 Rucha Datar1
Rucha Datar1 Andreu Coello Pelegrin1,2
Andreu Coello Pelegrin1,2 Sylvain Orenga1
Sylvain Orenga1 Valérie Chalansonnet1
Valérie Chalansonnet1 Caroline Mirande1Jill Dombrecht3
Caroline Mirande1Jill Dombrecht3 John D. Perry4
John D. Perry4 Audrey Perry4Herman Goossens2
Audrey Perry4Herman Goossens2 Alex van Belkum1*
Alex van Belkum1*- 1BioMérieux, La Balme les Grottes, France
- 2Laboratory of Medical Microbiology, Faculty of Medicine and Health Sciences, Vaccine and Infectious Disease Institute, University of Antwerp, Antwerp, Belgium
- 3Applied Maths, BioMérieux, Sint-Martens-Latem, Belgium
- 4Freeman Hospital, Newcastle upon Tyne, United Kingdom
Cystic fibrosis (CF) represents one of the major genetic and chronic lung diseases affecting Caucasians of European descent. Patients with CF suffer from recurring infections that lead to further damage of the lungs. Pulmonary infection due to Pseudomonas aeruginosa is most prevalent, further increasing CF-related mortality. The present study describes the phenotypic and genotypic variations among 36 P. aeruginosa isolates obtained serially from a non-CF and five CF patients before, during and after lung transplantation (LTx). The classical and genomic investigation of these isolates revealed a common mucoid phenotype and only subtle differences in the genomes. Isolates originating from an individual patient shared ≥98.7% average nucleotide identity (ANI). However, when considering isolates from different patients, substantial variations in terms of sequence type (ST), virulence factors and antimicrobial resistance (AMR) genes were observed. Whole genome multi-locus sequence typing (MLST) confirmed the presence of unique STs per patient regardless of the time from LTx. It was supported by the monophyletic clustering found in the genome-wide phylogeny. The antibiogram shows that ≥91.6% of the isolates were susceptible to amikacin, colistin and tobramycin. For other antibiotics from the panel, isolates frequently showed resistance. Alternatively, a comparative analysis of the 36 P. aeruginosa isolates with 672 strains isolated from diverse ecologies demonstrated clustering of the CF isolates according to the LTx patients from whom they were isolated. We observed that despite LTx and associated measures, all patients remained persistently colonized with similar isolates. The present study shows how whole genome sequencing (WGS) along with phenotypic analysis can help us understand the evolution of P. aeruginosa over time especially its antibiotic resistance.
Introduction
Cystic fibrosis (CF) is one of the most common and ultimately lethal autosomal recessive diseases, which primarily affects Caucasians of European descent (Cutting, 2015). Lung diseases are the main cause of morbidity and mortality among CF patients, especially those leading to respiratory failure caused by extensive lung damage, such as bronchiectasis or chronic endobronchial infections (Sanders and Fink, 2016). For patients with end-stage CF, lung transplantation (LTx) is the only viable option, as there is a 50% survival rate 10-year post-LTx (Snell et al., 2017).
The respiratory tract microbiota of CF is diverse and the most prevalent pathogens include Pseudomonas aeruginosa, Staphylococcus aureus, Haemophilus influenzae, Burkholderia cepacia complex (BCC), Stenotrophomonas maltophilia, Achromobacter xylosoxidans, and non-tuberculous mycobacteria (NTM). The exact incidence for each bacterial species has changed over the past years and continues to change, sometimes according to the geographic locale of the patients (Salsgiver et al., 2016). Frequent antibiotic therapies for patients with CF tend to select for antibiotic resistant variants. Multidrug-resistant (MDR) P. aeruginosa is one of the major end-stage bacterial species among CF patients, as even after antibiotic treatment it frequently recurs and establishes chronic infection. Such strains also display evolution of phenotypic and genomic characteristics over time (Winstanley et al., 2016; Stefani et al., 2017).
As P. aeruginosa is highly versatile in its pathogenicity and has significant genomic plasticity, assessing the whole genome sequence and identifying antimicrobial resistance (AMR) genes could be very useful to gain insights into the emergence of resistance against currently used antimicrobials. Population-level analysis and comparative genomics can help us to identify the co-existence of diverse strains during chronic illnesses and how they differ genotypically (Chung et al., 2012; Celli et al., 2015). High resolution genomics can enable us to better understand the high-risk clones (HRC) and segregate them as MDR or extensively-drug resistant or pan-drug resistant (Magiorakos et al., 2012). Amongst the many P. aeruginosa HRC studied, sequence type (ST) 235 is the most widespread lineage across the world.
The aim of this study is to evaluate how whole genome sequencing (WGS) and bio-informatic data analysis could assist in evaluating the impact of LTx on the diversity and antibiotic resistance profile of resident P. aeruginosa isolates. We studied culture based phenotypes of the isolates in combination with WGS to analyze the local epidemiology, evolution and resistome of P. aeruginosa isolates. They were serially obtained from five CF patients and a non-CF bronchiectasis patient, who all underwent LTx. Genomic data were analyzed versus a large control panel of P. aeruginosa strains (n = 672) from different ecological and clinical niches.
Materials and Methods
Sample Collection and Isolation of Pseudomonas aeruginosa
For the investigation of isolates from the same LTx patients, obtained at different time points, before, during and after LTx, serial peri-lung transplantation (SPLT) P. aeruginosa isolates, data from the six patients who underwent LTx during the years 2006–2017 were retrieved from the Freeman Hospital in Newcastle upon Tyne (United Kingdom). These patients were in the age group of 24–42 years at the time of LTx. For each patient, sampling time range from 6 to 22 months. At various intervals (i.e., before, during and after LTx) either broncho-alveolar lavage (BAL) or sputum specimens were collected for microbiological assessment at the Freeman Hospital. We selected all colony variants (mucoid, non-mucoid, large, small, etc.) isolated at a particular time interval. If only one isolate was selected for testing, then it means that all colonies of P. aeruginosa showed a consistent appearance. We cannot of course discount the possibility that colonies that look identical may have different phenotypes in terms of, for example, antimicrobial susceptibility. In total, 36 P. aeruginosa isolates belonging to five CF patients and a non-CF bronchiectasis patient, were isolated and preserved at −80°C until further analysis (Table 1). A sub-selection of 12 isolates, one pre- and one post-LTx from each of the six patients was used for full antibiotic susceptibility characterization (star marked in Table 1). Three isolates were used as quality controls throughout the study: one CLSI/EUCAST quality control strain (ATCC: 27853) and 2 isolates resistant to carbapenems by impermeability (BioMérieux collection numbers: API: 1508289 and API: 9208013).
