 Soledad Martos1*
Soledad Martos1* Sílvia Busoms1
Sílvia Busoms1 Laura Pérez-Martín1
Laura Pérez-Martín1 Mercè Llugany1
Mercè Llugany1 Catalina Cabot2
Catalina Cabot2 Charlotte Poschenrieder1
Charlotte Poschenrieder1- 1Plant Physiology Laboratory, Bioscience Faculty, Universitat Autònoma de Barcelona, Bellaterra, Spain
- 2Department of Biology, Universitat de les Illes Balears, Palma, Spain
Noccaea brachypetala is a close relative of Noccaea caerulescens, a model plant species used in metal hyperaccumulation studies. In a previous survey in the Catalan Pyrenees, we found two occidental and two oriental N. brachypetala populations growing on non-metalliferous soils, with accumulated high concentrations of Cd and Zn. Our hypothesis was that the microbiome companion of the plant roots may influence the ability of these plants to absorb metals. We performed high-throughput sequencing of the bacterial and fungal communities in the rhizosphere soil and rhizoplane fractions. The rhizobiomes and shoot ionomes of N. brachypetala plants were analyzed along with those from other non-hyperaccumulator Brassicaceae species found at the same sampling locations. The analyses revealed that microbiome richness and relative abundance tended to increase in N. brachypetala plants compared to non-hyperaccumulator species, regardless of plant location. We confirmed that the root compartment is a key factor in describing the community composition linked to the cohabiting Brassicaceae species, and the rhizoplane fraction contained the specific and rare taxa associated with each species. N. brachypetala plants harbored a similar relative abundance of fungi compared to the other plant hosts, but there was a notable reduction in some specific taxa. Additionally, we observed an enrichment in the hyperaccumulator rhizoplane of previously described metal-tolerant bacteria and bacteria involved in nitrogen cycling. The bacteria involved in the nitrogen cycle could contribute indirectly to the hyperaccumulator phenotype by improving soil quality and fertility. Our results indicate that N. brachypetala captures a particular prokaryotic community from the soil. This particular prokaryotic community may benefit the extraction of metal ions and/or improve plant nutrition. Our research identified satellite groups associated with the root niche of a hyperaccumulator plant that may assist in improving biological strategies in heavy metal remediation.
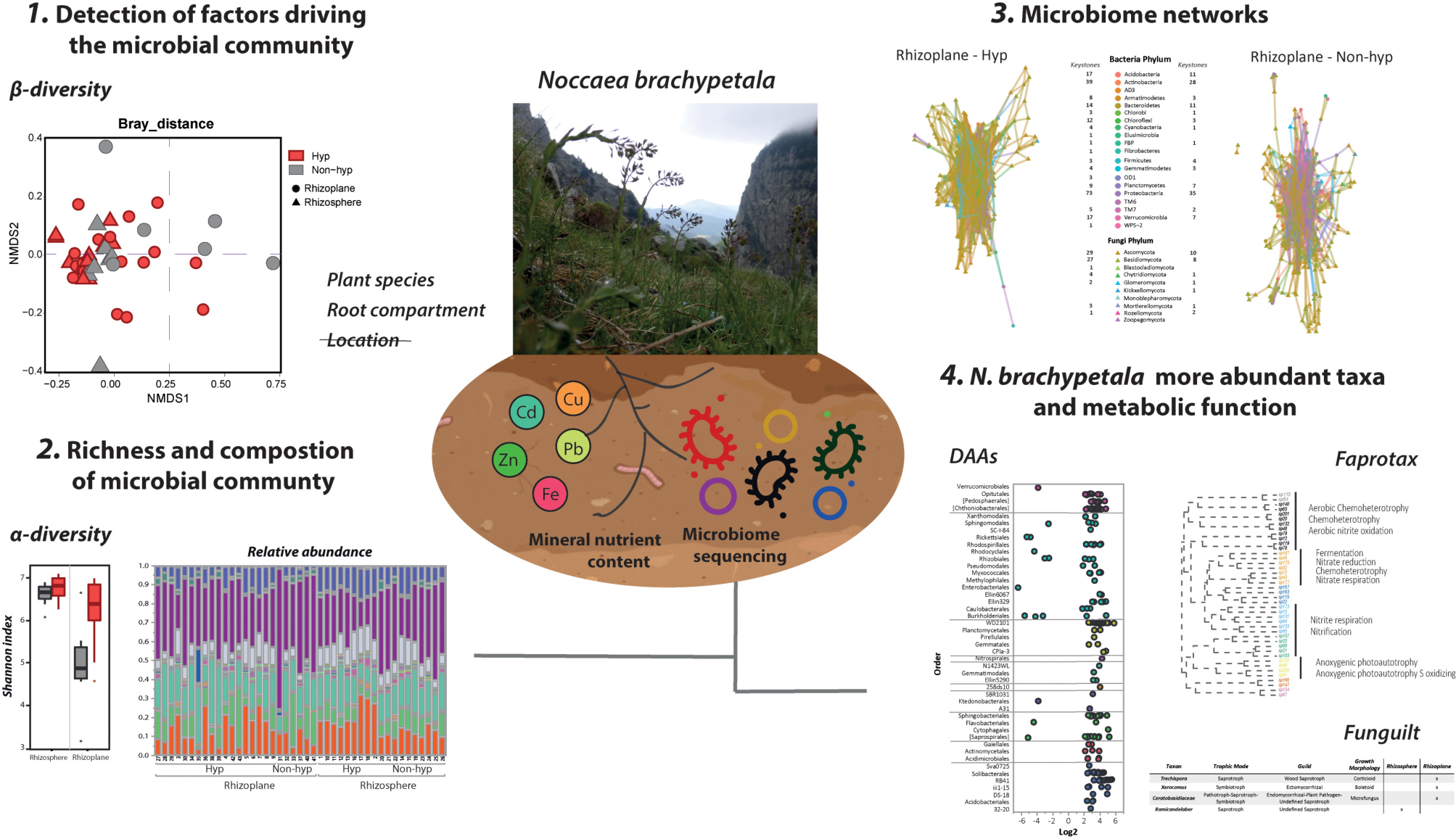
Graphical Abstract.
Introduction
Metal-rich soils are harsh environments for establishing life. However, metallophytes have evolved different strategies to survive under these restrictive growth conditions. Among these plant species, hyperaccumulators harbor metal concentrations within aerial tissues at levels far exceeding those present in their native soil. Without showing significant signs of toxicity, they can achieve 100-fold higher shoot metal concentrations than non-hyperaccumulator plants (Van der Ent et al., 2013). Hyperaccumulation appears to have evolved several times within vascular plants, and the Brassicaceae family contains a high percentage of the total hyperaccumulating taxa (Kramer, 2010). Noccaea caerulescens (formerly Thlaspi caerulescens) belongs to this family and is one of the best-known hyperaccumulators of Cd, Ni, and Zn (Assunçao et al., 2003). This genus contains several species, including Noccaea praecox (Likar et al., 2010), Noccaea camlikensis (Aksoy et al., 2015), Noccaea brachypetala (Martos et al., 2016), Noccaea kovatsii (Misljenovic et al., 2020), and Noccaea japonica (Nishida et al., 2020), which have also been described as metal hyperaccumulators. Interestingly, metallophytes are not restricted to metalliferous soils, and it has been suggested that Zn/Cd/Pb hyperaccumulation can evolve on non-metalliferous lands (Besnard et al., 2009). When growing on common soils, the metal concentrations in their shoots can achieve unusually high levels. The most popular theory suggests the evolutionary advantage of this behavior is an increased resistance to biotic stresses (Poschenrieder et al., 2006; Cappa and Pilon-Smits, 2014).
The ability to mobilize, chelate, and transport metals is directly linked to soil metal bioavailability and the genetic background of the plant. However, recent research highlights that plant-associated microorganisms can significantly enhance metal availability. The soil microbial community may greatly contribute to the hyperaccumulator phenotype (Visioli et al., 2015a). Various studies have reported a high diversity of microbes in the rhizosphere and endosphere (Thijs et al., 2017). Under unfavorable conditions of metalliferous soils, rhizosphere microorganisms may increase or decrease metal availability (Farinati et al., 2011). Previous studies have demonstrated both effects in Noccaea with root-bacteria favoring metal uptake (Whiting et al., 2001), whereas fungal mycorrhiza reduced plant absorption (Vogel-Mikus et al., 2006). However, a proper cohort of microbes may increase the potential of hyperaccumulators to extract metals. Various studies have confirmed that specific bacteria promote the uptake of Ni in N. caerulescens (Aboudrar et al., 2013; Visioli et al., 2014, 2015b). Therefore, rhizosphere-associated microbes from hyperaccumulators are a potential source of genotypes for biotechnological applications in phytoremediation and agriculture (Rajkumar et al., 2012). Characterization of the rhizobiome of hyperaccumulators is needed to enable the development of these applications. Despite this, a limited number of hyperaccumulator species have been investigated (Alford et al., 2010; Ma et al., 2011; Sessitsch et al., 2013). Within the genus Noccaea, the rhizosphere bacteria of Noccaea goesingense were characterized by Idris et al. (2004), and mycorrhizal colonization in N. praecox has been investigated (Pongrac et al., 2007). The microbiomes of other Noccaea hyperaccumulator species remain unknown.
Recent metagenomic studies revealed that soil properties are the major drivers for the differentiations in the assembly of root communities in Arabidopsis thaliana and in some related species, including the Cd-Zn hyperaccumulator, Arabidopsis halleri (Bulgarelli et al., 2012; Lundberg et al., 2012; Schlaeppi et al., 2014; Glynou et al., 2018). Plant genotype research remains conflicted. Some authors affirm that plant genotype is a key factor in microbiome assembly (Burns et al., 2015; Li et al., 2018); others do not consider this essential (Glynou et al., 2018; Macia-Vicente et al., 2020). Proximity to the root is also considered a relevant parameter that increases soil microbiota diversity and structure (Martinez-Diz et al., 2019; Macia-Vicente et al., 2020; Thiergart et al., 2020). Several studies have shown that plant exudates can favor specific microbiome communities mutualistically (Bulgarelli et al., 2013; Stringlis et al., 2018). Exudated organic compounds nurture rhizomicrobiota, allowing the root communities to modulate the plant phenotype. A well-reported case revealed an increased metal tolerance in plants associated with a metal-tolerant microbiome (Luo et al., 2014; Visioli et al., 2015a). However, the potential role of microbiota from non-metalliferous soils to the hyperaccumulation phenotype remains unknown. In the last few years, vast progress has been made in elucidating the composition and dynamics of root communities in the Brassicaceae family. However, additional studies in natural conditions are required. We investigated the impact of plant identity, location, and root compartment on the microbial diversity and community structure of the hyperaccumulator N. brachypetala and other Brassicaceae species (non-hyperaccumulators) coinhabiting undisturbed and non-metalliferous soils of the Catalan Pyrenees.
Materials and Methods
Plant and Soil Sampling
Plant and soil materials were collected from four sampling sites in the Catalan Pyrenees. Two sites were located in the Pallars region (Mauri and Alòs), and the other two were in the Ripollès region (Freser and Núria). All sites were located in the alpine biome of the Catalan Pyrenees (Figure 1A). Plants from Mauri and Aneu were grouped as Western populations (WPs), and Freser and Nuria plants were grouped as the Eastern populations (EPs). Both locations are separated by more than 150 km. The geographic coordinates of each sampling site are listed in Table 1.
