovich, M. L., Chaudhary, A., Wright, D., Murphy, B., Wells, G., et al. (2018). Metagenomics reveals the impact of wastewater treatment plants on the dispersal of microorganisms and genes in aquatic sediments. Appl. Environ. Microbiol. 84:e02168-17. doi: 10.1128/AEM.02168-17
Dick, L. K., Bernhard, A. E., Brodeur, T. J., Santo Domingo, J. W., Simpson, J. M., Walters, S. P., et al. (2005). Host distributions of uncultivated fecal Bacteroidales bacteria reveal genetic markers for fecal source identification. Appl. Environ. Microbiol. 71:3184. doi: 10.1128/AEM.71.6.3184-3191.2005
Dridi, B., Fardeau, M.-L., Ollivier, B., Raoult, D., and Drancourt, M. (2012). Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 62(Pt 8), 1902–1907. doi: 10.1099/ijs.0.033712-0
Dyksma, S., Bischof, K., Fuchs, B. M., Hoffmann, K., Meier, D., Meyerdierks, A., et al. (2016). Ubiquitous Gammaproteobacteria dominate dark carbon fixation in coastal sediments. ISME J. 10:1939. doi: 10.1038/ismej.2015.257
Feng, B., Cheng, A., and Wang, M. (2012). “Bioinformatics analysis of the complete nucleotide sequence of the Riemerella anatipestifer D15 gene,” in Proceedings of the 2012 5th International Conference on BioMedical Engineering and Informatics, Chongqing, 1171–1175. doi: 10.1109/BMEI.2012.6513130
Fernández-Gomez, B., Richter, M., Schüler, M., Pinhassi, J., Acinas, S. G., González, J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7:1026. doi: 10.1038/ismej.2012.169
Gujer, W., and Zehnder, A. J. B. (1983). Conversion processes in anaerobic digestion. Water Sci. Technol. 15, 127–167. doi: 10.2166/wst.1983.0164
Gupta, R. S. (2004). The phylogeny and signature sequences characteristics of Fibrobacteres, Chlorobi, and Bacteroidetes. Crit. Rev. Microbiol. 30, 123–143. doi: 10.1080/10408410490435133
Huber, O., and Sumper, M. (1994). Algal-CAMs: isoforms of a cell adhesion molecule in embryos of the alga Volvox with homology to Drosophila fasciclin I. EMBO J. 13, 4212–4222. doi: 10.1002/j.1460-2075.1994.tb06741.x
Kamagata, Y. (2015). Keys to cultivating uncultured microbes: elaborate enrichment strategies and resuscitation of dormant cells. Microbes Environ. 30:289. doi: 10.1264/jsme2.ME3004rh
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Kim, N.-K., Oh, S., and Liu, W.-T. (2016). Enrichment and characterization of microbial consortia degrading soluble microbial products discharged from anaerobic methanogenic bioreactors. Water Res. 90, 395–404. doi: 10.1016/j.watres.2015.12.021
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187:6258. doi: 10.1128/JB.187.18.6258-6264.2005
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lagkouvardos, I., Lesker, T. R., Hitch, T. C. A., Gálvez, E. J. C., Smit, N., Neuhaus, K., et al. (2019). Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 7:28. doi: 10.1186/s40168-019-0637-2
Ley, R. E., Harris, J. K., Wilcox, J., Spear, J. R., Miller, S. R., Bebout, B. M., et al. (2006). Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl. Environ. Microbiol. 72, 3685–3695. doi: 10.1128/AEM.72.5.3685-3695.2006
Li, H. (2015). BFC: correcting Illumina sequencing errors. Bioinformatics 31, 2885–2887. doi: 10.1093/bioinformatics/btv290
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Buchner, A., et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
McInerney, M. J. (1988). “Anaerobic hydrolysis of fats and protein,” in Biol Anaerobic Microorganisms. New York: John Wiley & Sons, Inc, 373–415.
