 Yi-ting Wang1†Rui-xin Shen1,2†Dan Xing1†Chen-pei Zhao1,3He-ting Gao1,4Jia-hong Wu2Ning Zhang1Heng-duan Zhang1Yan Chen2Tong-yan Zhao1*
Yi-ting Wang1†Rui-xin Shen1,2†Dan Xing1†Chen-pei Zhao1,3He-ting Gao1,4Jia-hong Wu2Ning Zhang1Heng-duan Zhang1Yan Chen2Tong-yan Zhao1* Chun-xiao Li1*
Chun-xiao Li1*- 1State Key Laboratory of Pathogen and Biosecurity, Beijing Key Laboratory of Vector Borne and Natural Focus Infectious Disease, Institute of Microbiology and Epidemiology, Beijing, China
- 2School of Basic Medical Sciences, Guizhou Medical University, Guiyang, China
- 3College of Life Sciences, Ludong University, Yantai, China
- 4College of Life Science and Technology, Beijing University of Chemical Technology, Beijing, China
Midgut microbiota can participate in the detoxification and metabolism processes in insects, but there are few reports on the relationship between midgut microbiota and insecticide resistance in mosquitoes. In this study, we performed metagenomic sequencing on a susceptible strain (SS), a field-collected Hainan strain (HN), and a deltamethrin-resistant strain (RR) of Culex pipiens quinquefasciatus to understand the diversity and functions of their midgut microbiota. The results revealed differences in midgut microbiota among the three strains of Cx. pipiens quinquefasciatus. At the phylum level, Proteobacteria was the most prominent, accounting for nearly 70% of their midgut microbes. At the genus level, Aeromonas made up the highest proportion. In addition, Aeromonas, Morganella, Elizabethkingia, Enterobacter, Cedecea, and Thorsellia showed significant differences between strains. At the species level, Bacillus cereus, Enterobacter cloacae complex sp. 4DZ3-17B2, Streptomyces sp. CNQ329, and some species of Pseudomonas and Wolbachia were more abundant in the two resistant strains. Principal component analysis (PCA) showed that the SS strain had significantly different metagenomic functions than the two deltamethrin-resistant strains (HN and RR strain). The HN and RR strains differed from the SS strain in more than 10 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The analysis of species abundance and functional diversity can provide directions for future studies.
Introduction
Microorganisms are an important part of the ecosystem and are intimately involved in the physiology of plants, animals, and humans (Engel and Moran, 2013). Insects are the most diverse animal group known on earth. Insects can maintain a population advantage in a complex ecological environment and depends largely on the various symbiotic microorganisms in their bodies (Tang et al., 2012). The microorganisms in insects mainly reside in their midgut and include bacteria, fungi, viruses, and archaea. These symbiotic microorganisms directly or indirectly regulate the growth, reproduction, immune defense, and other physiological activities of insects (Crotti et al., 2012). The midgut microbiota is divided into autochthonous and allochthonous communities. The autochthonous communities are the main microbes involved in the physiological functions of insects.
Recently, studies have shown that there is a relationship between insect midgut symbiotic bacteria and its resistance to insecticides in insects (Xia et al., 2013; Kach et al., 2018). Midgut microbiota mediate resistance mainly in two ways. One is the direct metabolism of chemical insecticides by symbiotic bacteria. Kikuchi et al. (2012) found that Burkholderia mediated the resistance of Riptortus pedestris to an organophosphorus insecticide, fenitrothion, by directly metabolizing the insecticide. Citrobacter sp. CF-BD plays a key role in the degradation of trichlorfon by Bactrocera dorsalis (Cheng et al., 2017). In addition, midgut microbiota indirectly affect insect resistance to insecticides by increasing the activity of detoxification enzymes or enhancing their gene expression. Pang et al. (2018) found that the symbiotic bacterium Arsenophonus strain (S-type) enhanced the resistance of Nilaparvata lugens to imidacloprid by increasing this insect’s P450s enzyme activity and UGT gene expression.
Culex pipiens quinquefasciatus is an important mosquito vector throughout the world. This species can transmit a variety of vector-borne infectious diseases, including Bancroft’s filariasis and Japanese encephalitis, and is a potential vector for West Nile virus and Western equine encephalitis virus (Nuss et al., 2018; Xu et al., 2019). Controlling mosquitoes is the main way to prevent and control mosquito-borne infectious diseases (Liu, 2015). At present, chemical control is the most important measures for the prevention and control of Cx. pipiens quinquefasciatus. However, due to the long-term use of large quantities of chemical insecticides, the insecticide resistance of Cx. pipiens quinquefasciatus has become increasingly prominent (Reid et al., 2018), and the extensive use of insecticides has also had a great impact on the environment. Deltamethrin is the most widely used pyrethroid in the world. The resistance of mosquitoes to it has become a very serious problem (Liu, 2015).
Most previous studies on mosquito resistance to insecticides have focused on their insecticide resistance genes. In recent years, the relationship between the insecticide resistance of mosquitoes and their symbiotic bacteria has attracted increasing attention. Symbiotic bacteria can enhance the metabolic activity of Anopheles stephensi in response to organophosphate insecticides, thus promoting resistance to these insecticides (Soltani et al., 2017). Hong et al. (2014) found that the expression of Wolbachia was higher in the deltamethrin-resistant strains of Cx. pipiens pallens, showing that Wolbachia is related to deltamethrin resistance in mosquitoes. Dada et al. (2018) reported that in fenitrothion-resistant strains of An. albimanus, metagenomic sequencing revealed enrichment of Klebsiella pneumoniae. These results showed that there is a certain correlation between the midgut microbiota and the insecticide resistance of mosquitoes.
For many years, the only way to identify microorganisms in the natural environment was to separate and cultivate them with traditional methods. However, due to technical limitations, traditional methods of separation and culturing can no longer meet the requirements of current research on insect midgut microbiota. With the rapid development of high-throughput sequencing, metagenomic sequencing has shown great advantages in the study of midgut microbiota (Mou et al., 2008). Using metagenomic sequencing, we can analyze and study the population diversity, biological activity, and functional roles of midgut microbiota. In this study, a susceptible strain (SS), a field Hainan strain (HN), and a deltamethrin-resistant strain (RR) of Cx. pipiens quinquefasciatus were selected for metagenomic sequencing. The microbial diversity and metabolic functions of different strains were analyzed in order to clarify the relationship between the symbiotic midgut microbiota of Cx. pipiens quinquefasciatus and its resistance to deltamethrin and to provide a new target for the biological control of Cx. pipiens quinquefasciatus.
Materials and Methods
Mosquito Strains
Three strains of Cx. pipiens quinquefasciatus were used in this study. The SS strain is a laboratory strain, which was originally from Guangzhou and has been kept in the laboratory for more than 10 years without exposure to any insecticides. The Hainan strain (HN strain) was collected from Haikou City, Hainan Province, in 2013, and was reared in the laboratory until this study. The RR strain was obtained by exposing the HN strain to deltamethrin in the laboratory for 30 generations. The three strains have been kept in the same laboratory and given the same feeding regimes. The mosquitoes were reared at 26 ± 1°C, 75 ± 5% relative humidity, and a light/dark schedule of 14 h:10 h. The adult mosquitoes from the three strains were fed with 8% sugar water and 3–5 days after emergence, fed with blood meal to breed the next generation. Before metagenomic sequencing, the larvae of these three strains were bioassayed, and the LC50 value for deltamethrin of the SS, HN, and RR strains were 0.0000029, 0.014, and 0.572 μg/ml, respectively. The LC50 values of the HN strain and RR strain are much higher than that of SS strain (Shen et al., 2020).
DNA Extraction of Mosquito Midgut
The midguts of the mosquitoes were removed from adult female Cx. pipiens quinquefasciatus at 3–5 days after emergence. Seventy female Cx. pipiens quinquefasciatus were rinsed with 75% ethanol for 90 s and then rinsed with sterile water three times. Each mosquito midgut was gently pulled out under a dissecting microscope, excess tissue was removed, and the midgut was rinsed with 1 × phosphate-buffered saline (PBS), placed in 100 μ l 1 × PBS, and frozen at −80°C. Three replicates were performed. The microbial genomic DNA of the sample was extracted with the QIAamp DNA Microbiome Kit (Shanghai, China). The concentration, total amount, purity, and degradation of the samples were tested, and the samples were then sent to Biomark Technologies (Beijing, China) for metagenomic sequencing.
Library Construction and Sequencing
After the genomic DNA of the sample passed the quality test, the DNA was fragmented by mechanical shearing (ultrasonication). Then, the fragmented DNA was purified, end-repaired, 3′-adenylated, ligated to the sequencing adapters, and then electrophoresed in agarose gel to perform fragment size selection. PCR amplification was conducted to form a sequencing library. The constructed library was first subjected to library quality inspection, and the Illumina sequencing platform was used for metagenomic sequencing of the qualifying library.
Bioinformatics Analysis
Quality control and host filtering were conducted on the original reads obtained by sequencing. After obtaining valid data (clean reads), the metagenome was assembled, and the unused reads from each sample were combined to form a mixed assembly to detect low-abundance species in the samples. Next, gene prediction, species annotation, and common-function database annotation were conducted. Taxonomic analysis was conducted using clean reads, and the species composition and abundance information of the samples were calculated.
Statistical Analysis
The metagenomeSeq of R software (Segata et al., 2011) was used to screen different species at the genus and species level, the DESeq2 package in R software was used to screen different KEGG Orthology (KO) pathways, and we used | Log2FoldChange| ≥ 2, padj ≤ 0.05 as a condition to screen different species and KO pathways. Then, we used the R package pheatmap to draw the heat map of the KO pathways. According to the previous literature, we identified the species and KO pathways related to resistance. We used STAMP software to screen the different species and pathways according to the principal component analysis (PCA) method and Welch’s t test (two-side) method under the condition of p < 0.05. After picking out the relevant species and functional pathways, we drew the extended error bar diagram. SPSS Statistics 21 was used to perform ANOVA, Dunnett t test was used to analyze the abundance of microbiota, the p value was Bonferroni corrected, and p < 0.05 was considered statistically significant.
