 Lucie Gebauer1
Lucie Gebauer1 Marie-Lara Bouffaud1
Marie-Lara Bouffaud1 Minh Ganther1Bunlong Yim2
Minh Ganther1Bunlong Yim2 Doris Vetterlein1,3
Doris Vetterlein1,3 Kornelia Smalla2
Kornelia Smalla2 François Buscot1,4
François Buscot1,4 Anna Heintz-Buschart1,4
Anna Heintz-Buschart1,4 Mika T. Tarkka1,4*
Mika T. Tarkka1,4*- 1Helmholtz Centre for Environmental Research, Halle, Germany
- 2Julius Kühn-Institute, Braunschweig, Germany
- 3Institute of Agricultural and Nutritional Sciences, Martin Luther University Halle-Wittenberg, Halle, Germany
- 4German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig, Leipzig, Germany
Preservation of the phytostimulatory functions of plant growth-promoting bacteria relies on the adaptation of their community to the rhizosphere environment. Here, an amplicon sequencing approach was implemented to specifically target microorganisms with 1-aminocyclopropane-1-carboxylate deaminase activity, carrying the acdS gene. We stated the hypothesis that the relative phylogenetic distribution of acdS carrying microorganisms is affected by the presence or absence of root hairs, soil type, and depth. To this end, a standardized soil column experiment was conducted with maize wild type and root hair defective rth3 mutant in the substrates loam and sand, and harvest was implemented from three depths. Most acdS sequences (99%) were affiliated to Actinobacteria and Proteobacteria, and the strongest influence on the relative abundances of sequences were exerted by the substrate. Variovorax, Acidovorax, and Ralstonia sequences dominated in loam, whereas Streptomyces and Agromyces were more abundant in sand. Soil depth caused strong variations in acdS sequence distribution, with differential levels in the relative abundances of acdS sequences affiliated to Tetrasphaera, Amycolatopsis, and Streptomyces in loam, but Burkholderia, Paraburkholderia, and Variovorax in sand. Maize genotype influenced the distribution of acdS sequences mainly in loam and only in the uppermost depth. Variovorax acdS sequences were more abundant in WT, but Streptomyces, Microbacterium, and Modestobacter in rth3 rhizosphere. Substrate and soil depth were strong and plant genotype a further significant single and interacting drivers of acdS carrying microbial community composition in the rhizosphere of maize. This suggests that maize rhizosphere acdS carrying bacterial community establishes according to the environmental constraints, and that root hairs possess a minor but significant impact on acdS carrying bacterial populations.
Introduction
Maize (Zea mays L.) is one of the most important staple crops worldwide in terms of agronomy and global economic impact (Food and Agriculture Organization of the United Nations, 2019). Various maize cultivars are grown in different climatic zones, and maize is used as fodder, for human consumption, or for industrial products such as biofuel. Maize cultivation requires intensive management and high levels of resources, and there is a growing need to find sustainable practices (Akinnifesi et al., 2010; Shiferaw et al., 2011; Sauerbrei et al., 2014).
Research on the development of maize root system and rhizosphere microbiome is steadily increasing, since improving root growth and the efficiency of plant beneficial microbes can support maize growth under normal and stress conditions (Lynch, 2015; Brisson et al., 2019). In order to support phytostimulatory plant-microbe interactions for sustainable maize production, the first step is to characterize maize rhizosphere communities. Investigations on the bacterial 16S rRNA microbiome indicate that like in other plants, maize rhizosphere bacterial community composition differs from that in bulk soil (Bouffaud et al., 2014). They also indicate that field conditions and crop rotation (Benitez et al., 2017), pathogens (Saravanakumar et al., 2017), soil properties and plant age (Bouffaud et al., 2012; Peiffer et al., 2013) interact to drive the maize rhizosphere microbiome composition. Further modifications are caused by maize genotypes (Bouffaud et al., 2014; Walters et al., 2018) and rhizodeposition (Cotton et al., 2019).
Plant growth promoting rhizobacteria (PGPR) are a ubiquitous and species-rich group of bacteria (Antoun and Prévost, 2005; Renoud et al., 2020) that have a considerable potential to support agricultural practice by their positive influence on plant nutrition, growth, abiotic stress and disease resistance (Egamberdiyeva, 2007; Tak et al., 2013; Timmusk et al., 2017). Enriching PGPR populations represents an attractive means for future agricultural technologies to maintain yields while reducing mineral fertilization (Gamalero and Glick, 2015; Brisson et al., 2019). Rhizosphere PGPR community is a subset of the bulk soil community (Li et al., 2012), and its structure, characterized by barcoding their 16S rRNA, is also modulated by the drivers described earlier for whole bacterial microbiome (Roesch et al., 2008; Coelho et al., 2009; Bouffaud et al., 2018; Long et al., 2018; Renoud et al., 2020). Variable efficacy of PGPR populations under field conditions, where several environmental factors act simultaneously and host plant species vary, indicates that limited knowledge on their community response to the environment and the organismal interactions in the root-soil interface are perhaps the main factors that hampers the widespread use of the PGPR (Timmusk et al., 2017).
Many PGPR have the potential to support growth of plants by altering their hormonal balance. This takes place either by affecting plant hormone production, exuding plant hormones or producing hormone precursor degrading enzymes (Shahzad et al., 2013; Rozier et al., 2017). Plant hormone related PGPR activities can support root growth, since root system architecture is adapted through the modulation of phytohormone levels (Vacheron et al., 2013) in response to environmental cues. For example, auxins such as indole-3-acetic acid modulate root elongation and induce lateral root formation, and indeed, the PGPR may alter root systems by either producing auxins, or inducing in planta auxin production (Sukumar et al., 2013). The effect of the plant hormone ethylene on root growth is largely mediated through interactions with auxin (Ivanchenko et al., 2008), via the regulation of auxin biosynthesis or local auxin distribution (Růžička et al., 2007), and increased ethylene levels essentially lead to the inhibition of root elongation and increased production of root hairs (Tanimoto et al., 1995). In this context, some bacteria deaminate the precursor of ethylene, 1-aminocyclopropane-1-carboxylate (ACC), and the ACC deaminase activity lowers ethylene levels and alleviates the repressive effect of ethylene on root elongation that leads to improved nutrient acquisition and water uptake capacity. Due to these effects on the host plant, the ACC deaminase positive bacteria support plants grown under fair as well as stressful conditions (Glick, 2005; Jaemsaeng et al., 2018; Yoolong et al., 2019).
Our earlier data from a maize field revealed, that the bacteria carrying the acdS gene (acdS+ bacteria) are more abundant in the rhizosphere than in the bulk soil, and amplicon sequencing analysis of the acdS+ community composition revealed that it is affected by maize genotype (Bouffaud et al., 2018). It was also identified, that maize growth stage further affects acdS+ community composition, and it represents an interacting driver to the field site. During the flowering stage, the acdS+ diversity level and community composition varied across three field sites with luvisol, fluvic cambisol, and calcisol soil types, but not during the early, six leaf stage of maize development (Renoud et al., 2020). It is not yet established whether depth within the soil or the production of root hairs affect the structure of acdS+ communities. Soil depth in the field affects the distribution of available nutrients, water and oxygen, and subsequently, different soil depths in the field show distinct assemblies of bacteria (Fierer et al., 2003). Root hairs are protrusions of epidermal cells that extend to the rhizosphere and change rhizosphere properties by taking up nutrients and by exudation of organic compounds and enzymes (Marschner et al., 2011). By using barley root hair formation and elongation mutants and applying 16S rRNA gene amplicon sequencing, Robertson-Albertyn et al. (2017) associated the presence of the root hairs to the depletion of the relative abundances of some and the increase of other bacterial taxa, and observed that the root hair mutants triggered a pronounced effect on the bacterial community in a substrate with lower organic matter content.
In the frame of the German Science Foundation’s priority program “Rhizosphere Spatiotemporal Organization” (Vetterlein et al., 2020a) we implemented a soil column experiment (Vetterlein et al., 2020b) in loam and sand substrates to assess the effects of substrate, the depths where the samples were taken, i.e., depth, and root hair presence/absence on acdS+ community composition. Three hypotheses were stated: (i) the relative phylogenetic distribution of the acdS+ community structure is affected by substrate, root hair formation, and depth, (ii) acdS+ genera that are differentially abundant between loam and sand will include genera whose relative abundance have been shown to be affected by field conditions, and, (iii) the influence of root hair presence/absence is different in loam than sand.
Materials and Methods
Soil Preparation and Plant Material
The design of the soil column experiments was described in detail by Vetterlein et al. (2020b). Briefly, loamy soil (L) substrate, which was classified as Haplic Phaeozem, consisted of 32.5% sand, 47.9% silt, and 19.5% clay (Supplementary Table 1). Inorganic carbon was not detectable; therefore, total carbon was assumed to represent the organic carbon content with 8.6 g kg–1 Corg. The nitrogen content was 0.84 g kg–1 Norg, resulting in a C:N ratio of 10.2. Soil pH (CaCl2) was 6.4. The substrate sand (S) was obtained by mixing L with quartz sand in 83.3%:16.7% ratio. The reason for mixing loamy soil (L) substrate with quartz sand was to yield two substrates with different textures but the same microbial inoculate (of the original soil). To enable similar nutrient uptake of maize on both substrates, loam and sand were fertilized differently according to Vetterlein et al. (2020b). Like for barley, root hair formation, and elongation mutants have also been constructed for maize (Hochholdinger and Tuberosa, 2009), which we used here to investigate maize root hair formation induced changes in rhizosphere acdS community composition. Zea mays root hair defective mutant rth3 and the corresponding B73 wildtype (Wen and Schnable, 1994; Hochholdinger et al., 2008) seeds were propagated at the experimental station Endenich of the Faculty of Agriculture of the University of Bonn. The monogenic mutant rth3 is transposon induced and shows normal root hair initiation but disturbed elongation (Hochholdinger et al., 2008).
