 Matthieu Pot1*
Matthieu Pot1* Yann Reynaud1
Yann Reynaud1 David Couvin1
David Couvin1 Célia Ducat1Séverine Ferdinand1
Célia Ducat1Séverine Ferdinand1 François Gravey2
François Gravey2 Gaëlle Gruel1François Guérin2,3Edith Malpote4
Gaëlle Gruel1François Guérin2,3Edith Malpote4 Sébastien Breurec1,5,6Antoine Talarmin1
Sébastien Breurec1,5,6Antoine Talarmin1 Stéphanie Guyomard-Rabenirina1
Stéphanie Guyomard-Rabenirina1- 1Transmission, Reservoir and Diversity of Pathogens Unit, Pasteur Institute of Guadeloupe, Les Abymes, France
- 2GRAM 2.0, Normandie University, UNICAEN, UNIROUEN, Caen, France
- 3Department of Clinical Microbiology, Caen University Hospital, Caen, France
- 4Laboratory of Clinical Microbiology, University Hospital of Guadeloupe, Pointe-à-Pitre/Les Abymes, France
- 5Faculty of Medicine Hyacinthe Bastaraud, University of the Antilles, Pointe-à-Pitre, France
- 6Centre for Clinical Investigation 1424, INSERM, Pointe-à-Pitre/Les Abymes, France
Species belonging to Enterobacter cloacae complex have been isolated in numerous environments and samples of various origins. They are also involved in opportunistic infections in plants, animals, and humans. Previous prospection in Guadeloupe (French West Indies) indicated a high frequency of E. cloacae complex strains resistant to third-generation cephalosporins (3GCs) in a local lizard population (Anolis marmoratus), but knowledge of the distribution and resistance of these strains in humans and the environment is limited. The aim of this study was to compare the distribution and antibiotic susceptibility pattern of E. cloacae complex members from different sources in a “one health” approach and to find possible explanations for the high level of resistance in non-human samples. E. cloacae complex strains were collected between January 2017 and the end of 2018 from anoles, farm animals, local fresh produce, water, and clinical human samples. Isolates were characterized by the heat-shock protein 60 gene-fragment typing method, and whole-genome sequencing was conducted on the most frequent clusters (i.e., C-VI and C-VIII). The prevalence of resistance to 3GCs was relatively high (56/346, 16.2%) in non-human samples. The associated resistance mechanism was related to an AmpC overproduction; however, in human samples, most of the resistant strains (40/62) produced an extended-spectrum beta-lactamase. No relation was found between resistance in isolates from wild anoles (35/168) and human activities. Specific core-genome phylogenetic analysis highlighted an important diversity in this bacterial population and no wide circulation among the different compartments. In our setting, the mutations responsible for resistance to 3GCs, especially in ampD, were diverse and not compartment specific. In conclusion, high levels of resistance in non-human E. cloacae complex isolates are probably due to environmental factors that favor the selection of these resistant strains, and this will be explored further.
Introduction
Bacteria in the Enterobacter cloacae complex (ECC) are widely distributed in numerous terrestrial and aquatic environments, and also in air and space equipment (Singh et al., 2018; Davin-Regli et al., 2019; Uchida et al., 2020). ECC are found in the gut microbiota of animals, including reptiles, mammals, and humans, and some studies have reported that the complex is endophytic (Liu et al., 2012; Singh et al., 2013; Davin-Regli et al., 2019). This bacterial complex also includes phytopathogenic clones (Humann et al., 2011), infection-causing strains in wild fauna and domestic animals (Haenni et al., 2016; Goldberg et al., 2019), and opportunistic pathogens that are involved in a wide variety of human infections, especially those associated with health care (Garinet et al., 2018).
The denomination “complex” refers to different Enterobacter species and subspecies, which are difficult to discriminate clearly only with phenotypic approaches. The current classification of ECC members is based on DNA analysis and specifically on partial sequence comparisons of heat-shock protein 60-gene (hsp60) fragments (Davin-Regli et al., 2019). The population structure of this complex was initially divided into 12 genetic clusters (C-I to C-XII) and a loosely knit group (C-xiii; Hoffmann and Roggenkamp, 2003). More recently, use of whole-genome sequencing (WGS) revealed a total of 22 phylogenetic clades (A–V), further illustrating the complex taxonomy of this genus (Chavda et al., 2016; Sutton et al., 2018). In addition, some phylogenetic clades are associated with previous hsp60 clusters, and a novel cluster was reported (Beyrouthy et al., 2018).
The distribution of the ECC clusters was analyzed in studies related to clinical isolates (Kremer and Hoffmann, 2012; Garinet et al., 2018). Strains belonging to C-III, -VI, and -VIII usually predominated and were found to carry various determinants of antibiotic resistance (Stock et al., 2001; Hoffmann et al., 2005; Kremer and Hoffmann, 2012; Peirano et al., 2018). These clusters appeared to be adapted to the hospital environment (Paauw et al., 2008), and geographic specificities were recently reported (Zhou et al., 2018). Few studies, however, have provided information about the distribution of ECC members in animals and other sources, which limits comparisons and impedes understanding of their global epidemiology (Hoffmann and Roggenkamp, 2003; Kämpfer et al., 2008; Haenni et al., 2016).
Enterobacter cloacae complex belongs to the ESKAPE group which referred to Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp., and is recognized as a “priority pathogen” due to its clinical relevance and association with antibiotic resistance and virulence genes (Rice, 2008; World Health Organization, 2017). All members of this complex have similar susceptibility to antibiotics (Davin-Regli et al., 2019), except fosfomycin (Stock et al., 2001). They are intrinsically resistant to aminopenicillin, the combination amoxicillin–clavulanic acid, and the first two generations of cephalosporins because they express an inducible ampC cephalosporinase. Under selection pressure, derepressed mutants often emerge, which overproduce cephalosporinase, conferring a high level of resistance to third-generation cephalosporins (3GCs) and increasing the minimum inhibitory concentration required for the fourth generation, such as cefepime (Guérin et al., 2015; Kohlmann et al., 2019; Mizrahi et al., 2020). Since the emergence of extended-spectrum beta-lactamase (ESBL), plasmidic acquisition of ESBL determinants has become an important mechanism in 3GCs resistance (3GC-R), especially among clinical ECC strains, as for most Enterobacteriaceae. These bacteria are also capable of acquiring genes that encode for carbapenemases, further restricting therapeutic management (Peirano et al., 2018). ESBL acquisition or cephalosporinase overproduction (CoP) appear to be distributed differently inside hsp60 clusters, as illustrated by a higher frequency of ESBL production in C-VI and -VIII isolates (Stock et al., 2001; Garinet et al., 2018). As in other high resource countries, community resistance of ECCs to 3GCs is rare in Guadeloupe because of lower selection pressure (Guyomard-Rabenirina, 2016).
Although these bacteria have been described in the environment, the susceptibility of environmental strains is not well understood. There is little information in Guadeloupe, as in other South American countries, on the distribution of ECC members or on the resistance to antibiotics of strains isolated in the environment and in clinical samples. Resistance of Enterobacteriaceae to 3GCs was rare in community-acquired urinary tract infections (4.0%), due mainly to ESBL production (Guyomard-Rabenirina, 2016), in contrast to the high frequency of ESBL in clinical ECC (19.7%) from the University Hospital of Guadeloupe (S. Breurec, personal communication). In the local environment, we observed a high prevalence of 3GC-R Enterobacteriaceae carriage in feces of Anolis marmoratus (89/234, 38.0%), a small endemic lizard, and ECC members were the most prevalent among isolated strains (57/115, 49.5%; Guyomard-Rabenirina, 2016).
We conducted this study to investigate the distribution of ECC hsp60 clusters isolated from various sources in Guadeloupe and to characterize their antibiotic susceptibility patterns in a “one health” approach. Genomic analysis was conducted on the main clusters in clinical samples (C-VI and C-VIII) and in different biotopes to compare the strains and to investigate the high level of 3GC-R in non-human samples.
Materials and Methods
Collection From Human
Between January and December 2018, clinical ECC isolates obtained during routine bacteriological diagnostics were collected prospectively from patients admitted to the University Hospital of Guadeloupe, a 900-bed teaching hospital in Pointe-à-Pitre/Les Abymes. The date and nature of the sample, and the results of antibiotic susceptibility analysis were recorded anonymously. Isolates were considered to be hospital-acquired if there were collected from patients hospitalized for more than 48 h after admission. The others were notified as to be community-acquired. Human samples were taken in accordance with the requirements of the local ethics committee and did not interfere with laboratory organization (reference A5_19_12_05_TRAMID).
Sampling for Animal and Environmental Isolates
Between January 2017 and December 2018, ECC strains were isolated from water catchment areas, local fruits and vegetables, and fresh fecal samples from A. marmoratus and farm animals sampled at different sites in Guadeloupe.
Overall, 168 free-living adult lizards were caught and sampled at 17 sampling sites throughout the island (Supplementary Table 1). All the procedures were approved by the regional environment, planning, and housing agency and by the Guadeloupe National Park. The project was also approved by the Committee for Ethics in animal experiments of the French West Indies and Guyana (reference 971-2016-12-20-001). Animals were cared for and used according to French decree No. 2013-118 of 1 February 2013 on the protection of animals, which meets European Union Directive 2010/63 on the protection of animals used for experimental and other scientific purposes (Guyomard-Rabenirina et al., 2020). To study a possible association between ECC carriage and the degree of human activity at the site at which wild individuals were caught, the sampling location and the type of environment (urban, coastline, and mountain forest) were recorded (Supplementary Table 1). Urban sites were considered to be associated with moderate to high human activity, and the coastline and mountain forest environments with limited human impact. Fresh fecal samples from 34 pigs and 28 beef cattle were collected at the only slaughterhouse in Guadeloupe, located in Le Moule. The municipality of origin of the sampled animal was recorded (Supplementary Table 1). No information was available on antibiotic treatment. A total of 76 samples of fresh, locally produced fruits and vegetables were collected aseptically at four local markets (Bergevin, Convenance, Gourdeliane, and Saint-Jules). The market and the farm of origin were recorded, as well as the type of fertilizer used (organic or chemical) on each identified farm (n = 27 in Supplementary Table 1). A total of 40 raw water samples, which corresponded to drinking water before treatment, were collected at 24 catchment points, in collaboration with the regional health agency and the hygiene laboratory of the Pasteur Institute of Guadeloupe. Most of these sampling points were located in Basse-Terre. All samples were transported rapidly to the laboratory, stored at 5 ± 3°C and analyzed within 4 h.
