 Soon-Hwan Oh1
Soon-Hwan Oh1 Allyson Isenhower2
Allyson Isenhower2 Rubi Rodriguez-Bobadilla2
Rubi Rodriguez-Bobadilla2 Brooke Smith2
Brooke Smith2 Jillian Jones2
Jillian Jones2 Vit Hubka3,4
Vit Hubka3,4 Christopher Fields5
Christopher Fields5 Alvaro Hernandez5
Alvaro Hernandez5 Lois L. Hoyer1*
Lois L. Hoyer1*- 1Department of Pathobiology, College of Veterinary Medicine, University of Illinois at Urbana-Champaign, Urbana, IL, United States
- 2Department of Biology, Millikin University, Decatur, IL, United States
- 3Department of Botany, Faculty of Science, Charles University, Prague, Czechia
- 4Laboratory of Fungal Genetics and Metabolism, Institute of Microbiology, Czech Academy of Sciences, Prague, Czechia
- 5Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign, Urbana, IL, United States
The agglutinin-like sequence (ALS) gene family encodes cell-surface adhesins that interact with host and abiotic surfaces, promoting colonization by opportunistic fungal pathogens such as Candida tropicalis. Studies of Als protein contribution to C. tropicalis adhesion would benefit from an accurate catalog of ALS gene sequences as well as insight into relative gene expression levels. Even in the genomics era, this information has been elusive: genome assemblies are often broken within ALS genes because of their extensive regions of highly conserved, repeated DNA sequences and because there are many similar ALS genes at different chromosomal locations. Here, we describe the benefit of long-read DNA sequencing technology to facilitate characterization of C. tropicalis ALS loci. Thirteen ALS loci in C. tropicalis strain MYA-3404 were deduced from a genome assembly constructed from Illumina MiSeq and Oxford Nanopore MinION data. Although the MinION data were valuable, PCR amplification and Sanger sequencing of ALS loci were still required to complete and verify the gene sequences. Each predicted Als protein featured an N-terminal binding domain, a central domain of tandemly repeated sequences, and a C-terminal domain rich in Ser and Thr. The presence of a secretory signal peptide and consensus sequence for addition of a glycosylphosphatidylinositol (GPI) anchor was consistent with predicted protein localization to the cell surface. TaqMan assays were designed to recognize each ALS gene, as well as both alleles at the divergent CtrALS3882 locus. C. tropicalis cells grown in five different in vitro conditions showed differential expression of various ALS genes. To place the C. tropicalis data into a larger context, TaqMan assays were also designed and validated for analysis of ALS gene expression in Candida albicans and Candida dubliniensis. These comparisons identified the subset of highly expressed C. tropicalis ALS genes that were predicted to encode proteins with the most abundant cell-surface presence, prioritizing them for subsequent functional analysis. Data presented here provide a solid foundation for future experimentation to deduce ALS family contributions to C. tropicalis adhesion and pathogenesis.
Introduction
Adhesion and subsequent colonization provide the opportunity for microbial pathogens to cause disease. Identification of adhesin-encoding genes is facilitated by the availability of genome sequences. An accurate catalog of potential adhesins focuses efforts to characterize protein function and develop therapeutic approaches that could be effective against multiple microbial species. Anti-adhesion therapies have been hailed as superior to traditional antimicrobials because their mechanism of action does not promote antimicrobial resistance (Krachler and Orth, 2013). This report describes characterization of genes encoding the agglutinin-like sequence (Als) family of adhesins in the fungal pathogen Candida tropicalis. The work highlights how emerging advances in DNA sequencing technology were used to fill key knowledge gaps that have persisted for many years.
Agglutinin-like sequence (ALS) genes encode cell-surface adhesins that recognize a broad variety of peptide ligands (Lin et al., 2014). The gene family was characterized initially in Candida albicans with investigations into protein structure and activity also focused on this species (reviewed in Hoyer and Cota, 2016). Starting in the pre-genome era and extending over a period of approximately 20 years, projects moved from identification of the first ALS gene, to the recognition that the gene was part of a larger family, to characterization of the family, and the relative abundance of specific Als proteins on the cell surface under a variety of growth conditions. More recently, the structure of the N-terminal Als protein adhesive domain was solved (NT-Als; reviewed in Hoyer and Cota, 2016). Experimental progress to define the C. albicans ALS family was slow because detection of additional ALS genes relied on techniques such as the Southern blotting of genomic DNA (Hoyer et al., 1995, 1998a,b). Locating ALS genes in draft genome sequences would accelerate progress substantially.
As emerging genome sequencing technologies were applied to pathogenic fungal species, the C. albicans ALS sequences were used to recognize potential ALS genes in draft genomes (Butler et al., 2009). ALS gene hallmarks such as the 5′ end sequences that encode the adhesive domain, central regions of a conserved 108-bp tandemly repeated sequence, and the 3′ end sequences that include a glycosylphosphatidylinositol (GPI) anchor addition site were clues for identifying possible ALS loci. While these techniques could quickly identify potential ALS genes, the large ALS gene size and considerable sequence identity across multiple physical loci in the same species complicated accurate assembly. Computational assemblies broke within the conserved tandemly repeated sequences in the middle of the coding region, or mistakenly placed the 5′ end of one gene onto the 3′ end of another. The research community was left without accurate knowledge of the ALS gene number or sequence in common fungal pathogens.
Like C. albicans, C. tropicalis causes superficial mucosal infections, as well as disseminated and deep-seated infections; the rising incidence of azole-resistant C. tropicalis is causing clinical concern (reviewed in de Oliveira et al., 2020). Previous reports about C. tropicalis ALS genes suggested that the family is larger in this species than in other common fungal pathogens (Butler et al., 2009; Jackson et al., 2009), representing perhaps the most challenging ALS family puzzle to solve. Short-read sequence technologies were not helpful in this regard because the read lengths often did not include unique information that could place ALS sequences in the correct physical location.
One key technological development that appeared at the start of this project was the ability to derive long-read DNA sequences. Here, we describe combining short-read (Illumina MiSeq) and long-read (Oxford Nanopore MinION) datasets to develop a C. tropicalis genome assembly that served as the foundation for characterization of the 13 ALS loci in strain MYA-3404. Accurate gene sequences facilitated design of a specific TaqMan assay for each ALS gene. The assays were used to assess relative gene expression under a variety of growth conditions to reveal which ALS genes were likely to produce the most abundant Als cell-surface proteins, thereby prioritizing them for further functional analysis toward the development of anti-adhesion therapies.
Materials and Methods
Fungal Strains
Table 1 shows the fungal strains used in this study. C. tropicalis strains were authenticated using several methods (data not shown). First, each isolate was streaked on CHROMagar Candida medium (Becton Dickinson) to ensure that it produced the expected blue colony color. Molecular verification of strain identification used PCR primers ITS4 (5′ TCCTCCGCTTATTGATATGC 3′) and ITS5 (5′ GGAAGTAAAAGTCGTAACAAGG 3′; White et al., 1990). PCR products were Sanger sequenced at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. DNA sequences were confirmed as C. tropicalis by comparison to the non-redundant nucleotide database at the National Center for Biotechnology Information using the blastn algorithm1. Karyotypes for the C. tropicalis strains were also investigated using contour-clamped homogeneous electrical field (CHEF) gels and found to be similar across the isolates; methods for the karyotype analysis and examples of the results were published previously (Hoyer et al., 2001).
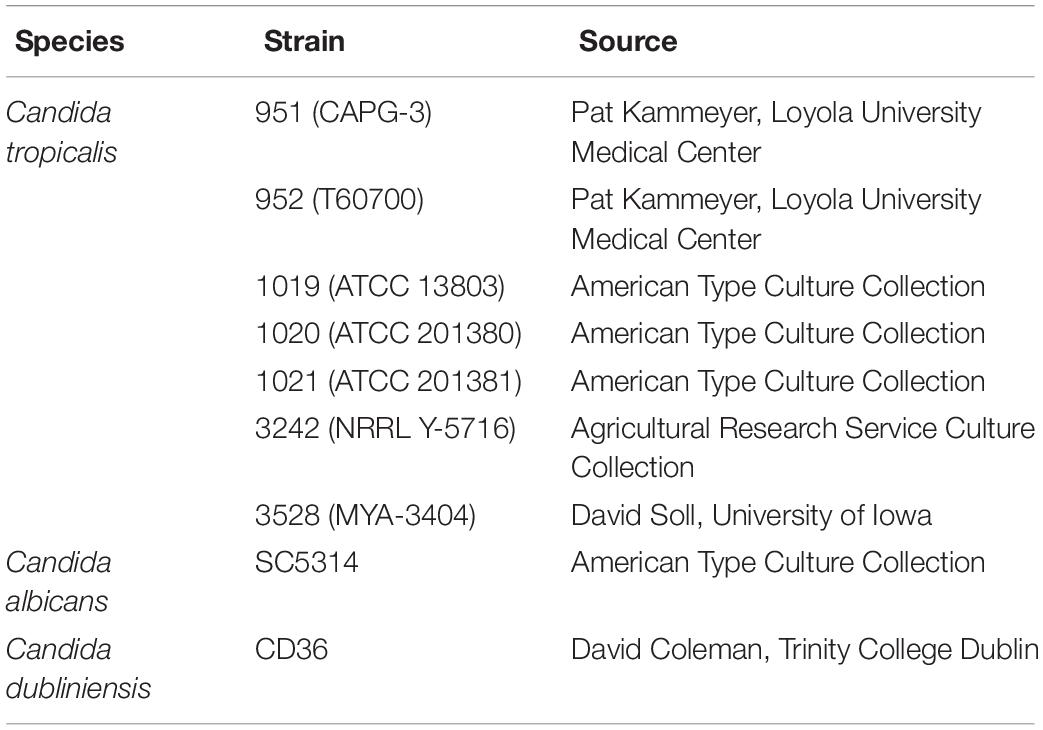
Table 1. Fungal strains and sources.
C. tropicalis Genome Sequences
A previously published C. tropicalis genome sequence was used as the basis for initial identification of putative ALS genes (strain MYA-3404; ASM633v3; GCA_000006335.3; Butler et al., 2009). A new genome sequence was generated for C. tropicalis strain MYA-3404. Methods for this effort were similar to the methods for generating a genomic sequence for Candida metapsilosis (Oh et al., 2019) and were reproduced here for the reader’s convenience.
Cells were grown in YPD (per liter: 10 g yeast extract, 20 g Bacto peptone, 20 g dextrose) to saturation (approximately 16 h at 37°C and 200 r/min shaking). Genomic DNA was isolated according to Sherman et al. (1986). The method involved zymolyase spheroplasting of cells, sodium dodecyl sulfate lysis, phenol extraction, DNA precipitation with isoproposal, and Proteinase K treatment of the final preparation. Gentle mixing and pipetting with wide-bore tips were used to minimize DNA shearing. High-molecular-weight DNA was visualized by agarose gel electrophoresis and ethidium-bromide staining prior to further processing.
Strain MYA-3404 libraries were constructed and sequenced at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. Data were derived using Illumina (short-read) and Oxford Nanopore (long-read) methods. MiSeq shotgun libraries were prepared with the Hyper Library construction kit (Kapa Biosystems). The library was quantitated by qPCR and sequenced on one MiSeq flowcell for 151 cycles from each end of the fragment using a MiSeq 300-cycle sequencing kit (version 2). FASTQ files were generated and demultiplexed with the bclfastq Conversion Software (Illumina, version 2.17.1.14). MiSeq reads were quality trimmed using Trimmomatic (Bolger et al., 2014) with the parameters “LEADING:30 TRAILING:30” prior to assembly.
For Oxford Nanopore long-read sequencing, 1 μg of genomic DNA was sheared in a gTube (Covaris, Woburn, MA, United States) for 1 min at 6,000 r/min in a MiniSpin plus microfuge (Eppendorf, Hauppauge, NY, United States). The sheared DNA was converted to a shotgun library with the LSK-108 kit from Oxford Nanopore, following the manufacturer’s instructions. The library was sequenced on a SpotON R9.4 flowcell for 48 h using a MinION MK 1B sequencer.
