 Satu Olkkola1
Satu Olkkola1 Mirko Rossi2,3Anniina Jaakkonen1
Mirko Rossi2,3Anniina Jaakkonen1 Maria Simola1Jouni Tikkanen1Marjaana Hakkinen1Pirkko Tuominen1Otso Huitu4Jukka Niemimaa4
Maria Simola1Jouni Tikkanen1Marjaana Hakkinen1Pirkko Tuominen1Otso Huitu4Jukka Niemimaa4 Heikki Henttonen4
Heikki Henttonen4 Rauni Kivistö2*
Rauni Kivistö2*- 1Finnish Food Authority, Helsinki, Finland
- 2Department of Food Hygiene and Environmental Health, Faculty of Veterinary Medicine, University of Helsinki, Helsinki, Finland
- 3European Food Safety Authority (EFSA), Parma, Italy
- 4Natural Resources Institute Finland (Luke), Helsinki, Finland
Small mammals are known to carry Campylobacter spp.; however, little is known about the genotypes and their role in human infections. We studied intestinal content from small wild mammals collected in their natural habitats in Finland in 2010–2017, and in close proximity to 40 pig or cattle farms in 2017. The animals were trapped using traditional Finnish metal snap traps. Campylobacter spp. were isolated from the intestinal content using direct plating on mCCDA. A total of 19% of the captured wild animals (n = 577) and 41% of the pooled farm samples (n = 227) were positive for C. jejuni, which was the only Campylobacter species identified. The highest prevalence occurred in yellow-necked mice (Apodemus flavicollis) and bank voles (Myodes glareolus) which carried Campylobacter spp. in 66.3 and 63.9% of the farm samples and 41.5 and 24.4% of individual animals trapped from natural habitats, respectively. Interestingly, all house mouse (Mus musculus) and shrew (Sorex spp.) samples were negative for Campylobacter spp. C. jejuni isolates (n = 145) were further characterized by whole-genome sequencing. Core genome multilocus sequence typing (cgMLST) clustering showed that mouse and vole strains were separated from the rest of the C. jejuni population (636 and 671 allelic differences, 94 and 99% of core loci, respectively). Very little or no alleles were shared with C. jejuni genomes described earlier from livestock or human isolates. FastANI results further indicated that C. jejuni strains from voles are likely to represent a new previously undescribed species or subspecies of Campylobacter. Core-genome phylogeny showed that there was no difference between isolates originating from the farm and wild captured animals. Instead, the phylogeny followed the host species-association. There was some evidence (one strain each) of livestock-associated C. jejuni occurring in a farm-caught A. flavicollis and a brown rat (Rattus norvegicus), indicating that although small mammals may not be the original reservoir of Campylobacter colonizing livestock, they may sporadically carry C. jejuni strains occurring mainly in livestock and be associated with disease in humans.
Introduction
Campylobacter spp., especially C. jejuni and C. coli, are common causes of gastroenteritis in humans globally, campylobacteriosis being the most frequently reported zoonosis in the EU area (EFSA and ECDC, 2019). Many food-producing animal species are known to be asymptomatic carriers of campylobacters, and undercooked chicken is considered to be the main source for human disease, but also other livestock such as bovines have been associated with human cases (Cody et al., 2019; Joensen et al., 2020). However, the original reservoir and mechanisms of spread of these pathogens on livestock farms are not well understood.
Campylobacters are also known to commonly inhabit the digestive tracts of many other warm-blooded animals, including wild birds and small mammals (Meerburg et al., 2006; Backhans et al., 2013; Llarena et al., 2015; Kovanen et al., 2019). Rodents, among other potential hosts, have been suggested to spread Campylobacter spp. on farms (Meerburg and Kijlstra, 2007). Previous research has found a possible link between the presence of rodents, or the absence of control thereof, and higher Campylobacter prevalence in chicken flocks (Kapperud et al., 1993; Berndtson et al., 1996; Sommer et al., 2013; Agunos et al., 2014; Allain et al., 2014; Torralbo et al., 2014). Several rodent species such as the bank vole (Myodes glareolus), yellow-necked mouse (Apodemus flavicollis), house mouse (Mus musculus), and brown rat (Rattus norvegicus) have been shown to carry Campylobacter species in their intestinal tracts (Fernie and Healing, 1976; Healing and Greenwood, 1991; Meerburg et al., 2006; Backhans et al., 2013; Lõhmus and Albihn, 2013). Of these, in particular, M. musculus and R. norvegicus are species that commonly live in close proximity to human habitation, but also M. glareolus and A. flavicollis frequently invade human settlements at the onset of winter.
Little research exists on the prevalence and genotypes of Campylobacter spp. from small mammals in general and especially from those found on swine, beef or dairy farms. A study from Netherlands found approximately 10% of M. musculus and 12.5% (1/8) R. norvegicus from organic swine farms campylobacter-positive, but no shared genotypes with those from pig manure were detected with AFLP typing. No other rodent species or shrews were found to carry campylobacters (Meerburg et al., 2006). In contrast, a study from New Zealand found 11% of the rodents from a dairy farm to harbor campylobacters and identified several shared genotypes between rodent and cattle feces with pulsed-field gel electrophoresis (PFGE) (Adhikari et al., 2004). In Sweden, C. jejuni was more commonly detected from rodents captured in chicken farms while C. coli was more often found from rodents in pig farms, suggesting possible transmission from food-producing animals, but no genotyping was done to support this hypothesis (Backhans et al., 2013). Williams et al. (2010) studied M. glareolus and wood mice (Apodemus sylvaticus) for the presence of Campylobacter spp. both in woodland habitats and on cattle farms in the United Kingdom. They found 41 and 18% of M. glareolus from the woodlands and on farms to carry Campylobacter spp., respectively, while only 1% of the A. sylvaticus, and only on the farms, were positive. Furthermore, all isolates from M. glareolus represented a novel C. jejuni clone, multilocus sequence type (ST) 3704, which was also identified from a calf. This clone was also detected in three of the six positive A. sylvaticus while the other positive A. sylvaticus harbored isolates with ST 61 and ST 583, which were also identified from cattle.
While traditional multilocus sequence typing (MLST), based on seven house-keeping genes, has been widely used for the typing of Campylobacter isolates from various sources, more powerful typing methods based on whole-genome sequencing (WGS) give us considerably higher discriminatory power for phylogenetic analyses and epidemiologic and source attribution studies (Llarena et al., 2017). Also, antimicrobial resistance phenotypes can be predicted based on WGS data in Campylobacter species (Zhao et al., 2015). Only a few studies exist worldwide that utilize new methods such as WGS-based typing on Campylobacter isolates from small wild mammals (Kim et al., 2020). Their role as a source of Campylobacter spp. for food-producing animals in Finland has not been explored previously.
Our aims were (i) to explore the occurrence, genotypes, and resistance markers of Campylobacter spp. isolates from small mammals, mainly rodents and shrews, caught from 40 Finnish beef, dairy, and swine farms, and from natural habitats throughout Finland, (ii) to compare them to isolates collected from other sources, and (iii) to estimate their role as vectors for Campylobacter spp. for Finnish livestock and humans.
Materials and Methods
Sampling
During October and November 2017, rodent traps were set on 40 farms in western and south-western Finland. Of these, 18 were beef cattle farms, two were dairy farms and 20 were swine farms. Both live and snap traps (traps that instantly kill the rodent) – 100 traps in total – were set at and near production buildings and at the edges of the farmyard areas. The small mammals (n = 442) were caught by Natural Resources Institute Finland. The trapping was done over two consecutive nights at every farm and the traps were checked in each day. These were all included as farm samples in the successive analyses.
Additionally, small mammals were collected using snap traps in the national regulatory monitoring of the vole population by the Natural Resources Institute Finland in June 2015 (n = 128), and in May–June and September–November 2017 (n = 384). The trapping sites were located throughout Finland in forest and field habitats (Korpela et al., 2013). These constituted the samples from the wild.
A sample (n = 65) of A. flavicollis trapped inside office and storage buildings in southern Finland in 2010–2015 was also included for comparison.
Sample Preparation and Isolation of Campylobacter spp.
Most of the animals, 86%, were dissected on the day they were trapped. The whole intestines were frozen in dry ice for transport and stored at −80°C in the laboratory. Samples were thawed at room temperature for 1–2 h before starting analysis. For practical reasons, a portion of the animals, 14%, were first stored in dry ice in the field, and then at −80°C as whole animals. After thawing, the intestines were dissected in a similar way to the freshly prepared animals and analyzed immediately.
Sample preparation was performed following the ISO-6887/6 (2013): The intestines and stomachs were first cut into small pieces with a sterile scalpel. Then, the subsamples were pooled so that one pool contained subsamples from a maximum of ten individuals of the same species, caught from a single farm on the same night. After pooling, 1:1 (w/w) buffered peptone water was added to the pooled sample. The sample was cultivated following the ISO-10272-1 (2017) standard Detection procedure C. The colonies were confirmed as C. jejuni using MALDI-TOF (Maldi® Biotyper, Bruker Daltonics GmbH, Germany, reference library version 8.0.0.0) with cut-off at 2.0. The colonies identified as C. jejuni with a score lower than 2.0 were further confirmed with microscopy. After identification, the isolates were stored in media containing brain heart infusion broth and 15% glycerol at −80°C.
Similarly, the intestines from individual animals trapped in their natural habitats were homogenized using a cotton swab dipped in sterile buffered peptone water and subsequently plated on mCCDA plates incubated under microaerobic conditions (5% ± 2% O2, 10% ± 3% CO2, ≤10% H2, balanced with N2; Anoxomat System, Mart Microbiology, Netherlands or ThermoForma Series 2 Water Jacketed Incubator (HEPA filter) Model 3131, Thermo Fisher Scientific, United States) at 41.5 ± 1.0°C for 48–72 h. One typical colony was confirmed as C. jejuni by the lack of aerobic growth on blood agar at 25°C, microscopy, and using species-specific PCR (Denis et al., 1999).