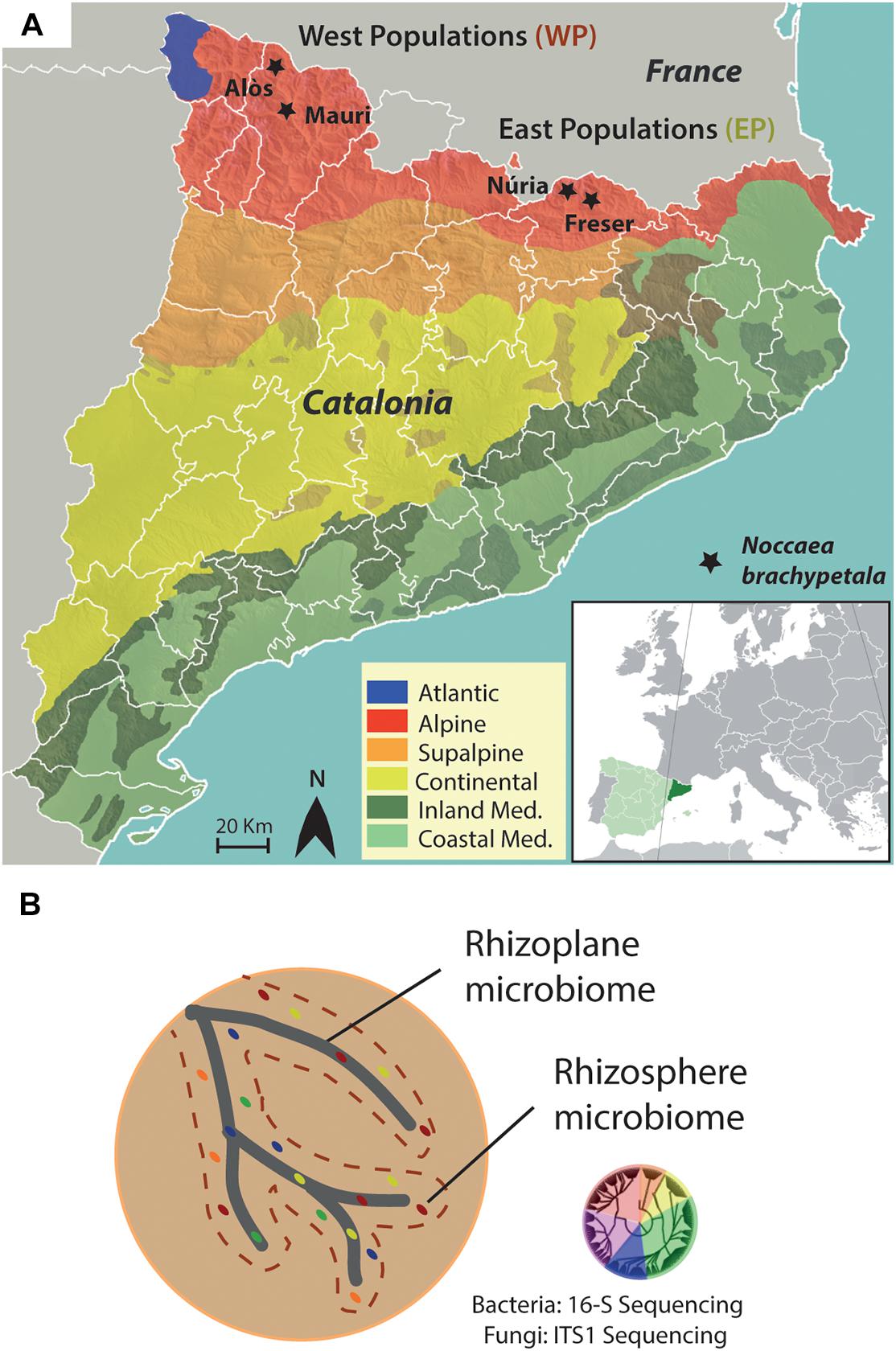
Figure 1. Experimental design. (A) Climatic map of Catalonia with the sampling locations of plants and soils indicated by stars. Western populations are composed form locations of Alòs and Mauri, whereas Eastern populations are from Núria and Freser. (B) Schematic drawing of the analyzed compartments of the root. DNA of the microbiome from the rhizoplane and rhizosphere was extracted followed by 16S and ITS sequencing of the bacteria and fungi, respectively.
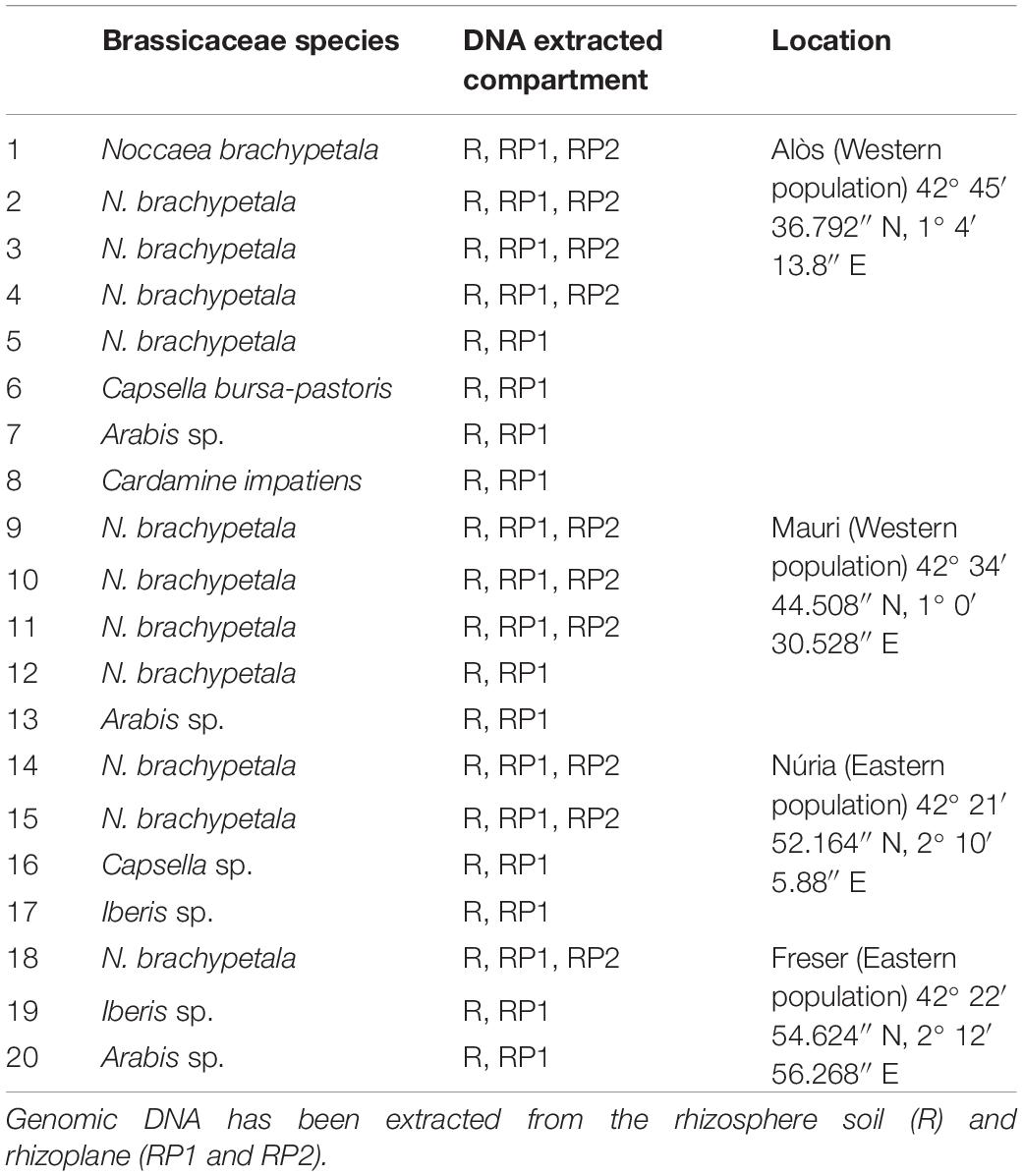
Table 1. Sampled plants for the rhizosphere microbioma sequencing.
At each site, N. brachypetala specimens [previously confirmed as Cd/Zn hyperaccumulators (Martos et al., 2016)] and other non-hyperaccumulator Brassicaceae species growing beside the specimens were also collected. All collectors wore gloves. A list of sampled species is provided in Table 1. Selected individuals were excavated with their rhizosphere soil without disturbing the plant root system. Plants were immediately transferred into polyethylene bags to avoid desiccation. From each site, bulk soil (five replicates per site) exempt from root or plant debris was collected at a depth of 10 cm using a 7-cm diameter Edelman drill (Eijkelkamp, Giesbeek, Netherlands). All collected plant and soil samples were refrigerated in a portable icebox and immediately transported to the laboratory for further processing.
Analysis of Metal Content From Soil and Plant Samples
Field-collected plants were separated into aerial and below ground tissue. All aerial tissue was carefully washed with distilled water and oven-dried at 60°C for 2 days. Dried shoots (three replicates/plant species) were finely powdered using a mortar and pestle. In parallel, collected bulk soils (five replicates per sampling site) were homogenized through a 2-mm sieve. Approximately 1 g of sieved bulk soil and 0.1 g of powdered shoots were acid-digested (HNO3: H2O2 69%: 30%, 5:2 vol/vol) in a hot-block digestion system (SC154-54-Well Hot BlockTM, Environmental Express, SC, United States). The concentrations of the selected metal elements (Cd, Cu, Pb, and Zn) and additional elements (Ca, Co, Fe, K, Mg, Mn, Mo, Na, P, and S) were determined by inductively coupled plasma (ICP) mass spectrometry (Perkin Elmer Inc., ELAN 6000, MA, United States) or ICP–optical emission spectrometry (Thermo Jarrell-Ash, model 61E Polyscan, England) (Bech et al., 2012). Internal references of the certified material BCR 62 Olea europaea and CRM 142 R Light Sandy Soil were included for quality control.
Statistical differences among groups in the element content of the soils or leaves were assessed using analysis of variance (two-way ANOVA). Normal distribution, homoscedasticity, and the independence of errors were checked prior to analyses. Data were log-transformed when necessary. Multiple comparisons of group means were analyzed by Tukey honestly significant difference using JMP® (version 13.0, SAS Institute Inc., 1989-2019).
Sampling of Rhizosphere and Rhizoplane Compartments
The soil adhering to the roots of each sampled specimen was separated into two fractions: rhizosphere and rhizoplane (Figure 1B). The sampling method for the rhizosphere and rhizoplane compartments followed the protocols described by Barillot et al. (2013) and Edwards et al. (2015), with minor modifications. In brief, the rhizosphere fraction was manually removed by vigorously shaking for 2 min until approximately 1 mm of soil was firmly attached to the roots. The rhizosphere fraction was suspended in 50 mL of 0.9% NaCl solution. Roots were then placed into sterile 100-mL tubes containing 50 mL of 0.9% NaCl solution and washed by manually shaking for 2 min (RP1 fraction). Roots were subsequently immersed in 0.9% NaCl solution and 0.01% Tween 80 and then washed by manually shaking for 2 min (RP2 fraction). Rhizosphere and rhizoplane RP1 and RP2 suspensions were then homogenized with gentle agitation (300g, OVAN Orbital Midi OM10E shaker, 25°C, 90 min) followed by centrifugation (5,000g, 25°C, 10 min) in sterile 100-mL tubes to concentrate soil particles into a pellet. Then, the supernatant was filtered through a 1-mm sieve to remove suspended residuals. Fractions were frozen in liquid nitrogen and stored at −80°C until required for DNA extraction.
Library Generation and Sequencing of Bacterial and Fungal DNA Regions
The total genomic DNA from the rhizosphere and rhizoplane compartments (approximately 200 mg) of the N. brachypetala specimens and the non-hyperaccumulator Brassicaceae species was extracted using the NucleoSpin® Soil kit (Macherey-Nagel GmbH & Co. KG, Düren, Germany), according to the manufacturer’s instructions. Extracted DNA was purified using the QIAamp DNA Stool Mini Kit (Qiagen, West Sussex, United Kingdom) and then quantified using the PicofluorTM method (PicoGreen dsDNA Quantitation Reagent, Molecular Probes, Inc., Eugene, OR, United States). Quality was measured using Nanodrop 2000 (Thermo Fisher Scientific, DE, United States) and confirmed by the OD A260/A280 between 1.8 and 2.0.
The prokaryotic 16S ribosomal RNA gene (16S rRNA) and the internal transcribed spacer (ITS) were the barcodes used for the metagenomic study of the bacteria and fungi, respectively. Microbial populations were obtained by high-throughput sequencing, targeting the V3–V4 region of 16S rRNA and ITS1 of the ITS region, using the MiSeq® Reagent Kit v2 (500 cycle) (MiSeq, Illumina, San Diego, CA, United States). The V3–V4 region was amplified using primers 341F (5′-CCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). The ITS1 region was amplified using primers ITSF (5′-CTTGGTCATTTAGAGGA AGTAA-3′) and ITS2 (5′-GCTGCGTTCTTCATCGATGC-3′).