Basecalling and demultiplexing were performed in real time with the Metrichor Agent V2.45.3 using the 1D BaseCalling plus Barcoding for FLO-MIN_106_450bp workflow. Removed from both ends of each Oxford Nanopore read were 60 nt, followed by additional trimming using a Github checkout (commit 92c0b65f) of Porechop (Wick et al., 2017) to remove reads with potential internal barcodes which were likely chimeric. Only reads longer than 800 nt were used in the final assembly. Canu v1.4 (Koren et al., 2017) was used for assembly with the following parameters: “canu -p asm -d C_trop_default genomeSize = 14m useGrid = false -nanopore-raw c_tropicalis.qualtrim.clean.fastq.gz.”
Oxford Nanopore reads were then aligned against the assembly using bwa mem (Li, 2018) with parameters “bwa mem -x ont2d C_tropicalisCanuAsm.fasta reads.fa”, and the alignment was then used to polish the assembly using nanopolish v 0.6.0 (Senol Cali et al., 2018). Quality-trimmed MiSeq data were used to polish the assembly using Pilon v1.21 for error correction (Walker et al., 2014). Supplementary File S1 includes details regarding the computational analyses and characteristics of the resulting C. tropicalis MYA-3404 genome sequence. The sequence was deposited in the NCBI database (ASM694213v1; GCA_006942135.1).
Ambiguities in the genome sequence data were resolved by PCR amplification of the region and Sanger sequencing of the product. Primer design was aided by the PrimerQuest Tool (Integrated DNA Technologies). Supplementary Table S1 lists PCR primer sequences that were used for amplification and/or Sanger sequencing for the various ALS loci.
During finalization of this manuscript, a new C. tropicalis MYA-3404 genome sequence was noted in the NCBI database (ASM1317755v1; GCA_013177555.1; Guin et al., 2020). The sequence was generated with a combination Illumina HiSeq and PacBio Sequel technology and had the same number of contigs as C. tropicalis has chromosomes. This sequence was used for comparative analysis of ALS sequences derived from ASM694213v1.
Identification of ALS Genes and Predicted Als Protein Features
Methods for identifying C. tropicalis ALS genes and deducing predicted protein features were identical to those reported for analysis of C. metapsilosis (Oh et al., 2019). Details were reproduced here for the reader’s convenience. BLAST2 was used to identify potential ALS genes and Als proteins in the genome sequences. Query sequences included all C. albicans ALS genes as reported by Oh et al. (2019). As new ALS/Als sequences were identified, they were also used as BLAST queries until search reports failed to reveal new sequences. SignalP Server3 (Nielsen, 2017) was used to locate putative secretory signal peptides. The big-PI Predictor4 (Eisenhaber et al., 1999) identified potential GPI anchor addition sites. European Bioinformatics Institute (EMBL-EBI) tools were used for translating nucleotide sequences, sequence alignment, and other general processes5 (Cook et al., 2017).
Phylogenetics Analysis
Phylogeny of the ALS family was estimated using sequences from the 5′ domain that is present in each gene (see Supplementary File S2). Because of the large sequence divergences within the 5′ domain, nucleotide sequences were translated using the alternative yeast nuclear code and the resulting amino acid sequences aligned with PROMALS3D (Pei et al., 2008). Poorly aligned regions were eliminated using Gblocks v 0.91b with default settings (Castresana, 2000). There were 227 positions in the final alignment. Model selection was performed using ModelFinder (Kalyaanamoorthy et al., 2017) implemented in IQ-TREE (Nguyen et al., 2015); LG+I+G4 was chosen as a best-fit model according to the Bayesian information criterion. The maximum likelihood tree was constructed with IQ-TREE v. 1.6.12 with nodal support determined by non-parametric bootstrapping with 500 replicates. Bayesian posterior probabilities were calculated using MrBayes 3.2.6 (Ronquist et al., 2012). The analysis ran for 3 × 106 generations. Two parallel runs were used with four chains each, sample frequency of 100 generations, and 25% burn-in.
Fungal Growth Conditions for Gene Expression Analysis
C. tropicalis, C. albicans, and Candida dubliniensis isolates were grown using the same methods except where noted below. All fungal isolates were stored as 30% glycerol stocks at −80°C and streaked to a YPD plate prior to use in an experiment. YPD stock plates were incubated for approximately 24 h at 37°C and then kept at 4°C for no more than 1 week. A starter culture was prepared by inoculating one representative colony from the YPD plate into 20 ml of liquid YPD in a 50-ml flask. The flask was incubated at 30°C and 200 r/min shaking for 16 h. All cells were collected by centrifugation and washed twice in sterile MilliQ water. Cell number was calculated using a hemacytometer. A small portion of the 16 h culture was flash frozen in dry ice/ethanol and duplicate samples stored at −80°C for RNA extraction to measure relative gene expression in a saturated culture.
Growth conditions were selected from previous analyses of ALS gene expression and Als protein production in cultured C. albicans cells. Growth in RPMI 1640 medium (Gibco; 11875-135) induces C. albicans germ tube formation and its associated differential Als protein production (Coleman et al., 2009). For growth in RPMI 1640, washed cells from the starter culture were used to inoculate 20 ml of the medium at a density of 5 × 107 cells/ml in a 50-ml flask. Flasks were incubated at 37°C and 200 r/min shaking for 1 h. The culture was divided into two equal portions, and cells were collected by filtration over a sterile 0.45-μm pore-size membrane (GVS Life Sciences; 1213776). Filters were flash frozen in dry ice/ethanol and stored at −80°C.
Analysis of ALS gene expression in cells from an early-growth-stage culture used YPD (rich) medium. Two identical 250-ml flasks were filled with 100 ml of YPD and inoculated at a cell density of 1 × 106 per ml. Flasks were incubated for 1 h at either 37°C (C. tropicalis) or 30°C (C. albicans and C. dubliniensis) and 200 r/min shaking. Cells were collected by filtration as detailed above and filters flash frozen and stored at −80°C for RNA extraction.
Additional growth conditions for C. tropicalis were intended to examine ALS gene expression during morphological change. Lackey et al. (2013) described growth in synthetic defined medium [SD; 6.7 g/l yeast nitrogen base without amino acids (BD Biosciences)] supplemented with 50% fetal bovine serum (FBS) for this purpose. Early (2 h) and late (24 h) time points were evaluated to assess differential gene expression over the course of a growth curve. Washed cells from the 16-h YPD starter culture were resuspended in 15 ml of SD medium at an OD600 of approximately 1.7. Ten milliliters of the SD culture were added to 10 ml of 100% FBS and incubated at 30°C and 200 r/min shaking for 2 h. The culture was divided in half, cells collected by centrifugation, flash frozen in dry ice/ethanol, and stored at −80°C. The remaining 5 ml of the 15 ml SD starter culture was combined with 5 ml of 100% FBS and incubated for 24 h at 30°C and 200 r/min shaking. Cells were collected and stored as described above.
Cultures were prepared on three different days. Each experimental day had daily replication by creating two unique cultures from two different colonies on the original agar plate.
TaqMan Assays for Analysis of ALS Gene Expression
TaqMan assays were designed to specifically detect each of the C. tropicalis ALS genes, as well as to differentiate between alleles of CtrALS3882. To place C. tropicalis gene expression data into a larger context, TaqMan assays were also designed and validated for each ALS gene in C. albicans. TaqMan assays were also designed for C. dubliniensis ALS genes because the literature lacked information about ALS gene expression in this species. Methods for designing and validating TaqMan assays were detailed by Oh et al. (2019). Although an amplicon size of 140 bp was targeted, some assays required a shorter or longer product to ensure specificity of detection. Amplicon sizes ranged from 106 to 213 bp; PCR efficiencies ranged from 95 to 102% (Supplementary Table S2).
TaqMan assay specificity was validated carefully using cloned control gene fragments (DNA templates; Supplementary Table S3). Detailed methods and examples of acceptable results were described previously (Oh et al., 2019). TaqMan control reactions targeted ACT1 and TEF1 using primer/probe sets capable of recognizing all species in the study (Supplementary Table S2).
Methods for RNA extraction, genomic DNA removal, cDNA synthesis, and TaqMan assays were described previously (Oh et al., 2019). Experiments in this report were completed entirely on a QuantStudio 3 Real-Time PCR System. Statistical significance was assessed using a mixed-model analysis of variance (PROC MIXED in SAS 9.4; SAS Institute Inc., Cary, NC, United States). LSMEANS was used for separation of means.
Results
Generation of a Novel Genome Sequence for C. tropicalis MYA-3404
The major goal of this work was to define the ALS gene family in C. tropicalis. At the time the project began, the best genome assembly available was accession ASM633v3, initially deposited in 2005. The dataset had 128 contigs, assembled into 24 scaffolds that, in places, were spliced together by strings of “NNN” to indicate sequence ambiguity. Putative ALS loci were located using BLAST. The fragmented loci were documented in Supplementary Figure S1 and the predicted partial proteins in Supplementary Figure S2.
Long-read sequencing using the Oxford Nanopore MinION technology was just emerging at the start of this project in 2017. The potential for long-read sequencing to span sometimes-lengthy repeated regions within ALS open reading frames led to the development of a new genome assembly that used Illumina MiSeq and Oxford Nanopore MinION data (ASM694213v1; GCA_006942135.1). The accurate Illumina data were included to correct the MinION data, which were already recognized as error prone (de Lannoy et al., 2017). The assembly included 29 contigs and no Ns (Supplementary File S1). Although the long-read data provided an improved assembly compared to ASM633v3, completion of the ALS loci still required additional considerable effort.
The overall strategy for accurate assembly of the C. tropicalis ALS family involved identifying unique genomic landmarks to anchor specific ALS loci in their proper physical location. PCR primers were designed to amplify regions that required improvement. Sanger sequencing was used to generate the final data. Figures 1, 2 display the assembled ALS loci and predicted proteins, respectively. Table 2 lists the assembled ALS genes and places them into the context of the original MYA-3404 genome assembly (ASM633v3) as well as previous publications that mentioned the C. tropicalis ALS family.