Experimental Design and Conduction of the Experiment
The basic design of the soil column experiment consisted of three factors and six replications, referring to six planted soil columns per treatment. The experimental factors included the substrate represented by loam (L) and sand (S) and the maize genotype represented by wild type (WT) and rth3. This resulted in 24 columns. The third experimental factor did not affect column replicate number; it was the depth of the column at three equally spaced layers (D1, D2, and D3) (Supplementary Figure 1). Substrates L and S were sieved to a particle size of ≤1 mm and fertilized with 50 mg N (NH4NO3), 50 mg K (K2SO4), 25 mg Mg (MgCl2 × 6 H2O), and 40 mg P (CaHPO4) per kg substrate dry mass for loam, but with 100 mg N (NH4NO3), 100 mg K (K2SO4), 50 mg Mg (MgCl2 × 6 H2O), 80 mg P (CaHPO4), 100 mg CaSO4 × 2 H2O, 3.25 mg MnSO4 × H2O, 0.79 mg Zn(NO3)2 × 4 H2O, 0.5 mg CuSO4 × 5 H2O, 0.17 mg H3BO3 and 3.25 mg FeEDTA for sand. Substrates were filled into acrylic columns (25 cm height, 7 cm inner diameter, 0.5 cm wall thickness) with a bulk density of 1.26 g cm–3. Maize seeds were surface-sterilized using 10 min 10% H2O2, 5 min H2O, followed by soaking in saturated CaSO4 for 3 h. The surface sterilized seeds were sown 1 cm below the substrate surface and covered with coarse gravel to reduce evaporation. Aluminum foil was placed around the columns to prevent algal growth, and irrigation was implemented from top and bottom to maintain a water content of 22% (v/v) throughout the growth period. The plants were grown for 22 days in a growth chamber with the following settings: 12 h light period at 350 μmol m–2 s–1 photosynthetically active radiation, 22°C/18°C day/night temperature and 65% humidity.
Cultivation was terminated at 3 weeks age of the plants as the growth of the maize plants is restricted in soil columns, which might affect root distribution and physiology (Poorter et al., 2012). Shoot height and leaf number were measured at harvest. Plant growth was not affected by substrate, but rth3 plant height and water consumption were lower than that of WT (Supplementary Figure 1). The harvest was implemented at three depths by horizontal cuts through the whole column. Maize roots were collected from soil slices of approximately 1.6 cm thickness at 4.5–6.1 cm (depth 1, D1), 9.0–10.6 cm (depth 2, D2), and 13.5–15.1 cm (depth 3, D3) below the soil surface (Supplementary Figure 1). Roots with adhering soil were picked up with tweezers. To obtain the material for the analyses of the rhizosphere microbiome, these roots were gently shaken and briefly submerged and shaken in sterile 0.3% NaCl [1:10 (w/v) dilution, i.e., 1 g root fresh mass + 9 mL NaCl]. The rhizosphere suspension was centrifuged at 5,000 × g for 30 min at 4°C and the rhizosphere pellets were stored at −20°C before DNA extraction.
acdS Illumina Library Preparations
Microbial composition was analyzed for six rhizosphere samples corresponding to six individual soil columns per treatment and sampling depth. Total DNA was extracted from the rhizosphere pellets (about 500 mg fresh mass) using FastDNA Spin Kit and Geneclean Spin Kit for soil following the manufacturer’s instructions (MP Biomedicals, United States). Partial acdS gene was amplified in duplicate using the primers acdSF5 and acdSR8 (Bouffaud et al., 2018). Primers were equipped with Illumina adapter sequences (Nextera XT Index Kit, Illumina, San Diego, CA, United States). To obtain error-poor amplifications, PCR was performed using Kapa Hifi HotStart ReadyMix (Kapa Hifi HotStart, KAPA-Biosystems, Wilmington, MA, United States). The PCR was performed in a S1000 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, United States) and the cycling conditions were 30 cycles of 65°C for 10 s, 72°C for 10 s. PCR products were purified using AMPure XP beads and, to assign the sequences to the respective samples, an index PCR was performed using the Illumina Nextera XT Index Kit and Kapa Hifi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA, United States). The PCR was performed in a S1000 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, United States) and the cycling conditions were 8 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s. The index PCR products were purified with AMPure XP beads and the PCR products were quantified with Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, Life Technologies, Carlsbad, CA, United States) following manufacturer’s instructions and using Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies, Santa Clara, CA, United States) with an excitation wavelength of 480 nm and an emission wavelength of 520 nm. The size and the quality of the pooled PCR products were analyzed using Agilent 2100 Bioanalyzer (Agilent). The acdS libraries were sequenced on an Illumina MiSeq system to 2 × 150 nucleotide lengths. Project design is described at NCBI under the Bioproject PRJNA666771, and raw sequences are placed in Short Reads Archive under the accessions SRX9227754-SRX9227812.
Sequencing Processing and Statistics
Only reads with the adequate primers were kept. The obtained sequences were analyzed using dadasnake (v. 0.7; Weißbecker et al., 20201) which uses the open source program R’s (v. 3.6.1; R Core Team 2017) DADA2 package (Callahan et al., 2016). acdS gene amplicon reads were cut until the lowest possible quality of a base was 11 and truncated to 100 and 90 nt for the forward and reverse reads overall length. The overall maximum expected error was constrained to 0.7. Read pairs were merged with zero mismatches, and exact sequence variants were determined to be used as ASVs (Amplicon Sequence Variants). Chimeric reads were removed using the DADA2 “consensus” algorithm (Callahan et al., 2016). The acdS sequences were aligned against an acdS in-house database extracted from FunGene (Fish et al., 2013) using BLASTn (v. 2.7.1) according to Bouffaud et al. (2018), and the ASVs not assigned taxonomically using BLASTn were removed. Two samples which were represented by less reads after quality control, ASV generation, chimera removal and taxonomic assignment steps than others were excluded from further analyses. These were one sample for loam WT maize at sampling depth D2 and one sample for sand rth3 maize at sampling depth D3 (Supplementary Table 1 and Supplementary Figure 2). After this, all treatments were represented by five replicates except for loam WT maize at sampling depth D2 and sand rth3 maize at sampling depth D3, for which only four replicates were further analyzed. A phylogenetic tree was constructed from a multiple alignment of the ASVs using mafft (v7.455; Katoh et al., 2002) by running fasttreeMP (v. 2.1.10) with the gamma option. Tips with more than 98% sequence identity were collapsed for visualization and renamed with the most abundant ASV or a random reference (if there were no ASVs in the set of collapsed tips). The tree was visualized in iroki (Moore et al., 2020). Analysis scripts are accessible at https://github.com/a-h-b/maize-acdS.
All statistical analyses and visualizations were performed in R. ASV patterns were cross-compared with permutational multivariate analysis of variance (PERMANOVA) using the “vegan” package. Since strongly skewed datasets were identified, apart from using ANOVA and Tukey’s tests, Kruskal–Wallis test followed by post hoc Dunn-Bonferroni test were carried out with “PMCMRplus.” The effect of watering during the conditioning phase was tested for each acdS genus, comparing the farming system and plant factor independently, using DESeq2 (Love et al., 2014) and visualized using pheatmap (Kolde and Kolde, 2015; Kolde, 2019).
Results
Estimation of acdS Species Richness and Diversity
Rhizosphere DNA was subjected to acdS amplicon sequencing. The total number of 3,838,082 reads clustered into 4,083 ASVs. The sequences were rarefied to the smallest read number per sample, 39,398 reads. At this level, all rarefaction curves reached their plateau, showing that this normalization was not at the expense of a satisfying sequencing depth (Supplementary Figure 2). Substrate and maize genotype affected the richness of observed ASVs. Higher average numbers of ASVs (ANOVA, P = 0.03) were detected in loam than in sand, and in loam, the average number of ASVs for rth3 was higher than for WT maize seedlings (P = 0.0005). The numbers of ASVs neither varied significantly between the root hair genotypes in sand nor between the depths where the samples were taken (Supplementary Table 2). Shannon index, a diversity measure combining the number of taxa and their evenness, was similar for loam (Figure 1A) and sand (Figure 1B) and for the three depths. Nevertheless, the Shannon index of WT in D1 was lower than that of rth3 in D1 and D2 in loam, and the index of WT in D3 was higher than rth3 in D1 in sand. Variation in Shannon indices was particularly high for rth3 in D3.