ECC Isolation and Antibiotic Susceptibility Analysis
All non-human samples were enriched. Animal stools, fruits, and vegetables were mixed with buffered peptone water. For water samples, 100 mL of serially diluted samples were filtered through a 0.45-μm membrane filter (Millipore, Guyancourt, France), and the membranes were placed in 9 mL of buffered peptone water solution. The non-selective enrichment broth for all samples was incubated for 16–20 h at 37°C. Then, 100 μL were inoculated onto chromogenic agar (CCA, CHROMagar, Paris, France). In addition, to increase the chances of detection of 3GC-R strains in the bacterial population, the same medium supplemented with ceftriaxone at 4 mg/L was also inoculated. Plates were incubated for 16–20 h at 37°C. This antibiotic was selected as it is considered to be a weak inducer of CoP as other 3GCs (Mizrahi et al., 2020). A maximum of five presumptive ECC colonies from each plate were isolated and identified by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (Shimadzu Biotech, Kyoto, Japan) and associated software.
Susceptibility to ampicillin (10 μg), amikacin (30 μg), amoxicillin–clavulanic acid (20–10 μg), aztreonam (30 μg), cefepim (30 μg), cefotaxime (5 μg), cefoxitin (30 μg), ceftazidime (10 μg), ciprofloxacin (5 μg), ertapenem (10 μg), gentamicin (10 μg), nalidixic acid (30 μg), temocillin (30 μg), ticarcillin (75 μg), tigecycline (15 μg), and trimethoprim–sulfamethoxazole (1.25–23.75 μg) was determined for all ECC strains by the disk diffusion method on Mueller-Hinton agar (Bio-Rad, Marnes-la-Coquette, France). Isolates were classified as resistant, intermediate, or susceptible according to the 2018 guidelines of CA-SFM/EUCAST1. Isolates of intermediate susceptibility were grouped with resistant isolates for data analysis (Supplementary Table 2). Production of ESBL was detected with the double-disk synergy test, according to CA-SFM/EUCAST recommendations.
DNA Extraction
Total bacterial DNA was initially extracted from pure cultures with the Qiagen QIAamp DNA minikit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions.
ESBL Resistance Gene Screening
Extended-spectrum beta-lactamase-encoding genes were screened by polymerase chain reaction (PCR). On the basis of previous epidemiological evidence for ECC in the community, PCR was performed only for blaCTX–M group 1 (Dallenne et al., 2010; Guyomard-Rabenirina, 2016). Amplicons were sequenced at Eurofins (Eurofins Genomic SAS, Les Ullis, France). Resistance genes were identified from the ResFinder database (Zankari et al., 2012).
ECC hsp60 Typing
As ECC members cannot be differentiated reliably with classic identification methods, we conducted sequence analysis of the partial hsp60 gene, as described previously on 313 local strains (Hoffmann and Roggenkamp, 2003). PCR products were sequenced at Eurofins, and DNA sequences and chromatograms were analyzed with ApE software2. Maximum likelihood phylogenetic reconstruction was performed with RAxML in 100 replications (Stamatakis, 2014). The tree was drawn with iTOL (Letunic and Bork, 2019) and rooted with Klebsiella aerogenes hsp60 partial sequence (AB008141.1; Hoffmann and Roggenkamp, 2003). Accession numbers for previously identified hps60 cluster sequences are listed in Supplementary Figure 1. To complete the analysis, hsp60 partial sequences were extracted from the assembled genomes of ECC ST873 strains, characterized, and added as a new ECC cluster (C-XIV – Clade S; Beyrouthy et al., 2018). Similar initial bioinformatic analysis was conducted on a selection of different ECC strains used and identified by Chavda et al. (2016), and Sutton et al. (2018; Supplementary Figure 1). We also included hsp60 partial sequences of recently named strains: E. wuhouensis, E. quasihormaechei (Wang et al., 2020), E. huaxiensis, E. chuandaensis (Wu et al., 2019), and E. oligotrophicus (formerly E. oligotrophica) (Akita et al., 2019). The collected strains were assigned to a cluster on the basis of the reference data set used and bootstrap values (Supplementary Figure 1). When multiple strains were found in the same sample, only one in each cluster or resistance phenotype profile against beta-lactam antibiotics was conserved for the analysis, to avoid duplicates.
Core-Genome Phylogenetic Analyses
To better describe possible circulation of ECC lineages collected from animals, fresh food, raw water, and humans and to gain understanding of the emergence of 3GC-R ECC from non-human samples, WGS was conducted on randomly selected strains in the two most prevalent hsp60 clusters: C-VI (n = 45) and C-VIII (n = 86). Their origins were: 47 from Anolis, 5 from domestic animals, 14 from fresh produce, 10 from raw water samples, and 55 from human isolates. WGS was performed at the “Plateforme de microbiologie mutualisée” of the Pasteur International Bioresources network (Institut Pasteur, Paris, France). The method and software used for sequencing, quality checking and core-genome extraction were described previously (Guyomard-Rabenirina et al., 2020). Raw reads were trimmed and filtered with AlienTrimmer (Criscuolo and Brisse, 2014). Genomes were assembled with SPAdes software (Bankevich et al., 2012), and final quality was appreciated with QUAST and BUSCO score (Gurevich et al., 2013; Simão et al., 2015).
Total core single nucleotide polymorphisms (SNPs) were detected with Snippy software version 4.4.5, with GENC200 strain as reference for C-VI isolates and GENC071 strain for C-VIII3. Recombination elements were removed from the global core genome alignment with ClonalFrameML software (Didelot and Wilson, 2015), The two maximum likelihood phylogenetic reconstructions were performed with RAxML software in the GTR-CAT model and 1000 bootstrap replicates, and the trees were drawn with iTOL. Multilocus sequence typing was performed in silico with mlst software4 against the PubMLST database (Jolley et al., 2018), and virulence gene factors were identified with Abricate software5 associated with the Virulence Factor Database with a threshold of 95% coverage and 75% nucleotide identity (Chen et al., 2016). Abricate was used with these parameters to assess plasmid replicon and antibiotic resistance gene content associated with PlasmidFinder and ResFinder databases, respectively (Supplementary Table 3; Zankari et al., 2012; Carattoli et al., 2014).
Focus on Cephalosporinase Genes and Mutations
In the ECC collection, 11 wild-type (WT)/CoP pairs from the same sample were analyzed for mutations. Each strain pair differed by fewer than 45 SNPs (Supplementary Table 4). Nine pairs belonging to ECC C-VIII and two to C-VI were selected. Most of the strains (14/22) were isolated from reptiles. Mutation analyses were performed with Snippy software, and the corresponding GenBank flat file format for each gene was retrieved from the NCBI website6 (Benson et al., 2018). The accession numbers of the sequences used as references were: NZ_CP012165.1 (E. hormaechei subsp. oharae strain 34978, complete genome) and NC_014121.1 (E. cloacae subsp. cloacae ATCC 13047 chromosome, complete genome). All genes which could be involved in CoP and 3GC-R phenotype were analyzed and listed in Supplementary Table 4 (see Reference.gbk). An in-house Perl script was used to extract results from files generated by Snippy and to create a table of the numbers of non-synonymous and synonymous gene mutations in the ECC sequences retrieved from the same host but with different antibiotic resistance profiles.
Statistical Analyses
The analyses and data collection were performed with Microsoft Access 2003. Pearson’s χ2 or Fisher’s exact test was used. P values < 0.05 were considered significant.
Results
Between January 2017 and December 2018, 313 unique ECC strains were isolated from various sample types, after removal of duplicates (Table 1). In non-human isolates, ECC strains (WT and 3GC-R) were isolated from 57.9% (44/76) of fresh produce and 42.9% (72/168) of anole samples; half of the raw water samples (20/40) were positive, while ECC strains were isolated from only 29.0% of livestock samples (18/62). Overall, resistance to 3GCs was found in 16.2% of non-human samples (56/346) and was due only to CoP. Most of these 3GC-R strains exhibited a 6 mm inhibition diameter on Mueller-Hinton agar plate for cefotaxime and ceftazidime antibiotics (data not shown). The prevalence of CoP was higher in anole samples (35/168, 20.8%) than in vegetable (13/76, 17.1%), livestock (6/62, 9.7%), or water isolates (2/40, 5.0%). Most of the anoles were trapped in areas impacted by human activities (n = 112, 66.7%); however, no significant difference was found in the rates of CoP by degree of human activity (Table 2). A positive association was found between the prevalence of positive for ECC and use of organic fertilizer (11/11) rather than chemical fertilizer (4/12; P = 0.001) from the 27 identified farms, but a similar association was not found for the CoP rate (P = 0.15, i.e., 4/11 and 1/12; Supplementary Table 1).