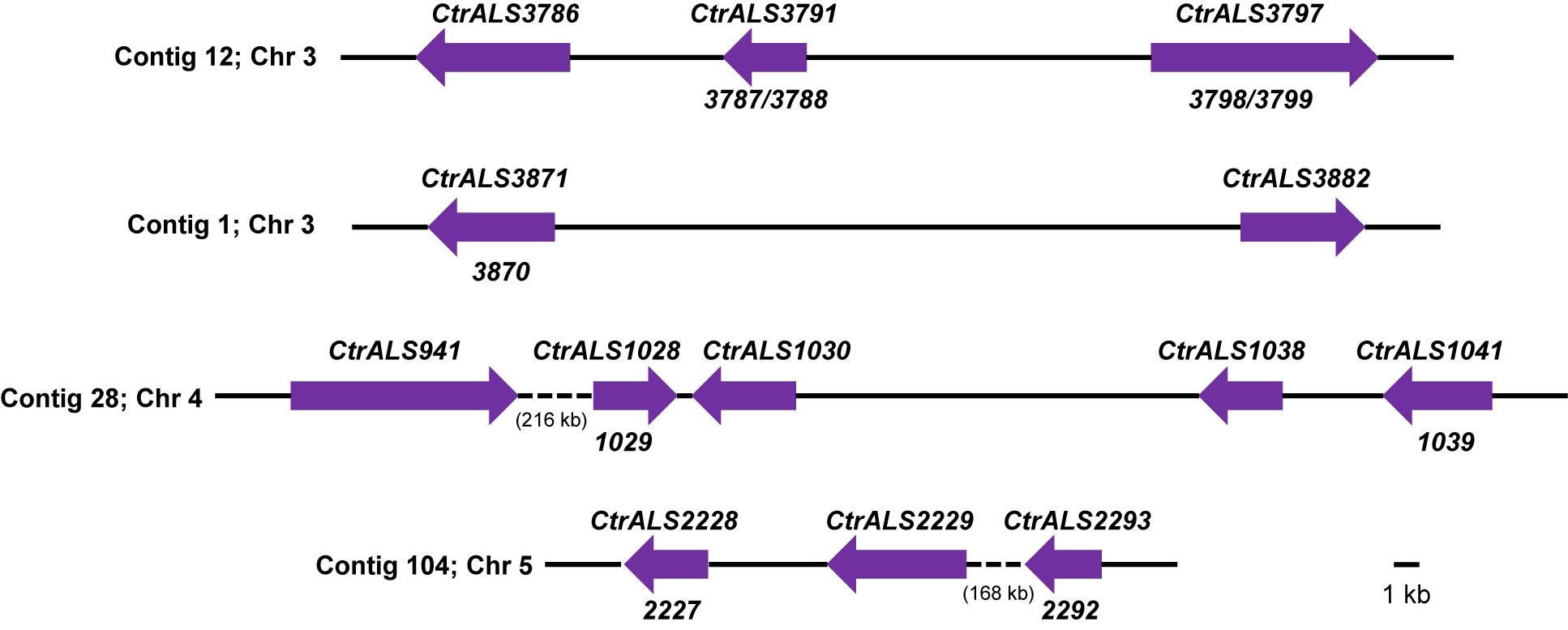
Figure 1. Schematic of ALS loci in Candida tropicalis MYA-3404 as deduced using assembly ASM694213v1, PCR amplification, and Sanger sequencing. Contig numbers from assembly ASM694213v1 were noted at the left of each diagram; chromosome assignments included in assembly ASM1317755v1 (Guin et al., 2020) were also noted. Arrows were drawn to scale and represent the size of one allele at each ALS locus. Because ALS sequences were finalized using PCR amplification for Sanger sequencing, the alleles shown here were likely biased toward the smaller allele when one existed. Final gene names were indicated above each locus; ASM633v3 open reading frames (ORFs) that were combined into the final gene were designated below. Intergenic distances were drawn to scale except where quite large and then indicated by a dashed line and approximate length (in kb). Sequences were deposited in GenBank and accession numbers listed in Table 2. In some cases where the number of copies of the tandem repeat sequence was exceptionally large, unique primers for PCR amplification and Sanger sequencing could not be designed. Instead, the reported sequence was the consensus from the 105× coverage of the Illumina MiSeq/Oxford Nanopore MinION data.
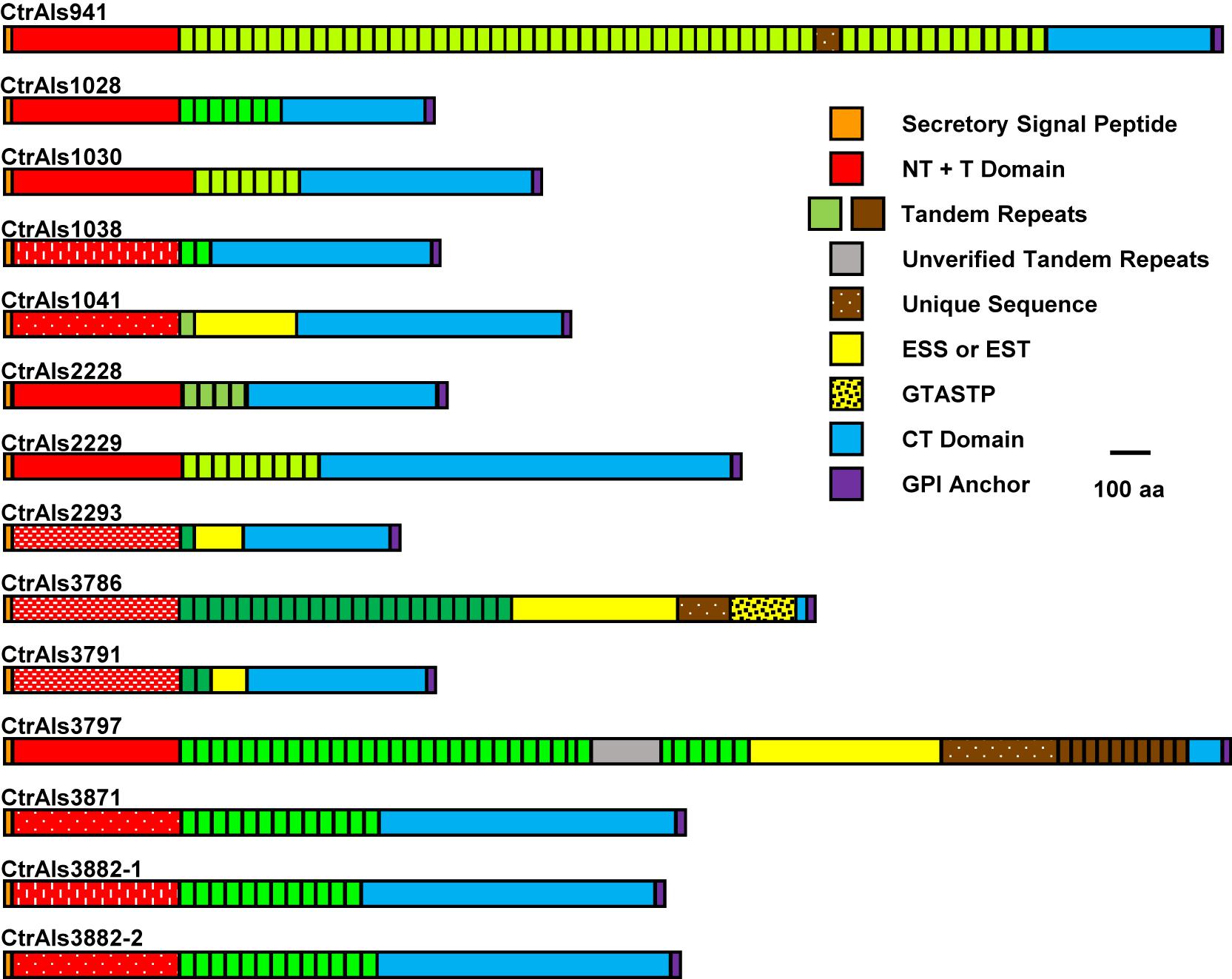
Figure 2. Schematic of predicted C. tropicalis MYA-3404 Als proteins drawn to scale. Protein sequences showed common Als features (reviewed in Hoyer and Cota, 2016) including a secretory signal sequence, NT + T domain, copies of tandemly repeated sequences, and a C-terminal (CT) domain that was rich in Ser/Thr. C. tropicalis Als proteins had some novel repeated sequences such as those rich in ESS or EST, or the GTASTP motif. Protein features were color coded to recognize similarity across the family. Red blocks that represent NT + T domains were further modified to denote similarity between sequences (e.g., speckles or dashed lines as reflected by percent identity values in Figure 3). Blocks denoting tandemly repeated sequences were in different shades of green to indicate sequences with greater similarity to each other. CtrAls2293 was drawn to include a predicted GPI anchor domain, although the putative signal is much weaker in this sequence than others in the family. Extremely long tandem-repeat domains (e.g., CtrALS941 and CtrALS3797) could not be verified by PCR amplification and Sanger sequencing because highly conserved repeated units did not permit the design of unique primers. The reported sequence relied on the 105× coverage from assembly ASM694213v1, which for CtrALS3797 required insertion of “XXX” (gray color) to make the size of the gene match the fragment sizes generated by PCR.
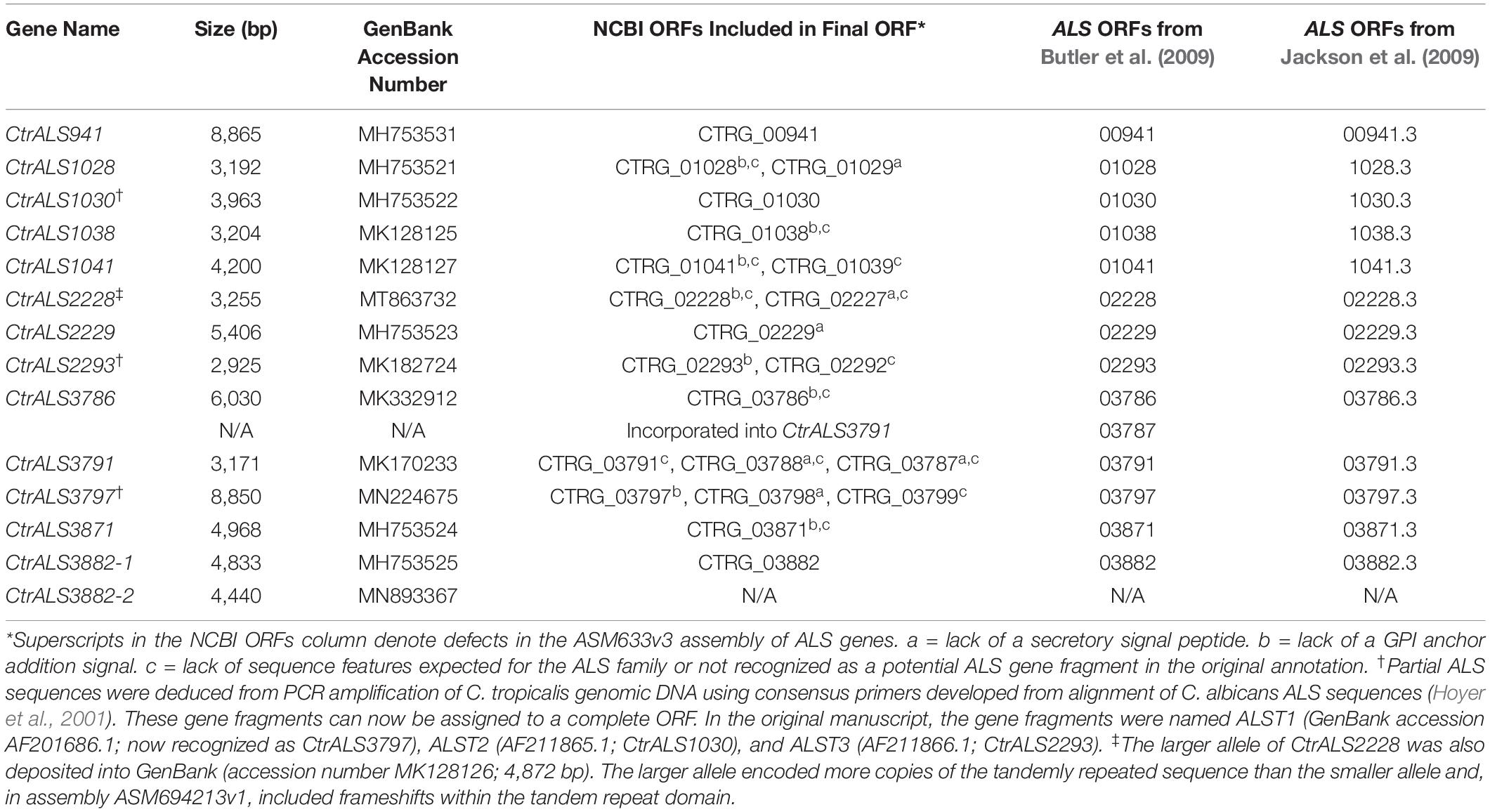
Table 2. C. tropicalis MYA-3404 ALS genes from ASM694213v1 and their corresponding open reading frames (ORFs) in previously published work.
In May 2020, a new C. tropicalis MYA-3404 genome assembly became available in the NCBI database (ASM1317755v1; GCA_01317755.1; Guin et al., 2020). This assembly was generated using PacBio Sequel and Illumina HiSeq data. It was notable because the number of contigs equaled the number of C. tropicalis chromosomes, signaling the highest-quality assembly available to date. A comparison between our carefully curated ALS genes and the new assembly is presented in Table 3. Overall, the new Sequel assembly had the correct number of ALS genes, placed in the proper physical locations. Some of the Sequel ALS ORFs were identical in length to our hand-curated data, although many were longer. Nucleotide polymorphisms existed between our gene sequences and those in the Sequel assembly. For three genes, these polymorphisms resulted in frameshifts and/or premature stop codons, thus an incomplete ALS ORF in the Sequel data.