Quality Control of Sequencing Data
Sequence reads of both datasets (16S and ITS) from the MiSeq Illumina® system were processed using QIIME v.1.9.1 (Caporaso et al., 2010). The quality filter of the already demultiplexed sequences was performed at a maximum unacceptable Phred quality score of Q20. The resulting reads were clustered into operational taxonomic units (OTUs) using uclust with 97% sequence similarity and the subsampling pick open reference method (Rideout et al., 2014) at 10% of the subsampled sequences. Seven samples with number of reads fewer than 5,000 or a percentage of chimeras higher than 70% were removed from the analysis (Supplementary Dataset 1). Rarefaction curves showed that 90.7% of the sequenced samples achieved a plateau zone, indicating that no additional sequencing effort was required (Supplementary Figure 1). After filtering, a total of 4,752,917 high-quality sequences were obtained from 43 samples: 2,442,668 from the 16S region (bacteria) and 2,310,249 sequences from the ITS region (fungi) (Supplementary Dataset 1). The media range of sequence reads per sample was 56,806 for bacteria and 53,726 for fungi.
Representative sequences were assigned to taxonomic groups using the bacterial 16S GreenGenes v.13.8 reference database at a 90% confidence threshold and sequence alignment using QIIME. The taxonomy of fungi/oomycetes was assigned to full ITS sequences from the UNITE_v2020 database (Nilsson et al., 2018) using the DADA2 software package (Callahan et al., 2016). To reduce artifacts in the dataset, we removed singletons, OTUs with relative abundances across all samples less than 0.005% (Bokulich et al., 2012), and OTUs assigned as chloroplasts or mitochondria. The OTU table was resampled to the minimum number of sequences per sample (Weiss et al., 2017), leading to a total of 3,110 OTUs in the bacterial domain and 1,060 OTUs in the fungal domain.
Diversity Metrics and Relative Abundance
Output data from QIIME and DADA2 were merged and used to obtain an OUT Phyloseq object generated with R (McMurdie and Holmes, 2012). The α diversity was calculated using the Shannon index. The Kruskal–Wallis significance test was used for all pairwise combinations (between root compartments and plant species).
Based on the OTU relative abundance, a β-diversity assessment was performed using Bray–Curtis metrics. The resulting distance/dissimilarity matrices were subjected to ordination using principal coordinate analysis.
Non-metric multidimensional scaling was analyzed based on the Bray–Curtis distances between samples using the Vegan package (Oksanen et al., 2013). The significance of each factor [root compartment, location, plant species, and extraction methodology (RP1, RP2)] and all the edaphological variables (nutrient mineral content) in the microbial composition were assessed by permutational multivariate ANOVA (PERMANOVA) using the Adonis function (R package vegan). Prior to analysis, edaphological data were standardized (Z scores) to avoid bias caused by the order of magnitude. All factors were independently analyzed. Factors that did not condition the microbiome of any analyzed group were not further analyzed.
The assessment of the taxonomic composition was performed at the phylum level using staked plots. To detect differences in bacterial and fungal community composition, we performed ANOVA pairwise comparisons between root compartments and plant species.
Co-occurrence Network
The OTU phyloseq object was converted into an adjacency matrix based on covariance by the spiec.easi() function of the SpiecEasi package (version 1.0.7) and using MB’s neighborhood selection and minimum λ threshold set to 0.01, for all networks. Bacteria and fungi domains were analyzed together, and independent networks were constructed for each root compartment and plant species. Fundamental OTU networks (keystones) were the highest “betweenness” and “node count” in more than 50% of the distribution. Each adjacency matrix was then converted into an igraph object and visualized as a network using the adj2igraph() and plot network() functions in SpiecEasi.
Differential Abundance Analysis
To establish which taxa were associated more commonly with N. brachypetala compared to non-hyperaccumulator species in each compartment, a differential abundance analysis (DAA) was performed using the DESeq2 package. This method models the observed abundances using negative binomial distribution after normalizing data with the corresponding scaling methods to account for differences in sampling fractions, recommended for small datasets (< 20 samples per group) (Paulson et al., 2013; Weiss et al., 2017; Lin and Peddada, 2020). Plot visualization was performed using the ggplot package.
Metabolic and Functional Analysis
To determine the metabolic potential of the N. brachypetala bacterial communities that were differentially abundant, a predictive functional analysis of bacterial communities was performed using FAPROTAX software (Louca et al., 2016). Similarly, functional annotation of the N. brachypetala–specific fungal communities was obtained using FUNGuild (Nguyen et al., 2016).
Results
Soil and Leaf Mineral Content
The analyses of the metal concentrations catalogued all the soil samples as non-metalliferous. The bulk soil collected with the different plant species revealed slight differences in the metal contents according to their location (EP/WP) (Figure 2A and Supplementary Dataset 2). For example, WPs of N. brachypetala accumulated higher levels of Cd and Zn compared to the EPs. However, the principal component analysis (PCA) of the soil samples did not differentiate the samples into location clusters (Figure 2B). The PCA of the ionomic data identified the hyperaccumulation trait as the key clustering factor in the samples of N. brachypetala with the other Brassicaceae species in independent groups (Figure 2C). The metal content in the leaves of N. brachypetala was 200 times higher for Cd and more than 100 times higher for Zn than for the non-hyperaccumulator species (Figure 2A and Supplementary Dataset 2). The data confirmed the Cd and Zn hyperaccumulator ability of N. brachypetala. Other elements, such as K, Cu, P, and Pb, were also increased in N. brachypetala leaves but at lower levels (from 1.5 to 10 times higher levels, Supplementary Dataset 2).
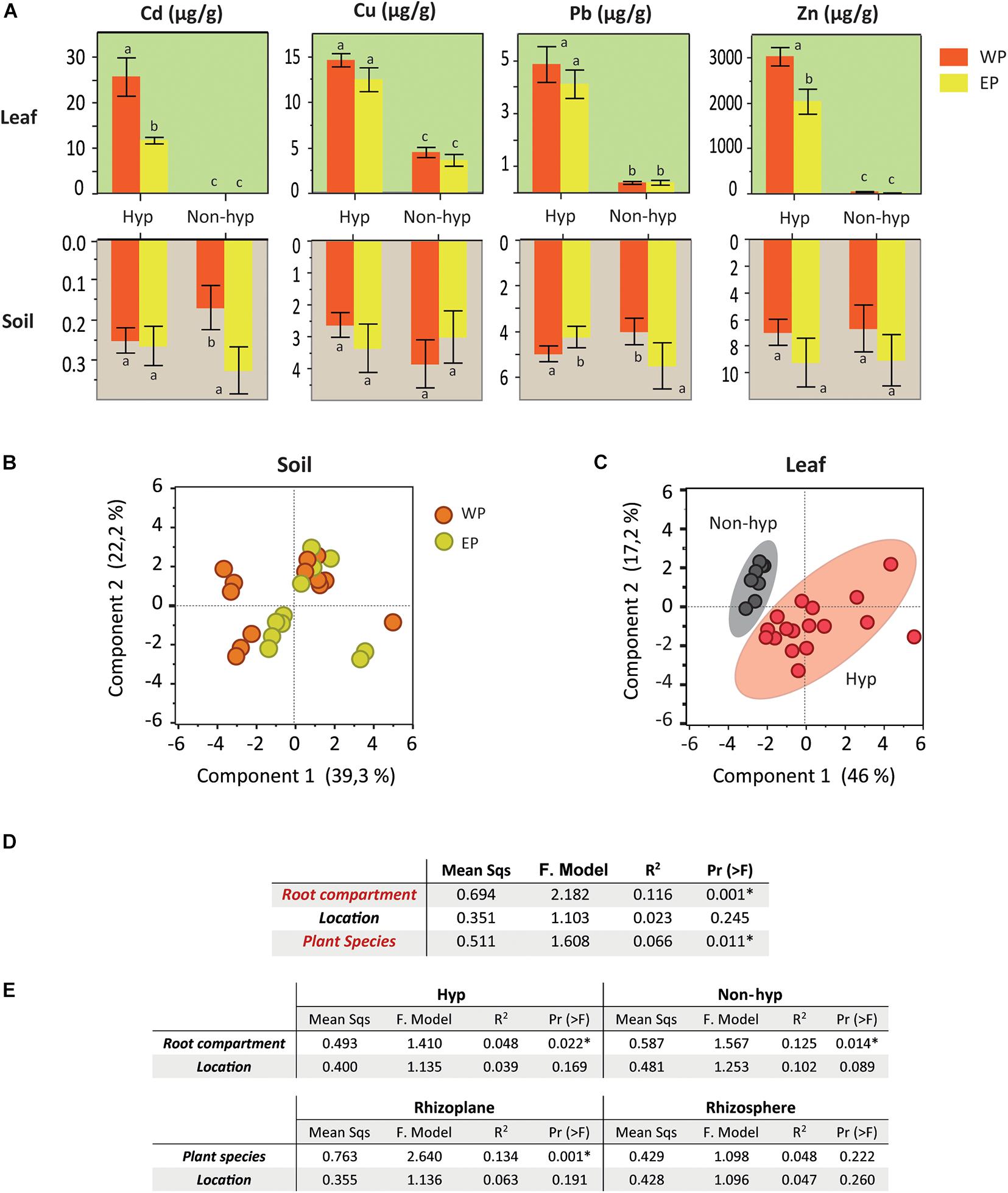
Figure 2. Soil and leaf nutrient mineral content and factors potentially influencing the microbiome. (A) Bar plots for soil and leaf metal content of hyperaccumulator and non-hyperaccumulator plants from Western (WP in orange) and Eastern populations (EP in yellow). Error bars correspond to the standard error. Different letters indicate statistically significant differences among treatments at P < 0.05. (B) Principal component analysis of the soil mineral content in the Western (orange dots) and Eastern populations (yellow dots). (C) Principal component analysis of the leaf mineral content. Hyperaccumulator plants are clustered in red and non-hyperaccumulator species in gray. (D) Summarized table of the PERMANOVA on the effect of the soil mineral content, root compartment, plant species, and location on the microbiome community (grouping bacteria and fungi). (E) Summarized table of the PERMANOVA on the effects of location and root compartment or plant species on the microbiome community (grouping bacteria and fungi).
Root Compartment and Plant Species Condition the Microbial Composition
The statistical analysis (PERMANOVA) of the OTUs from the bacteria and fungi domains revealed that the root compartment and plant species were relevant factors in explaining the variability of microbiome data (Figure 2D). However, the weight of the factor “plant location” was not significant. The effect of location was irrelevant even when plant species and root compartments were analyzed separately (Figure 2E).
Additionally, we evaluated the effect of the extraction method on the rhizoplane. The multivariate ANOVA confirmed no statistically significant difference between the extraction methods (RP1 and RP2) (Supplementary Datasets 1,2). Therefore, we selected root compartment and plant species as factors to investigate their effects on microbial diversity, structure, and composition.
High Influence of Root Compartment and Plant Species in the Bacterial Community
Root-associated microbes were divided into bacteria and fungi to investigate factors that could drive the microbial community within each domain (Figures 3A–C). The root compartment and plant species in the bacterial composition possessed larger proportions of variation (19%, P < 0.01, and 12%, P < 0.05, respectively) (Figures 3A,C). In contrast, the Bray–Curtis analysis indicated that the root compartment was a unique and significant factor describing the fungal composition, whereas the plant species was not relevant (Figures 3B,D).