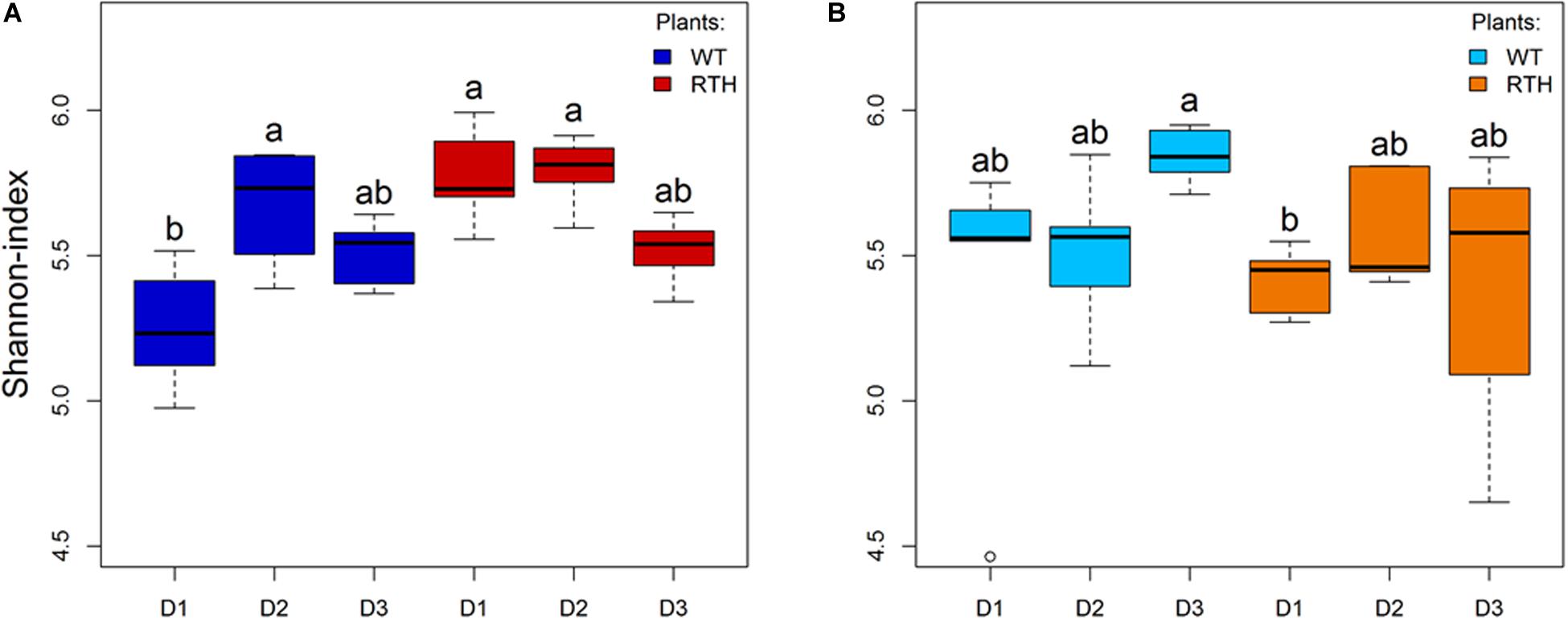
Figure 1. Shannon indices of the acdS sequences. Loam (A) and sand (B) substrate data of wild type (WT) and rth3 (RTH) maize seedlings from three depths, with uppermost sampling depth D1, middle depth D2, and lowest depth D3, were examined by Kruskal–Wallis test followed by post hoc Dunn-Bonferroni test. Different letters indicate significant differences (p < 0.05).
Factors Shaping acdS Carrying Community Composition
Soil texture was the main factor influencing the composition of acdS carrying (acdS+) bacterial communities, followed by the depth, and the effect of the maize root hair genotype according to a PERMANOVA analysis (Table 1 and Supplementary Figure 3). The acdS+ communities from the loam and sand substrates were separately analyzed in order to visualize the plant genotype and depth effects (Figure 2 and Table 1). In both substrates, the depth had a major impact on the acdS+ community. The impact of genotype was stronger in loam than in sand, and particularly strong in loam at the uppermost depth D1.
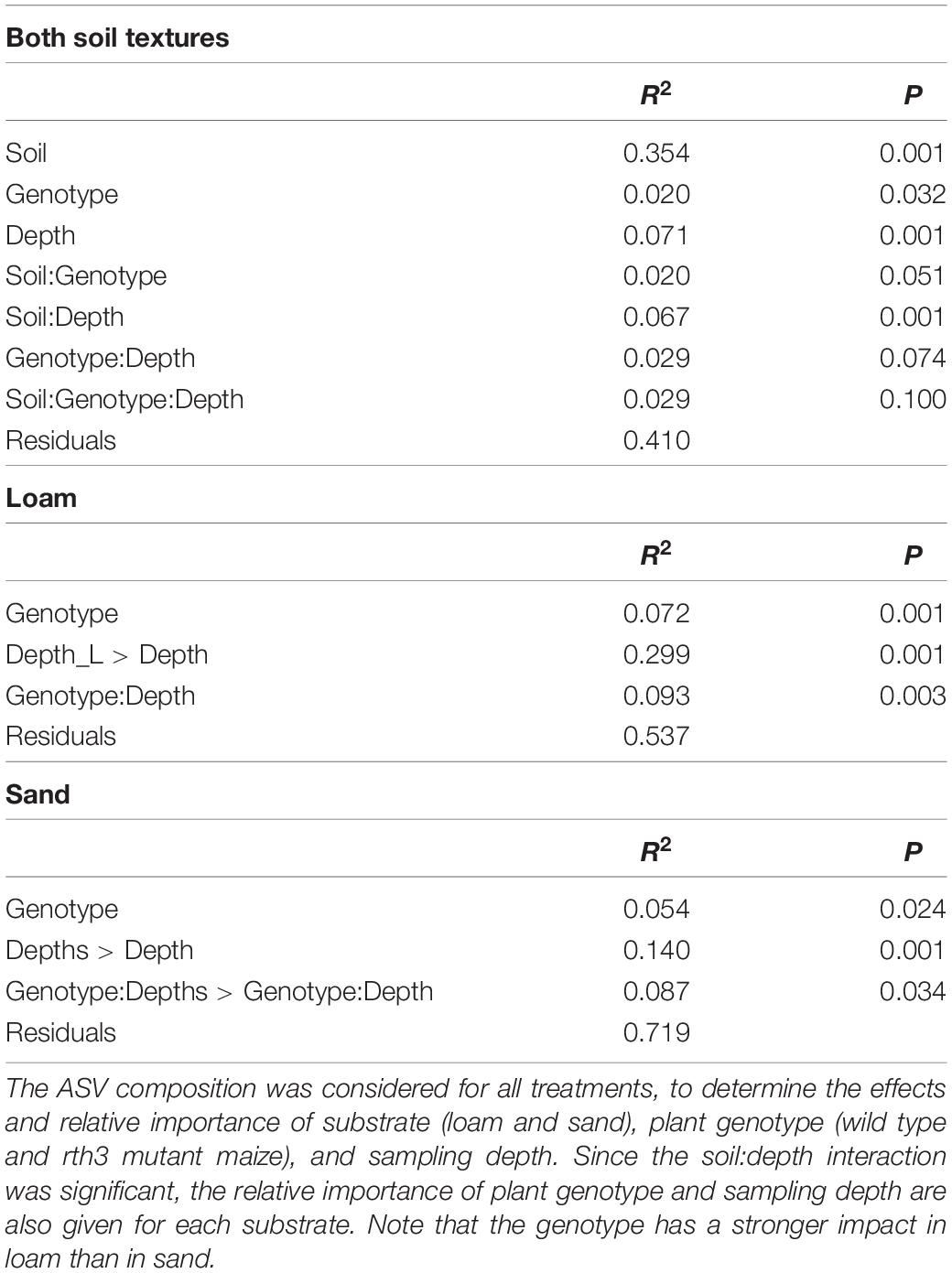
Table 1. PERMANOVA of the 1-aminocyclopropane-1-carboxylate deaminase gene (acdS) community composition.
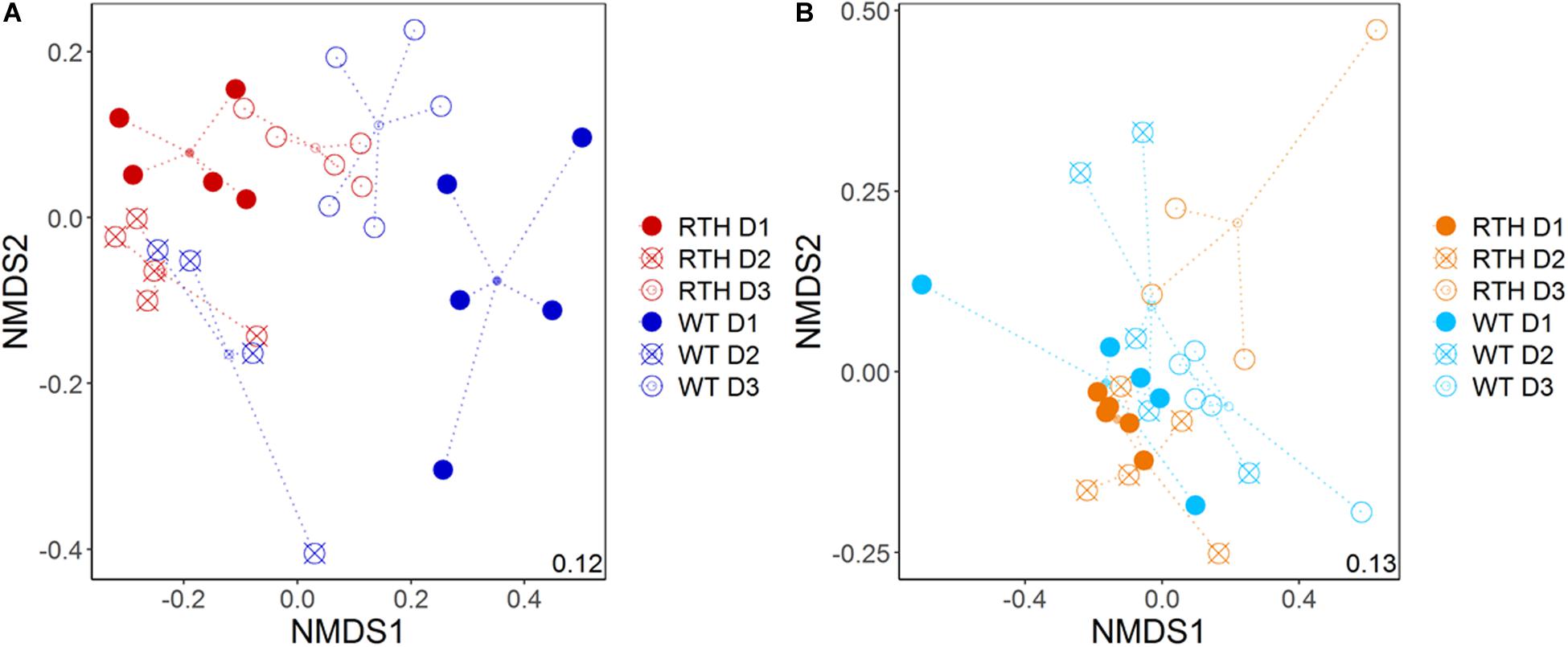
Figure 2. Non-metric dimensional scaling (NMDS) plot of acdS sequence distribution for maize rhizospheres in loam (A) and sand (B). WT, wild type; RTH, rth3 mutant; D1, uppermost sampling depth; D2, middle depth; D3, lowest depth.