Bacterial Strains, DNA Extraction, and Whole-Genome Sequencing
A selection of C. jejuni isolates from the farm samples (see below) and all 102 isolates from the wild animal samples were subjected to WGS (of which 99 were included in the cgMLST, phylogenetic, ANI, and AMR analyses (Supplementary Datasets), and two additional strains were included only in the MLST and AMR analyses (Table 1); one strain failed at the sequence assembly and QC stage. Of the farm samples, one isolate per farm and per animal species was selected, giving a total of 46 strains. Genomic DNA was extracted with PureLink Genomic DNA kit (Thermo Fisher Scientific, United States) or DNeasy Blood and Tissue kit (Qiagen, United States), according to the manufacturer’s instructions. The purity of DNA samples was tested using NanoDrop-apparatus (Thermo Fisher Scientific, United States), accepting the extraction if the A260/280-ratio exceeded 1.8 and the A260/230 ratio exceeded 2.0. DNA concentration was measured using Qubit-fluorometer (Thermo Fisher Scientific, United States) and Qubit dsDNA Broad Range -kit (Thermo Fisher Scientific, United States). The extracted DNA was stored at −20°C.
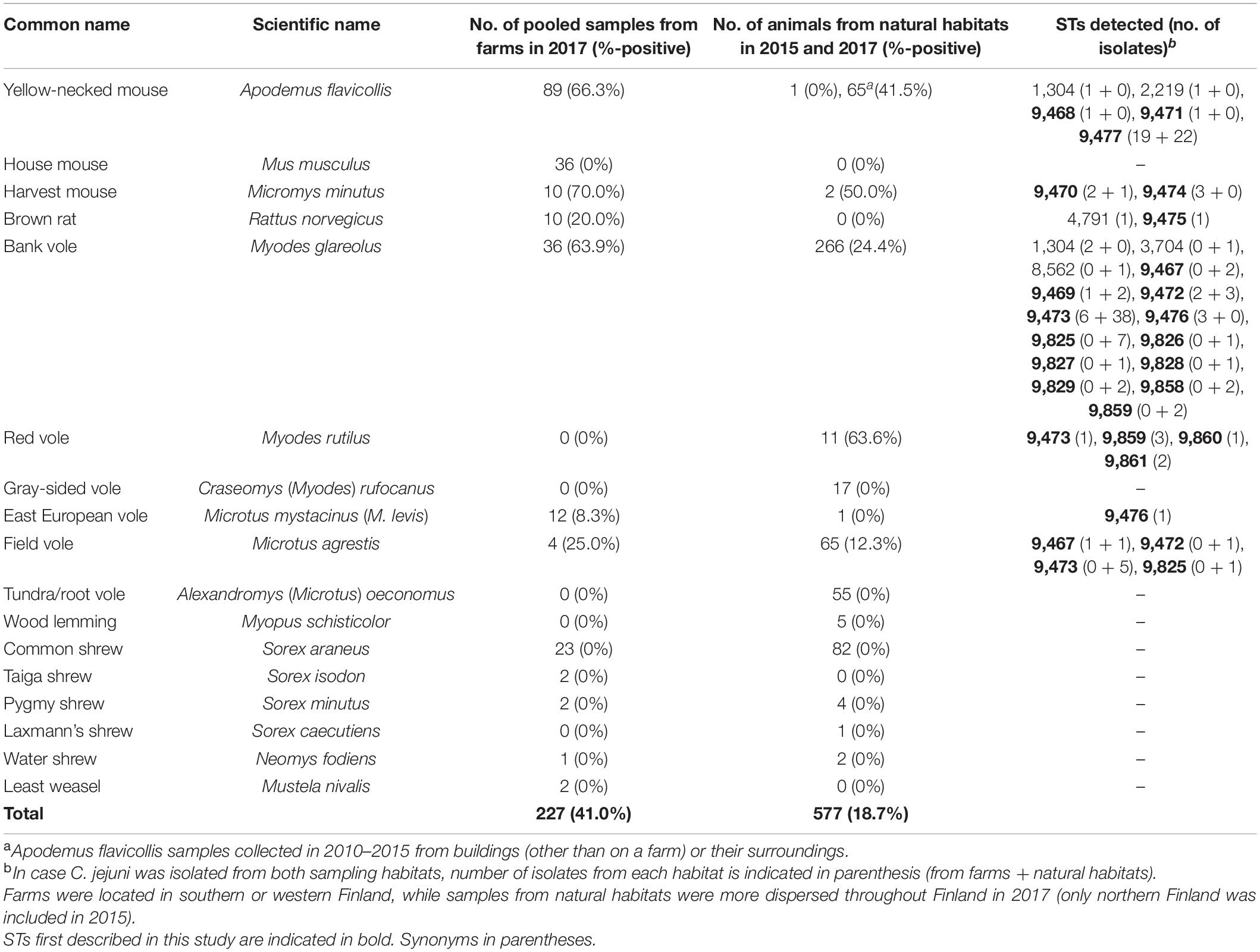
Table 1. Prevalence of Campylobacter jejuni in different animal species and sampling habitats, and sequence types (STs) detected.
WGS of the farm isolates was outsourced to a commercial company (CeGat GmbH, Tuebingen, Germany). The sequencing of the wild animal isolates was performed by Istituto Zooprofilattico Sperimentale Dell’Abruzzo e Molise, Teramo, Italy under a Memorandum of Intent between the faculty and the institution. Sequencing libraries were prepared with a Nextera XT Library Prep kit (Illumina, San Diego, CA, United States) and sequenced on the Illumina NovaSeq6000 or NextSeq platform (Illumina, United States), with a 2 × 100-bp paired-ended protocol according to the manufacturer’s instructions, aiming at >100X coverage.
Assembly, Multilocus Sequence Typing, and Core Genome MLST Analysis
Draft genome sequences were assembled using a docker image of INNUca v3.1 (Machado et al., 2017) available at1. In brief, after calculating if the sample raw data fulfils the expected coverage (min. 15x), INNUca checks read quality using FastQC2, and processes the reads using Trimmomatic (Bolger et al., 2014). Then, INNUca proceeds to de novo draft genome assembly with SPAdes 3.11 (Bankevich et al., 2012), followed by checking the assembly depth of coverage (min. 30x) and improving assembly using Pilon (Walker et al., 2014). Finally, 7-gene MLST sequence type (ST) is automatically assigned with the mlst software (Seemann, 2017). This analysis made use of the PubMLST website3 developed by Keith Jolley (Jolley and Maiden, 2010), sited at the University of Oxford. The development of this website was funded by the Wellcome Trust. Assembly statistics are available in Supplementary Dataset 1.
Core genome MLST analysis was performed using a docker image of the chewBBACA suite (Silva et al., 2018) available at4 and the INNUENDO cgMLST schema available in Zenodo5 (Rossi et al., 2018), consisting of 678 loci. In addition to 145 sequenced C. jejuni genomes from rodents and shrews, the cgMLST analysis included 6,526 genomes belonging to the reference INNUENDO dataset (Rossi et al., 2018) and other genomes previously sequenced (Kovanen et al., 2019), 6,830 genomes in total. The genomes had no more than 2% of missing loci. Metadata, including year of isolation, country, 7-gene MLST ST, and source for all the strains, are available in Supplementary Dataset 2. The allelic profiles were clustered using the globally optimal eBURST (goeBURST) algorithm (Francisco et al., 2009) in PHYLOViZ V 2.0 software (Francisco et al., 2012), and clusters were generated at all possible thresholds.
Allele Segregation
To analyze allele segregation according to source, the cgMLST profiles of a total of 4,323 strains for which source information was available were analyzed using proCompare.py (Pinho et al., 2016) available at6. Briefly, the software performs a pairwise comparison searching those loci with shared (or exclusive) alleles between two groups.
Average Nucleotide Identity Calculations
To evaluate the taxonomical position of the newly described C. jejuni genomes, all-against-all pairwise average nucleotide identity (ANI) values for the mouse and vole strains were calculated using FastANI (v1.0) with default parameters (Jain et al., 2018). In addition, ANI values were calculated between each strain and the following reference genomes: C. jejuni subsp. jejuni NCTC 11168 (GenBank Accession number: NC_002163.1), C. jejuni subsp. jejuni RM1221 (NC_003912.7), C. jejuni subsp. doylei 269.97 (CP000768.1), C. coli 76339 (NC_022132.1), and C. coli OR12 (NZ_CP019977.1).
Phylogenomics
A phylogenetic tree was constructed based on the core genomes of 1,406 C. jejuni strains selected among the 6,830 genomes, as described above, excluding the samples isolated from humans and those without information on source (Supplementary Dataset 2). The genomes were annotated using a docker image of prokka 1.12 (Seemann, 2014) available at7, and pan-genome analysis was performed using a docker image of Roary 3.7.0 with default settings (Page et al., 2015) available at8. The jobs were run in parallel using GNU Parallel (Tange, 2011). FastTree 2.153 (Price et al., 2009, 2010) was used with the Jukes-Cantor model of nucleotide evolution for building the approximation of a maximum-likelihood (ML) phylogenetic tree based on the core genome alignment (99% shared loci in Roary analysis, including 864 core genes). iTOL54 v4.2.3 (Letunic and Bork, 2019) was used for visualization. The tree was rooted at midpoint.
For improving the resolution of the phylogeny of the vole and mice specific lineages, ad hoc pangenome analysis was performed. The resulting nucleotide multiple core genome alignment files were used as an input for phylogenetic inference using maximum likelihood methodology implemented in iqtree (Nguyen et al., 2015), applying model selection (Kalyaanamoorthy et al., 2017) and ultrafast bootstrapping (Hoang et al., 2018). The core-genome alignments and the ML trees generated by iqtree were used for the subsequent assessment of recombination using ClonalFrameML (Didelot and Wilson, 2015) ignoring sites with any ambiguous bases. The trees were rooted at midpoint.
Antimicrobial Resistance
The antimicrobial resistance determinants of the isolates were searched from the quality-controlled assemblies using default settings in ResFinder 3.2 (Zankari et al., 2012), accessed April 24th and 25th, 2020. The resistance gene sequences were aligned using Clustal Omega9 and blasted in the NCBI Blast nucleotide10 against the nucleotide collection (nr/nt) (retrieved September 25th, 2020).