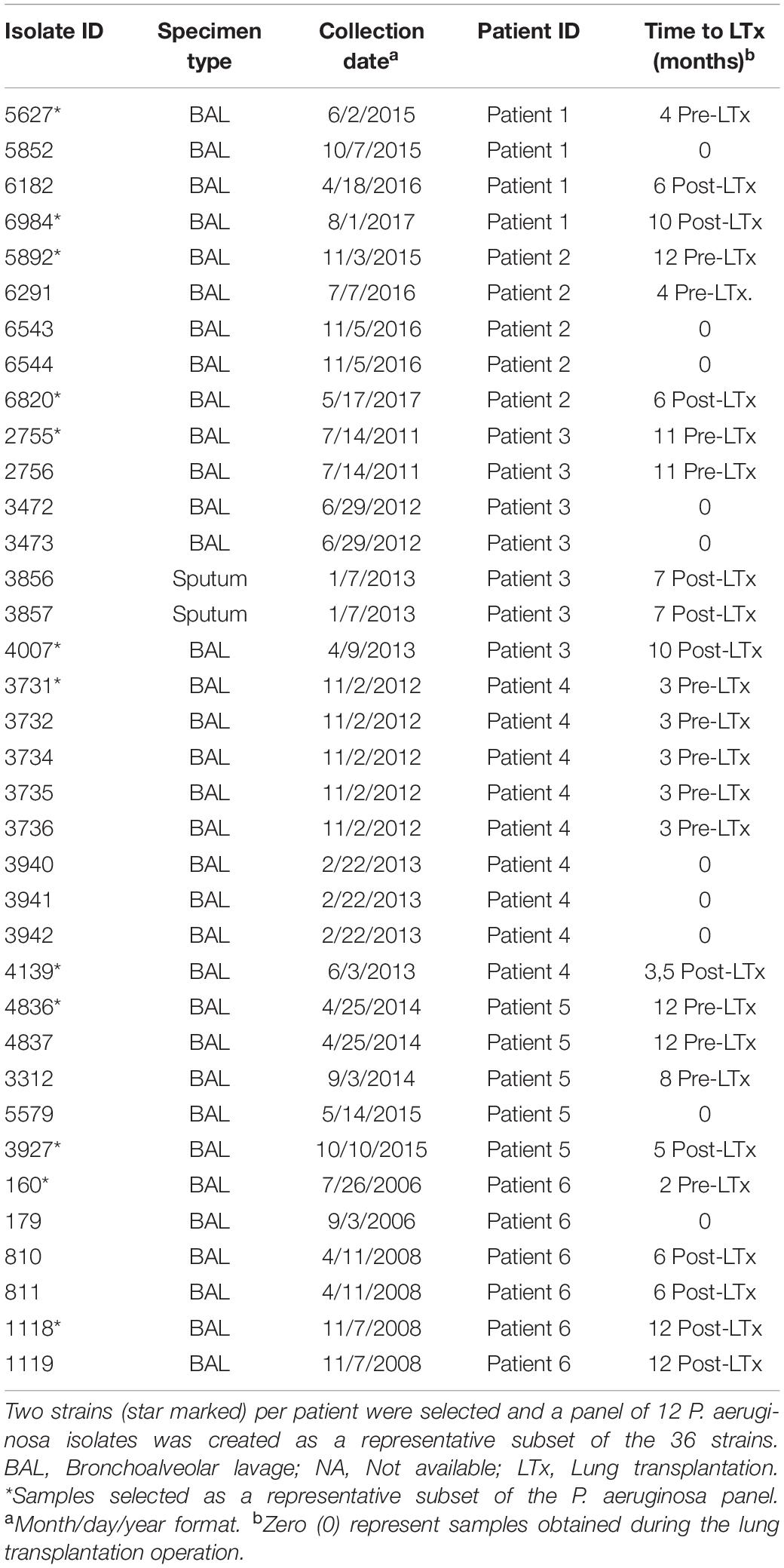
Table 1. The 36 Pseudomonas aeruginosa isolates represented on the basis of the patient number.
Phenotypic Characterization of Serial Peri-Lung Transplantation Pseudomonas aeruginosa Isolates
Identification of SPLT Pseudomonas aeruginosa Isolates
The 36 P. aeruginosa isolates obtained were revived from glycerol stocks by streaking on Columbia agar + 5% sheep blood (COS agar) and incubated at 37°C for 24 h. The identity of the isolates was validated using mass spectrometry (Matrix Assisted Laser Desorption Ionization–Time of Flight MS) using VITEK® MS (BioMérieux).
Antibiotic Susceptibility Testing Profiles of SPLT Pseudomonas aeruginosa Isolates
The 36 isolates were tested for their resistance/susceptibility profile against an array of antimicrobials by disk diffusion assay (Oxoid) according to CLSI (2017) guidelines. Additionally, minimum inhibitory concentrations (MIC) of a sub-selection of 12 isolates were defined by broth micro-dilution (BMD) assays to validate the results from disk diffusion assays. The antibiotic susceptibility testing (AST) profiling by BMD involved assessment using eight antibiotics viz. amikacin (AK), ciprofloxacin (CIP), levofloxacin (LEV), tobramycin (TOB), colistin (COL), imipenem (IMP), aztreonam (ATM), and ceftolozane-tazobactam (CZT). For BMD, the antibiotics were diluted using Mueller Hinton Broth (MHB) forming a concentration gradient in 96 well plates. Concentrations were chosen based on the upper limit of each antibiotic defined by the CLSI. The bacterial suspension prepared as per CLSI guidelines was incubated at 37°C for 24 h. The MICs were recorded after 24-h incubation by visual examination. For both methods, the isolates were classified as resistant, intermediate or susceptible based on CLSI breakpoints. The antibiotics chosen in this study are commonly used for treating P. aeruginosa infections.
Genomic Characterization of SPLT Isolates Pseudomonas aeruginosa
Multi-locus sequence typing (MLST) and construction of a core genome-based phylogenetic tree using bio-informatic tools was employed to define the genomic relatedness between the 36 serial P. aeruginosa isolates and a control panel of 672 P. aeruginosa strains.
Genomic DNA from all 36 isolates was extracted using the UltraClean® microbial DNA isolation kit (Qiagen, Netherlands) as per the manufacturer’s instructions. The qualitative and quantitative estimation of the obtained DNA was carried out using the LabChip® GX TouchTM nucleic acid analyzer (PerkinElmer, United States) and Qubit dsDNA BR assay kits (Thermo Fisher Scientific, Waltham, MA, United States). The high-quality DNA was used for whole genome library preparation using S4 Reagent Kit v1.5 by following manufacturer’s instructions. The resultant libraries were sequenced on the NovaSeq 6000 platform (Illumina Inc.) using 2 × 150 bp chemistry at CeGaT (Tübingen, Germany).
Bio-Informatic Data Processing and Analysis
Raw reads were quality filtered and assembled using the A5-MiSeq pipeline. The assembled contigs were used for further downstream analysis viz. annotation and phylogenetic analysis. The potential for AMR was inferred by annotating the genomes using the PATRIC3.6.2 server. The virulome and resistome were analyzed using the Virulence Factors Database (VFDB) and Comprehensive Antibiotic Resistance Database (CARD) database on March 12, 2020 and September 9, 2019, respectively (Chen et al., 2005; Alcock et al., 2020). The genomic antibiogram was validated using the BioMérieux EPISEQ® CS tool (v1.0.2)1. The genomic antibiogram depicted gene abundance. Gene abundance is the number of times the genes involved in the particular function (e.g., efflux pump conferring resistance) appears in the whole genome sequence. As these genes encoding resistance are usually part of a group, presence or absence of one gene from the group could impact the level of resistance. A core genome-based phylogenetic tree using the FAST tree method was constructed to quantify the genomic relatedness level between the SPLT isolates. This tree was constructed using the parsnp tool (v1.2; Treangen et al., 2014) and was curated using iTOL (v5; Letunic and Bork, 2016). Alternatively, the 7-loci MLST of the 36 P. aeruginosa isolates was performed using PubMLST2 and BioMérieux EPISEQ® CS to decipher STs and to define relatedness among the isolates (Jolley et al., 2018).
In case of disparity in results between PubMLST and BioMérieux EPISEQ® CS, sequence reads were aligned with the housekeeping gene reference alleles using the BWA MEM algorithm from the Burrows-Wheeler Alignment Tool (BWA; v0.7.17). We used SAMtools (v1.7) to obtain BAM files of the alignments, which were later visualized using the integrative genomics viewer (IGV; ∗v2.4.1; Li et al., 2009; Li and Durbin, 2010; Robinson et al., 2011).
