 Öncü Maraci1*
Öncü Maraci1* Anna Antonatou-Papaioannou2,3
Anna Antonatou-Papaioannou2,3 Sebastian Jünemann4,5Omar Castillo-Gutiérrez4,5
Sebastian Jünemann4,5Omar Castillo-Gutiérrez4,5 Tobias Busche5
Tobias Busche5 Jörn Kalinowski5
Jörn Kalinowski5 Barbara A. Caspers1
Barbara A. Caspers1- 1Department of Behavioural Ecology, Bielefeld University, Bielefeld, Germany
- 2Evolutionary Biology, Bielefeld University, Bielefeld, Germany
- 3Institute of Biology-Zoology, Freie Universität Berlin, Berlin, Germany
- 4Faculty of Technology, Bielefeld University, Bielefeld, Germany
- 5Center for Biotechnology (CeBiTec), Bielefeld University, Bielefeld, Germany
Microbial communities residing in the gastrointestinal tracts of animals have profound impacts on the physiological processes of their hosts. In humans, host-specific and environmental factors likely interact together to shape gut microbial communities, resulting in remarkable inter-individual differences. However, we still lack a full understanding of to what extent microbes are individual-specific and controlled by host-specific factors across different animal taxa. Here, we document the gut microbial characteristics in two estrildid finch species, the Bengalese finch (Lonchura striata domestica) and the zebra finch (Taeniopygia guttata) to investigate between-species and within-species differences. We collected fecal samples from breeding pairs that were housed under strictly controlled environmental and dietary conditions. All individuals were sampled at five different time points over a range of 120 days covering different stages of the reproductive cycle. We found significant species-specific differences in gut microbial assemblages. Over a period of 3 months, individuals exhibited unique, individual-specific microbial profiles. Although we found a strong individual signature in both sexes, within-individual variation in microbial communities was larger in males of both species. Furthermore, breeding pairs had more similar microbial profiles, compared to randomly chosen males and females. Our study conclusively shows that host-specific factors contribute structuring of gut microbiota.
Introduction
Animal bodies are inhabited by billions of microorganisms, which are collectively termed microbiota. The majority of these microorganisms reside in the gastrointestinal tract (gut) of the animals (Qin et al., 2010). A strong body of evidence has left no doubt that several physiological processes of the animal hosts, which had been previously attributed to the host itself, are functionally influenced by microbial activities (Tremaroli and Bäckhed, 2012; McFall-Ngai et al., 2013; Kohl and Carey, 2016). Consequently, the microbial symbionts influence several aspects of the host biology, such as host energy metabolism (Gill et al., 2006), immunity (Round and Mazmanian, 2009; Kohl, 2012; Maynard et al., 2012; Kamada et al., 2013), development (Borre et al., 2014; Videvall et al., 2019; Kirschman et al., 2020), behavior (Archie and Theis, 2011; Ezenwa et al., 2012; Lim et al., 2016; Davidson et al., 2018, 2020; Johnson and Foster, 2018; Sherwin et al., 2019), and evolution (Moeller and Sanders, 2020). Therefore, taxonomic diversity of the microbial species contained in the gut, as well as their relative abundance is likely deterministic for the host’s fitness (Gould et al., 2018; Videvall et al., 2020).
The composition and the structure of gut microbial communities are shaped by the combination of extrinsic and host-specific factors in both stochastic and deterministic ways. Animals acquire their microbes vertically from their parents (Palmer et al., 2007; Jiménez et al., 2008; Collado et al., 2012; DiGiulio, 2012; Jeurink et al., 2013; Trevelline et al., 2018) or horizontally from the physical and social environment, first in the postnatal phases and then through their lives (Tung et al., 2015; Moeller et al., 2018; Chen et al., 2020). Therefore, the environmental pool of microorganisms and ecological parameters driving the distribution of microbial communities have non-negligible roles in shaping host-associated microbial communities (Candela et al., 2012; Rothschild et al., 2018). Besides, gut microbiota, – at least to some extent – is an entity, regulated by the host itself in a deterministic way. When an animal host cross paths with a microorganism, the host either selectively establishes symbiotic associations or eliminates the survival and reproduction of the microorganism. Although the exact mechanism of these complex processes is still largely elusive, it probably involves selective pressures imposed by distinct features of the gastrointestinal environment, physiology, immunity, and dietary adaptations of the host (Ley et al., 2008; Muegge et al., 2011; Kubinak et al., 2015; Stagaman et al., 2017). Consequently, it is feasible to assume that each individual harbors a unique microbial assemblage reflecting both, the signatures of their interactions with the environment (Engel et al., 2020) and host-specific factors. At the same time, considering the vital functions of the microbes in several physiological processes of the hosts, the gut microbiota, or at least a subset of microorganisms should be conserved among the members of the same species. In line with these assumptions, human microbiota consists of some temporally stable microbial species that are highly adapted to their host, along with some transient taxa that respond quite rapidly to the dietary, developmental, and physiological alterations (Turnbaugh et al., 2007; Lozupone et al., 2012; Lloyd-Price et al., 2017). Interestingly, in this rapidly growing field, we are still lacking the knowledge to what extent microbial communities are similar between conspecifics, what drives inter-individual differences and if and how stable are communities across the distinct life-history stages of individual hosts in non-human species.
This information gap is specifically striking for avian taxa (Maraci et al., 2018). Existing knowledge indicates that the gut microbiota of birds is shaped primarily by external factors such as diet (Blanco et al., 2006; Waite and Taylor, 2014; Bodawatta et al., 2018; Kohl et al., 2018; Michel et al., 2018; Teyssier et al., 2020), local habitat (Hird et al., 2014; Waite and Taylor, 2014), or nesting environment (Teyssier et al., 2018; van Veelen et al., 2020). Gut microbial species can be transferred between parents and offsprings (Chen et al., 2020) and breeding pairs (Kreisinger et al., 2015; Ambrosini et al., 2019). Individuals from the same nest have a similar gut microbial composition (Lombardo et al., 1996; Ambrosini et al., 2019). However, it is unclear whether these congruences are originating from genetic relatedness or similarities in diet or environmental conditions. Although the link between the gut microbial profile and host taxonomy (Ruiz-Rodríguez et al., 2009, 2018; Waite and Taylor, 2014; Hird et al., 2015) and genetics (Banks et al., 2009; Zhao et al., 2013) have been demonstrated, evidence for the strength of these host-specific factors is equivocal. This can be explained by the fact that to date, most avian studies that investigate the determinants of the gut microbiota have been carried out on wild animals in natural conditions, except for the poultry. While undoubtedly important, the findings of those studies on host-specific factors are clearly mitigated by strong environmental and dietary effects. In this regard, studies conducted under captivity conditions where the diet and environmental conditions are standardized would allow us to understand how much of the microbial diversity is driven by deterministic host-specific factors.
The present study aimed to document interspecies and intraspecies differences in gut microbiota in two estrildid finches, the zebra finch (Taeniopygia guttata) and the Bengalese finch (Lonchura striata domestica), under strictly controlled environmental and dietary conditions to determine whether gut microbial diversity is influenced by host-specific factors. If the gut microbiota is influenced by host taxonomy and genetics, we expect that (i) individuals of the same species should harbor more similar microbial profiles compared to members of different species, (ii) there should be consistent between-individual variations, and (iii) at least some proportion of the microbial species harbored by the hosts should be conserved over time, even throughout the different stages of the reproductive cycle. To test these assumptions and explore the impact of sex and social transmission, we characterized gut microbial profiles in breeding pairs of the zebra finches and the Bengalese finches at five different time points, over a period of 120 days.
Materials and Methods
Ethics Statement
Housing and breeding of birds were approved by the Gesundheits-, Veterinär-und Lebensmittelüberwachungsamt der Stadt Bielefeld (#530.421630–1,18.4.2002). All birds remained in the aviary stock after experimentation. All experiments were performed following the animal experimentation guidelines and laws of Germany.
Study Organisms and Sampling
Between January and August 2017, we examined gut microbial profiles of 42 individuals of two captive bird species, the zebra finches (ZF; N = 28, 14 females, 14 males, referred to as “Bielefeld” in Forstmeier et al. (2007) and the Bengalese finches (BF; N = 14, 7 females, 7 males), from the laboratory stock at the Bielefeld University. The zebra finches were transferred from single-sexed indoor aviaries (2.30 × 2.90 × 3.30 m) to indoor cages (0.80 × 0.30 × 0.40 m) as pairs. The Bengalese finches were transferred from mixed-sex indoor aviaries (2.30 × 2.90 × 3.30 m) to indoor cages (0.80 × 0.30 × 0.40 m). After a week of habituation, nesting material (coconut fiber) was provided and a wooden nest box (15 × 15 × 15 cm) was attached at each cage. The pairs were kept in these cages until their youngest offspring reached nutritional independence (approximately 35 days after hatching). After this point, all birds were transferred into mixed-sexed indoor aviaries (2.30 × 2.90 × 3.30 m), where they were kept with other conspecifics from this study. All birds kept under a 14:10 h light/dark cycle with a temperature range of 24.5–25.5°C. They were fed the standard diet, comprising of seeds ad libitum, a vitamin–mineral supplement and additional egg food (Tropical Finches, CéDé, Evergreen, Belgium) and germinated seeds every day.
Previous studies showed that fecal sampling is a non-lethal and non-invasive method that can successfully capture gut microbial structure in different bird species (for example Videvall et al., 2018), including the zebra finches (Berlow et al., 2020). Therefore, we used community profiles from fecal samples as a proxy for the gut microbial communities. To collect feces, we transferred the birds to sampling cages (30 × 40 × 30 cm) where they were kept individually for 30 min. Before the sampling, cages were cleaned using 78% ethanol and the bottom of cages were covered with sterile aluminum plates. After 30 min, we transferred the fecal materials from the aluminum plates to sterile 2 ml Eppendorf tubes using a sterile pipet tip. The samples were kept at ice during the collection and directly transferred to −80 within the first hour after the collection, where they were stored until further processing. The sampling equipment was always handled using nitrile gloves sterilized by 78% ethanol. We collected samples at five different time-points during the breeding period (over 120 days). The first samples were collected during incubation, approximately 7 days after the completion of the clutch. The second and third samples were collected during the chick-rearing period, 5 and 10 days after hatching of the youngest chick, respectively. The fourth samples were collected when the juveniles reached nutritional independence, 35 days after hatching of the youngest chick. The fifth samples were collected after the reproductive phase, when the juveniles reached sexual maturity, 100 days after hatching of the youngest chick. In total, we collected 210 samples from 42 birds.