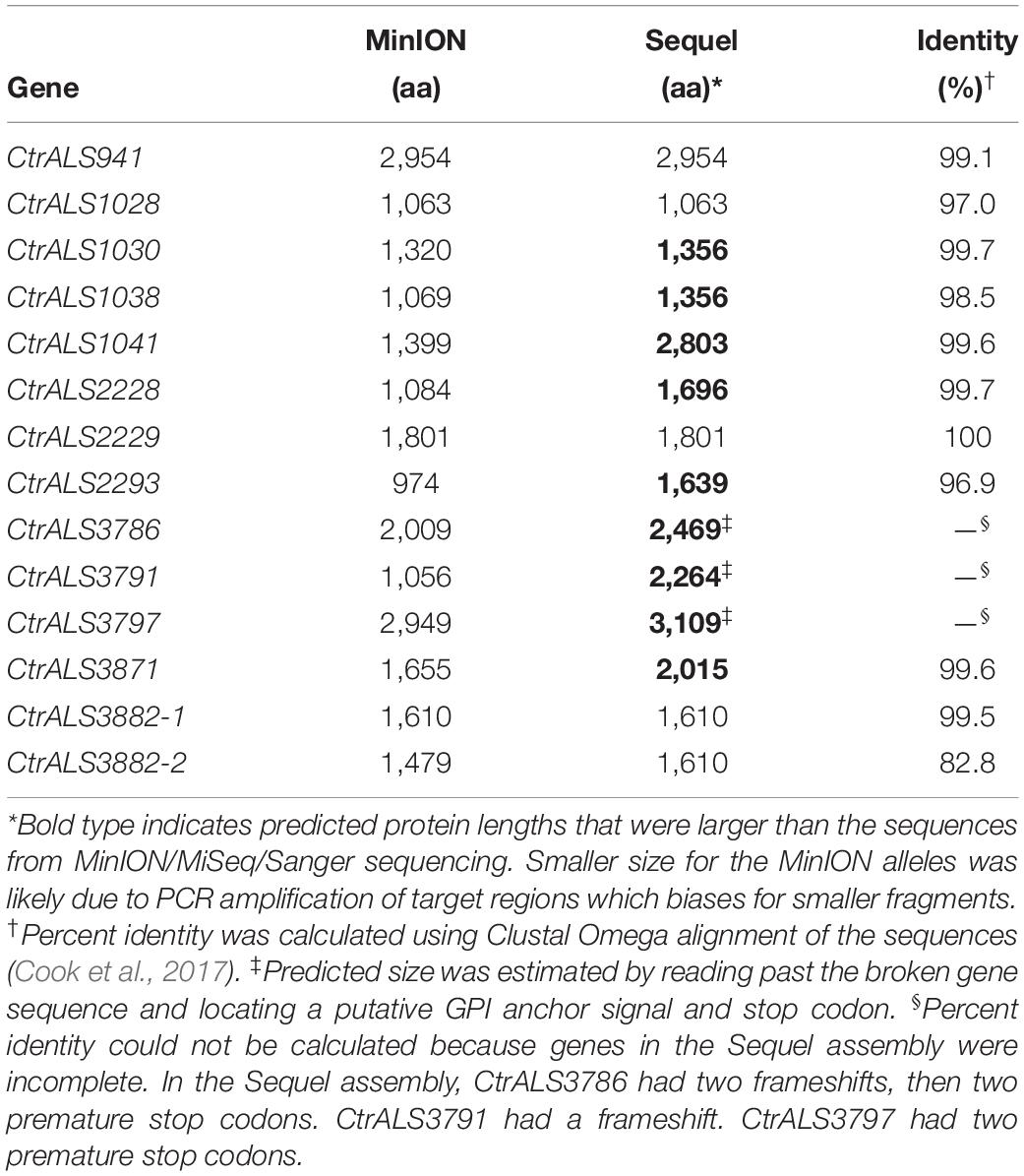
Table 3. Comparison between ALS sequences in C. tropicalis MYA-3404 genome assemblies ASM694213v1 (MiSeq/MinION) and ASM1317755v1 (HiSeq/Sequel).
The C. tropicalis ALS/Als Family
The 13 distinct physical loci in the C. tropicalis ALS family (Figure 1) predicted proteins (Figure 2) with Als features including a secretory signal peptide and a GPI anchor addition site that target the mature protein toward linkage to β-1,6-glucan in the fungal cell wall (Lu et al., 1994). Each predicted protein had a domain structure characteristic of Als proteins (Hoyer and Cota, 2016) including an NT-Als adhesive domain (Lin et al., 2014), followed by a Thr-rich (T) sequence and at least one copy of a sequence that resembled an Als tandem repeat. Like previously characterized Als sequences, those in C. tropicalis were increasingly rich in Ser and Thr residues toward the C-terminal end (reviewed in Hoyer et al., 2008; Lombardi et al., 2019; Oh et al., 2019).
Closer examination of these features suggested that the predicted NT-Als adhesive domain of each C. tropicalis Als protein was only approximately 50% identical to C. albicans NT-Als3 for which the molecular structure is known (Lin et al., 2014; Figure 3). Percent sequence identity among the predicted NT-Als domains in C. tropicalis was also in the 30–50% range for most comparisons, even between proteins encoded by the CtrALS3882 alleles, which were only 52% identical (Figure 4). A high degree of sequence conservation was noted among some NT-Als domains including CtrAls2293/CtrAls3786/CtrAls3791, which were 88–93% identical (Figure 3). CtrAls3882-2 NT-Als was 88% identical to CtrAls3871, while CtrAls3882-1 NT-Als more closely resembled CtrAls1038 (73%). CtrAls1041 NT-Als was 80% identical to the same region in CtrAls3871. Nearly all NT-Als domains predicted from DNA sequence data encoded eight conserved Cys residues that direct folding of the adhesive domain (Lin et al., 2014); CtrAls1030 had only six Cys residues and CtrAls3871 had 10.
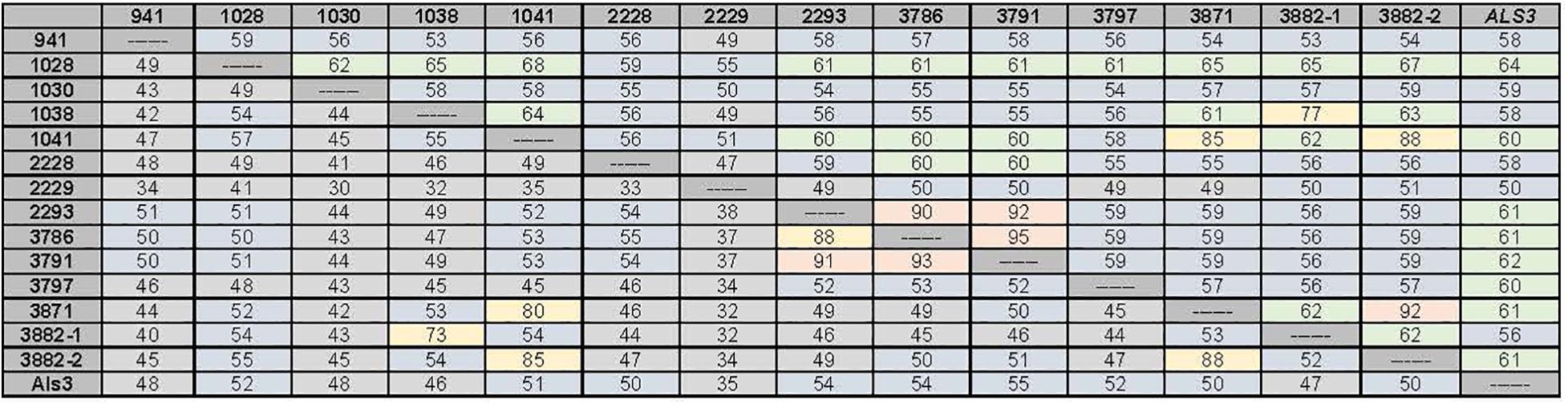
Figure 3. Percent identity values between the nucleotide sequences for the 5′ domain of the C. tropicalis ALS genes (upper diagonal) and their predicted amino acid sequences (lower diagonal). The region used for comparisons corresponds to the NT-Als domain of each protein. C. albicans Als3 (GenBank AY223552.1) was included for comparison. Boxes were shaded to indicate overall percent identity with hotter colors (red, yellow) used for higher percent identity than cooler colors (green, blue, and gray). Both alleles of CtrALS3882 were included in the diagram to highlight lack of identity to each other, but high sequence conservation with other loci.
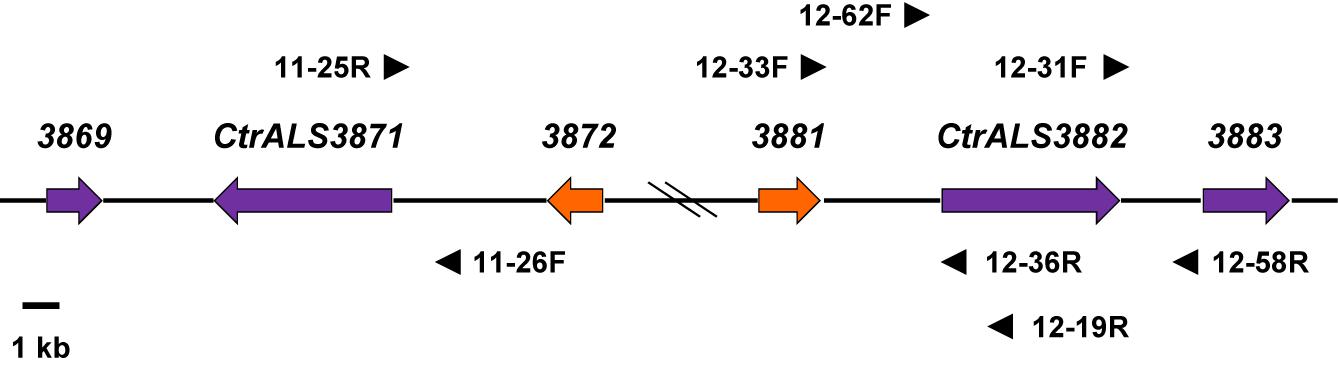
Figure 4. Schematic of the CtrALS3882 locus to support the conclusion that CtrALS3882-1 and CtrALS3882-2 occupied the same relative physical location on the diploid chromosomes of C. tropicalis MYA-3404. Hash marks in the diagram indicated an approximate distance; an accurate distance was shown in Figure 1. CtrALS3882-1 and CtrALS3882-2 were only 52% identical in the 5′ domain of the ORF (Figure 3). ORFs were represented by arrows that indicate the orientation of each gene. Orange arrows indicate CTRG_03872 and CTRG_03881, which were 97% identical. Primers used to demonstrate the physical location of each ORF are shown; primer sequences are recorded in Supplementary Table S1 to indicate whether they recognized both alleles or were specific for CtrALS3882-1 or CtrALS3882-2.