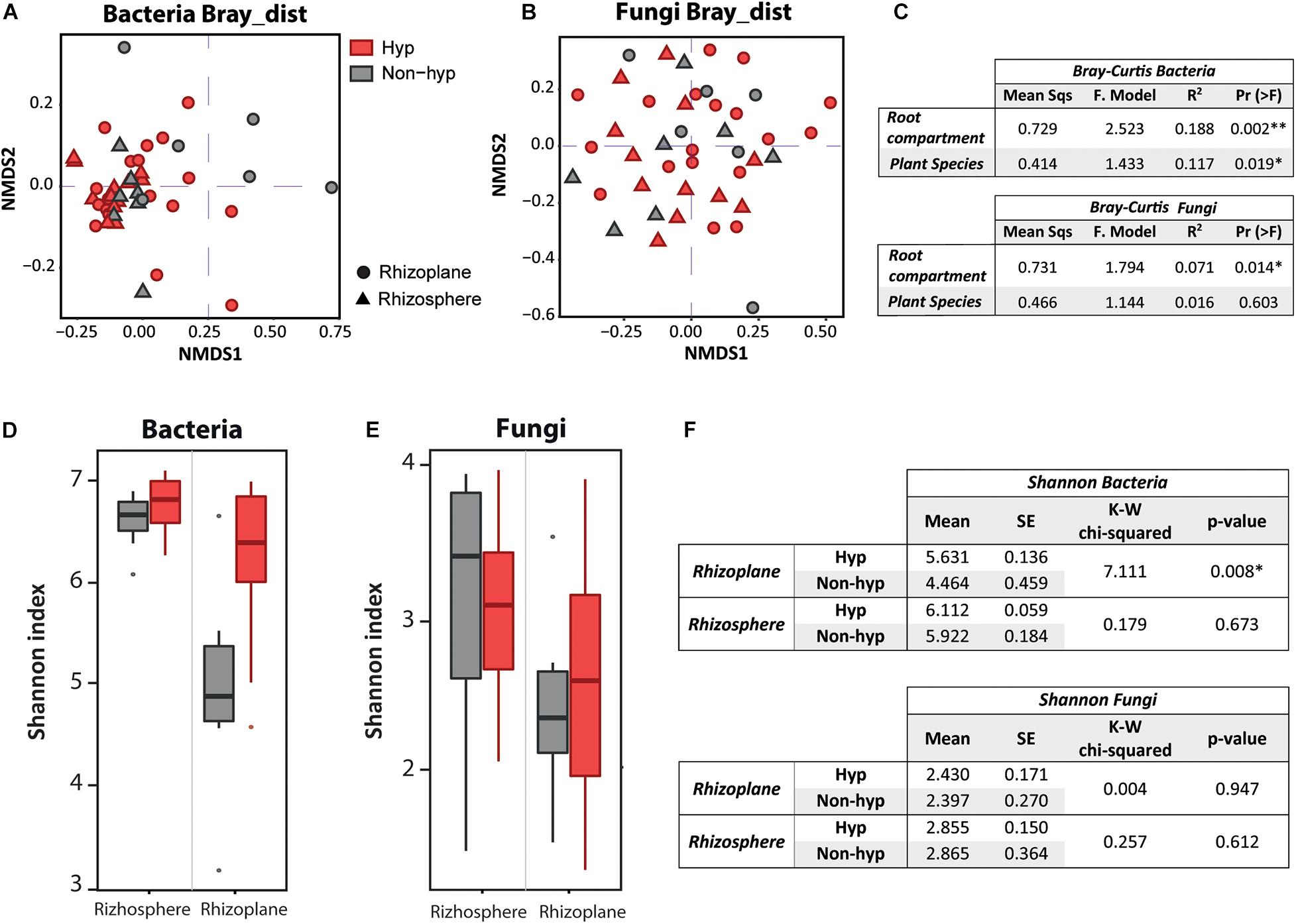
Figure 3. Factors influencing the bacterial and fungal community and diversity. Non-metric multidimensional scaling visualization of the Bray–Curtis results of the bacterial domain (A) and fungal domain (B). Plant species are differentiated by color (red is the hyperaccumulator; gray is the non-hyperaccumulator), and root compartments by shape (dots represent the rhizoplane; triangles represent the rhizosphere). (C) Summarized table of the PERMANOVA of the Bray–Curtis distances on the effect of root compartment and plant species for the bacterial and fungal domain, separately. (D) α Diversity boxplot of the bacterial domain. (E) α Diversity boxplot of the fungal domain. The Shannon index divided by root compartment and plant genotype. The x axis indicates root compartment, whereas plant genotype is differentiated by color (red is N. brachypetala; gray represents non-hyperaccumulator species). (F) Summarized table of the Kruskal–Wallis analysis from the Shannon index on the effect of root compartment of the bacterial and also the fungal domain.
The α-diversity boxplots (using the Shannon index) showed higher diversity in the rhizosphere than in the rhizoplane compartment for both microbial groups (Figures 3D,E). Moreover, rhizoplane diversity in the bacteria community was increased in N. brachypetala (hyperaccumulator) compared to the non-hyperaccumulator species (P < 0.01, Figure 3F). Fungal diversity did not vary between plant species (P > 0.05, Figure 3F).
More Complex Network in the Rhizoplane of N. brachypetala
The interaction patterns of the overall microbiome were analyzed and revealed that the hyperaccumulator networks were globally more interconnected than the non-hyperaccumulator networks (Figures 4A–D). N. brachypetala established more connections among phyla, as indicated by the increased number of total edges when compared to the other species (Figure 4E). Interestingly, the rhizoplane of the hyperaccumulator displayed the most complex interaction network with more nodes and edges (Figures 4A,E). In all the represented networks, the central module was composed of bacterial phyla (circles) surrounded by fungal phyla (triangles) (Figures 4A–D).
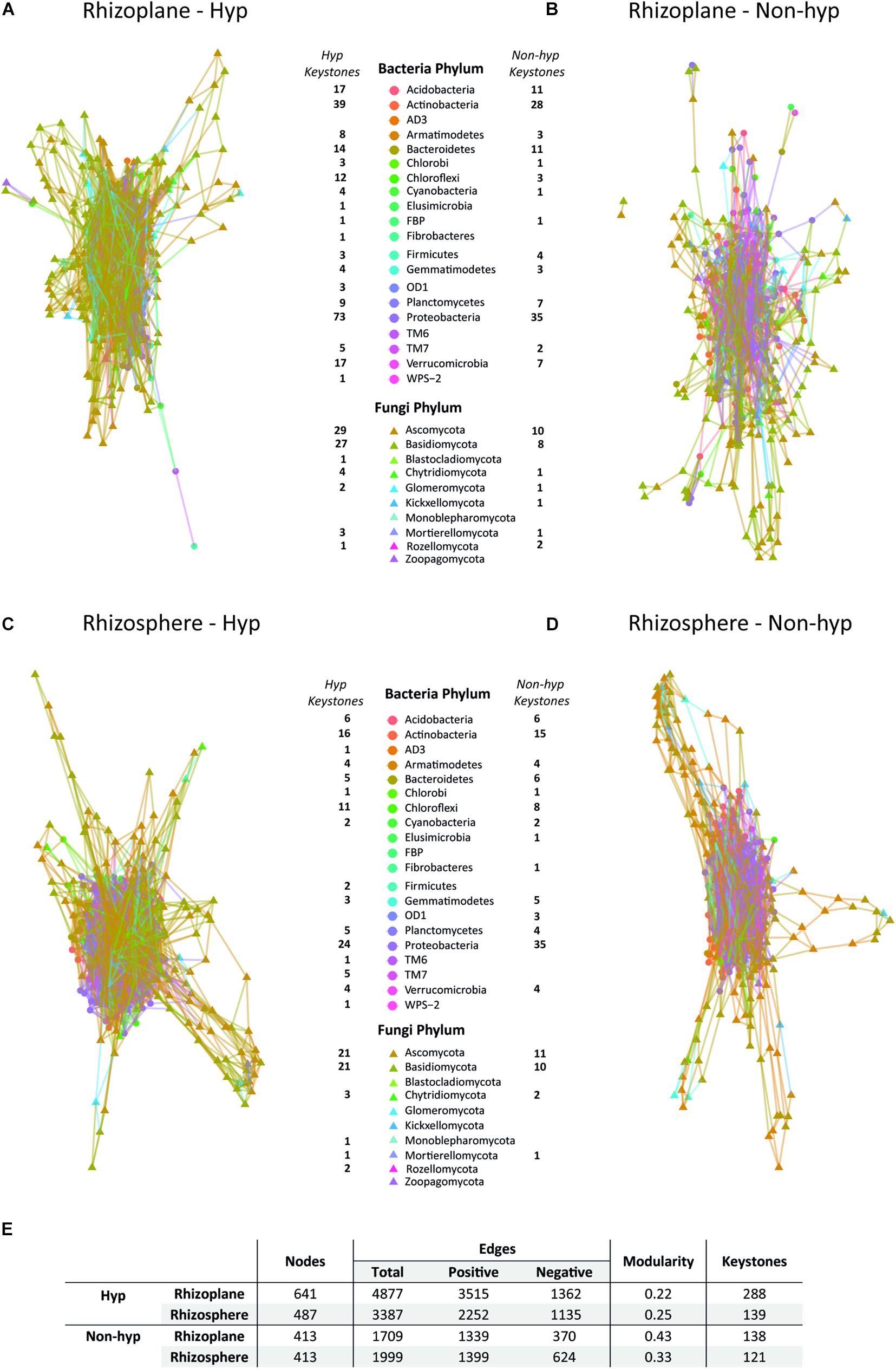
Figure 4. Co-occurrence network. Potential relationship among the bacterial and fungal OTUs in the rhizoplane (A,B) and the rhizosphere (C,D) of N. brachypetala (Hyp) (A,C), and the non-hyperaccumulator species (Non-hyp) (B,D). (E) Table with the main parameters of the co-occurrence networks.
The most important OTUs for the overall structure (keystones) were identified. The N. brachypetala rhizoplane presented the highest number of keystones. Proteobacteria and Actinobacteria species and Ascomycota and Basidiomycota species may play an important role in plant-microbial feedback as they possess the largest number of keystones in the four networks analyzed (Figures 4A–D).
Higher Relative Abundance of Bacteria and Fungi in N. brachypetala With Some Exclusive Taxa
To detect differences in the composition of bacterial and fungal communities, the absolute abundance was transformed into relative values. Data were clustered by phyla and grouped by hyperaccumulator/non-hyperaccumulator and rhizosphere/rhizoplane (Figure 5). The relative abundance of each sample is shown in Supplementary Figure 3. The bacterial domain was dominated by six major phyla: Acidobacteria, Actinobacteria, Bacteroidetes, Planctomycocetes, Proteobacteria, and Verrucomicrobia, comprising 90.98% of the total relative abundance (Figures 5A,B). The remaining identified phyla (22) showed a relative abundance lower than 3% and were considered rare phyla. Confronting the relative abundance between plant groups, four phyla (Fibrobacteres, Gemmatimodetes, Planctomycetes, and Verrumicrobia) showed significantly higher relative abundance in the rhizoplane of the hyperaccumulator compared to the non-hyperaccumulators (Figure 5A). Additionally, two phyla (BRC1 and OP11) were absent on the non-hyperaccumulator rhizoplane but were present on the hyperaccumulator. Hereafter, we refer to these as “exclusive taxa” of N. brachypetala (Figure 5A). Three more phyla (Acidobacteria, Elusimicrobia, and WS3) were more abundant in the rhizosphere of the hyperaccumulator. In contrast, the phyla Fibrobacteres and Proteobacteria exhibited increased abundance in the non-hyperaccumulator rhizosphere and rhizoplane, respectively. The phylum Fibrobacteres was in a higher abundance in the rhizosphere of the non-hyperaccumulator and, at the same time, had a larger presence in the rhizoplane of the hyperaccumulator. To sum up, six rare and three dominant phyla differed between N. brachypetala and the non-hyperaccumulators in both root compartments.