DNA Extraction and Library Preparation
Microbial DNA was extracted from 0.02 grams of the fecal sample using the QIAamp PowerFecal DNA Kit (Qiagen), according to the manufacturer’s instructions. We prepared the16S rRNA gene libraries following the Illumina 16S Metagenomic Library Preparation Guide, 15044223-B. We targeted hypervariable V3–V4 region of the 16S ribosomal RNA (rRNA) genes by using the primers 5′-CCTACGGGNGGCWGCAG-3′ and 5′-GACTACHVGGGTATCTAATCC-3′ (Klindworth et al., 2013). The Illumina overhang adapters attached to the primers were as follows:
Forward: 5′-TCGTCGGCAGCGTCAGATGTGTATAAG AGACAG-3′,
Reverse: 5′-GTCTCGTGGGCTCGGAGATGTGTATAAG AGACAG-3′.
The first polymerase chain reactions (PCR) was performed in a 25 μL reaction volume containing 5 μL DNA, 12.5 μL KAPA HiFi HotStart ReadyMix (KAPA Biosystems, MA, United States), 2.5 μL of each primer (2 μM) and 5 μL of PCR grade water. The amplification conditions were as follows: an initial denaturation step at 95°C for 3 min, followed by 25 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 30 s, with a final extension step of 5 min at 72°C. To remove free primers and primer dimers and PCR products were subsequently purified using the Agencourt AMpure XP PCR purification system (Beckman Coulter, Brea, CA, United States) as described in the manufacturer’s protocol. To be able to multiplex the libraries, Dual Illumina indices (The Nextera XT Index Kit, Illumina, Inc., San Diego, CA, United States) were attached to the PCR products were by another PCR which was performed in a 50 μL volume containing 5 μL of the purified amplicon PCR product, 25 μL KAPA HiFi HotStart ReadyMix, 5 μL of each index primer, 10 μL of PCR grade water. The temperature profile of the amplification was as follows: an initial denaturation step at 95°C for 5 min, followed by eight cycles of 30 s at 95°C, 30 s at 55°C 30 s at 72°C with a final extension step of 5 min at 72°C. Two blank controls for PCR amplification and five replicates of the same sample were also included in the sequencing. The amplified fragments were subjected to another purification step using the Agencourt AMpure XP PCR purification system. The size of the amplified fragments was verified by running 1 μl of 1:50 dilutions of the final libraries on a Bioanalyzer DNA 1000 chip (Agilent Technologies, Palo Alto, CA, United States). Then, the concentration of the libraries was quantified by PicoGreen dsDNA Assay on a TECAN Infinite Reader M200 instrument. Seven samples were discarded due to unsuccessful amplification. Three negative control of extraction, PCR and clean-up steps and two technical replicated of three samples were included in the final pool. Accordingly, the final library contained equal concentrations of 209 uniquely barcoded amplicons. Sequencing of the final library was performed using paired-end mode (2 × 300 sequencing cycles) on the Illumina MiSeq system (Illumina, Inc., San Diego, CA, United States) at the CeBiTec, Bielefeld University.
Bioinformatics Processing
Processing of raw MiSeq forward and reverse PE reads were done as described by Engel et al. (2018) with the following minor adjustments to individual bioinformatic steps. To achieve a higher assembly rate, we assembled Miseq PE reads in an iterative manner using Flash v1.2.11 (Magoč and Salzberg, 2011). All reads failing the first round of read assembly were clipped to a q20 average quality threshold using sickle v1.33 (Joshi and Fass, 2011) and re-submitted to flash. This process was consecutively repeated while increasing the quality clipping threshold by three up to the point where either all reads could be assembled or a maximum quality clipping threshold of q35 was reached. All other steps, i.e., (i) adapter clipping with cutadapt v1.18 (Martin, 2011), (ii) de-replication, alignment, filtering, and de-noising with mothur v1.41.3 (Schloss et al., 2009), (iii) chimera checking and operational taxonomic unit (OTU) clustering with USEARCH v8.0.1477 (Edgar, 2010), and (iv) taxonomically classification based on the full SILVA database v138 (Quast et al., 2013) was carried out as described in detail previously (Engel et al., 2018) but without performing a length trimming step after primer clipping. The reason for employing the OTU approach is that we are mainly interested in structural community differences or conformities rather the detection of extremely rare and low abundant community members. Therefore, the finer taxonomic resolution provided by ASVs over OTUs does not further our understanding.
Statistical Analyses
We conducted all consecutive statistical analyses in R version 4.0.0 (R Core Team, 2020) and Primer-e software version 7 (Clarke et al., 2014). The code used in this study is provided in the GitHub repository at https://github.com/AnnaAntonatouPap/Microbiota-of-estrildid-finches-.git. We excluded the samples with less than 10,000 total read counts (n = 5). After this filtering step, 204 samples were retained in the dataset. We filtered out all the OTUs that could not be classified at phylum level or that were classified as mitochondria or chloroplasts (n = 44) as those are very likely to be issued by sequencing errors.
To account for the potential bias due to the uneven sequencing depth across the samples, we rarefied OTU read count data to the lowest read count observed in the dataset (12,472) and estimated alpha diversity based on this dataset. We computed Shannon’s diversity index, which accounts for both abundance and evenness of the taxa present (Shannon, 1997). We investigated the drivers of alpha diversity using a linear mixed model, as implicated in the lme4 package version 1.1−15 (Bates et al., 2015). We used untransformed Shannon’s diversity index as the response variable; the host ID and couple ID as random effects; species, sex, and sampling time as fixed effects. We also included the interaction between sex and sampling time as fixed effects to account for potential sex-specific changes in alpha diversity over time. Residuals of the models were inspected visually. To visualize the taxonomic and compositional structure of the microbial communities in the zebra finches and the Bengalese finches based on the non-rarefied dataset, we produced stacked bar plots, based on the family level taxonomy using ggplot2 version 3.3.2 (Wickham, 2009).
To estimate between-group diversity, first, the filtered dataset was subjected to Cumulative Sum Scaling (CSS) normalization (Paulson et al., 2013a) using the r package metagenomeseq version 1.30.0 (Paulson et al., 2013b) to deal with unequal sequence coverage. Subsequently, to account for compositional variations within the data, we Log (x + 0.0001)−transformed the data and later corrected the transformed values by subtracting the log of the pseudo count as recommended by Thorsen et al. (2016). Then, we computed a dissimilarity matrix based on Jaccard (1912), Bray–Curtis (Bray and Curtis, 1957), unweighted UniFrac (Lozupone and Knight, 2005), and weighted UniFrac (Lozupone et al., 2007) distances. To visualize the dissimilarities between the zebra finches and the Bengalese finches, we used a principal coordinate analysis (PCoA) as implemented by the function “ordinate” in vegan package version 2.5-6 (Oksanen et al., 2019). Based on PCoA plots, we selected the two dissimilarity measures explaining the highest variation (Bray–Curtis and weighted UniFrac) and used these distance matrices in all further analyses. We statistically tested the differences in the gut microbial assemblages in a priori defined groups by non-parametric analysis of similarities (ANOSIM) (Clarke, 1999) with 999 permutations in Primer 7. We analyzed the differences in microbial communities between the host species, performing one-way ANOSIM. We tested whether the samples collected from the same individual are more similar to each other compared to the samples collected from different individuals using two-way nested ANOSIM (individuals nested in species). To further evaluate whether the length of the period between the sample collection has any effect on microbial composition, we calculated a distance matrix that incorporated the time between the sample collection (setting the first sampling = 0). Then we performed a Mantel-like test (RELATE) to analyze the correlation between the distance matrix based on weighted UniFrac and the distance matrix of the time between sample collection by correcting for host ID, using Primer 7. We analyzed whether the individuals of the same sex have more similar microbial communities using a two-way crossed ANOSIM. Next, to address the potential sex-specific temporal differences in the community structure, we subset the samples collected from a given sex of each species. In each group, we tested whether the samples collected at a particular time point are more similar then the samples collected at different time points using one-way ANOSIM. Finally, to evaluate whether the mating pairs have more similar microbial communities compared to randomly chosen males and females, we first merged the samples collected from the same individuals by averaging taxonomic representatives from these samples to prevent any bias due to the use of repeated sampling of the same bird. Second, we performed a two-way nested ANOSIM (couples nested in species).
To identify specific OTUs potentially shaping the differences among species, we analyzed differentially abundant OTUs. We estimated logarithmic fold changes between groups by a negative binomial Wald test as implemented in DESeq2 version 1.12.4 extension (Love et al., 2014) of the Phyloseq package version 1.32.0 (McMurdie and Holmes, 2013). As a significance threshold for p-values, we used a 0.01 threshold after a Benjamini and Hochberg (1995) false−discovery rate correction.
Results
We sequenced the hypervariable V3–V4 region of the 16S rRNA gene from 210 gut microbial community samples originating from 42 individual birds. After the bioinformatics processing, our dataset consisted of 209 samples, 626 different OTUs with a total read count of 14,821,860 (mean: 72,656). After the filtering process, our dataset contained 204 samples, 582 OTUs with a total read count of 14,639,903 (Mean = 71,764.23).
We identified 20 microbial phyla, with the domination of Firmicutes (79.15%), Campilobacterota (14.07%), Proteobacteria (4.36%), and Actinobacteria (2.40%) (the numbers indicate the total abundance; the mean values and standard deviations are provided in Supplementary Table 1). At a finer taxonomic scale, identified taxa corresponded to 199 microbial families, 96.26% of which is constituted by Lactobacillaceae (76.10%), Campylobacteraceae (13.63%), Enterobacteriaceae (3.03%), Leuconostocaceae (1.88%), and Bifidobacteriaceae (1.62%) (Supplementary Table 2). Although the most abundant families remained the same through our study, we observed some compositional alterations among the species and across time (Figure 1).