Statistical Analysis
Statistical significance of the observed differences in Campylobacter prevalence between different sampling times and locations were tested using the Chi-square or Fisher’s Exact tests in Microsoft Excel 2016. Statistically significant differences were considered for p-values below 0.05.
Results
A. flavicollis, M. musculus, Micromys minutus (harvest mouse), and R. norvegicus were almost exclusively caught on-farm (year 2017) or inside buildings (years 2010–2015, A. flavicollis collection) (Table 1). The distribution of red voles (Myodes rutilus), tundra/root voles (Alexandromys oeconomus), and gray-sided voles (Craseomys rufocanus) are limited to northern parts of Finland and accordingly, these species were not caught on farms in south-western or western Finland. M. glareolus, Sorex araneus (common shrew), and Microtus agrestis (field vole) were common among both in-farm caught samples and those collected from natural habitats throughout Finland.
Prevalence of Campylobacter spp. Was Highest in A. flavicollis
A total of 41% (93/227) of the pooled samples from farms, and 19% (75/384 in 2017, 6/128 in 2015, and 27/65 for A. flavicollis) of the individual animals captured from office and storage buildings were positive for C. jejuni (Table 1), which was the only Campylobacter species identified. The highest prevalence of C. jejuni occurred in A. flavicollis, M. minutus, and M. glareolus, M. rutilus, and M. agrestis. Interestingly, all shrew, Alexandromys (Microtus) oeconomus (tundra/root vole), and M. musculus samples were negative for Campylobacter species.
M. glareolus from natural habitats was more likely to carry Campylobacter when caught from fields (60%) compared to forests (18%) (p < 0.0001) in 2017. The samples from fields were comparable in Campylobacter prevalence with those from farms. In natural habitats, M. glareolus was significantly more often Campylobacter-positive in southern Finland (42%) compared to Lapland (19%) in northern Finland (p < 0.0001) in 2017. In Lapland, Campylobacter prevalence in M. glareolus was significantly higher in 2017 (19%) compared to 2015 (7%) (p = 0.0001). For other parts of the country (excluding Lapland), we had samples only from 2017.
MLST Analysis Revealed Genotypes Differing From Livestock and Humans
Only seven out of the 147 C. jejuni isolates from mice, rats, or voles matched a previously defined 7-gene MLST profile, most of which were associated with a clonal complex (CC): ST 1304 (n = 3, ST-1304 CC), ST 2219 (n = 1, ST-45 CC), ST 3704 (n = 1), ST 4791 (n = 1, ST-45 CC), or ST 8562 (n = 1) (Supplementary Dataset 1). For the rest of the dataset, new alleles or new combinations of alleles were detected (Table 1). All the isolates with STs belonging to ST-45 CC or ST-1304 CC (n = 5) were from rodents captured from farms. ST-45 CC was isolated from A. flavicollis from a pig farm (ST 2219) and from R. norvegicus from a cattle farm (ST 4791). ST 1304 was isolated from A. flavicollis from a cattle farm and from two M. glareolus from pig and cattle farms. Two other previously identified STs, ST 3704 and ST 8562, were both isolated from M. glareolus from a forest in central Finland and from northern Finland, respectively.
Novel STs that were most prominent in the dataset included ST 9473 from M. glareolus, M. agrestis and M. rutilus, and ST 9477 only identified in A. flavicollis. ST 9473 was identified among C. jejuni isolates from M. glareolus from both on-farm (pig and cattle farms) and natural habitats in both 2015 and 2017. ST 9477 was identified among C. jejuni isolates from A. flavicollis from both on-farm (pig and cattle farms) and other buildings (office and storage buildings) in southern Finland from 2010 to 2017. In addition, ST 9470 was isolated from M. minutus and ST 9467 from M. agrestis caught both on-farm and from natural habitats. Furthermore, the same STs occurred in C. jejuni isolates from both M. agrestis and M. glareolus.
cgMLST and Allele Segregation Analyses Further Highlighted the Differences
Of the 138 C. jejuni strains isolated from voles and mice with previously undefined ST designation, the cgMLST based on 678 loci yielded a total of 105 unique profiles. This revealed that the C. jejuni strains isolated from these animals form distinct populations that diverge largely from the ones described so far. Changes in the composition of clusters was investigated at all goeBURST thresholds. At the threshold of 671 allelic differences (99.0% of the core loci), the C. jejuni strains from voles formed two separate clusters from the rest of the strains. At 636 allelic differences (93.8% of core genes), the C. jejuni strains from mice separated from the larger cluster (composed by strains isolated from livestock, humans, and wild birds), which was split further into two sub-clusters at 531 allelic differences (78.3% of core genes). One contained strains isolated exclusively from A. flavicollis and one was composed of strains isolated exclusively from M. minutus (Supplementary Dataset 3). Descriptive statistics of the pairwise distance among strains of each group are summarized in Table 2.

Table 2. Pairwise allelic distance within each goeBURST group defined at threshold 531 allelic differences based on core genome multilocus sequence typing (cgMLST) schema composed by 678 loci.
Considering the large diversity in the cgMLST profiles observed between mouse and vole strains and the rest of the C. jejuni population, we investigated how many core loci alleles were shared between C. jejuni isolates obtained from different taxa. To perform this study, we searched how many times an allele detected in a C. jejuni strain isolated from one mouse or vole taxon was also detected in a C. jejuni strain isolated from another taxon. For this analysis, seven strains isolated from mice and voles but not belonging to the four goeBURST groups described above were excluded. Supplementary Dataset 4 shows the results of the pairwise comparison. The C. jejuni isolates obtained from A. flavicollis shared 184 loci with strains isolated from M. minutus and a median of 2 (max 6) and 53 (max 141) with voles and other taxa, respectively. Similar results were obtained for strains isolated from M. minutus. Very few alleles were shared between strains isolated from voles and any other taxa, 18 being the maximum number of shared alleles detected. Overall, this analysis confirmed the divergent nature of the C. jejuni population circulating both in mice and voles.
FastANI and Phylogenetic Analyses Revealed That Vole Isolates Probably Form a Novel (Sub-)Species of Campylobacter
The little or no sharing of alleles between the C. jejuni isolated from voles and other taxa is an indication that these strains might form a different Campylobacter species or subspecies. To verify this hypothesis, we calculated the ANI percentage between mouse and vole strains and reference strains from C. jejuni subspecies and C. coli (Table 3). Compared with the reference genomes of C. jejuni and C. coli, the ANI values calculated for vole strains were significantly lower than the ones calculated for the mouse strains (unpaired t-test; P < 0.001). The mouse strains had >95% ANI versus C. jejuni subspecies and <87% ANI versus C. coli. On the contrary, the vole strains had on average 90.9% ANI versus C. jejuni subspecies and <84% ANI versus C. coli. Both Campylobacter strains from voles and mice formed cohesive groups with ANI average at 99% and min. 96%. This data confirmed the hypothesis that Campylobacter strains from voles might form a different taxonomic group. The maximum likelihood phylogenetic tree based on the alignment of 864 core genes supports the ANI results (Figure 1). The vole strains formed a distinct clade clearly separated from C. jejuni, while the strains isolated from mice grouped monophyletically within the diversity of the C. jejuni population.
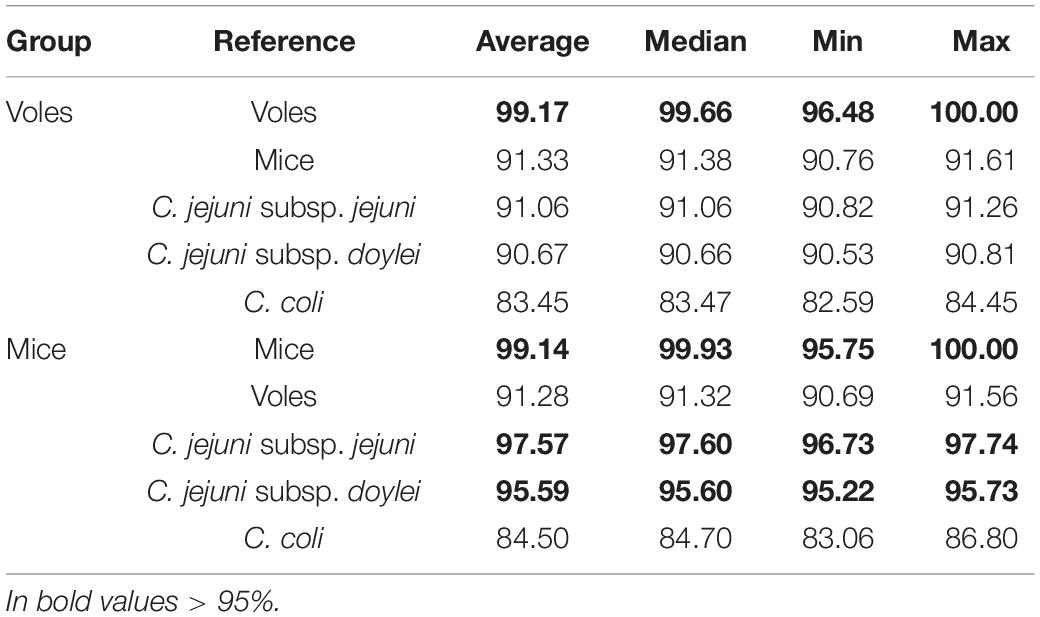
Table 3. All-versus-all average nucleotide identity (ANI%).