Taxonomic Affiliations of the acdS Sequences and Factors Affecting Their Distribution
The acdS amplicons were largely associated with two phyla, Actinobacteria and Proteobacteria. As a demonstration of the phylogenetic classification of the acdS amplicons, we determined a tree (Supplementary Figure 4) using the ASVs and published reference sequences. There were two major clusters in the phylogenetic tree, according to the main represented phyla Actinobacteria and Proteobacteria. At the order level, Proteobacteria corresponded mainly to Burkholderiales, whereas the Actinobacteria were dominated by the Streptomycetales and Micrococcales, and at a lesser extent by the Pseudonocardiales (Figure 3; statistical differences related to substrate, plant genotype, and/or sampling depth in Supplementary Table 3). Sequences from eukaryotic microorganisms of the fungal order Sordariales were also detected. The acdS sequences from the 20 most abundant genera in maize rhizospheres represented 91% of the total number of reads (Figure 4A), and the dominant distribution of Actinobacteria and Proteobacteria ASVs was preserved. Except for the genera Pseudomonas and Methylobacterium, the other six genera from the Proteobacteria corresponded to the order Burkholderiales from Betaproteobacteria, and among the Actinobacteria, the ten genera were distributed in five different orders. Variovorax, Acidovorax, and Burkholderia of Burkholderiales, as well as Streptomyces of Streptomycetales and Tetrasphaera of Micrococcales were among the most numerous acdS+ genera.
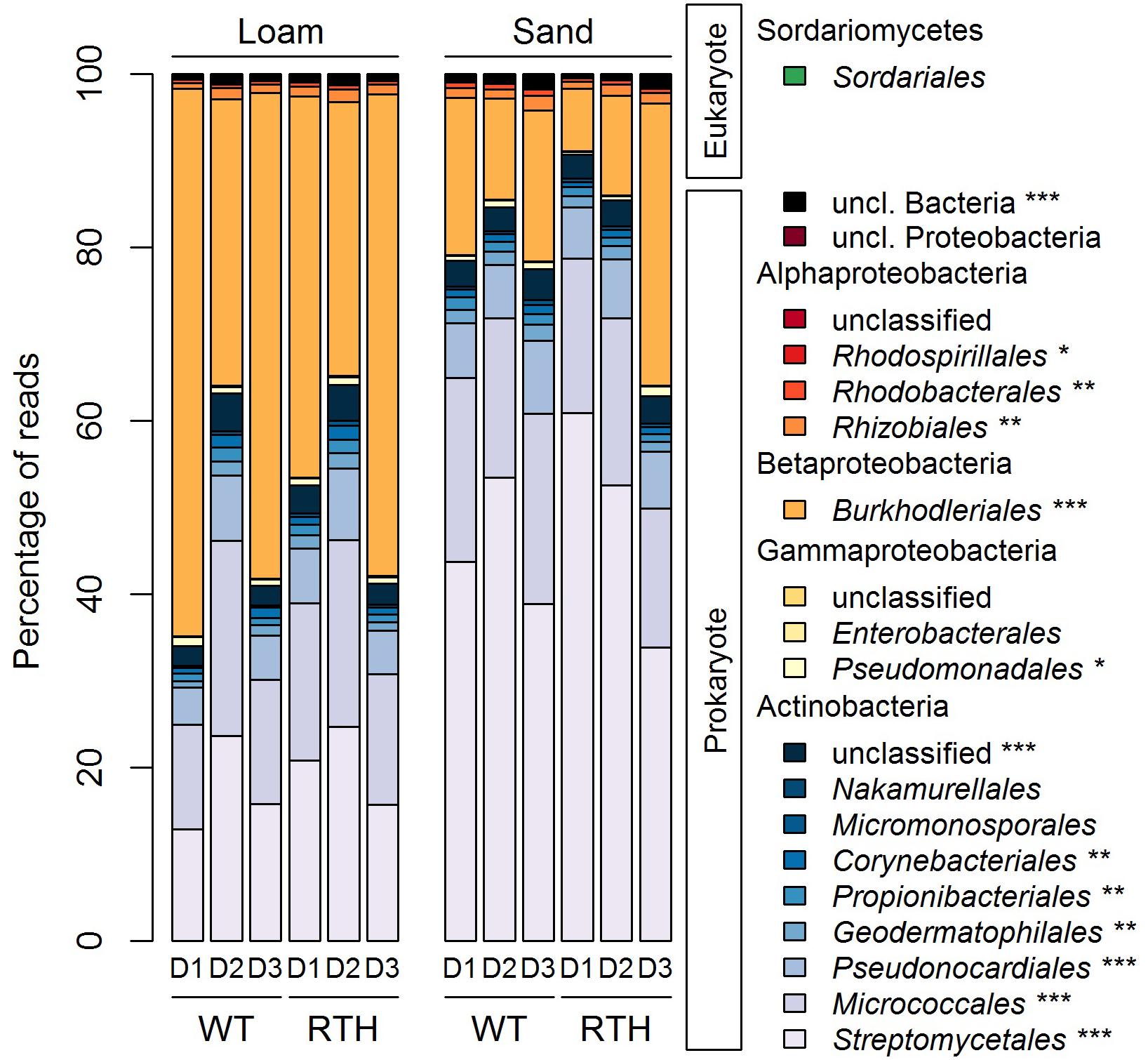
Figure 3. Relative abundance of acdS sequences in the different orders in columns with loam (L) and sand (S) of wild type B73 (WT) and rth3 (RTH) maize rhizosphere in three sampling depths (D1, D2, and D3). Different relative abundances of acdS sequences at the order level according to Kruskal–Wallis and post hoc Dunn-Bonferroni test are marked by asterisks (***P < 0.001, **P < 0.01, *P < 0.05). Statistical differences indicated in this figure are related to substrate, plant genotype, and/or sampling depth, and they are described in Supplementary Table 2.