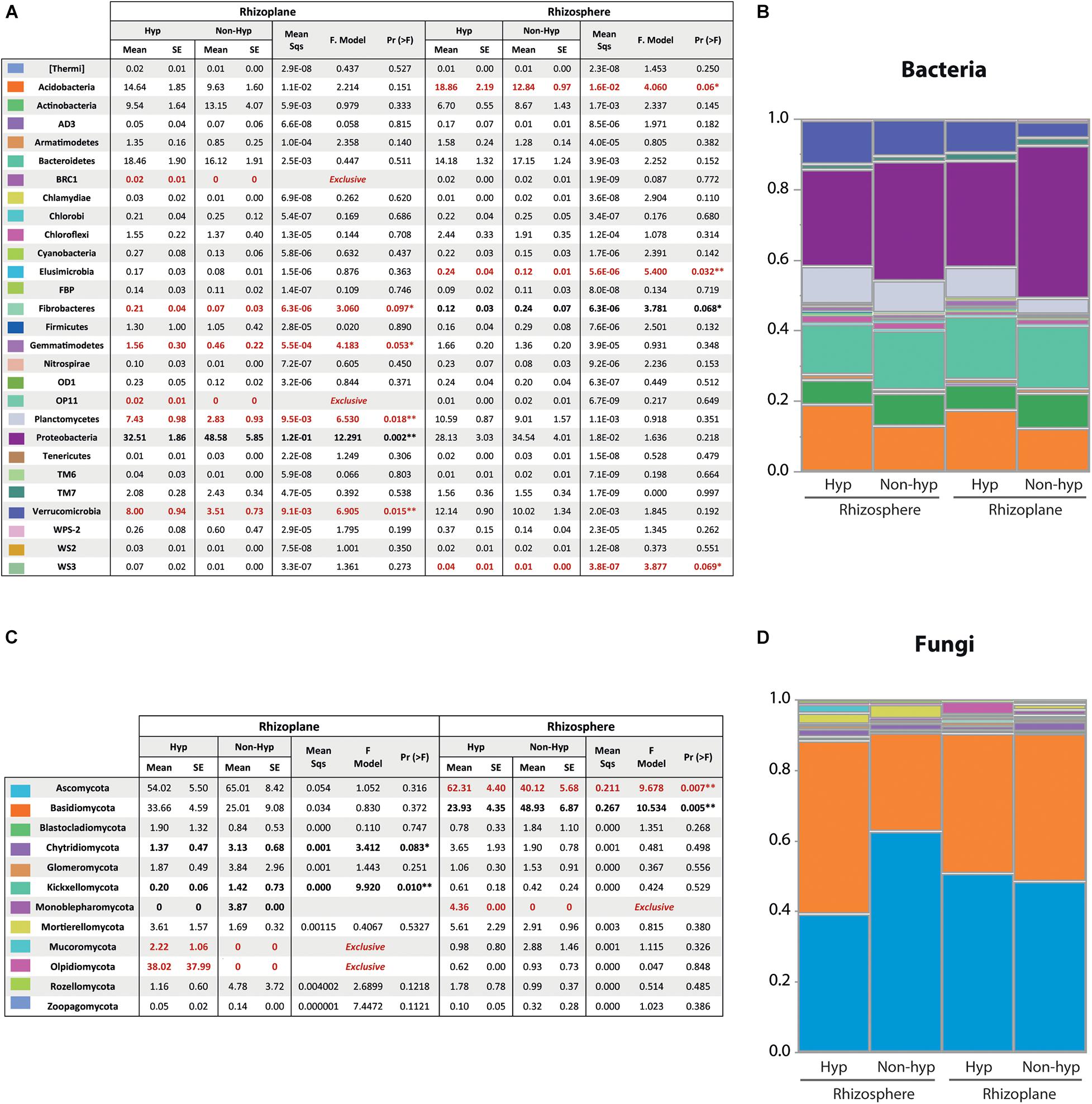
Figure 5. Bacterial and fungal community composition and relative abundance. (A) Means and standard errors (SE) of the relative abundances of the bacteria domain at phylum level. Differences and summarized results of an ANOVA pairwise comparison between N. brachypetala and non-hyperaccumulators at each soil compartment were calculated by the log2 ratio. (B) Bacterial relative abundance at the phylum level in a staked plot. The x axis indicates root compartment and plant genotype. (C) Means and standard errors (SE) of the relative abundances at phylum level on the fungi domain. Differences and summarized results of an ANOVA pairwise comparison between N. brachypetala and the non-hyperaccumulators at each soil compartment calculated by the log2 ratio. (D) Fungal relative abundance (at the phylum level) in a staked plot. The x axis indicates the root compartment and plant genotype.
Regarding the fungal domain, the dominant phyla were Ascomycota (53%) and Basidiomycota (36%) (Figures 5C,D). The remaining identified phyla (10) had a combined relative abundance of less than 3% (Figure 5C). The relative abundance of each sample is provided in Supplementary Figure 3. The hyperaccumulator harbored two exclusive fungal phyla (Mucoromycota and Olpidiomycota) in the rhizoplane and one (Monoblepharomycota) in the rhizosphere (Figure 5C). The major phyla were in different composition according to the plant host. Ascomycota were more abundant in the rhizosphere of the hyperaccumulator, whereas Basidiomycota had a larger presence in the rhizosphere of the non-hyperaccumulators. Conversely, in the non-hyperaccumulators, three phyla were more abundant in their rhizoplane (Chytridiomycota, Kickxellomycota, and Monoblepharomycota) and one (Basidiomycota) in the rhizosphere. Additionally, six minor phyla were composed differently between the plant species.
Generally, the taxa abundance by prevalence revealed that the most abundant bacteria were also the most prevalent. In contrast, the most abundant species in the fungal domain exhibited a lower prevalence (Supplementary Figure 2).
The Bacterial Community of N. brachypetala Is Globally More Diverse With Higher Relative Abundance in the Rhizoplane Compartment
To gain further insight into the taxa distribution, the bacterial OTUs were subjected to a DAA comparing N. brachypetala with the non-hyperaccumulator species (Figure 6). The DAA results were presented at the taxonomic level of order, the lowest level that was visually informative. The bacterial analyses of the rhizoplane showed an important accumulation of dots with positive log2 values, indicating a higher abundance of bacteria (by order) in N. brachypetala (Figure 6A). The highest number of differentially abundant bacteria belonged to five dominant phyla (Acidobacteria, Bacteroidetes, Planctomycetes, Proteobacteria, and Verrucomicrobia). Four rare phyla (Chloroflexi, Fibrobacteres, Gemmatimonadetes, and Nitrospirae) were also abundant in the hyperaccumulator rhizoplane. At the taxonomic level of family, N. brachypetala presented 108 families in the rhizoplane and 112 in the rhizosphere with a higher relative abundance than in the non-hyperaccumulator species (Supplementary Dataset 3). The number of more abundant families associated with the non-hyperaccumulator species was in an order of magnitude lower, of 20 and 15 in the rhizoplane and rhizosphere, respectively. The most prevalent taxa appearing repetitively were Candidatus Solibacter and the genera Flavobacterium, Koribacter, Opitutus, Planctomyces, Pseudomonas, and Streptomyces (Supplementary Dataset 3).
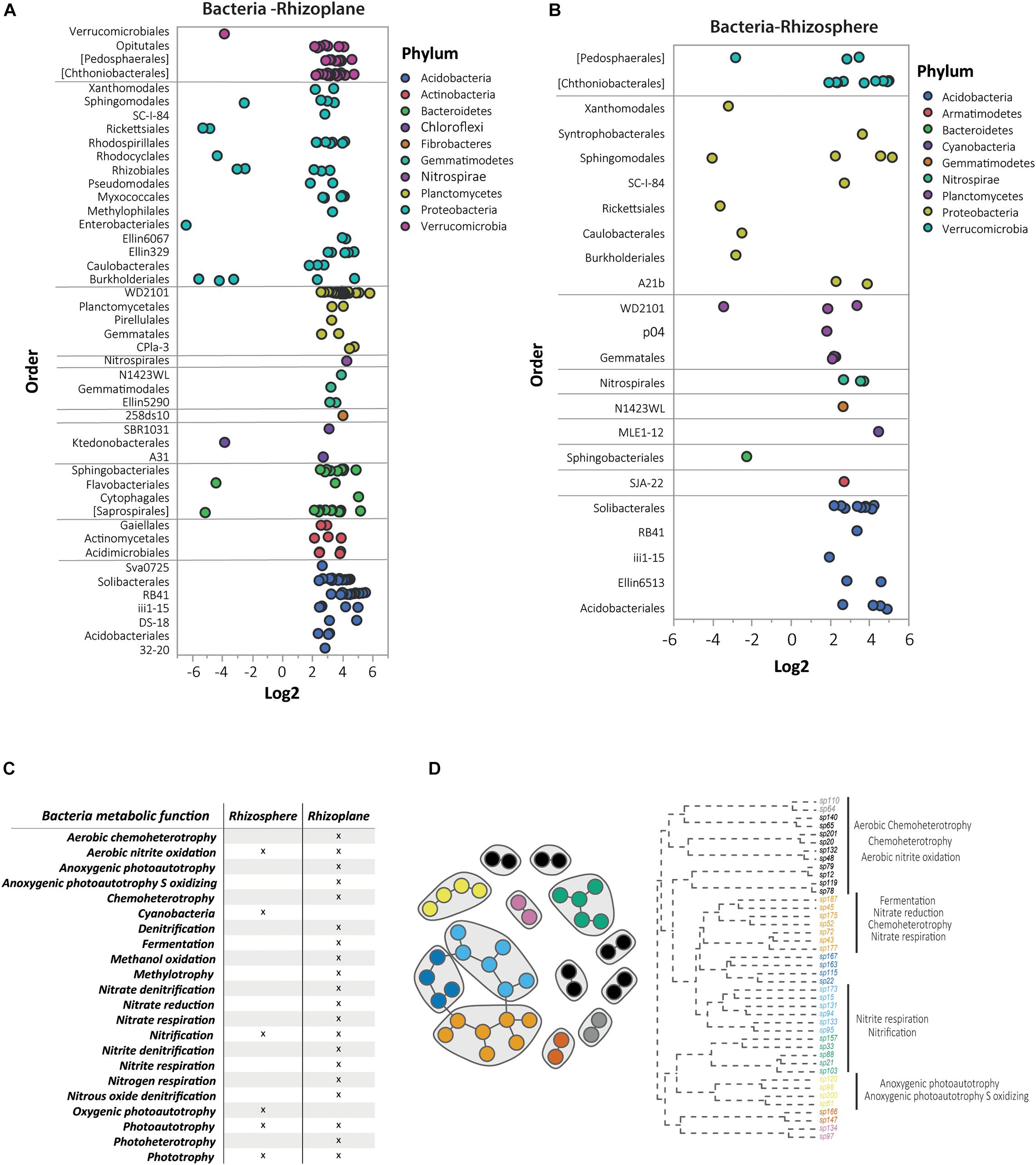
Figure 6. Differential abundance analysis (DAA) and metabolic functions of bacterial OTUs in N. brachypetala and non-hyperaccumulator species. Bubble plots of the differential abundant bacterial OTUs on the rhizoplane (A) and rhizosphere (B). The x axis indicates the log2 ratio (hyperaccumulators vs. non-hyperaccumulators). The y axis indicates bacteria at the taxonomical level of order. Bubble colors indicate the associated phylum. (C) The co-occurrence network of the DAA bacterial OTUs in the rhizoplane of N. brachypetala and a dendrogram with the annotated functions. (D) Table of metabolic functions of the DAA bacteria in N. brachypetala in each root compartment.
The DA OTUs from N. brachypetala were explored to identify their metabolic functions (Figures 6C,D). Functional analysis of the hyperaccumulator bacteria identified 22 metabolic functions. The most representative functions were those related to the nitrogen cycle (Figure 6C). The rhizoplane with 20 of 22 obtained functions was the most diverse functional root compartment. Rhizoplane bacteria were involved in functions related to the nitrogen cycle, whereas the rhizosphere bacteria were exclusively linked to cyanobacteria and oxygenic photautotrophy (Figure 6C). The interaction network in the DA bacteria of the rhizoplane and the associated functions are provided in Figure 6D. The observed pattern revealed that interconnected bacteria were involved in several metabolic functions. Aerobic nitrite oxidation was assigned to isolated bacteria (black and gray dots), with the nitrification and nitrite respiration assigned to non-connected bacteria (blue and green dots). Therefore, the functions were not diversified within the connected groups.
The Fungal Community of N. brachypetala Presents a Reduced Differential Abundance Compared to the Non-hyperaccumulator Species
The DAA of the fungi revealed more abundant fungi (by order) in both root compartments of the non-hyperaccumulator species (Figures 7A,B). Some of the limited fungal species in the rhizoplane of N. brachypetala were Candida railenensis, Clarireedia bennettii, Oidiodendron chlamydosporicum, Trechispora byssinella, and Acremonium rutilum (Supplementary Dataset 3).