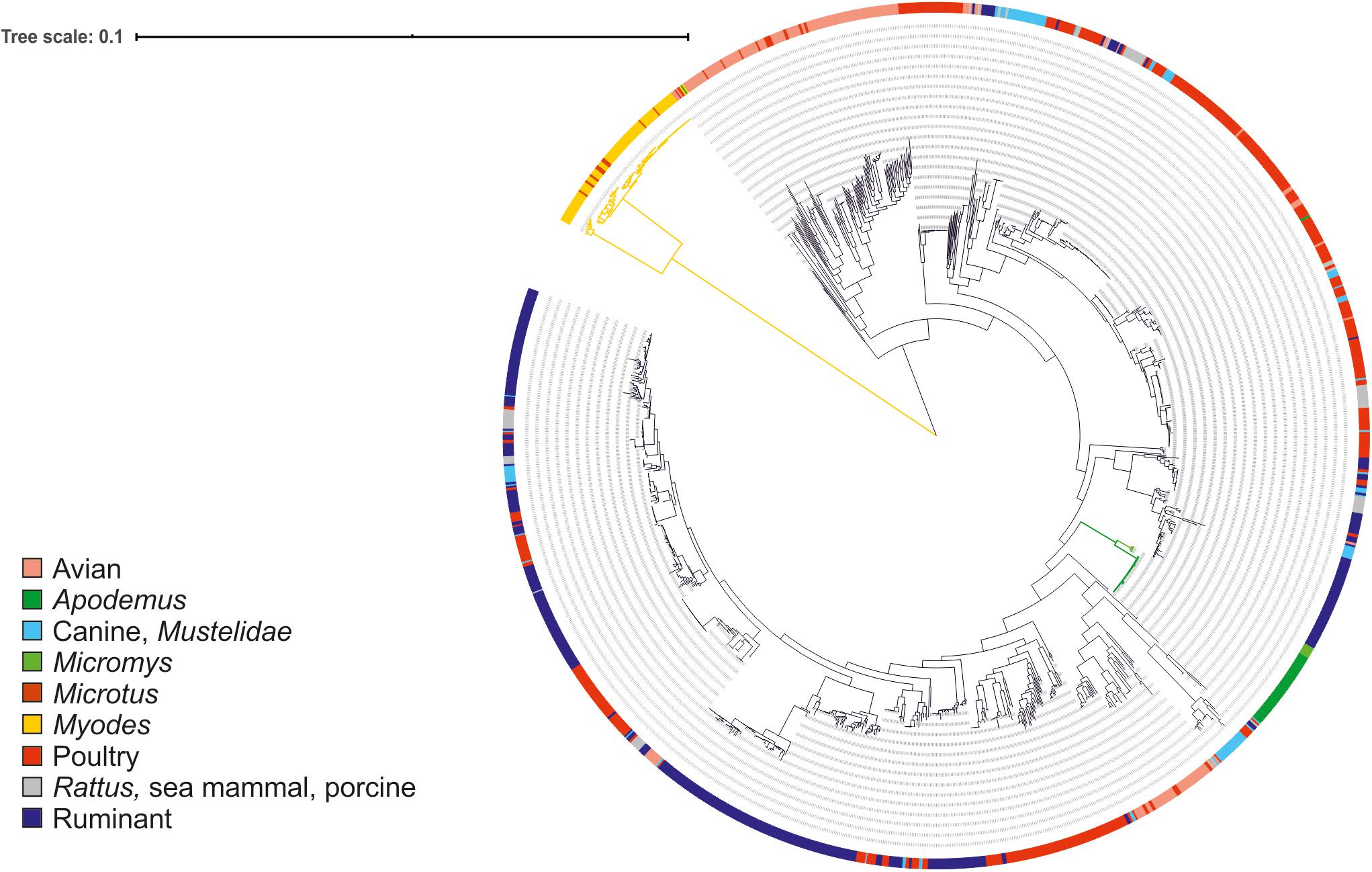
Figure 1. Maximum likelihood phylogenetic tree based on the alignment of 864 core genes of 1,406 C. jejuni strains from various sources. The tree was rooted at mid-point. The clade associated with voles is shown in yellow and the clades associated with mice in green. Color in the external ring indicates the source of the strain (see legend).
For investigating the presence of phylogeographical or temporal signals and to see whether there were differences between strains isolated from animals captured in the proximity of the farm or not, we reconstructed the genealogy of the mouse and vole specific lineages based on ad hoc core-genome alignment. The phylogenetic reconstructions were based on a core genome alignment of 1,238 and 1,402 genes for the vole and the mouse lineages, respectively. For the vole lineages, ClonalFrameML detected a total of 1,059 recombination events, 337 on terminal branches and the rest on internal nodes, and the imported sizes ranged between 2 and 103,802 bp (median 419). For the mouse lineages, 225 recombination events were detected, 32 on terminal branches and the rest on internal nodes, and the imported sizes ranged between 2 and 6,597 bp (median 712). The posterior mean calculated for the ratio of recombination events compared to mutation (rho/theta) was 0.342058 (posterior variance 1.82762 × 10–05) for the vole and 0.111041 (posterior variance 2.78479 × 10–05) for the mice. This analysis showed that mutation was approximately 3 and 9 times more frequent than recombination in the vole and mouse lineages, respectively.
Geographical and Temporal Differences Between Genotypes Were Small
Figures 2, 3 show the genealogies of the vole and mouse lineages, respectively, alongside the information concerning the site and time of capture of the animals. The Campylobacter strains did not segregate clearly according to space or time, and no major differences were observed between animals captured within farms and those captured in the wild. However, the smaller cluster of Campylobacter strains from voles, which diverged significantly from the main population (Figure 2), consisted of samples collected only from one location in Lapland and represented all except one isolate from M. rutilus (orange taxon) and few isolates from M. glareolus (yellow taxon). Furthermore, for the Campylobacter isolates from mice, the smaller cluster consisted of strains from M. minutus (orange taxon) and the larger one from A. flavicollis (purple taxon) (Figure 3). The Campylobacter strains from A. flavicollis showed some minor clustering according to location.
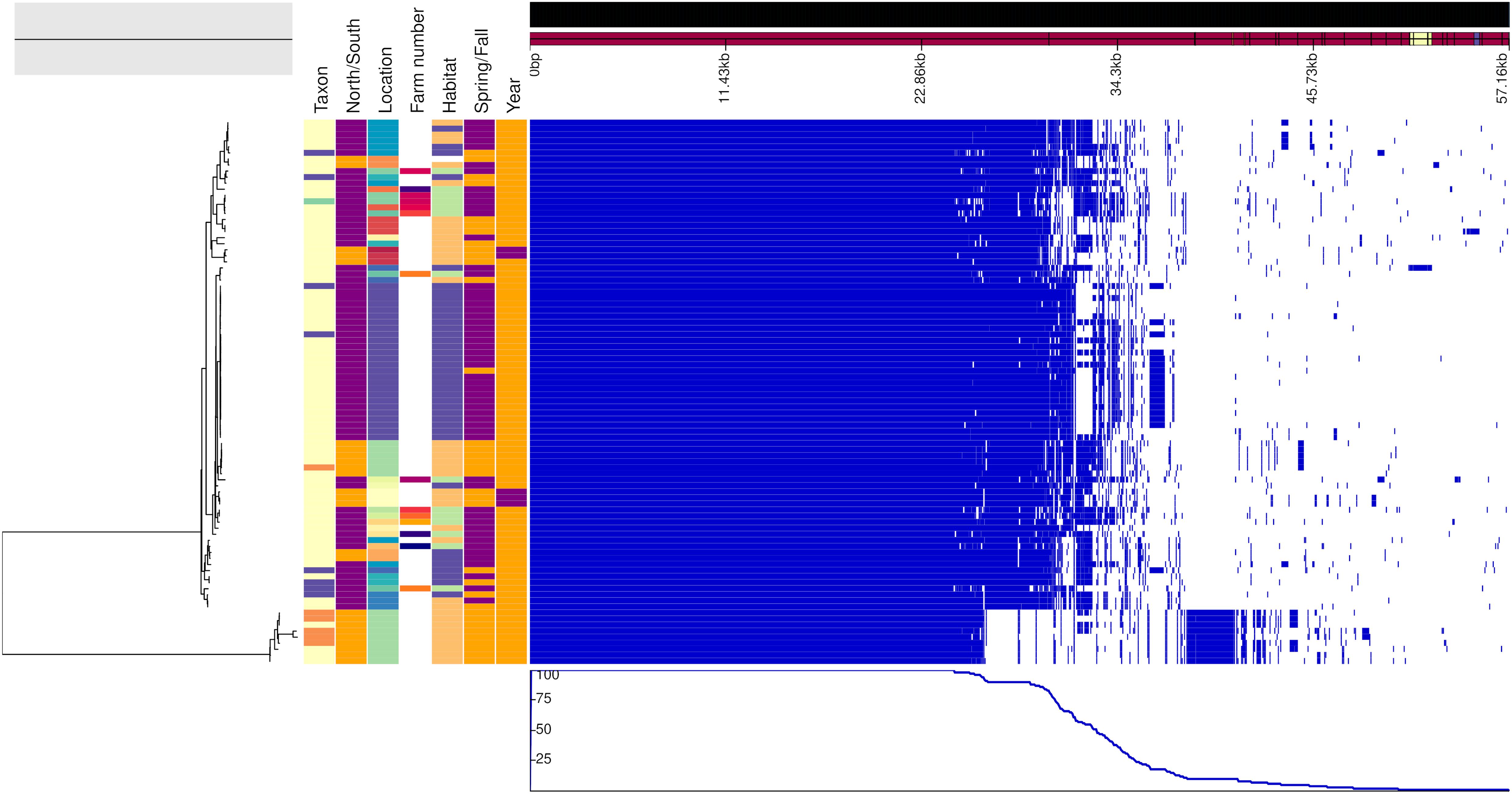
Figure 2. Genealogy of vole-associated Campylobacter lineage (left) with associated metadata, i.e., taxon (M. glareolus in light yellow, M. rutilus in orange, M. agrestis in violet, and M. mystacinus in turquoise), geographical area (north Finland in orange and south in purple), location, farm number (if applicable), habitat (field in blue, forest in orange, and farm in green), time (spring in orange and fall in purple) and year (2015 in purple and 2017 in orange), displayed alongside the gene presence (blue)/absence (white) plot from the Roary pangenome analysis. The figure was drawn using the phandango.net web application (Hadfield et al., 2017). The phylogeny based on 1,238 core genes was reconstructed using ClonalFrameML (Didelot and Wilson, 2015) and rooted at mid-point.
