 Meshack Juma1†
Meshack Juma1† Arun Sankaradoss2†Redcliff Ndombi1Patrick Mwaura1Tina Damodar2Junaid Nazir2Awadhesh Pandit2
Arun Sankaradoss2†Redcliff Ndombi1Patrick Mwaura1Tina Damodar2Junaid Nazir2Awadhesh Pandit2 Rupsy Khurana2
Rupsy Khurana2 Moses Masika1Ruth Chirchir1John Gachie1Sudhir Krishna2,3
Moses Masika1Ruth Chirchir1John Gachie1Sudhir Krishna2,3 Ramanathan Sowdhamini2
Ramanathan Sowdhamini2 Omu Anzala1*
Omu Anzala1* Iyer S. Meenakshi2*
Iyer S. Meenakshi2*- 1KAVI Institute of Clinical Research, University of Nairobi, Nairobi, Kenya
- 2National Centre for Biological Sciences, Tata Institute of Fundamental Research (TIFR), Bengaluru, India
- 3School of Interdisciplinary Life Sciences, Indian Institute of Technology Goa, Ponda, India
Background: Africa has one of the highest incidences of gonorrhea. Neisseria gonorrhoeae is gaining resistance to most of the available antibiotics, compromising treatment across the world. Whole-genome sequencing (WGS) is an efficient way of predicting AMR determinants and their spread in the population. Recent advances in next-generation sequencing technologies like Oxford Nanopore Technology (ONT) have helped in the generation of longer reads of DNA in a shorter duration with lower cost. Increasing accuracy of base-calling algorithms, high throughput, error-correction strategies, and ease of using the mobile sequencer MinION in remote areas lead to its adoption for routine microbial genome sequencing. To investigate whether MinION-only sequencing is sufficient for WGS and downstream analysis in resource-limited settings, we sequenced the genomes of 14 suspected N. gonorrhoeae isolates from Nairobi, Kenya.
Methods: Using WGS, the isolates were confirmed to be cases of N. gonorrhoeae (n = 9), and there were three co-occurrences of N. gonorrhoeae with Moraxella osloensis and N. meningitidis (n = 2). N. meningitidis has been implicated in sexually transmitted infections in recent years. The near-complete N. gonorrhoeae genomes (n = 10) were analyzed further for mutations/factors causing AMR using an in-house database of mutations curated from the literature.
Results: We observe that ciprofloxacin resistance is associated with multiple mutations in both gyrA and parC. Mutations conferring tetracycline (rpsJ) and sulfonamide (folP) resistance and plasmids encoding beta-lactamase were seen in all the strains, and tet(M)-containing plasmids were identified in nine strains. Phylogenetic analysis clustered the 10 isolates into clades containing previously sequenced genomes from Kenya and countries across the world. Based on homology modeling of AMR targets, we see that the mutations in GyrA and ParC disrupt the hydrogen bonding with quinolone drugs and mutations in FolP may affect interaction with the antibiotic.
Conclusion: Here, we demonstrate the utility of mobile DNA sequencing technology in producing a consensus genome for sequence typing and detection of genetic determinants of AMR. The workflow followed in the study, including AMR mutation dataset creation and the genome identification, assembly, and analysis, can be used for any clinical isolate. Further studies are required to determine the utility of real-time sequencing in outbreak investigations, diagnosis, and management of infections, especially in resource-limited settings.
Introduction
Gonorrhea is one of the most common sexually transmitted infections (STIs), which is caused by Neisseria gonorrhoeae (gonococci). It is estimated to have affected 87 million people (between the ages 15 and 49) worldwide, 11.4 million in the African region, in 2016 [World Health Organisation (WHO), 2018]. Gonorrhea can cause epididymitis in men and pelvic inflammatory disease in women, which can result in infertility and ectopic pregnancies. It is also implicated in neonatal ophthalmia, which can result in blindness, and increased acquisition and transmission of other STIs (Unemo and Dillon, 2011). WHO included N. gonorrhoeae in its list of AMR “priority pathogens” in 2017 [World Health Organisation (WHO), 2017] and has projected a 90% reduction in global gonorrhea incidence by 2030 [World Health Organisation (WHO), 2018]. Although N. gonorrhoeae infections are treated with antibiotics, gonococci have acquired resistance to most of the antibiotics used to treat gonorrhea, and despite two clinical trials, no vaccines are available (Wi et al., 2017). Increasing antimicrobial resistance threatens effective treatment and control.
Many strains are developing resistance to ceftriaxone and azithromycin, which are used as the last options of the first-line therapies of gonorrhea (Unemo and Nicholas, 2012). However, with isolates showing increased MICs to both these antibiotics, this is not viable as a long-term treatment (Papp et al., 2017). Many countries in the African region have reported decreased susceptibility to drugs like ceftriaxone, azithromycin, or ciprofloxacin (Wi et al., 2017). Gentamycin has been used as an alternative therapy in some places in Africa, although a few cases of resistance have been reported in other parts of the world (Chisholm et al., 2011). Whole-genome sequencing (WGS) allows for timely detection and elucidation of AMR determinants in bacteria (Collineau et al., 2019). High-throughput sequencing has many advantages, like long read lengths, short time, and reduction of bias introduced through amplification by PCR steps (Goldstein et al., 2019). WGS has been used to investigate quinolone-resistant gonorrhea outbreaks (Duncan et al., 2011; Kivata et al., 2019) in Kenya. It has also been used to analyze isolates resistant to ciprofloxacin, azithromycin, and ESCs in other countries (Chaudhry et al., 2002; Eyre et al., 2018; Golparian et al., 2018).
MinION, a mobile sequencing device from ONT (Oxford, United Kingdom), sequences DNA by monitoring the transfer of individual DNA molecules through various types of pores, resulting in very long and unbiased sequence reads, as there is no amplification or chemical reactions used during sequencing (Golparian et al., 2018). The mobile nature of the MinION offers many advantages and the sequencer has been evaluated for use in resource-limited settings and on the International Space Station (ISS) (Castro-Wallace et al., 2017; Elliott et al., 2020). MinION has also been shown to be effective for diagnostics, high-quality assemblies, and AMR surveillance in bacteria. However, the high error rate for the sequencer limits its use. Golparian and co-workers sequenced 14 clinical isolates of N. gonorrhoeae and evaluated multiple methods for genome assemblies using MinION with Illumina reads (Golparian et al., 2018). Street and co-workers used MinION to obtain a de novo assembly of N. gonorrhoeae isolated from patient urine samples (Street et al., 2020). Zhang and co-workers showed that AMR-profiling results for N. gonorrhoeae were comparable when using only MinION-based assemblies or MinION-Illumina hybrid assemblies (Zhang et al., 2020). Naidenov and co-workers used Nanopore-only sequencing to obtain complete genomes and carried out AMR profiling of two new strains from the Elizabethkingia genus (Naidenov et al., 2019). Sanderson and co-workers used nanopore sequencing data and showed that it is possible to generate accurate consensus bacterial genomes from metagenomic sequencing data without using Illumina short reads (Sanderson et al., 2020).
In a previous study from Kenya, 22 multi-drug-resistant gonococcal isolates from heterosexual patients (2 females and 20 males) were sequenced using Illumina MiSeq for understanding gyrA and parC mutations in ciprofloxacin resistance (Kivata et al., 2019). Cehovin et al. sequenced 103 genomes from 73 patients from coastal Kenya, mainly men who had sex with men, using Illumina HiSeq and analyzed the plasmids conferring antibiotic resistance (Cehovin et al., 2018). These studies in Kenya have used the Illumina platform and mainly focused on isolates from male patients. We sequenced 10 isolates derived from women who visited STI clinics in Nairobi between 2012 and 2017, using only MinION sequencing in Kenya and evaluated methods for assembly and AMR profiling of genomes. We have also carried out phylogenies with previously deposited sequences in PubMLST, from Kenya, and other countries. Here, we assess the possibility of using MinION data alone for WGS, assembly, and comparative analysis. This study demonstrates the utility of the portable MinION sequencer for genome-based analysis in regular clinical setups.
Materials and Methods
Bacterial Isolates and Culture Conditions
Neisseria gonorrhoeae isolates were derived from high vaginal swabs collected between January 2012 and December 2017. The stocked organisms were stored at –70°C and retrieved and cultured on Modified Thayer–Martin agar. The plates were incubated at 37°C and 5% carbon dioxide (under raised CO2) as per the standard protocol.
Isolates were identified by Gram stain, Catalase test, Oxidase test, and carbohydrate-utilization studies of glucose, maltose, fructose, and sucrose, and confirmed by analytical profile index testing kit (API NH, bioMérieux). All gonococcus isolates were re-stocked in 20% glycerol-tryptic soy broth for long-term storage at –70°C for external quality control, sequencing, and molecular characterization. Standard N. gonorrhoeae isolates, i.e., WHO K, WHO P, WHO O, WHO R, and WHO M (Unemo et al., 2016), were used to test the media for viability and colonial characteristics. Phenotypic characterization was performed by characterizing lactamase production determined using nitrocefin solution. In the present study, clinical isolates from 10 patients were sequenced (Supplementary Table 1).