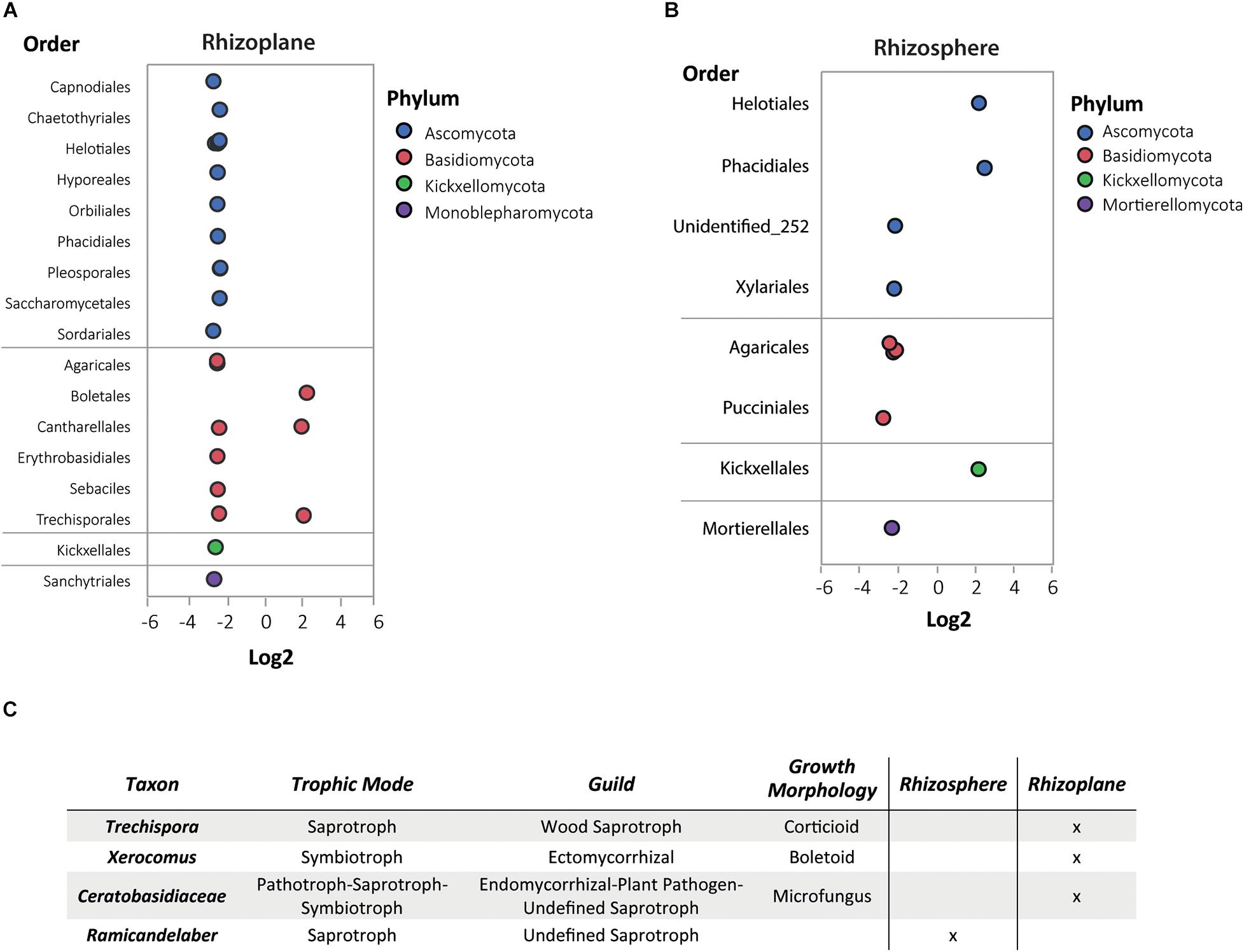
Figure 7. Differential abundance analysis (DAA) and guilds of fungal OTUs in N. brachypetala and non-hyperaccumulator species. Bubble plots of the differential abundant fungal OTUs in the rhizoplane (A) and rhizosphere (B). The x axis indicates the log2 ratios (hyperaccumulators vs. non-hyperaccumulators). The y axis indicates fungi at the taxonomical level of order. Bubble colors indicate the associated phylum. (C) Table of trophic mode and guilds of DAA fungi in N. brachypetala in each root compartment.
Conversely, we also detected seven fungi OTUs exclusive to the N. brachypetala plants (Supplementary Dataset 3). To our knowledge, there are no tools available to classify fungal communities into metabolic functional groups, but FUNGuild (Nguyen et al., 2016) predicts function at the guild level. Figure 7C indicates that the four fungal OTUs exhibit different trophic strategies. Among them, the genus Xerocomus deserves highlighting because it is a symbiont described as an ectomycorrhiza.
Discussion
Microbiota associated with plant roots are considered important mediators of plant–soil interactions. In the case of metal hyperaccumulator plants, rhizosphere bacteria have been associated with positive impacts on plant growth and increased metal accumulation (Visioli et al., 2015a). Therefore, the root microbiome of hyperaccumulators has attracted increased attention for potential biotechnological applications in phytoremediation. Despite the interest, there is little information on the root–microbiota communities and their associated functions in most hyperaccumulators (Thijs et al., 2017). Additionally, attention has been focused on hyperaccumulators from metalliferous soils, and those inhabiting non-polluted soils remain practically unexplored. Our study aimed to identify the bacterial and fungal taxa and their functional attributes associated with N. brachypetala plants from non-metalliferous soils. Microbia identification of field samples was performed using high-throughput sequencing in order to identify novel taxa that could be involved in metal hyperaccumulation in a cultivation-independent manner.
According to our results, the hyperaccumulator, N. brachypetala, exhibited an increased richness in the bacterial community compared to the companion non-hyperaccumulator Brassicaceae species. This hyperaccumulator also presented the most complex and highly interconnected interaction network. In our study, the rhizoplane of N. brachypetala harbored a higher relative abundance of bacterial OTUs, with distinct rare taxa. Low-abundance taxa have been recognized to provide critical functions in ecosystems and represent a vast functional gene pool (Jousset et al., 2017; Compant et al., 2019; Xiong et al., 2020). Therefore, the identification of satellite groups associated with the root niche of a hyperaccumulator plant growing on natural and undisturbed soil is highly interesting. For example, Nitrospiraceae and Gemmatimodaceae (which are more abundant in the rhizoplane of N. brachypetala) have been previously associated with the rhizosphere of the Cd/Zn hyperaccumulator A. halleri (Muehe et al., 2015). The metal(loid)-tolerant phylum Chlamydiae (Schneider et al., 2017) also increased in the rhizoplane of our hyperaccumulator. Additionally, the rhizosphere of N. brachypetala includes Thermogemmatisporaceae, which was formerly detected in high concentrations in the reclaimed soil of an ancient mine (Pershina et al., 2020), and Moraxellaceae, which includes heavy metal resistant species (Chen et al., 2015).
Ultimately, there is a substantial variation in the consortia of microbes in the taxonomically related plants that cohabit the undisturbed soils, but differ in the hyperaccumulator phenotype. Considering that the cited microbial taxa are much less abundant in the soil fraction associated with non-hyperaccumulator plants, our results suggest a selection of metal-tolerant bacteria exist in the root fractions of N. brachypetala. The metal tolerance of microbia colonizing the rhizosphere of hyperaccumulators in common soils was not the focus of this research. Therefore, it is an interesting topic for future studies on microbial cultivation.
Our results from the DAA identified Candidatus Solibacter (Acidobacteria) as one of the most prevalent bacteria in the rhizoplane of N. brachypetala growing on non-metalliferous soil. C. Solibacter has been identified in the core of the bacterial community in soils polluted with heavy metals (Golebiewski et al., 2014; Wang et al., 2018). Kaistobacter (which is associated with hyperaccumulators) has been similarly described in contaminated soils (Wu et al., 2019). Another abundant genus was Streptomyces, which has been previously affiliated with the plant Sedum alfredii in Cd-polluted soil (Hou et al., 2018). We also identified Flavobacterium with a high DAA ratio, which has been associated with metal accumulation in different hyperaccumulators (Idris et al., 2004; Luo et al., 2011, 2017; Zhang et al., 2012). Any of the differentially abundant bacteria of N. brachypetala may be assigned as metal solubilizers because the applied software (FAPROTAX) did not include this functional category.
A distinctive functional trait of the bacterial communities in N. brachypetala is their role in the nitrogen cycle. This function is not a direct mechanism of metal acquisition or tolerance. However, increased nitrogen availability can be advantageous for plants growing in metal-polluted soils. Nitrate assimilation is reduced under moderate metal exposure, confirming that metal toxicity alters nitrogen uptake by plants (Wang et al., 2008). Interestingly, co-cropping of legumes with hyperaccumulators has been reported to improve soil quality and fertility and increase metal yield (Saad et al., 2016, 2021). Our results provide a potential specialized rhizosphere bacterial community that facilitates N recycling. The plants in our study inhabited non-polluted sites; therefore, bacterial selection should occur regardless of the soil metal content. To confirm this hypothesis, it will be necessary to study the microbial community of N. brachypetala under metal exposure.
Our results identified bacteria contrasts with the DAA ratios for mycobiota between hyperaccumulator and non-hyperaccumulator species. Fungal species were generally much more enriched in the non-hyperaccumulator species than in the hyperaccumulator. However, it cannot be concluded that N. brachypetala was globally deficient in fungi, as the relative abundance for all plant species was high. Hyperaccumulator plants harbored seven exclusive fungal taxa. Nonetheless, certain specific fungal biota were absent in the hyperaccumulator. If we assume that the distribution of the root-associated microorganisms is not a hazardous process, we can speculate a potential positive selection in the rhizoplane of N. brachypetala toward certain bacteria with a detrimental effect on specific fungal groups. Among the exclusive fungi in the hyperaccumulator, Xerocomus could positively affect the functioning of N. brachypetala as it is a symbiotroph annotated as ectomycorrhizal species. Ectomycorrhizal fungi can help the hyperaccumulator phenotype through chemical immobilization in the cell wall or complexation with organic exudations (Luo et al., 2014). The Brassicaceae family has a high percentage of non-mycorrhizal species (Wang and Qiu, 2006), but with exceptions such as N. praecox (a Zn, Cd, and Pb hyperaccumulator), which, under field conditions, is colonized by mycorrhiza during the developmental stages that are coincident with low glucosinolate levels (Vogel-Mikus et al., 2005; Pongrac et al., 2008). Our DA analysis revealed that Xerocomus subtomentosus was 100 times more abundant in the rhizoplane of N. brachypetala. However, the confirmation of functional root colonization requires further analysis. To the best of our knowledge, any of these increased fungal taxa have been previously related to metal-polluted soils or hyperaccumulator species. Only A. rutilum and O. chlamydosporicum have been described as tolerant to abiotic stresses such as salinity and acidic pH, respectively (Fritze and Baath, 1993; Georgieva et al., 2012). These taxa deserve further investigation for their potential contributions to the hyperaccumulation phenotype.
Our results demonstrated the root compartment (rhizosphere vs. rhizoplane) and plant host (N. brachypetala vs. non-hyperaccumulators) were the major factors influencing the microbiome diversity, structure, and composition. Previous studies have revealed that microbial communities are strongly influenced by plant roots increasing toward the root (Bulgarelli et al., 2012; Lundberg et al., 2012; Martinez-Diz et al., 2019; Thiergart et al., 2020). In contrast, the host plant is suggested to be a weaker predictor of root–microbiota composition (Schlaeppi et al., 2014; Glynou et al., 2018; Macia-Vicente et al., 2020). However, host influence has mainly been evaluated among genotypes of a specific plant species. Consequently, studies addressing the role of host identity in harboring different microbial species are required. Recent field research indicates that host identity in comparing different plant species is a strong variable in the determination of microbial assembly (Burns et al., 2015; Borymski et al., 2018; Li et al., 2018; Wang et al., 2020).
Previous studies have indicated that the composition of root-inhabiting microbiome communities is highly influenced by soil type (Bulgarelli et al., 2012; Lundberg et al., 2012; Schlaeppi et al., 2014) and geographical location (Glynou et al., 2018; Lin et al., 2020). Nonetheless, both factors affect the root-associated microbes in a domain-dependent manner, with a stronger effect of soil properties on bacteria and the geographic location of fungi (Thiergart et al., 2020). In our research, the study location was not a significant factor in the microbial community variations. Further large-scale research with more populations from distant locations should be conducted to confirm this result. The Pyrenees are the southern limit of N. brachypetala, where the species is rare and in regression (Martos et al., 2016); future studies should consider researching Northern populations.
Root–microbial communities play a pivotal role in plant performance by improving mineral nutrition (Jacoby et al., 2017). Bacteria and fungi have been reported to contribute to the use of major nutrients such as N, P, and S (Hong et al., 2015; Almario et al., 2017; Perez-Izquierdo et al., 2019; Zhang et al., 2019). Microbes have also been involved in the cycling of some micronutrients, mainly Fe, Zn, and Mn (Alegria Terrazas et al., 2016). In our study, some soil elements (Ca and K) made a minor but significant contribution to the microbial assembly. To the best of our knowledge, the role of these elements in the composition and structure of root microbes has yet to be investigated.