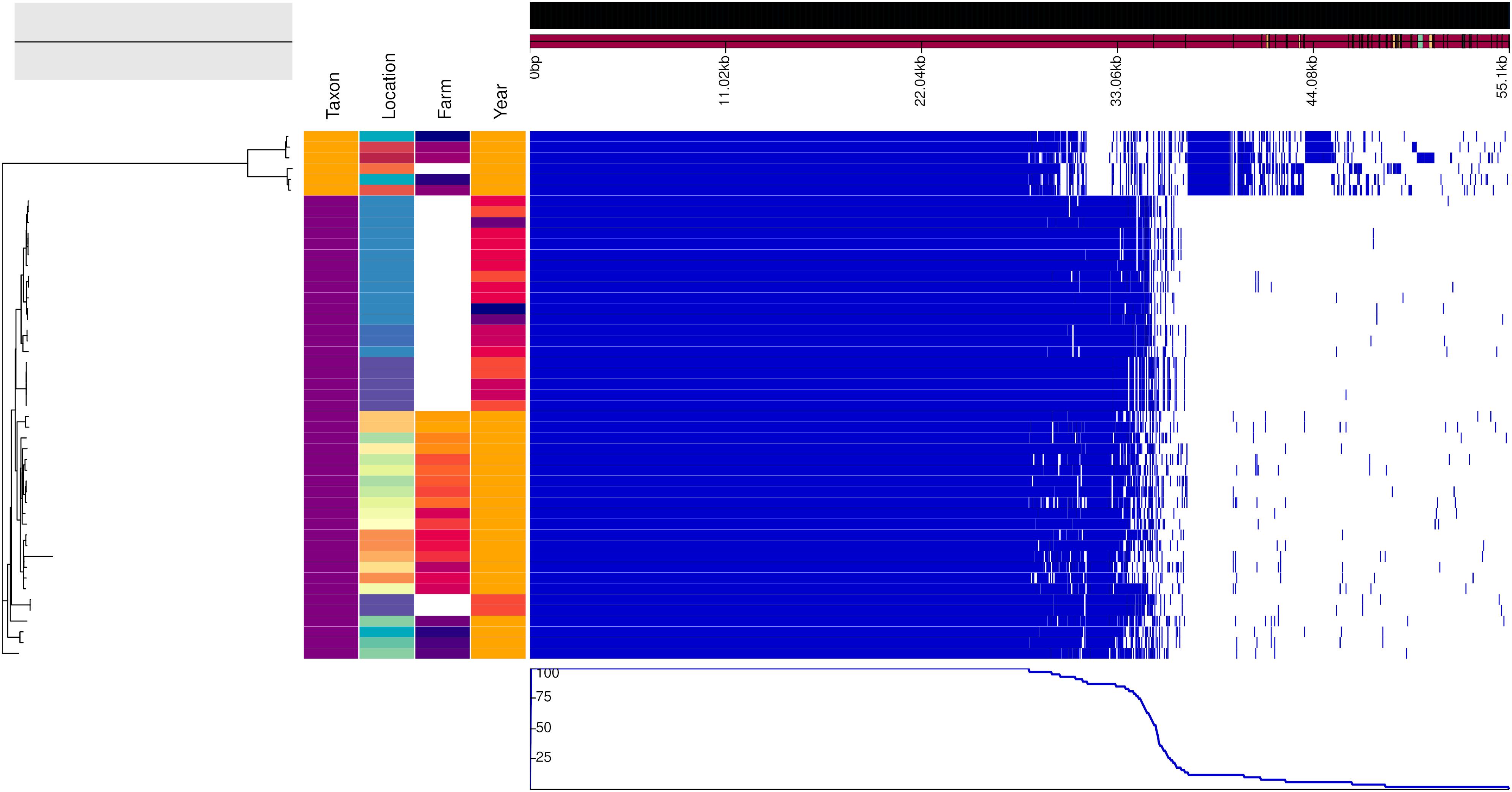
Figure 3. Genealogy of mice-associated Campylobacter lineage (left) with associated metadata, i.e., taxon (A. flavicollis in purple and M. minutus in orange), location, farm (if applicable) and year (2017 in orange, 2015 in dark orange, 2014 in light red, 2013 in dark red, 2011 in violet, and 2010 in blue), displayed alongside the gene presence (blue)/absence (white) plot from the Roary pangenome analysis. The figure was drawn using the phandango.net web application (Hadfield et al., 2017). The phylogeny based on 1,402 core genes was reconstructed using ClonalFrameML (Didelot and Wilson, 2015) and rooted at mid-point.
Antimicrobial Resistance Markers
We found a nucleotide substitution resulting in P104S amino acid change in GyrA in 62.6% (92/147) of the C. jejuni isolates (Table 4). No other known resistance-associated mutations were identified using Resfinder 3.2. The remaining isolates apart from one, 36.7% (54/147) harbored a beta-lactamase gene, the type of which was associated with the ST. Of these, 79.6% (43/54) had a nucleotide (T) deletion at position 69 of the betalactamase gene resulting in a frameshift and premature stop codon at amino acid 35 and most likely a non-functional gene product (all from A. flavicollis, mainly ST-9477). Only one isolate had a beta-lactamase gene with 100% nucleotide identity to that previously deposited to Genbank. Not surprisingly, this isolate, derived from A. flavicollis trapped on a swine farm, had a previously recognized ST (ST 2219, ST-45 CC). All isolates that had the same ST carried identical resistance markers. Both the gyrA mutation and beta-lactamase genes were never present in the same isolates, and only one isolate [from R. norvegicus trapped from a cattle farm having ST 4791 (ST-45 CC)] had neither.
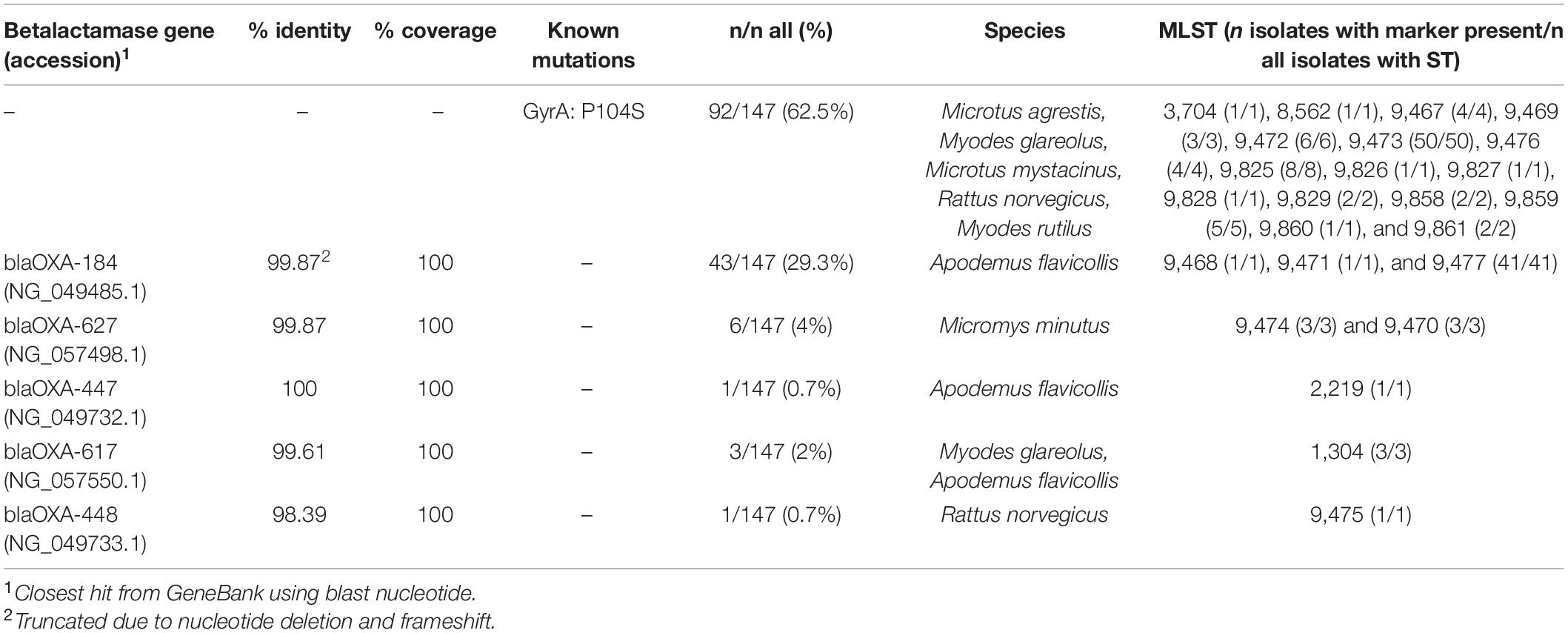
Table 4. Detected resistance markers.
Discussion
Prevalence of C. jejuni in Rodents and Shrews
Campylobacter jejuni occurred in nearly all animal species sampled, even those collected from the less populated northern parts of Finland (including red, tundra, and gray-sided voles). The prevalence of Campylobacter was slightly higher among animals trapped on farms compared to those caught in their natural habitats. However, this is likely due to the fact that the farm samples were pooled from 1–10 individual animals compared to the animals from natural habitats which were studied individually. A. flavicollis were almost exclusively (except for one) caught near or inside buildings and mainly in the fall, and showed the highest prevalence (42% for individual animals trapped from natural habitats, 66% for pooled farm samples) of Campylobacter. Previously, C. jejuni has rarely been isolated from A. flavicollis (1/45, 2%), from human dwellings during the cold season in rural areas around Uppsala, Sweden (Lõhmus and Albihn, 2013). In another study from Sweden, C. jejuni was isolated from 5/18 (28%) and C. upsaliensis from 2/18 (11%) A. flavicollis, respectively, from pig farms, chicken farms, and non-farm locations in Sweden (Backhans et al., 2013), showing more comparable results with our study, which included a total of 155 samples from this species. However, all the isolates from our study were identified as C. jejuni. In this study, no Campylobacter spp. were detected in A. flavicollis caught in one location for three successive years. Among the other locations and years studied, the prevalence of Campylobacter ranged from 20 to 86%.