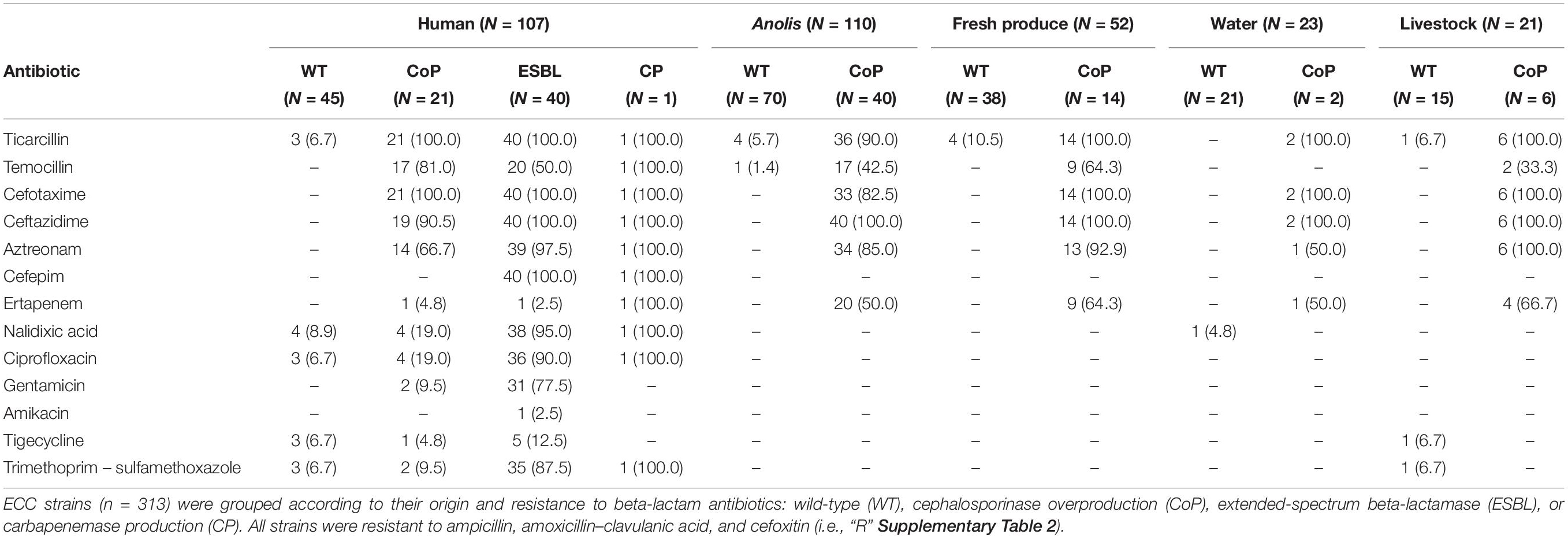
Table 1. Prevalence of resistance to each tested antibiotic of E. cloacae complex (ECC) isolates.

Table 2. Carriage of E. cloacae complex and third-generation cephalosporin-resistant (3GC-R) strains in the Anolis population, according to degree of human activity.
Most of human isolates were hospital-acquired (95/107) and 3GC-R were mainly associated to ESBL production (40/62; Table 1 and Supplementary Table 2). Genes encoding for ESBL were identified in all clinical strains positive in the double-disk synergy test (40/107, 37.4%). Amplicon sequencing revealed a blaCTX–M–15 gene in 38 isolates (95.0%). In addition, WGS allowed us to identify a blaGES–7 gene on the two last ESBL producers (GENC084, GENC220). One strain of human origin carried a blaOXA–48 gene (GENC133).
Co-resistance against beta-lactams (including 3GCs) and other antibiotic families was observed mainly in human associated strains and was usually to fluoroquinolones (41/62, 66.2%; Table 1). Co-resistance to gentamicin and trimethoprim–sulfamethoxazole was also found frequently, in 29 3GC-R clinical strains (46.8%) but not in samples from other sources. Only one strain from a raw water sample was resistant to nalidixic acid (ECC403), and one strain from livestock was notified resistant to trimethoprim–sulfamethoxazole and tigecycline (ECC408).
Distribution of hsp60 Clusters
Strain diversity was first investigated with hsp60 typing. A neighbor-joining tree was constructed with 129 alignment patterns of 313 partial hsp60 sequences from the different isolates, comprising 131 strains from the animal collection (110 from anole, 9 from pig, and 12 from beef cattle), 23 from water, 52 from fresh produce, and 107 from clinical samples (Table 3 and Supplementary Figure 1). All Enterobacter Hoffman clusters except C-VII (E. hormaechei subsp. hormaechei) were represented in our study, including C-XIV (n = 2) in human and domestic animal samples (Hoffmann and Roggenkamp, 2003; Beyrouthy et al., 2018); 28 strains, mainly human isolates (14/28), did not correspond to any of the 14 previously defined hsp60 clusters. They were grouped into six undefined hsp60 clusters (recorded as UD1–6), which included ECC clades recently identified by WGS (K,L,P,N,T) and E. oligotrophica (Table 3, Supplementary Table 2, and Supplementary Figure 1; Chavda et al., 2016; Sutton et al., 2018; Akita et al., 2019).
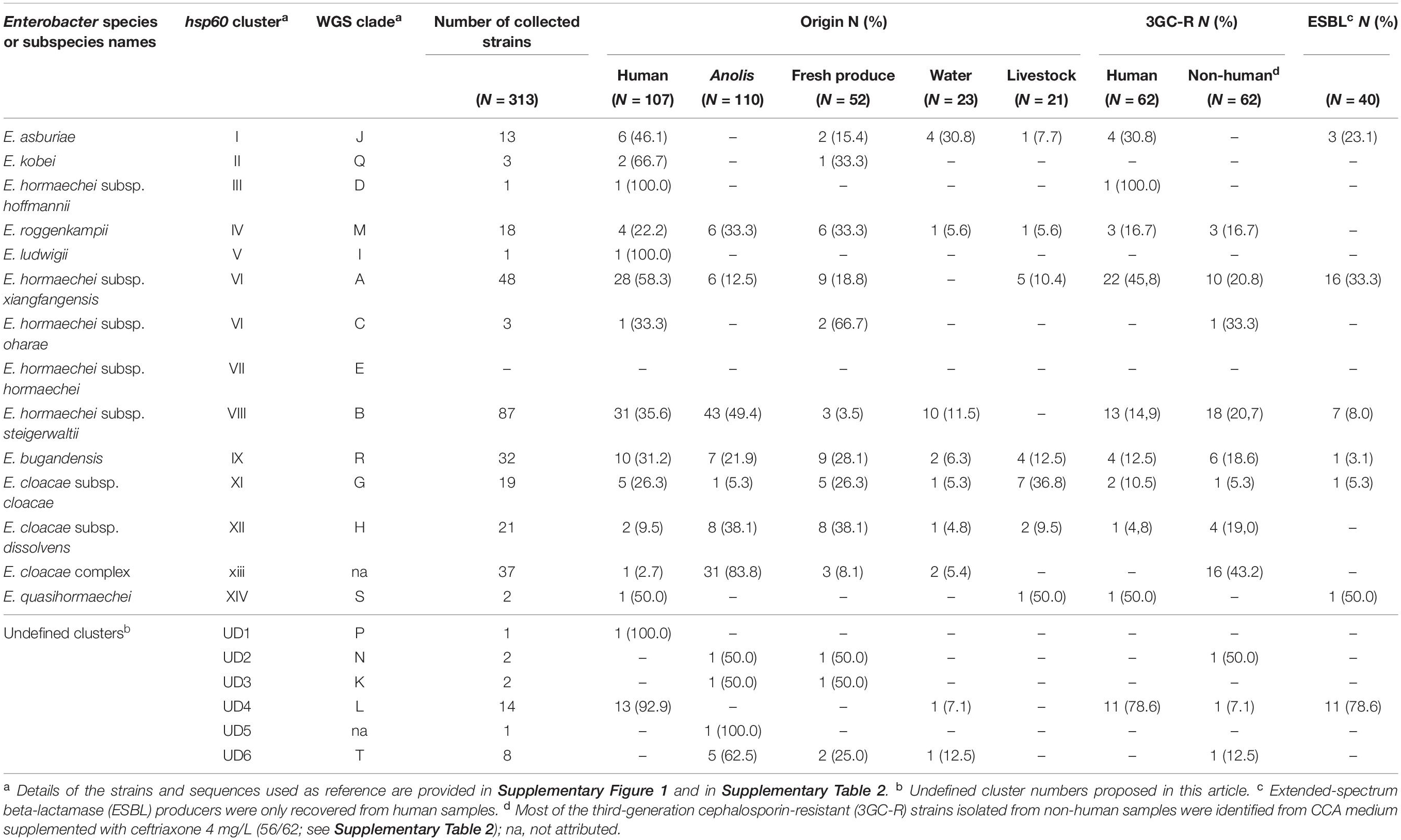
Table 3. Distribution of E. cloacae complex (ECC) members in samples of different origin.
Cluster VIII was best represented, except in livestock and fresh produce. C-VI was the second most frequent in clinical strains, whereas C-xiii was over-represented among anole isolates (28.2%), and E. asburiae was more frequent in water samples (C-I, 17.4%). In livestock, C-XI, -VI, and -IX predominated (Table 3 and Supplementary Figure 1). CoP strains were found in nearly all clusters except C-II, -V, and UD1, 3 and 5. The highest rate of CoP strains was found in cluster VIII (Table 3).