Comparative Genome Analysis of SPLT Pseudomonas aeruginosa Isolates
A core genome single nucleotide polymorphism (SNP)-based approach using the parsnp tool was used to compare the sequence of SPLT P. aeruginosa isolates to diverse strains of P. aeruginosa (n = 672) to define the genomic variation between P. aeruginosa strains from different niches and geographies. The 672 P. aeruginosa strains were obtained from three collections: the private BioMérieux collection (n = 219; van Belkum et al., 2015); the Kos collection (n = 390; Kos et al., 2015); and the Pirnay collection (n = 63; Pirnay et al., 2009). Further, the phylogeny was reconstructed based on the core genomes.
Results
Identification and MLST Profiling of the SPLT Pseudomonas aeruginosa Isolates
On COS agar plates, the isolates were mucoid and slow growing. Despite the aberrant growth patterns for the strains, the verification of the identification of bacterial isolates using VITEK® MS (BioMérieux) confirmed that all isolates were P. aeruginosa. The WGS of the CF isolates generated an average of 50,043,100 ± 7,423,658 (average ± SD) pair-end reads per genome (Supplementary Table 1). De novo assembly revealed a mean genome size of 6.36 ± 0.17 mb (mean ± SD) with average coverage of 591× (±91×). Both MLST profiling methods traced four STs covering 18 genomes. However, the remaining 18 genomes (the majority belonging to patients 1, 2, 5, and 6) could not be assigned to any known ST and, hence, represent novel STs. LTx had no impact on the changes in the ST of the isolates. For patient 3, all of the isolates belonged to the same known ST (146), while for patients 1 and 5 they all belonged to novel STs. For patients 2 and 6, some of the isolates pre- and post-transplantation belonged to the same known ST (ST1029 and ST1567, respectively) while others belonged to novel STs. Finally for patient 4, some of the isolates at the time of transplant belonged to the same known sequence type (ST882) as one of the earlier isolates, while others belong to a novel ST identical to the post-LTx isolate (Supplementary Table 1).
Through the BioMérieux EPISEQ® CS analysis, ST235 was assigned to isolates 810, 1,118, and 1,119 which were isolated from patient 6 after LTx. MLST performed by aligning the reads to the reference sequence analysis showed mutation in the acsA allele in isolate 1,118. However, no mutation was found in the isolates 810 and 1,119. For our specific set of P. aeruginosa isolates, BioMérieux EPISEQ® CS proved to be a more robust MLST tool as it could assign STs to a larger number of isolates.
Comparison of Pseudomonas aeruginosa From Different Collections of Isolates
The genomic comparison of the isolates under investigation with the genomes of 672 isolates depicted unique clustering of the SPLT isolates in seven different clades spanning across the range of isolates isolated from diverse niches (Figure 1). However, the SPLT isolates were found to be conserved at the individual patient level, forming a monophyletic clade per patient, except for the case of patient 6 where isolates formed two distinct clades. Clade I contained the isolates 160, 179, and 811 with ST1567 whilst the other clade (clade II) was comprised of isolates 810, 1,118, and 1,119, with ST235 according to BioMérieux EPISEQ CS tool.
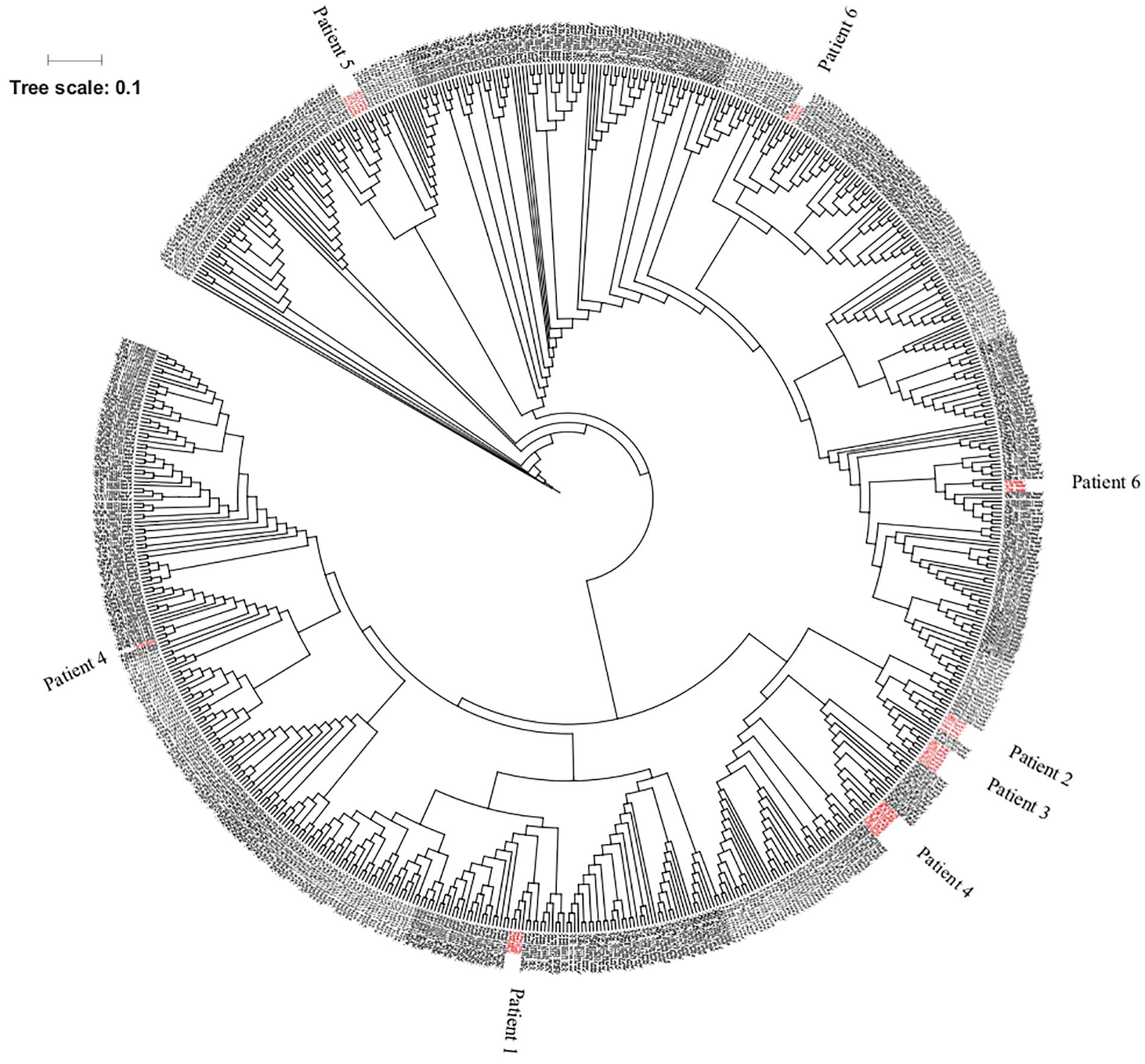
Figure 1. Genotypic similarities between 708 Pseudomonas aeruginosa strains at core genome SNP level and genotypic similarities/differences in a SNP based tree. Out of the 708 strains, 36 isolates were from lung transplant patients (in red) and the others belong to BioMérieux collection, Kos collection, and Pirnay collection.
Genomic Analysis and Antibiotic Resistance
The genomic investigation of the 36 P. aeruginosa isolates using WGS revealed differences in the genome architecture sharing ≥98.7% average nucleotide identity (ANI; Supplementary Table 1). Further, a core genome phylogeny developed using the FAST tree method suggested formation of six different clades representing isolates from six different patients. We found intergroup variability with respect to the origin of the isolates. However, clustering did not correlate with time of isolation of the isolates around LTx (Figure 2).