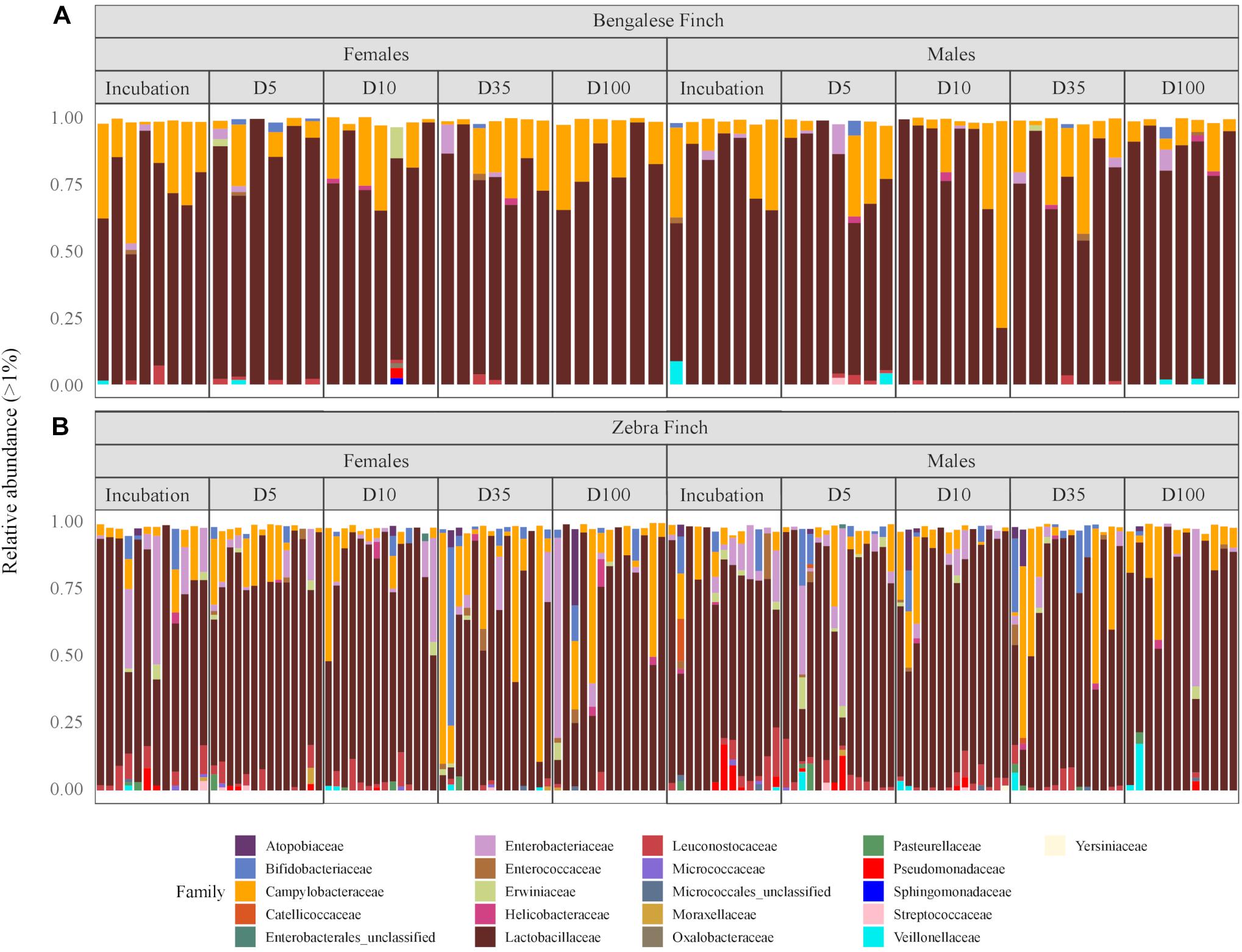
Figure 1. The relative abundance of microbial families in gut samples from (A) the Bengalese Finches and (B) the zebra finches through the different phases of the reproductive cycle: at incubation, 5 days (D5), 10 days (D10), 35 days (D35), and 100 days (D100) after hatching the youngest chick, respectively. Rare phyla with relative abundances below 1% are not shown.
Structure of the Gut Microbial Communities in Relation to Host Taxonomy
We found pronounced differences in the gut microbial structure of the zebra finches and the Bengalese finches. Based on the linear mixed model, which explained a considerable variation in diversity (R2-marginal = 0.117, R2-conditional = 0.261), we found a significant difference in the alpha diversity estimates between the two bird species: the zebra finches exhibited higher diversity based on Shannon diversity index (β = 0.19 ± 0.09, 95% CI [0.02 − 0.35], p = 0.028; Figure 2A).
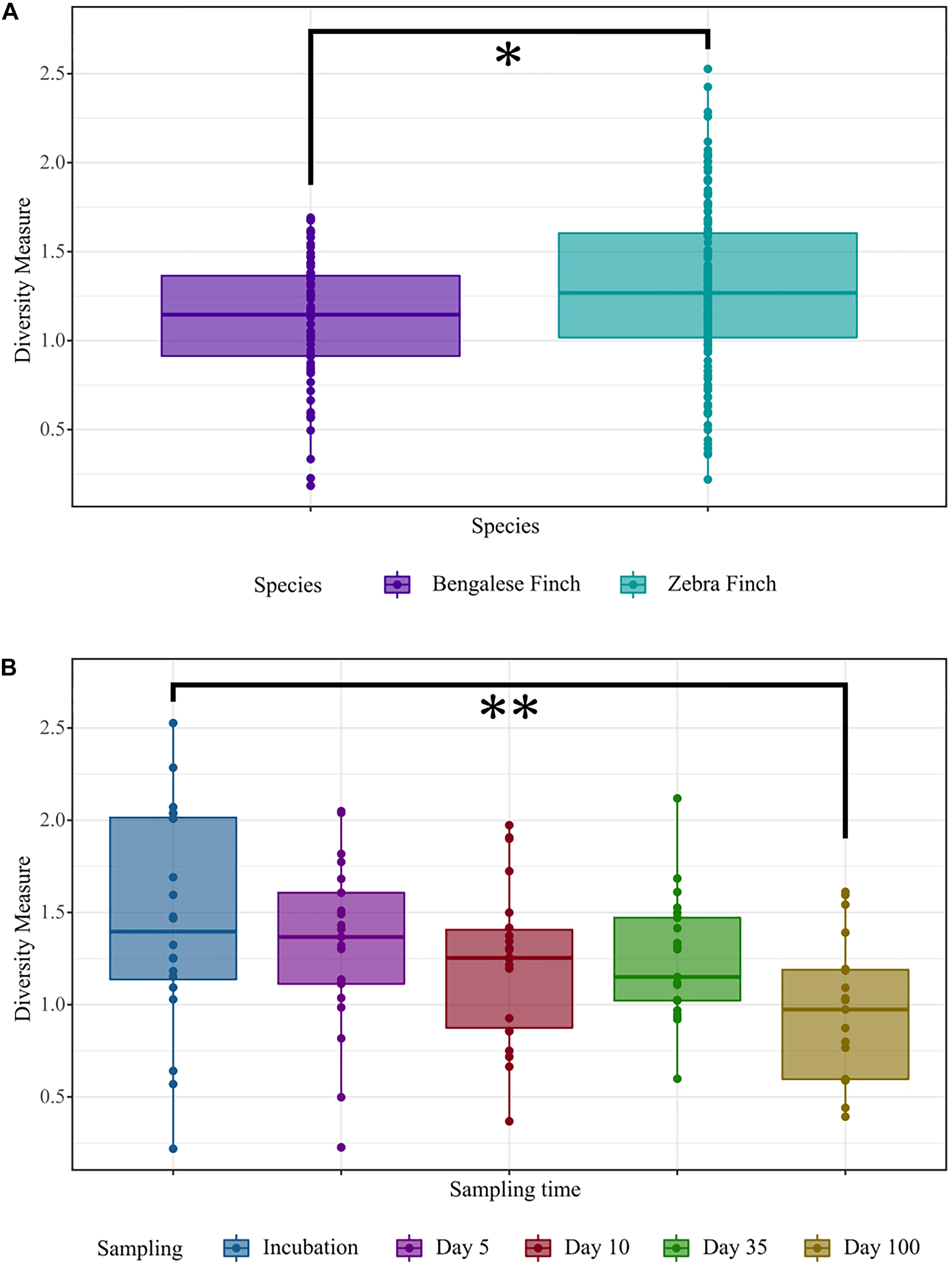
Figure 2. Comparisons of Shannon’s diversity index. (A) Among the host species (Bengalese Finches and zebra finches). (B) In males of both species across five different sampling times: at incubation, 5 days (D5), 10 days (D10), 35 days (D35), and 100 days (D100) after hatching of the youngest chick, respectively. The significance was determined based on the linear mixed model, at p-values ≤ 0.05 (*), p ≤ 0.01 (**), and p ≤ 0.001 (***). In the box plots, the line within indicates the median and the lower and upper boundary of the boxes indicates the 25th and 75th percentile, respectively. Whiskers above and below the boxes correspond to the range of 1.5 times the inter-quartile range (IQR) above and below the 25th and 75th percentile, respectively.
We visualized the dissimilarities in microbial communities among the species using a PCoA based on Jaccard (Figure 3A), Bray–Curtis (Figure 3B), unweighted (Figure 3C), and weighted UniFrac distances (Figure 3D). The microbial communities harbored by each species clustered together in all plots. As the variation explained by the first two axes was higher in the PCoA plots based on Bray–Curtis (Axis1: 17.1%, Axis2: 9.9%) and weighted UniFrac distances (Axis1: 32%, Axis2: 15.2), compared to Jaccard (Axis1: 11.2%, Axis2: 7.2%) and unweighted UniFrac (Axis1: 15%, Axis2: 10.6%) dissimilarity, we used the former two when computing ANOSIM to statistically test for potential differences between the two species. The ANOSIM confirmed a statistical difference in the microbial communities between the two species, which was already revealed by PCoA (based on Bray–Curtis; one-way ANOSIM; factor species: global R = 0.247, p = 0.0001; based on weighted UniFrac, one-way ANOSIM; factor species: global R = 0.230, p = 0.0001).