Results
Basic Statistics
Metagenomic sequencing of the SS, HN, and RR strains was conducted on the Illumina sequencing platform. After host sequence removal and quality control, the sequencing depths of nine samples were all approximately 10G. The number of reads of the nine samples was between 33,090,131 and 39,863,554. The Q20 values were all above 96%, the Q30 values were all above 90%, and the N50 values were all above 900 bp, indicating that the gene assembly had high integrity and good quality, ensuring the credibility of the subsequent analysis (Supplementary Table 1).
A total of 3,807 species, including 95 Archaea, 3,362 Bacteria, 1 Eukaryota, 780 Fungi, 4 Metazoa, and 157 Viruses (Supplementary Table 2), were annotated by the sequencing. Among them, 3,398 (89.26%) were shared by all three strains. There were 174, 24, and 13 species unique to the midguts of the SS, HN, and RR strain, respectively, accounting for 4.57, 0.63, and 0.34% of the total number of species, respectively. Sixty-six species were shared by the HN strain and the RR strain but not the SS strain, accounting for 1.73% of the total number of species (Figure 1). The species accumulation curve revealed that as the sample size increased, the curve rose sharply, indicating that many species were discovered, and then, the curve tended to be flattened, indicating that the number of species had reached saturation and that we had enough samples to cover most of the species (Figure 2).
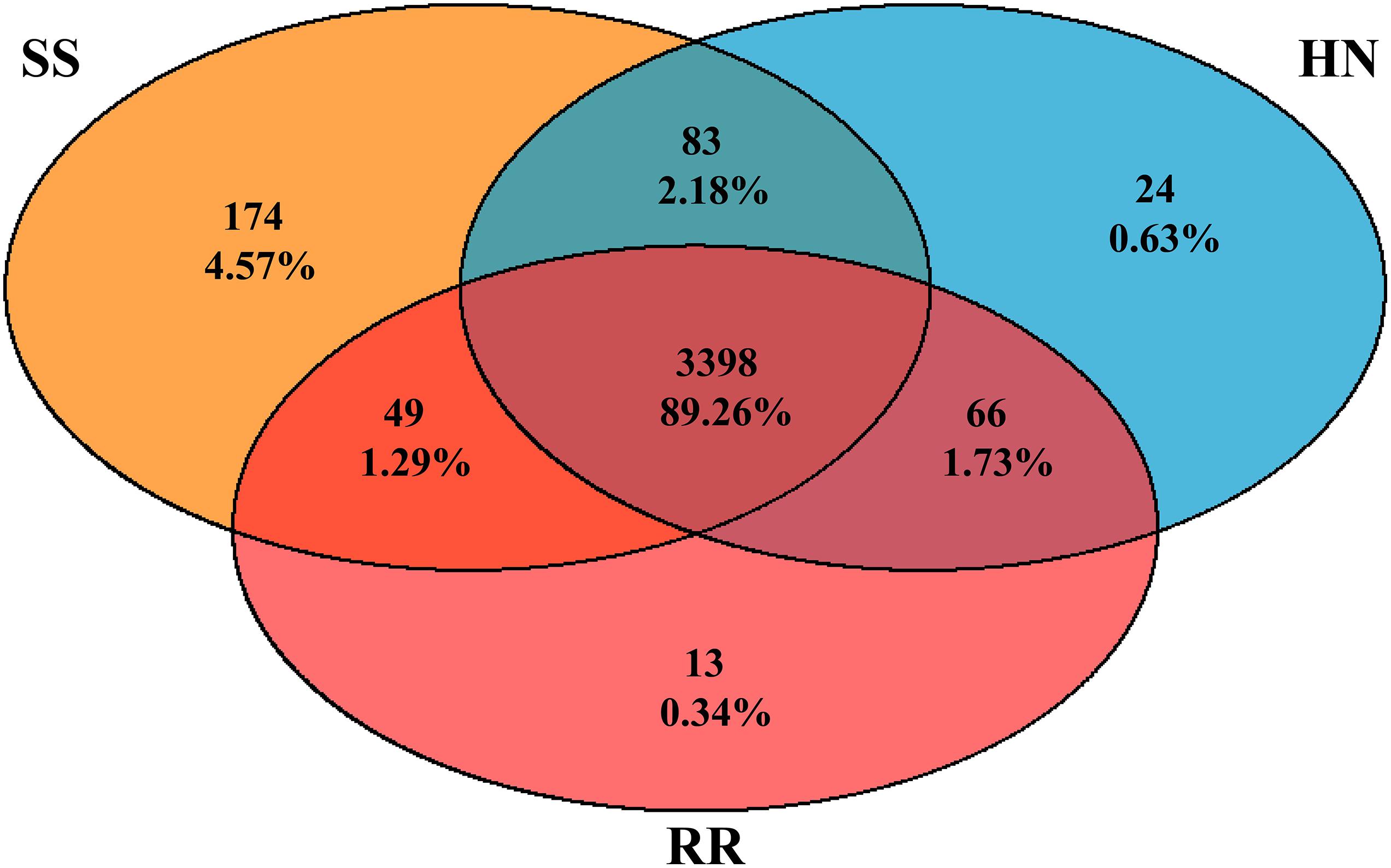
Figure 1. Venn diagram of the number of shared and unique bacteria genera among the microbiota of the SS strain (orange), the HN strain (blue), and the RR strain (red).
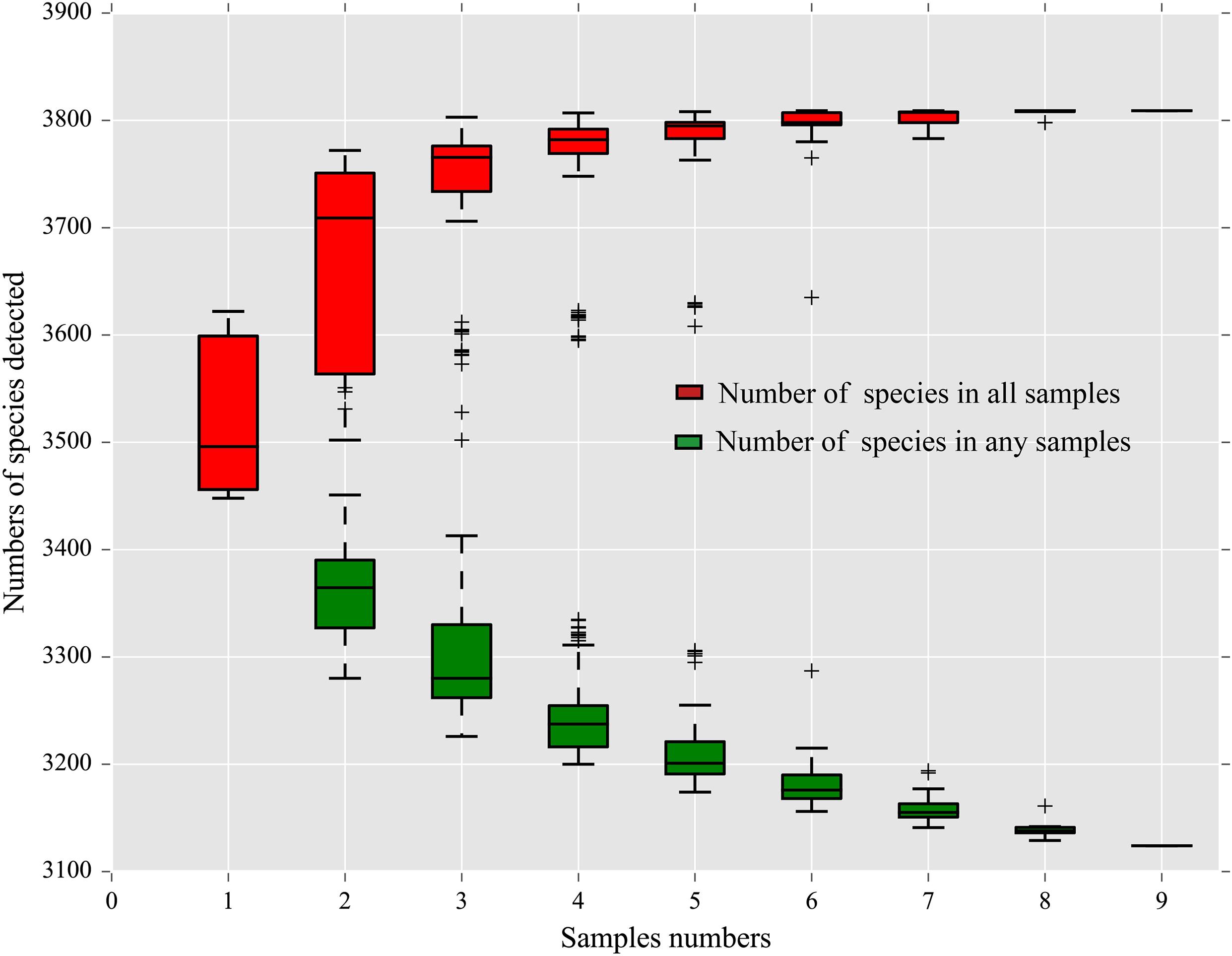
Figure 2. Box plot of species accumulation curves.
The Composition of Midgut Microbiota in Cx. pipiens quinquefasciatus
At the phylum level (Supplementary Figure 1), the top 10 phyla accounted for more than 95% of all phyla, including five bacterial phyla (Proteobacteria, Bacteroidetes, Actinobacteria, Firmicutes, and Planctomycetes) and five fungal phyla (Mucoromycota, Ascomycota, Chytridiomycota, Basidiomycota, and Zoopagomycota). Among them, Proteobacteria predominated, accounting for nearly 70% of the microbiota phyla. There was no significant difference among strains at the phylum level. At the genus level, the top 30 genera accounted for more than 77%; among them, Aeromonas dominated, accounting for nearly 20%; the six genera Asaia, Morganella, Elizabethkingia, Wolbachia, Enterobacter, and Serratia each accounted for 5–10%; and the remaining genera each accounted for <2% (Figure 3A). At the species level, the top 30 species accounted for more than 55%, of which Aeromonas veronii had the highest abundance, accounting for nearly 14%; Morganella morganii and Enterobacter sp. Ag1 were also relatively rich, accounting for approximately 6 and 5%, respectively. The remaining species accounted for <5% (Figure 3B).