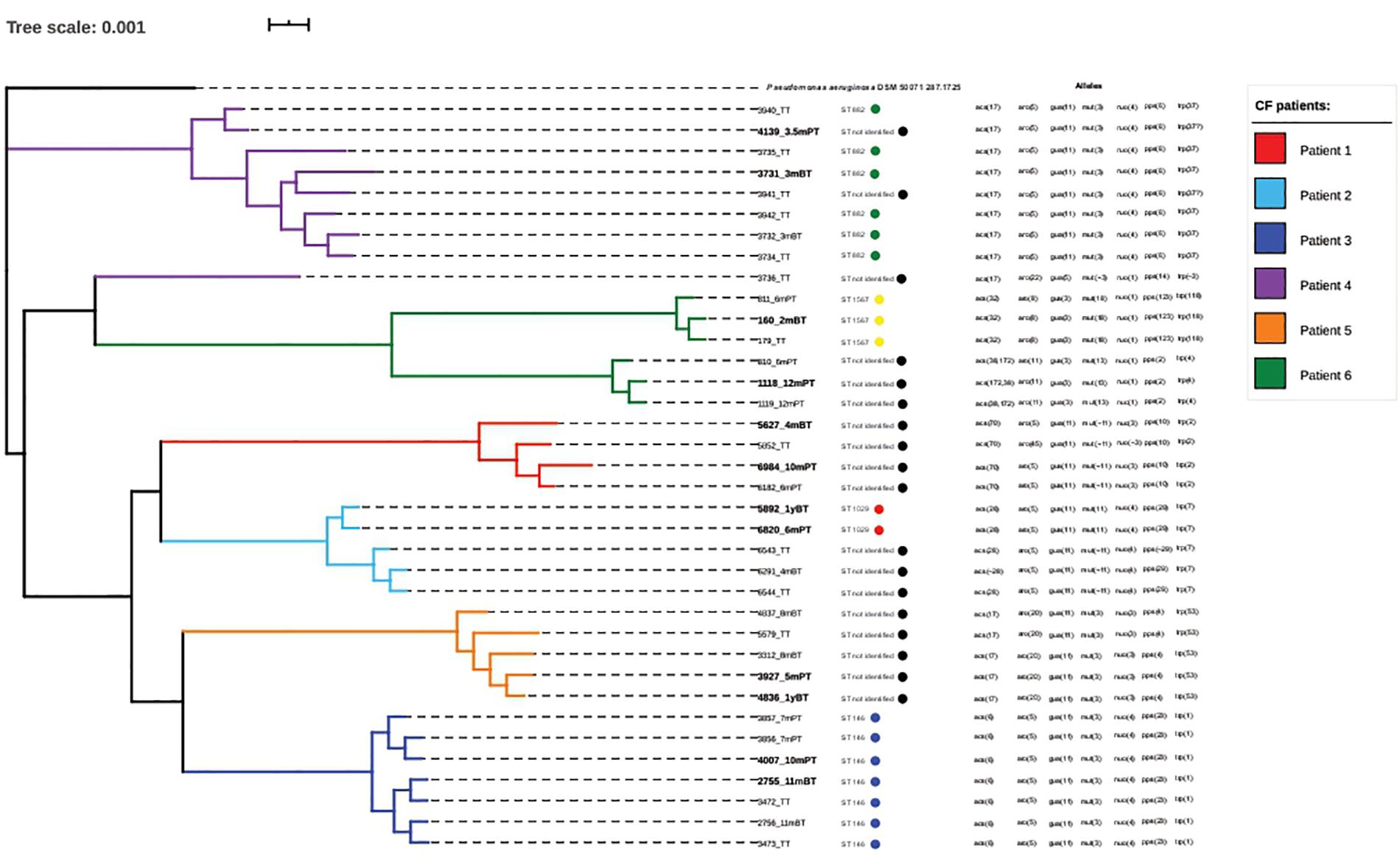
Figure 2. The phylogenetic tree constructed using the whole genome sequence (WGS) of 36 clinical Pseudomonas aeruginosa isolates demonstrates the variability between the isolates. Each clade represents one patient. The isolates selected for phenotypic analysis using broth micro-dilution (BMD) assay (in bold) are from two different time points: before and after transplant. The figure also depicts the sequence types and allelic differences among the 36 P. aeruginosa isolates from lung transplantation (LTx) patients.
The nucleotide sequences of the small subunit (SSU) 16S rRNA gene of all the isolates were extracted and taxonomic identity was reconfirmed using a BLAST search. Further, we noted the presence of 6,150 ± 193 (mean ± SD) coding sequences (CDS) per genome. A subset of the CDSs accounting for conferring antibiotic resistance was derived using CARD, which was in the range of 55 ± 1 resistance CDS per isolate. The genes retrieved from the CARD analysis were mostly classified as beta-lactam resistance genes (n = 6), efflux pumps (n = 37), an aminoglycoside resistance gene (n = 1), other antibiotic inactivation enzymes (n = 8), antibiotic resistance gene variants or mutants (n = 4), a gene modulating bacterial permeability to antibiotics (n = 1), genes altering cell wall charge conferring antibiotic resistance (n = 2) and an antibiotic target protection protein gene (n = 1; Figure 3).

Figure 3. Heatmap-dendrogram analysis of 36 Pseudomonas aeruginosa isolates and reference stains depicting the taxonomic relatedness of the isolates isolated across three time points: before LTx, at the time of LTx and after LTx. Furthermore, the qualitative estimates of antibiotic resistance genes (ARGs) were compared against the observed antimicrobial resistance (AMR) phenotypes. AST results are shown by color code red (resistant) and green (susceptible).
BioMérieux EPISEQ® CS analysis showed the presence of various antibiotic resistance genes (ARGs) in P. aeruginosa isolates. We noted different frequencies of genes conferring resistance against drugs or drug families such as aminocoumarin, aminoglycoside (100%), polymyxin B (100%), chloramphenicol (100%), elfamycin (88.89%), sulfonamide, sulfone (100%), fosfomycin (100%), azithromycin, ciprofloxacin, erythromycin, novobiocin, tetracycline (oprM gene, 2.78%), beta-lactam, carbapenem, cephalosporin, cephamycin, penicillin (from 97.22% for OXA-50 to 8.33% for PDC-2 or PDC-3), and fluoroquinolone (100%). For strain 3,473 (belonging to patient 3), we noted the prevalence of genes responsible for resistance against the antibiotics azithromycin, chloramphenicol, ciprofloxacin, erythromycin, novobiocin and tetracycline. Interestingly, in contrast to all other isolates, isolate 4,837 (deriving from patient 5) lacked the presence of genes providing resistance against the drug family “beta-lactam, carbapenem, cephalosporin, cephamycin, and penicillin” (Figure 4).