Antibiotic Susceptibility Testing
Ciprofloxacin, spectinomycin, cefixime, ceftriaxone, gentamycin, and cefoxitin strips were used to set E-test according to the Clinical and Laboratory Standards Institute (CLSI). An antimicrobial gradient diffusion (E-test) method was used to determine the minimum inhibitory concentration (MIC) of N. gonorrhoeae, and interpretation of susceptibilities for selected antimicrobial agents was carried out as recommended by CLSI (Supplementary Table 2).
Isolation of Genomic DNA
All the colonies from Thayer–Martin agar (37°C in a humidified 5% CO2 environment for 36 h) with the phenotypic characteristics of N. gonorrhoeae were picked up to create a culture suspension in 1X PBS. The genomic DNA was isolated using the Qiagen DNA isolation kit using the manufacturer’s specifications. Extracted DNA was purified using AMPure XP beads (Beckman Coulter, United Kingdom), eluted in 50 μl of nuclease-free water, and quantified using a QiaXpert (Qiagen). Pipetting was minimized to reduce shearing of the DNA prior to sequencing.
ONT Library Preparation and MinION Sequencing
DNA library preparation for Nanopore sequencing was carried out using Ligation Kit SQK-LSK109 (ONT). Fragmented DNA was repaired and dA-tailed using the NEBNext FFPE DNA Repair Mix and NEBNext Ultra II End Repair/dA-Tailing Module (New England BioLabs). An individual barcode was added to dA-tailed DNA by using the barcoding extension kit EXP-NPB104 in accordance with the ONT protocols with NEB Blunt/TA Ligase Master Mix (New England BioLabs). Each barcoded DNA was pooled in equimolar amounts, and an adapter was attached using the NEBNext Quick Ligation Module (New England BioLabs). The MinION flow cell and reagents were shipped from Bangalore, India, to Nairobi at 4°C. The number of active pores was checked before loading. The samples were pooled using equimolar pooling, the library was loaded into the SpotON flow cell R9.5 (FLO-MIN106), and sequencing was carried out on MinKNOW using the 48-h script.
Base-Calling, Read Trimming, and Processing
Guppy (v 3.2.2) (ONT) was used for base-calling of fast5 files. Reads with a mean q score (quality) greater than 7 and a read length greater than 500 bp were used and trimmed for adaptor sequences and barcodes using qcat (v1.1.0)1 from ONT. The files corresponding to each barcode (sample) were analyzed as follows.
Assembly and Assessment
The selected raw reads were corrected, trimmed, and assembled using Canu (v1.8)2 using the parameters genome Size = 2.1 m and minReadLength = 500 (Koren et al., 2017). Minimap (v 2.17)3 and Miniasm (v0.3)4 were used for the self-mapping of the raw reads and the concatenation of the alignments to get the de novo assembly, respectively (Li, 2016). Two rounds of error correction of this draft assembly were carried out with Racon (v1.4.3)5 using raw reads (Vaser et al., 2017). The above assemblies were polished using the raw nanopore reads with Nanopolish (v0.11.1)6 (Loman et al., 2015).
Species identification was carried out using the Ribosomal Multilocus Sequence Typing rMLST (Jolley et al., 2012) approach from BIGSdb and PubMLST to identify the causative organism and co-infections (Jolley et al., 2018). We used the de novo assemblies for the above step. The database uses 53 genes encoding the bacterial ribosome protein subunits (rps genes) for species assignment. We also used blastn with 16S rRNA and 23S rRNA as the queries for species confirmation. The plasmid sequences were identified from the de novo assemblies. The plasmid sequences were used for detecting the AMR determinants.
We also carried out a guided genome mapping using reference genome N. gonorrhoeae FA1090 (NCBI:txid485). The trimmed raw reads were aligned to the reference genome using the bwa-mem option from bwa (v0.7.12)7 (Li and Durbin, 2010). Samtools (v1.9)8 was used for deriving the bam alignment file and the consensus genome (bcftools), after normalizing the indels and variant calling (Li et al., 2009). The commands samtools mpileup with the options -Ou -f and bcftools calls with the options -mv -Oz --ploidy 1 were used for variant calling. The options bcftools filter --IndelGap 5 and bcftools consensus -H ‘‘A’’ were used for deriving the consensus genome. Mummer (v3.9.4)9 (Kurtz et al., 2004) and bedtools (v2.25.0)10 (Quinlan and Hall, 2010) were used for calculating the correspondence of the assemblies with the reference genome, genome coverage, and sequencing depth. The base positions with zero coverage were extracted using awk commands, and bedtools maskfasta option was used to derive the draft genomes; missing nucleotides were marked in the genome as “N.”
Annotation
The gene prediction and annotation were carried out using the RAST server11 using the reference genome FA1090 for guiding the gene prediction (Overbeek et al., 2013).
Genome-Based MLST Analysis
Gene profiles for sequence typing of N. gonorrhoeae were downloaded from different databases like NG-STAR (N. gonorrhoeae Sequence Typing for Antimicrobial Resistance)12 (Demczuk et al., 2017), PubMLST (Public databases for multi-locus sequence-typing)13 (Bennett et al., 2007), and NG-MAST (N. gonorrhoeae multi-antigen sequence typing)14 (Martin et al., 2004). NG-MAST uses two highly polymorphic loci, the outer-membrane porin (por) and transferrin binding protein β-subunit tbpB, for sequence typing. NG-STAR uses variants in seven antimicrobial resistance determinants (penA, mtrR, porB, ponA, gyrA, parC, and 23S rRNA) to three classes of antibiotics (cephalosporins, macrolides, and fluoroquinolones), and MLST (database PubMLST) uses seven housekeeping genes (abcZ, adk, aroE, fumC, gdh, pdhC, and pgm) for sequence typing. Profiles were created and blastn (E-value 10−7) from standalone BLAST+ 15 (Altschul, 2005) was used to assign the genes to the different alleles for MLST genes.
Identification of Mutations Causing AMR
Around 85 mutations in 14 genes reported to be involved in AMR were checked for in the sequenced strains, including promoter mutations resulting in overexpression of efflux proteins (Unemo and Shafer, 2011). Additionally, we checked for the presence of four drug efflux pumps, two genes from conjugative plasmids, and plasmid-mediated AMR determinants (Supplementary File). Mutations were manually screened for, in the protein sequences identified using the RAST server in all the genomes. Sequencing depth at the corresponding gene loci was calculated from the results of the samtools depth command. We compared our results with that of three publicly available databases for AMR, CARD (Comprehensive Antibiotic Resistance Database), ARIBA (Antimicrobial Resistance Identification By Assembly) (Hunt et al., 2017), and Pathogenwatch (Balloux et al., 2018; Alcock et al., 2019). Resistance genes from plasmids were identified using blastn (E-value 10−7) against NCBI Bacterial Antimicrobial Resistance Reference Gene Database16 (NCBI Resource Coordinators, 2017).
Homology Modeling
To understand the basis of antibiotic resistance, wild-type and mutant proteins implicated in AMR were modeled using templates from other bacteria. Coordinates of heteroatoms like antibiotics and DNA are present in the templates used. Hence, the heteroatom modeling module from Modeler (v9.23) (Sánchez and Sali, 2000) was used to build a multi-chain model with symmetry restraints. Clustal Omega17 was used for the alignment of query protein sequences with the template protein sequence, with manual correction of the alignment (Sievers and Higgins, 2014). The residues in different chains were separated using “/” and the symbol “.” was used to indicate the ligand molecules in the alignment.
Multiple-Sequence Alignment and Phylogenetics
Phylogenetic tree analysis was carried out using 123 previously sequenced N. gonorrhoeae strains from Kenya and an additional 130 strains from different countries across the world, standard WHO strains, and the reference genome. The Genome Comparator module from Bacterial Isolate Genome Sequence Database (BIGSdb)18 was used to obtain the sequences and generate core genome alignments (cgMLST1649-) using the MAFFT module within BIGSdb (Jolley et al., 2018). We used a dataset of strains from different WHO geographical regions including strains from Kenya and WHO reference strains. The parameters used were minimum percentage identity 70%, minimum percentage alignment 50%, and blastn search to annotate loci in genomes with ≥50% loci (word size of 20). Incomplete loci were not used for pairwise comparison and pairwise missing loci and paralogous loci were excluded from the analysis. The N. meningitidis strain (PubMLST ID: 12672| 053442, cc4821) was included as an outgroup. The alignments were checked for redundancy in sequences of core genes; the sequences with 100% redundancy and the sequences missing all the gene loci analyzed were removed. Phylogenetic trees were derived based on Maximum likelihood approach using RAxML software (v8.2.12) using GTR + GAMMA substitution model with 1000 bootstraps (Kozlov et al., 2019). The best ML search trees were identified for both the datasets (only Kenyan strains and combined dataset of all 255 strains) annotated with details like WHO geographical region and antibiotic sensitivity from BIGSdb and Cehovin et al. (2018) using iTOL (Letunic and Bork, 2016).