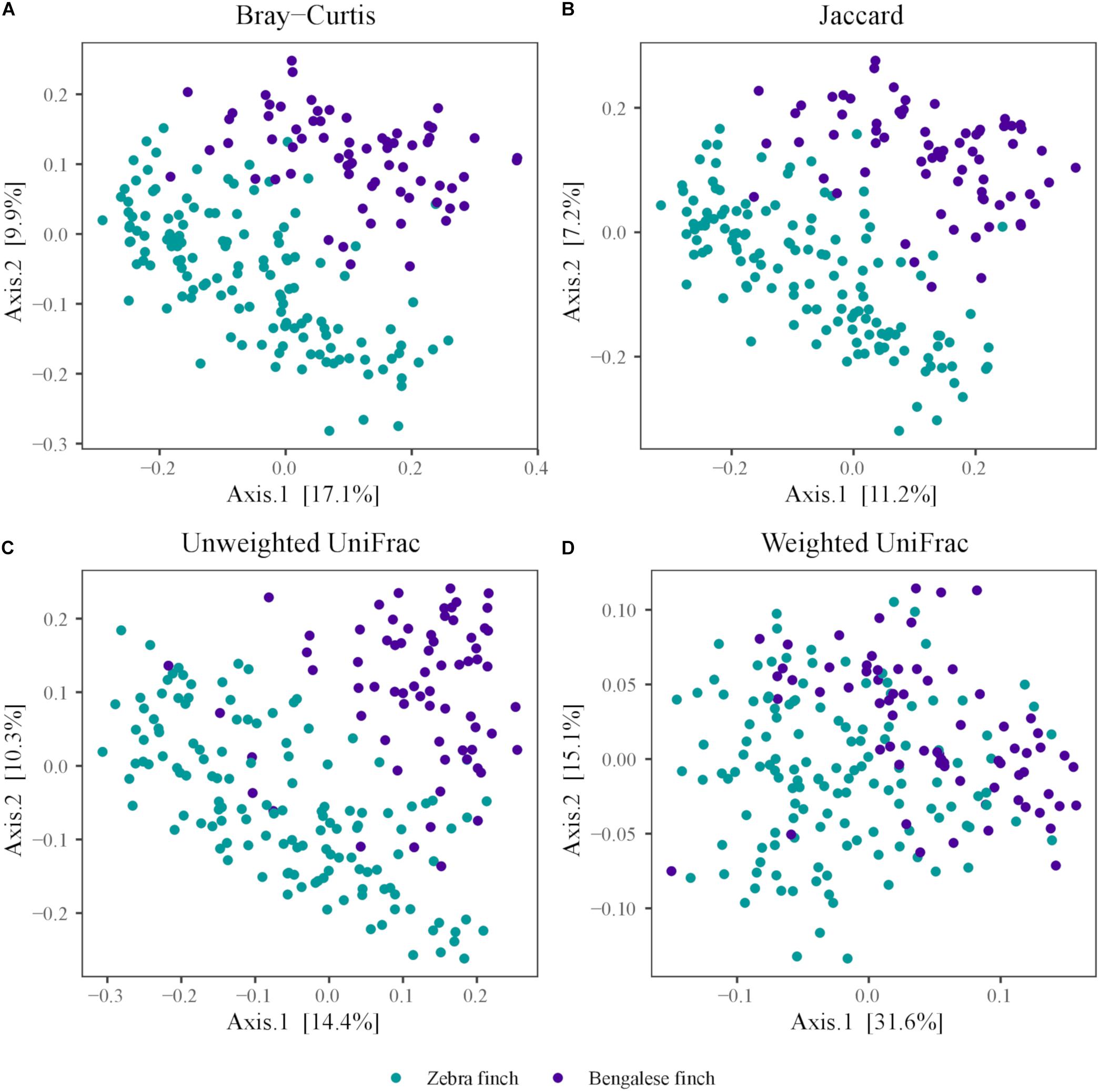
Figure 3. Principal coordinate analysis plots of the dissimilarities of zebra Finch and the Bengalese finch gut microbiota. Distances were computed using the (A) Jaccard and (B) Bray–Curtis dissimilarity index, and the (C) unweighted (D) weighted UniFrac distance metric.
We determined the differentially abundant OTUs underlying the observed differences among the species using the DESeq2 method. We provided detailed information on differentially abundant OTUs in the Supplementary Table 3. Overall, we found 81 significantly differentially abundant OTUs, which constituted for 13.91% of all OTUs (Figure 4). Of these, seven were significantly more abundant in the Bengalese finches and 74 were significantly more abundant in the zebra finches. Expect three of those differentially abundant OTUs, all belonged to three microbial phyla: Proteobacteria (n = 32), Firmicutes (n = 29), and Actinobacteriota (n = 17).
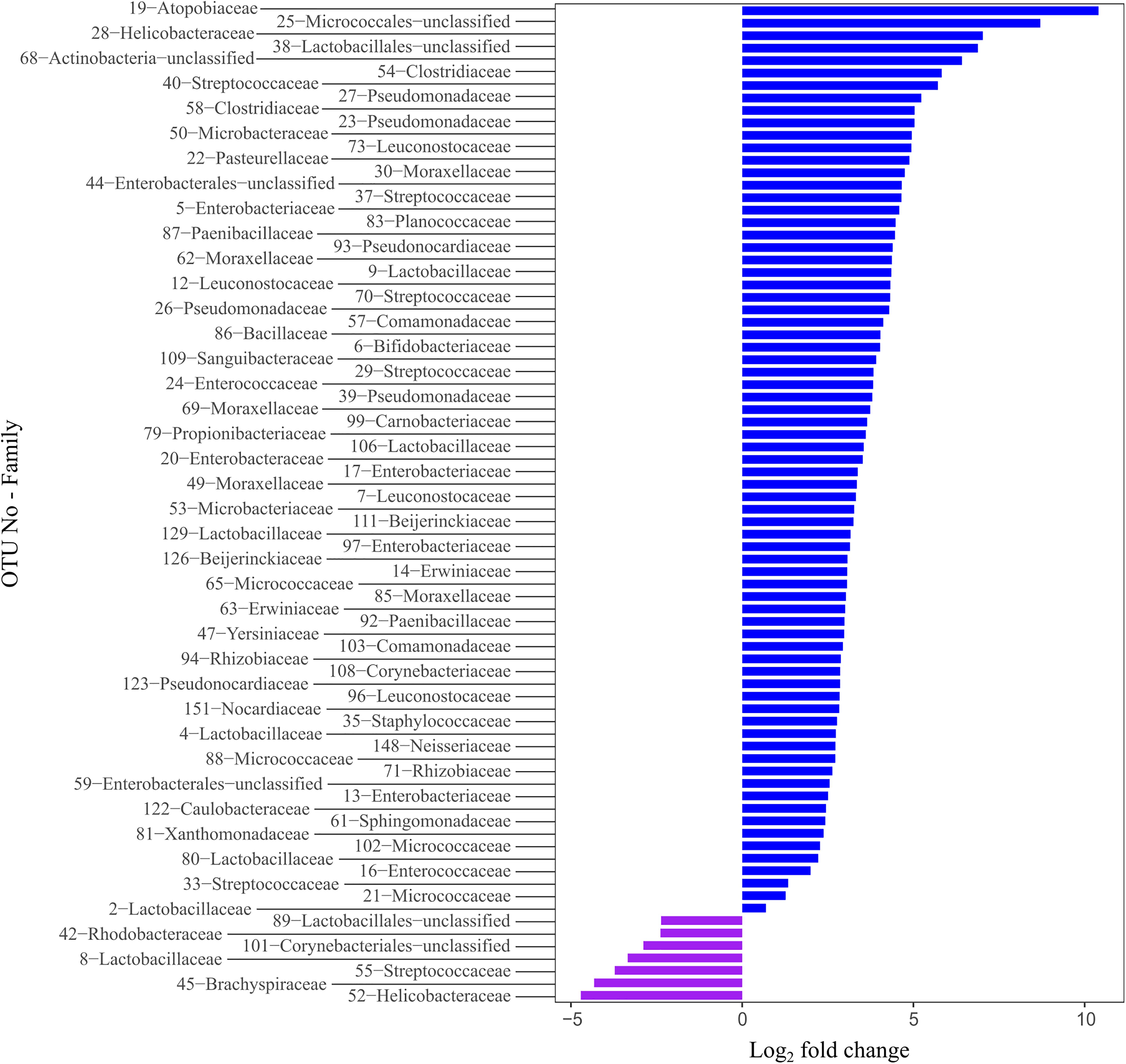
Figure 4. The differentially abundant phyla between the zebra finches and the Bengalese finches. Bars represent OTUs that are significantly differentially abundant between the two host species. OTUs with a log2–fold–change larger than zero are more abundant in the zebra finches (blue bars), while the OTUs with a log2–fold–change smaller than zero are more abundant in the Bengalese finches (purple bars).
Structure of the Gut Microbial Communities in Relation to Host ID
The microbial composition of samples collected from the same individual (Defined by the Host ID) at different time points were on average more similar than those of the samples collected from different individuals (two-way nested ANOSIM Bray–Curtis; Individuals nested in species; factor species: global R = 0.442, p = 0,001; factor Host ID: global R = 0.273, p = 0.001; Weighted UniFrac, factor species: global R = 0.527, p = 0.001; factor Host ID: global R = 0.096, p = 0.001; Supplementary Figure 1A). Furthermore, in zebra finches the length of the period between the sample collection negatively correlated with the microbial similarity: the smaller the sampling periods, the more similar the microbial communities are (Mantel test, Rho: 0.142, p = 0.038). However, this correlation was not evident in the Bengalese finches (Mantel test, Rho: 0.085, p = 0.181).
Sex-Specific Differences in Diversity and Structure of the Gut Microbial Communities
When we analyzed the alpha diversity in relation to host sex, we did not find any evidence for sex-specific differences (the linear mixed model, β = 0.04 ± 0.13, 95% CI [−0.21 to 0.30], p > 0.5). Furthermore, no significant differences were found in the microbial communities between males and females of a given species (two-way crossed ANOSIM; Bray–Curtis; factor Species: global R = 0.246, p = 0.001; factor Sex: global R = 0.004, p = 0.263; Weighted UniFrac; factor Species: global R = 0.229, p = 0.001; factor Sex: global R = −0.004, p = 0.673). However, as the linear mixed model showed a reduction in alpha diversity over time (β = −0.25 ± 0.12, 95% CI [−0.49 to −0.01], p = 0.038), we partitioned the diversity across sexes. In males, the microbial diversity decreased over time with a significant difference between incubation and day 100 (β = −0.48 ± 0.12, 95% CI [−0.87 to −0.08], p = 0.005; Supplementary Figure 1B). By contrast, we did not observe any significant diversity change across sampling times in the females. When comparing the gut microbial communities across the time within a given sex of each species, we found that both taxonomic composition and relative abundance of the microbial assemblages is changing over time in the zebra finch males (ANOSIM; Bray–Curtis; global R = 0.115 p = 0.0003; Weighted UniFrac; global R = 0.067, p = 0.009; Supplementary Figure 1B). In the Bengalese finch males, the alterations between the sampling times were only significant when using the Bray–Curtis measure (global R = 0.118 p = 0.009; Supplementary Figure 1B). No significant change was observed in females.
Microbial Similarity Between Mating Pairs
Mating pairs (defined by the Couple ID) had more similar microbial assemblages compared to randomly chosen males and females (two-way nested ANOSIM; Couples nested in species, Bray–Curtis; factor species: global R = 0.679, p = 0.001; factor Couple ID: global R = 0.555, p = 0.001; Weighted UniFrac; factor species: global R = 0.54, p = 0.001; factor Couple ID: global R = 0.115, p = 0.001; Supplementary Figure 1C).