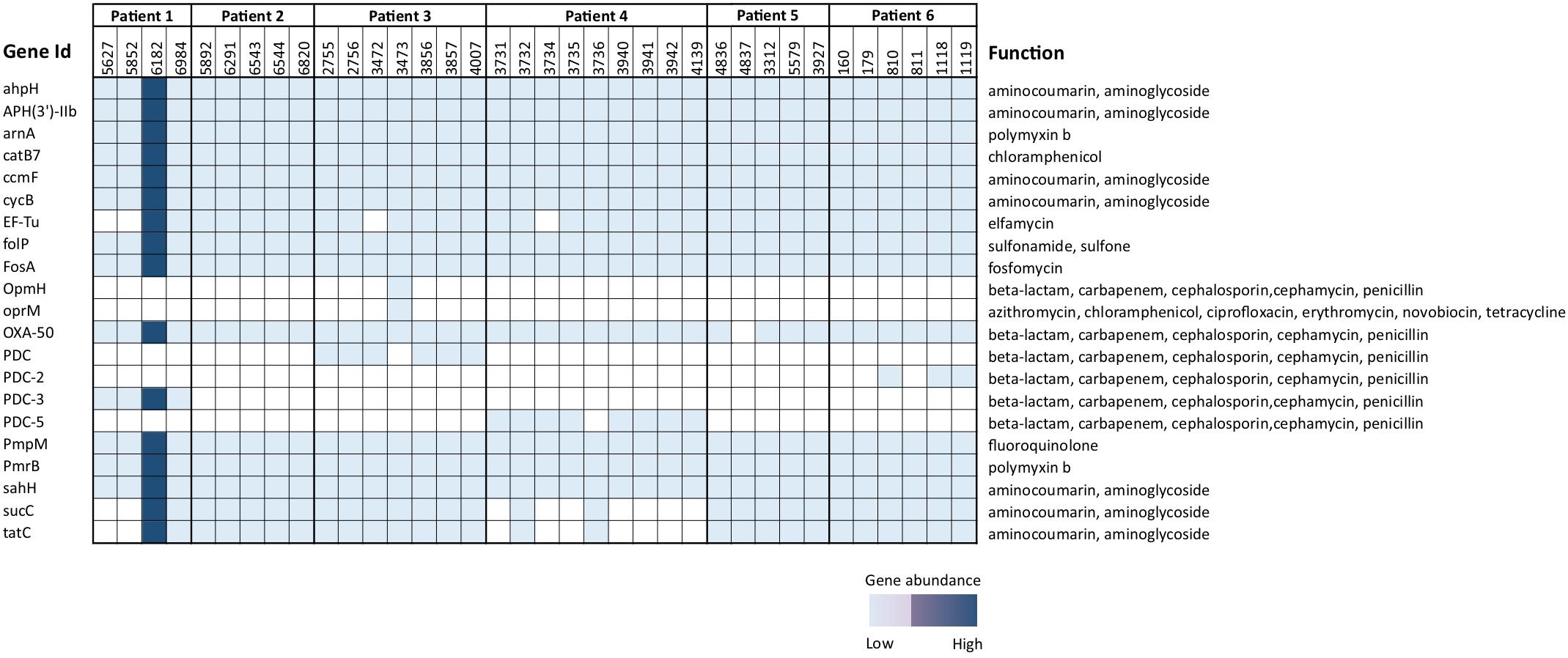
Figure 4. Presence and absence of antimicrobial resistance genes in 36 Pseudomonas aeruginosa isolates. Varied degree of presence of genes conferring resistance against drug families was noted. The analysis was done using EPISEQ.
Virulence Genes
The investigation of virulence factors in P. aeruginosa isolates using VFDB revealed isolates containing various virulence genes ranging from 236 to 266 in number. Genes with no known functionality in the VFDB database were eliminated. The remaining 28 genes were found to be differentially abundant across the genomes off all isolates in the study. The major functional attributes of these 28 genes were adherence (17.8%, relative abundance) and Type VI secretion (14.2%, relative abundance; Figure 5).
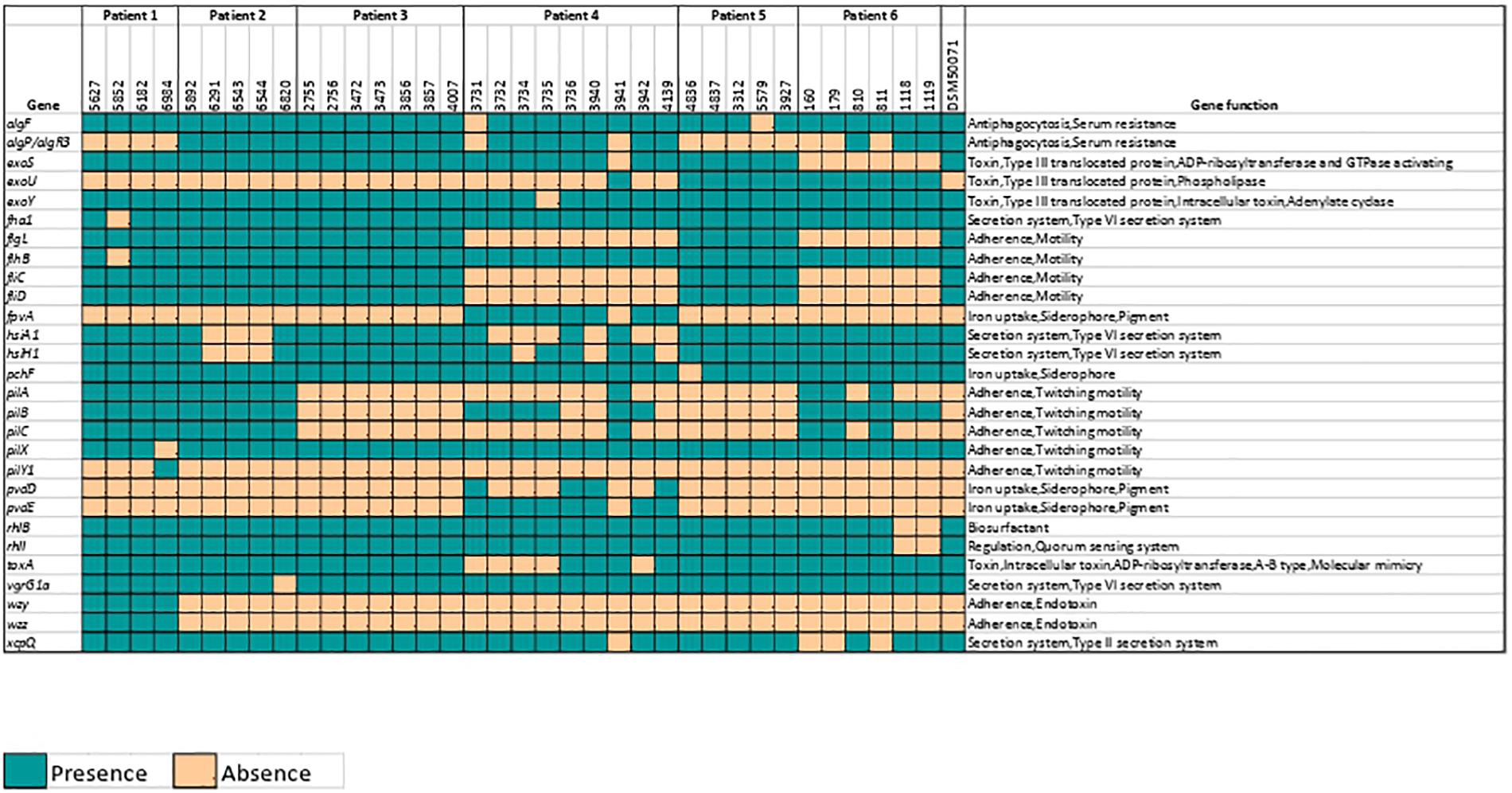
Figure 5. Presence of various virulence factors among 36 Pseudomonas aeruginosa isolates. Yellow square indicates absence while green square indicates presence of virulence genes identified using Virulence Factors Database (VFDB).
Antibiotic Susceptibility Testing
More than 75% of the isolates were found to be resistant to at least half of the antibiotics under study (Table 2). Isolates 810 (patient 6) and 4,139 (patient 4) were found to be the most susceptible. Tobramycin was found to be effective against 64% of the isolates. Additionally, isolates 3,735 and 3,736 (patient 4), 6,291 and 6,543 (patient 2), 5,852 (patient 1), and 3,473, 3,472, 2,755 and 2,756 (patient 3) were found to be resistant to all the antibiotics tested. Interestingly, all the isolates of an ST belonging to one patient did not always show the same susceptibility pattern (Figure 3).