The workflow for consensus genome used in the study has been illustrated in Supplementary Figure 1A, and the subsequent analysis workflow has been shown in Supplementary Figure 1B. The workflow for the de novo assemblies has been shown in Supplementary Figure 1C.
Results
Overview of Sequenced Data
We obtained between 534 and 2,387 Mb of reads from the MinION sequencing runs for each sample. The longest reads ranged from 156 to 406 kb with an average length of 1.5–2.7 kb (Table 1). We sequenced clinical isolates from 14 patients and obtained 12 N. gonorrhoeae genome sequences. Out of these, 10 genomes were near-complete with a good sequencing depth and were used for subsequent analysis.
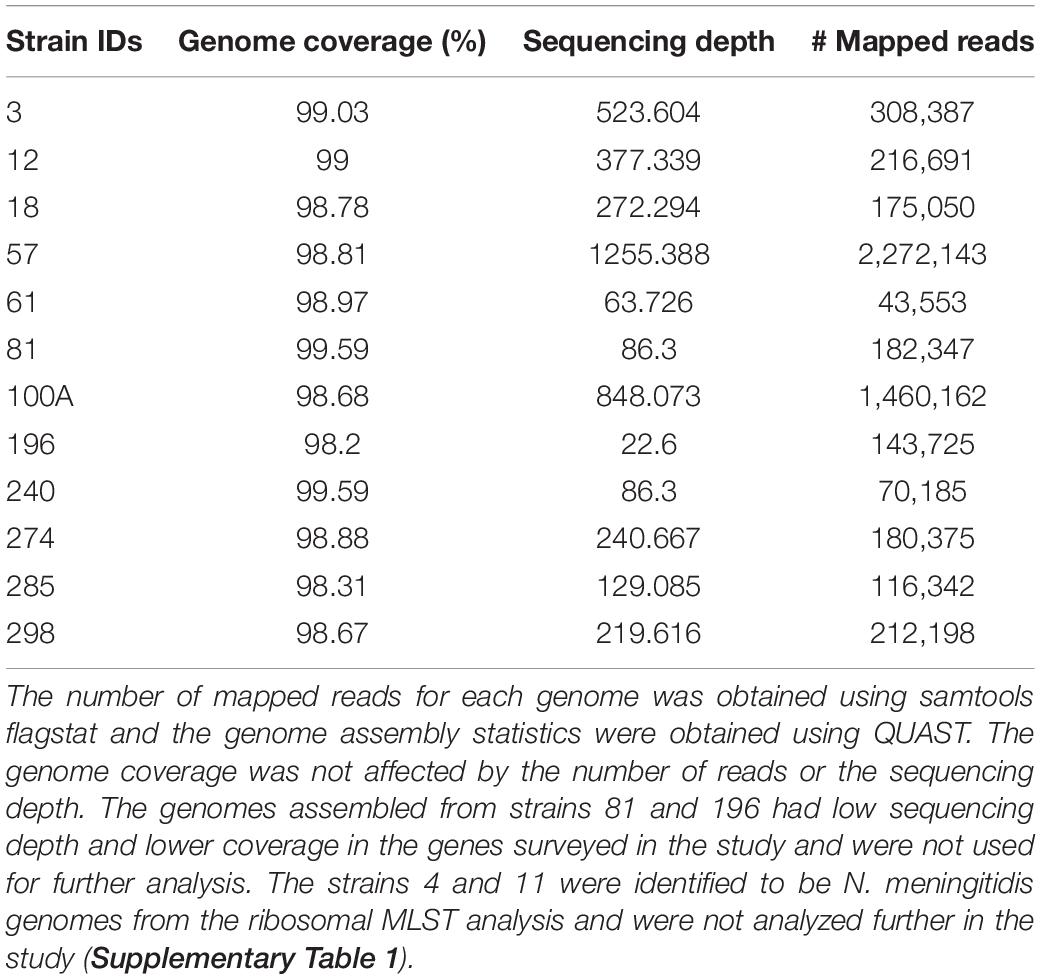
Table 1. Read and assembly statistics.
Genome Assembly Statistics and Species Identification
Overview of the mappability of the reads from different strains to the reference genome has been depicted in Supplementary Figure 2. Assemblies were compared and selected based on the following criteria: the lowest number of mismatches, misassemblies, contigs, and the highest fraction of genome coverage using QUAST (Gurevich et al., 2013) (Supplementary Tables 3A–C). The length of the assemblies and the number of CDS identified were fairly the same across different methods. No plasmid or transposon gene insertions were observed in the genomes assembled de novo and the consensus genome. Similar to previous reports, we observed frameshifts with the de novo assemblies (Golparian et al., 2018). The consensus genome (FA1090 was used for read mapping) was used for further analysis as the genome coverage, number of complete genomic features, number of indels, and number of mismatches were better.
Among the 14 samples, 2 samples showed the presence of N. meningitidis, 9 samples showed the presence of N. gonorrhoeae, and 3 samples showed the presence of both N. gonorrhoeae and M. osloensis (Supplementary Table 1). N. meningitidis has been implicated in STI in recent years, while M. osloensis is also a commensal of the urogenital tract (Slack, 2010; Johnson et al., 2016).
Although high error rates have been reported with the use of the MinION sequencer, we have used long sequencing time (48 h) and obtained high depths of sequencing (63-1255X; Table 1). Previous studies have shown that sequencing using MiNION for longer time duration increases the sequencing output and the error rate can be minimized by using depth and consensus (Tyler et al., 2018; Plesivkova et al., 2019).
Sequence Typing of the Strains
The alleles used in sequence typing (MLST, NG-MAST, and NG-STAR) were assigned to the genes from different strains, and we identified few novel alleles. The novel allele sequences have been deposited in the PubMLST database and the assigned allele numbers have been mentioned (Supplementary Tables 4A–C). The sequencing depth at each of these loci was found to be very high (>50–600) (Supplementary Table 4D). This is higher than the recommended depth for SNP detection using Nanopore sequencing by Sanderson and co-workers (20X) (Sanderson et al., 2020).
Antibiotic Resistance and Mutations in AMR Genes
The MIC results with six different antibiotics have been provided in Table 2.
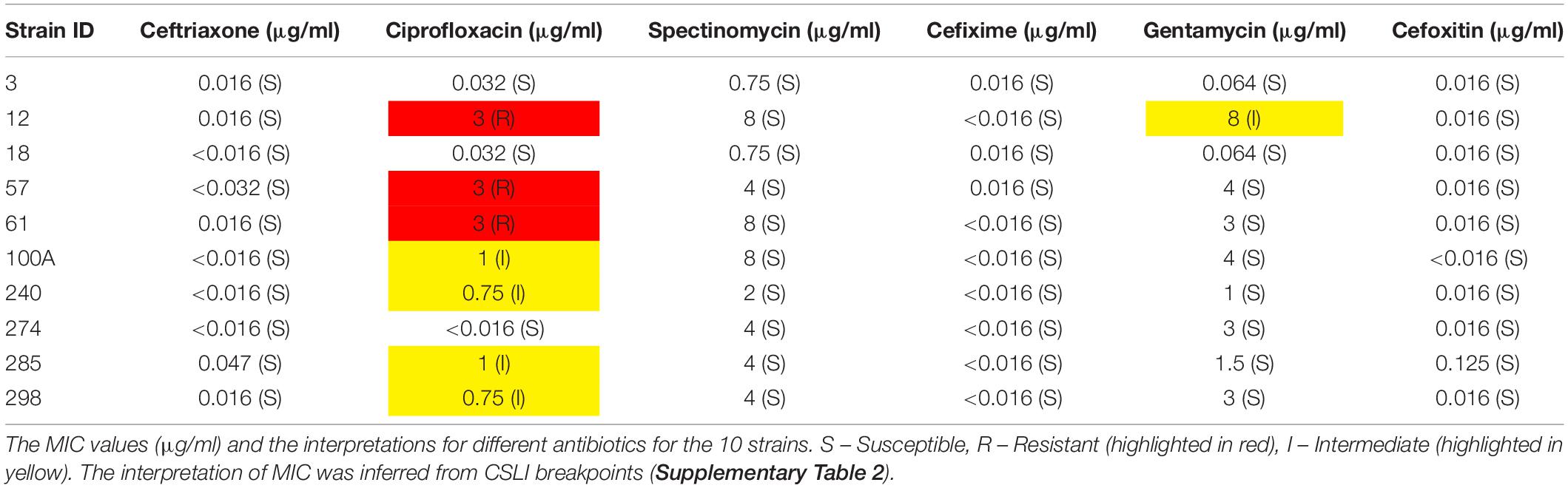
Table 2. MIC values for different antibiotics.