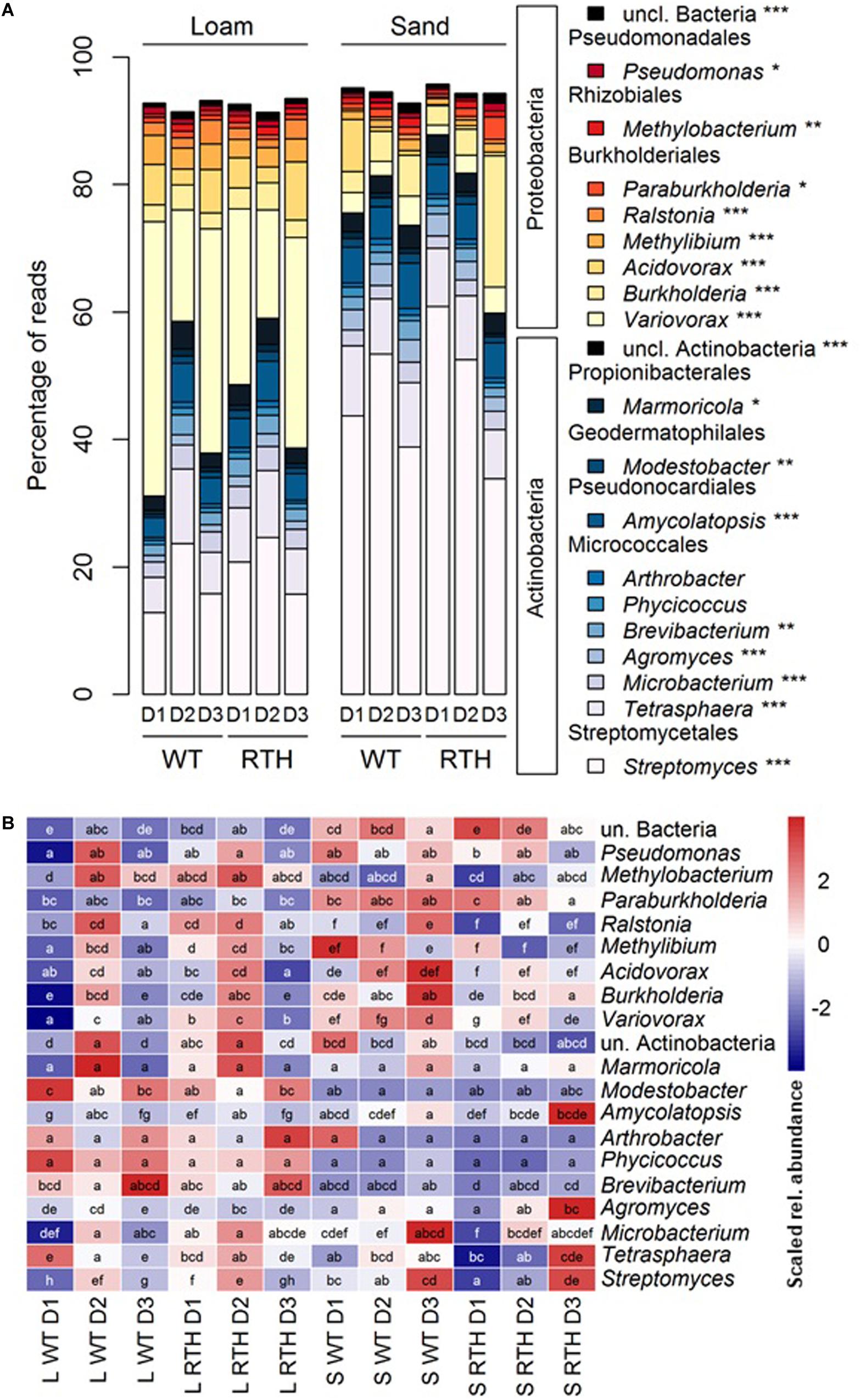
Figure 4. Effects of substrate, maize genotype, and sampling depth on the 20 most-abundant genera according to acdS analysis. Relative abundances of the 20 most abundant genera in maize rhizosphere in loam and sand with wild type B73 (WT) and rth3 (RTH) maize and three sampling depths (D1, D2, and D3). (A) Distribution of the genera in Actinobacteria and Proteobacteria. Asterisks mark differential distribution across the treatments according to Kruskal–Wallis test followed by Dunn post hoc test. (B) Statistics of the relative abundances across treatments. Red color indicates high relative mean number of sequences and blue color low relative mean number of sequences of a genus level. Different letters indicate significant differences according to Kruskal–Wallis test followed by Dunn post hoc test.
As indicated by the PERMANOVA, the substrate had a strong effect on acdS sequence composition. Compared to loam, sand showed lower relative abundance of acdS carrying Proteobacteria (34–72% vs. 8.6–53.9%), and higher abundance of Actinobacteria (28–65% vs. 46–92%), caused predominantly by lower relative abundance of Burkholderiales but higher relative abundance of Streptomycetales acdS sequences in sand (Figure 3; statistical differences at order level are described in Supplementary Table 3). Abundance of acdS reads from the genera Streptomyces and Agromyces was higher in the sand samples, whereas in loam Variovorax, Acidovorax, Ralstonia, and Methylibium were more dominant (Figure 4).
The phyla displayed marked differences in their relative abundances between the three depths of the column (descending order D1, D2, and D3). Higher relative abundances of Actinobacteria sequences were evident in the D2 than in D1 or D3, in particular in loam. Differences across the treatments were found, e.g., for Micrococcales, Propionibacteriales, Pseudonocardiales, and unclassified Actinobacteria with particularly high abundance in D2 in loam. By contrast, the acdS sequences from Burkholderiales displayed lower relative abundances in D2 than D1 or D3 in loam (Figure 3 and Supplementary Table 3). Depth also influenced the abundance of acdS from several genera (Figure 4). In loam, the effect of depth was evident on the level of genera Streptomyces, unclassified Actinobacteriaceae, Tetrasphaera, and Amycolatopsis acdS, which were more prevalent in D2. In sand, Paraburkholderia, Burkholderia, and Variovorax acdS were more abundant in D3.
Finally, maize genotype affected some acdS+ phyla as well, particularly in the uppermost layer D1. Regardless of substrate, Burkholderia acdS at order level and Variovorax acdS at genus level were more abundant in WT rhizosphere. Furthermore, for loam we observed higher abundance of and higher abundance of Methylibium acdS in WT rhizosphere. By contrast, Streptomycetales acdS at order level and Streptomyces acdS at genus level were more abundant in the rth3 rhizosphere regardless of substrate, and in loam, same pattern occurred for Microbacterium and Modestobacter acdS.
Discussion
General Differences in acdS+ Community Structure Across Soil Textures
Glick (2014) argued that the more precisely the drivers of PGPR distribution and activity are researched, the more likely it is that novel means can be developed to support plant growth in the changing environment. Contributing to this aim we investigated acdS carrying (acdS+) microbial community composition in maize rhizosphere in a multifactorial design. We observed that the main drivers in descending order of impact were substrate, depth and maize genotype. These observations essentially confirmed our first hypothesis, and suggest that in the frame of functional redundancy among ACC deaminase phyla, maize rhizosphere selects rapidly a suitable acdS+ bacterial community that maintains ACC deaminase activity in the rhizosphere.
acdS+ Communities of Maize Rhizospheres
The maize cultivar of this study, B73, is a cultivar of the Corn Belt dent genetic group of maize (Labate et al., 2003). For Mo17 and FV252, which are representatives of the same genetic group, Bouffaud et al. (2018) found a similar phylogenetic distribution of the 20 most abundant acdS+ bacterial genera to the current study: All genera correspond to Proteobacteria and Actinobacteria, contributing over 90% of the sequences, including acdS+ bacterial genera Burkholderia, Streptomyces, Acidovorax, Variovorax, and Methylibium. Of these, members of Streptomyces have been characterized as efficient biocontrol agents through production of antimicrobials, but also as plant growth and health stimulators (Merriman et al., 1974; Xiao et al., 2002; Schrey and Tarkka, 2008). ACC deminase activity plays a crucial role in this process. For instance, inoculation with Streptomyces sp. GMKU 336 stimulates plant shoot and root growth, enhances chlorophyll levels but suppresses formation of ethylene and reactive oxygen species - effects which are absent during inoculation with an ACC deaminase-deficient strain (Jaemsaeng et al., 2018). Similarly, an ACC deaminase overproducing strain of Streptomyces venezuelae ATCC 10712 stimulated shoot growth and elongation more strongly than the wild type strain (Yoolong et al., 2019).
Substrate: The Main Driver of the acdS+ Bacterial Community
The acdS+ substrate responder genera in this experiment, i.e., those with a differences in their relative abundances between loam and sand, included several genera with substrate-dependent relative abundance patterns at the 6 leaves stage of maize identified by Bouffaud (Renoud et al., 2020). Growth stage of the present experiment (BBCH14 stadium) corresponds to the 6 leaves stage. This confirms our second hypothesis on the relation between the substrate and different types of field soil used by Renoud et al. (2020). Out of the 20 most abundant acdS+ genera in this study, the relative abundances of Burkholderia, Ralstonia, and Variovorax were altered by substrate, and they belonged to the genera with differences in distribution between three field sites representing luvisol, fluvic cambisol and calcisol substrates that were studied by Renoud et al. (2020). Of note, acdS gene is not only confined to PGPR, since some members of these genera include plant pathogens, e.g., R. solanacearum of Ralstonia and P. syringae of Pseudomonas, and they both possess the acdS gene. Other members of Pseudomonas are reported to exhibit a wide range of plant growth-promoting traits. This comprises ACC deaminase activity, which is widespread also among the cultivated members of Pseudomonas for maize rhizosphere (Vacheron et al., 2016b). acdS+ Pseudomonas isolates promote maize growth (Shaharoona et al., 2006) and may reduce stress-induced ethylene levels of maize. For instance under salt stress, acdS+ Pseudomonas fluorescens strain enhanced N, P, and K uptake and increased maize biomass and grain yield over the control (Nadeem et al., 2009). Substrate may not only modify the abundance but it may also affect the relative distribution patterns of acdS+ Pseudomonas strains between rhizosphere and bulk soil. Vacheron et al. (2016b) observed that the proportion of acdS+ Pseudomonas can be higher, lower, or unchanged in maize rhizosphere than bulk soil, depending on substrate. It has also been described that the relative expression of the Pseudomonas acdS gene is modulated by maize genotype, and Pseudomonas acdS- mutant analyses suggested that the ACC deaminase activity of P. fluorescens contributes to either root branching or stress alleviation, depending on maize genotype (Vacheron et al., 2016a).