Genetic Analysis of ECC Clusters VI and VIII Populations
The WGS generated a mean of 149.87 bp paired-end reads, with an estimate coverage of 77.851-fold (AlienTrimmer; Criscuolo and Brisse, 2014). Quality of the assembly indicated a mean N50 of 309532 (minimum 43293, maximum 782933), and a mean single-copy BUSCO score of 98.6% completeness (Gurevich et al., 2013; Simão et al., 2015). Of the 45 sequenced isolates assigned to C-VI, 42 belonged to E. hormaechei subsp. xiangfangensis (clade A), and only three were identified as E. hormaechei subsp. oharae (clade C; Table 3; Sutton et al., 2018). These clade C strains were isolated from fresh produce and human samples (ECC 312, ECC336, and GENC003) and were not conserved for further analysis. Maximum likelihood phylogenetic analysis of E. hormaechei subsp. xiangfangensis strains indicated wide diversity among isolates (mean SNP of all isolates n = 17973, minimum n = 4, and maximum n = 22821). Fifteen strains were not assigned to a well-known sequence type (ST) or identified as new, whereas the others belonged to nine clearly identified sequence types (Figure 1). The largest cluster contained 11 strains in the international clone ST114 (mean SNP between isolates n = 935, minimum n = 4, and maximum n = 2122), which were isolated only from humans. Most of the strains (8/11) were ESBL producers. Some genetically related clusters with isolates of different origins were observed, comprising a cluster with three ST98 strains (two from humans and one from a cucumber; SNP mean = 484, minimum = 68) and a second with three vegetables strains belonging to ST344. The samples, taken at the same time at Bergevin market on 12 January 2018 (Supplementary Table 1), were from two farms, had the same antibiotic resistance profile (3GC-R), and differed by a mean of 45 SNPs (minimum = 36). One strain from Anolis clustered with an isolate from fresh produce, with only 65 SNPs difference (Figure 1).
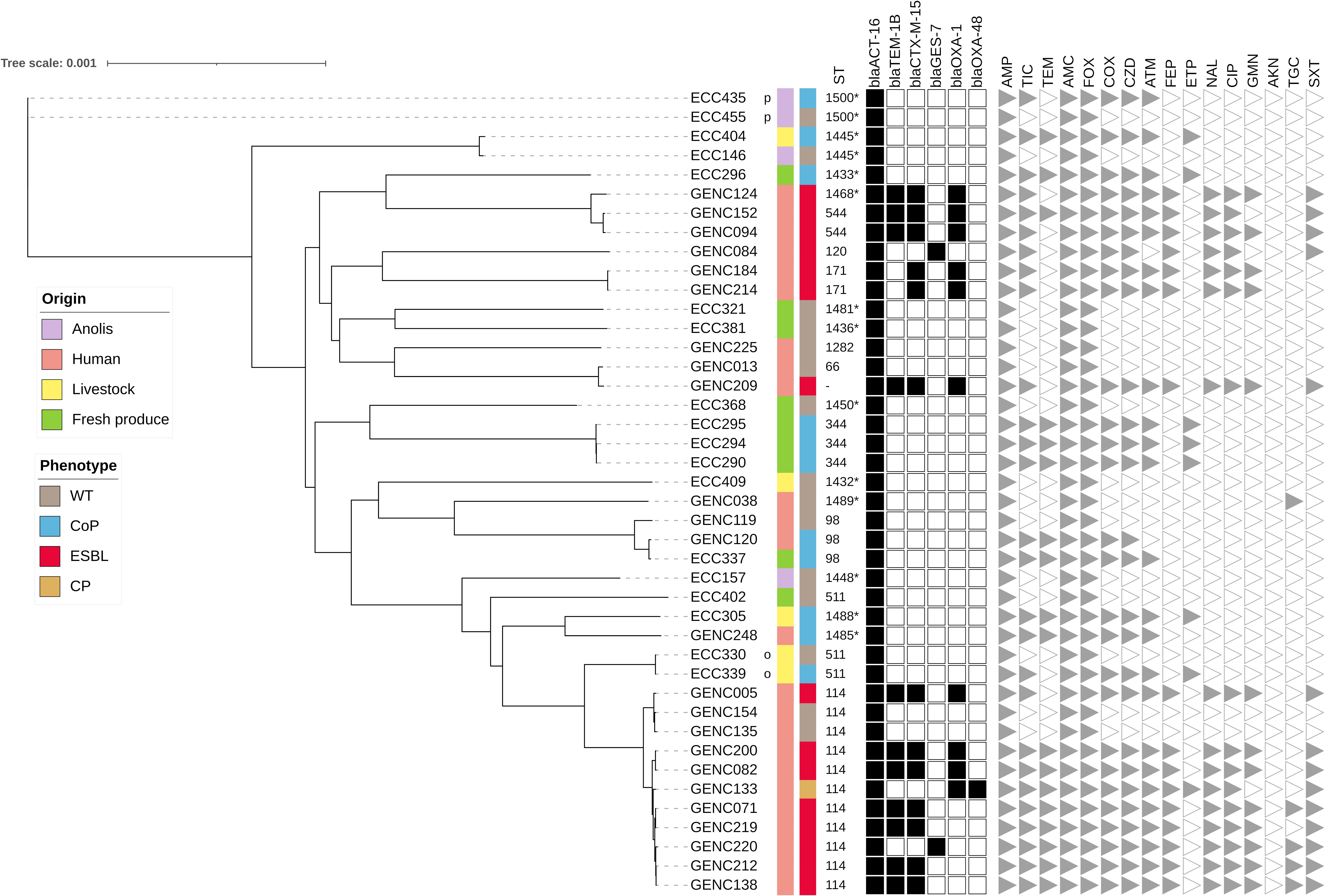
Figure 1. Maximum likelihood phylogenetic tree of E. cloacae complex C-VI – clade A isolates recovered in Guadeloupe (n = 42). Maximum likelihood phylogenetic reconstructions were performed with RAxML software (1000 bootstrap replicates), and the tree was drawn with iTOL. Hosts and phenotypes are indicated by vertical colored strips. The letters indicate specific wild-type and cephalosporinase overproduction pairs in the same sample. New sequence types (STs) identified in this study are indicated by a star, while unknown ST is denoted by a dash. Only genes that confer resistance to beta-lactam antibiotics were included. They were characterized by ResFinder and are indicated by black squares; all genetic details are provided in Supplementary Table 3. Antibiotic resistance profiles are indicated by gray triangles; AKN, amikacin; AMC, amoxicillin–clavulanic acid; AMP, ampicillin; ATM, aztreonam; CIP, ciprofloxacin; COX, cefotaxime; CZD, ceftazidime; ETP, ertapenem; FEP, cefepim; FOX, cefoxitin; GMN, gentamicin; NAL, nalidixic acid; TEM, temocillin; TGC, tigecycline; TIC, ticarcillin; and SXT, trimethoprim–sulfamethoxazole.
Phylogenetic analysis of C-VIII revealed greater diversity, especially among anole strains (mean SNP for all isolates n = 23125, minimum n = 8, and maximum n = 27680). Various lineages were observed in the same sample (samples a, e, and f in Figure 2 and Supplementary Table 4). In silico analyses revealed the presence of two main groups in this second tree. The first (C-VIII-A, n = 32) consisted mainly of clinical isolates (20/32; mean SNP among isolates n = 26166, minimum n = 8, and maximum n = 31449), while reptile strains predominated in the second one (C-VIII-B, 31/49). ESBL producers were found only in C-VIII-A and, as expected, only in human isolates. Like ST114 in ECC C-VI, ST113 was well represented in human samples for C-VIII (n = 9), and four were ESBL producers. This ST was not found in other biotopes (Supplementary Table 3).
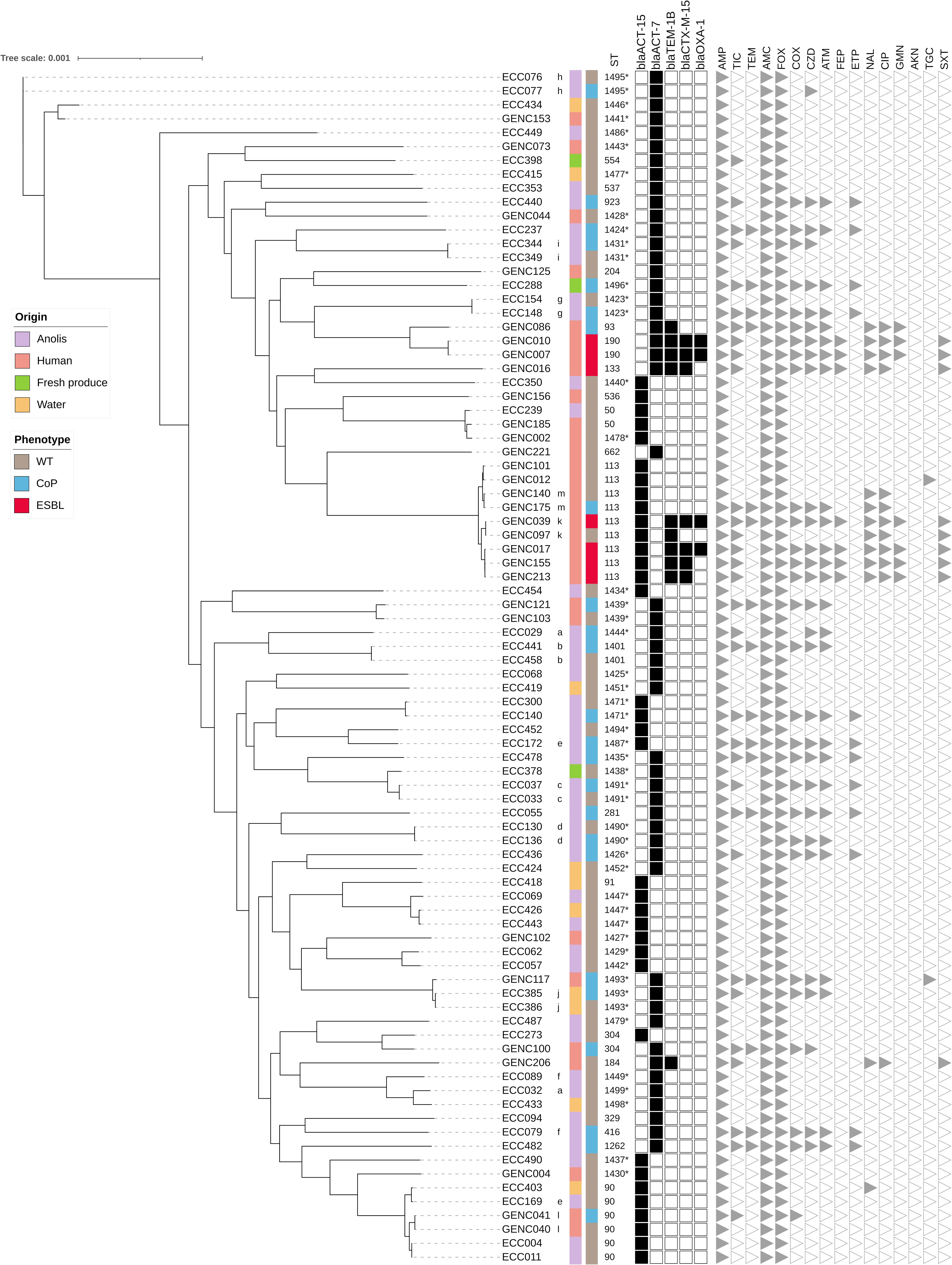
Figure 2. Maximum likelihood phylogenetic tree of E. cloacae complex C-VIII isolates recovered in Guadeloupe (n = 86). Maximum likelihood phylogenetic reconstructions were performed with RAxML software (1000 bootstrap replicates), and the tree was drawn with iTOL. Hosts and phenotypes are indicated by vertical colored strips. The letters indicate specific wild-type and cephalosporinase overproduction pairs in the same sample. New sequence types (STs) identified in this study are indicated by a star. Only genes that confer resistance to beta-lactam antibiotics were included. They were characterized by ResFinder and are indicated by black squares; all genetic details are provided in Supplementary Table 3. Antibiotic resistance profiles are indicated by gray triangles; AKN, amikacin; AMC, amoxicillin–clavulanic acid; AMP, ampicillin; ATM, aztreonam; CIP, ciprofloxacin; COX, cefotaxime; CZD, ceftazidime; ETP, ertapenem; FEP, cefepim; FOX, cefoxitin; GMN, gentamicin; NAL, nalidixic acid; TEM, temocillin; TGC, tigecycline; TIC, ticarcillin; and SXT, trimethoprim–sulfamethoxazole.