A dataset of AMR targets was identified using literature (Supplementary Table 5A). These included mutations in the promoter region of repressors resulting in overexpression of efflux proteins, mutations in drug targets that result in higher MICs for the drugs either individually or in combination with other mutations, plasmids for drug resistance, and mosaic alleles in genes and promoter regions reported from drug-resistant strains of N. gonorrhoeae from across the world. Out of these, 28 mutations (in 11 genes) were observed in the strains (Table 3). The sequencing depth of gyrA, parC, mtrR, ponA1, penA, porB, folP, rplD, pilQ, rpoB, and rpsJ genes was high (Supplementary Tables 4D, 5B) and there were no missing bases in these genes. Other databases used for AMR profiling (Pathogenwatch, ARIBA, and CARD) missed a few of the mutations we identified (Supplementary Table 5C). The ARIBA database has not been updated; hence, it may be missing some recently identified AMR-associated mutations (Hunt et al., 2017). CARD collects AMR determinants from several bacteria and is not specific to N. gonorrhoeae; it may include mutations causing AMR in other bacteria but not gonococci (Alcock et al., 2019), whereas compared to these databases, Pathogenwatch is frequently updated; we identified the mutation rplD G70D (implicated in macrolide resistance) in three strains, but this mutation has been missed by Pathogenwatch (Ma et al., 2020b).
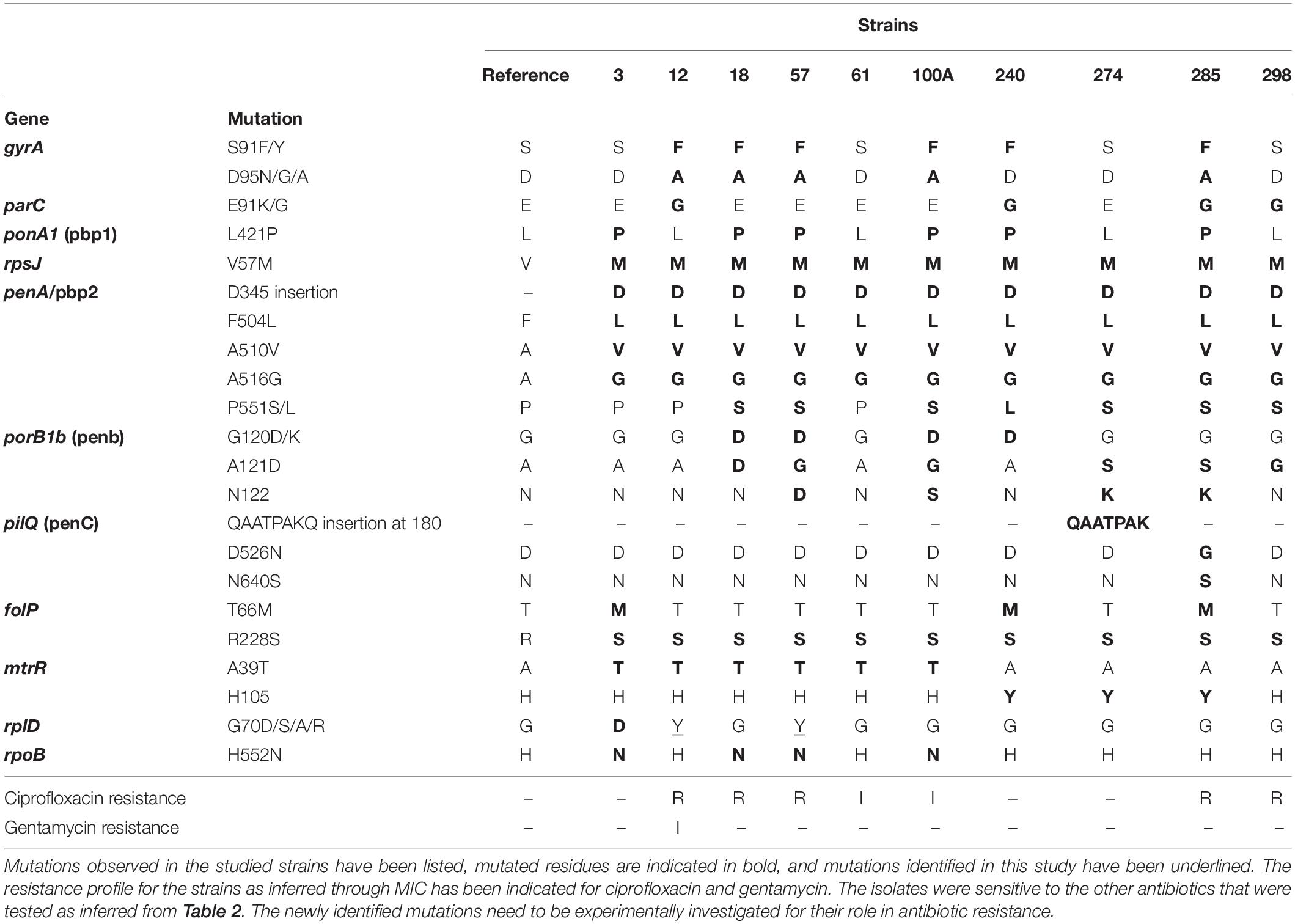
Table 3. AMR gene profile of the 10 strains.
We found six isolates carrying AMR mutations in penA C-terminal region (penicillin and cephalosporin resistance determinant) and four isolates with mutations in ponA1 (penicillin and cephalosporin resistance determinant), respectively. We detected the D345 insertion and F504L mutation in penA (Zapun et al., 2016), which could be involved in penicillin and cephalosporin resistance (Table 3). Five isolates harbored gyrA resistance mutations, two of which also had a parC mutation. One strain had a mutation in only the parC gene; however, this is a silent mutation that was among the set of mutations observed in a cephalosporin-resistant isolate (Chaudhry et al., 2002). One of the ciprofloxacin-resistant strain contained a gyrA mutation, consistent with previous observations of these mutations in susceptible (Tanaka et al., 2000; Chaudhry et al., 2002) isolates. These mutations, in the Quinolone Resistance Determining Region (QRDR) of both the proteins, have been shown to contribute to the high MICs for ciprofloxacin (Belland et al., 1994). Interestingly, one of the gyrA mutations we observed, D95A/G, and the mutation E91G in ParC have been reported to be specific to Kenyan isolates (Kivata et al., 2019). Specific mutations in porB1b (penB AMR determinant), implicated in decreased influx of antimicrobials through the porin PorB (Lee et al., 2010; Chen and Seifert, 2013), were identified in 6 of the 10 isolates. All 10 strains harbored the V57M mutation in RpsJ, which has been shown to contribute to tetracycline resistance and R228S mutation in FolP, which results in sulfonamide resistance (Yun et al., 2012). Three strains harbored mutations in rplD gene, which is implicated in azithromycin resistance (Ma et al., 2020b). Eight strains had mutations in pilQ gene (PenC), implicated in penicillin, ESC, and tetracycline resistance, while four strains had the H522N mutation in rpoB, which is implicated in rifampicin resistance. Plasmid-mediated AMR determinants [high-level resistance to benzylpenicillin and tetracycline -pblaTEM and ptet(M)] were detected in all strains except one where we detected only the tet(M)-containing plasmid. We did not identify mef or erm containing conjugative plasmids/transposons, which are reported to cause resistance to macrolides (Roberts et al., 1999) or resistance-conferring mutations for spectinomycin, in either the de novo assemblies or consensus genome.
Understanding the Basis of Antibiotic Resistance
We investigated the basis of drug resistance for the mutations in the proteins GyrA, ParC, FolP, porB1b, PonA1, and RpoB (Supplementary Figures 3–8, template details in Supplementary Table 6) through homology modeling using templates with co-crystallized antibiotic structures. The results have been described in Supplementary File (Section 8).
Phylogenetic Relationship With Other N. gonorrhoeae Genomes From Kenya, Other WHO Geographical Regions, and WHO Strains
To understand the genetic relatedness of isolates from Kenya with isolates from other geographical regions and WHO reference isolates, we constructed a phylogenetic tree using the alignments derived from the core genome MLST (cgMLST). After filtering for sequence redundancy and loci completeness, we retained 238 sequences from different geographical regions across the world (Americas, Europe, Western Pacific, Africa and WHO reference strains) for the phylogenetic analysis. The metadata for the strains like geographical region and antibiotic resistance details have been indicated in the phylogenetic trees (details in Supplementary Table 7).
We observed eight clusters of sequences from across the world (clusters I–VIII). Clades I and II consisted of only Kenyan sequences, while sequences from different geographical regions and Kenya co-clustered in the rest of the clades (Figure 1). Cehovin et al. had observed in an earlier study that strains from Kenya were distinct from strains sequenced from USA and the UK (Cehovin et al., 2018). Consistent with this observation, we see Kenyan sequence-specific clades, but we also observe clades where sequences from Kenya are mixed from sequences from other geographical regions. The clades were more or less consistent in the phylogenetic tree constructed using sequences from all over the world and using sequences from only Kenya (Figure 2). Out of the strains sequenced in the study, strain 3 clustered in clade I; strain 298 in clade V; clade 274 in clade VI; strains 12, 18, 61, 57, and 100A in clade VII; and strains 240 and 285 in clade VIII. Each cluster consisted of strains belonging to different multi-locus sequence types (MLSTs). Strains belonging to certain STs were specific to a cluster (ST1588 – cluster VII, ST1893 – cluster II) while strains from some other STs were found in multiple clusters (ST1599 in cluster III, IV, V, and VIII and ST1902 in clusters III and VIII) (metadata in Supplementary Table 7). Broader sampling and analysis of clinical isolates from Kenya can help us determine the extent of genetic diversity within the N. gonorrhoeae strains.