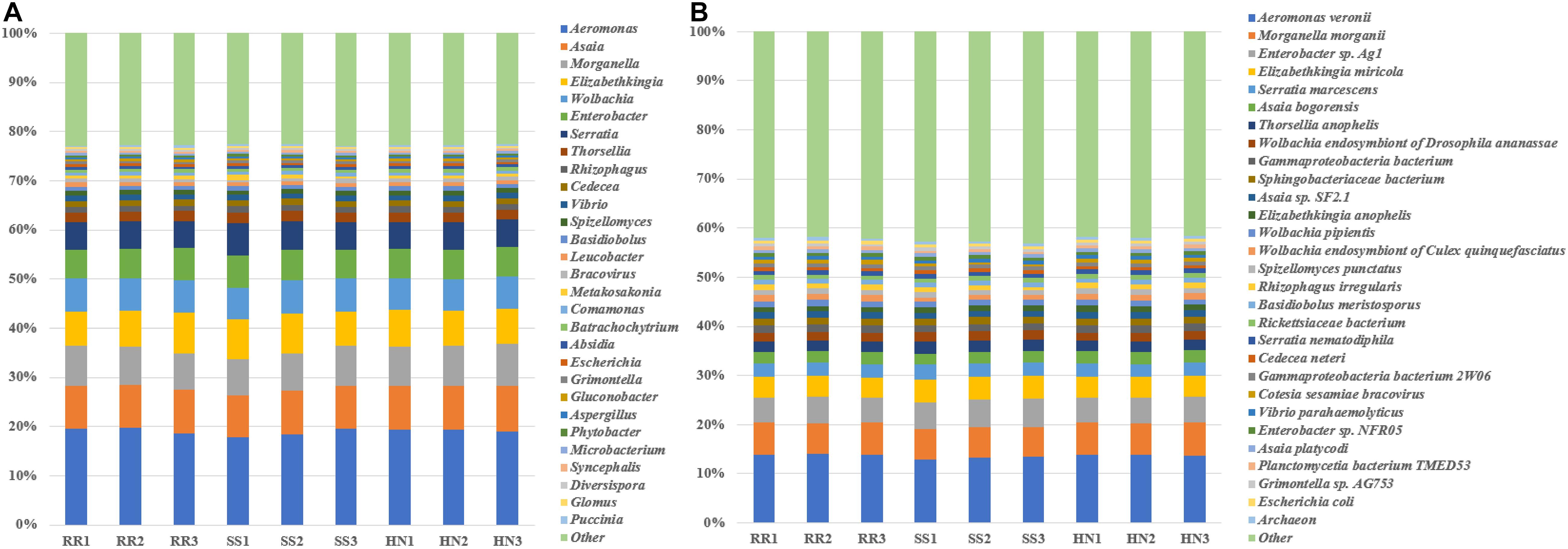
Figure 3. The taxonomic composition of the dominant microorganisms of the three strains of Cx. pipiens quinquefasciatus. (A) The average taxonomic composition of the dominant genera of the three strains of Cx. pipiens quinquefasciatus. (B) The average taxonomic composition of the dominant species of the three strains of Cx. pipiens quinquefasciatus.
At the genus level, we conducted a difference analysis for species with relatively high abundance; Aeromonas and Morganella were more abundant in the HN strain and the RR strain than in the SS strain, while Elizabethkingia, Enterobacter, and Thorsellia were significantly more abundant in the SS strain (p < 0.01). The levels of Wolbachia, Serratia, and Asaia in the three strains were not significantly different (Figure 4).
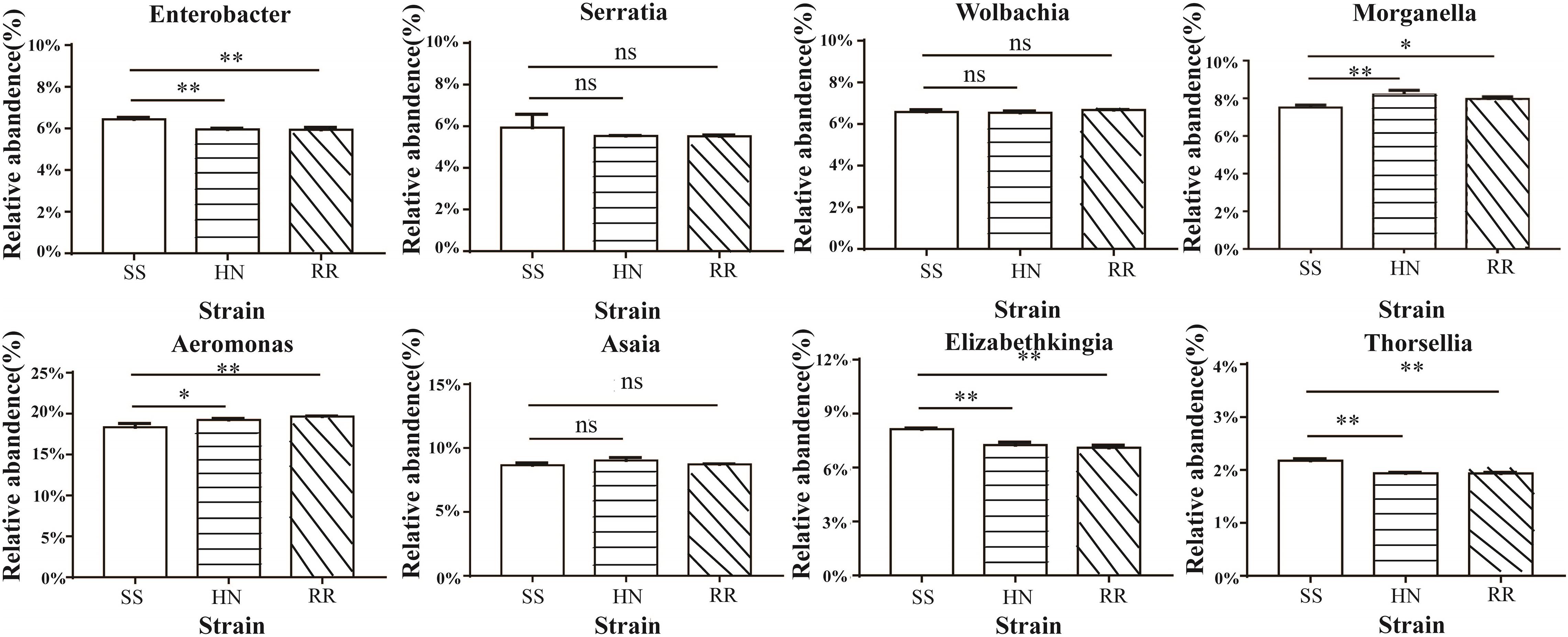
Figure 4. Histogram of midgut microbiota in the SS, HN, and RR strains of Cx. pipiens quinquefasciatus (*p < 0.05, **p < 0.01; ns, no significant difference).
The metagenomeSeq tool was used for screening at the genus level. The results showed that the two deltamethrin-resistant strains (HN and RR) had eight microbial genera that were more abundant than they were in strain SS: Acaricomes, Desertifilum, Claussenomyces, Oleiagrimonas, Citromicrobium, Mizugakiibacter, Aciduliprofundum, and Chitinimonas. The HN and RR strains had 10 microbial genera with lower abundance than in the SS strain, including Flexivirga, Thalassotalea, Porphyromonas, Phormidesmis, Methylomicrobium, Verminephrobacter, Basfia, Palleronia, Halogeometricum, and Candidatus Thioglobus (Supplementary Table 3).
The metagenomeSeq tool was also used for screening at the species level, and it showed that strains HN and RR had 17 species with greater abundance than in the SS strain, including Aciduliprofundum boonei, Desertifilum sp. IPPAS B-1220, Phycisphaerales bacterium, Phialocephala subalpina, Claussenomyces sp., Oleiagrimonas soli, and Arthrobacter luteolus. The HN and RR strains had 31 microorganisms with lower abundance than in the SS strain, including Citrobacter braakii, Citrobacter amalonaticus, Streptococcus mitis, Bacteroides thetaiotaomicron, Paenibacillus thiaminolyticus, Sphingomonas haloaromaticamans, Halogeometricum borinquense, and Proteus penneri (Supplementary Table 4).
At the species level, PCA was conducted for abundance analysis. The results showed that there were significant differences between the SS strain and the HN strain as well as between the SS strain and the RR strain (Figure 5). PCA1 explained 95.8% of the variance. Based on Welch’s t test, comparing the RR strain and the SS strain, a total of 1,172 species with significant differences were screened (p < 0.05). Among them, 598 strains were more abundant in the RR strain, and the remaining 574 strains were more abundant in the SS strain. Comparing the HN strain and the SS strain, a total of 1,007 strains with significant differences were screened (p < 0.05). Among them, 491 strains were more abundant in the HN strain, and the remaining 516 strains were more abundant in the SS strain. Among the above-mentioned different bacteria, we searched through the literature to screen out strains with large differences between the mosquito strains. The strains that were upregulated (p < 0.05) in the two resistant strains were Pseudomonas (Pseudomonas sp. 57B-090624, Pseudomonas sp. FeS53a, Pseudomonas sp. M30-35, Pseudomonas luteoia), Aspergillus (Aspergillus calidoustus, Aspergillus nidulans, Aspergillus sclerotialis, Wolbachia endosymbiont of Culex quinquefasciatus, Streptomyces sp. JS01, Bacillus sp. FJAT-42F376, Bacillus sp. 14578, Bacillus sp. FJAT-29814), Flavobacterium sp. JRM, Acinetobacter sp. NIPH 236, E. cloacae complex sp. 4DZ3-17B2, and Acidomonas (Acidomonas methanolica). The strains that were downregulated (p < 0.05) in the two resistant strains include Bacteroides caccae, Streptomyces violens, Oscillatoria acuminate, Microbotryum intermedium, Pseudovibrio sp. Ad46, Phaeomoniella chlamydospora, Rahnella woolbedingensis, and Fibrisoma limi. The strains only upregulated in the RR strain were Aspergillus (Aspergillus lentulus), Wolbachia pipientis, Streptomyces exfoliates, Streptomyces chattanoogensis, Bacillus enclensis, B. cereus, Bacillus megaterium, Flavobacterium sp. CJ74, Citrobacter sp. MH181794, and Stenotrophomonas rhizophila. The strains only upregulated in the HN strain were P. pachastrellae, Streptomyces sp. CNQ329, Bacillus sp. FJAT-18017, Flavobacterium johnsoniae, intestinal Enterobacter cloacae complex sp. CH23B, and Sphingobacterium sp. 1.A.5 (Figure 6).