As observed in the C-VI phylogenetic tree, some strains of different origins were genetically related. ST90 was recovered from three different A. marmoratus, one water sample, and one human (two strains from the same patient, l in Figure 2 and Supplementary Table 4; mean SNP among isolates n = 1283, minimum n = 8, and maximum n = 2221). Most of these strains presented a CoP (5/6). Two other STs found in wild fauna and human samples clustered together: ST50, (ECC239–GENC185, 225 SNPs) and ST304 (ECC273–GENC100, 7698 SNPs). The human strain GENC117 shared the same target sequence of seven housekeeping genes with ECC386 isolated from water and a difference of 123 SNPs (i.e., new ST1493). One strain from Anolis clustered with an isolate from water, with a difference of only 101 SNPs (ECC443–ECC426). Two strains isolated from two Anolis 28 km apart clustered, with a difference of only 81 SNPs (ECC140–ECC300).
Few different resistance gene types were shared by ECC isolates from the 5 origins (Supplementary Table 3). The genes belonged to blaACT, and one gene conferred resistance to fosfomycin (fosA). Genes encoding for efflux pumps (oqxA–oqxB and mdfA) were observed in human and non-human isolates. Genes encoding for cephalosporinase were specific for each cluster. E. hormaechei subsp. xiangfangensis was related to the blaACT–16 gene type, while blaACT–15 and blaACT–7 were associated with E. hormaechei subsp. steigerwaltii. Human strains belonging to C-VI and C-VIII expressed more resistance genes (mean, nine) than those from other compartments (mean, three; Supplementary Table 3). All sequenced and analyzed strains resistant to fluoroquinolones (28/128; except GENC200) harbored quinolone resistance genes (qnrB1, qnrB19 or qnrS2). Regarding the plasmid distribution, 50.0% (64/128) of the sequenced C-VI and C-VIII carried at least one identifiable replicon type. Among them Col (48/64), IncFII (n = 24), IncHI2 (n = 21), and IncFIB (n = 20) were the most often identified. Only 24.3% (18/74) of non-human strains presented at least one replicon. This prevalence was higher in the human collection (46/54, 85.2%), and whole-genome sequenced ESBL-producing ECC were especially associated with an IncHI2 signature (19/22, 86.4%; Supplementary Table 3). We also identified genes associated with virulence in C-VI and C-VIII populations in both human and non-human strains, which are involved in bacterial adherence, iron uptake, motility, or toxin production (Supplementary Table 3). Globally, human and non-human isolates expressed a similar number of virulence genes (mean twenty-one). All sequenced C-VIII strains (n = 86) harbored genes involved in the salmochelin siderophore system (iroB, iroC, iroD, iroE, and iroN), while this system was not found in the C-VI population. The yersiniabactin system (fyuA, irp1, irp2, ybtA, ybtE, ybtP, ybtQ, ybtS, ybtT, ybtU, and ybtX) was identified in two non-human strains belonging to ST90 (ECC169 and ECC403).
Cephalosporinase Genes Mutation Profile Analysis
In isolates analyzed for cephalosporinase mutation, alignment of the AmpD protein sequences belonging to the WT ECC showed relatively good conservation (with an overall identity of 94.65%). Few mismatches were found (Supplementary Table 4). Comparison of 3GC-R with WT isolates from the same sample indicated that differences in mutations were found mainly in the ampD gene. Significant mutations are listed in the last column of Supplementary Table 4. Furthermore, other genes such as dacB and ramR could play a non-negligible role in the differentiation of resistant and susceptible strains. Our analysis indicates non-synonymous mutations different from those was observed previously, although Ala60Val substitution in ampD was also detected in ECC033 and ECC037 (Flury et al., 2016).
Discussion
This study of ECC diversity in samples from different compartments in Guadeloupe showed by hsp60 typing analysis that six clusters (C-IV, -VI, -VIII, -IX, -XI, and -XII) are identified in more than two thirds of the strains (228/313, 72.8%), and most are present in all sample types. Although this typing method is limited and focus on only one gene (Hoffmann and Roggenkamp, 2003; Wu et al., 2020), it indicates a high degree of diversity of this bacterial complex which is present in a wide variety of compartments.
The E. hormaechei metacluster predominated especially with C-VI (51/313, 16.3%) and C-VIII (87/313, 27.8%), which were also the most frequent clusters in human infections (27.1 and 29.0%, respectively). Surprisingly, C-III was rare in Guadeloupe, with only one strain in a human sample, whereas this E. hormaechei subspecies is one of the most frequently reported in clinical studies (Hoffmann and Roggenkamp, 2003; Kremer and Hoffmann, 2012; Garinet et al., 2018). In contrast, we notified the high prevalence of the hsp60 UD4 cluster in our set of clinical isolates which should referred to ECC clade L (Sutton et al., 2018), and a possible new successful ESBL-producing lineage.
Among other important clusters, E. bugandensis (C-IX) accounted for 10 of the 107 clinical strains, and 22 isolates were recovered from non-clinical samples. This species was found mainly in fresh produce (9/22) and reptile feces (7/22). It has been reported in wild fauna, livestock, and the environment in only a few studies (Khanna et al., 2013; Singh et al., 2018; Matteoli et al., 2019; Wu et al., 2020). E. bugandensis has been described as the most virulent and pathogenic of all ECC strains (Doijad et al., 2016; Pati et al., 2018), which raises concern, as fresh produce may be the origin of dissemination of virulent pathogens in the community, as for some Escherichia coli lineages (Luna-Guevara et al., 2019).
As C-VI and -VIII were the most prevalent hsp60 clusters in this collection, the WGS analysis was conducted to investigate their potential clone diffusion among human, animal, and environmental samples. Among C-VI, E. hormaechei subsp. xiangfangensis was overrepresented in comparison to the other related subspecies E. hormaechei subsp. oharae (3/45) as reported in previous studies (Peirano et al., 2018; Sutton et al., 2018). Core-genome analysis showed that clonal spread at hospital level was limited to a few genetic backgrounds which were previously found in human infections (i.e., ST113 and ST114; Peirano et al., 2018; Siebor et al., 2019). In accordance with our observations, they have rarely been observed in samples of other origins, except from companion and wild animals (Haenni et al., 2016; Harada et al., 2017; Goldberg et al., 2019). Different sample types shared a few lineages, one being ST90 (n = 5), which is also involved in human and animals infections (Peirano et al., 2018; Zhu et al., 2020). Moreover, two ST90 strains isolated from an anole and a raw water sample harbored genes encoding for the yersiniabactin system. The presence of this highly pathogenic island has previously been described in other Enterobacteriaceae species and was associated with a specific E. hormaechei clone that caused a hospital outbreak in Netherlands (Paauw et al., 2009). Another ST previously found in a clinical sample (ST98) was present in both a human collection and fresh produce (Izdebski et al., 2015). ST344 clones were found in three fresh products but from two different market stands, suggesting manual strain circulation.
This study also showed a high frequency of 3GC-R ECC members in non-human samples such as livestock and fresh local produce, and confirmed previous observations on the local anole population (Guyomard-Rabenirina, 2016). Three hypotheses have been proposed to explain this high prevalence of CoP ECC strains in these compartments. The first one was a human origin of these resistant strains, with the spread of successful lineages among compartments; however, hsp60 clusters distribution within each sample type and the WGS analysis of C-VI and -VIII, indicated a wide diversity, which was not in favor of exchanges. Although some whole-genomes sequenced strains isolated from humans, lizards, and other origins were genetically close, most of them were grouped separately.