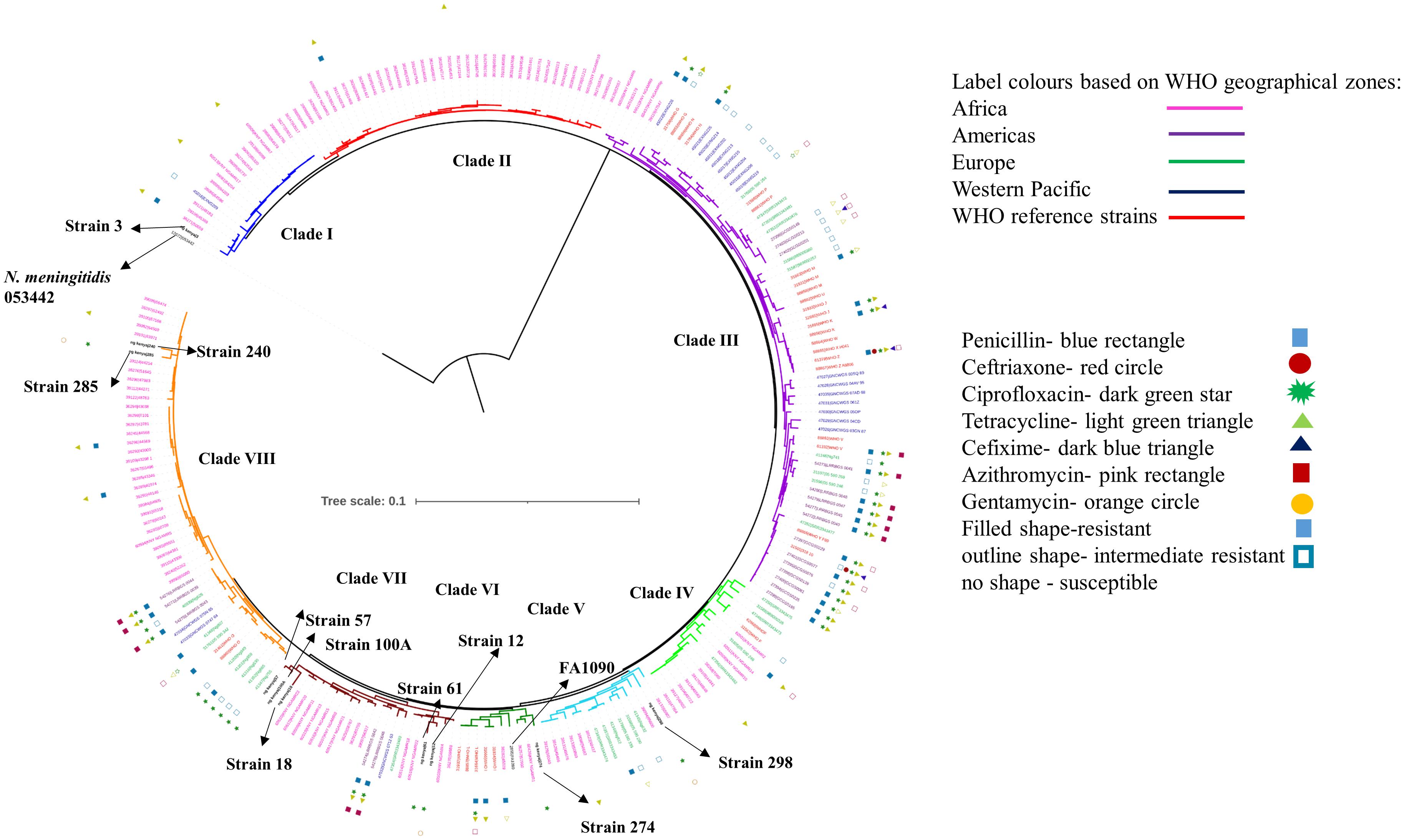
Figure 1. Phylogenetic tree of the sequenced strains with strains from WHO geographical regions and WHO reference strains. The strains sequenced in this study have been indicated in black, bold text in the tree. The antibiotic resistance profile, wherever available, has been indicated as metadata (Penicillin – blue rectangle, Ceftriaxone – red circle, Ciprofloxacin – dark green star, Tetracycline – right-pointing light green triangle, Cefixime – left-pointing dark blue triangle, Azithromycin – pink rectangle, Gentamycin – orange circle, Filled shape – resistant, outline shape – intermediate resistant, no shape – susceptible). The clades inferred from the phylogeny analysis have been indicated in different colors and numbered I–VIII. The strains from different WHO geographical regions have been indicated in different color labels (Africa – Pink, Americas – Magenta, Europe – Green, Western Pacific – Blue, WHO reference strains – Red) The strains sequenced in this study, the reference strain for N. gonorrhoeae (FA1090), and an N. meningitidis strain (PubMLST id:053442) used as an outgroup for the analysis have been labeled and indicated with arrows.
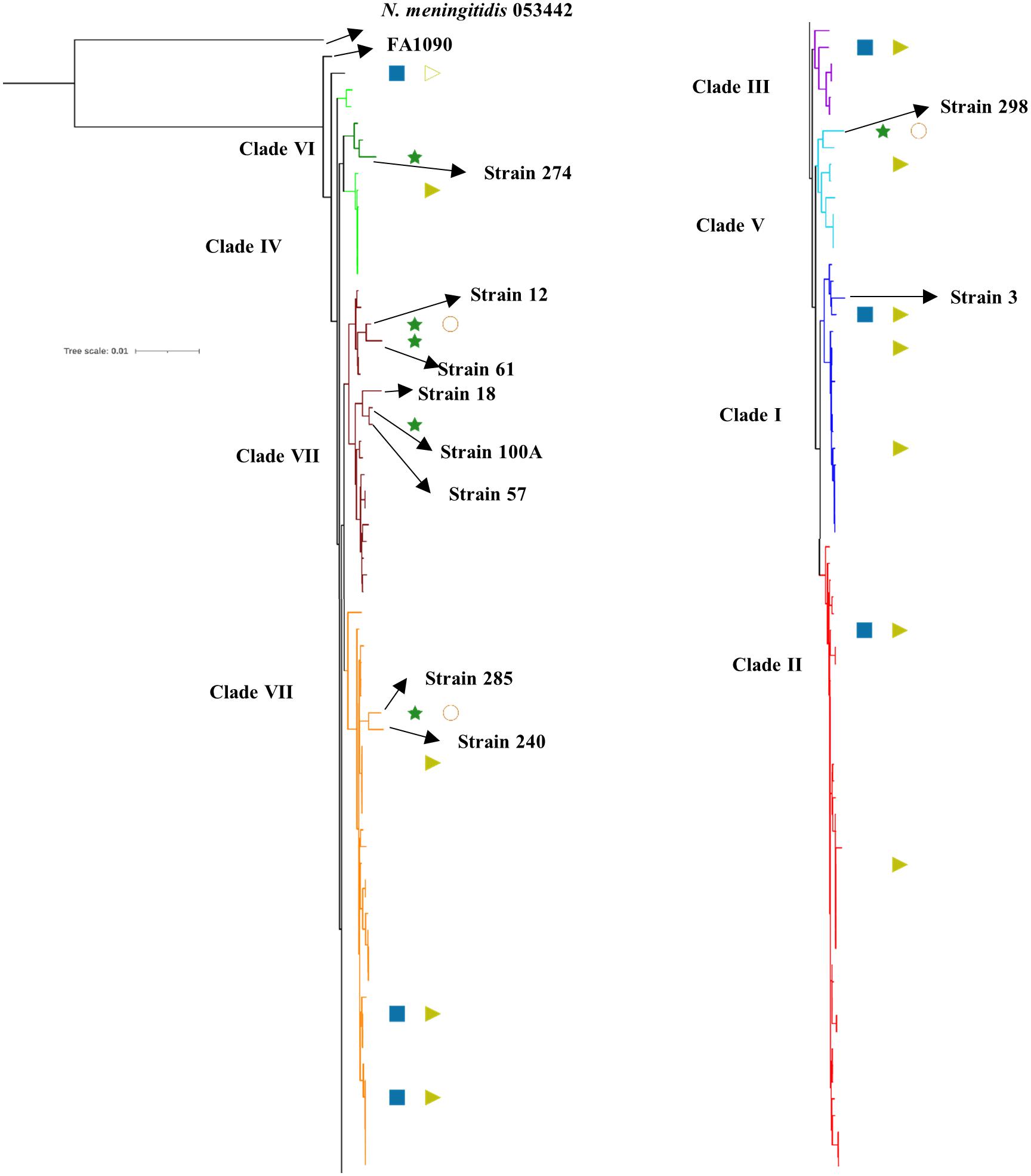
Figure 2. Phylogenetic tree of the sequenced strains with previously reported strains from Kenya. The antibiotic resistance profile, wherever available, has been indicated as metadata (Penicillin – blue rectangle, Ciprofloxacin – dark green star, Tetracycline – right-pointing light green triangle, Gentamycin – orange circle, Filled shape – resistant, outline shape – intermediate resistant, no shape – susceptible). The clades inferred from the phylogeny analysis have been indicated in different colors and numbered I–VIII as per Figure 1. The strains sequenced in this study, the reference strain for N. gonorrhoeae (FA1090), and an N. meningitidis strain (PubMLST id:053442) used as an outgroup for the analysis have been labeled and indicated with arrows.