Discussion
The relevance of symbiotic microorganisms for host ecology and evolution has become increasingly evident. However, we still lack a full understanding of the factors shaping microbial assemblages. Although studies are accumulating, the knowledge on microbial symbiosis in birds is still far behind compared to mammalian species. Furthermore, a huge proportion of the avian studies are conducted on wild populations in their natural habitats (reviewed in Maraci et al., 2018), where several environmental parameters and host-specific factors interact together to shape patterns of microbial colonization and maintenance. Although these studies are very important, they provide little insight into the relative contribution of host-specific factors. Controlled studies enable us to comprehend to what extent hosts can control their microbial assemblages by minimizing the impact of host diet and other extrinsic factors. However, such studies are scarce in avian species. To fill this gap, we characterized gut microbial assemblages of the zebra finches and the Bengalese finches under strictly controlled conditions. As expected, we found prominent interspecific differences in the structure of gut microbial communities. Although the individuals exhibited unique, individual-specific microbial patterns over 3 months, males of both species manifest higher within-individual variations, compared to females. Our study conclusively shows that gut microbes are host-specific, providing essential insights on the host characteristics shaping gut microbial communities.
Taxonomic Composition of the Gut Microbial Communities
Microbial assemblages of the zebra finches and the Bengalese finches were dominated by four microbial phyla: Firmicutes, Campilobacterota, Proteobacteria, and Actinobacteria. The microbial structure recovered in our study was remarkably different from that of wild birds (Hird et al., 2015; Grond et al., 2018). Firmicutes were overrepresented, while there is a deficit of proteobacteria in our study, which is consistent with the findings of previous studies carried out under captivity (Xie et al., 2016). In our study, the second most abundant phylum was Campilobacterota, which is not a usual phylum reported by avian studies conducted under natural or captivity conditions. However, the phylum Campilobacterota was introduced with the SILVA database v138 release, replacing the phylum Epsilonbacteraeota, which was indeed among the abundant phyla reported in previous avian studies (Zhao et al., 2019). This change to the SILVA reference taxonomy, among other taxonomic changes on various levels (York, 2018), was done as SILVA now adopts to the Genome Taxonomy Database (GTDB), a new and more precise taxonomy based on phylogeny inferred from the concatenation of 120 ubiquitous single-copy proteins (Parks et al., 2018). Although some members of this phylum can potentially cause diseases in wild and domestic animals (Bull et al., 2008), they are considered to be non-pathogenic in bird species (Oakley et al., 2014) and are frequently isolated from healthy zebra finches (Benskin et al., 2010; Chen et al., 2020).
Species-Specific Differences in Community Diversity and Composition
A substantial body of evidence has revealed that host taxonomy and evolutionary history are among the main determinants of the gut microbial communities harbored by mammals (Ochman et al., 2010; Phillips et al., 2012; Moeller et al., 2014; Knowles et al., 2019; Song et al., 2020). However, the avian studies investigating interspecific differences in host-associated microbial communities revealed contrasting findings. Some studies failed to show a link between host-taxonomy and gut microbial structuring (Ruiz-Rodríguez et al., 2009, 2018; Hird et al., 2014). On contrary, some comparative studies have shown remarkable species-specific differences (Waite and Taylor, 2014; Hird et al., 2015; Michel et al., 2018; Fu et al., 2020; Lee et al., 2020). It is, however, important to note that all these studies have been conducted using natural populations. As different bird species have distinct dietary preferences and occupy different habitats with altering ecological variables it is not clear whether the detected patterns reflect a true host-specificity.
In the present study, although some proportion of the gut microbiota was conserved between the zebra finches and the Bengalese finches, we also found remarkable differences in the gut microbial communities between these two closely related bird species. Zebra finches harbored more diverse microbial colonies compared to the Bengalese finches. The microbial communities of the two species exhibited variations in terms of taxonomic diversity, relative abundance, and phylogenetic distance of the harbored species. As the diet and other environmental parameters were strictly controlled in our experiment and the birds were housed in the same environment and consequently exposed to the same microbial reservoir, these findings likely underpin the differences in host biology. Although Bengalese finches and zebra finches are widely used in the ecological investigations, interestingly the studies investigating biological differences between these two species are limited to vocal communication, impeding any conclusive inference on what drives species-specific differences in microbial communities. However, one potential explanation for the observed changes is interspecific differences in physiology and anatomy of the digestive system. Although the gross anatomy of the digestive system is expected to be similar in these two bird species, there can be fine-scale anatomical and physiological differences which were indicated by two studies showing that mechanics of drinking movement (Heidweiller and Zweers, 1990) and water retention efficiency (Gere, 1977) differs between Bengalese finches and zebra finches. Alternatively, given the fact that host genetic variation, especially the allelic diversity of immune genes such as immunoglobulin genes, major histocompatibility (MHC) genes, toll-like receptors and cytokines, affects diversity and community structure of host-associated microbes (Bolnick et al., 2014; Blekhman et al., 2015), it is feasible to assume that observed microbial differentiation might reflect prominent genetic differences among the species. Considering the ancestral species White-rumped munia (Lonchura striata acuticauda) and wild zebra finch (T. guttata) originate from finches coming from two geographically distinct locations – China and Australia, respectively – presumably, they experienced differential selection pressures exposed by coevolving pathogens, which in turn result in variations in immune genes. Another possibility is that the strength of artificial selection during the domestication was different for these two species resulting in differential patterns of genetic variation. These predictions partially align with the existing literature: a comparative study investigating the genetic variability between captive and wild populations of zebra finches revealed that domestication led to the loss of some genetic variation but not a severe bottleneck (Forstmeier et al., 2007). Furthermore, domesticated zebra finches also exhibited high allelic diversity for the MHC complex class I genes which play a crucial role in adaptive immunity (Newhouse and Balakrishnan, 2015). To the best of our knowledge, there is no study investigating the loss of genetic diversity in immune genes due to the domestication Bengalese finches. However, considering during the domestication of Bengalese finches from White-rumped munias, breeders selected some specific traits such as good parenting behavior and white color morphs (Takahasi and Okanoya, 2010), presumably domestication led to a substantial decrease in host genetic diversity. However, all these potential interactions warrant further testing.
Individual-Specificity and Stability of Microbial Communities
In humans, tremendous inter-individual variations in microbial communities were documented (Turnbaugh et al., 2009; Lozupone et al., 2012; Kolde et al., 2018) and some of these variations were shown to be stable over time (Lozupone et al., 2012; Faith et al., 2013) leading to the conclusion that individuals have unique microbial profiles. Interestingly, these inter-individual variations in the gut microbial repertoire are prominent shortly after birth (Dominguez-Bello et al., 2010; Raveh-Sadka et al., 2015), while the individuals have very limited interactions with their environment, suggesting the role of vertical transmission and host genetics in shaping individual-specific microbial profiles. Nevertheless, far less is known about the individual-specificity of microbial communities in non-human species.
Temporal stability of gut microbes has been studied in zebra finches using temperature gradient gel electrophoresis (Benskin et al., 2010). However, to the best of our knowledge, individual specificity, and long-term stability of gut microbiota in avian species under controlled conditions were investigated using a metagenomic approach for the first time in our study. We found significant inter-individual variations in microbial communities. The samples collected from the same individual were more similar to one another than those from different individuals. Although we observed some temporal variations in the composition of gut microbial communities in males (as explained in more detail in the next section), a considerable proportion of the gut microbes harbored by individuals was conserved throughout our study. These findings are consistent with the previous study (Benskin et al., 2010) and emphasize the idea that some microbial species are highly adapted to their host and resilient to perturbations, while others are more flexible (Lozupone et al., 2012; Ursell et al., 2012). Accordingly, it is feasible to conclude that hosts can, at least to some extent, control their microbial communities. This further indicates that host genetics has a function in structuring gut microbial communities. However, the exact genetic pathways involved in the regulation of host-symbiont interactions and consequences of these inter-individual variations warrants further investigation.
The Effect of Sex and Sex-Specific Temporal Alterations in Community Diversity and Composition
As males and females of a given species have some differences in their behavior, hormonal profiles and physiology (Tarka et al., 2018), which might influence their symbiotic interactions, we would expect to observe some microbial differences among the sexes of the same species. However, the evidence for the impact of sex on microbial communities is equivocal. In some species, there are sex-specific differences (Leclaire et al., 2014; Elderman et al., 2018; Stoffel et al., 2020), while in others no sexual dimorphism was detected (Benskin et al., 2010; Tung et al., 2015; Bennett et al., 2016; Ren et al., 2017).
In our study, there were no differences in alpha and beta diversity between males and females of the host species, when analyzed regardless of sampling time. However, when we examined the temporal changes in alpha and beta diversity across sexes, we observed sex-specific differences: the males exhibit temporal alterations in both alpha and beta diversity indices, while these metrics remained the same in females, over time. The observed sexual dimorphism can be explained by the plastic alterations in gut microbial communities in response to fluctuating testosterone levels during reproduction. Our sampling times coincide with the different stages of the reproductive cycle, covering all phases between incubation and post-reproduction. Testosterone mediates several reproductive behaviors (Adkins-Regan, 2005) and suppresses the immune system (Folstad and Karter, 1992). This suppression can allow the opportunistic microorganisms to colonize different habitats in the animal body and lead to an increased microbial diversity (Escallón et al., 2017). In socially monogamous birds with biparental care, like the zebra finches or the Bengalese finches, testosterone levels peak during the mating phase declines during the parental care phase with fluctuations and reaches lowest levels during the non-breeding period after reproduction (Hill et al., 2005; Schwabl et al., 2005). In our study, alpha diversity followed the same pattern in males. They were highest during incubation, the phase just after mating, decreased during parental care phase and lowest during non-breeding. Similarly, in rufous-collared sparrows (Zonotrichia capensis), cloacal microbial communities exhibited sexual dimorphism. In males, the diversity decreased during reproduction and increased again as they transitioned to non-breeding condition, while this pattern has not been observed in females (Escallón et al., 2019). However, as we did not measure the testosterone levels in our study, this interaction remains to be investigated.
Microbial Similarity Between the Mating Pairs
We observed similarities between the microbial compositions of mating pairs. This finding supports the existing body of literature on transmission of cloacal bacteria among sexual partners in birds (Kulkarni and Heeb, 2007; White et al., 2010; Escallón et al., 2019). A birds’ cloaca performs multiple functions for the digestive, urinary, and reproductive system. During sexual intercourse, it allows the transfer of microbes between the pairs, from these different sources. Although this kind of microbial exchange can lead to the transmission of diseases, it can also ensure the transfer of beneficial microbes between sexual partners (Smith and Mueller, 2015). Potential fitness benefits provided by sexually transmitted microbes and involvement of these microbes into mate choice decisions should be further studied.