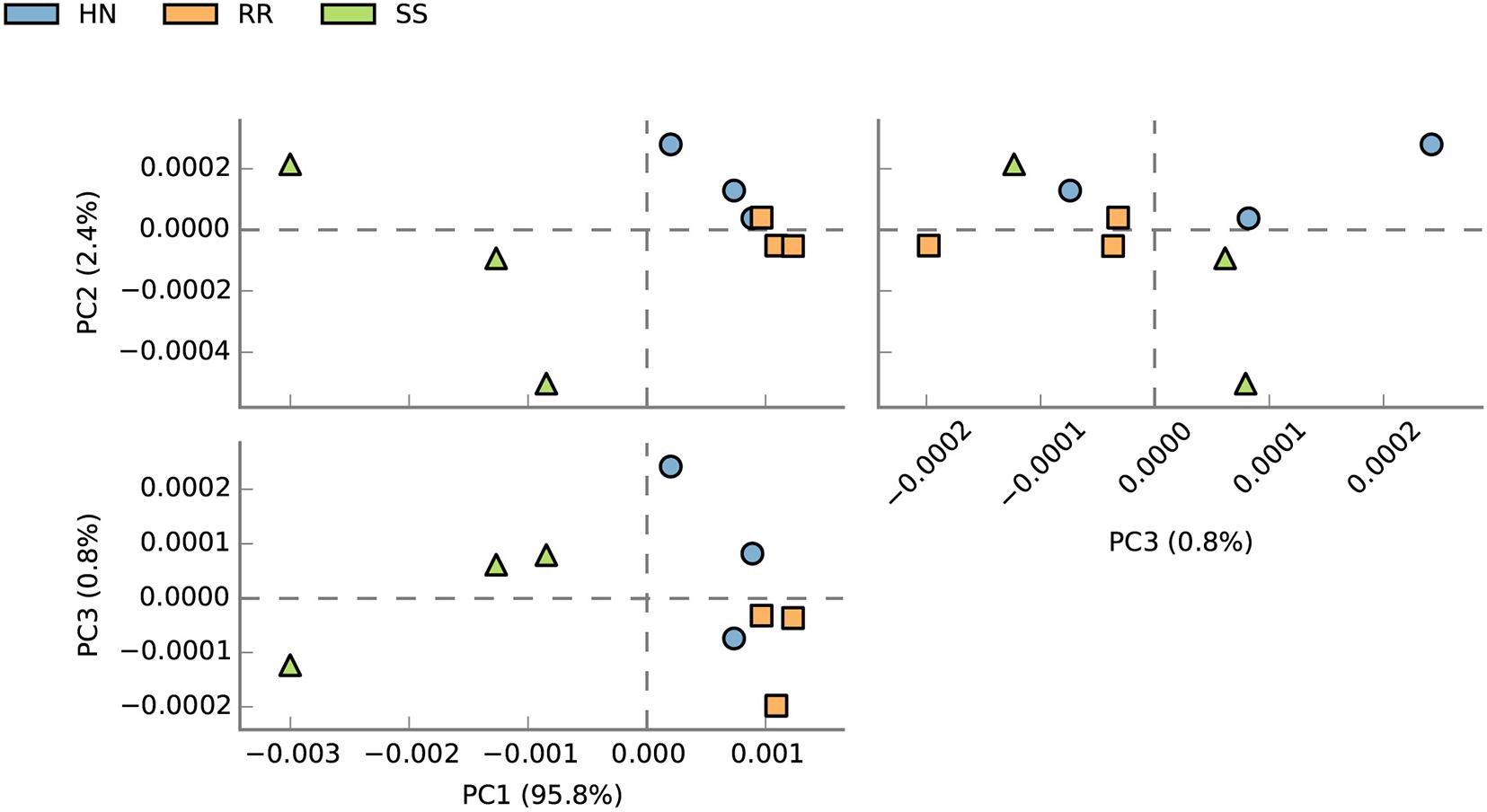
Figure 5. Principal component analysis (PCA) of the abundance of midgut microbial species in the SS, HN, and RR strains of Cx. pipiens quinquefasciatus.
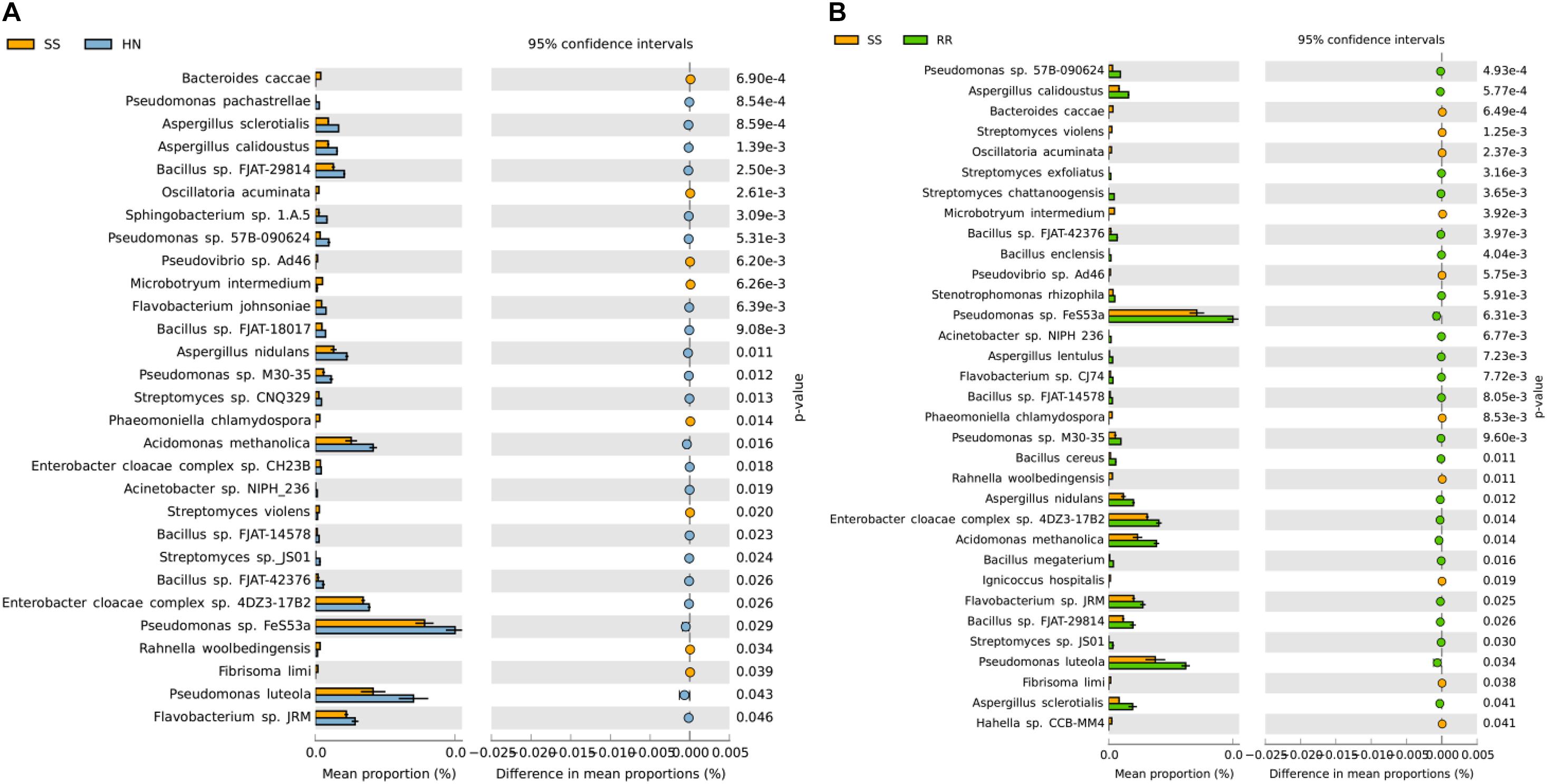
Figure 6. The differentially expressed bacteria screened by Welch’s t test. (A) Comparative analysis of the relative abundance of species between the SS strain and the HN strain. (B) Comparative analysis of the relative abundance of species between the SS strain and the RR strain.
Species Random Forest Regression and Prediction Analysis
Random forest analysis can efficiently and quickly select the most important species for classification (Figure 7). We selected the 30 most important species at the species level for analysis and found that the 10 most important species were Cotesia congregata bracovirus, Melampsora larici-populina, F. bacterium A100, Mamestra configurata nucleopolyhedrovirus B, Candidatus Pacearchaeota archaeon, Aeromonas sp. L1B53, Asaia sp. SF2.1, Deltaproteobacteria bacterium, M. caseolyticus, and UR2 sarcoma virus. By querying the correspondence between species and function, we found that the annotation results of M. caseolyticus were related to the functions of P450 enzymes and glutathione S-transferase. The annotation results of Candidatus Pacearchaeota archaeon and Kluyvera sp. Nf5 were related to the functions of sodium-dependent transport and insecticide metabolism enzymes, respectively. F. bacterium A100 is a bacterium belonging to the genus Flavobacteria, and the specific function of the annotation result is not yet clear. Serratia sp. FDAARGOS506 and P. syringae belong to the genera Serratia and Pseudomonas, respectively, and were very important for the classification of the three strains. Other species may be involved in some life processes and may be related to the functions of enzymes involved in the life activities of mosquitoes, such as nucleic acid synthesis, molting, information processing, and synthesis of amino acids and proteins.