Discussion
In this study, we assessed the feasibility of nanopore sequencing and AMR gene annotation in a clinical microbiology laboratory setting. We found that DNA extraction followed by MinION sequencing facilitates efficient detection of chromosomal and plasmid-mediated AMR genes in clinical isolates. From the 14 isolates sequenced in this work, we obtained 12 N. gonorrhoeae genomes, out of which 10 had more than 98% coverage with high depth in the AMR and MLST gene regions analyzed in the study. We were able to detect mutations in multiple genes involved in AMR and we also focused on investigating the structure–function aspects of drug resistance mutations in some proteins.
The consensus genome mapping obtained using bwa-mem was chosen for analysis, due to higher genome coverage and lower number of indels and misassemblies compared to the reference genome. Although earlier studies have shown that bacterial MinION sequences show high mapping rates to the reference genome, with fewer indel rates, bwa-mem has found limited application with long error-prone reads (Finn et al., 2014; Laver et al., 2015; Gerstmans et al., 2020; Nguyen et al., 2020). We show that it is possible to obtain assemblies with good genome coverage for AMR determinant detection by using nanopore-only reads with a consensus genome mapping strategy. Previous studies have obtained hybrid genome assembly for gonococcal isolates by combining short-read (Illumina) and long-read (ONT) sequence data (Eyre et al., 2018; Golparian et al., 2018). This is another approach for deriving a consensus genome using MinION sequencing and, accordingly, an alternative to de novo assembly.
Although the error rate for single nanopore reads is higher, consensus approaches can result in highly reliable assemblies. Assemblies derived from MinION-1D sequencing have shown a good agreement between AMR testing and AMR prediction results (Lemon et al., 2017; Sanderson et al., 2020). Studies on comparison of assemblies derived from Nanopore-only, Illumina, and hybrid assemblies have shown that the AMR prediction is comparable across all three methods (Chen et al., 2020). Recent advances in Nanopore sequencing and analysis workflows have demonstrated the utility of ONT sequencers for metagenomic analysis (Sanderson et al., 2020; Street et al., 2020). Additionally, there are workflows available for real-time sequence analysis that make the process faster.
Databases like PubMLST, CARD, ARIBA, and Pathogenwatch include profiles for resistance mediators like penA, mtrR, penB, ponA, 23S rDNA, gyrA, and tetM and do not include other determinants in their gonococcal AMR characterization module. We identified a set of AMR determinants from literature, including chromosomal gene mutations, promoter mutations, and the presence of plasmids with AMR genes. Plasmids containing genes encoding beta-lactamase and tetracycline resistance mediators [tet(M) and blaTEM] were identified in all but one strain (strain 12 contained only a TetM containing plasmid). This is consistent with a previous study that the plasmids are almost ubiquitous in Kenyan strains, thought to be because of the overuse of doxycycline for treating STIs (Cehovin et al., 2018). Among the reported mutations, we observed S91F and D95Y in the QRDR (residues 55–110) of GyrA and E91G in the ParC QRDR (residues 66–119). We also observed the silent mutations Y104 (codon change TAC − > TAT seen in all isolates except 298) and L131 (codon change of CTG − > CTA seen in isolates 3 and 240) in ParC reported to be associated with ciprofloxacin-resistant strains (Chaudhry et al., 2002). Novel mutations, V68A in the QRDR region of ParC (strain 285), E79 insertion in FolP (all isolates), and G70Y in RplD (strains 12 and 57), were also observed. However, whether these mutations have any effect on antibiotic resistance has to be experimentally validated. We were able to explain the basis of ciprofloxacin resistance in all strains except one, strain 298, which showed no mutations in gyrA, but had the E91G mutation in parC. We did not identify plasmids containing aminoglycoside resistance determinants like aminoglycoside N-acetyltransferases (aat) for gentamycin resistance (Garneau-Tsodikova and Labby, 2016).
Based on homology modeling using templates crystalized with antibiotics, we see that the mutations in GyrA disrupt the hydrogen bonding with ciprofloxacin, and mutations in ParC occur very close to Ser 88, which is involved in hydrogen bonding with moxifloxacin. We also observe that mutations in FolP at the substrate-binding site could affect the interaction with the antibiotic, as the residue at R228 interacts with sulfonamide. A phylogenetic study with previously sequenced Kenyan strains clustered our strains in the previously described cluster. From phylogenetic analysis using sequences from different countries, we observed five major clusters, with most of the Kenyan sequences (including the sequences from this study) clustered together with strains from different geographical regions.
We anticipate that the nanopore sequencing technology will expand the scope for rapid genome sequencing, which can help us understand the mechanistic basis of drug resistance and clinical management. AMR in N. gonorrhoeae is a result of a single to multiple mutations in same or different genes acting synergistally and additional unknown mutations/genes may be involved (Harrison et al., 2016; Ma et al., 2020a). Hence, there is a need for updated resistance gene databases and more studies on AMR genotype–phenotype correlations (Lemon et al., 2017). Recent advances have also shown promise for identifying clinical isolates and detecting AMR elements in real time (Schmidt et al., 2017; Sanderson et al., 2018). However, the read error rates mandate a consensus approach for read assembly and AMR allele detection. With future improvements in read accuracy and computational tools for base-calling, we believe that AMR detection can happen in clinical settings within hours of sequencing (Lemon et al., 2017).
Conclusion
Using a literature mining approach, we have predicted ciprofloxacin resistance using mutations in gyrA/parC in strains showing decreased susceptibility to ciprofloxacin. However, few of the gyrA mutations can also occur in susceptible strains, and molecular tests using gene-based PCR or WGS can be used to complement culture-based antibiotic resistance testing. Culture-based testing can reflect mutations in unknown antibiotic targets, but cannot predict if a patient can develop resistance later on, based on pre-existing mutations.
In conclusion, in the first consensus genome for gonococci using only MinION, we show that using this approach, we can obtain near-complete genomes that were effectively used for AMR and phylogenetic analysis. We also show that currently available tools for AMR analysis of gonococci are not able to capture many mutations listed in the literature. We have also provided a dataset of over 100 existing mutations in different genes implicated in AMR, plasmids, and efflux pumps, which can be used by researchers across the world. This list will be continuously updated to keep up with the identification of new AMR targets/mutations.
Here, we demonstrate the potential of using MinION in resource-limited settings where NGS facility is unavailable. This can be used in settings with concerns about the export of samples/DNA for WGS to other countries. This approach can also be used across a range of bacterial genomes, even in case of metagenomic samples, where reads from a bacterial genome can be separated from one another, for subsequent analysis. Testing in clinical datasets can potentially provide a new approach to complement traditional methods for diagnosis, AMR surveillance, and public health management of N. gonorrhoeae. Further work is required in evaluating the MinION sequencer for outbreak prediction and clinical diagnosis of bacterial infection.
Data Availability Statement
The raw reads generated for this study have been deposited in BioProject Accession: PRJNA660404 (https://www.ncbi.nlm.nih.gov). The biosample details are available under the IDs SAMN15960547, SAMN15960548, SAMN15960549, SAMN15960550, SAMN15960551, SAMN15960552, SAMN15960553, SAMN15960554, and SAMN15960555. The corresponding genomes and annotation files are available under the IDs CP061491, CP061490, CP061498, CP061488, CP061487, CP061486, CP061492, CP061485, CP061484, and CP061483.
Ethics Statement
The ethics approval for the study was obtained from the Kenyatta National Hospital – University of Nairobi Ethics and Research Committee, Nairobi, Kenya, for the use of anonymized samples collected from female patients attending local Sexually Transmitted Disease (STD) clinics.
Author Contributions
MJ collected the isolates and carried out characterization, culturing, and sequencing. AS, TD, AP, and RK planned and carried out the sequencing. IM conceived and performed the bioinformatics analysis, and wrote the manuscript with inputs from SK, AS, MM, and MJ. RN, PM, MM, RC, JN, and JG performed the experiments. SK, RS, and OA provided intellectual support and valuable discussions. All authors listed have made a substantial contribution to the work and approved it for publication.