The second hypothesis was an impact of human activities, which could exert selective pressure for resistant ECC strains, as few international ST were found in non-clinical isolates (ST90, ST98; Izdebski et al., 2015; Peirano et al., 2018). No significant difference was found between Anolis individuals sampled near or far from areas of human activity, which is congruent with the results of a study on 3GC-R E. coli carriage in lizard in Guadeloupe (Guyomard-Rabenirina et al., 2020). To go further in a recent survey, Anolis (n = 20) and other wild animals (n = 67; rat, bird, toad, and cockroach) living near the hospital sewers and at the associated wastewater treatment plant were sampled. Among them, 21.8% (19/87) carried 3GC-R ECC including one Anolis. These strains were exclusively ESBL-producers, and WGS analysis indicated the presence of a ST114 lineage closely related to human samples and the dissemination of an IncHI2/blaCTX–M–15 plasmid. Taken together, these results suggested that only specific polluted environments associated with an important selective pressure are in favor of a large dissemination and maintenance of human related resistant strains in the wild fauna compartment (Pot et al., 2021). This second hypothesis is less well supported for fruits and vegetables, as the origin of the resistant clones in such samples could be multifactorial (Hölzel et al., 2018). Overall, the prevalence of 3GC-R clones in fresh produce was higher than in a previous larger collection, but only for CoP clones (van Hoek et al., 2015), in contrast to other published reports of ESBL producers (van Hoek et al., 2015; Hölzel et al., 2018). Our results suggest that organic fertilization is associated with a higher load of ECC strains in fresh produce but is not correlated with higher counts of CoP ECC, as described previously (Marti et al., 2013).
As it has been suggested that ECC members have higher mutation rates due to derepression than other genera with constitutive ampC (Kohlmann et al., 2018), we explored a third hypothesis, that non-human strains have a greater ability to acquire specific mutations in genes encoding for AmpC. This hypothesis was rapidly excluded by analysis of 11 WT/CoP pairs, in which most mutations were shared by strains from the different compartments and especially mutations in ampD, which has been suggested to be the leading mechanism in ECC with CoP in human samples (Guérin et al., 2015).
Our results increase the understanding of reservoirs definition and sources of ECC infections in a tropical setting. Several limitations should be pointed in this study. First, human isolates were recovered from only infectious sites and were mostly specified to be hospital-acquired. Nevertheless, since ECC members are opportunistic pathogens and belong to Enterobacteriaceae, most of infections came from endogenous colonizing strains (Gorrie et al., 2018). As we used selective medium to facilitate resistance detection, it could led to the overrepresentation of 3GC-R ECC in non-human isolates. This selection bias was limited by using ceftriaxone as selective antibiotic, described as a weak CoP inducer (Mizrahi et al., 2020). Moreover, previous authors indicated a relatively low rate of derepressed mutant in ECC population (3 × 10–8; Kohlmann et al., 2018), although we did not estimate the mutation rate in our non-human samples due to the presence of various cultivable genus and species in agar plates.
Conclusion
Our findings highlight the widely diverse distribution of ECC members in non-human and human samples. We found a high prevalence of 3GC-R ECC in non-human samples, due exclusively to CoP. None of our hypotheses could explain this prevalence, and higher mutation rate is not excluded. These results suggest that this characteristic confers a selective advantage for these strains. Unknown persistent environmental factors, which should be further explored, may favor such overproduction.
Data Availability Statement
The 313 partial hsp60 sequences are available in Supplementary Table 2 with strain details, while the whole-genome sequences of C-VI, clade A (n = 42) and C-VIII (n = 86) are deposited in GenBank under BioSample accession numbers SAMN15680734 to SAMN15680861 (Supplementary Table 3).
Ethics Statement
The parts of this project involving human participants were reviewed and approved by Commission de recherche éthique, Direction de la recherche et de l’innovation, CHU de la Guadeloupe (A5_19_12_05_TRAMID). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. The animal study was reviewed and approved by the Committee for Ethics in animal experiments of the French West Indies and Guyana (reference 971-2016-12-20-001). Written informed consent for participation was not obtained from the owners because feces were collected from livestock animals at the slaughterhouse, just before slaughter. No data concerning farm was avalaible. Only the municipality origin of screened animals batchs was recorded.
Author Contributions
MP, SG-R, and AT conceived and designed the study. MP, SB, CD, SF, GG, SG-R, EM, and AT collected biological samples, isolates, and epidemiological data. MP, DC, FGu, SG-R, FGr, AT, and YR analyzed the data. MP, SB, DC, SG-R, and AT wrote the manuscript. All the authors critically revised and approved the final version of the manuscript.
Funding
This work was supported by a European Regional Development Fund grant, financed by the European Union and Guadeloupe Region (Grant ID: 2018-FED-1084).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to all the technicians and students for their participation in this project at the ARS (Agence Régionale de Santé), the University Hospital, the Pasteur Institute of Guadeloupe and the “Plateforme de microbiologie mutualisée.” We thank the curator team of the E. cloacae typing PubMLST database for their co-operation. We extend special thanks to E. Heseltine for editorial assistance. We also express our gratitude to the employees of the slaughterhouse in Le Moule and all the farmers and fruit and vegetable merchants for their collaboration.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.628058/full#supplementary-material
Supplementary Figure 1 | Maximum likelihood phylogenetic tree based on the partial sequence hsp60 gene of E. cloacae complex (ECC) isolates recovered in Guadeloupe (n = 313). Maximum likelihood phylogenetic reconstructions were performed with RAxML software (100 bootstrap replicates), and the tree rooted with Klebsiella aerogenes hsp60 sequence (AB008141.1) was drawn with iTOL. All the sequences used as reference in the tree have a corresponding GenBank accession number, and only sequences extracted from whole-genome of ECC strains were labeled with current names. Each hsp60 cluster is identified by a specific color, and bootstrap values ≥ 60 are indicated. Hsp60 sequences from C-X, which correspond to Lelliottia nimipressuralis (formerly Enterobacter nimipressuralis) are inserted (AJ567887.1, AJ567900.1). For better readability, C-IV nodes are grouped. The hosts of each local strain are specified by a circle (human), a square (livestock), a star (fresh produce), a right-pointing triangle (water), or a left-pointing triangle (Anolis). Phenotypes are indicated by a white form for wild-type against beta-lactam antibiotics, and a black form for third-generation cephalosporin-resistant strains. The global distribution is provided in Table 3, and all hsp60 partial sequences are available in Supplementary Table 2. List of ECC partial hps60 sequences included in this figure (associated cluster: C or UD, and accession number): C-I: AJ417140.1, AJ567846.1, AJ567893.1, FXLQ00000000; C-II: AJ567849.1, AJ567862.1, AJ567886.1, AJ567888.1, AJ567899.1, CP017181.1, C-III: AJ543781.1, AJ543789.1, AJ567871.1, AJ567872.1, AJ567877.1, AJ567880.1, CP017186.1; C-IV: AJ543784.1, AJ543806.1, AJ543807.1, AJ543867.1, AJ543877.1, AJ543889.1, AJ543893.1, CP017184.1; C-V: AJ417114.1, AJ862859.1, AJ862861.1, AJ862862.1, AJ862863.1, CP017279.1; C-VI: AJ543778.1, AJ543782.1, AJ567878.1, AJ866507.1, CP017180.1, CP017183.1; C-VII: AJ417108.1, AJ862866.1, AJ862867.1, AJ866491.1, CAI28773.1, MKEQ00000000.1; C-VIII: AJ543821.1, AJ543849.1, AJ543857.1, AJ543908.1, AJ567884.1, AJ567889.1, AJ567890.1, AJ567892.1, CAD66305, CAI28810.1, CP017179.1; C-IX: AJ543819.1, AJ543820.1, AJ543878.1, AJ543881.1, CAD66281.1, CAD66282.1, LT992502; C-XI: AJ417139.1, AJ417142.1, AJ543768.1, AJ543855.1, NC014121.1; C-XII: AJ417143.1, AJ543817.1, AJ543847.1, AJ862872.1, CP003678.1; C-xiii: AJ417128.1, AJ543837.1, AJ543870.1, AJ543872.1, CAD66299.1, CAD66332.1, CAD66334.1, CAD99100.1, LVUF00000000, FYBB00000000, SJOO00000000, AEXB00000000, QZCT01000000, FYBA00000000; C-XIV: ERS2281247, ERS2281248, LXPT00000000.1, LZEN00000000, NSIZ00000000.1, SJON00000000.1; UD1: JDWG00000000.1, JDWH00000000.1, JUZJ00000000.1, JUZQ00000000; UD2: BBUQ00000000.1, CP021851.1, JZYX00000000.1, NPNR00000000; UD3: AZXZ00000000.1, FCNO00000000.1, JACW00000000.1, JWAU00000000; UD4: CP043318.1, JVIL00000000.1, JZKT00000000.1, LEDN00000000; UD5: AP019007.1; and UD6: MTKD00000000, QZCS00000000.
Supplementary Table 1 | Details of non-clinical samples: sampling date, location, type of environment, farm characteristics.
Supplementary Table 2 | Details of strains: hsp60 partial sequence and results for antibiotic resistance. Resistance profiles were obtained against 16 antibiotics. Intermediate or resistant results are labeled “R”; “s” corresponds to a susceptible phenotype.
Supplementary Table 3 | Details of genetic content of sequenced E. hormaechei subsp. xiangfangensis (C-VI – clade A; n = 42) and E. hormaechei subsp. steigerwaltii (C-VIII – clade B; n = 86).