In conclusion, our results comparing non-hyperaccumulator and hyperaccumulator species indicate that the root compartment and the plant host are key factors influencing the diversity, structure, and composition of root-associated bacteria. The fungal community was mainly determined by the root niche, but not by the host. Conversely, location was an irrelevant factor in any microbial group variation. We observed a global reduction in microbial α diversity in the rhizoplane. N. brachypetala was a notable exception, presenting a prominent diversity of bacteria forming a complex interaction network in the closest-root compartment. N. brachypetala harbored a large group of differentially abundant bacteria, whereas fungi were more dominantly associated with the non-hyperaccumulator species. Even though both the non-hyperaccumulator and the hyperaccumulator inhabited non-metalliferous soils, we detected enriched levels in the N. brachypetala root compartments of bacteria previously described as metal-tolerant or formerly associated with hyperaccumulators in metal-rich soils. In contrast, the hyperaccumulator was impoverished in certain fungi. Interestingly, we found that most of the differentially abundant bacteria in the hyperaccumulator were involved in the nitrogen cycle. Our results suggest a potential selection of beneficial taxa in the soil fractions influenced by the roots of N. brachypetala that can favor N availability to the plants, indirectly contributing to their hyperaccumulator phenotype.
Data Availability Statement
All sequence data are freely available in the European Nucleotide Archive database (Project PRJEB41937) and will be available upon publication.
Author Contributions
CP conceived the study and revised the manuscript. SM designed the study and wrote the manuscript. SB, ML, and CC performed the field sampling work. LP-M and SB analyzed the data. All authors approved the final manuscript.
Funding
This study was funded by the Spanish Government grant references BFU2016-75176-R and PID2019-104000-R.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Rosa Padilla for processing the ionome data and Carol Diaz-Gutierrez for her help with microbiota DNA extraction. We would also like to thank Raquel Egea for the first analysis of the bacterial dataset. We are grateful to Llorenç Saez and Iñigo Granzow-de la Cerda for their botanical advice during plant sampling.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.639997/full#supplementary-material
Supplementary Figure 1 | Rarefaction curves.
Supplementary Figure 2 | Prevalence for bacteria and fungi.
Supplementary Figure 3 | Relative abundance per sample.
Supplementary Dataset 1 | Sample information and sequencing quality. Stats for RP1 and RP2.
Supplementary Dataset 2 | Soil and plant leaf ions composition and statistics.
Supplementary Dataset 3 | List of bacterial and fungal taxa according to the differential abundance analysis (DAA) within each root compartment.
References
Aboudrar, W., Schwartz, C., Morel, J. L., and Boularbah, A. (2013). Effect of nickel-resistant rhizosphere bacteria on the uptake of nickel by the hyperaccumulator Noccaea caerulescens under controlled conditions. J. Soils Sediments 13, 501–507. doi: 10.1007/s11368-012-0614-x
Aksoy, A., Leblebici, Z., and Prasad, M. N. V. (2015). Metal-accumulating plants from serpentine habitats of Kizildag, Konya Province, Turkey. Austral. J. Bot. 63, 372–378 doi: 10.1071/bt14354
Alegria Terrazas, R., Giles, C., Paterson, E., Robertson-Albertyn, S., Cesco, S., and Mimmo, T. (2016). Plant-Microbiota interactions as a driver of the mineral turnover in the Rhizosphere. Adv. Appl. Microbiol. 95, 1–67 doi: 10.1016/bs.aambs.2016.03.001
Alford, E. M., Pilon-Smits, E. A. H., and Paschke, M. W. (2010). Metallophytes—a view from the rhizosphere. Plant Soil 337, 33–50 doi: 10.1007/s11104-010-0482-3
Almario, J., Jeena, G., Wunder, J., Langen, G., Zuccaro, A., and Coupland, G. (2017). Root-associated fungal microbiota of nonmycorrhizal Arabis alpine and its contribution to plant phosphorus nutrition. Proc. Natl. Acad. Sci. U.S.A. 114, E9403–E9412
Assunçao, A. G. L., Schat, H., and Aarts, M. G. M. (2003). Thlaspi caerulescens, an attractive model species to study heavy metal hyperaccumulation in plants. New Phytol. 159, 351–360 doi: 10.1046/j.1469-8137.2003.00820.x
Barillot, C. D. C., Sarde, C. O., Bert, V., Tarnaud, E., and Cochet, N. (2013). A standardized method for the sampling of rhizosphere and rhizoplan soil bacteria associated to a herbaceous root system. Ann. Microbiol. 63, 471–476. doi: 10.1007/s13213-012-0491-y
Bech, J., Corrales, I., Tume, P., Barcelo, J., Duran, P., Roca, N., et al. (2012). Accumulation of antimony and other potentially toxic elements in plants around a former antimony mine located in the Ribes Valley (Eastern Pyrenees). J. Geochem. Explor. 113, 100–105 doi: 10.1016/j.gexplo.2011.06.006
Besnard, G., Basic, N., Christin, P. A., Savova-Bianchi, D., and Galland, N. (2009). Thlaspi caerulescens (Brassicaceae) population genetics in western Switzerland: is the genetic structure affected by natural variation of soil heavy metal concentrations? New Phytol. 181, 974–984 doi: 10.1111/j.1469-8137.2008.02706.x
Bokulich, N. A., Joseph, C. M. L., Allen, G. R., Benson, A., and Mills, D. A. (2012). Next-generation sequencing reveals significant bacterial diversity of botrytized wine. PLoS One 7:e36357 doi: 10.1371/journal.pone.0036357
Borymski, S., Cycon, M., Beckmann, M., Mur, L. A. J., and Piotrowska-Seget, Z. (2018). Plant species and heavy metals affect biodiversity of microbial communities associated with metal-tolerant plants in metalliferous soils. Front. Microbiol. 9:1425
Bulgarelli, D., Rott, M., Schlaeppi, K., Ver Loren van Themaatm, E., and Ahmadinejad, N. (2012). Revealing structure and assembly cues for Arabidopsis root inhabiting bacterial microbiota. Nature 488, 91–95 doi: 10.1038/nature11336
Bulgarelli, D., Schlaeppi, K., Spaepen, S., Ver Loren van Themaat, E., and Schulze-Lefert, P. (2013). Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 64, 807–838 doi: 10.1146/annurev-arplant-050312-120106
Burns, J. H., Anacker, B. L., Strauss, S. Y., and Burke, D. J. (2015). Soil microbial community variation correlates most strongly with plant species identity, followed by soil chemistry, spatial location and plant genus. AoB Plants 7:lv030
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336
Cappa, J. J., and Pilon-Smits, E. A. H. (2014). Evolutionary aspects of elemental hyperaccumulation. Planta 239, 267–275 doi: 10.1007/s00425-013-1983-0
Chen, B., Ma, X., Liu, G., Xu, X., Pan, F., and Zhang, J. (2015). An endophytic bacterium Acinetobacter calcoaceticus Sasm3-enhanced phytoremediation of nitrate–cadmium compound polluted soil by intercropping Sedum alfredii with oilseed rape. Environ. Sci. Pollut. Res. 22, 17625–17635 doi: 10.1007/s11356-015-4933-5
Compant, S., Samad, A., Faist, H., and Sessitsch, A. (2019). A review on the plant microbiome: ecology, functions, and emerging trends in microbial application. J. Adv. Res. 19, 29–37. doi: 10.1016/j.jare.2019.03.004
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., and Bhatnagar, S. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 112, E911–E920.
Farinati, S., DalCorso, G., Panigati, M., and Furini, A. (2011). Interaction between selected bacterial strains and Arabidopsis halleri modulates shoot proteome and cadmium and zinc accumulation. J. Exp. Bot. 62, 3433–3447. doi: 10.1093/jxb/err015
Fritze, H., and Baath, E. (1993). Microfungal species composition and fungal biomass in a coniferous forest soil polluted by alkaline deposition. Microb. Ecol. 25, 83–92.
Georgieva, M. L., Lebedeva, M. P., and Bilanenko, E. N. (2012). Mycelial fungi in saline soils of the western Transbaikal region. Eurasian Soil Sci. 45, 1159–1168. doi: 10.1134/s1064229312120058
Golebiewski, M., Deja-Sikora, E., Cichosz, M., Tretyn, A., and Wrobel, B. (2014). 16S rDNA pyrosequencing analysis of bacterial community in heavy metals polluted soils. Microb. Ecol. 67, 635–647. doi: 10.1007/s00248-013-0344-7
Glynou, K., Thines, M., and Macia-Vicente, J. G. (2018). Host species identity in annual Brassicaceae has a limited effect on the assembly of root-endophytic fungal communities. Plant Ecol. Divers. 11, 569–580. doi: 10.1080/17550874.2018.1504332
Hong, Y., Liao, D., Hu, A., Wang, H., Chen, J., and Khan, S. (2015). Diversity of endophytic and rhizoplane bacterial communities associated with exotic Spartina alterniflora and native mangrove using Illumina amplicon sequencing. Can. J. Microbiol. 61, 723–733. doi: 10.1139/cjm-2015-0079
Hou, D. D., Lin, Z., Wang, R. Z., Ge, J., Wei, S., Xie, R. H., et al. (2018). Cadmium exposure-sedum alfredii planting interactions shape the bacterial community in the hyperaccumulator plant rhizosphere. Appl. Environ. Microbiol. 84:e02797-17. doi: 10.1128/AEM.02797-17
Idris, R., Trifonova, R., Puschenreiter, M., Wenzel, W. W., and Sessitsch, A. (2004). Bacterial communities associated with flowering plants of the ni hyperaccumulator Thlaspi goesingense. Appl. Environ. Microbiol. 70, 2667–2677 doi: 10.1128/aem.70.5.2667-2677.2004
Jacoby, R., Peukert, M., Succurro, A., Koprivova, A., and Kopriva, S. (2017). The Role of soil microorganisms in plant mineral nutrition—current knowledge and future directions. Front. Plant Sci. 8:1617.
Jousset, A., Bienhold, C., Chatzinotas, A., Gallien, L., Gobet, A., and Kurm, V. (2017). Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J. 11, 853–862. doi: 10.1038/ismej.2016.174
Kramer, U. (2010). Metal hyperaccumulation in plants. Annu. Rev. Plant Biol. 61, 517–534. doi: 10.1146/annurev-arplant-042809-112156
Li, Y., Wu, X., Chen, T., Wang, W., Liu, G., and Zhang, W. (2018). Plant phenotypic traits eventually shape its microbiota: a common garden test. Front. Microbiol. 9:2479
Likar, M., Pongrac, P., Vogel-Mikus, K., and Regvar, M. (2010). Molecular diversity and metal accumulation of different Thlaspi praecox populations from Slovenia. Plant Soil 330, 195–205. doi: 10.1007/s11104-009-0192-x
Lin, H., and Peddada, S. D. (2020). Analysis of microbial compositions: a review of normalization and differential abundance analysis. npj Biofilms Microbiomes? 6:60.