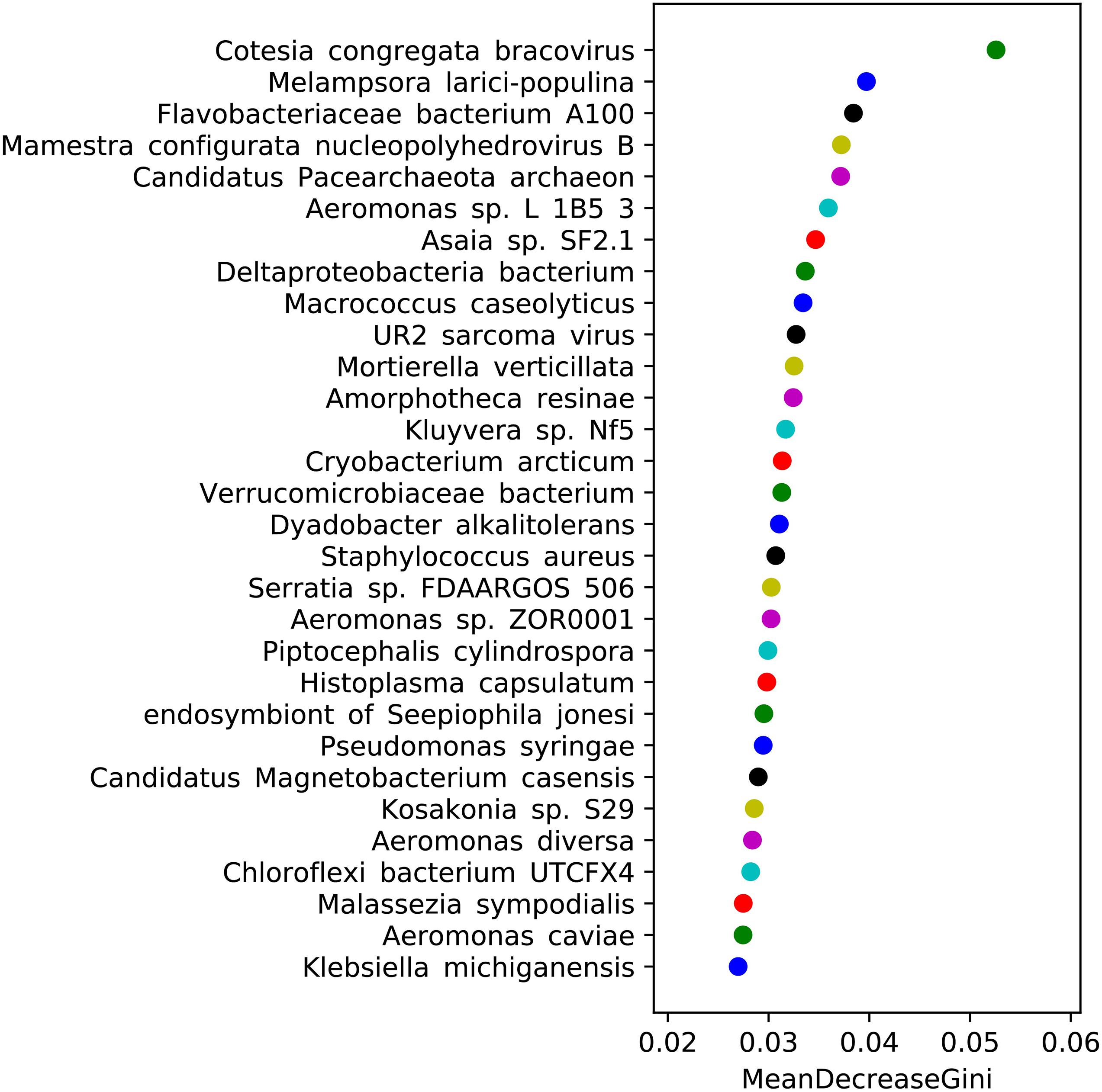
Figure 7. Species importance ranking map produced by random forest analysis. (The abscissa is the measure of species importance, and the ordinate is the species name sorted by importance).
Metagenomic Functional Analysis
Analysis of genes of the differentially expressed microbiota revealed that the annotated genes of microbiota with higher abundance in the deltamethrin-resistant strains primarily coded for enzymes involved in biological processes, such as bilirubin, nucleic acid, amino acid, and glyoxylic acid cycle processes and transmembrane protein synthesis. The upregulation of these enzymes increases the activity of these processes, which, in turn, provide more nutrients and energy to the mosquitoes. In addition, the annotation results of the microbiota with reduced abundance in the deltamethrin-resistant strains were related to the functions of proteins or enzymes such as ATP-binding proteins, ribosomal proteins, transfer proteins, transport proteins and transferases, ligases, phosphatases, oxidase, reductase, dehydrogenase, peptidase, and hydrolase (Supplementary Table 3).
STAMP software was used to analyze the sequencing results, and the relative abundances of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways related to midgut microbiota in the SS, HN, and RR strains were obtained (Supplementary Table 5). PCA showed that the SS strain had a different functional spectrum than the two deltamethrin-resistant strains: the SS strain had significant separation characteristics and a PCA1-explained variance of 90.1% (Figures 8A–C). Welch’s t-test (p < 0.05) was used to identify the KEGG pathways with significant differences between the two deltamethrin-resistant strains and the SS strain, including the metabolic and synthetic pathways involved in transcription, translation signal transduction, folding, classification, and cell motility. Among them, pathways related to substance synthesis, transportation, and catabolism were more abundant in the two deltamethrin-resistant strains, while pathways involved in the metabolism of amino acids, nucleotides, coenzymes, and vitamins were more abundant in the SS strain.
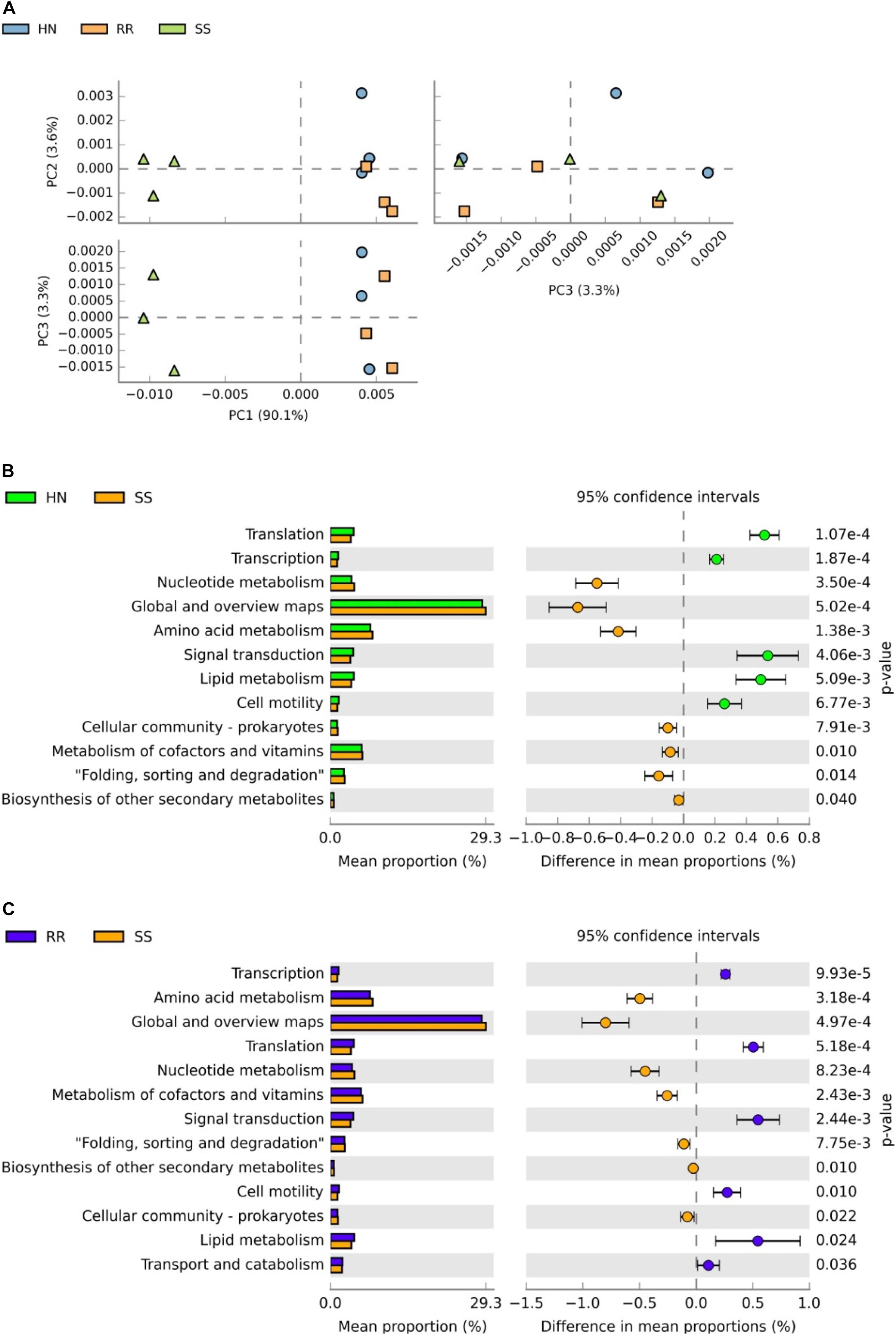
Figure 8. Analysis of differentially expressed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. (A) Principal component analysis of KEGG pathways in the three mosquito strains. (B) Comparative analysis of the relative abundance of KEGG pathways between the SS strain and HN strain. (C) Comparative analysis of the relative abundance of KEGG pathways.