Funding
This work was supported by the capacity component of the “Indo Africa dengue sequencing to vaccine” grant from Narayana Murthy (Infosys) and the NCBS core funds to SK. We would like to acknowledge KAVI-ICR for the initial funding for the project. We thank NCBS (TIFR) and KAVI-ICR for infrastructural and financial support. RS acknowledges funding and support provided by JC Bose Fellowship (SB/S2/JC-071/2015) from Science and Engineering Research Board, India and Bioinformatics Centre Grant funded by Department of Biotechnology, India (BT/PR40187/BTIS/137/9/2021).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.647565/full#supplementary-material
Footnotes
- ^ https://github.com/nanoporetech/qcat
- ^ https://github.com/marbl/canu
- ^ https://github.com/lh3/minimap2
- ^ https://github.com/lh3/miniasm
- ^ https://github.com/isovic/racon
- ^ https://github.com/jts/nanopolish
- ^ http://bio-bwa.sourceforge.net/
- ^ http://www.htslib.org/
- ^ http://mummer.sourceforge.net/
- ^ https://bedtools.readthedocs.io/en/latest/
- ^ https://rast.nmpdr.org/
- ^ https://ngstar.canada.ca/
- ^ https://pubmlst.org/
- ^ http://www.ng-mast.net/
- ^ https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/
- ^ https://www.ncbi.nlm.nih.gov/bioproject/PRJNA313047/
- ^ https://www.ebi.ac.uk/Tools/msa/clustalo/
- ^ https://pubmlst.org/software/database/bigsdb/
References
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2019). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525.
Altschul, S. F. (2005). “BLAST Algorithm,” in Encyclopedia of Life Sciences, eds R. Bridgewater. (New York: John Wiley and Sons)
Balloux, F., Brønstad Brynildsrud, O., van Dorp, L., Shaw, L. P., Chen, H., Harris, K. A., et al. (2018). From Theory to Practice: translating Whole-Genome Sequencing (WGS) into the Clinic. Trends Microbiol. 26, 1035–1048. doi: 10.1016/j.tim.2018.08.004
Belland, R. J., Morrison, S. G., Ison, C., and Huang, W. M. (1994). Neisseria gonorrhoeae acquires mutations in analogous regions of gyrA and parC in fluoroquinolone-resistant isolates. Mol. Microbiol. 14, 371–380. doi: 10.1111/j.1365-2958.1994.tb01297.x
Bennett, J. S., Jolley, K. A., Sparling, P. F., Saunders, N. J., Hart, C. A., Feavers, I. M., et al. (2007). Species status of Neisseria gonorrhoeae: evolutionary and epidemiological inferences from multilocus sequence typing. BMC Biol. 5:35. doi: 10.1186/1741-7007-5-35
Castro-Wallace, S. L., Chiu, C. Y., John, K. K., Stahl, S. E., Rubins, K. H., McIntyre, A. B. R., et al. (2017). Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Sci. Rep. 7:18022. doi: 10.1038/s41598-017-18364-0
Cehovin, A., Harrison, O. B., Lewis, S. B., Ward, P. N., Ngetsa, C., Graham, S. M., et al. (2018). Identification of Novel Neisseria gonorrhoeae Lineages Harboring Resistance Plasmids in Coastal Kenya. J. Infect. Dis. 218, 801–808. doi: 10.1093/infdis/jiy240
Chaudhry, U., Ray, K., Bala, M., and Saluja, D. (2002). Mutation patterns in gyrA and parC genes of ciprofloxacin resistant isolates of Neisseria gonorrhoeae from India. Sex. Transm. Infect. 78, 440–444. doi: 10.1136/sti.78.6.440
Chen, A., and Seifert, H. S. (2013). Structure-function studies of the Neisseria gonorrhoeae major outer membrane porin. Infect. Immun. 81, 4383–4391. doi: 10.1128/iai.00367-13
Chen, Z., Kuang, D., Xu, X., González-Escalona, N., Erickson, D. L., Brown, E., et al. (2020). Genomic analyses of multidrug-resistant Salmonella Indiana, Typhimurium, and Enteritidis isolates using MinION and MiSeq sequencing technologies. PLoS One 15:e0235641. doi: 10.1371/journal.pone.0235641
Chisholm, S. A., Quaye, N., Cole, M. J., Fredlund, H., Hoffmann, S., Jensen, J. S., et al. (2011). An evaluation of gentamicin susceptibility of Neisseria gonorrhoeae isolates in Europe. J. Antimicrob. Chemother. 66, 592–595. doi: 10.1093/jac/dkq476
Collineau, L., Boerlin, P., Carson, C. A., Chapman, B., Fazil, A., Hetman, B., et al. (2019). Integrating Whole-Genome Sequencing Data Into Quantitative Risk Assessment of Foodborne Antimicrobial Resistance: a Review of Opportunities and Challenges. Front. Microbiol. 10:1107. doi: 10.3389/fmicb.2019.01107
Demczuk, W., Sidhu, S., Unemo, M., Whiley, D. M., Allen, V. G., Dillon, J. R., et al. (2017). Neisseria gonorrhoeae Sequence Typing for Antimicrobial Resistance, a Novel Antimicrobial Resistance Multilocus Typing Scheme for Tracking Global Dissemination of N. gonorrhoeae Strains. J. Clin. Microbiol. 55, 1454–1468. doi: 10.1128/jcm.00100-17
Duncan, S., Thiong’o, A. N., Macharia, M., Wamuyu, L., Mwarumba, S., Mvera, B., et al. (2011). High prevalence of quinolone resistance in Neisseria gonorrhoeae in coastal Kenya. Sex,. Transm. Infect. 87:231. doi: 10.1136/sti.2010.048777
Elliott, I., Batty, E. M., Ming, D., Robinson, M. T., Nawtaisong, P., de Cesare, M., et al. (2020). Oxford Nanopore MinION Sequencing Enables Rapid Whole Genome Assembly of Rickettsia typhi in a Resource-Limited Setting. Am. J. Trop. Med. Hyg. 102, 408–414. doi: 10.4269/ajtmh.19-0383
Eyre, D. W., Sanderson, N. D., Lord, E., Regisford-Reimmer, N., Chau, K., Barker, L., et al. (2018). Gonorrhoea treatment failure caused by a Neisseria gonorrhoeae strain with combined ceftriaxone and high-level azithromycin resistance, England, February 2018. Euro Surveill. 23:1800323.
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230.
Garneau-Tsodikova, S., and Labby, K. J. (2016). Mechanisms of Resistance to Aminoglycoside Antibiotics: overview and Perspectives. Medchemcomm 7, 11–27. doi: 10.1039/c5md00344j
Gerstmans, H., Grimon, D., Gutiérrez, D., Lood, C., Rodríguez, A., van Noort, V., et al. (2020). A VersaTile-driven platform for rapid hit-to-lead development of engineered lysins. Sci. Adv. 6:eaaz1136. doi: 10.1126/sciadv.aaz1136
Goldstein, S., Beka, L., Graf, J., and Klassen, J. L. (2019). Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genomics. 20:23. doi: 10.1186/s12864-018-5381-7
Golparian, D., Donà, V., Sánchez-Busó, L., Foerster, S., Harris, S., Endimiani, A., et al. (2018). Antimicrobial resistance prediction and phylogenetic analysis of Neisseria gonorrhoeae isolates using the Oxford Nanopore MinION sequencer. Sci. Rep. 8:17596. doi: 10.1038/s41598-018-35750-4
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Harrison, O. B., Clemence, M., Dillard, J. P., Tang, C. M., Trees, D., Grad, Y. H., et al. (2016). Genomic analyses of Neisseria gonorrhoeae reveal an association of the gonococcal genetic island with antimicrobial resistance. J. Infect. 73, 578–587. doi: 10.1016/j.jinf.2016.08.010
Hunt, M., Mather, A. E., Sánchez-Busó, L., Page, A. J., Parkhill, J., Keane, J. A., et al. (2017). ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microb. Genom. 3:e000131.
Johnson, L., Weberman, B., Parker, N., and Hackert, P. (2016). Neisseria meningitidis: an Emerging Sexually Transmitted Infection. Open Forum Infect. Dis. 3:1301. doi: 10.1093/ofid/ofw172.1004
Jolley, K. A., Bliss, C. M., Bennett, J. S., Bratcher, H. B., Brehony, C., Colles, F. M., et al. (2012). Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158, 1005–1015. doi: 10.1099/mic.0.055459-0
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Kivata, M. W., Mbuchi, M., Eyase, F. L., Bulimo, W. D., Kyanya, C. K., Oundo, V., et al. (2019). gyrA and parC mutations in fluoroquinolone-resistant Neisseria gonorrhoeae isolates from Kenya. BMC Microbiol. 19:76. doi: 10.1186/s12866-019-1439-1
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12.
Laver, T., Harrison, J., O’Neill, P. A., Moore, K., Farbos, A., Paszkiewicz, K., et al. (2015). Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 3, 1–8. doi: 10.1016/j.bdq.2015.02.001
Lee, S.-G., Lee, H., Jeong, S. H., Yong, D., Chung, G. T., Lee, Y. S., et al. (2010). Various penA mutations together with mtrR, porB and ponA mutations in Neisseria gonorrhoeae isolates with reduced susceptibility to cefixime or ceftriaxone. J. Antimicrob. Chemother. 65, 669–675. doi: 10.1093/jac/dkp505
Lemon, J. K., Khil, P. P., Frank, K. M., and Dekker, J. P. (2017). Rapid Nanopore Sequencing of Plasmids and Resistance Gene Detection in Clinical Isolates. J. Clin. Microbiol. 55, 3530–3543. doi: 10.1128/jcm.01069-17
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, 242–245.