Conclusion and Outlook
Symbiotic microorganisms influence nearly all aspects of an animals’ biology. However, we still know very little about the role of host characteristics in the regulation of these complex systems, especially in avian taxa. This is partly because most studies have been conducted on wild populations, and in such systems, host-specific factors can be masked by spatially varying environmental factors. On the contrary, our study was conducted under fully controlled conditions to minimize interference of dietary and environmental variations and therefore has specific importance in terms of understanding the effect of host-specific factors in sculpturing symbiotic microbial assemblages. Our study convincingly shows that in zebra finches and Bengalese finches gut microbes are species-specific, unique to individuals, considerably stable over-time (with some lesser extent fluctuations in males across the different stages of the reproductive cycle) and exhibit similarities between mating pairs. This descriptive study is an important first step in elucidating the function of host-microbe interactions in avian ecology and evolution.
Our findings have naturally raised some further questions to be investigated. For example, an investigation of heritable components of the microbiota and the relative importance of different transmission routes can improve our understanding of the species-specificity of gut microbes. Furthermore, the exact mechanisms enabling the individual hosts to regulate this highly complex system warrant further research. In this regard, the investigation of host immune genes can broaden our insights about the colonization dynamics. Lastly, the interaction between reproductive hormones and gut microbial composition should be further studied.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: European Nucleotide Archive, Project ID: PRJEB41573, accession number ERP125369.
Ethics Statement
The animal study was reviewed and approved by Gesundheits-, Veterinär-und Lebensmittelüberwachungsamt der Stadt Bielefeld (#530.421630–1,18.4.2002).
Author Contributions
ÖM and BC conceptualized the research idea and planned the experiments. ÖM carried out the experiments. TB contributed to the sample preparation. JK contributed to the analytical tools. SJ performed the bioinformatic analyses. AA-P and OC-G carried out the statistical analysis with the supervision of BC and ÖM. ÖM wrote the manuscript in consultation with BC. All authors approved the final version of the manuscript.
Funding
This study was financially supported by a Freigeist Fellowship from the Volkswagen Foundation to BC and the Bielefeld University Young Researchers Fund to ÖM. SJ received financial support by the German Federal Ministry of Education and Research (BMBF) as part of the project “Bielefeld-Gießen Center for Microbial Bioinformatics – BiGi” (Grant number 031A533) within the German Network for Bioinformatics Infrastructure (de.NBI).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Elke Hippauf, Anika Winkler, Yvonne Kutter, and Katharina Hanuschka for technical support during the laboratory procedures. We express our gratitude to Ursula Kodytek, for taking care of the birds. Furthermore, we thank Kathrin Engel and Verena Schöler for their help during the breeding experiment and sampling procedures. We express our gratitude to Alfredo Sanchez-Tojar, Meinolf Ottensmann, Tony Rinaud, and Hugo de Eira Pereira for their feedbacks during the statistical analyses. We acknowledge the financial support of the German Research Foundation (DFG) and the Open Access Publication Fund of Bielefeld University for the article processing charge. We thank the reviewers for their valuable comments and suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.619141/full#supplementary-material
Supplementary Figure 1 | Principal coordinate analysis plots computed using Bray–Curtis dissimilarity index visualized by (A) Species and host ID. (B) Sex and sampling time. (C) Species and couple ID. In the three different versions of the same PCoA plot, different color, and shape schemes indicate different variables influencing beta diversity. In plots (A) and (C) the host species are represented by different shapes (zebra finches are represented by the circles and the Bengalese finches are represented by triangles) while color codes are used to indicate individual ID and couple ID, respectively. In plot (B), host sexes are represented by different shapes (females are represented by squares and males are represented by stars) and the samples are color coded according to the sampling time.
Supplementary Table 1 | The total abundance, the mean abundance and the standard deviation of the identified microbial phyla. The numbers represent the percentages.
Supplementary Table 2 | The total abundance, the mean abundance and the standard deviation of the identified microbial families. The numbers represent the percentages.
Supplementary Table 3 | Detailed information on the differentially abundant microbial taxa in the zebra finches and the Bengalese finches.
References
Adkins-Regan, E. (2005). Hormones and Animal Social Behavior. New Jersey, NJ: Princeton University Press.
Ambrosini, R., Corti, M., Franzetti, A., Caprioli, M., Rubolini, D., Motta, V. M., et al. (2019). Cloacal microbiomes and ecology of individual barn swallows. FEMS Microbiol. Ecol. 95:fiz061. doi: 10.1093/femsec/fiz061
Archie, E. A., and Theis, K. R. (2011). Animal behaviour meets microbial ecology. Anim. Behav. 82, 425–436. doi: 10.1016/j.anbehav.2011.05.029
Banks, J. C., Cary, S. C., and Hogg, I. D. (2009). The phylogeography of Adelie penguin faecal flora. Environ. Microbiol. 11, 577–588. doi: 10.1111/j.1462-2920.2008.01816.x
Bates, D., Mächler, M., Bolker, B. M., and Walker, S. C. (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48. doi: 10.18637/jss.v067.i01
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bennett, G., Malone, M., Sauther, M. L., Cuozzo, F. P., White, B., Nelson, K. E., et al. (2016). Host age, social group, and habitat type influence the gut microbiota of wild ring-tailed lemurs (Lemur catta). Am. J. Primatol. 78, 883–892. doi: 10.1002/ajp.22555
Benskin, C. M. W. H., Rhodes, G., Pickup, R. W., Wilson, K., and Hartley, I. R. (2010). Diversity and temporal stability of bacterial communities in a model passerine bird, the zebra finch. Mol. Ecol. 19, 5531–5544. doi: 10.1111/j.1365-294X.2010.04892.x
Berlow, M., Kohl, K. D., and Derryberry, E. P. (2020). Evaluation of non-lethal gut microbiome sampling methods in a passerine bird. Ibis 162, 911–923. doi: 10.1111/ibi.12807
Blanco, G., Lemus, J. A., and Grande, J. (2006). Faecal bacteria associated with different diets of wintering red kites: influence of livestock carcass dumps in microflora alteration and pathogen acquisition. J. Appl. Ecol. 43, 990–998. doi: 10.1111/j.1365-2664.2006.01200.x
Blekhman, R., Goodrich, J. K., Huang, K., Sun, Q., Bukowski, R., Bell, J. T., et al. (2015). Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 16:191. doi: 10.1186/s13059-015-0759-1
Bodawatta, K. H., Sam, K., Jønsson, K. A., and Poulsen, M. (2018). Comparative analyses of the digestive tract microbiota of New Guinean passerine birds. Front. Microbiol. 9:1830. doi: 10.3389/fmicb.2018.01830
Bolnick, D. I., Snowberg, L. K., Caporaso, J. G., Lauber, C., Knight, R., and Stutz, W. E. (2014). Major histocompatibility complex class IIb polymorphism influences gut microbiota composition and diversity. Mol. Ecol. 23, 4831–4845. doi: 10.1111/mec.12846
Borre, Y. E., O’Keeffe, G. W., Clarke, G., Stanton, C., Dinan, T. G., and Cryan, J. F. (2014). Microbiota and neurodevelopmental windows: implications for brain disorders. Trends Mol. Med. 20, 509–518. doi: 10.1016/j.molmed.2014.05.002
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
Bull, S. A., Thomas, A., Humphrey, T., Ellis-Iversen, J., Cook, A. J., Lovell, R., et al. (2008). Flock health indicators and Campylobacter spp. in commercial housed broilers reared in Great Britain. Appl. Environ. Microbiol. 74, 5408–5413. doi: 10.1128/AEM.00462-08
Candela, M., Biagi, E., Maccaferri, S., Turroni, S., and Brigidi, P. (2012). Intestinal microbiota is a plastic factor responding to environmental changes. Trends Microbiol. 20, 385–391. doi: 10.1016/j.tim.2012.05.003
Chen, C. Y., Chen, C. K., Chen, Y. Y., Fang, A., Shaw, G. T. W., Hung, C. M., et al. (2020). Maternal gut microbes shape the early-life assembly of gut microbiota in passerine chicks via nests. Microbiome 8:129. doi: 10.1186/s40168-020-00896-9
Clarke, K. R. (1999). Nonmetric multivariate analysis in community-level ecotoxicology. Environ. Toxicol. Chem. 18:118. doi: 10.1897/1551-50281999018<0118:NMAICL<2.3.CO;2
Clarke, K. R., Gorley, R., Somerfield, P., and Warwick, R. (2014). Change in Marine Communities: an Approach to Statistical Analysis and Interpretation, 3rd Edn. Primer-E Ltd: Plymouth.