The DESeq2 package was used to screen and obtain the heat map of the differentially expressed KO pathways (Figure 9A and Supplementary Table 6). The heat map showed that the identified microbiota of each strain had good reproducibility, and color comparison showed that the two deltamethrin-resistant strains (HN and RR) were very different from the SS strain. Based on the detoxification enzymes related to deltamethrin resistance reported in the literature (Figure 9B), including glutathione S-transferase (K00799, K07393, and K11208) (Li et al., 2018) and cytochrome P450s (K07408, K07418, K12664, and K15001) (Chandor-Proust et al., 2013), we selected the different KO pathways. In the HN and RR strains, the KO pathways K07393, K07418, and K15001were more abundant than in the SS strain, putatively recognized as the glutathione S-transferase, cytochrome P450 family 2 subfamily J, and cytochrome P450 family pathways, respectively. Pathways K00799, K11208, K07408, and K12664 were less abundant in the HN and RR strains than in the SS strain, representing the glutathione S-transferase, GST-like protein, cytochrome P450 family 1 subfamily A polypeptide 1, and cytochrome P450 family 26 subfamily B pathway, respectively. K00799, K07393, K07418, and K12664 had significantly different abundances in the SS strain than in the two deltamethrin-resistant strains (p < 0.05), while the abundance of the K15001 pathway differed only between the SS and RR strains.
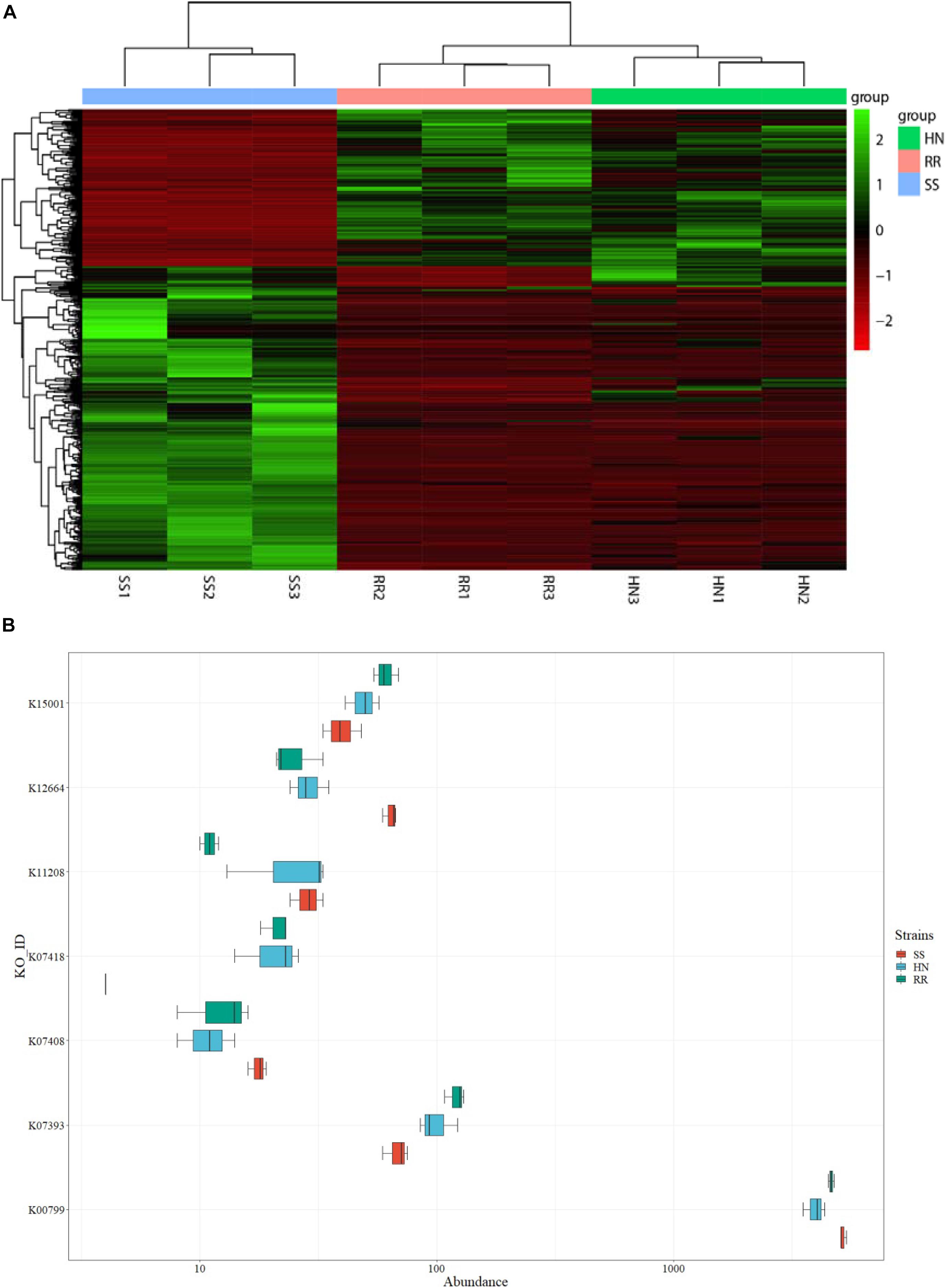
Figure 9. Analysis of differentially expressed Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) pathways in the three strains of Cx. pipiens quinquefasciatus. (A) KO pathway heat map of all differentially expressed microorganisms. (B) Box plot of differentially expressed KO pathways.
Discussion
The composition of the mosquito midgut microbiota varies with the species, growth stage, and geographical area. Food and water are the main sources of midgut microbiota in mosquitoes and are also the main reasons for the differences in the composition of the midgut microbiota among different populations of mosquitoes. The three strains of Cx. pipiens quinquefasciatus in this study had been reared in the laboratory for many years; therefore, the consistency of their midgut microbiota was very high. In this situation, the microbiota identified by differential analysis would be more likely to be related to insecticide resistance.
Proteobacteria is the dominant phylum in Cx. pipiens quinquefasciatus, and Firmicutes is also found in Cx. pipiens quinquefasciatus (Rani et al., 2009; Gusmao et al., 2010; Boissiere et al., 2012). In this study, 76.4% of microorganisms sequenced by metagenomics were bacteria. Among them, Proteobacteria accounted for the largest proportion, at 70%, which is consistent with previous studies. The 10 genera accounting for the largest proportions of microorganisms in adult mosquitoes were Aeromonas, Asaia, Morganella, Elizabethkingia, Wolbachia, Enterobacter, Serratia, Thorsellia, Rhizophagus, and Cedecea. Asaia can synthesize vitamin B to provide nutrition for mosquitoes (Crotti et al., 2010; Chouaia et al., 2012), and Wolbachia can stimulate mosquitoes to produce reactive oxygen species to activate the immune system, leading to the production of antimicrobial peptides to fight viruses (Pan et al., 2012). The high levels of Enterobacter and Serratia are also notable. These genera can produce hemolytic enzymes and promote the digestion of food after blood sucking (Gusmao et al., 2010; Gaio Ade et al., 2011), and they play key roles in the growth, development, and life cycle of mosquitoes (Xia et al., 2013; Zhang et al., 2019).
Bacteria and fungi that play important roles in insecticide resistance in the life cycle of Cx. pipiens quinquefasciatus may be present throughout the lives of mosquitoes. A study of Cx. pipiens quinquefasciatus larvae found bacteria represented by Bacillus sp. and Pseudomonas sp. in the midgut as well as the fungus Aspergillus (Aspergillus sp.) and the actinomycete Streptomyces (Streptomyces sp.) (Vasanthi and Hoti, 1992). The above-mentioned bacteria and fungi were also found in the adult mosquitoes in the present study.
B. cereus (Zhang et al., 2016), Acidomonas sp. (Paingankar et al., 2005), Streptomyces aureus (Chen et al., 2011), P. fluorescens, Enterobacter cloacae (Hao et al., 2018), and Aspergillus niger (Liang et al., 2005) can degrade deltamethrin. Whether other species of Bacillus, Acidomonas sp., Pseudomonas sp., Streptomyces, Enterobacter sp., and Aspergillus play a role in metabolism needs to be investigated further. The metagenomic sequencing of this study found that B. cereus and E. cloacae complex sp. 4DZ3-17B2 in the RR strain was significantly more abundant than in the SS strain, indicating that they may metabolize deltamethrin in Cx. pipiens quinquefasciatus. The annotation result of Streptomyces sp. CNQ329 showed that it was a cytochrome P450 gene. P450s participate in the detoxification metabolism of deltamethrin. This study showed that the abundance of Streptomyces sp. CNQ329 in the HN strain was higher than that in the SS strain, suggesting that Streptomyces sp. CNQ329 may contribute to the resistance of Cx. pipiens quinquefasciatus to deltamethrin.
Tago et al. (2006) and Singh (2009) have shown that the application of fenitrothion sharply increases the abundance of Pseudomonas and Flavobacterium, which can degrade fenitrothion to 3-methyl-4-nitrophenol, a compound with no insecticide activity, and then further metabolize 3-methyl-4-nitrophenol into a carbon source for its growth (Kikuchi et al., 2012). Hong et al. (2014) found that Wolbachia was more abundant in deltamethrin-resistant strains of Cx. pipiens pallens, suggesting that Wolbachia contributes to deltamethrin resistance. Pseudomonas plays a role in the metabolism of organophosphates and pyrethroids (Zhang et al., 2018). This study showed that the abundance of a Wolbachia and four Pseudomonas strains in the two resistant mosquito strains was higher than that in the SS strains, and there were significant differences (p < 0.05), suggesting that the resistance of Cx. pipiens quinquefasciatus to deltamethrin may be closely related to the activity of Pseudomonas and Wolbachia. This finding shows that microorganisms not only have a metabolic effect on an insecticide and that the metabolism of the insecticides may furthermore require the synergistic effect of multiple microorganisms, and the development of insecticide resistance may also be the result of the synergistic effect of multiple mechanisms.