Mei, R., Narihiro, T., Nobu, M. K., Kuroda, K., and Liu, W. T. (2016). Evaluating digestion efficiency in full-scale anaerobic digesters by identifying active microbial populations through the lens of microbial activity. Sci. Rep. 6:34090. doi: 10.1038/srep34090
Mei, R., Nobu, M. K., and Liu, W.-T. (2020). Identifying anaerobic amino acids degraders through the comparison of short-term and long-term enrichments. Environ. Microbiol. Rep. 12, 173–184. doi: 10.1111/1758-2229.12821
Mei, R., Nobu, M. K., Narihiro, T., Yu, J., Sathyagal, A., Willman, E., et al. (2018). Novel Geobacter species and diverse methanogens contribute to enhanced methane production in media-added methanogenic reactors. Water Res. 147, 403–412. doi: 10.1016/j.watres.2018.10.026
Nagano, K., Murakami, Y., Nishikawa, K., Sakakibara, J., Shimozato, K., and Yoshimura, F. (2007). Characterization of RagA and RagB in Porphyromonas gingivalis: study using gene-deletion mutants. J. Med. Microbiol. 56, 1536–1548. doi: 10.1099/jmm.0.47289-0
Narihiro, T., Nobu, M. K., Bocher, B. T. W., Mei, R., and Liu, W. T. (2018). Co-occurrence network analysis reveals thermodynamics-driven microbial interactions in methanogenic bioreactors. Environ. Microbiol. Rep. 10, 673–685. doi: 10.1111/1758-2229.12689
Narihiro, T., Nobu, M. K., Kim, N. K., Kamagata, Y., and Liu, W. T. (2015). The nexus of syntrophy-associated microbiota in anaerobic digestion revealed by long-term enrichment and community survey. Environ. Microbiol. 17, 1707–1720. doi: 10.1111/1462-2920.12616
Nguyen, T. T. H., Myrold, D. D., and Mueller, R. S. (2019). Distributions of extracellular peptidases across prokaryotic genomes reflect phylogeny and habitat. Front. Microbiol. 10:413. doi: 10.3389/fmicb.2019.00413
Nobu, M. K., Narihiro, T., Kuroda, K., Mei, R., and Liu, W. T. (2016). Chasing the elusive Euryarchaeota class WSA2: genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J. 10, 2478–2487. doi: 10.1038/ismej.2016.33
Nobu, M. K., Narihiro, T., Mei, R., Kamagata, Y., Lee, P. K. H., Lee, P.-H., et al. (2020). Catabolism and interactions of uncultured organisms shaped by eco-thermodynamics in methanogenic bioprocesses. Microbiome 8:111. doi: 10.1186/s40168-020-00885-y
Nobu, M. K., Narihiro, T., Rinke, C., Kamagata, Y., Tringe, S. G., Woyke, T., et al. (2015). Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor. ISME J. 9, 1710–1722. doi: 10.1038/ismej.2014.256
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Ormerod, K. L., Wood, D. L. A., Lachner, N., Gellatly, S. L., Daly, J. N., Parsons, J. D., et al. (2016). Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 4:36. doi: 10.1186/s40168-016-0181-2
Orsi, W. D., Richards, T. A., and Francis, W. R. (2018). Predicted microbial secretomes and their target substrates in marine sediment. Nat. Microbiol. 3:32. doi: 10.1038/s41564-017-0047-9
Ozer, E. A., Allen, J. P., and Hauser, A. R. (2014). Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics 15:737. doi: 10.1186/1471-2164-15-737
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P.-A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2:1533. doi: 10.1038/s41564-017-0012-7
Pikuta, E. V., Lyu, Z., Hoover, R. B., Liu, Y., Patel, N. B., Busse, H. J., et al. (2017). Williamwhitmania taraxaci gen. nov., sp. nov., a proteolytic anaerobe with a novel type of cytology from Lake Untersee in Antarctica, description of Williamwhitmaniaceae fam. nov., and emendation of the order Bacteroidales Krieg 2012. Int. J. Syst. Evol. Microbiol. 67, 4132–4145. doi: 10.1099/ijsem.0.002266
Ramayo-Caldas, Y., Mach, N., Lepage, P., Levenez, F., Denis, C., Lemonnier, G., et al. (2016). Phylogenetic network analysis applied to pig gut microbiota identifies an ecosystem structure linked with growth traits. ISME J. 10, 2973–2977. doi: 10.1038/ismej.2016.77
Rawlings, N. D., Barrett, A. J., Thomas, P. D., Huang, X., Bateman, A., and Finn, R. D. (2017). The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632. doi: 10.1093/nar/gkx1134
Reeves, A. R., Wang, G. R., and Salyers, A. A. (1997). Characterization of four outer membrane proteins that play a role in utilization of starch by Bacteroides thetaiotaomicron. J. Bacteriol. 179:643. doi: 10.1128/JB.179.3.643-649.1997
Reichardt, N., Vollmer, M., Holtrop, G., Farquharson, F. M., Wefers, D., Bunzel, M., et al. (2017). Specific substrate-driven changes in human faecal microbiota composition contrast with functional redundancy in short-chain fatty acid production. ISME J. 12, 610–622. doi: 10.1038/ismej.2017.196
Rosenberg, E., DeLong, E. F., Lory, S., Stackebrandt, E., and Thompson, F. (2014). The Prokaryotes. Berlin: Springer. doi: 10.1007/978-3-642-39044-9
Saier, M. H. Jr., Reddy, V. S., Tsu, B. V., Ahmed, M. S., Li, C., and Moreno-Hagelsieb, G. (2015). The transporter classification database (TCDB): recent advances. Nucleic Acids Res. 44, D372–D379. doi: 10.1093/nar/gkv1103