Supplementary Table 4 | Cephalosporinase gene complex mutation. ∗Clone numbers refer to letters used in Figures 1, 2 and correspond to two strains from the same sample with different resistance profile against beta-lactam antibiotics: wild-type or cephalosporinase overproduction. Each strain is considered to be a clone that differs by < 45 single nucleotide polymorphisms (SNPs). na: not attributed. The abbreviation “N/S” after the gene names indicates the number of non-synonymous mutations (N) versus the number of synonymous mutations (S) in comparison to the reference data. Please note that only the different non-synonymous mutations between strains from the same sample have been conserved in the observation column.
Abbreviations
3GC, third-generation cephalosporin; 3GC-R, third-generation cephalosporin resistant; ECC, Enterobacter cloacae complex; ESBL, extended-spectrum beta-lactamase; ESKAPE, Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.; CoP, cephalosporinase overproduction; hsp60, heat-shock protein 60 gene-fragment; SNP, single nucleotide polymorphism; ST, sequence type; UD, undefined cluster; WGS, whole-genome sequencing; WT, wild-type.
Footnotes
- ^ http://www.sfm-microbiologie.org
- ^ https://jorgensen.biology.utah.edu/wayned/ape/
- ^ https://github.com/tseemann/snippy
- ^ https://github.com/tseemann/mlst
- ^ https://github.com/tseemann/abricate
- ^ https://www.ncbi.nlm.nih.gov/genbank/
References
Akita, H., Matsushika, A., and Kimura, Z. (2019). Enterobacter oligotrophica sp. nov., a novel oligotroph isolated from leaf soil. MicrobiologyOpen. 8, 1–11. doi: 10.1002/mbo3.843
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Ostell, J., Pruitt, K. D., et al. (2018). GenBank. Nucleic Acids Res. 46, D41–D47. doi: 10.1093/nar/gkx1094
Beyrouthy, R., Barets, M., Marion, E., Dananché, C., Dauwalder, O., Robin, F., et al. (2018). Novel Enterobacter lineage as leading cause of nosocomial outbreak involving carbapenemase-producing strains. Emerg Infect Dis. 24, 1505–1515. doi: 10.3201/eid2408.180151
Carattoli, A., Zankari, E., García-Fernández, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Chavda, K. D., Chen, L., Fouts, D. E., Sutton, G., Brinkac, L., Jenkins, S. G., et al. (2016). Comprehensive genome analysis of carbapenemase-producing Enterobacter spp.: New insights into phylogeny, population structure, and resistance mechanisms. MBio. 7, 1–16. doi: 10.1128/mBio.02093-16
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis – 10 years on. Nucleic Acids Res. 44, 694–697. doi: 10.1093/nar/gkv1239
Criscuolo, A., and Brisse, S. (2014). AlienTrimmer removes adapter oligonucleotides with high sensitivity in short-insert paired-end reads. Commentary on Turner (2014) Assessment of insert sizes and adapter content in FASTQ data from NexteraXT libraries. Front Genetics. 5:130. doi: 10.3389/fgene.2014.00130
Dallenne, C., Da Costa, A., Decre, D., Favier, C., and Arlet, G. (2010). Development of a set of multiplex PCR assays for the detection of genes encoding important β-lactamases in Enterobacteriaceae. J Antimicrob Chemother. 65, 490–495. doi: 10.1093/jac/dkp498
Davin-Regli, A., Lavigne, J. P., and Pagès, J. M. (2019). Enterobacter spp.: Update on taxonomy, clinical aspects, and emerging antimicrobial resistance. Clin Microbiol Rev. 32, 1–32. doi: 10.1128/CMR.00002-19
Didelot, X., and Wilson, D. J. (2015). ClonalFrameML: Efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol. 11:e1004041. doi: 10.1371/journal.pcbi.1004041
Doijad, S., Imirzalioglu, C., Yao, Y., Pati, N. B., Falgenhauer, L., Hain, T., et al. (2016). Enterobacter bugandensis sp. nov., isolated from neonatal blood. Int J Syst Evol Microbiol. 66, 968–974. doi: 10.1099/ijsem.0.000821
Flury, B. B., Ellington, M. J., Hopkins, K. L., Turton, J. F., Doumith, M., Loy, R., et al. (2016). Association of novel nonsynonymous single nucleotide polymorphisms in AmpD with cephalosporin resistance and phylogenetic variations in AmpC, AmpR, OmpF, and OmpC in Enterobacter cloacae isolates that are highly resistant to carbapenems. Antimicrob Agents Chemother. 60, 2383–2390. doi: 10.1128/AAC.02835-15
Garinet, S., Fihman, V., Jacquier, H., Corvec, S., Le Monnier, A., Guillard, T., et al. (2018). Elective distribution of resistance to beta-lactams among Enterobacter cloacae genetic clusters. J Infect. 77, 178–182. doi: 10.1016/j.jinf.2018.05.005
Goldberg, D. W., Fernandes, M. R., Sellera, F. P., Costa, D. G. C., Loureiro Bracarense, A. P., and Lincopan, N. (2019). Genetic background of CTX-M-15-producing Enterobacter hormaechei ST114 and Citrobacter freundii ST265 co-infecting a free-living green turtle (Chelonia mydas). Zoonoses Public Health 66, 1–6. doi: 10.1111/zph.12572
Gorrie, C. L., Mirceta, M., Wick, R. R., Judd, L. M., Wyres, K. L., Thomson, N. R., et al. (2018). Antimicrobial-resistant Klebsiella pneumoniae carriage and infection in specialized geriatric care wards linked to acquisition in the referring hospital. Clin Infect Dis. 67, 161–170. doi: 10.1093/cid/ciy027
Guérin, F., Isnard, C., Cattoir, V., and Giard, J. C. (2015). Complex regulation pathways of AmpC-mediated β-lactam resistance in Enterobacter cloacae complex. Antimicrob Agents Chemother. 59, 7753–7761. doi: 10.1128/AAC.01729-15
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Guyomard-Rabenirina, S. (2016). Résistance aux antibiotiques des Entérobactéries en Guadeloupe: Importance en milieu communautaire et diffusion environnementale. Thesis. Saint-Claude: Université des Antilles.
Guyomard-Rabenirina, S., Reynaud, Y., Pot, M., Albina, E., Couvin, D., Ducat, C., et al. (2020). Antimicrobial resistance in wildlife in Guadeloupe (French West Indies): Distribution of a single blaCTX–M–1 Incl1/ST3 among humans and wild animals. Front Microbiol. 11:1524. doi: 10.3389/fmicb.2020.01524
Haenni, M., Saras, E., Ponsin, C., Dahmen, S., Petitjean, M., Hocquet, D., et al. (2016). High prevalence of international ESBL CTX-M-15-producing Enterobacter cloacae ST114 clone in animals. J Antimicrob Chemother. 71, 1497–1500. doi: 10.1093/jac/dkw006
Harada, K., Shimizu, T., Mukai, Y., Kuwajima, K., Sato, T., Kajino, A., et al. (2017). Phenotypic and molecular characterization of antimicrobial resistance in Enterobacter spp. Isolates from companion animals in Japan. PLoS One. 12:e0174178. doi: 10.1371/journal.pone.0174178
Hoffmann, H., and Roggenkamp, A. (2003). Population genetics of the nomenspecies Enterobacter cloacae. Appl Environ Microbiol. 69, 5306–5318. doi: 10.1128/AEM.69.9.5306-5318.2003
Hoffmann, H., Stindl, S., Ludwig, W., Stumpf, A., Mehlen, A., Monget, D., et al. (2005). Enterobacter hormaechei subsp. oharae subsp. nov., E. hormaechei subsp. hormaechei comb. nov., and E. hormaechei subsp. steigerwaltii subsp. nov., three new subspecies of clinical importance. J Clin Microbiol. 43, 3297–3303. doi: 10.1128/JCM.43.7.3297-3303.2005
Hölzel, C. S., Tetens, J. L., and Schwaiger, K. (2018). Unraveling the role of vegetables in spreading antimicrobial-resistant bacteria: A need for quantitative risk assessment. Foodborne Pathogens Dis. 15, 671–688. doi: 10.1089/fpd.2018.2501
Humann, J. L., Wildung, M., Cheng, C. H., Lee, T., Stewart, J. E., Drew, J. C., et al. (2011). Complete genome of the onion pathogen Enterobacter cloacae EcWSU1. Stand Genomic Sci. 5, 279–286. doi: 10.4056/sigs.2174950
Izdebski, R., Baraniak, A., Herda, M., Fiett, J., Bonten, M. J. M., Carmeli, Y., et al. (2015). MLST reveals potentially high-risk international clones of Enterobacter cloacae. J Antimicrob Chemother. 70, 48–56. doi: 10.1093/jac/dku359
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications [version 1; referees: 2 approved]. Wellcome Open Res. 3, 124. doi: 10.12688/wellcomeopenres.14826.1
Kämpfer, P., Nienhüser, A., Packroff, G., Wernicke, F., Mehling, A., Nixdorf, K., et al. (2008). Molecular identification of coliform bacteria isolated from drinking water reservoirs with traditional methods and the Colilert-18 system. Int J Hyg Environ Health. 211, 374–384. doi: 10.1016/j.ijheh.2007.07.021
Khanna, N., Kumar Ghosh, A., Huntemann, M., Deshpande, S., Han, J., Chen, A., et al. (2013). Complete genome sequence of Enterobacter sp. IIT-BT 08?: A potential microbial strain for high rate hydrogen production. Stand Genomic Sci. 9, 359–369. doi: 10.4056/sigs.4348035