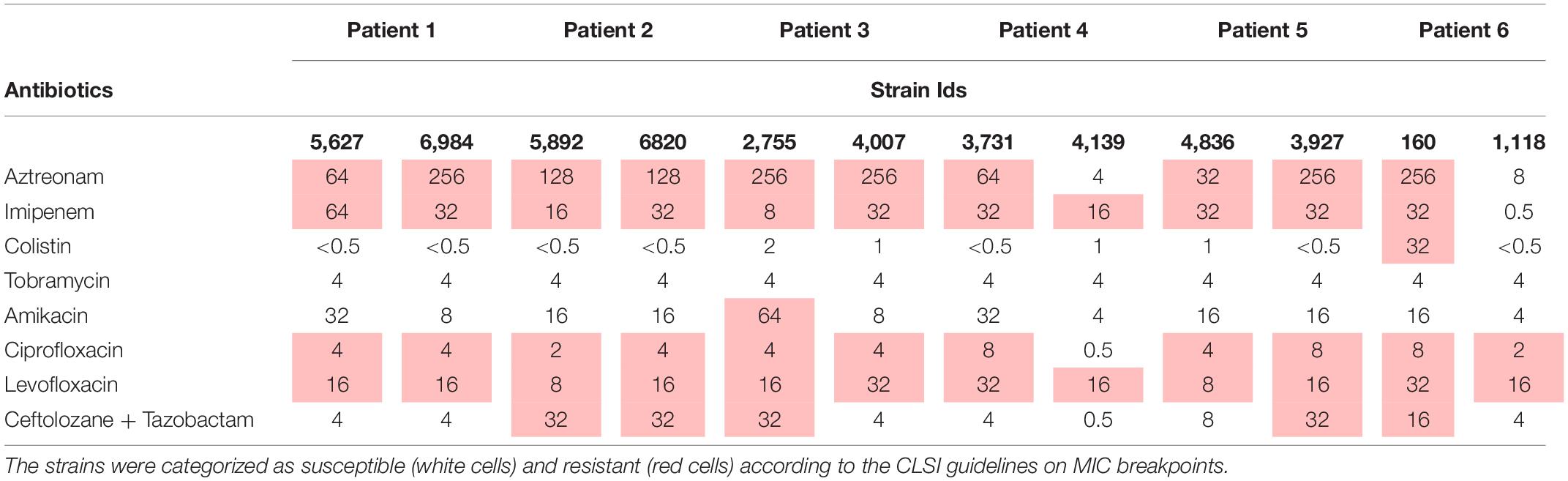
Table 2. The MICs (mg/L) of the eight antibiotics against the panel of 12 P. aeruginosa isolates by broth micro-dilution (BMD).
We tried to correlate the phenotypic antibiograms to the AMR gene patterns obtained using the CARD analysis, but there were inconsistencies in the results. In patient 3, isolates 4,007 and 3,856 were susceptible to tobramycin, piperacillin and amikacin in contrast to other genotypically similar isolates from the same patient (Figure 3). We noted the unique presence of a β-lactam resistance gene blaPDC–2 for isolates 810, 1,118, and 1,119, isolated post-transplant from patient 6. Similarly strain 3,312 (8 months before LTx) was the only strain containing a gene for the antibiotic target protection protein rpsJ. Interestingly, 5 months after the LTx this gene seemed to be lost from isolate 3,927. Unlike others, isolates 6,291, 6,543, and 6,544 (4 months before LTx and during LTx, respectively) isolated from patient 2 lacked the gene modulating permeability to antibiotics. The isolates from different time points from patient 4 lacked the presence of mexZ gene, which codes for a regulator of an important efflux pump responsible for imparting resistance to aminoglycosides (Figure 3).
Discussion
Our analysis shows that P. aeruginosa isolates from different time points, but from the same patient, contained identical STs suggesting lack of cross infection in the patients under study. These isolates showed resistance to antibiotics in comparison to the susceptible reference isolates. Correct treatment depends on continuous and serial assessment of resistance features. Moreover, in the majority of recurrent infections patients tend to harbor genotypically similar P. aeruginosa isolates (Workentine et al., 2013). P. aeruginosa is a tough bug to kill and it persists even after prolonged antibiotic treatment as well as lung transplantation (Walter et al., 1997; Mainz et al., 2012; Beaume et al., 2017; Parkins et al., 2018). Therefore, a more frequent follow-up of infection after LTx is necessary to see how the same strain evolves over the course of time.
We determined the taxonomic identity and genomic relatedness of the P. aeruginosa isolates. We observed significant genomic homology for the isolates from the same CF patients. Furthermore the MLST typing of the 36 SPLT isolates demonstrated characteristic patient specific unique STs. This observation is supported by the difference seen in the Canadian MLST typing of P. aeruginosa (Jeukens et al., 2019). They noted a significant heterogeneity and a need of personalized patient care in the context of CF Core genome phylogeny of the 36 SPLT isolates depicted six clades, one per patient, establishing the fact that patient specific genotypic diversity exists for the P. aeruginosa populations during chronic CF infection (Fothergill et al., 2007; Mena et al., 2008; Bragonzi et al., 2009; Wilder et al., 2009; Huse et al., 2010; Mowat et al., 2011; Chung et al., 2012; Workentine et al., 2013; Jiricny et al., 2014). The genotypic and phenotypic divergence among the isolates from different patients could be a result of antibiotic treatment driven evolution of the P. aeruginosa populations. Alternative reasons for such evolution could be selective stress due to nutrient and oxygen deficiency, impaired immune response and barrier function of the mucosal linings (Feliziani et al., 2014; Lozano et al., 2018). In CF patients, P. aeruginosa causes infections through two ways, i.e., using both adaptive and mutational mechanisms (Lister et al., 2009). The study highlights how isolates from the same patients evolve over time and how it is difficult to correlate the increased MIC with the genotype as there tends to be no clear differences at the genomic level. The resistant nature of the strains could also be related to mutations. Thus, identification of the mutations along with the resistance genes could aid our understanding of increased antibiotic resistance (Khaledi et al., 2019).
Likewise, the assessment of genomic similarity between the SPLT isolates from the other clinical and environmental P. aeruginosa strains does not hinder the branching pattern of the SPLT isolates under study except for strains from patients 4 and 6. It is supported by a previous report focusing on indels, recombination or SNPs for definition of inter-patient variability and intra-patient similarity (Huse et al., 2010). Interestingly in patient 6, two clades were formed. One clade consisted of strains before LTx while the other consisted of strains after LTx, which shows how the genotype can alter overtime. Additionally, we saw the spread of the SPLT isolates throughout the phylogenetic tree based on core genes, still showing patient specific clustering. This observation was found to be coherent with the noted difficulties in discriminating CF isolates based on their geography and origin (Jeukens et al., 2019; Pincus et al., 2020).The latter demands further understanding of the emergence of resistance and virulence among these P. aeruginosa to avoid or manage lung infections (Cho et al., 2014; Jaillard et al., 2017).
Our investigation of genomic features revealed various AMR and virulence genes. The main characteristics of the virulence genes were related to adhesion and secretion thereby suggesting that these isolates could be biofilm producers. Similarly, the isolates displayed a varied response to the array of antibiotics even though the isolates have homologous MLST types found during the analysis. This might mean that the resistance profile might also be related to host-related factors, which are largely unknown. There have been extensive studies on increased antibiotic resistance in P. aeruginosa. Also, the microbes have an ability to integrate exogenous DNA to withstand the selective pressure of antibiotics. Thus, in order to understand these dynamics, there have been various efforts to understand the correlation of genotype and phenotype (e.g., response to antibiotics) by several researchers (Mena et al., 2008; Hazan et al., 2014; Macia et al., 2014; Sousa and Pereira, 2014; Kos et al., 2015; Jaillard et al., 2017; Lozano et al., 2018; Garcia-Nuñez et al.,2020). Furthermore, the MLST profiles derived from two different tools-PubMLST and EPISEQ and the phenotypic antibiotic susceptibility profiles shows disparity. This finding highlights the limitations of the tools and databases and recognizes the impact this could have on predicting resistance and treating infections (Ng et al., 2021). As this study has focused on a very specific set of isolates, from respiratory tract specimens of a few patients from the same hospital, those observed advantages for each tool may be different with a different set of strains.