M. glareolus, which was common among both samples from farms and from natural habitats, also showed a high prevalence of Campylobacter (24% of individuals, 64% of pooled farm samples). Another common mammal that is widespread in Europe, M. agrestis, was less often (12% of individuals, 25% from pooled farm samples) positive for Campylobacter than M. glareolus. Previous studies have reported contradictory results concerning the occurrence of campylobacter in M. glareolus. While research from the United Kingdom and Sweden showed a prevalence of 18–77% (Fernie and Healing, 1976; Williams et al., 2010; Lõhmus and Albihn, 2013), a study from Norway (Rosef et al., 1983) found no positive animals, suggesting possible geographical differences in the occurrence, even though methodological variation might also have influenced the outcomes. Furthermore, Campylobacter spp. have not been isolated from M. agrestis either in the United Kingdom or in Sweden (Fernie and Healing, 1976; Meerburg et al., 2006), which also suggests geographical differences.
In Lapland (Pallasjärvi, Muonio), M. glareolus had a clearly higher prevalence in 2017 than 2015. A continuous long-term monitoring of vole dynamics has been running in this collection area since 1970 (Henttonen et al., 1987; Henttonen, 2000). Consequently, we can compare the campylobacter infection parameters with the vole densities. The spring–fall density indices (animals per 100 trap nights) of M. glareolus during 2013–2018 were: 2013: 0.3–5.1; 2014: 5.0–25.3; 2015: 11.1–31.3; 2016: 12.6–23.6; 2017: 5.2–3.8; 2018: 0.1–4.4. Thus, sampling in 2015 was in the middle of an extended high-density period while in 2017 it was in the decline–low phase. Prevalence was not directly density-dependent, rather there seemed to be a delayed density-dependent pattern. Parasitological parameters in small mammals depend largely on the population structure of the sample (sex, age structure, breeding or not, etc.) and we emphasize that this comparison was made between similar samples, i.e., breeding bank voles in early summer. Even if the exact reason for the great difference between 2 years is not known, it is important to realize generally in epidemiological sampling that temporal differences can be pronounced.
M. musculus and S. araneus were consistently Campylobacter negative despite being quite common among the studied samples. A similar finding concerning M. musculus was presented recently from South Korea (Kim et al., 2020) involving 49 M. musculus trapped on sesame fields. In another study conducted on pig farms, chicken farms, and at non-farm locations in Sweden, however, C. jejuni was isolated from 2%, C. coli from 12% and C. upsaliensis from 2% of the M. musculus samples, respectively (Backhans et al., 2013). The authors concluded that C. jejuni was more common on chicken farms and C. coli on pig farms, suggesting that rodents are not the original source of Campylobacter on farms but rather become carriers through contact with the feces of farm animals. However, in France C. jejuni, instead of C. coli, was identified from 12% of the M. musculus batches on pig farms (LeMoine et al., 1987). In another study, one (7%) C. hyointestinalis, two (13%) C. coli, and three (20%) C. jejuni strains were isolated from 15 M. musculus caught on a Dutch organic pig farm (Meerburg et al., 2006). Campylobacter spp. were, however, not isolated from the pig manure collected at the same farm. In the same study, on two other farms C. coli was isolated from 1/6 (16.7%) and Campylobacter spp. from 1/30 (3.3%) of M. musculus, respectively.
Concerning S. araneus, our results were in line with a previous study in which all shrews (10 S. araneus common shrews and 119 Crocidura russula greater white-toothed shrews), were negative for Campylobacter species (Meerburg et al., 2006). Another study, however, reported isolation of C. jejuni from the spleens of one water (Neomys fodiens) and one common shrew (S. araneus), even though they were not isolated from the gut, in the United Kingdom (Healing and Greenwood, 1991). Thus, it is also possible that the spleens of the animals collected in our study would have been positive for Campylobacter, but this was not tested.
Rattus norvegicus are widespread throughout most of the world and have viable populations in Finland. They are especially adapted to living in close proximity to human habitation and they may cause large economic losses by destroying materials, eating and defecating on food and feed, and by spreading disease. In this study, C. jejuni was identified in 20% (2/10) of the pooled R. norvegicus samples from farms. Previous studies have also shown that R. norvegicus may be carriers of Campylobacter. C. jejuni was isolated from three and C. coli from nine percent of R. norvegicus, respectively, collected from pig farms, chicken farms, and non-farm locations in Sweden (Backhans et al., 2013). In France, C. jejuni was identified from 40% of R. norvegicus collected from pig farms (LeMoine et al., 1987). Another study identified C. coli from 12.5% of R. norvegicus caught on a pig farm in Netherlands (Meerburg et al., 2006).
Genotyping
For WGS, we chose farm isolates that represented all the different animal species in each sampling and, whenever possible, animals caught inside or in close proximity to production or storage buildings. In addition, all isolates from animals trapped in natural habitats were included for WGS analysis. The MLST types that occurred among the Campylobacter isolates differed mostly from the ones previously described from livestock and humans, and the majority of the STs were novel. A previously defined ST was available for only seven Campylobacter isolates out of all the 147 typed isolates in our study. Markedly, the majority (5/7) of these were isolated from on-farm pooled samples. These STs have previously been isolated from, e.g., wild birds in Sweden and New Zealand (ST 1304, ST-1304 CC), chicken offal or meat in Denmark (ST 4791, ST-45 CC), and various sources including chicken, cattle, dog, and human gastroenteritis worldwide (ST 2219, ST-45 CC). ST 2219 was isolated from A. flavicollis (one strain isolated from a pig farm sample), and the other STs from R. norvegicus, M. glareolus, and A. flavicollis from on-farm samples. Our findings are in agreement with previous studies finding also similar genotypes from small mammals and livestock (Adhikari et al., 2004; Williams et al., 2010), underlining the need for stringent biosecurity measures on farms. Unfortunately, food-producing animals were not sampled for Campylobacter in our study and thus we were unable to test the hypothesis that the C. jejuni isolates having ST-45 CC or other known STs originated from the farm animals. Furthermore, only one colony per pooled sample was analyzed and it is possible that an even higher proportion of livestock-associated STs might have been detected if several colonies were picked.
To our knowledge, MLST or WGS has only previously been used for typing Campylobacter isolates from small mammals in a South Korean study investigating isolates from M. minutus trapped in sesame fields (Kim et al., 2020), and in a United Kingdom study targeting M. glareolus and A. sylvaticus caught from a woodland habitat and six cattle farms (Williams et al., 2010). Our isolates, including those from M. minutus, differed in all seven loci of the MLST scheme from those reported from South Korea. The Campylobacter strains from the Finnish M. minutus trapped on-farm and from natural habitats in different locations, however, shared the same ST and the strains were very clonal, suggesting recent clonal expansion of the population in Finland. Williams et al. (2010) reported a single clonal C. jejuni population from M. glareolus with all the isolates representing the same ST 3704 regardless of whether they were caught from woodland or on farms. In contrast, we found only one C. jejuni having this ST from M. glareolus trapped in the woods while the remaining isolates had other, mainly novel sequence types. This finding most likely reflects geographical and temporal differences in the M. glareolus populations and expands our knowledge of this species as a carrier of different C. jejuni strains. However, we also found the same STs regardless of the trapping site (woods, field, or farm), further suggesting C. jejuni lineages specific to M. glareolus.
Concerning R. norvegicus, the Dutch study identified C. coli from 1/8 (12.5%) R. norvegicus caught on a pig farm in Netherlands (Meerburg et al., 2006). Amplified fragment length polymorphism (AFLP) typing, however, showed that the genotype differed significantly from that isolated from pig manure on the same farm (Meerburg et al., 2006). In another study, 86.7% of rats collected at a duck farm were Campylobacter positive (Kasrazadeh and Genigeorgis, 1987). The authors concluded that the most probable source of colonization of the ducks by C. jejuni were the rats and mice found in abundance on the premises, since rat and mice droppings were found in the duck feeding and watering troughs in that study. In our study, however, very few rats were caught, which is likely due to their neophobic behavior and the short sampling period on the farms. Thus, we could not thoroughly evaluate their role as a reservoir for Campylobacter. Our results, however, suggest that R. norvegicus may carry STs also identified in livestock, making them a possible vector of Campylobacter on farms. Since rats are prevalent in urban and rural settings, live in sewers and are in contact with human and livestock wastes, they would be an interesting subject for further studies.
cgMLST clustering showed that the strains from mice and voles were quite separate from the rest of the C. jejuni population (over 600 allele difference). The majority of the cgMLST alleles were segregated according to species (A. flavicollis and M. minutus) or group (vole). Very few or no alleles were shared with livestock or human Campylobacter strains. Core-genome phylogeny of the animal-associated strains confirmed the separation of the lineages as observed in cgMLST. The strains from voles formed a distinct clade that was clearly separate from C. jejuni, while the strains isolated from mice grouped monophyletically within the diversity of the C. jejuni population. Moreover, there was no difference between Campylobacter strains from farm versus wild captured animals, whose phylogeny followed the animal species-association indicating adaptation to different host species. The only major exceptions to this were the clustering of different Campylobacter strains from M. glareolus, M. agrestis, and M. mystacinus (syn. M. levis) isolated from different locations throughout Finland (both on-farm and natural habitats), and M. glareolus and M. rutilus isolated from northern Finland. The overlap in habitat selection among vole species and the commonness of M. glareolus could explain the spread of Campylobacter from M. glareolus to other sympatric vole species. The larger cluster of vole strains also contained the bank vole having ST 3704, the genotype that was previously also identified in the United Kingdom (Williams et al., 2010). The smaller cluster of isolates from voles was, however, particularly associated with M. rutilus in northern parts of Finland and seems more likely to have spread from this closely-related species to M. glareolus. The new strain/variant of campylobacter found in the red vole, M. rutilus, is interesting since this vole species has a wide northern distribution in the Holarctic; from northernmost Fennoscandia over north Russia and Siberia to northern North America, which may suggest that this strain could be widely spread in northern regions.