Random forest analysis helped us identify the 30 most important species in the Cx. pipiens quinquefasciatus midgut, whose corresponding annotated proteins were related to the classification of the three strains of mosquitoes. The three mosquito strains were basically the same except for their collection location and insecticide exposure conditions. Among these 30 species, Flavobacteriaceae bacterium A100 is a bacterium belonging to the genus Flavobacterium. Flavobacteria can degrade organophosphorus insecticides (Kikuchi et al., 2012). The annotation results of differentially expressed genes of M. caseolyticus revealed P450s and glutathione S-transferase. These two detoxification enzymes are associated with deltamethrin resistance (Chandor-Proust et al., 2013; Li et al., 2018). Therefore, we speculate that M. caseolyticus may contribute to the resistance of mosquitoes to deltamethrin. The annotation result of Candidatus Pacearchaeota archaeon revealed a sodium-dependent transporter, which may be related to Na+ ion channels. The pyrethroid resistance of mosquitoes is related to the insensitivity of sodium ion channels (Narahashi, 1988). Therefore, Candidatus Pacearchaeota archaeon may also participate in the production of deltamethrin resistance. The annotation result of Kluyvera sp. Nf5 revealed an enzyme related to insecticide metabolism, which may be involved in the metabolic process of insecticides or other substances. Serratia sp. FDAARGOS506 and P. syringae were also very important for the classification of the three strains. Serratia sp. and Pseudomonas can degrade insecticides (Xia et al., 2018). We speculate that these two bacteria may be involved in the deltamethrin resistance of Cx. pipiens quinquefasciatus.
The upregulated and downregulated bacterial strains were screened using the metagenomeSeq tool. Among them, A. boonei, an upregulated strain, can utilize many enzymes encoding proteolytic or peptide hydrolysis activity and use peptides in their main metabolic pathway (Reysenbach and Flores, 2008). Campoletis sonorensisichnovirus can change the growth, development, and immunity of its host (Fath-Goodin et al., 2006). The function of Phycisphaerales bacterium is to participate in the nitrogen and carbon cycles (Fukunaga et al., 2010). Mesorhizobium alhagi can use mannitol as a carbon source to produce many extracellular polysaccharides, which can affect cellular Na+ content and antioxidase activity (Liu et al., 2017). Among the downregulated bacteria, Kluyvera georgiana is attractive for its biodegradation of acrylamide and other aliphatic amides in the environment (Thanyacharoen et al., 2012). C. amalonaticus can utilize multiple carbon sources and can use sucrose as a substrate to achieve high succinic acid production, indicating that reduced carbon substrates help maximize the redox potential (Amulya and Mohan, 2019). B. thetaiotaomicron digests a variety of complex carbohydrates and produces short-chain carbohydrates and organic acids that the host can absorb as energy sources (Porter et al., 2018). Paenibacillus thiaminolyticus can use phenol (Chandra et al., 2011), and S. haloaromaticamans can degrade o-phenol (Perruchon et al., 2017). The endosymbiotic bacterium Buchnera aphidicola is essential to the nutrient metabolism and normal development of the host (Nakabachi and Ishikawa, 1997). In general, these bacteria directly or indirectly provide the host with nitrogen sources, carbon sources, amino acids, vitamins, and energy and affect the host’s life activities.
After the analysis of differentially expressed KO pathways, the IDs were searched against the P450 database, and it was found that the K07408, K07418, K12664, and K15001 pathways were related to the CYP303, CYP18, CYP301, and CYP4 superfamilies, respectively. The CYP4 (K15001) superfamily is related to the development of deltamethrin resistance in mosquitoes (Chang et al., 2014). K15001 is only significantly different in the RR strain, which may be the reason why the RR strain is more resistant to deltamethrin than the HN strain. We speculate that the CYP4 superfamily of the midgut microbiota may play a role in the production of Cx. pipiens quinquefasciatus resistance to deltamethrin. Although the CYP303, CYP18, and CYP301 superfamilies have not been reported to be related to deltamethrin resistance, there were differences in the KO pathways between deltamethrin-resistant and susceptible strains, and attention should be given to these superfamilies in future research.
In this study, we identified mosquito midgut microbiota through metagenomic sequencing, annotated their functions, explored the relationships between some of the midgut microbiota and insecticide resistance, and identified some new bacteria, fungi, and viruses that may be related to this resistance. In future research, certain target midgut microbes should be cultivated or antibiotics could be applied to remove them from the mosquito midgut to further clarify their functions.
Data Availability Statement
The data presented in the study are deposited in the NCBI BioProject repository, accession number PRJNA680753.
Author Contributions
Y-tW, Rx-S, and DX conducted the analysis of the sequencing data. Y-tW and Rx-S wrote the manuscript. C-pZ participated in the analysis of processes and performed the software mapping work with H-tG. J-hW and H-dZ completed the manuscript and initial proofreading of the data. NZ participated in the preparation of the samples. YC, T-yZ, and C-xL reviewed and provided suggestions for the manuscript. All authors contributed to the writing and approved the final version of the manuscript.
Funding
This work was funded by grants from the National Science and Technology Major Project of China (No. 2017ZX10303404).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Biomarker Technologies for their valuable technical help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.625539/full#supplementary-material
Supplementary Figure 1 | The taxonomic composition of the dominant phyla of the three strains of Cx. pipiens quinquefasciatus.
Supplementary Table 1 | Statistics of metagenomic sequencing data.
Supplementary Table 2 | All microorganisms were annotated by the sequencing in Cx. pipiens quinquefasciatus.
Supplementary Table 3 | Different bacteria at the genus level screened by metagenomeSeq method.
Supplementary Table 4 | Different bacteria at the species level screened by metagenomeSeq method.
Supplementary Table 5 | The differentially expressed KEGG pathways in the three strains of Cx. pipiens quinquefasciatus.
Supplementary Table 6 | The differentially expressed KEGG Orthology (KO) pathways in the three strains of Cx. pipiens quinquefasciatus.
References
Amulya, K., and Mohan, S. V. (2019). Fixation of CO2, electron donor and redox microenvironment regulate succinic acid production in Citrobacter amalonaticus. Sci. Total Environ. 695:133838. doi: 10.1016/j.scitotenv.2019.133838
Boissiere, A., Tchioffo, M. T., Bachar, D., Abate, L., Marie, A., Nsango, S. E., et al. (2012). Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 8:e1002742. doi: 10.1371/journal.ppat.1002742
Chandor-Proust, A., Bibby, J., Regent-Kloeckner, M., Roux, J., Guittard-Crilat, E., Poupardin, R., et al. (2013). The central role of mosquito cytochrome P450 CYP6Zs in insecticide detoxification revealed by functional expression and structural modelling. Biochem. J. 455, 75–85. doi: 10.1042/bj20130577
Chandra, R., Yadav, S., Bharagava, R. N., and Rai, V. (2011). Phenol degradation by Paenibacillus thiaminolyticus and Bacillus cereus in axenic and mixed conditions. World J. Microbiol. Biotechnol. 27, 2939–2947. doi: 10.1007/s11274-011-0777-4
Chang, X., Zhong, D., Fang, Q., Hartsel, J., Zhou, G., Shi, L., et al. (2014). Multiple resistances and complex mechanisms of Anopheles sinensis mosquito: a major obstacle to mosquito-borne diseases control and elimination in China. PLoS Negl. Trop Dis. 8:e2889. doi: 10.1371/journal.pntd.0002967
Chen, S., Lai, K., Li, Y., Hu, M., Zhang, Y., and Zeng, Y. (2011). Biodegradation of deltamethrin and its hydrolysis product 3-phenoxybenzaldehyde by a newly isolated Streptomyces aureus strain HP-S-01. Appl. Microbiol. Biotechnol. 90, 1471–1483. doi: 10.1007/s00253-011-3136-3
Cheng, D., Guo, Z., Riegler, M., Xi, Z., Liang, G., and Xu, Y. (2017). Gut symbiont enhances insecticide resistance in a significant pest, the oriental fruit fly Bactrocera dorsalis (Hendel). Microbiome 5:13.
Chouaia, B., Rossi, P., Epis, S., Mosca, M., Ricci, I., Damiani, C., et al. (2012). Delayed larval development in Anopheles mosquitoes deprived of Asaia bacterial symbionts. BMC Microbiol. 12 (Suppl. 1):S2.
Crotti, E., Balloi, A., Hamdi, C., Sansonno, L., Marzorati, M., Gonella, E., et al. (2012). Microbial symbionts: a resource for the management of insect-related problems. Microb. Biotechnol. 5, 307–317. doi: 10.1111/j.1751-7915.2011.00312.x
Crotti, E., Rizzi, A., Chouaia, B., Ricci, I., Favia, G., Alma, A., et al. (2010). Acetic acid bacteria, newly emerging symbionts of insects. Appl. Environ. Microbiol. 76, 6963–6970. doi: 10.1128/aem.01336-10
Dada, N., Sheth, M., Liebman, K., Pinto, J., and Lenhart, A. (2018). Whole metagenome sequencing reveals links between mosquito microbiota and insecticide resistance in malaria vectors. Sci. Rep. 8:2084.
Engel, P., and Moran, N. A. (2013). The gut microbiota of insects – diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. doi: 10.1111/1574-6976.12025
Fath-Goodin, A., Gill, T. A., Martin, S. B., and Webb, B. A. (2006). Effect of Campoletis sonorensis ichnovirus cys-motif proteins on Heliothis virescens larval development. J. Insect. Physiol. 52, 576–585. doi: 10.1016/j.jinsphys.2006.02.005
Fukunaga, Y., Higashino, M., Tanimura, S., Takemura, M., Fujiwara, Y., and Osugi, H. (2010). Laparoscopic surgery for stage IV colorectal cancer. Surg. Endosc. 24, 1353–1359. doi: 10.1007/s00464-009-0778-7
Gaio Ade, O., Gusmao, D. S., Santos, A. V., Berbert-Molina, M. A., Pimenta, P. F., and Lemos, F. J. (2011). Contribution of midgut bacteria to blood digestion and egg production in aedes aegypti (diptera: culicidae) (L.). Parasit. Vect. 4:105. doi: 10.1186/1756-3305-4-105
Gusmao, D. S., Santos, A. V., Marini, D. C., Bacci, M. Jr., Berbert-Molina, M. A., and Lemos, F. J. (2010). Culture-dependent and culture-independent characterization of microorganisms associated with Aedes aegypti (Diptera: Culicidae) (L.) and dynamics of bacterial colonization in the midgut. Acta Trop. 115, 275–281. doi: 10.1016/j.actatropica.2010.04.011
Hao, X., Zhang, X., Duan, B., Huo, S., Lin, W., Xia, X., et al. (2018). Screening and genome sequencing of deltamethrin-degrading bacterium ZJ6. Curr. Microbiol. 75, 1468–1476. doi: 10.1007/s00284-018-1546-5
Hong, S. C., Yuan, L., Fang, F. J., and Zhu, C. L. (2014). Presumptive role of Wolbachia in deltamethrin resistance of Culex pipiens pallens. Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi 26, 672–674, 680.