Pseudomonas aeruginosa is a key organism in cases of bronchiectasis leading to increased mortality, as documented in Europe (Ringshausen et al., 2013). The observation that the isolates are genetically alike but show AMR heterogeneity is evident through the recent increase in cases of bronchiectasis by P. aeruginosa. Moreover, studies focusing on the United Kingdom population suggested approximately 29% mortality in the bronchiectasis patients (Loebinger et al., 2009). Thus, the British Thoracic Society emphasizes microbiological assessment and personalized treatment to mitigate drug resistance during bronchiectasis due to the multi-drug resistance profile of P. aeruginosa (Pasteur et al., 2010).
Conclusion
Our study sheds light on the lack of intra-patient diversity and phenotypic plasticity of P. aeruginosa isolates associated with CF patients undergoing LTx. Most patients with CF carry similar P. aeruginosa isolates before, during and after LTx. Genomic similarity between these isolates is observed but they show phenotypic variation. Thus, despite continuous P. aeruginosa colonization and infection by a single genotype, these isolates still show varied responses to antibiotics. Our study equally endorses careful assessment for antibiotic susceptibility and high-throughput genomic-level monitoring of antibiotic therapy against P. aeruginosa in patients with CF.
Data Availability Statement
Whole genome sequences for all samples used in this study have been deposited in National Centre for Biotechnology Information (NCBI) and are available under BioProject ID: PRJNA630383 here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA630383. The isolates for which sequence type were not assigned have been submitted to PubMLST database under the ID: BIGSdb_20200507140502_147974_63511.
Ethics Statement
All isolates examined in this study were recovered as a result of routine diagnostic procedures. No additional human samples were collected for the purposes of this study. The researchers were supplied with samples of Pseudomonas from an archived frozen collection. Researchers were not given access to any data that would allow identification of a particular patient. As the project did not require the use of any human material or any personal data, approval of the local ethics committee was not required.
Author Contributions
AB, SO, and JP conceived the study. JP and AP collected P. aeruginosa isolates from the patients serially and gathered the metadata. RD conducted the microbiological experimentation for P. aeruginosa isolates, interpreted the sequence data and wrote the first version of the manuscript. AC, RD, and HG carried out the whole genome sequencing studies. JD assisted in the WGS analysis. All authors discussed the results and edited the manuscript.
Funding
This work was funded by the European Union’s Horizon 2020 research and innovation programs: New Diagnostics for Infectious Diseases (ND4ID) under Marie Skłodowska-Curie Grant Agreement No. 675412 and Viral and Bacterial Adhesin Network Training (ViBrANT) under Marie Skłodowska-Curie Grant Agreement No. 765042.
Conflict of Interest
During this study AB, SO, VC, RD, AP, CM, and JD were employes of BioMérieux, a company designing, developing, and marketing tests in the domain of infectious diseases. The company was not involved in the design of the current study and the opinions expressed are those of the authors and may be different from formal company opinions and policies.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.604555/full#supplementary-material
Supplementary Figure 1 | Genomic similarity among the strain of P. aeruginosa isolated from LTx patients, represented by the average nucleotide identity (ANI) as present inter-genomic distances.Supplementary Table 1 | Genomic characteristics of P. aeruginosa isolates isolated from LTx patients. It includes the attributes of sequencing, MLST types and genomic features viz. ARGs and virulence genes.
Footnotes
References
Alcock, B. P., Raphenya, A. R., Lau, T. T., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Beaume, M., Köhler, T., Greub, G., Manuel, O., Aubert, J. D., Baerlocher, L., et al. (2017). Rapid adaptation drives invasion of airway donor microbiota by Pseudomonas after lung transplantation. Sci. Rep. 7, 1–10. doi: 10.1038/srep40309
Bragonzi, A., Paroni, M., Nonis, A., Cramer, N., Montanari, S., Rejman, J., et al. (2009). Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am. J. Respir. Crit. Care Med. 180, 138–145. doi: 10.1164/rccm.200812-1943OC
Celli, B. R., Decramer, M., Wedzicha, J. A., Wilson, K. C., Agustí, A., Criner, G. J., et al. (2015). An official American Thoracic Society/European Respiratory Society statement: research questions in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 191, e4–e27. doi: 10.1183/09031936.00009015
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33(Suppl._1), D325–D328. doi: 10.1093/nar/gki008
Cho, H. H., Kwon, K. C., Kim, S., and Koo, S. H. (2014). Correlation between virulence genotype and fluoroquinolone resistance in carbapenem-resistant Pseudomonas aeruginosa. Ann. Lab. Med. 34, 286–292. doi: 10.3343/alm.2014.34.4.286
Chung, J. C., Becq, J., Fraser, L., Schulz-Trieglaff, O., Bond, N. J., Foweraker, J., et al. (2012). Genomic variation among contemporary Pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J. Bacteriol. 194, 4857–4866. doi: 10.1128/JB.01050-12
Clinical and Laboratory Standards Institute (2017). Performance Standards for Antimicrobial Susceptibility Testing. CLSI supplement M100. Wayne: CLSI.
Cutting, G. R. (2015). Cystic fibrosis genetics: from molecular understanding to clinical application. Nat. Rev. Genet. 16, 45–56. doi: 10.1038/nrg3849
Feliziani, S., Marvig, R. L., Lujan, A. M., Moyano, A. J., Di Rienzo, J. A., Johansen, H. K., et al. (2014). Coexistence and within-host evolution of diversified lineages of hypermutable Pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet. 10:e1004651. doi: 10.1371/journal.pgen.1004651
Fothergill, J. L., Panagea, S., Hart, C. A., Walshaw, M. J., Pitt, T. L., and Winstanley, C. (2007). Widespread pyocyanin over-production among isolates of a cystic fibrosis epidemic strain. BMC Microbiol. 7:45. doi: 10.1186/1471-2180-7-45
Garcia-Nuñez, M., Garcia-Gonzalez, M., Pomares, X., Montón, C., Millares, L., Quero, S., et al. (2020). The respiratory microbiome in cystic fibrosis: compartment patterns and clinical relationships in early stage disease. Front. Microbiol. 11:1463. doi: 10.3389/fmicb.2020.01463
Hazan, R., Maura, D., Que, Y. A., and Rahme, L. G. (2014). “Assessing Pseudomonas aeruginosa persister/antibiotic tolerant cells,” in Pseudomonas Methods and Protocols, eds A. Filloux and J. L. Ramos (New York, NY: Humana Press), 699–707. doi: 10.1007/978-1-4939-0473-0_54
Huse, H. K., Kwon, T., Zlosnik, J. E., Speert, D. P., Marcotte, E. M., and Whiteley, M. (2010). Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. mBio 1:e00199-10. doi: 10.1128/mBio.00199-10
Jaillard, M., van Belkum, A., Cady, K. C., Creely, D., Shortridge, D., Blanc, B., et al. (2017). Correlation between phenotypic antibiotic susceptibility and the resistome in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 50, 210–218. doi: 10.1016/j.ijantimicag.2017.02.026