The divergent sequences in the 5′ domain of CtrALS3882-1 and CtrALS3882-2 complicated the assembly and definition of the C. tropicalis ALS family. The presence of divergent sequences was first noted during attempts to amplify the 5′ domain of CtrALS3882 for Sanger sequencing. Resulting chromatograms yielded overlapping peaks that suggested a diploid sequence (data not shown). Subsequent efforts focused on designing PCR primers that anchored CtrALS3882 to an exact physical location, then demonstrating that both alleles occupied the same location, presumably on sister chromosomes (Figure 4). The effort was made more difficult by additional sequence conservation in the region, including the closely related genes CTRG_03872 and CTRG_03881, which were 97% identical. DNA fragments were amplified, cloned, and Sanger sequenced to reveal unique areas to which additional primers could be designed. For example, primers 12-62F and 12-58R were used with Q5 polymerase (New England Biolabs) to specifically amplify CtrALS3882-2. The physical location of each allele was validated by amplifying upstream and downstream fragments for Sanger sequence analysis. Examples include the use of primers 12-33F/12-36R and 12-31F/12-58R that amplified the regions upstream and downstream of CtrALS3882-2, respectively, amplification of the region upstream of CtrALS3882-1 with primers 12-33F/12-19R, and amplification of the region upstream of CtrALS3871 with primers 11-26F/11-25R. Sequence polymorphisms unique to each physical location were deduced by comparison among the PCR products and available genome assemblies. Each CtrALS3882 allele was detected in all six C. tropicalis isolates studied suggesting that heterozygosity at this locus is reasonably common within the species. DNA sequences for each allele in the various isolates are included in Supplementary File S3.
Tandem repeat copy number in some C. tropicalis MYA-3404 ALS alleles was large (e.g., CtrALS941 and CtrALS3797) and in others was limited to a single unit (CtrALS1041 and CtrALS2293). The consensus sequence derived from the main, central domain of tandem repeats was similar to those found in previously characterized Als proteins (Figure 5A). Repeat unit length generally was 36 amino acids, although CtrAls1030 contained multiple copies of a 37-amino-acid repeated sequence, and other alleles contained irregular repeat lengths (i.e., individual units of 35 or 40 amino acids). Novel repeat sequences were present such as short motifs (ESS, EST, GTASTP) or a 30-amino acid tandem motif in CtrAls3797 (Figure 5B).

Figure 5. Analysis of tandem repeat consensus sequences to assess similarity among repeat units in Als proteins from various fungal species. Most repeat units were 36 amino acids, although exceptions were noted. Consensus sequences required that 80% or more of the amino acids were identical at each position. (A) Consensus sequences derived from protein translation of C. tropicalis ALS sequences identified in Table 2. The repeat unit in CtrAls1030 was 37 amino acids. Asterisks marked sequences for which only one to two tandem repeat copies were present. Lack of tandem repeat copies in these proteins provided the potentially false impression of a high level of sequence conservation. ALS tandem repeat consensus sequences for C. albicans (Ca; Oh et al., 2019), C. dubliniensis (Cd), and the C. parapsilosis species complex (CpCoCm; Oh et al., 2019) were included for comparison. (B) Consensus sequence for the novel tandem repeat from the C-terminal domain of CtrAls3797 (see Figure 2).
Phylogenetic analysis was conducted using the amino acid sequences from the NT-Als adhesive (functional) domain from each Als protein in C. albicans (Ca), C. dubliniensis (Cd), C. tropicalis (Ctr), Candida parapsilosis (Cp), Candida orthopsilosis (Co), and C. metapsilosis (Cm; Figure 6). Sequences beyond this region could not be included because of their high level of divergence. Nucleotide sequences encoding NT-Als were also too divergent, so amino acid sequences were aligned and variable regions extracted as described in Section “Materials and Methods.” Results showed three phylogenetically distant groups: Ca/Cd, Cp/Co/Cm, and Ctr. Orthologs were apparent in the Ca/Cd group (e.g., CaAls9/CdAls64220, CaAls4/CdAls64610, CaAls6/CdAls86290, and CaAls7/CdAls86150). A previous publication (Jackson et al., 2009) offered evidence to support the orthology of CaAls1/CdAls64210. Within the Cp/Co/Cm group, orthologs CpAls660/CoAls800/CmAls800 were evident. Paralogous sequences, most likely resulting from duplication events and generating contiguous genes within a species included CoAls4210/CoAls4220 and CpAls4770/CpAls4780/CpAls4800. The NT-Als sequences from C. tropicalis were relatively distant phylogenetically. The larger number of ALS genes in C. tropicalis compared to the other species likely arose from gene duplication. Potential paralogs included CtrAls1041/CtrAls3871/CtrAls3882-2, CtrAls1038/CtrAls3882-1, and CtrAls3791/CtrAls2293/CtrALS3786. CtrAls3797 grouped more closely to the Cp/Co/Cm sequences. This trend was also apparent for CpAls4790/CmAls4220 that grouped more closely with the Ca/Cd sequences suggesting a potential ortholog within that group. Functional information for each protein would aid interpretation of these distinctions.
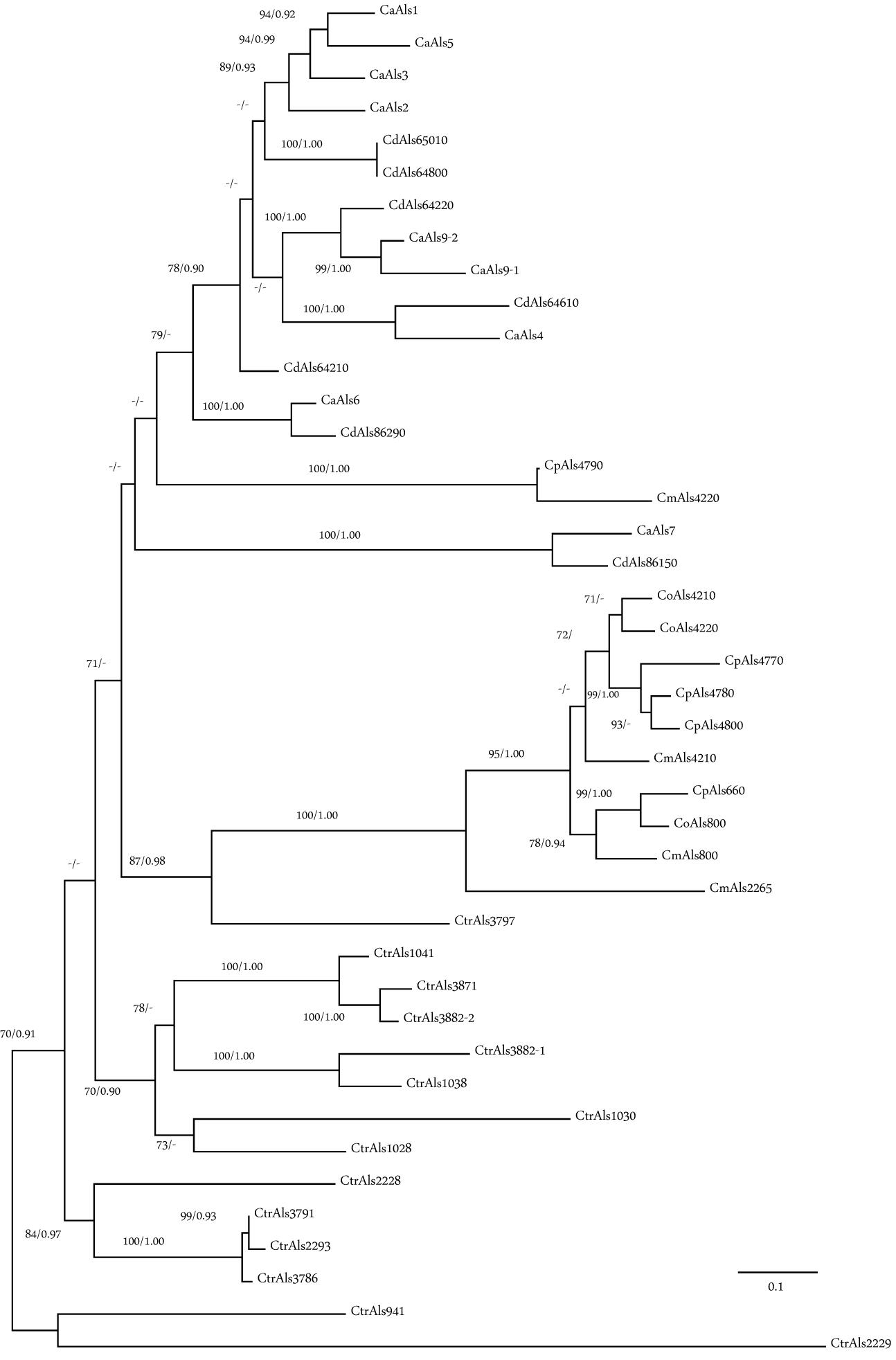
Figure 6. Phylogeny of Als N-terminal domain sequences from C. albicans (Ca), C. dubliniensis (Cd), C. tropicalis (Ctr), C. parapsilosis (Cp), C. orthopsilosis (Co), and C. metapsilosis (Cm). The best-scoring Maximum likelihood tree is shown with maximum likelihood bootstrap values and Bayesian posterior probabilities at each node; only support values greater than 70% and 0.90, respectively, were shown. Branch lengths were proportional to evolutionary change and measured in substitutions per site.
Relative Expression Levels of Candida tropicalis ALS Genes
Real-time RT-PCR analysis of ALS gene expression was pursued to determine if the family was differentially expressed by growth condition and cellular morphology. High gene expression levels might also predict which Als proteins were present in the greatest abundance on the C. tropicalis cell surface, thereby positioned to contribute most to the adhesive function. A unique TaqMan assay was designed for each ALS gene and to differentiate between the alleles of CtrALS3882. Assay primers and probe sequences were placed at a similar location within the 5′ domain for each gene. C. tropicalis MYA-3404 cells were harvested from five different culture conditions, RNA extracted, and cDNA synthesized for TaqMan analysis.
Color coding in Figure 7 denotes high (red) and low (purple) expression levels based on the threshold (ΔCt) values relative to the ACT1 and TEF1 control genes. With Ct values that lagged only one to two cycles behind the control gene expression, CtrALS1028 showed high expression levels in RPMI medium and from a 2-h culture of SD + FBS. These expression levels were not significantly different from each other (p = 0.1276). Expression levels were lower in cells from a 16-h YPD culture and the 24-h SD + FBS condition; these values were not significantly different from each other either (p = 0.1245). All other comparisons were significantly different suggesting differential expression of CtrALS1028 by stage of culture growth (1 vs. 16 h in YPD; p < 0.0001; 2 h vs. 24 h in SD + FBS; p < 0.0001) with higher expression levels in cells that were more recently transferred to fresh growth medium. In contrast, CtrALS941 expression was lower at early stages of culture growth and significantly higher as the culture reached saturation (16 h vs. 1 h in YPD; p = 0.0007; 24 vs. 2 h in SD + FBS; p < 0.0001). CtrALS3791 showed yet another type of differential expression pattern with its highest expression in the 1 h YPD culture and significantly lower values in the early-stage growth in other media (p < 0.0001). Many genes had an expression pattern like CtrALS1030, expressed at only moderate levels in all five growth conditions. For CtrALS1030, none of the statistical comparisons were significant (p > 0.05) suggesting lack of differential expression for the growth conditions tested.