At the level of 16S rRNA gene amplicon sequencing, it is established that soil physics and chemistry are often the strongest factors in shaping the total rhizospheric bacterial community structure and function, in particular soil particle size and pore distribution, moisture, pH and nutrient availability (Berg and Smalla, 2009; Fierer, 2017; Wang et al., 2019). In this work, we compared plants grown in loam (haplic phaeozem) and sand (ratio between quartz sand and haplic phaeozem 83.3 to 16.7%), the substrates of the central experiments of the priority program Rhizosphere Spatiotemporal Organization – PP 2089 (Vetterlein et al., 2020a). To achieve similar plant nutrition and achieve similar water distribution in the soil columns, nutrient differences were equilibrated by the different fertilizer regimes for the two substrates, and watering was carried out from the top and the bottom of the columns (Vetterlein et al., 2020b). The fact that the impact of the substrate on the acdS+ community was highly significant implies that soil texture has a major impact on rhizosphere properties and plant-microbe interactions. Feedback processes between the plant and the microorganisms should not be neglected, since soil texture also influences root architecture and root physiology (Rich and Watt, 2013; Rogers et al., 2016; D. V., Eva Lippold, Maxime Phalempin, Steffen Schlüter, unpublished data). Fischer et al. (2010) observed in a similar approach than the one used here, i.e., fewer sorption sites in the mixture than in the original soil, that higher concentrations of leached substances and increased maize root exudation was observed in the soil-sand mixture. In the maize system, higher maize secondary metabolism (phenylpropanoid and terpenoid metabolism) and plant immunity (pathogenesis related protein genes) gene expression was observed in sand than in loam, as well as increased water channel expression (M. G., unpublished data). This could indicate that the plant exerts a stronger selective influence on the acdS+ community in sand than in loam.
The Relation Between Depth and acdS+ Community Composition
Community composition of acdS+ bacteria was strongly affected by the depth of the soil column, with increased relative abundances of the members of acdS+ Burkholderiales (Paraburkholderia, Burkholderia, and Variovorax) in D3, but acdS+ Actinobacteria (Streptomyces, Tetrasphaera, and Amycolatopsis) in D2. Of these, a widespread ACC deaminase activity in Burkholderia species has been indicated by a cultivation based analysis. From a Burkholderia collection, 18 out of 20 species (90%) exhibited ACC deaminase activity (Onofre-Lemus et al., 2009). Enhancement of growth and salt tolerance of rice seedlings by ACC deaminase-producing Burkholderia strain was not supported by ACC deaminase mutant that was not able to reduce stress ethylene, confirming that the plant beneficial impact was associated with a reduction in ethylene level (Sarkar et al., 2018). And, Huang et al. (2017) reported that among rhizosphere bacterial isolates of plants subjected to water limitation, Burkholderia isolate was among the two most powerful PGPR. That particular Burkholderia strain exhibited ACC deaminase activity and promoted drought tolerance by decreasing plant ethylene levels. Preliminary results from the analysis of the root systems from a comparable experiment (D. V., Eva Lippold, Maxime Phalempin, Steffen Schlüter, unpublished) indicates that roots grow fast through the substrate and that the density of young roots (0–7 days old) in the lower section of the columns is higher than on the top at this growth stage. This indicates that changes in acdS+ taxa distribution between different depths could be, at least in part, explained by changes in the distribution of young, active roots. Resource quality and distribution might play a role in the differences between the enrichment patterns of Burkholderiales and actinobacteria, since whereas the first have often been affiliated as copiotrophs, highly responsive to easily degradable carbon and other resources upon availability (Padmanabhan et al., 2003; Fierer et al., 2007; reviewed in Ho et al., 2017), actinomycetes commonly contribute to decomposition through active breakdown of plant biomass (Lewin et al., 2016). To test this hypothesis, rhizodeposition at different depths of the column should be investigated.
The roothairless3 Mutant
Despite their importance for plant growth (Bates and Lynch, 2000) and nutrient acquisition (Zhu et al., 2010), information on the role of the root hairs in rhizosphere microbiome assembly is limited. Root hairs extend into the rhizosphere and alter its spatial dimensions. They take up nutrients and modulate resource distribution in the rhizosphere, and thus generate micro-niches that could enrich some but deplete other members of the rhizosphere microbiota (Pascale et al., 2020). The genotype effects on the maize rhizosphere microbiota included an increase in the relative abundances of acdS+ Actinomycetes; Streptomyces in both substrates, but Microbacterium and Modestobacter only in loam in rth3, compared to an increase of the Burkholderiales acdS+ genera in WT, Variovorax in both substrates, but Methylibium only in loam. This observation confirmed our third hypothesis, stating that the effect of plant genotype differs between loam and sand. In this respect, it is in accordance with the rhizosphere 16S rRNA gene amplicon sequencing results of WT and root hair mutant barley (Robertson-Albertyn et al., 2017). Interestingly, while maize acdS+ bacteria predominantly differed in their responses to rth3 in loam, the barley total bacterial community composition responded stronger to plant genotype in a substrate with lower organic matter content (Robertson-Albertyn et al., 2017). Higher richness of acdS+ ASVs of rth3 maize than the WT was also in contrast to barley total bacterial OTU richness, which showed no obvious differences between WT than root hair mutant plants (Robertson-Albertyn et al., 2017). Ultimately, the comparison between maize acdS and total bacterial community studies (K. S. and B. Y., unpublished data) will determine the correspondence between the acdS and the 16S rRNA gene-based community composition patterns, and the relatedness between the maize and barley results. The current study indicates that the importance of the rth3 mutation, or the association between elongated root hairs and the structure of the rhizosphere microbiome, is higher in the upper (D1) than lower (D2 and D3) area of the soil column. Our preliminary results (Nina Naderi, Sophie Wachter, M. T. T., and D. V., unpublished data) suggest that the general occurrence of root hairs is similar in loam and sand in the column system, but we have not yet investigated if their density or elongation are affected by depth.
Perspectives
Following the findings in this study, we anticipate that the spatial distribution of the acdS microorganisms is likely a vital feature of bacterium-plant interactions. Our recently conducted experiment (Ganther et al., 2020) confirmed that X-ray computed tomography is compatible with a subsequent microbiome analysis, facilitating further research on the spatial distribution of the acdS microorganisms. Future work should also target the temporal, plant age associated variation in acdS abundance and population structure.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, Bioproject PRJNA666771.
Author Contributions
MG, MT, and M-LB designed the experiment. LG performed and analyzed the experiment with input from AH-B, M-LB, and MT. BY prepared rhizosphere DNA. AH-B provided assistance for the bioinformatics pipeline and data analysis. LG, M-LB, and MT wrote the manuscript with contributions from all authors. All authors contributed to the article and approved the submitted version.
Funding
This research was conducted within the research program “Rhizosphere Spatiotemporal Organization – a Key to Rhizosphere Functions” of the German Science Foundation (project numbers 403637238, 403640293, and 403641192). AH-B was supported by the German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig, funded by the German Research Foundation (DFG-FZT 118 and 202548816).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to Beatrix Schnabel (UFZ Halle) for the excellent Illumina sequencing. We would like to acknowledge the contributions from the UFZ/iDiv high performance cluster administration team and Matthias Bernt. The access to unpublished data by Eva Lippold, Maxime Phalempin, Steffen Schlüter, Nina Naderi, and Sophie Wachter and to maize seeds produced by Frank Hocholdinger’s laboratory is gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.616828/full#supplementary-material
Supplementary Figure 1 | Experimental design, maize growth, and water consumption.
Supplementary Figure 2 | Rarefaction curves for partial acdS sequences.
Supplementary Figure 3 | Distribution of the acdS sequences is the most strongly affected by substrate.
Supplementary Figure 4 | Tree of amplicon sequence variants and reference sequences.
Supplementary Table 1 | Summary of substrate characteristics for loam and sand.
Supplementary Table 2 | Frequency of acdS amplicon sequencing variants in maize rhizosphere.
Supplementary Table 3 | Relative abundances of acdS sequences affiliated to the order level across all treatments.
Footnotes
References
Akinnifesi, F. K., Ajayi, O. C., Sileshi, G., Chirwa, P. W., and Chianu, J. (2010). Fertiliser trees for sustainable food security in the maize-based production systems of East and Southern Africa. A review. Agron. Sustain. Dev. 30, 615–629. doi: 10.1051/agro/2009058
Antoun, H., and Prévost, D. (2005). “Ecology of plant growth promoting rhizobacteria,” in PGPR: Biocontrol and Biofertilization, (Dordrecht: Springer), 1–38.