Jeukens, J., Freschi, L., Kukavica-Ibrulj, I., Emond-Rheault, J. G., Allard, C., Barbeau, J., et al. (2019). The Pseudomonas aeruginosa population among Cystic Fibrosis patients in Quebec, Canada: a disease hotspot without known epidemic isolates. J. Clin. Microbiol. 57:e02019-18. doi: 10.1128/JCM.02019-18
Jiricny, N., Molin, S., Foster, K., Diggle, S. P., Scanlan, P. D., Ghoul, M., et al. (2014). Loss of social behaviours in populations of Pseudomonas aeruginosa infecting lungs of patients with cystic fibrosis. PLoS One 9:e83124. doi: 10.1371/journal.pone.0083124
Jolley, K. A., Bray, J. E., and Maiden, M. C. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST. org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Khaledi, A., Weimann, A., Schniederjans, M., Asgari, E., Kuo, T. H., Oliver, A., et al. (2019). Fighting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. BioRxiv [Preprint]. doi: 10.15252/emmm.201910264
Kos, V. N., Deraspe, M., McLaughlin, R. E., Whiteaker, J. D., Roy, P. H., Alm, R. A., et al. (2015). The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob. Agents Chemother. 59, 427–436. doi: 10.1128/AAC.03954-14
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lister, P. D., Wolter, D. J., and Hanson, N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22, 582–610. doi: 10.1128/CMR.00040-09
Loebinger, M. R., Wells, A. U., Hansell, D. M., Chinyanganya, N., Devaraj, A., Meister, M., et al. (2009). Mortality in bronchiectasis: a long-term study assessing the factors influencing survival. Eur. Respir. J. 34, 843–849. doi: 10.1183/09031936.00003709
Lozano, C., Azcona-Gutiérrez, J. M., Van Bambeke, F., and Sáenz, Y. (2018). Great phenotypic and genetic variation among successive chronic Pseudomonas aeruginosa from a cystic fibrosis patient. PLoS One 13:e0204167. doi: 10.1371/journal.pone.0204167
Macia, M. D., Rojo-Molinero, E., and Oliver, A. (2014). Antimicrobial susceptibility testing in biofilm-growing bacteria. Clin. Microbiol. Infect. 20, 981–990. doi: 10.1111/1469-0691.12651
Magiorakos, A. P., Srinivasan, A., Carey, R. B., Carmeli, Y., Falagas, M. E., Giske, C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Mainz, J. G., Hentschel, J., Schien, C., Cramer, N., Pfister, W., Beck, J. F., et al. (2012). Sinonasal persistence of Pseudomonas aeruginosa after lung transplantation. J. Cystic Fibrosis 11, 158–161. doi: 10.1016/j.jcf.2011.10.009
Mena, A., Smith, E. E., Burns, J. L., Speert, D. P., Moskowitz, S. M., Perez, J. L., et al. (2008). Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J. Bacteriol. 190, 7910–7917. doi: 10.1128/JB.01147-08
Mowat, E., Paterson, S., Fothergill, J. L., Wright, E. A., Ledson, M. J., Walshaw, M. J., et al. (2011). Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am. J. Respir. Crit. Care Med. 183, 1674–1679. doi: 10.1164/rccm.201009-1430OC
Ng, R. N., Tai, A. S., Chang, B. J., Stick, S. M., and Kicic, A. (2021). Overcoming challenges to make bacteriophage therapy standard clinical treatment practice for cystic fibrosis. Front. Microbiol. 11:3389. doi: 10.3389/fmicb.2020.593988
Parkins, M. D., Somayaji, R., and Waters, V. J. (2018). Epidemiology, biology, and impact of clonal Pseudomonas aeruginosa infections in cystic fibrosis. Clin. Microbiol. Rev. 31:e00019-18. doi: 10.1128/CMR.00019-18
Pasteur, M. C., Bilton, D., and Hill, A. T. (2010). British thoracic Society guideline for non-CFbronchiectasis. Thorax 65(Suppl. 1), i1–i58. doi: 10.1136/thx.2010.142778
Pincus, N. B., Ozer, E. A., Allen, J., Nguyen, M., Davis, J. J., Winter, D. R., et al. (2020). A genome-based model to predict the virulence of Pseudomonas aeruginosa isolates. mBio 11:e01527-20. doi: 10.1128/mBio.01527-20
Pirnay, J. P., Bilocq, F., Pot, B., Cornelis, P., Zizi, M., Van Eldere, J., et al. (2009). Pseudomonas aeruginosa population structure revisited. PLoS One 4:e7740. doi: 10.1371/journal.pone.0007740
Ringshausen, F. C., de Roux, A., Pletz, M. W., Hämäläinen, N., Welte, T., and Rademacher, J. (2013). Bronchiectasis-associated hospitalizations in Germany, 2005–2011: a population-based study of disease burden and trends. PLoS One 8:e71109. doi: 10.1371/journal.pone.0071109
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Salsgiver, E. L., Fink, A. K., Knapp, E. A., LiPuma, J. J., Olivier, K. N., Marshall, B. C., et al. (2016). Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis. Chest 149, 390–400. doi: 10.1378/chest.15-0676
Sanders, D. B., and Fink, A. K. (2016). Background and epidemiology. Pediatr. Clin. 63, 567–584. doi: 10.1016/j.pcl.2016.04.001
Snell, G., Reed, A., Stern, M., and Hadjiliadis, D. (2017). The evolution of lung transplantation for cystic fibrosis: a 2017 update. J. Cystic Fibrosis 16, 553–564. doi: 10.1016/j.jcf.2017.06.008
Sousa, A. M., and Pereira, M. O. (2014). Pseudomonas aeruginosa diversification during infection development in cystic fibrosis lungs—a review. Pathogens 3, 680–703. doi: 10.3390/pathogens3030680
Stefani, S., Campana, S., Cariani, L., Carnovale, V., Colombo, C., Lleo, M. M., et al. (2017). Relevance of multidrug-resistant Pseudomonas aeruginosa infections in cystic fibrosis. Int. J. Med. Microbiol. 307, 353–362. doi: 10.1016/j.ijmm.2017.07.004
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
van Belkum, A., Soriaga, L. B., LaFave, M. C., Akella, S., Veyrieras, J. B., Barbu, E. M., et al. (2015). Phylogenetic distribution of CRISPR-Cas systems in antibiotic-resistant Pseudomonas aeruginosa. mBio 6:e01796-15. doi: 10.1128/mBio.01796-15
Walter, S., Gudowius, P., Boßhammer, J., Römling, U., Weißbrodt, H., Schürmann, W., et al. (1997). Epidemiology of chronic Pseudomonas aeruginosa infections in the airways of lung transplant recipients with cystic fibrosis. Thorax 52, 318–321. doi: 10.1136/thx.52.4.318
Wilder, C. N., Allada, G., and Schuster, M. (2009). Instantaneous within-patient diversity of Pseudomonas aeruginosa quorum-sensing populations from cystic fibrosis lung infections. Infect. Immun. 77, 5631–5639. doi: 10.1128/IAI.00755-09
Winstanley, C., O’Brien, S., and Brockhurst, M. A. (2016). Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 24, 327–337. doi: 10.1016/j.tim.2016.01.008
Keywords: Pseudomonas aeruginosa, lung transplantation, antimicrobial resistance, cystic fibrosis, whole genome sequencing
Citation: Datar R, Coello Pelegrin A, Orenga S, Chalansonnet V, Mirande C, Dombrecht J, Perry JD, Perry A, Goossens H and van Belkum A (2021) Phenotypic and Genomic Variability of Serial Peri-Lung Transplantation Pseudomonas aeruginosa Isolates From Cystic Fibrosis Patients. Front. Microbiol. 12:604555. doi: 10.3389/fmicb.2021.604555
Received: 09 September 2020; Accepted: 15 March 2021;
Published: 07 April 2021.
Edited by:
Rustam Aminov, University of Aberdeen, United KingdomReviewed by:
Barbara C. Kahl, University of Münster, GermanyKumar Siddharth Singh, Helmholtz Center for Infection Research, Helmholtz Association of German Research Centers (HZ), Germany
Kunal Jani, University of Chemistry and Technology in Prague, Czechia
Copyright © 2021 Datar, Coello Pelegrin, Orenga, Chalansonnet, Mirande, Dombrecht, Perry, Perry, Goossens and van Belkum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alex van Belkum, YWxleC52YW5iZWxrdW1AYmlvbWVyaWV1eC5jb20=