FastANI further indicated that the vole strains might form a different species or sub-species of Campylobacter. Usually a species is defined to include strains that share ≥95% ANI (Jain et al., 2018). However, C. coli is known to form lineages (clades 1, 2, and 3) that are almost as separate as different species. The ANI value for C. coli clade 3 versus clade 1, which included most of the human and farm animal isolates (Sheppard et al., 2013; Skarp-de Haan et al., 2014), for example, is ∼92%. The Campylobacter strains from voles showed 90 to 91% ANI with C. jejuni and <85% ANI with C. coli (same with C. hepaticus), while strains from mice shared ≥95% ANI with both C. jejuni subspecies, clearly indicating that the mice strains belong to C. jejuni, more specifically C. jejuni subsp. jejuni. The isolates from voles tested hippurate positive in the phenotypic hippurate hydrolysis test. However, the majority of them were only weakly identified as C. jejuni using MALDI-TOF-MS, further suggesting they may represent a novel sub-species.
Antimicrobial Resistance Markers
Clinically relevant resistant markers were not found in our study. The only known mutation that was identified was that leading to the amino acid change P104S in the Gyrase subunit A, which has been linked to fluoroquinolone resistance only in connection with other mutations in gyrA (Hakanen et al., 2002; Piddock et al., 2003; Payot et al., 2006). It is therefore unlikely that the isolates in this study would be resistant to quinolones. Additionally, slightly more than one-third of the isolates harbored a beta-lactamase gene. This proportion is lower compared to some previous studies that report even 80–90% of C. jejuni strains isolated from various sources producing beta-lactamases and/or harboring beta-lactamase genes (Lachance et al., 1991; Tajada et al., 1996; Marotta et al., 2019). Although the presence of beta-lactamases has been shown to lower the MICs for certain beta-lactams in Campylobacter spp., campylobacters are generally considered intrinsically resistant to many agents in this group and beta-lactams are not recommended in the treatment of campylobacteriosis (Aarestrup and Engberg, 2001; Griggs et al., 2009; Wieczorek and Osek, 2013). The type of resistance marker present in the isolates was linked with the ST, with all the isolates of the same ST harboring identical markers. This likely reflects the clonality of the isolates, rather than any external selective pressure.
Conclusion
Rodents, especially A. flavicollis and M. glareolus, were frequent carriers of Campylobacter. However, the majority of the detected genotypes differed markedly from those circulating in humans and livestock. Occasionally rodents, especially the species living in close connection to humans, may also carry strains associated with colonization in livestock and disease in humans. How transient the colonization is, remains an open question. It is possible that rodents participate in the maintenance of bacteria on farms, even though they are not the original source of the Campylobacter strains circulating in livestock. Further studies should be conducted to confirm this.
Data Availability Statement
The raw sequence data produced in this study was submitted to the European Nucleotide Archive (ENA) database and is publicly available under the project accession number PRJEB37870.
Ethics Statement
Animal trapping was carried out according to stipulations of national animal welfare and environmental legislature (permits from Ministry of Agriculture and Forestry of Finland and Ministry of the Environment of Finland). The Animal Ethics Council of the State Provincial Office of Southern Finland has prior to this study ruled that terminal snap trapping of small rodents does not constitute an animal experiment and therefore does not require an animal experimentation permit. No human studies are presented in this manuscript. No potentially identifiable human images or data is presented in this study.
Author Contributions
RK, MR, MH, PT, MS, OH, SO, JN, and HH contributed to the conception and design of the study. JN, OH, and HH collected and prepared all the animal samples. MR, JT, MS, AJ, SO, and RK analyzed the data. MR performed the phylogenomic analyses. MR and RK interpreted the results. RK performed the statistical analysis. MR wrote the first draft of the manuscript. RK, SO, and MS wrote sections of the manuscript. All authors made substantial intellectual contributions, revised the manuscript critically, and approved the final version of the manuscript.
Funding
This study was funded by the Finnish Foundation of Veterinary Research (SELS/RK/2017), the Walter Ehrström Foundation (WE 2018/RK), and the Ministry of Agriculture and Forestry of Finland (MMM Dnro 1524/03.01.02/2016). Open access funding was provided by the University of Helsinki.
Disclaimer
MR is employed with the European Food Safety Authority (EFSA) in the Unit BIOCONTAM that provides scientific and administrative support to EFSA’s Scientific Activities in the area of Rapid Outbreak Assessment and molecular typing of foodborne pathogens. However, the present article is published under the sole responsibility of MR and may not be considered as an EFSA scientific output. The positions and opinions presented in this article are those of the author/s alone and are not intended to represent the views/any official position or scientific works of EFSA. To know about the views or scientific outputs of EFSA, please consult its website under http://www.efsa.europa.eu.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Noora Harju, Urszula Hirvi, Maaret Hyppönen, Suvi Wallgren, Anne Wilén, and Meeri Ylänen for their excellent technical assistance. The authors wish to acknowledge CSC – IT Center for Science, Finland, for computational resources. This publication made use of the Campylobacter Multi Locus Sequence Typing website (https://pubmlst.org/campylobacter/) sited at the University of Oxford (Jolley et al. Wellcome Open Res 2018, 3:124). The development of this site has been funded by the Wellcome Trust.
Correction Note
A correction has been made to this article. Details can be found at: 10.3389/fmicb.2025.1656693.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.621490/full#supplementary-material
Supplementary Dataset 1 | Assembly statistics.
Supplementary Dataset 2 | Metadata used in the cgMLST analysis including year of isolation, country, 7-gene MLST sequence type, and source for all strains.
Supplementary Dataset 3 | goeBURST groups based on the cgMLST schema (678 loci), at different thresholds of shared loci.
Supplementary Dataset 4 | Allele segregation analysis showing shared loci according to source calculated using the cgMLST profiles of 4,323 strains for which source information was available (Supplementary Dataset 2).
Footnotes
- ^ https://hub.docker.com/r/ummidock/innuca:3.2
- ^ https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ https://pubmlst.org/
- ^ https://hub.docker.com/r/mickaelsilva/chewbbaca_py3
- ^ http://doi.org/10.5281/zenodo.1322564
- ^ https://github.com/cimendes/proCompare
- ^ https://hub.docker.com/r/ummidock/prokka
- ^ https://hub.docker.com/r/sangerpathogens/roary
- ^ https://www.ebi.ac.uk/Tools/msa/clustalo/
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi
References
Aarestrup, F. M., and Engberg, J. (2001). Antimicrobial resistance of thermophilic Campylobacter. Vet. Res. 32, 311–321. doi: 10.1051/vetres:2001127
Adhikari, B., Connolly, J. H., Madie, P., and Davies, P. R. (2004). Prevalence and clonal diversity of Campylobacter jejuni from dairy farms and urban sources. N. Z. Vet. J. 52, 378–383. doi: 10.1080/00480169.2004.36455
Agunos, A., Waddell, L., Léger, D., and Taboada, E. (2014). A systematic review characterizing on-farm sources of Campylobacter spp. for broiler chickens. PLoS One 9:e104905. doi: 10.1371/journal.pone.0104905
Allain, V., Chemaly, M., Laisney, M. J., Rouxel, S., Quesne, S., and Le Bouquin, S. (2014). Prevalence of and risk factors for Campylobacter colonisation in broiler flocks at the end of the rearing period in France. Br. Poult. Sci. 55, 452–459. doi: 10.1080/00071668.2014.941788
Backhans, A., Jacobson, M., Hansson, I., Lebbad, M., Lambertz, S. T., Gammelgard, E., et al. (2013). Occurrence of pathogens in wild rodents caught on Swedish pig and chicken farms. Epidemiol. Infect. 141, 1885–1891. doi: 10.1017/S0950268812002609
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Berndtson, E., Danielsson-Tham, M. L., and Engvall, A. (1996). Campylobacter incidence on a chicken farm and the spread of Campylobacter during the slaughter process. Int. J. Food Microbiol. 32, 35–47. doi: 10.1016/0168-1605(96)01102-6
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cody, A. J., Maiden, M. C., Strachan, N. J., and McCarthy, N. D. (2019). A systematic review of source attribution of human campylobacteriosis using multilocus sequence typing. Euro Surveill. 24:1800696. doi: 10.2807/1560-7917.ES.2019.24.43.1800696
Denis, M., Soumet, C., Rivoal, K., Ermel, G., Blivet, D., Salvat, G., et al. (1999). Development of a m-PCR assay for simultaneous identification of Campylobacter jejuni and C. coli. Lett. Appl. Microbiol. 29, 406–410. doi: 10.1046/j.1472-765x.1999.00658.x
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
EFSA, and ECDC (2019). The european union one health 2018 zoonoses report. EFSA J. 17:e05926. doi: 10.2903/j.efsa.2019.5926
Fernie, D. S., and Healing, T. D. (1976). Wild bank voles (Clethrionomys glariolus) are possibly a natural reservoir of campylobacters (microaerophilic vibrios). Nature 263:496. doi: 10.1038/263496a0
Francisco, A. P., Bugalho, M., Ramirez, M., and Carrico, J. A. (2009). Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics 10:152. doi: 10.1186/1471-2105-10-152
Francisco, A. P., Vaz, C., Monteiro, P. T., Melo-Cristino, J., Ramirez, M., and Carrico, J. A. (2012). PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinformatics 13:87. doi: 10.1186/1471-2105-13-87
Griggs, D. J., Peake, L., Johnson, M. M., Ghori, S., Mott, A., and Piddock, L. J. (2009). Beta-lactamase-mediated beta-lactam resistance in Campylobacter species: prevalence of Cj0299 (bla OXA-61) and evidence for a novel beta-Lactamase in C. jejuni. Antimicrob. Agents Chemother. 53, 3357–3364. doi: 10.1128/AAC.01655-08
Hadfield, J., Croucher, N. J., Goater, R. J., Abudahab, K., Aanensen, D. M., and Harris, S. R. (2017). Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293. doi: 10.1093/bioinformatics/btx610
Hakanen, A., Jalava, J., Kotilainen, P., Jousimies-Somer, H., Siitonen, A., and Huovinen, P. (2002). gyrA polymorphism in Campylobacter jejuni: detection of gyrA mutations in 162 C. jejuni isolates by single-strand conformation polymorphism and DNA sequencing. Antimicrob. Agents Chemother. 46, 2644–2647. doi: 10.1128/aac.46.8.2644-2647.2002
Healing, T. D., and Greenwood, M. H. (1991). Frequency of isolation of Campylobacter spp., Yersinia spp. and Salmonella spp. from small mammals from two sites in Southern Britain. Int. J. Environ. Health Res. 1, 54–62. doi: 10.1080/09603129109356704
Henttonen, H. (2000). Long-term dynamics of the bank vole Clethrionomys glareolus at Pallasjärvi, northern Finnish taiga. In: Bujalska, G. & Hansson, L. (eds.). Bank vole biology: Recent advances in the population biology of a model species. Polish J. Ecol. 48, 87–96.