Li, H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110. doi: 10.1093/bioinformatics/btw152
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735. doi: 10.1038/nmeth.3444
Ma, K. C., Mortimer, T. D., Duckett, M. A., Hicks, A. L., Wheeler, N. E., Sánchez-Busó, L., et al. (2020b). Increased power from conditional bacterial genome-wide association identifies macrolide resistance mutations in Neisseria gonorrhoeae. Nat. Commun. 11:5374. doi: 10.1038/s41467-020-19250-6
Ma, K. C., Mortimer, T. D., Duckett, M. A., Hicks, A. L., Wheeler, N. E., Sánchez-Busó, L., et al. (2020a). Increased power from bacterial genome-wide association conditional on known effects identifies Neisseria gonorrhoeae macrolide resistance mutations in the 50S ribosomal protein L4. bioRxiv [preprint].
Martin, I. M. C., Ison, C. A., Aanensen, D. M., Fenton, K. A., and Spratt, B. G. (2004). Rapid sequence-based identification of gonococcal transmission clusters in a large metropolitan area. J. Infect. Dis. 189, 1497–1505. doi: 10.1086/383047
Naidenov, B., Lim, A., Willyerd, K., Torres, N. J., Johnson, W. L., Hwang, H. J., et al. (2019). Pan-Genomic and Polymorphic Driven Prediction of Antibiotic Resistance in Elizabethkingia. Front. Microbiol. 10:1446. doi: 10.3389/fmicb.2019.01446
NCBI Resource Coordinators (2017). Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 45, D12–D17.
Nguyen, S. H., Cao, M. D., and Coin, L. (2020). Real-time resolution of short-read assembly graph using ONT long reads. bioRxiv [preprint].
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2013). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Papp, J. R., Abrams, A. J., Nash, E., Katz, A. R., Kirkcaldy, R. D., O’Connor, N. P., et al. (2017). Azithromycin Resistance and Decreased Ceftriaxone Susceptibility in Neisseria gonorrhoeae, Hawaii, USA. Emerg. Infect. Dis. 23, 830–832. doi: 10.3201/eid2305.170088
Plesivkova, D., Richards, R., and Harbison, S. (2019). A review of the potential of the MinIONTM single-molecule sequencing system for forensic applications. Wiley Interdiscip. Rev. Forensic. Sci. 1:e1323. doi: 10.1002/wfs2.1323
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Roberts, M. C., Chung, W. O., Roe, D., Xia, M., Marquez, C., Borthagaray, G., et al. (1999). Erythromycin-resistant Neisseria gonorrhoeae and oral commensal Neisseria spp. carry known rRNA methylase genes. Antimicrob. Agents Chemother. 43, 1367–1372. doi: 10.1128/aac.43.6.1367
Sánchez, R., and Sali, A. (2000). Comparative protein structure modeling. Introduction and practical examples with modeller. Methods Mol. Biol. 143, 97–129. doi: 10.1385/1-59259-368-2:97
Sanderson, N. D., Street, T. L., Foster, D., Swann, J., Atkins, B. L., Brent, A. J., et al. (2018). Real-time analysis of nanopore-based metagenomic sequencing from infected orthopaedic devices. BMC Genom. 19:714. doi: 10.1186/s12864-018-5094-y
Sanderson, N. D., Swann, J., Barker, L., Kavanagh, J., Hoosdally, S., Crook, D., et al. (2020). High precision Neisseria gonorrhoeae variant and antimicrobial resistance calling from metagenomic Nanopore sequencing. Genome Res. 30, 1354–1363. doi: 10.1101/gr.262865.120
Schmidt, K., Mwaigwisya, S., Crossman, L. C., Doumith, M., Munroe, D., Pires, C., et al. (2017). Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J. Antimicrob. Chemother. 72, 104–114. doi: 10.1093/jac/dkw397
Sievers, F., and Higgins, D. G. (2014). Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 1079, 105–116. doi: 10.1007/978-1-62703-646-7_6
Slack, M. P. E. (2010). “Chapter 172 - Gram-negative coccobacilli,” in Powderly WGBT-ID (Third E), eds J. Cohen and S. M. Opal (London: Content Repository Only), 1738–1756. doi: 10.1016/b978-0-323-04579-7.00172-6
Street, T. L., Barker, L., Sanderson, N. D., Kavanagh, J., Hoosdally, S., Cole, K., et al. (2020). Optimizing DNA Extraction Methods for Nanopore Sequencing of Neisseria gonorrhoeae Directly from Urine Samples. J. Clin. Microbiol. 58, e01822–19.
Tanaka, M., Nakayama, H., Haraoka, M., and Saika, T. (2000). Antimicrobial resistance of Neisseria gonorrhoeae and high prevalence of ciprofloxacin-resistant isolates in Japan, 1993 to 1998. J. Clin. Microbiol. 38, 521–525. doi: 10.1128/jcm.38.2.521-525.2000
Tyler, A. D., Mataseje, L., Urfano, C. J., Schmidt, L., Antonation, K. S., Mulvey, M. R., et al. (2018). Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 8:10931. doi: 10.1038/s41598-018-29334-5
Unemo, M., and Dillon, J.-A. R. (2011). Review and international recommendation of methods for typing neisseria gonorrhoeae isolates and their implications for improved knowledge of gonococcal epidemiology, treatment, and biology. Clin. Microbiol. Rev. 24, 447–458. doi: 10.1128/cmr.00040-10
Unemo, M., Golparian, D., Sánchez-Busó, L., Grad, Y., Jacobsson, S., Ohnishi, M., et al. (2016). The novel 2016 WHO Neisseria gonorrhoeae reference strains for global quality assurance of laboratory investigations: phenotypic, genetic and reference genome characterization. J. Antimicrob. Chemother. 71, 3096–3108. doi: 10.1093/jac/dkw288
Unemo, M., and Nicholas, R. A. (2012). Emergence of multidrug-resistant, extensively drug-resistant and untreatable gonorrhea. Future Microbiol. 7, 1401–1422. doi: 10.2217/fmb.12.117
Unemo, M., and Shafer, W. M. (2011). Antibiotic resistance in Neisseria gonorrhoeae: origin, evolution, and lessons learned for the future. Ann. N. Y. Acad. Sci. 1230, E19–E28.
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Wi, T., Lahra, M. M., Ndowa, F., Bala, M., Dillon, J.-A. R., Ramon-Pardo, P., et al. (2017). Antimicrobial resistance in Neisseria gonorrhoeae: global surveillance and a call for international collaborative action. PLoS Med. 14:e1002344. doi: 10.1371/journal.pmed.1002344
World Health Organisation (WHO) (2017). Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. 74. Available at: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed January 12, 2021).
World Health Organisation (WHO) (2018). Report On Global Sexually Transmitted Infection Surveillance. 74. Available Online at: https://www.who.int/reproductivehealth/publications/stis-surveillance-2018/en/ (accessed January 12, 2021).
Yun, M.-K., Wu, Y., Li, Z., Zhao, Y., Waddell, M. B., Ferreira, A. M., et al. (2012). Catalysis and Sulfa Drug Resistance in Dihydropteroate Synthase. Science 335, 1110–1114. doi: 10.1126/science.1214641
Zapun, A., Morlot, C., and Taha, M.-K. (2016). Resistance to β-Lactams in Neisseria ssp Due to Chromosomally Encoded Penicillin-Binding Proteins. Antibiotics 5:35. doi: 10.3390/antibiotics5040035
Keywords: nanopore sequencing, whole-genome sequencing, antimicrobial resistance, AMR prediction, sequence-typing
Citation: Juma M, Sankaradoss A, Ndombi R, Mwaura P, Damodar T, Nazir J, Pandit A, Khurana R, Masika M, Chirchir R, Gachie J, Krishna S, Sowdhamini R, Anzala O and Meenakshi IS (2021) Antimicrobial Resistance Profiling and Phylogenetic Analysis of Neisseria gonorrhoeae Clinical Isolates From Kenya in a Resource-Limited Setting. Front. Microbiol. 12:647565. doi: 10.3389/fmicb.2021.647565
Received: 12 January 2021; Accepted: 31 May 2021;
Published: 27 July 2021.
Edited by:
Guido Werner, Robert Koch Institute (RKI), GermanyReviewed by:
Leonor Sánchez-Busó, Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunitat Valenciana (FISABIO), SpainSebastian Banhart, Robert Koch Institute (RKI), Germany
Copyright © 2021 Juma, Sankaradoss, Ndombi, Mwaura, Damodar, Nazir, Pandit, Khurana, Masika, Chirchir, Gachie, Krishna, Sowdhamini, Anzala and Meenakshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iyer S. Meenakshi, bWVlbmFrc2hpc0BuY2JzLnJlcy5pbg==; Omu Anzala, b2FuemFsYUBrYXZpdW9uLm9yZw==
†These authors share first authorship