Lin, M., Xiong, H., Xiang, X., Zhou, Z., Liang, L., and Mei, Z. (2020). The effect of plant geographical location and developmental stage on root-associated microbiomes of Gymnadenia conopsea. Front. Microbiol. 11:1257
Louca, S., Parfrey, L. W., and Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277 doi: 10.1126/science.aaf4507
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., and Malfatti, S. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Luo, J., Tao, Q., Wu, K., Li, J., Qian, J., and Liang, Y. (2017). Structural and functional variability in root-associated bacterial microbiomes of Cd/Zn hyperaccumulator Sedum alfredii. Appl. Microbiol. Biotechnol. 101, 7961–7976. doi: 10.1007/s00253-017-8469-0
Luo, S., Chen, L., Chen, J., Xiao, X., Xu, T., and Wan, Y. (2011). Analysis and characterization of cultivable heavy metal-resistant bacterial endophytes isolated from Cdhyperaccumulator Solanum nigrum L. and their potential use for phytoremediation. Chemosphere 85, 1130–1138. doi: 10.1016/j.chemosphere.2011.07.053
Luo, Z. B., Wu, C., Zhang, C., Li, H., Lipka, U., and Polle, A. (2014). The role of ectomycorrhizas in heavy metal stress tolerance of host plants. Environ. Exp. Bot. 108, 47–62, doi: 10.1016/j.envexpbot.2013.10.018
Ma, Y., Prasad, M. N. V., Rajkumar, M., and Freitas, H. (2011). Plant growth promoting rhizobacteria and endophytes accelerate phytoremediation of metalliferous soils. Biotechnol. Adv. 29, 248–258. doi: 10.1016/j.biotechadv.2010.12.001
McMurdie, P. J., and Holmes, S. (2012). Phyloseq: a bioconductor package for handling and analysis of high-throughput phylogenetic sequence data. Pac. Symp. Biocomput. 2012, 235–246
Macia-Vicente, J. G., Nam, B., and Thines, M. (2020). Root filtering, rather than host identity or age, determines the composition of root-associated fungi and oomycetes in three naturally co-occurring Brassicaceae. Soil Biol. Bioch. 146:107806. doi: 10.1016/j.soilbio.2020.107806
Martinez-Diz, M. P., Andres-Sodupe, M., Bujanda, R., Diaz-Losada, E., Eichmeier, A., and Gramaje, D. (2019). Soil-plant compartments affect fungal microbiome diversity and composition in grapevine. Fungal Ecol. 41, 234–244 doi: 10.1016/j.funeco.2019.07.003
Martos, S., Gallego, B., Saez, L., Lopez-Alvarado, J., Cabot, C., and Poschenrieder, C. (2016). Characterization of Zinc and Cadmium Hyperaccumulation in three Noccaea (Brassicaceae) populations from non-metalliferous sites in the Eastern Pyrenees. Front. Plant Sci. 7:128
Misljenovic, T., Jovanovic, S., Mihailovic, N., Gajic, B., Tomovic, G., and Baker, A. J. M. (2020). Natural variation of nickel, zinc and cadmium (hyper)accumulation in facultative serpentinophytes Noccaea kovatsii and N. praecox. Plant Soil 447, 475–495 doi: 10.1007/s11104-019-04402-5
Muehe, E. M., Weigold, P., Adaktylou, I. J., Planer-Friedrich, B., Kraemer, U., and Kappler, A. (2015). Rhizosphere microbial community composition affects cadmium and zinc uptake of the metal-hyperaccumulating plant Arabidopsis halleri. Appl. Environ. Microbiol. 81, 2173–2181 doi: 10.1128/aem.03359-14
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., and Menke, J. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Nilsson, R. H., Larsson, K. H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., and Schigel, D. (2018). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264
Nishida, S., Tanikawa, R., Ishida, S., Yoshida, J., Mizuno, T., and Nakanishi, H. (2020). Elevated expression of vacuolar nickel transporter gene IREG2 is associated with reduced root-to-shoot nickel translocation in Noccaea japonica. Front. Plant Sci. 11:610
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., and O’hara, R. (2013). Package ‘vegan’. Community Ecology Package, Version
Paulson, J. N., Pop, M., and Bravo, H. C. (2013). MetagenomeSeq: statistical analysis for sparse high-throughput sequencing. Bioconductor Package 1:191.
Perez-Izquierdo, L., Zabal-Aguirre, M., Gonzalez-Martinez, S. C., Buee, M., Verdu, M., and Rincon, A. (2019). Plant intraspecific variation modulates nutrient cycling through its below-ground rhizospheric microbiome. J. Ecol. 107, 1594–1605 doi: 10.1111/1365-2745.13202
Pershina, E., Ivanova, E., Kimeklis, A., Zverev, A., Kichko, A., and Aksenova, T. (2020). Soil microbiomes of reclaimed and abandoned mines of the Yamal region. Polish Polar Res. 41, 95–114
Pongrac, P., Vogel-Mikus, K., Regvar, M., Tolra, R., Poschenrieder, C., and Barcelo, J. (2008). Glucosinolate profiles change during the life cycle and mycorrhizal colonization in a Cd/Zn hyperaccumulator Thlaspi praecox (Brassicaceae). J. Chem. Ecol. 34, 1038–1044 doi: 10.1007/s10886-008-9502-7
Pongrac, P., Vogel-Mikus, K., Kump, P., Necemer, M., Tolra, R., and Poschenrieder, C. (2007). Changes in elemental uptake and arbuscular mycorrhizal colonisation during the life cycle of Thlaspi praecox Wulfen. Chemosphere 69, 1602–1609 doi: 10.1016/j.chemosphere.2007.05.046
Poschenrieder, C., Tolra, R., and Barcelo, J. (2006). Can metals defend plants against biotic stress? Trends Plant Sci. 11, 288–295 doi: 10.1016/j.tplants.2006.04.007
Rajkumar, M., Sandhya, S., Prasad, M. N. V., and Freitas, H. (2012). Perspectives of plant-associated microbes in heavy metal phytoremediation. Biotechnol. Adv. 30, 1562–1574. doi: 10.1016/j.biotechadv.2012.04.011
Rideout, J. R., He, Y., Navas-Molina, J. A., Walters, W. A., Ursell, L. K., and Gibbons, S. M. (2014). Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2:e545 doi: 10.7717/peerj.545
Saad, R., Echevarria, G., Rodriguez-Garrido, B., Kidd, P., and Benizri, E. (2021). ∗A two-year field study of nickel-agromining using Odontarrhena chalcidica co-cropped with a legume on an ultramafic soil: temporal variation in plant biomass, nickel yields and taxonomic and bacterial functional diversity. Plant Soil. doi: 10.1007/s11104-021-04834-y
Saad, R., Kobaissi, A., Robin, C., Echevarria, G., and Benizri, E. (2016). Nitrogen fixation and growth of Lens culinaris as affected by nickel availability: a pre-requisite for optimization of agromining. Environ. Exp. Bot. 131, 1–9 doi: 10.1016/j.envexpbot.2016.06.010
Schlaeppi, K., Dombrowski, N., Garrido-Oter, R., van Themaat, E. V. L., and Schulze-Lefert, P. (2014). Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. PNAS 111, 585–592 doi: 10.1073/pnas.1321597111
Schneider, A. R., Gommeaux, M., Duclercq, J., Fanin, N., Conreux, A., and Alahmad, A. (2017). Response of bacterial communities to Pb smelter pollution in contrasting soils. Sci. Total Environ. 605, 436–444 doi: 10.1016/j.scitotenv.2017.06.159
Sessitsch, A., Kuffner, M., Kidd, P., Vangronsveld, J., Wenzel, W. W., and Fallman, K. (2013). The role of plant-associate bacteria in the mobilization and phytoextraction of trace elements in contaminated soils. Soil Biol. Biochem. 60, 182–194 doi: 10.1016/j.soilbio.2013.01.012
Stringlis, I. A., Yu, K., Feussner, K., de Jonge, R., Van Bentum, S., and Van Verk, M. C. (2018). MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA 115, E5213–E5222
Thiergart, T., Duran, P., Ellis, T., Vannier, N., Garrido-Oter, R., and Kemen, E. (2020). Root microbiota assembly and adaptive differentiation among European Arabidopsis populations. Nat. Ecol. Evol. 4, 122–131 doi: 10.1038/s41559-019-1063-3
Thijs, S., Langill, T., and Vangronsveld, J. (2017). The bacterial and fungal microbiota of hyperaccumulator plants: small organisms, large influence. Adv. Bot. Res. 83, 43–86 doi: 10.1016/bs.abr.2016.12.003
Van der Ent, A., Baker, A. J. M., Reeves, R. D., Pollard, A. J., and Schat, H. (2013). Hyperaccumulators of metal and metalloid trace elements: facts and fiction. Plant Soil 362, 319–334 doi: 10.1007/s11104-012-1287-3
Visioli, G., D’Egidio, S., Vamerali, T., Mattarozzi, M., and Sanangelantoni, A. M. (2014). Culturable endophytic bacteria enhance Ni translocation in the hyperaccumulator Noccaea caerulescens. Chemosphere 117, 538–544 doi: 10.1016/j.chemosphere.2014.09.014
Visioli, G., D’Egidio, S., and Sanangelantoni, A. M. (2015a). The bacterial rhizobiome of hyperaccumulators: future perspectives based on omics analysis and advanced microscopy. Front. Plant Sci. 5:752
Visioli, G., Vamerali, T., Mattarozzi, M., Dramis, L., and Sanangelantoni, A. M. (2015b). Combined endophytic inoculants enhance nickel phytoextraction from serpentine soil in the hyperaccumulator Noccaea caerulescens. Front. Plant Sci. 6:638
Vogel-Mikus, K., Drobne, D., and Regvar, M. (2005). Zn, Cd and Pb accumulation and arbuscular mycorrhizal colonisation of pennycress Thlaspi praecox Wulf. (Brassicaceae) from the vicinity of a lead mine and smelter in Slovenia. Environ. Pollut. 133, 233–242. doi: 10.1016/j.envpol.2004.06.021
Vogel-Mikus, K., Pongrac, P., Kump, P., Necemer, M., and Regvar, M. (2006). Colonisation of a Zn, Cd and Pb hyperaccumulator Thlaspi praecox Wulfen with indigenous arbuscular mycorrhizal fungal mixture induces changes in heavy metal and nutrient uptake. Environ. Pollut. 139, 362–371 doi: 10.1016/j.envpol.2005.05.005
Wang, H., Zeng, Y., Guo, C., Bao, Y., Lu, G., and Reinfelder, J. R. (2018). Bacterial, archaeal, and fungal community responses to acid mine drainage-laden pollution in a rice paddy soil ecosystem. Sci. Total Environ. 616–617, 107–116 doi: 10.1016/j.scitotenv.2017.10.224
Wang, L., Zhou, Q. X., Ding, L. L., and Sun, Y. B. (2008). Effect of cadmium toxicity on nitrogen metabolism in leaves of Solanum nigrum L. as a newly found cadmium hyperaccumulator. J. Hazard. Mater. 154, 818–825. doi: 10.1016/j.jhazmat.2007.10.097
Wang, Y. L., Gao, C., Chen, L., Ji, N. N., Wu, B. W., and Lu, P. P. (2020). Community assembly of endophytic fungi in ectomycorrhizae of Betulaceae plants at a regional scale. Front. Microb. 10:3105
Wang, B., and Qiu, Y. L. (2006). Phylogenetic distribution and evolution of mycorrhizas in land plants. Mycorrhiza 16, 299–363 doi: 10.1007/s00572-005-0033-6
Weiss, S., Xu, Z. Z., Peddada, S., Amir, A., Bittinger, K., and Gonzalez, A. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5:27
Whiting, S. N., Souza, M. P., and Terry, N. (2001). Rhizosphere bacteria mobilize Zn for Hyperaccumulation by Thlaspi caerulescens. Environ. Sci. Technol. 35, 3144–3150 doi: 10.1021/es001938v
Wu, Z., Gao, G., and Wang, Y. (2019). Effects of soil properties, heavy metals, and PBDEs on microbial community of e-waste contaminated soil. Ecotox. Environ. Saf. 180, 705–714. doi: 10.1016/j.ecoenv.2019.05.027
Xiong, C., He, J. Z., Singh, B. K., Zhu, Y. G., Wang, J. T., and Li, P. P. (2020). Rare taxa maintain the stability of crop mycobiomes and ecosystem functions. Environ Microbiol. 23, 1907–1924 doi: 10.1111/1462-2920.15262
Zhang, J., Liu, Y., Zhang, N., Hu, B., Jin, T., and Xu, H. (2019). NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotech. 37, 676–684 doi: 10.1038/s41587-019-0104-4
Keywords: high-throughput sequencing (deep sequencing), Noccaea brachypetala, root-associated microbial communities, Zn/Cd hyperaccumulator, non-metalliferous soil
Citation: Martos S, Busoms S, Pérez-Martín L, Llugany M, Cabot C and Poschenrieder C (2021) Identifying the Specific Root Microbiome of the Hyperaccumulator Noccaea brachypetala Growing in Non-metalliferous Soils. Front. Microbiol. 12:639997. doi: 10.3389/fmicb.2021.639997
Received: 11 December 2020; Accepted: 06 April 2021;
Published: 14 May 2021.
Edited by:
Michel Chalot, Université Bourgogne Franche-Comté, FranceReviewed by:
Shuo Jiao, Northwest A and F University, ChinaAlison Bennett, The Ohio State University, United States
Kristian Harris, The Ohio State University, United States, in collaboration with reviewer AB
Copyright © 2021 Martos, Busoms, Pérez-Martín, Llugany, Cabot and Poschenrieder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Soledad Martos, c29sZWRhZC5tYXJ0b3NAdWFiLmNhdA==