Kach, H., Mathe-Hubert, H., Dennis, A. B., and Vorburger, C. (2018). Rapid evolution of symbiont-mediated resistance compromises biological control of aphids by parasitoids. Evol. Appl. 11, 220–230. doi: 10.1111/eva.12532
Kikuchi, Y., Hayatsu, M., Hosokawa, T., Nagayama, A., Tago, K., and Fukatsu, T. (2012). Symbiont-mediated insecticide resistance. Proc. Natl. Acad. Sci. U.S.A. 109, 8618–8622. doi: 10.1073/pnas.1200231109
Li, Y., Xu, J., Zhong, D., Zhang, H., Yang, W., Zhou, G., et al. (2018). Evidence for multiple-insecticide resistance in urban Aedes albopictus populations in southern China. Paras. Vec. 11:4.
Liang, W. Q., Wang, Z. Y., Li, H., Wu, P. C., Hu, J. M., Luo, N., et al. (2005). Purification and characterization of a novel pyrethroid hydrolase from Aspergillus niger ZD11. J. Agric. Food Chem. 53, 7415–7420. doi: 10.1021/jf051460k
Liu, N. (2015). Insecticide resistance in mosquitoes: impact, mechanisms, and research directions. Annu. Rev. Entomol. 60, 537–559. doi: 10.1146/annurev-ento-010814-020828
Liu, X., Luo, Y., Li, Z., Wang, J., and Wei, G. (2017). Role of exopolysaccharide in salt stress resistance and cell motility of Mesorhizobium alhagi CCNWXJ12-2(T). Appl. Microbiol. Biotechnol. 101, 2967–2978. doi: 10.1007/s00253-017-8114-y
Mou, X., Sun, S., Edwards, R. A., Hodson, R. E., and Moran, M. A. (2008). Bacterial carbon processing by generalist species in the coastal ocean. Nature 451, 708–711. doi: 10.1038/nature06513
Nakabachi, A., and Ishikawa, H. (1997). Differential display of mRNAs related to amino acid metabolism in the endosymbiotic system of aphids. Insect. Biochem. Mol. Biol. 27, 1057–1062. doi: 10.1016/s0965-1748(97)00092-1
Narahashi, T. (1988). Role of various ion channels in insecticide action. Abstr. Pap. Am. Chem. Soc. 196, 26-AGRO.
Nuss, A. B., Brown, M. R., Murty, U. S., and Gulia-Nuss, M. (2018). Insulin receptor knockdown blocks filarial parasite development and alters egg production in the southern house mosquito, Culex quinquefasciatus. PLoS Negl. Trop Dis. 12:e0006413. doi: 10.1371/journal.pntd.0006413
Paingankar, M., Jain, M., and Deobagkar, D. (2005). Biodegradation of allethrin, a pyrethroid insecticide, by an acidomonas sp. Biotechnol. Lett. 27, 1909–1913. doi: 10.1007/s10529-005-3902-3
Pan, X., Zhou, G., Wu, J., Bian, G., Lu, P., Raikhel, A. S., et al. (2012). Wolbachia induces reactive oxygen species (ROS)-dependent activation of the Toll pathway to control dengue virus in the mosquito Aedes aegypti. Proc. Natl. Acad. Sci. U.S.A. 109, E23–E31.
Pang, R., Chen, M., Yue, L., Xing, K., Li, T., Kang, K., et al. (2018). A distinct strain of Arsenophonus symbiont decreases insecticide resistance in its insect host. PLoS Genet 14:e1007725. doi: 10.1371/journal.pgen.1007725
Perruchon, C., Vasileiadis, S., Rousidou, C., Papadopoulou, E. S., Tanou, G., Samiotaki, M., et al. (2017). Metabolic pathway and cell adaptation mechanisms revealed through genomic, proteomic and transcription analysis of a Sphingomonas haloaromaticamans strain degrading ortho-phenylphenol. Sci. Rep. 7:6449.
Porter, N. T., Luis, A. S., and Martens, E. C. (2018). Bacteroides thetaiotaomicron. Trends Microbiol. 26, 966–967.
Rani, A., Sharma, A., Rajagopal, R., Adak, T., and Bhatnagar, R. K. (2009). Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi-an Asian malarial vector. BMC Microbiol. 9:96. doi: 10.1186/1471-2180-9-96
Reid, W. R., Zhang, L., Gong, Y., Li, T., and Liu, N. (2018). Gene expression profiles of the Southern house mosquito Culex quinquefasciatus during exposure to permethrin. Insect. Sci. 25, 439–453. doi: 10.1111/1744-7917.12438
Reysenbach, A. L., and Flores, G. E. (2008). Electron microscopy encounters with unusual thermophiles helps direct genomic analysis of Aciduliprofundum boonei. Geobiology 6, 331–336. doi: 10.1111/j.1472-4669.2008.00152.x
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60.
Shen, R. X., Wang, Y. T., Li, C. X., Wu, J. H., Zhao, T. Y., and Chen, Y. (2020). Analysis of deltamethrin resistance-related genes based on the transcriptome of Culex pipiens quinquefasciatus. Chin. J. Parasitol. Parasit. Dis. 38, 453–463.
Singh, B. K. (2009). Organophosphorus-degrading bacteria: ecology and industrial applications. Nat. Rev. Microbiol. 7, 156–164. doi: 10.1038/nrmicro2050
Soltani, A., Vatandoost, H., Oshaghi, M. A., Enayati, A. A., and Chavshin, A. R. (2017). The role of midgut symbiotic bacteria in resistance of Anopheles stephensi (Diptera: Culicidae) to organophosphate insecticides. Pathog. Glob. Health 111, 289–296. doi: 10.1080/20477724.2017.1356052
Tago, K., Yonezawa, S., Ohkouchi, T., Hashimoto, M., and Hayatsu, M. (2006). Purification and characterization of fenitrothion hydrolase from Burkholderia sp. NF100. J. Biosci. Bioeng. 101, 80–82. doi: 10.1263/jbb.101.80
Tang, X., Freitak, D., Vogel, H., Ping, L., Shao, Y., Cordero, E. A., et al. (2012). Complexity and variability of gut commensal microbiota in polyphagous lepidopteran larvae. PLoS One 7:e36978. doi: 10.1371/journal.pone.0036978
Thanyacharoen, U., Tani, A., and Charoenpanich, J. (2012). Isolation and characterization of Kluyvera georgiana strain with the potential for acrylamide biodegradation. J. Environ. Sci. Health A Tox. Hazard Subst. Environ. Eng. 47, 1491–1499.
Vasanthi, V., and Hoti, S. L. (1992). Microbial flora in gut of Culex quinquefasciatus breeding in cess pits. South. Asia. J. Trop. Med. Public Health 23, 312–317.
Xia, X., Sun, B., Gurr, G. M., Vasseur, L., Xue, M., and You, M. (2018). Gut microbiota mediate insecticide resistance in the diamondback moth, Plutella xylostella (L.). Front. Microbiol. 9:25.
Xia, X., Zheng, D., Zhong, H., Qin, B., Gurr, G. M., Vasseur, L., et al. (2013). DNA sequencing reveals the midgut microbiota of diamondback moth, Plutella xylostella (L.) and a possible relationship with insecticide resistance. PLoS One 8:e68852. doi: 10.1371/journal.pone.0068852
Xu, N., Sun, X. H., Liu, Z. H., Xu, Y., Sun, Y., Zhou, D., et al. (2019). Identification and classification of differentially expressed genes in pyrethroid-resistant Culex pipiens pallens. Mol. Genet. Genomics 294, 861–873. doi: 10.1007/s00438-018-1521-7
Zhang, H., Zhang, Y., Hou, Z., Wang, X., Wang, J., Lu, Z., et al. (2016). Biodegradation potential of deltamethrin by the Bacillus cereus strain Y1 in both culture and contaminated soil. Int. Biodeterior. Biodegrad. 106, 53–59. doi: 10.1016/j.ibiod.2015.10.005
Zhang, J. H., Yu, N., Xu, X. X., and Liu, Z. W. (2019). Community structure, dispersal ability and functional profiling of microbiome existing in fat body and ovary of the brown planthopper, Nilaparvata lugens. Insect. Sci. 26, 683–694. doi: 10.1111/1744-7917.12575
Keywords: Culex pipiens quinquefasciatus, metagenomics, gut microbiota, deltamethrin, resistance
Citation: Wang Y-t, Shen R-x, Xing D, Zhao C-p, Gao H-t, Wu J-h, Zhang N, Zhang H-d, Chen Y, Zhao T-y and Li C-x (2021) Metagenome Sequencing Reveals the Midgut Microbiota Makeup of Culex pipiens quinquefasciatus and Its Possible Relationship With Insecticide Resistance. Front. Microbiol. 12:625539. doi: 10.3389/fmicb.2021.625539
Received: 06 November 2020; Accepted: 25 January 2021;
Published: 25 February 2021.
Edited by:
Yoshitomo Kikuchi, National Institute of Advanced Industrial Science and Technology (AIST), JapanReviewed by:
Stefanie Christine Becker, University of Veterinary Medicine Hannover, GermanyMonica Rosenblueth, National Autonomous University of Mexico, Mexico
Copyright © 2021 Wang, Shen, Xing, Zhao, Gao, Wu, Zhang, Zhang, Chen, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tong-yan Zhao, dG9uZ3lhbnpoYW9AMTI2LmNvbQ==; Chun-xiao Li, YWVkZXNAMTI2LmNvbQ==
†These authors share first authorship