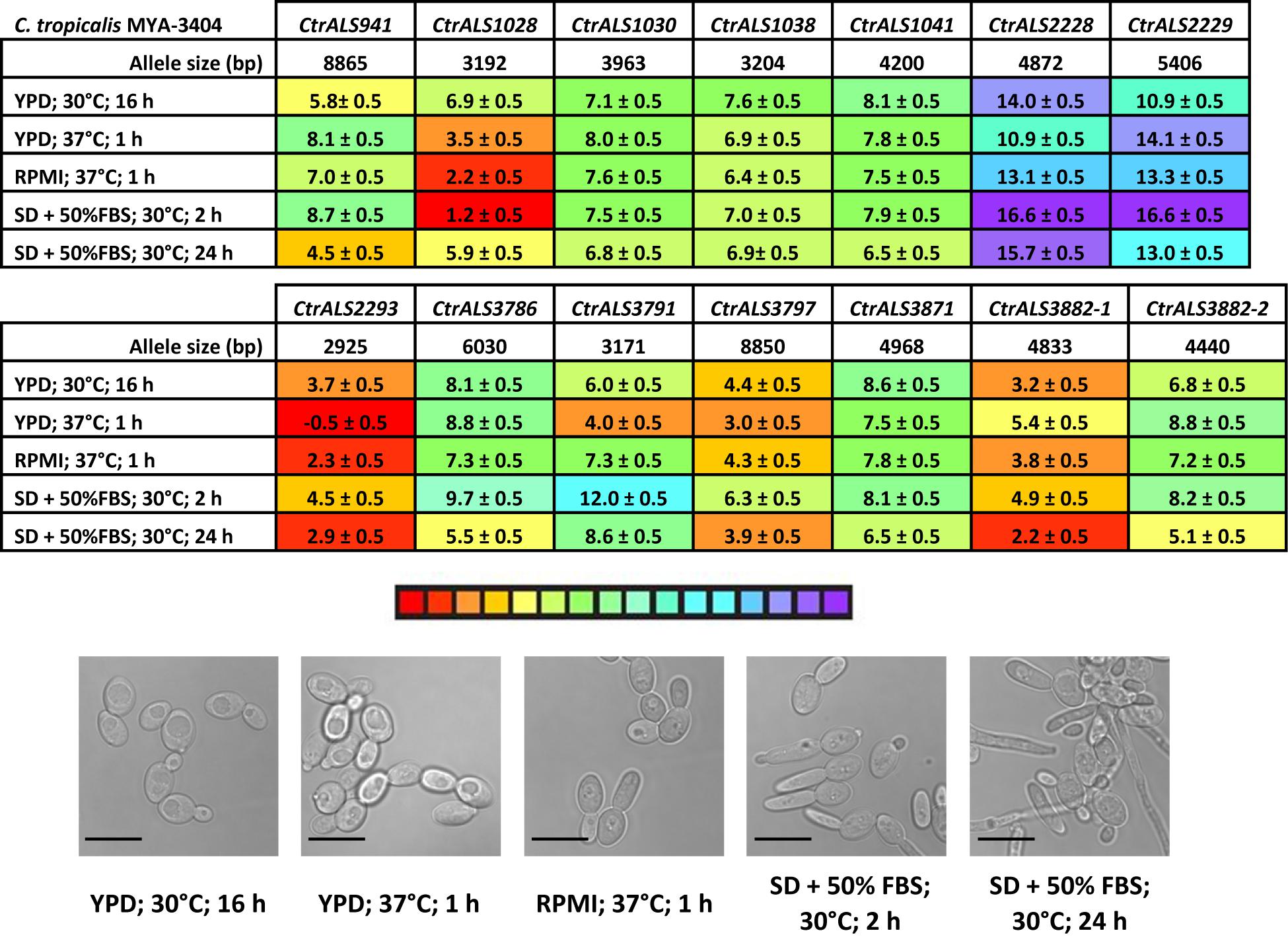
Figure 7. Relative expression of C. tropicalis ALS genes as measured by TaqMan assays. Expression of the 13 C. tropicalis ALS genes was measured for strain MYA-3404 grown in five different culture conditions. Micrographs of representative cells from each culture condition were captured to show cellular morphology that corresponded to the gene expression data (scale bar = 10 μm). ΔCt values and standard errors of the mean were reported. The color bar below the data tables was shaded to represent high (red) to low (purple) expression levels.
The examples above provided a context for statistical significance for differences between the gene expression values. Coupled with the color coding in Figure 7, many other comparisons can be made visually and quickly evaluated for their potential statistical significance. However, the data did not provide a biological context for the relationship between TaqMan ΔCt values and detectable Als protein on the C. tropicalis cell surface. A potential context was provided by designing TaqMan assays for C. albicans ALS genes (Supplementary Table S2) and using them to assess gene expression in growth conditions for which the association between differential gene expression and protein abundance on the fungal cell surface was known. The rationale for this approach was that use of monoclonal antibodies specific for individual Als proteins demonstrated that CaAls1, CaAls3, and CaAls4 can be visualized readily by immunofluorescent labeling on the C. albicans surface, while CaAls5 and CaAls6 cannot (Coleman et al., 2009, 2010, 2012). CaAls5 could be detected in Western blots of C. albicans cell wall extracts, while the CaAls6 signal remained negative (Zhao et al., 2011). These previous observations corresponded well with the TaqMan ΔCt values (Figure 8A) and suggested that a ΔCt value of approximately 7 might serve as a potential cutoff between detectable and non-detectable cell-surface protein.

Figure 8. TaqMan measurement of ALS gene expression in C. albicans (A) and C. dubliniensis (B). C. albicans SC5314 and C. dubliniensis CD36 were grown in various culture conditions known to display differential expression of C. albicans ALS genes. Images were captured for representative cells from each culture (scale bar = 10 μm). ΔCt values and standard errors of the mean were reported. The color-coded scale bar ranged from high expression (red) to low (purple). Two C. dubliniensis ALS loci had identical sequences and were detected by the same TaqMan assay (CdALS64800 and CdALS65010). C. dubliniensis ALS genes were orthologous to C. albicans ALS loci as designated in (B). Prior to availability of a C. dubliniensis genome sequence, ALS genes were identified using consensus PCR primers derived from C. albicans ALS sequences (Hoyer et al., 2001). These were designated ALSD1 (now recognized as CdALS86290), ALSD2 (CdALS64210), and ALSD3 (CdALS64610).
Revisiting Figure 7 with this insight suggested that although most comparisons between TaqMan ΔCt values for CtrALS2228 and CtrALS2229 were statistically significant, neither gene was transcribed at a level that was likely to produce detectable protein on the C. tropicalis cell surface. In each growth condition assayed, several other genes were transcribed at a level that was also unlikely to produce detectable protein (i.e., CtrALS1030, CtrALS1038, CtrALS1041, and CtrALS3871). Some genes were differentially expressed at a sufficient level to produce detectable protein in specific growth conditions (e.g., CtrALS3791 in 1 h YPD), while others appeared to produce the proteins that might have the greatest cell-surface presence. For example, CtrALS2293 was transcribed highly in all growth conditions tested. In a 24-h SD + FBS culture, it was likely that CtrAls2293 and CtrAls3882-1 would dominate the cell surface. In each growth condition tested, CtrALS3882-1 was more highly expressed than its allele CtrALS3882-2 (p < 0.0001).
One larger goal of our work was to understand the cell-surface Als presence on various fungal species that cause candidiasis. In this regard, little attention has been given to the expression of C. dubliniensis ALS genes. C. dubliniensis and C. albicans are closely related species in which ALS genes occupy similar physical loci (Jackson et al., 2009). However, C. dubliniensis does not encode an ALS3 ortholog, and sequences for two of the C. dubliniensis ALS loci (Cd64800 and Cd65010) are 100% identical within the 5′ domain of the gene. TaqMan assays were designed and used to analyze ALS gene expression in cells from strain CD36 grown using the same conditions applied to C. tropicalis and C. albicans; a single assay detected transcription from Cd64800 and Cd65010 (Figure 8B). Some C. dubliniensis ALS genes were capable of high expression levels (e.g., Cd64610, Cd64800/Cd65010), while others were barely transcribed (Cd86150). Differential expression was observed among the growth conditions tested (Cd64210, Cd86290).
The newly validated TaqMan assays were a tool that others may use to explore various hypotheses regarding the C. tropicalis ALS family. One potential experimental question was whether ALS expression patterns varied across C. tropicalis clinical isolates. DNA sequences were derived for the 5′ domain of each ALS gene in the six C. tropicalis isolates in our laboratory collection (Supplementary File S3). While many sequences showed a perfect match to TaqMan assay primers and probes, sequence variation was noted in some instances. These target sequences were cloned and verified; dilutions of purified DNA were used as the TaqMan assay template. Figure 9 shows an array of these examples, selected to titrate the degree of sequence mismatch that might result in underestimated or false-negative assay results. For example, one nucleotide mismatch in the middle of one primer was not sufficient to interfere with assay function (Figure 9A); however, accumulation of more mismatches, especially in key primer or probe positions led to falsely low estimates of transcript abundance (Figures 9B,C). Some mismatches between the TaqMan primers/probe and target sequences were so extensive that the assay was unable to detect the sequence (Figure 9D). Examination of the sequence data for the collection of six C. tropicalis isolates (Supplementary File S3) suggested that the TaqMan assays matched target sequences in nearly all instances. Strain 951 was the most divergent with three genes at risk of falsely low estimates of expression level (CtrALS1038, CtrALS3797, and CtrALS3882-2); sequence mismatches for CtrALS3797 in three additional strains (1019, 1020, 3242) suggested the potential for redesign of that TaqMan assay depending on the strains and biological questions addressed.
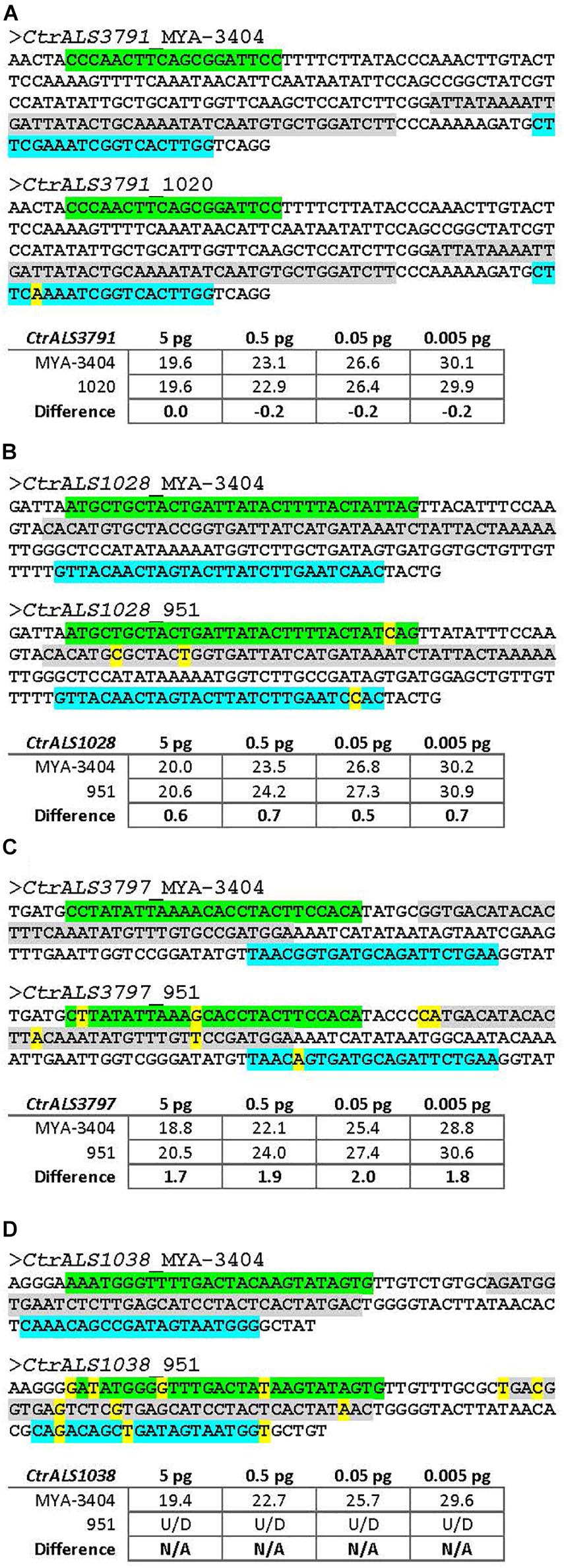
Figure 9. Effect of nucleotide sequence mismatch on TaqMan assay measurement of nucleic acid abundance. TaqMan assays were designed using gene sequences from C. tropicalis strain MYA-3404. Selection of unique target regions that distinguished among genes in the ALS family was the primary consideration in assay design. Sequencing of the 5′ domain of each ALS gene in six other C. tropicalis isolates revealed some mismatches in the TaqMan primer and/or probe sequences. Examples were selected to titrate the effect of an increasing number of mismatches, some in key primer/probe positions. Sequences in each panel were taken from Supplementary File S3. The forward primer was highlighted in green, reverse primer in blue, and probe in gray. Mismatches were noted with yellow color. Target sequences in each strain examined were PCR amplified, cloned, and Sanger sequence verified to ensure the mismatches were present. Cloned DNA was purified, diluted, and used in the assays reported here. (A) The sequence of the 5′ domain of CtrALS3791 in strain 1020 showed a single nucleotide mismatch in the middle of the reverse primer. Ct values from TaqMan assays with 10-fold dilutions of cloned construct showed almost an equal ability for the assay to detect each strain (e.g., no difference for 5 pg DNA and negligible differences for subsequent dilutions). (B) C. tropicalis strain 951 showed one to two mismatches each in the primer and probe sequences for gene CtrALS1028. Ct values suggested that detection of strain 951 lagged approximately 0.6 cycle behind detection of strain MYA-3404, providing an underestimate of DNA abundance. (C) Increasing numbers of mismatches between primer and probe sequences resulted in greater underestimates of DNA abundance for strain 951, this time in CtrALS3797 where nearly a two-cycle difference in Ct was observed. (D) Mismatches for CtrALS1038 in strain 951 were so marked that the TaqMan assay was unable to detect genomic DNA, even at the highest concentration. These data provide the foundation necessary to adapt use of the TaqMan assays to C. tropicalis isolates in which primer and probe mismatches may exist.