Collado, M. C., Cernada, M., Baüerl, C., Vento, M., and Pérez-Martínez, G. (2012). Microbial ecology and host-microbiota interactions during early life stages. Gut Microbes 3, 352–365. doi: 10.4161/gmic.21215
Davidson, G. L., Cooke, A. C., Johnson, C. N., and Quinn, J. L. (2018). The gut microbiome as a driver of individual variation in cognition and functional behaviour. Philos. Trans. R. Soc. B Biol. Sci. 373:20170286. doi: 10.1098/rstb.2017.0286
Davidson, G. L., Raulo, A., and Knowles, S. C. L. (2020). Identifying microbiome-mediated behaviour in wild vertebrates. Trends Ecol. Evol. 35, 972–980. doi: 10.1016/j.tree.2020.06.014
DiGiulio, D. B. (2012). Diversity of microbes in amniotic fluid. Semin. Fetal Neonatal Med. 17, 2–11. doi: 10.1016/j.siny.2011.10.001
Dominguez-Bello, M. G., Costello, E. K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U.S.A. 107, 11971–11975. doi: 10.1073/pnas.1002601107
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Elderman, M., Hugenholtz, F., Belzer, C., Boekschoten, M., van Beek, A., de Haan, B., et al. (2018). Sex and strain dependent differences in mucosal immunology and microbiota composition in mice. Biol. Sex Differ. 9:26. doi: 10.1186/s13293-018-0186-6
Engel, K., Pankoke, H., Jünemann, S., Brandl, H. B., Sauer, J., Griffith, S. C., et al. (2020). Family Matters: Skin Microbiome Reflects the Social Group and Spatial Proximity in Wild Zebra Finches. Researc Square (Preprint). doi: 10.21203/rs.3.rs-24732/v2 CrossRef Full TextAvailable Online at: https://www.researchsquare.com/article/rs-24732/v2 (Accessed at October 15, 2020)
Engel, K., Sauer, J., Jünemann, S., Winkler, A., Wibberg, D., Kalinowski, J., et al. (2018). Individual-and species-specific skin microbiomes in three different Estrildid Finch species revealed by 16S amplicon sequencing. Microb. Ecol. 76, 518–529. doi: 10.1007/s00248-017-1130-8
Escallón, C., Becker, M. H., Walke, J. B., Jensen, R. V., Cormier, G., Belden, L. K., et al. (2017). Testosterone levels are positively correlated with cloacal bacterial diversity and the relative abundance of Chlamydiae in breeding male rufous-collared sparrows. Funct. Ecol. 31, 192–203. doi: 10.1111/1365-2435.12696
Escallón, C., Belden, L. K., and Moore, I. T. (2019). The cloacal microbiome changes with the breeding season in a wild bird. Integr. Org. Biol. 1, 1–16. doi: 10.1093/iob/oby009
Ezenwa, V. O., Gerardo, N. M., Inouye, D. W., Medina, M., and Xavier, J. B. (2012). Animal behavior and the microbiome. Science 338, 198–199. doi: 10.1126/science.1227412
Faith, J. J., Guruge, J. L., Charbonneau, M., Subramanian, S., Seedorf, H., Goodman, A. L., et al. (2013). The long-term stability of the human gut microbiota. Science 341:1237439. doi: 10.1126/science.1237439
Folstad, I., and Karter, A. J. (1992). Parasites, bright males, and the immunocompetence handicap. Am. Nat. 139, 603–622. doi: 10.1086/285346
Forstmeier, W., Segelbacher, G., Mueller, J. C., and Kempenaers, B. (2007). Genetic variation and differentiation in captive and wild zebra finches (Taeniopygia guttata). Mol. Ecol. 16, 4039–4050. doi: 10.1111/j.1365-294X.2007.03444.x
Fu, R., Xiang, X., Dong, Y., Cheng, L., and Zhou, L. (2020). Comparing the intestinal bacterial communies of sympatric wintering Hooded Crane (Grus monacha) and Domestic Goose (Anser anser domesticus). Avian Res. 11:13. doi: 10.1186/s40657-020-00195-9
Gere, G. (1977). No Beiträge zur Untersuchung des Wasserhaushaltes des Zebrafinken (Taeniopygia guttata Vieill.) und des Japanischen Mövchens (domestizierte Form von Lonchura striata (L.)). Opusc. zool. Budapest X 111, 1–2. doi: 10.1159/000404845
Gill, S. R., Pop, M., Deboy, R. T., Eckburg, P. B., Turnbaugh, P. J., Samuel, B. S., et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359. doi: 10.1126/science.1124234
Gould, A. L., Zhang, V., Lamberti, L., Jones, E. W., Obadia, B., Korasidis, N., et al. (2018). Microbiome interactions shape host fitness. Proc. Natl. Acad. Sci. U.S.A. 115, E11951–E11960. doi: 10.1073/pnas.1809349115
Grond, K., Sandercock, B. K., Jumpponen, A., and Zeglin, L. H. (2018). The avian gut microbiota: community, physiology and function in wild birds. J. Avian Biol. 49:e01788. doi: 10.1111/jav.01788
Heidweiller, J., and Zweers, G. A. (1990). Drinking mechanisms in the zebra finch and the bengalese finch. Condor 92, 1–28. doi: 10.2307/1368379
Hill, W. L., Ballard, S., Coyer, M. J., and Rowley, T. (2005). The interaction of testosterone and breeding phase on the reproductive behavior and use of space of male zebra finches. Horm. Behav. 47, 452–458. doi: 10.1016/j.yhbeh.2004.11.016
Hird, S. M., Carstens, B. C., Cardiff, S. W., Dittmann, D. L., and Brumfield, R. T. (2014). Sampling locality is more detectable than taxonomy or ecology in the gut microbiota of the brood-parasitic Brown-headed Cowbird (Molothrus ater). PeerJ 2:e321. doi: 10.7717/peerj.321
Hird, S. M., Sánchez, C., Carstens, B. C., and Brumfield, R. T. (2015). Comparative gut microbiota of 59 neotropical bird species. Front. Microbiol. 6:1403. doi: 10.3389/fmicb.2015.01403
Jaccard, P. (1912). The distribution of the flora in the alpine zone. New Phytol 11, 37–50. doi: 10.1111/j.1469-8137.1912.tb05611.x
Jeurink, P. V., van Bergenhenegouwen, J., Jiménez, E., Knippels, L. M. J., Fernández, L., Garssen, J., et al. (2013). Human milk: a source of more life than we imagine. Benef. Microbes 4, 17–30. doi: 10.3920/BM2012.0040
Jiménez, E., Marín, M. L., Martín, R., Odriozola, J. M., Olivares, M., Xaus, J., et al. (2008). Is meconium from healthy newborns actually sterile? Res. Microbiol. 159, 187–193. doi: 10.1016/j.resmic.2007.12.007
Johnson, K. V. A., and Foster, K. R. (2018). Why does the microbiome affect behaviour? Nat. Rev. Microbiol. 16, 647–655. doi: 10.1038/s41579-018-0014-3
Joshi, N., and Fass, J. (2011). Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33) [Software].
Kamada, N., Seo, S. U., Chen, G. Y., and Núñez, G. (2013). Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 13, 321–335. doi: 10.1038/nri3430
Kirschman, L. J., Khadjinova, A., Ireland, K., and Milligan-Myhre, K. C. (2020). Early life disruption of the microbiota affects organ development and cytokine gene expression in three spine stickleback. Integr. Comp. Biol. 60:icaa136. doi: 10.1093/icb/icaa136
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Knowles, S. C. L., Eccles, R. M., and Baltrûnaitë, L. (2019). Species identity dominates over environment in shaping the microbiota of small mammals. Ecol. Lett. 22, 826–837. doi: 10.1111/ele.13240
Kohl, K. D. (2012). Diversity and function of the avian gut microbiota. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 182, 591–602. doi: 10.1007/s00360-012-0645-z
Kohl, K. D., and Carey, H. V. (2016). A place for host–microbe symbiosis in the comparative physiologist’s toolbox. J. Exp. Biol. 219, 3496–3504. doi: 10.1242/jeb.136325
Kohl, K. D., Varner, J., Wilkening, J. L., and Dearing, M. D. (2018). Gut microbial communities of American pikas (Ochotona princeps): evidence for phylosymbiosis and adaptations to novel diets. J. Anim. Ecol. 87, 323–330. doi: 10.1111/1365-2656.12692
Kolde, R., Franzosa, E. A., Rahnavard, G., Hall, A. B., Vlamakis, H., Stevens, C., et al. (2018). Host genetic variation and its microbiome interactions within the human microbiome project. Genome Med. 10:6. doi: 10.1186/s13073-018-0515-8
Kreisinger, J., Čízková, D., Kropácková, L., and Albrecht, T. (2015). Cloacal microbiome structure in a long-distance migratory bird assessed using deep 16sRNA pyrosequencing. PLoS One 10:e0137401. doi: 10.1371/journal.pone.0137401
Kubinak, J. L., Stephens, W. Z., Soto, R., Petersen, C., Chiaro, T., Gogokhia, L., et al. (2015). MHC variation sculpts individualized microbial communities that control susceptibility to enteric infection. Nat. Commun. 6:8642. doi: 10.1038/ncomms9642
Kulkarni, S., and Heeb, P. (2007). Social and sexual behaviours aid transmission of bacteria in birds. Behav. Process. 74, 88–92. doi: 10.1016/j.beproc.2006.10.005
Leclaire, S., Nielsen, J. F., and Drea, C. M. (2014). Bacterial communities in meerkat anal scent secretions vary with host sex, age, and group membership. Behav. Ecol. 25, 996–1004. doi: 10.1093/beheco/aru074
Lee, C. Y., Peralta-Sánchez, J. M., Martínez-Bueno, M., Møller, A. P., Rabelo-Ruiz, M., Zamora-Muñoz, C., et al. (2020). The gut microbiota of brood parasite and host nestlings reared within the same environment: disentangling genetic and environmental effects. ISME J. 14, 2691–2702. doi: 10.1038/s41396-020-0719-y
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Lim, E. S., Wang, D., and Holtz, L. R. (2016). The bacterial microbiome and virome milestones of infant development. Trends Microbiol. 24, 801–810. doi: 10.1016/j.tim.2016.06.001
Lloyd-Price, J., Mahurkar, A., Rahnavard, G., Crabtree, J., Orvis, J., Hall, A. B., et al. (2017). Strains, functions and dynamics in the expanded human microbiome project. Nature 550, 61–66. doi: 10.1038/nature23889
Lombardo, M. P., Thorpe, P. A., Cichewicz, R., Henshaw, M., Millard, C., Steen, C., et al. (1996). Communities of cloacal bacteria in tree swallow families. Condor 98, 167–172. doi: 10.2307/1369521
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Lozupone, C. A., Hamady, M., Kelley, S. T., and Knight, R. (2007). Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. doi: 10.1128/AEM.01996-06
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K., and Knight, R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. doi: 10.1038/nature11550
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Maraci, Ö, Engel, K., and Caspers, B. A. (2018). Olfactory communication via microbiota: what is known in birds? Genes (Basel) 9:387. doi: 10.3390/genes9080387
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Maynard, C. L., Elson, C. O., Hatton, R. D., and Weaver, C. T. (2012). Reciprocal interactions of the intestinal microbiota and immune system. Nature 489, 231–241. doi: 10.1038/nature11551
McFall-Ngai, M. M., Hadfield, M. G., Bosch, T. T. C. G. T., Carey, H. V. H., Domazet-Lošo, T., Douglas, A. A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U.S.A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Michel, A. J., Ward, L. M., Goffredi, S. K., Dawson, K. S., Baldassarre, D. T., Brenner, A., et al. (2018). The gut of the finch: uniqueness of the gut microbiome of the Galápagos vampire finch. Microbiome 6:167. doi: 10.1186/s40168-018-0555-8
Moeller, A. H., Li, Y., Ngole, E. M., Ahuka-Mundeke, S., Lonsdorf, E. V., Pusey, A. E., et al. (2014). Rapid changes in the gut microbiome during human evolution. Proc. Natl. Acad. Sci. U.S.A. 111, 16431–16435. doi: 10.1073/pnas.1419136111
Moeller, A. H., and Sanders, J. G. (2020). Roles of the gut microbiota in the adaptive evolution of mammalian species. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 375:20190597. doi: 10.1098/rstb.2019.0597
Moeller, A. H., Suzuki, T. A., Phifer-Rixey, M., and Nachman, M. W. (2018). Transmission modes of the mammalian gut microbiota. Science 362, 453–457. doi: 10.1126/science.aat7164
Muegge, B. D., Kuczynski, J., Knights, D., Clemente, J. C., González, A., Fontana, L., et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974. doi: 10.1126/science.1198719
Newhouse, D. J., and Balakrishnan, C. N. (2015). High major histocompatibility complex class I polymorphism despite bottlenecks in wild and domesticated populations of the zebra finch (Taeniopygia guttata). BMC Evol. Biol. 15:265. doi: 10.1186/s12862-015-0546-3
Oakley, B. B., Lillehoj, H. S., Kogut, M. H., Kim, W. K., Maurer, J. J., Pedroso, A., et al. (2014). The chicken gastrointestinal microbiome. FEMS Microbiol. Lett. 360, 100–112. doi: 10.1111/1574-6968.12608
Ochman, H., Worobey, M., Kuo, C. H., Ndjango, J. B. N., Peeters, M., Hahn, B. H., et al. (2010). Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 8:e1000546. doi: 10.1371/journal.pbio.1000546
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., Mcglinn, D., et al. (2019). Package “Vegan” Title Community Ecology Package Version 2.5-6.