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Segata, N., Börnigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4:2304. doi: 10.1038/ncomms3304
Søndergaard, D., Pedersen, C. N. S., and Greening, C. (2016). HydDB: a web tool for hydrogenase classification and analysis. Sci. Rep. 6:34212. doi: 10.1038/srep34212
Song, W.-Z., and Thomas, T. (2017). Binning_refiner: improving genome bins through the combination of different binning programs. Bioinformatics 33, 1873–1875. doi: 10.1093/bioinformatics/btx086
Stams, A., and Hansen, T. (1984). Fermentation of glutamate and other compounds by Acidaminobacter hydrogenoformans gen. nov. sp. nov., an obligate anaerobe isolated from black mud. Studies with pure cultures and mixed cultures with sulfate-reducing and methanogenic bacteria. Arch. Microbiol. 137, 329–337. doi: 10.1007/BF00410730
Su, X.-L., Tian, Q., Zhang, J., Yuan, X.-Z., Shi, X.-S., Guo, R.-B., et al. (2014). Acetobacteroides hydrogenigenes gen. nov., sp. nov., an anaerobic hydrogen-producing bacterium in the family Rikenellaceae isolated from a reed swamp. Int. J. Syst. Evol. Microbiol. 64, 2986–2991. doi: 10.1099/ijs.0.063917-0
Tang, Y., Shigematsu, T., Morimura, S., and Kida, K. (2005). Microbial community analysis of mesophilic anaerobic protein degradation process using bovine serum albumin (BSA)-fed continuous cultivation. J. Biosci. Bioeng. 99, 150–164. doi: 10.1263/jbb.99.150
Tomazetto, G., Hahnke, S., Wibberg, D., Pühler, A., Klocke, M., and Schlüter, A. (2018). Proteiniphilum saccharofermentans str. M3/6T isolated from a laboratory biogas reactor is versatile in polysaccharide and oligopeptide utilization as deduced from genome-based metabolic reconstructions. Biotechnol. Rep. 18:e00254. doi: 10.1016/j.btre.2018.e00254
Urich, T., Lanzén, A., Stokke, R., Pedersen, R. B., Bayer, C., Thorseth, I. H., et al. (2014). Microbial community structure and functioning in marine sediments associated with diffuse hydrothermal venting assessed by integrated meta−omics. Environ. Microbiol. 16, 2699–2710. doi: 10.1111/1462-2920.12283
Vanwonterghem, I., Jensen, P. D., Rabaey, K., and Tyson, G. W. (2016). Genome−centric resolution of microbial diversity, metabolism and interactions in anaerobic digestion. Environ. Microbiol. 18, 3144–3158. doi: 10.1111/1462-2920.13382
Voorhies, A. A., Eisenlord, S. D., Marcus, D. N., Duhaime, M. B., Biddanda, B. A., Cavalcoli, J. D., et al. (2016). Ecological and genetic interactions between cyanobacteria and viruses in a low−oxygen mat community inferred through metagenomics and metatranscriptomics. Environ. Microbiol. 18, 358–371. doi: 10.1111/1462-2920.12756
Wang, Q., Liang, J., Zhan, Y., Yao, X., Liu, Z., Li, Q. X., et al. (2018). Treatment of petroleum wastewater using an up-flow anaerobic sludge blanket (UASB) reactor and turf soil as a support material. J. Chem. Technol. Biotechnol. 93, 3317–3325. doi: 10.1002/jctb.5694
Widmann, M., Juhl, P. B., and Pleiss, J. (2010). Structural classification by the Lipase Engineering database: a case study of Candida antarctica lipase A. BMC Genomics 11:123.
Woodcroft, B. J., Singleton, C. M., Boyd, J. A., Evans, P. N., Emerson, J. B., Zayed, A. A. F., et al. (2018). Genome-centric view of carbon processing in thawing permafrost. Nature 560, 49–54.
Wu, Y.-W., Simmons, B. A., and Singer, S. W. (2015). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607.
Yang, G., Zhang, P., Zhang, G., Wang, Y., and Yang, A. (2015). Degradation properties of protein and carbohydrate during sludge anaerobic digestion. Bioresource Technol. 192, 126–130. doi: 10.1016/j.biortech.2015.05.076
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K.-H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nature Rev. Microbiol. 12, 635–645.
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2013). The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Zealand, A. M., Mei, R., Papachristodoulou, P., Roskilly, A. P., Liu, W. T., and Graham, D. W. (2018). Microbial community composition and diversity in rice straw digestion bioreactors with and without dairy manure. Appl. Microbiol. Biotechnol. 102, 8599–8612.
Zhang, H., Yohe, T., Huang, L., Entwistle, S., Wu, P., Yang, Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101.
Keywords: metagenomics, uncultured, proteolytic, amino acid, anaerobic digester
Citation: Mei R, Nobu MK, Narihiro T and Liu W-T (2020) Metagenomic and Metatranscriptomic Analyses Revealed Uncultured Bacteroidales Populations as the Dominant Proteolytic Amino Acid Degraders in Anaerobic Digesters. Front. Microbiol. 11:593006. doi: 10.3389/fmicb.2020.593006
Received: 09 August 2020; Accepted: 13 October 2020;
Published: 30 October 2020.
Edited by:
Leonardo Erijman, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Seung Gu Shin, Pohang University of Science and Technology, South KoreaJohn Michael Regan, Pennsylvania State University (PSU), United States
Copyright © 2020 Mei, Nobu, Narihiro and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Tso Liu, d3RsaXVAaWxsaW5vaXMuZWR1