Bates, T. R., and Lynch, J. P. (2000). Plant growth and phosphorus accumulation of wild type and two root hair mutants of Arabidopsis thaliana (Brassicaceae). Am. J. Bot. 87, 958–963. doi: 10.2307/2656994
Benitez, M. S., Osborne, S. L., and Lehman, R. M. (2017). Previous crop and rotation history effects on maize seedling health and associated rhizosphere microbiome. Sci. Rep. 7:15709. doi: 10.1038/s41598-017-15955-9
Berg, G., and Smalla, K. (2009). Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere. FEMS Microbiol. Ecol. 68, 1–13. doi: 10.1111/j.1574-6941.2009.00654.x
Bouffaud, M. L., Kyselkova, M., Gouesnard, B., Grundmann, G., Muller, D., and Moenne-Loccoz, Y. (2012). Is diversification history of maize influencing selection of soil bacteria by roots? Mol. Ecol. 21, 195–206. doi: 10.1111/j.1365-294X.2011.05359.x
Bouffaud, M.-L., Poirier, M.-A., Muller, D., and Moënne-Loccoz, Y. (2014). Root microbiome relates to plant host evolution in maize and other Poaceae. Environ. Microbiol. 16, 2804–2814. doi: 10.1111/1462-2920.12442
Bouffaud, M. L., Renoud, S., Dubost, A., Moënne-Loccoz, Y., and Muller, D. (2018). 1-Aminocyclopropane-1-carboxylate deaminase producers associated to maize and other Poaceae species. Microbiome 6:114. doi: 10.1186/s40168-018-0503-7
Brisson, V. L., Schmidt, J. E., Northen, T. R., Vogel, J. P., and Gaudin, A. C. (2019). Impacts of maize domestication and breeding on rhizosphere microbial community recruitment from a nutrient depleted agricultural soil. Sci. Rep. 9, 1–14. doi: 10.1038/s41598-019-52148-y
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Coelho, M. R., Marriel, I. E., Jenkins, S. N., Lanyon, C. V., Seldin, L., and O’Donnell, A. G. (2009). Molecular detection and quantification of nifH gene sequences in the rhizosphere of sorghum (Sorghum bicolor) sown with two levels of nitrogen fertilizer. Appl. Soil Ecol. 42, 48–53. doi: 10.1016/j.apsoil.2009.01.010
Cotton, T. A., Pétriacq, P., Cameron, D. D., Al Meselmani, M., Schwarzenbacher, R., Rolfe, S. A., et al. (2019). Metabolic regulation of the maize rhizobiome by benzoxazinoids. ISME J. 13, 1647–1658. doi: 10.1038/s41396-019-0375-2
Egamberdiyeva, D. (2007). The effect of plant growth promoting bacteria on growth and nutrient uptake of maize in two different soils. Appl. Soil Ecol. 36, 184–189. doi: 10.1016/j.apsoil.2007.02.005
Fierer, N. (2017). Embracing the unknown: disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 15, 579–590. doi: 10.1038/nrmicro.2017.87
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Fierer, N., Schimel, J. P., and Holden, P. A. (2003). Variations in microbial community composition through two soil depth profiles. Soil Biol. Biochem. 35, 167–176. doi: 10.1016/S0038-0717(02)00251-1
Fish, J. A., Chai, B., Wang, Q., Sun, Y., Brown, C. T., Tiedje, J. M., et al. (2013). FunGene: the functional gene pipeline and repository. Front. Microbiol. 4:291. doi: 10.3389/fmicb.2013.00291
Fischer, H., Eckhardt, K. U., Meyer, A., Neumann, G., Leinweber, P., Fischer, K., et al. (2010). Rhizodeposition of maize: Short-term carbon budget and composition. J. Plant Nutr. Soil Sci. 173, 67–79. doi: 10.1002/jpln.200800293
Food and Agriculture Organization of the United Nations (2019). Availabel online at: http://www.fao.org/faostat/en/#data/QC/visualize
Gamalero, E., and Glick, B. R. (2015). Bacterial modulation of plant ethylene levels. Plant Physiol. 169, 13–22. doi: 10.1104/pp.15.00284
Ganther, M., Yim, B., Ibrahim, Z., Bienert, M. D., Lippold, E., Maccario, L., et al. (2020). Compatibility of X-ray computed tomography with plant gene expression, rhizosphere bacterial communities and enzyme activities. J. Exp. Bot. 71, 5603–5614. doi: 10.1093/jxb/eraa262
Glick, B. (2005). Modulation of plant ethylene levels by the bacterial enzyme ACC deaminase. FEMS Microbiol. Lett. 251, 1–7. doi: 10.1016/j.femsle.2005.07.030
Glick, B. R. (2014). Bacteria with ACC deaminase can promote plant growth and help to feed the world. Microbiol. Res. 169, 30–39. doi: 10.1016/j.micres.2013.09.009
Ho, A., Di Lonardo, D. P., and Bodelier, P. L. (2017). Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol. Ecol. 93:fix006. doi: 10.1093/femsec/fix006
Hochholdinger, F., and Tuberosa, R. (2009). Genetic and genomic dissection of maize root development and architecture. Curr. Opin. Plant Biol. 12, 172–177. doi: 10.1016/j.pbi.2008.12.002
Hochholdinger, F., Wen, T. J., Zimmermann, R., Chimot−Marolle, P., Da Costa e Silva, O., Bruce, W., et al. (2008). The maize (Zea mays L.) roothairless3 gene encodes a putative GPI−anchored, monocot−specific, COBRA−like protein that significantly affects grain yield. Plant J. 54, 888–898. doi: 10.1111/j.1365-313X.2008.03459.x
Huang, X. F., Zhou, D., Lapsansky, E. R., Reardon, K. F., Guo, J., Andales, M. J., et al. (2017). Mitsuaria sp. and Burkholderia sp. from Arabidopsis rhizosphere enhance drought tolerance in Arabidopsis thaliana and maize (Zea mays L.). Plant Soil 419, 523–539. doi: 10.1007/s11104-017-3360-4
Ivanchenko, M. G., Muday, G. K., and Dubrovsky, J. G. (2008). Ethylene-auxin interactions regulate lateral root initiation and emergence in Arabidopsis thaliana. Plant J. 55, 335–347. doi: 10.1111/j.1365-313X.2008.03528.x
Jaemsaeng, R., Jantasuriyarat, C., and Thamchaipenet, A. (2018). Molecular interaction of 1-aminocyclopropane-1-carboxylate deaminase (ACCD)-producing endophytic Streptomyces sp. GMKU towards salt-stress resistance of Oryza sativa L. cv. KDML105. Sci. Rep. 8, 1–15. doi: 10.1038/s41598-018-19799-9
Katoh, K., Misawa, K., Kuma, K. I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kolde, R. (2019). pheatmap: pretty heatmaps. R Package Version 1.0.12. Available online at: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf
Labate, J. A., Lamkey, K. R., Mitchell, S. E., Kresovich, S., Sullivan, H., and Smith, J. S. (2003). Molecular and historical aspects of corn belt dent diversity. Crop Sci. 43, 80–91. doi: 10.2135/cropsci2003.8000
Lewin, G. R., Carlos, C., Chevrette, M. G., Horn, H. A., McDonald, B. R., Stankey, R. J., et al. (2016). Evolution and ecol. of Actinobacteria and their bioenergy applications. Annu. Rev. Microbiol. 70, 235–254. doi: 10.1146/annurev-micro-102215-095748
Li, X., Penttinen, P., Gu, Y., and Zhang, X. (2012). Diversity of nifH gene in rhizosphere and non-rhizosphere soil of tobacco in Panzhihua. China. Ann. Microbiol. 62, 995–1001. doi: 10.1007/s13213-011-0339-x
Long, X. E., Yao, H., Huang, Y., Wei, W., and Zhu, Y. G. (2018). Phosphate levels influence the utilisation of rice rhizodeposition carbon and the phosphate-solubilising microbial community in a paddy soil. Soil Biol. Biochem. 118, 103–114. doi: 10.1016/j.soilbio.2017.12.014
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lynch, J. P. (2015). Root phenes that reduce the metabolic costs of soil exploration: opportunities for 21st century agriculture. Plant Cell Environ. 38, 1775–1784. doi: 10.1111/pce.12451
Marschner, P., Crowley, D., and Rengel, Z. (2011). Rhizosphere interactions between microorganisms and plants govern iron and phosphorus acquisition along the root axis-model and research methods. Soil Biol. Biochem. 43, 883–894. doi: 10.1016/j.soilbio.2011.01.005
Merriman, P. R., Price, R. D., Kollmorgen, J. F., Piggott, T., and Ridge, E. H. (1974). Effect of seed inoculation with Bacillus subtilis and Streptomyces griseus on the growth of cereals and carrots. Austr. J. Agric. Res. 25, 219–226. doi: 10.1071/AR9740219
Moore, R. M., Harrison, A. O., McAllister, S. M., Polson, S. W., and Wommack, K. E. (2020). Iroki: automatic customization and visualization of phylogenetic trees. PeerJ 8:e8584. doi: 10.7717/peerj.8584
Nadeem, S. M., Zahir, Z. A., Naveed, M., and Arshad, M. (2009). Rhizobacteria containing ACC-deaminase confer salt tolerance in maize grown on salt-affected fields. Can. J. Microbiol. 55, 1302–1309. doi: 10.1139/W09-092
Onofre-Lemus, J., Hernández-Lucas, I., Girard, L., and Caballero-Mellado, J. (2009). ACC (1-aminocyclopropane-1-carboxylate) deaminase activity, a widespread trait in Burkholderia species, and its growth-promoting effect on tomato plants. Appl. Environ. Microbiol. 75, 6581–6590. doi: 10.1128/AEM.01240-09
Padmanabhan, P., Padmanabhan, S., DeRito, C., Gray, A., Gannon, D., Snape, J. R., et al. (2003). Respiration of 13C-labeled substrates added to soil in the field and subsequent 16S rRNA gene analysis of 13C-labeled soil DNA. Appl. Environ. Microbiol. 69, 1614–1622. doi: 10.1128/AEM.69.3.1614-1622.2003