Discussion
Advances in DNA sequencing technology are providing long-sought information about the composition of the ALS family in fungal species. The availability of Oxford Nanopore MinION technology provided the initial opportunity to incorporate long-read DNA sequence data to resolve the C. tropicalis ALS genes. Despite the insights added by these long-read data, assembling accurate ALS sequences required considerable additional effort. The more recently released PacBio Sequel-based assembly (Guin et al., 2020) leverages technological improvements in DNA sequence accuracy that will continue to reduce the tedium associated with solving genomic puzzles that involve complex genes like those in the ALS family. The accurate list of C. tropicalis ALS loci provides an essential foundation for subsequent investigations into adhesion and pathogenesis of the species.
The C. tropicalis gene list (Table 2) also provides a standard set of names that can be used to derive and communicate unambiguous experimental results. Two published studies attempted to characterize C. tropicalis ALS gene expression without the benefit of a complete gene list (Yu et al., 2016; Galán-Ladero et al., 2019). Supplementary Table S4 shows that some of the PCR primers used in these studies are likely to amplify more than one C. tropicalis ALS gene, potentially complicating interpretation of results.
ALS gene names across publications can also be confusing. This confusion seems to originate from misinterpretation of Supplementary Table 23 from Butler et al. (2009). The Als family portion of the table is reproduced in Supplementary Table S5. Protein names in the left column match the entries in the C. albicans column but do not necessarily correspond to protein names in the other species. For example, the Candida Gene Order Browser6 (Fitzpatrick et al., 2010; Maguire et al., 2013) indicates that there is no positional ortholog for C. albicans ALS1 (orf19.5714) in C. tropicalis, so C. tropicalis CTRG_02293 is not “C. tropicalis ALS1” and is definitely not ALST1 as described by Hoyer et al. (2001; Table 2). This disambiguation of gene names is offered to assist readers with interpreting published information about the C. tropicalis ALS family.
The current literature is limited to two papers that examine C. tropicalis ALS gene expression (Yu et al., 2016; Galán-Ladero et al., 2019). The analyses in both papers compared C. tropicalis ALS gene expression in planktonic and sessile cells. Yu et al. (2016) grew sessile C. tropicalis on either a polystyrene surface or a human urinary bladder epithelial cell line. Nine clinical isolates and ATCC 750 were studied. Although results varied by strain, ALST1 (CtrALS3797) expression increased in sessile cells. ALST3 (which likely combines readings from CtrALS2293, CtrALS3786, and CtrALS3791) had the highest expression across the three experimental conditions, perhaps consistent with the high level of CtrALS2293 expression demonstrated here (Figure 7). Galán-Ladero et al. (2019) studied similar C. tropicalis properties, also using multiple clinical isolates. Although there was considerable strain variation, ALS gene expression generally increased in biofilm-grown cells. Interest in the role of ALS genes in C. tropicalis biofilm formation is stimulated by the key role that Als proteins play in C. albicans biofilms (reviewed in Lohse et al., 2018).
Gene expression patterns investigated here focus on the potential for differential ALS expression with growth stage or cellular morphology. Growth conditions (e.g., use of YPD and RPMI media) were selected deliberately to place C. tropicalis results into a well-characterized context that was developed for studies of ALS gene expression in C. albicans (reviewed in Hoyer et al., 2008). The ability of C. tropicalis to grow in a filamentous form (Lackey et al., 2013; Figure 7) was also leveraged to assess ALS gene expression changes associated with morphology. Comparative analyses were furthered by developing TaqMan assays for C. albicans and C. dubliniensis ALS genes. TaqMan assays correctly identified C. albicans ALS genes that were capable of high expression levels (CaALS1, CaALS3, and CaALS4) and those that were not (CaALS6, CaALS7; reviewed in Hoyer et al., 2008). Differential expression patterns such as upregulation of CaALS3 in growth conditions that promote germ tube and hyphal formation were also observed (Hoyer et al., 1998a). High levels of CaALS4 expression in saturated cultures were also noted using the TaqMan assay, as were the residual high level of mRNA in cells that were early in the growth stage in fresh medium that had been previously documented using Northern blots (Hoyer et al., 1998b). TaqMan-assessed expression of CaALS1 showed high levels in cells transferred to fresh medium and in cells transitioning to hyphal growth (Zhao et al., 2004). However, the CaALS1 expression level in saturated culture conditions was higher than anticipated.
Some of these C. albicans ALS gene expression themes were noted in C. tropicalis such as CtrALS1028 increased expression in cells placed into fresh growth medium and CtrALS941 increased expression in saturated cultures (Figure 7). None of the CtrALS genes showed increased expression in filamentous growth morphologies. Other genes, particularly CtrALS2293 and CtrAls3882-1, were highly expressed regardless of growth condition. The highly abundant CtrAls proteins are the strongest candidates for contributing to adhesive function in vitro. However, the C. albicans ALS gene expression is known to differ in vitro and in vivo (Coleman et al., 2012), so assaying in vivo-grown C. tropicalis would better predict the display of Als proteins on cells in the host environment. Understanding which Als proteins are prominent on the fungal cell surface is the first step toward developing adhesion inhibitors that will function across pathogenic fungal species.
Although strain differences in CtrALS gene expression were not systematically examined here, the works of Yu et al. (2016) and Galán-Ladero et al. (2019) suggest that they exist. Strain differences in ALS gene expression were also noted for C. parapsilosis and C. metapsilosis (Oh et al., 2019). Although there was some strain variation in C. orthopsilosis ALS gene expression, ALS expression increases in cells that are transferred to fresh culture media compared to saturated cultures (Lombardi et al., 2019). C. orthopsilosis ALS gene transcription also showed a reliable hierarchy of expression level with CoALS4220 > CoALS4210 > CoALS800 (Lombardi et al., 2019).
ALS genes in other species are known to have considerable allelic variation, even within the same strain (Zhang et al., 2003; reviewed in Hoyer et al., 2008). C. tropicalis genes likely have this large degree of allelic variation, as well. For example, examination of CtrALS1030 (called “ALS2” in the manuscript) in 68 C. tropicalis isolates revealed considerable sequence variation in the coding region, localized primarily to the central tandem repeat domain (Zhang et al., 2019). The present work with strain MYA-3404 resulted in two distinct CtrALS2228 sequences deposited in GenBank (Table 2) that primarily varied in the number of copies of the repeated sequence in the tandem repeat domain. DNA sequences in Supplementary File S3 document diploid nucleotide sequences and variation between strains within the 5′ domain of each C. tropicalis ALS gene reported here. Comparison between the MinION C. tropicalis ALS sequences and those derived from the Sequel assembly (Guin et al., 2020) reveals length variations that are probably the diploid alleles in strain MYA-3404. MinION data were validated by PCR amplification and Sanger sequencing, techniques that would preferentially amplify the shorter allele where one exists.
All six C. tropicalis strains examined encode the 13 ALS loci described here (Supplementary File S3). Genome sequencing would be helpful to determine if the strains encode additional ALS loci. The number of ALS loci varies among strains in some species. For example, examination of draft C. parapsilosis genome sequences available in GenBank revealed variable ALS gene numbers with some strains encoding only two and others having five (Oh et al., 2019). Intergenic recombination between the contiguous C. albicans ALS5 and ALS1 loci resulted in a reduction in the ALS gene number in approximately 5% of isolates tested (Zhao et al., 2011); heterozygosity at the locus was observed with some strains revealing recombination on one chromosome but not the other. In C. tropicalis, the recombination events that created the divergent 5′ domains of CtrALS3882-1 and CtrALS3882-2 are found in the six isolates examined. Divergent 5′ domain sequences were also noted for CaALS9, although the sequences were 89% identical compared to the 52% identity at the CtrALS3882 locus (Zhao et al., 2007). Analysis of the adhesive phenotype of strains in which C. albicans ALS9 was deleted and individual alleles reintegrated revealed the ability of CaALS9-2, but not CaALS9-1, to rescue the adhesion defect. It is possible that alleles of CtrALS3882 may make different contributions to C. tropicalis adhesion because of their sequence differences but also because of the greater representation of CtrALS3882-1 on the cell surface as predicted from the higher transcriptional activity from this allele (Figure 7).
Compared to discovery of the ALS family in C. albicans where genes were introduced into the literature over the period of a decade or more as they were detected using Southern blots (reviewed in Hoyer et al., 2008), advances in genome sequencing technology make it possible to detect all genes in the family at one time. Because of the complexity of the ALS genes in C. tropicalis, however, accurate assembly of the sequences was not attempted until the availability of long-read sequence technology. The Illumina MiSeq/Oxford Nanopore genome assembly (ASM694213v1) was completed during 2017 yet the large number of C. tropicalis ALS genes, lengthy tandem repeat regions, and unexpected allelic variation at the CtrALS3882 locus required considerably more effort to resolve. The recent PacBio Sequel/Illumina HiSeq assembly (ASM1317755v1; Guin et al., 2020) would have provided a great time savings and suggests that other fungal genomes should be re-sequenced as newer, more accurate, long-read technologies emerge. The relatively low cost of generating accurate long-read sequence data would more than pay for itself in savings of human effort to assemble the complex jigsaw puzzle for ALS sequences and other gene families that contain conserved, repeated sequences.
Data Availability Statement
The genome assembly for Candida tropicalis strain MYA-3404, generated using Illumina MiSeq and Oxford Nanopore MinION data, is available in GenBank under BioProject accession number PRJNA432250, BioSample accession number SAMN08439037, and Genome accession number PQTP00000000. Version 01 of the project has the accession number PQTP01000000 and consists of sequences PQTP01000001–PQTP01000029. All individual ALS gene sequences were deposited in GenBank. Accession numbers are noted throughout the manuscript.