Henttonen, H., Oksanen, T., Jortikka, A., and Haukisalmi, V. (1987). How much do weasels shape microtine cycles in the northern Fennoscandian taiga? Oikos 50, 353–365. doi: 10.2307/3565496
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Joensen, K. G., Kiil, K., Gantzhorn, M. R., Nauerby, B., Engberg, J., Holt, H. M., et al. (2020). Whole-genome sequencing to detect numerous Campylobacter jejuni outbreaks and match patient isolates to sources, Denmark, 2015-2017. Emerg. Infect. Dis. 26, 523–532. doi: 10.3201/eid2603.190947
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595.
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kapperud, G., Skjerve, E., Vik, L., Hauge, K., Lysaker, A., Aalmen, I., et al. (1993). Epidemiological investigation of risk factors for campylobacter colonization in Norwegian broiler flocks. Epidemiol. Infect. 111, 245–255. doi: 10.1017/s0950268800056958
Kasrazadeh, M., and Genigeorgis, C. (1987). Origin and prevalence of Campylobacter jejuni in ducks and duck meat at the farm and processing plant level. J. Food Prot. 50, 321–326. doi: 10.4315/0362-028X-50.4.321
Kim, J., Guk, J. H., Mun, S. H., An, J. U., Kim, W., Lee, S., et al. (2020). The wild mouse (Micromys minutus): reservoir of a novel Campylobacter jejuni strain. Front. Microbiol. 10:3066. doi: 10.3389/fmicb.2019.03066
Korpela, K., Delgado, M., Henttonen, H., Korpimäki, E., Koskela, E., Ovaskainen, O., et al. (2013). Nonlinear effects of climate on boreal rodent dynamics: mild winters do not negate high-amplitude cycles. Glob. Chang. Biol. 19, 697–710. doi: 10.1111/gcb.12099
Kovanen, S., Rossi, M., Pohja-Mykra, M., Nieminen, T., Raunio-Saarnisto, M., Sauvala, M., et al. (2019). Population genetics and characterization of Campylobacter jejuni isolates from Western Jackdaws and game birds in Finland. Appl. Environ. Microbiol. 85, e2365–e2318.
Lachance, N., Gaudreau, C., Lamothe, F., and Lariviere, L. A. (1991). Role of the beta-lactamase of Campylobacter jejuni in resistance to beta-lactam agents. Antimicrob. Agents Chemother. 35, 813–818. doi: 10.1128/aac.35.5.813
LeMoine, V., Vannier, P., and Jestin, A. (1987). Microbiological studies of wild rodents in farms as carriers of pig infectious agents. Prev. Vet. Med. 4:399. doi: 10.1016/0167-5877(87)90026-2
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res, 47, W256–W259. doi: 10.1093/nar/gkz239
Llarena, A. K., Skarp-de Haan, C. P., Rossi, M., and Hanninen, M. L. (2015). Characterization of the Campylobacter jejuni population in the barnacle geese reservoir. Zoonoses Public Health 62, 209–221. doi: 10.1111/zph.12141
Llarena, A. K., Taboada, E., and Rossi, M. (2017). Whole-genome sequencing in epidemiology of Campylobacter jejuni infections. J. Clin. Microbiol. 55, 1269–1275. doi: 10.1128/JCM.00017-17
Lõhmus, M., and Albihn, A. (2013). Gastrointestinal pathogens in rodents overwintering in human facilities around Uppsala, Sweden. J. Wildl. Dis. 49, 747–749. doi: 10.7589/2013-02-028
Machado, M. P., Halkilahti, J., Jaakkonen, A., Silva, D. N., Mendes, I., Nalbantoglu, Y., et al. (2017). INNUca. GitHub. Available online at: https://github.com/B-UMMI/INNUca (accessed on July 12, 2017)
Marotta, F., Garofolo, G., di Marcantonio, L., Di Serafino, G., Neri, D., Romantini, R., et al. (2019). Antimicrobial resistance genotypes and phenotypes of Campylobacter jejuni isolated in Italy from humans, birds from wild and urban habitats, and poultry. PLoS One 14:e0223804. doi: 10.1371/journal.pone.0223804
Meerburg, B. G., and Kijlstra, A. (2007). Role of rodents in transmission of Salmonella and Campylobacter. J. Sci. Food Agric. 87, 2774–2781. doi: 10.1002/jsfa.3004
Meerburg, B. G., Jacobs-Reitsma, W. F., Wagenaar, J. A., and Kijlstra, A. (2006). Presence of Salmonella and Campylobacter spp. in wild small mammals on organic farms. Appl. Environ. Microbiol. 72, 960–962. doi: 10.1128/aem.72.1.960-962.2006
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Payot, S., Bolla, J. M., Corcoran, D., Fanning, S., Megraud, F., and Zhang, Q. (2006). Mechanisms of fluoroquinolone and macrolide resistance in Campylobacter spp. Microbes. Infect. 8, 1967–1971. doi: 10.1016/j.micinf.2005.12.032
Piddock, L. J., Ricci, V., Pumbwe, L., Everett, M. J., and Griggs, D. J. (2003). Fluoroquinolone resistance in Campylobacter species from man and animals: detection of mutations in topoisomerase genes. J. Antimicrob. Chemother. 51, 19–26. doi: 10.1093/jac/dkg033
Pinho, M. D., Erol, E., Ribeiro-Goncalves, B., Mendes, C. I., Carrico, J. A., Matos, S. C., et al. (2016). Beta-hemolytic Streptococcus dysgalactiae strains isolated from horses are a genetically distinct population within the Streptococcus dysgalactiae taxon. Sci. Rep. 6:31736. doi: 10.1038/srep31736
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Rosef, O., Gondrosen, B., Kapperud, G., and Underdal, B. (1983). Isolation and characterization of Campylobacter jejuni and Campylobacter coli from domestic and wild mammals in Norway. Appl. Environ. Microbiol. 46, 855–859. doi: 10.1128/aem.46.4.855-859.1983
Rossi, M., Silva, M. S. D., Ribeiro-Gonçalves, B. F., Silva, D. N., Machado, M. P., Oleastro, M., et al. (2018). INNUENDO whole genome and core genome MLST schemas and datasets for Campylobacter jejuni (Version 1.0) [Data set]. Zenodo doi: 10.5281/zenodo.1322564
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seemann, T. (2017). mlst. GitHub. Available online at: https://github.com/tseemann/mlst (accessed on July 12, 2017)
Sheppard, S. K., Didelot, X., Jolley, K. A., Darling, A. E., Pascoe, B., Meric, G., et al. (2013). Progressive genome-wide introgression in agricultural Campylobacter coli. Mol. Ecol. 22, 1051–1064. doi: 10.1111/mec.12162
Silva, M., Machado, M. P., Silva, D. N., Rossi, M., Moran-Gilad, J., Santos, S., et al. (2018). chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 4:e000166. doi: 10.1099/mgen.0.000166
Skarp-de Haan, C. P., Culebro, A., Schott, T., Revez, J., Schweda, E. K., Hänninen, M. L., et al. (2014). Comparative genomics of unintrogressed Campylobacter coli clades 2 and 3. BMC Genomics 15:129. doi: 10.1186/1471-2164-15-129
Sommer, H. M., Heuer, O. E., Sørensen, A. I., and Madsen, M. (2013). Analysis of factors important for the occurrence of Campylobacter in Danish broiler flocks. Prev. Vet. Med. 111, 100–111. doi: 10.1016/j.prevetmed.2013.04.004
Tajada, P., Gomez-Graces, J. L., Alos, J. I., Balas, D., and Cogollos, R. (1996). Antimicrobial susceptibilities of Campylobacter jejuni and Campylobacter coli to 12 beta-lactam agents and combinations with beta-lactamase inhibitors. Antimicrob. Agents Chemother. 40, 1924–1925. doi: 10.1128/aac.40.8.1924
Torralbo, A., Borge, C., Allepuz, A., García-Bocanegra, I., Sheppard, S. K., Perea, A., et al. (2014). Prevalence and risk factors of Campylobacter infection in broiler flocks from southern Spain. Prev. Vet. Med. 114, 106–113. doi: 10.1016/j.prevetmed.2014.01.019
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wieczorek, K., and Osek, J. (2013). Antimicrobial resistance mechanisms among Campylobacter. Biomed. Res. Int. 2013:340605. doi: 10.1155/2013/340605
Williams, N. J., Jones, T. R., Leatherbarrow, H. J., Birtles, R. J., Lahuerta-Marin, A., Bennett, M., et al. (2010). Isolation of a novel Campylobacter jejuni clone associated with the bank vole, Myodes glareolus. Appl. Environ. Microbiol. 76, 7318–7321. doi: 10.1128/AEM.00511-10
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Campylobacter jejuni, phylogeny, comparative genomics, rodent, mouse, vole, shrew
Citation: Olkkola S, Rossi M, Jaakkonen A, Simola M, Tikkanen J, Hakkinen M, Tuominen P, Huitu O, Niemimaa J, Henttonen H and Kivistö R (2021) Host-Dependent Clustering of Campylobacter Strains From Small Mammals in Finland. Front. Microbiol. 11:621490. doi: 10.3389/fmicb.2020.621490
Received: 26 October 2020; Accepted: 17 December 2020;
Published: 13 January 2021;
Corrected: 04 August 2025.
Edited by:
Greta Gölz, Freie Universität Berlin, GermanyReviewed by:
Odile Tresse, INRA Centre Angers-Nantes Pays de la Loire, FranceYoung Min Kwon, University of Arkansas, United States
Copyright © 2021 Olkkola, Rossi, Jaakkonen, Simola, Tikkanen, Hakkinen, Tuominen, Huitu, Niemimaa, Henttonen and Kivistö. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rauni Kivistö, cmF1bmkua2l2aXN0b0BoZWxzaW5raS5maQ==