Palmer, C., Bik, E. M., DiGiulio, D. B., Relman, D. A., and Brown, P. O. (2007). Development of the human infant intestinal microbiota. PLoS Biol. 5:1556–1573. doi: 10.1371/journal.pbio.0050177
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P. A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Paulson, J. N., Colin Stine, O., Bravo, H. C., and Pop, M. (2013a). Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–1202. doi: 10.1038/nmeth.2658
Paulson, J. N., Olson, N. D., Braccia, D. J., Wagner, J., Talukder, H., Pop, M., et al. (2013b). metagenomeSeq: Statistical Analysis for Sparse High-Throughput Sequncing. Bioconductor package. Available online at: http://www.cbcb.umd.edu/software/metagenomeSeq.
Phillips, C. D., Phelan, G., Dowd, S. E., McDonough, M. M., Ferguson, A. W., Delton Hanson, J., et al. (2012). Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Mol. Ecol. 21, 2617–2627. doi: 10.1111/j.1365-294X.2012.05568.x
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Raveh-Sadka, T., Thomas, B. C., Singh, A., Firek, B., Brooks, B., Castelle, C. J., et al. (2015). Gut bacteria are rarely shared by co-hospitalized premature infants, regardless of necrotizing enterocolitis development. Elife 4:e05477. doi: 10.7554/eLife.05477
Ren, T., Boutin, S., Humphries, M. M., Dantzer, B., Gorrell, J. C., Coltman, D. W., et al. (2017). Seasonal, spatial, and maternal effects on gut microbiome in wild red squirrels. Microbiome 5:163. doi: 10.1186/s40168-017-0382-3
Rothschild, D., Weissbrod, O., Barkan, E., Kurilshikov, A., Korem, T., Zeevi, D., et al. (2018). Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215. doi: 10.1038/nature25973
Round, J. L., and Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323. doi: 10.1038/nri2515
Ruiz-Rodríguez, M., Lucas, F. S., Heeb, P., and Soler, J. J. (2009). Differences in intestinal microbiota between avian brood parasites and their hosts. Biol. J. Linn. Soc. 96, 406–414. doi: 10.1111/j.1095-8312.2008.01127.x
Ruiz-Rodríguez, M., Martín-Vivaldi, M., Martínez-Bueno, M., and Soler, J. J. (2018). Gut microbiota of great spotted cuckoo nestlings is a mixture of those of their foster magpie siblings and of cuckoo adults. Genes (Basel) 9:381. doi: 10.3390/genes9080381
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schwabl, H., Flinks, H., and Gwinner, E. (2005). Testosterone, reproductive stage, and territorial behavior of male and female European stonechats Saxicola torquata. Horm. Behav. 47, 503–512. doi: 10.1016/j.yhbeh.2004.08.003
Shannon, C. E. (1997). The mathematical theory of communication. M.D. Comput. 14, 306–317. doi: 10.2307/410457
Sherwin, E., Bordenstein, S. R., Quinn, J. L., Dinan, T. G., and Cryan, J. F. (2019). Microbiota and the social brain. Science 366:eaar2016. doi: 10.1126/science.aar2016
Smith, C. C., and Mueller, U. G. (2015). Sexual transmission of beneficial microbes. Trends Ecol. Evol. 30, 438–440. doi: 10.1016/j.tree.2015.05.006
Song, S. J., Sanders, J. G., Delsuc, F., Metcalf, J., Amato, K., Taylor, M. W., et al. (2020). Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. MBio 11:e02901. doi: 10.1128/mBio.02901-19
Stagaman, K., Burns, A. R., Guillemin, K., and Bohannan, B. J. M. (2017). The role of adaptive immunity as an ecological filter on the gut microbiota in zebrafish. ISME J. 11, 1630–1639. doi: 10.1038/ismej.2017.28
Stoffel, M. A., Acevedo-Whitehouse, K., Morales-Durán, N., Grosser, S., Chakarov, N., Krüger, O., et al. (2020). Early sexual dimorphism in the developing gut microbiome of northern elephant seals. Mol. Ecol. 29, 2109–2122. doi: 10.1111/mec.15385
Takahasi, M., and Okanoya, K. (2010). Song learning in wild and domesticated strains of white-rumped munia, Lonchura striata, compared by cross-fostering procedures: domestication increases song variability by decreasing strain-specific bias. Ethology 116, 396–405. doi: 10.1111/j.1439-0310.2010.01761.x
Tarka, M., Guenther, A., Niemelä, P. T., Nakagawa, S., and Noble, D. W. A. (2018). Sex differences in life history, behavior, and physiology along a slow-fast continuum: a meta-analysis. Behav. Ecol. Sociobiol. 72:132. doi: 10.1007/s00265-018-2534-2
Teyssier, A., Lens, L., Matthysen, E., and White, J. (2018). Dynamics of gut microbiota diversity during the early development of an avian host: evidence from a cross-foster experiment. Front. Microbiol. 9:1524. doi: 10.3389/fmicb.2018.01524
Teyssier, A., Matthysen, E., Hudin, N. S., de Neve, L., White, J., and Lens, L. (2020). Diet contributes to urban-induced alterations in gut microbiota: experimental evidence from a wild passerine. Proc. R. Soc. B Biol. Sci. 287:20192182. doi: 10.1098/rspb.2019.2182
Thorsen, J., Brejnrod, A., Mortensen, M., Rasmussen, M. A., Stokholm, J., Al-Soud, W. A., et al. (2016). Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies. Microbiome 4:62. doi: 10.1186/s40168-016-0208-8
Tremaroli, V., and Bäckhed, F. (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249. doi: 10.1038/nature11552
Trevelline, B. K., MacLeod, K. J., Knutie, S. A., Langkilde, T., and Kohl, K. D. (2018). In ovo microbial communities: a potential mechanism for the initial acquisition of gut microbiota among oviparous birds and lizards. Biol. Lett. 14:20180225. doi: 10.1098/rsbl.2018.0225
Tung, J., Barreiro, L. B., Burns, M. B., Grenier, J. C., Lynch, J., Grieneisen, L. E., et al. (2015). Social networks predict gut microbiome composition in wild baboons. Elife 4:e05224. doi: 10.7554/eLife.05224
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 454, 480–484. doi: 10.1038/nature07540
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449, 804–810. doi: 10.1038/nature06244
Ursell, L. K., Metcalf, J. L., Parfrey, L. W., and Knight, R. (2012). Defining the human microbiome. Nutr. Rev. 70(Suppl. 1), S38–S44. doi: 10.1111/j.1753-4887.2012.00493.x
van Veelen, H. P. J., Falcão Salles, J., Matson, K. D., van der Velde, M., and Tieleman, B. I. (2020). Microbial environment shapes immune function and cloacal microbiota dynamics in zebra finches Taeniopygia guttata. Anim. Microbiome 2:21. doi: 10.1186/s42523-020-00039-3
Videvall, E., Song, S. J., Bensch, H., Strandh, M., Engelbrecht, A., Serfontein, N., et al. (2020). Early-life gut dysbiosis linked to juvenile mortality in ostriches. BMC Microbiome 8:147. doi: 10.1186/s40168-020-00925-7
Videvall, E., Song, S. J., Bensch, H. M., Strandh, M., Engelbrecht, A., Serfontein, N., et al. (2019). Major shifts in gut microbiota during development and its relationship to growth in ostriches. Mol. Ecol. 28, 2653–2667. doi: 10.1111/mec.15087
Videvall, E., Strandh, M., Engelbrecht, A., Cloete, S., and Cornwallis, C. K. (2018). Measuring the gut microbiome in birds: comparison of faecal and cloacal sampling. Mol. Ecol. Resour. 18, 424–434. doi: 10.1111/1755-0998.12744
Waite, D. W., and Taylor, M. W. (2014). Characterizing the avian gut microbiota: membership, driving influences, and potential function. Front. Microbiol. 5:233. doi: 10.3389/fmicb.2014.00223
White, J., Mirleau, P., Danchin, E., Mulard, H., Hatch, S. A., Heeb, P., et al. (2010). Sexually transmitted bacteria affect female cloacal assemblages in a wild bird. Ecol. Lett. 13, 1515–1524. doi: 10.1111/j.1461-0248.2010.01542.x
Xie, Y., Xia, P., Wang, H., Yu, H., Giesy, J. P., Zhang, Y., et al. (2016). Effects of captivity and artificial breeding on microbiota in feces of the red-crowned crane (Grus japonensis). Sci. Rep. 6:33350. doi: 10.1038/srep33350
York, A. (2018). Next-generation bacterial taxonomy. Nat. Rev. Microbiol. 16:583. doi: 10.1038/s41579-018-0083-3
Zhao, L., Wang, G., Siegel, P., He, C., Wang, H., Zhao, W., et al. (2013). Quantitative genetic background of the host influences gut microbiomes in chickens. Sci. Rep. 3:1163. doi: 10.1038/srep01163
Keywords: gut microbiota, symbionts, birds, zebra finch, Bengalese finch, host-specific factors, inter-individual differences, temporal stability
Citation: Maraci Ö, Antonatou-Papaioannou A, Jünemann S, Castillo-Gutiérrez O, Busche T, Kalinowski J and Caspers BA (2021) The Gut Microbial Composition Is Species-Specific and Individual-Specific in Two Species of Estrildid Finches, the Bengalese Finch and the Zebra Finch. Front. Microbiol. 12:619141. doi: 10.3389/fmicb.2021.619141
Received: 19 October 2020; Accepted: 25 January 2021;
Published: 19 February 2021.
Edited by:
David William Waite, Ministry for Primary Industries, New ZealandReviewed by:
Alice Risely, Deakin University, AustraliaVeronika Gvozdikova Javurkova, Institute of Vertebrate Biology (ASCR), Czechia
Copyright © 2021 Maraci, Antonatou-Papaioannou, Jünemann, Castillo-Gutiérrez, Busche, Kalinowski and Caspers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Öncü Maraci, b25jdS5tYXJhY2lAdW5pLWJpZWxlZmVsZC5kZQ==