Pascale, A., Proietti, S., Pantelides, I. S., and Stringlis, I. A. (2020). Modulation of the root microbiome by plant molecules: the basis for targeted disease suppression and plant growth promotion. Front. Plant Sci. 10:1741. doi: 10.3389/fpls.2019.01741
Peiffer, J. A., Spor, A., Koren, O., Jin, Z., Tringe, S. G., Dangl, J. L., et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U.S.A. 110, 6548–6553. doi: 10.1073/pnas.1302837110
Poorter, H., Bühler, J., van Dusschoten, D., Climent, J., and Postma, J. A. (2012). Pot size matters: a meta-analysis of the effects of rooting volume on plant growth. Funct. Plant Biol. 39, 839–850. doi: 10.1071/FP12049
Renoud, S., Bouffaud, M. L., Dubost, A., Prigent-Combaret, C., Legendre, L., Moënne-Loccoz, Y., et al. (2020). Co-occurrence of rhizobacteria with nitrogen fixation and/or 1-aminocyclopropane-1-carboxylate deamination abilities in the maize rhizosphere. FEMS Microbiol. Ecol. 96:fiaa062. doi: 10.1093/femsec/fiaa062
Rich, S. M., and Watt, M. (2013). Soil conditions and cereal root system architecture: review and considerations for linking Darwin and Weaver. J. Exp. Bot. 64, 1193–1208. doi: 10.1093/jxb/ert043
Robertson-Albertyn, S., Alegria Terrazas, R., Balbirnie, K., Blank, M., Janiak, A., Szarejko, I., et al. (2017). Root hair mutations displace the barley rhizosphere microbiota. Front. Plant Sci. 8:1094. doi: 10.3389/fpls.2017.01094
Roesch, L. F. W., Camargo, F. A., Bento, F. M., and Triplett, E. W. (2008). Biodiversity of diazotrophic bacteria within the soil, root and stem of field-grown maize. Plant Soil 302, 91–104. doi: 10.1007/s11104-007-9458-3
Rogers, E. D., Monaenkova, D., Mijar, M., Nori, A., Goldman, D. I., and Benfey, P. N. (2016). X-ray computed tomography reveals the response of root system architecture to soil texture. Plant Physiol. 171, 2028–2040. doi: 10.1104/pp.16.00397
Rozier, C., Hamzaoui, J., Lemoine, D., Czarnes, S., and Legendre, L. (2017). Field-based assessment of the mechanism of maize yield enhancement by Azospirillum lipoferum CRT1. Sci. Rep. 7, 1–12. doi: 10.1038/s41598-017-07929-8
Růžička, K., Ljung, K., Vanneste, S., Podhorská, R., Beeckman, T., Friml, J., et al. (2007). Ethylene regulates root growth through effects on auxin biosynthesis and transport-dependent auxin distribution. Plant Cell 19, 2197–2212. doi: 10.1105/tpc.107.052126
Saravanakumar, K., Li, Y., Yu, C., Wang, Q. Q., Wang, M., Sun, J., et al. (2017). Effect of Trichoderma harzianum on maize rhizosphere microbiome and biocontrol of Fusarium Stalk rot. Sci. Rep. 7, 1–13. doi: 10.1038/s41598-017-01680-w
Sarkar, A., Pramanik, K., Mitra, S., Soren, T., and Maiti, T. K. (2018). Enhancement of growth and salt tolerance of rice seedlings by ACC deaminase-producing Burkholderia sp. MTCC 12259. J. Plant Physiol. 231, 434–442. doi: 10.1016/j.jplph.2018.10.010
Sauerbrei, R., Ekschmitt, K., Wolters, V., and Gottschalk, T. K. (2014). Increased energy maize production reduces farmland bird diversity. GCB Bioenergy 6, 265–274. doi: 10.1111/gcbb.12146
Schrey, S. D., and Tarkka, M. T. (2008). Friends and foes: streptomycetes as modulators of plant disease and symbiosis. Antonie Leeuwenhoek 94, 11–19. doi: 10.1007/s10482-008-9241-3
Shaharoona, B., Arshad, M., Zahir, Z. A., and Khalid, A. (2006). Performance of Pseudomonas spp. containing ACC-deaminase for improving growth and yield of maize (Zea mays L.) in the presence of nitrogenous fertilizer. Soil Biol. Biochem. 38, 2971–2975. doi: 10.1016/j.soilbio.2006.03.024
Shahzad, S. M., Arif, M. S., Riaz, M., Iqbal, Z., and Ashraf, M. (2013). PGPR with varied ACC-deaminase activity induced different growth and yield response in maize (Zea mays L.) under fertilized conditions. Eur. J. Soil Biol. 57, 27–34. doi: 10.1016/j.ejsobi.2013.04.002
Shiferaw, B., Prasanna, B., Hellin, J., and Bänziger, M. (2011). Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur. 3, 307–327. doi: 10.1007/s12571-011-0140-5
Sukumar, P., Legue, V., Vayssieres, A., Martin, F., Tuskan, G. A., and Kalluri, U. C. (2013). Involvement of auxin pathways in modulating root architecture during beneficial plant-microorganism interactions. Plant Cell Environ. 36, 909–919. doi: 10.1111/pce.12036
Tak, H. I., Ahmad, F., and Babalola, O. O. (2013). “Advances in the application of plant growth-promoting rhizobacteria in phytoremediation of heavy metals,” in Reviews of Environ.Cont. Toxicol, Vol. 223, ed. D. Whitacre (New York, NY: Springer), 33–52. doi: 10.1007/978-1-4614-5577-6_2
Tanimoto, M., Roberts, K., and Dolan, L. (1995). Ethylene is a positive regulator of root hair development in Arabidopsis thaliana. Plant J. 8, 943–948. doi: 10.1046/j.1365-313x.1995.8060943.x
Timmusk, S., Behers, L., Muthoni, J., Muraya, A., and Aronsson, A. C. (2017). Perspectives and challenges of microbial application for crop improvement. Front. Plant Sci. 8:49. doi: 10.3389/fpls.2017.00049
Vacheron, J., Desbrosses, G., Bouffaud, M. L., Touraine, B., Moënne-Loccoz, Y., Muller, D., et al. (2013). Plant growth-promoting rhizobacteria and root system functioning. Front. Plant Sci. 4:356. doi: 10.3389/fpls.2013.00356
Vacheron, J., Combes-Meynet, E., Walker, V., Gouesnard, B., Muller, D., Moënne-Loccoz, Y., et al. (2016a). Expression on roots and contribution to maize phytostimulation of 1-aminocyclopropane-1-decarboxylate deaminase gene acdS in Pseudomonas fluorescens F113. Plant Soil 407, 187–202. doi: 10.1007/s11104-016-2907-0
Vacheron, J., Moënne-Loccoz, Y., Dubost, A., Gonçalves-Martins, M., Muller, D., and Prigent-Combaret, C. (2016b). Fluorescent Pseudomonas strains with only few plant-beneficial properties are favored in the maize rhizosphere. Front. Plant Sci. 7:1212. doi: 10.3389/fpls.2016.01212
Vetterlein, D., Carminati, A., Kögel-Knabner, I., Bienert, G. P., Smalla, K., Oburger, E., et al. (2020a). Rhizosphere spatiotemporal organization-A key to rhizosphere functions. Front. Agronomy 2:8. doi: 10.3389/fagro.2020.00008
Vetterlein, D., Lippold, E., Schreiter, S., PHalempin, M., Steffen Schlüter, M., Fahrenkampf, T., et al. (2020b). Experimental platforms for the investigation of spatiotemporal patterns in the rhizosphere - laboratory and field scale. J. Plant Nutr. Soil Sci. doi: 10.1002/jpln.202000079 [Epub ahead of print].
Walters, W. A., Jin, Z., Youngblut, N., Wallace, J. G., Sutter, J., Zhang, W., et al. (2018). Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. U.S.A. 115, 7368–7373. doi: 10.1073/pnas.1800918115
Wang, H., Lu, L., Mao, D., Huang, Z., Cui, Y., Jin, S., et al. (2019). Dominance of electroactive microbiomes in bioelectrochemical remediation of hydrocarbon-contaminated soils with different textures. Chemosphere 235, 776–784. doi: 10.1016/j.chemosphere.2019.06.229
Weißbecker, C., Schnabel, B., and Heintz-Buschart, A. (2020). Dadasnake, a Snakemake implementation of DADA2 to process amplicon sequencing data for microbial ecology. GigaScience 9:giaa135. doi: 10.1093/gigascience/giaa135
Wen, T. J., and Schnable, P. S. (1994). Analyses of mutants of three genes that influence root hair development in Zea mays (Gramineae) suggest that root hairs are dispensable. Am. J. Bot. 81, 833–842. doi: 10.1002/j.1537-2197.1994.tb15564.x
Xiao, K., Kinkel, L. L., and Samac, D. A. (2002). Biological control of Phytophthora root rots on alfalfa and soybean with Streptomyces. Biol. Control 23, 285–295. doi: 10.1006/bcon.2001.1015
Yoolong, S., Kruasuwan, W., Ph. am, H. T. T., Jaemsaeng, R., Jantasuriyarat, C., and Thamchaipenet, A. (2019). Modulation of salt tolerance in Thai jasmine rice (Oryza sativa L. cv. KDML105) by Streptomyces venezuelae ATCC 10712 expressing ACC deaminase. Sci. Rep. 9, 1–10. doi: 10.1038/s41598-018-37987-5
Keywords: ethylene, rhizosphere, soil, root, plant-microbe interactions, PGPR, crop
Citation: Gebauer L, Bouffaud M-L, Ganther M, Yim B, Vetterlein D, Smalla K, Buscot F, Heintz-Buschart A and Tarkka MT (2021) Soil Texture, Sampling Depth and Root Hairs Shape the Structure of ACC Deaminase Bacterial Community Composition in Maize Rhizosphere. Front. Microbiol. 12:616828. doi: 10.3389/fmicb.2021.616828
Received: 13 October 2020; Accepted: 14 January 2021;
Published: 04 February 2021.
Edited by:
Aleksa Obradović, University of Belgrade, SerbiaReviewed by:
Clarisse Brígido, University of Évora, PortugalVasvi Chaudhry, University of Tübingen, Germany
Copyright © 2021 Gebauer, Bouffaud, Ganther, Yim, Vetterlein, Smalla, Buscot, Heintz-Buschart and Tarkka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mika T. Tarkka, bWlrYS50YXJra2FAdWZ6LmRl