Author Contributions
LLH conceptualized the study, acquired funding, and was in charge of project administration. VH, CF, and AH conducted formal analysis. S-HO, AI, RR-B, BS, JJ, CF, AH, and LLH performed the investigation. VH, CF, AH, and LLH developed the study methodology. S-HO, AI, RR-B, JJ, VH, CF, AH, and LLH wrote the original draft. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by R15 DE026401 from the National Institute of Dental and Craniofacial Research, National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AI, RR-B, BS, and JJ were part of the Undergraduate Program in Fungal Genomics, which is a collaboration between the University of Illinois at Urbana-Champaign and Millikin University. Other students who contributed to this effort included Kaia Ball, Madeline Batek, Anton Bershanskiy, Stephen DeMartini, Erica Forbes, Zeidy Garcia, Jessie Kirk, Mariah McNamer, Deniz Namik, and Quinn Nguyen. The authors thank Drs. Travis Wilcoxen and Laura Zimmerman, Millikin University Department of Biology, for coordinating student participation and supervising research credit for students in the program. The authors thank Pat Kammeyer (Loyola University Medical Center), David Coleman (Trinity College Dublin), and David Soll (University of Iowa) for providing the fungal isolates.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.594531/full#supplementary-material
Footnotes
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ http://www.cbs.dtu.dk/services/SignalP
- ^ https://mendel.imp.ac.at/gpi/gpi_server.html
- ^ https://www.ebi.ac.uk/services
- ^ http://cgob.ucd.ie/
References
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Butler, G., Rasmussen, M. D., Lin, M. F., Santos, M. A. S., Sakthikumar, S., Munro, C. A., et al. (2009). Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–662. doi: 10.1038/nature08064
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–542. doi: 10.1093/oxfordjournals.molbev.a026334
Coleman, D. A., Oh, S.-H., Manfra-Maretta, S. L., and Hoyer, L. L. (2012). A monoclonal antibody specific for Candida albicans Als4 demonstrates overlapping localization of Als family proteins on the fungal cell surface and highlights differences between Als localization in vitro and in vivo. FEMS Immunol. Med. Micobiol. 64, 321–333. doi: 10.1111/j.1574-695X.2011.00914.x
Coleman, D. A., Oh, S.-H., Zhao, X., and Hoyer, L. L. (2010). Heterogeneous distribution of Candida albicans cell-surface antigens demonstrated with an Als1-specific monoclonal antibody. Microbiology 156, 3645–3659. doi: 10.1099/mic.0.043851-0
Coleman, D. A., Oh, S.-H., Zhao, X., Zhao, H., Hutchins, J. T., Vernachio, J. H., et al. (2009). Monoclonal antibodies specific for Candida albicans Als3 that immunolabel fungal cells in vitro and in vivo and block adhesion to host surfaces. J. Microbiol. Methods 78, 71–78. doi: 10.1016/j.mimet.2009.05.002
Cook, C. E., Bergman, M. T., Cochrane, G., Apweiler, R., and Birney, E. (2017). The European Bioinformatics Institute in 2017: data coordination and integration. Nucleic Acids Res. 46, D21–D29. doi: 10.1093/nar/gkx1154
de Lannoy, C., de Ridder, D., and Risse, J. (2017). The long reads ahead: de novo genome assembly using the MinION. F1000Res. 6:1083. doi: 10.12688/f1000research.12012.2
de Oliveira, J. S., Pereira, V. S., Castelo-Branco, D. S. C. M., Cordeiro, R. A., Sidrim, J. J. C., Brilhante, R. S. N., et al. (2020). The yeast, the antifungal, and the wardrobe: a journey into the antifungal resistance mechanisms of Candida tropicalis. Can. J. Microbiol. 66, 377–388. doi: 10.1139/cjm-2019-0531
Eisenhaber, B., Bork, P., and Eisenhaber, F. (1999). Prediction of potential GPI-modification sites in proprotein sequences. Mol. Cell Biol. 292, 741–758. doi: 10.1006/jmbi.1999.3069
Fitzpatrick, D. A., O’Gaora, P., Byrne, K. P., and Butler, G. (2010). Analysis of gene evolution and metabolic pathways using the Candida Gene Order Browser. BMC Genomics 11:290. doi: 10.1186/1471-2164-11-290
Galán-Ladero, M. A., Blanco-Blanco, M. T., Fernández-Calderón, M. C., Lucio, L., Gutiérrez-Martín, Y., Blanco, M. T., et al. (2019). Candida tropicalis biofiom formation and expression levels of the CTRG ALS-like genes in sessile cells. Yeast 36, 107–115. doi: 10.1002/yea.3370
Guin, K., Chen, Y., Mishra, R., Muzaki, S. R. B., Thimmappa, B. C., O’Brien, C. E., et al. (2020). Spatial inter-centromeric interactions facilitated the emergence of evolutionary new centromeres. Elife 9:e58556. doi: 10.7554/eLife.58556
Hoyer, L. L., and Cota, E. (2016). Candida albicans agglutinin-like sequence (Als) family vignettes: a review of Als protein structure and function. Front. Microbiol. 7:280. doi: 10.3389/fmicb.2016.00280
Hoyer, L. L., Fundyga, R., Hecht, J. E., Kapteyn, J. C., Klis, F. M., and Arnold, J. (2001). Characterization of agglutinin-like sequence genes from non-albicans Candida and phylogenetic analysis of the ALS family. Genetics 157, 1555–1567.
Hoyer, L. L., Green, C. B., Oh, S.-H., and Hoyer, L. L. (2008). Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family – a sticky pursuit. Med. Mycol. 46, 1–15. doi: 10.1080/13693780701435317
Hoyer, L. L., Payne, T. L., Bell, M., Myers, A. M., and Scherer, S. (1998a). Candida albicans ALS3 and insights into the nature of the ALS gene family. Curr. Genet. 33, 451–459. doi: 10.1007/s002940050359
Hoyer, L. L., Payne, T. L., and Hecht, J. E. (1998b). Identification of Candida albicans ALS2 and ALS4 and localization of Als proteins to the fungal cell surface. J. Bacteriol. 180, 5334–5343. doi: 10.1128/JB.180.20.5334-5343.1998
Hoyer, L. L., Scherer, S., Shatzman, A. R., and Livi, G. P. (1995). Candida albicans ALS1: domains related to a Saccharomyces cerevisiae sexual agglutinin separated by a repeating motif. Mol. Microbiol. 15, 39–54. doi: 10.1111/j.1365-2958.1995.tb02219.x
Jackson, A. P., Gamble, J. A., Yeomans, T., Moran, G. P., Saunders, D., Harris, D., et al. (2009). Comparative genomics of the fungal pathogens Candida dubliniensis and Candida albicans. Genome Res. 19, 2231–2244. doi: 10.1101/gr.097501.109
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Krachler, K. M., and Orth, K. (2013). Targeting the bacteria-host interface: strategies in anti-adhesion therapy. Virulence 4, 284–294. doi: 10.4161/viru.24606
Lackey, E., Vipulananadan, G., Childers, D. S., and Kadosh, D. (2013). Comparative evolution of morphological regulatory functions in Candida species. Eukaryot. Cell 12, 1356–1368. doi: 10.1128/EC.00164-13
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Lin, J., Oh, S.-H., Jones, R., Garnett, J. A., Salgado, P. S., Rushnakova, S., et al. (2014). The peptide-binding cavity is essential for Als3-mediated adhesion of Candida albicans to human cells. J. Biol. Chem. 289, 18401–18412. doi: 10.1074/jbc.M114.547877
Lohse, M. B., Gulati, M., Johnson, A. D., and Nobile, C. J. (2018). Development and regulation of single- and multi-species Candida albicans biofilms. Nat. Rev. Microbiol. 16, 19–31. doi: 10.1038/nrmicro/2017.107
Lombardi, L., Zoppo, M., Rizzato, C., Bottai, D., Hernandez, A. G., Hoyer, L. L., et al. (2019). Characterization of the Candida orthopsilosis agglutinin-like sequence (ALS) genes. PLoS One 14:e0215912. doi: 10.1371/journal.pone.0215912
Lu, C. F., Kurjan, J., and Lipke, P. N. (1994). A pathway for cell wall anchorage of Saccharomyces cerevisiae alpha-agglutinin. Mol. Cell. Biol. 14, 4825–4833. doi: 10.1128/MCB.14.7.4825
Maguire, S. L., ÓhÉigeartaigh, S. S., Byrne, K. P., Schröder, M. S., O’Gaora, P., Wolfe, K. H., et al. (2013). Comparative genome analysis, and gene finding in Candida species using CGOB. Mol. Biol. Evol. 30, 1281–1291. doi: 10.1093/molbev/mst042
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nielsen, H. (2017). “Predicting secretory proteins with SignalP,” in Protein Function Prediction, Methods in Molecular Biology, Vol. 1611, ed. D. Kihara (New York, NY: Humana Press), 59–73. doi: 10.1007/978-1-4939-7015-5_6
Oh, S.-H., Smith, B., Miller, A. N., Staker, B., Fields, C., Hernandez, A., et al. (2019). Agglutinin-like sequence (ALS) genes in the Candida parapsilosis species complex: blurring the boundaries between gene families that encode cell-wall proteins. Front. Microbiol. 10:781. doi: 10.3389/fmicb.2019.00781
Pei, J., Kim, B.-H., and Grishin, N. V. (2008). PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300. doi: 10.1093/nar/gkn072
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Senol Cali, D., Kim, J. S., Ghose, S., Alkan, C., and Mutlu, O. (2018). Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief. Bioinform. 20, 1542–1559. doi: 10.1093/bib/bby017
Sherman, F., Fink, G. R., and Hicks, J. B. (1986). Laboratory Course Manual for Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Press.
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
White, T. J., Bruns, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungi ribosomal RNA genes for phylogenetics,” in PCR Protocols. A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (San Diego, CA: Academic Press), 315–322. doi: 10.1016/b978-0-12-372180-8.50042-1
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 3:e000132. doi: 10.1099/mgen.0.000132
Yu, S., Li, W., Liu, X., Che, J., Wu, Y., and Lu, J. (2016). Distinct expression levels of ALS, LIP, and SAP genes in Candida tropicalis with diverse virulent activities. Front. Microbiol. 7:1175. doi: 10.3389/fmicb.2016.01175
Zhang, L.-J., Yu, S.-B., Li, W.-G., Zhang, W.-Z., Wu, Y., and Lu, J.-X. (2019). Polymorphism analysis of virulence-related genes among Candida tropicalis isolates. Chinese Med. J. 132, 446–453. doi: 10.1097/CM9.0000000000000069
Zhang, N., Harrex, A. L., Holland, B. R., Fenton, L., Cannon, R. D., and Schmid, J. (2003). Sixty alleles of the ALS7 open reading frame in Candida albicans: ALS7 is a hypermutable contingency locus. Genome Res. 13, 2005–2017. doi: 10.1101/gr.1024903
Zhao, X., Oh, S.-H., Cheng, G., Green, C. B., Nuessen, J. A., Yeater, K., et al. (2004). ALS3 and ALS8 represent a single locus that encodes a Candida albicans adhesin; functional comparisons between Als3p and Als1p. Microbiology 150, 2415–2428. doi: 10.1099/mic.0.26943-0
Zhao, X., Oh, S.-H., Coleman, D. A., and Hoyer, L. L. (2011). ALS51, a newly discovered gene in the Candida albicans ALS family, created by intergenic recombination: analysis of the gene and protein, and implications for evolution of microbial gene families. FEMS Immunol. Med. Microbiol. 61, 245–257. doi: 10.1111/j.1574-695X.2010.00769.x
Keywords: genome, Candida tropicalis, ALS genes, gene expression, fungal adhesion
Citation: Oh S-H, Isenhower A, Rodriguez-Bobadilla R, Smith B, Jones J, Hubka V, Fields C, Hernandez A and Hoyer LL (2021) Pursuing Advances in DNA Sequencing Technology to Solve a Complex Genomic Jigsaw Puzzle: The Agglutinin-Like Sequence (ALS) Genes of Candida tropicalis. Front. Microbiol. 11:594531. doi: 10.3389/fmicb.2020.594531
Received: 13 August 2020; Accepted: 17 November 2020;
Published: 20 January 2021.
Edited by:
Lívia Kmetzsch, Federal University of Rio Grande do Sul, BrazilReviewed by:
Eugenio Mancera, Unidad Irapuato (CINVESTAV), MexicoBruno César Feltes, Federal University of Rio Grande do Sul, Brazil
Copyright © 2021 Oh, Isenhower, Rodriguez-Bobadilla, Smith, Jones, Hubka, Fields, Hernandez and Hoyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lois L. Hoyer, bGhveWVyQGlsbGlub2lzLmVkdQ==