Kohlmann, R., Bähr, T., and Gatermann, S. G. (2018). Species-specific mutation rates for ampC derepression in Enterobacterales with chromosomally encoded inducible AmpC β-lactamase. J Antimicrobial Chemother. 73, 1530–1536. doi: 10.1093/jac/dky084
Kohlmann, R., Bähr, T., and Gatermann, S. G. (2019). Effect of ampC derepression on cefepime MIC in Enterobacterales with chromosomally encoded inducible AmpC β-lactamase. Clin Microbiol Infect. 25, 1158. doi: 10.1016/j.cmi.2019.05.007
Kremer, A., and Hoffmann, H. (2012). Prevalences of the Enterobacter cloacae complex and its phylogenetic derivatives in the nosocomial environment. Eur J Clin Microbiol Infect Dis. 31, 2951–2955. doi: 10.1007/s10096-012-1646-2
Letunic, I., and Bork, P. (2019). Interactive tree of life (ITOL) v4: Recent updates and new developments. Nucleic Acids Res. 47, 256–259. doi: 10.1093/nar/gkz239
Liu, W. Y., Chung, K. M. K., Wong, C. F., Jiang, J. W., Hui, R. K. H., and Leung, F. C. C. (2012). Complete genome sequence of the endophytic Enterobacter cloacae subsp. cloacae strain ENHKU01. J Bacteriol. 194, 5965. doi: 10.1128/JB.01394-12
Luna-Guevara, J. J., Arenas-Hernandez, M. M. P., Martinez de la Peña, C., Silva, J. L., and Luna-Guevara, M. L. (2019). The role of pathogenic E. coli in fresh vegetables: Behavior, contamination factors, and preventive measures. Int J Microbiol. 2019, 2894328. doi: 10.1155/2019/2894328
Marti, R., Scott, A., Tien, Y. C., Murray, R., Sabourin, L., Zhang, Y., et al. (2013). Impact of manure fertilization on the abundance of antibiotic-resistant bacteria and frequency of detection of antibiotic resistance genes in soil and on vegetables at harvest. Appl Environ Microbiol. 79, 5701–5709. doi: 10.1128/AEM.01682-13
Matteoli, F. P., Passarelli-Araujo, H., Pedrosa-Silva, F., and Olivares, F. L. (2019). Genomics population structure and pangenome analysis of Enterobacter bugandensis uncover the presence of blaCTX–M–55, blaNDM–5 and blaIMI–1, along with sophisticated iron acquisition strategies. Genomics. 112, 1–10. doi: 10.1016/j.ygeno.2019.07.003
Mizrahi, A., Delerue, T., Morel, H., Le Monnier, A., Carbonnelle, E., Pilmis, B., et al. (2020). Infections caused by naturally AmpC-producing Enterobacteriaceae: Can we use third-generation cephalosporins? A narrative review. Int J Antimicrob Agents 55, 105834. doi: 10.1016/j.ijantimicag.2019.10.015
Paauw, A., Caspers, M. P. M., Schuren, F. H. J., Leverstein-van Hall, M. A., Delétoile, A., Montijn, R. C., et al. (2008). Genomic diversity within the Enterobacter cloacae complex. PLoS One. 3:e3108. doi: 10.1371/journal.pone.0003018
Paauw, A., Leverstein-van Hall, M. A., Van Kessel, K. P. M., Verhoef, J., and Fluit, A. C. (2009). Yersiniabactin reduces the respiratory oxidative stress response of innate immune cells. PLoS One. 4:0008240. doi: 10.1371/journal.pone.0008240
Pati, N. B., Doijad, S. P., Schultze, T., Mannala, G. K., Yao, Y., Jaiswal, S., et al. (2018). Enterobacter bugandensis: A novel enterobacterial species associated with severe clinical infection. Sci Rep. 8, 5392. doi: 10.1038/s41598-018-23069-z
Peirano, G., Matsumura, Y., Adams, M. D., Bradford, P., Motyl, M., Chen, L., et al. (2018). Genomic epidemiology of global carbapenemase-producing Enterobacter spp., 2008–2014. Emerg Infect Dis. 24, 2008–2014. doi: 10.3201/eid2406.171648
Pot, M., Guyomard-Rabenirina, S., Couvin, D., Ducat, C., Enouf, V., Ferdinand, S., et al. (2021). Dissemination of extended-spectrum-β-lactamase-producing Enterobacter cloacae complex from a hospital to the nearby environment in Guadeloupe (French West Indies): ST114 lineage coding for a successful IncHI2/ST1 plasmid. Antimicrob Agents Chemother. 65, 1–12. doi: 10.1128/AAC.02146-20
Rice, L. B. (2008). Federal funding for the study of antimicrobial resistance in nosocomial pathogens. No ESKAPE. J Infect Dis. 197, 1079–1081. doi: 10.1086/533452
Siebor, E., De Curraize, C., and Neuwirth, C. (2019). Identification of AGI1-A, a variant of Acinetobacter genomic island 1 (AGI1), in a French clinical isolate belonging to the Enterobacter cloacae complex. J Antimicrobial Chemother. 74, 311–314. doi: 10.1093/jac/dky442
Simão, A. F., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Singh, B. R., Singh, V., Ebibeni, N., and Singh, R. K. (2013). Antimicrobial and herbal drug resistance in enteric bacteria isolated from faecal droppings of common house lizard/gecko (Hemidactylus frenatus). Int J Microbiol. 2013, 340848. doi: 10.1155/2013/340848
Singh, N. K., Bezdan, D., Checinska Sielaff, A., Wheeler, K., Mason, C. E., and Venkateswaran, K. (2018). Multi-drug resistant Enterobacter bugandensis species isolated from the international space station and comparative genomic analyses with human pathogenic strains. BMC Microbiol. 18:175. doi: 10.1186/s12866-018-1325-2
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stock, I., Grüger, T., and Wiedemann, B. (2001). Natural antibiotic susceptibility of strains of the Enterobacter cloacae complex. Int J Antimicrob Agents. 18, 537–545. doi: 10.1016/S0924-8579(01)00463-0
Sutton, G. G., Brinkac, L. M., Clarke, T. H., and Fouts, D. E. (2018). Enterobacter hormaechei subsp. hoffmannii subsp. nov., Enterobacter hormaechei subsp. xiangfangensis comb. nov., Enterobacter roggenkampii sp. nov., and Enterobacter muelleri is a later heterotypic synonym of Enterobacter asburiae based on computational analysis of sequenced Enterobacter genomes. [version 2; referees: 2 approved]. F1000Research. 7, 521. doi: 10.12688/f1000research.14566.1
Uchida, A., Kim, H. L., Purbojati, R. W., Vettath, V. K., Gupta, A. B., Houghton, J. N. I., et al. (2020). Complete genome of Enterobacter sichuanensis strain SGAir0282 isolated from air in Singapore. Gut Pathog. 12, 12. doi: 10.1186/s13099-020-00350-z
van Hoek, A. H. A. M., Veenman, C., van Overbeek, W. M., Lynch, G., de Roda Husman, A. M., and Blaak, H. (2015). Prevalence and characterization of ESBL- and AmpC-producing Enterobacteriaceae on retail vegetables. Int J Food Microbiol. 204, 1–8. doi: 10.1016/j.ijfoodmicro.2015.03.014
Wang, C., Wu, W., Wei, L., Feng, Y., Kang, M., Xie, Y., et al. (2020). Enterobacter wuhouensis sp. nov. and Enterobacter quasihormaechei sp. nov. recovered from human sputum. Int J Syst Evol Microbiol. 70, 874–881. doi: 10.1099/ijsem.0.003837
World Health Organization. (2017). News release. WHO publishes list of bacteria for which new antibiotics are urgently needed. Geneva: World Health Organization.
Wu, W., Feng, F., and Zong, Z. (2020). Precise species identification for Enterobacter: A genome sequence-based study with reporting of two novel species, Enterobacter quasiroggenkampii sp. nov. and Enterobacter quasimori sp. nov. MSystems. 5, 1–16. doi: 10.1128/mSystems.00527-20
Wu, W., Wei, L., Feng, Y., Kang, M., and Zong, Z. (2019). Enterobacter huaxiensis sp. nov. and Enterobacter chuandaensis sp. nov., recovered from human blood. Int J Syst Evol Microbiol. 69, 708–714. doi: 10.1099/ijsem.0.003207
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Zhou, K., Yu, W., Cao, X., Shen, P., Lu, H., Luo, Q., et al. (2018). Characterization of the population structure, drug resistance mechanisms and plasmids of the community-associated Enterobacter cloacae complex in China. J Antimicrob Chemother. 73, 66–76. doi: 10.1093/jac/dkx361
Keywords: Anolis marmoratus, cephalosporinase overproduction, Enterobacter cloacae complex, ESBL, hsp60, one health, phylogeny, Caribbean
Citation: Pot M, Reynaud Y, Couvin D, Ducat C, Ferdinand S, Gravey F, Gruel G, Guérin F, Malpote E, Breurec S, Talarmin A and Guyomard-Rabenirina S (2021) Wide Distribution and Specific Resistance Pattern to Third-Generation Cephalosporins of Enterobacter cloacae Complex Members in Humans and in the Environment in Guadeloupe (French West Indies). Front. Microbiol. 12:628058. doi: 10.3389/fmicb.2021.628058
Received: 10 November 2020; Accepted: 31 May 2021;
Published: 25 June 2021.
Edited by:
Rustam Aminov, University of Aberdeen, United KingdomReviewed by:
Astrid Louise Wester, Norwegian Institute of Public Health (NIPH), NorwayAlain Hartmann, Institut National de Recherche pour l’Agriculture, l’Alimentation et l’Environnement (INRAE), France
Kai Zhou, First Affiliated Hospital of Southern University of Science and Technology, China
Copyright © 2021 Pot, Reynaud, Couvin, Ducat, Ferdinand, Gravey, Gruel, Guérin, Malpote, Breurec, Talarmin and Guyomard-Rabenirina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthieu Pot, bXBvdEBwYXN0ZXVyLWd1YWRlbG91cGUuZnI=