 Joshua Mbanga
Joshua Mbanga Daniel G. Amoako
Daniel G. Amoako Akebe L. K. Abia
Akebe L. K. Abia Mushal Allam
Mushal Allam Arshad Ismail
Arshad Ismail Sabiha Y. Essack
Sabiha Y. Essack- 1Antimicrobial Research Unit, College of Health Sciences, University of Kwazulu-Natal, Durban, South Africa
- 2Department of Applied Biology and Biochemistry, National University of Science and Technology, Bulawayo, Zimbabwe
- 3Sequencing Core Facility, National Institute for Communicable Diseases, National Health Laboratory Service, Johannesburg, South Africa
We investigated the antibiotic resistome, mobilome, virulome, and phylogenomic lineages of Enterococcus spp. obtained from a wastewater treatment plant and its associated waters using whole-genome sequencing (WGS) and bioinformatics tools. The whole genomes of Enterococcus isolates including Enterococcus faecalis (n = 4), Enterococcus faecium (n = 5), Enterococcus hirae (n = 2), and Enterococcus durans (n = 1) with similar resistance patterns from different sampling sites and time points were sequenced on an Illumina MiSeq machine. Multilocus sequence typing (MLST) analysis revealed two E. faecalis isolates that had a common sequence type ST179; the rest had unique sequence types ST841, and ST300. The E. faecium genomes belonged to 3 sequence types, ST94 (n = 2), ST361 (n = 2), and ST1096 (n = 1). Detected resistance genes included those encoding tetracycline [tet(S), tet(M), and tet(L)], and macrolides [msr(C), msr(D), erm(B), and mef(A)] resistance. Antibiotic resistance genes were associated with insertion sequences (IS6, ISL3, and IS982), and transposons (Tn3 and Tn6000). The tet(M) resistance gene was consistently found associated with a conjugative transposon protein (TcpC). A total of 20 different virulence genes were identified in E. faecalis and E. faecium including those encoding for sex pheromones (cCF10, cOB1, cad, and came), adhesion (ace, SrtA, ebpA, ebpC, and efaAfs), and cell invasion (hylA and hylB). Several virulence genes were associated with the insertion sequence IS256. No virulence genes were detected in E. hirae and E. durans. Phylogenetic analysis revealed that all Enterococcus spp. isolates were more closely related to animal and environmental isolates than clinical isolates. Enterococcus spp. with a diverse range of resistance and virulence genes as well as associated mobile genetic elements (MGEs) exist in the wastewater environment in South Africa.
Introduction
The efficiency of wastewater treatment plants (WWTPs) is critical to preventing the spread of antibiotic resistant bacteria (ARB) and antibiotic resistance genes (ARGs) into the environment (Karkman et al., 2017; Alexander et al., 2020). Although AMR surveillance in clinical settings and animals is well established in most developed and some developing countries, surveillance in the environment still lags behind (Huijbers et al., 2019). The emergence of ARB and ARGs in the water environment has become an important environmental health issue (Conte et al., 2017; Karkman et al., 2017; Alexander et al., 2020). Dissemination of ARGs is thought to occur in the environment mainly through the transfer of mobile genetic elements (MGEs) such as plasmids, transposons, integrons, gene cassettes, Integrative and conjugative elements (ICE), and insertion sequence common regions between bacterial species (Sanderson et al., 2020). The selection pressure in a given environment is crucial as it influences the spread and accumulation of ARGs some of which may be novel (Bengtsson-Palme et al., 2018). The risk of transfer of ARGs to pathogens increases in environments with a high fecal load and associated fecal bacteria (Huijbers et al., 2019).
Enterococcus species are Gram positive non-sporulating organisms that mainly exist as commensals in the intestinal flora of healthy animals and humans. They can thus be excreted into environmental sources including soil and surface water as fecal matter and are thus commonly used as indicator organisms in water environments (Berendonk et al., 2013; Karkman et al., 2018). Some like faecalis and Enterococcus faecium are opportunistic pathogens whilst other species such as Enterococcus hirae and Enterococcus durans are rarely pathogenic in humans (Bourafa et al., 2015; Ryu et al., 2019). Enterococcus spp. can easily acquire and disseminate resistance determinants (Medeiros et al., 2014) making them suitable for antibiotic resistance surveillance studies.
Whole-genome sequencing (WGS) is a highly discriminatory technique for studying bacterial species, including Enterococci. However, very few studies have used WGS to study environmental enterococcal isolates (Sanderson et al., 2020; Zaheer et al., 2020). The application of WGS to antibiotic resistance surveillance remains largely confined to clinical and animal settings, with very little attention given to the environment (Hendriksen et al., 2019; Su et al., 2019; WHO, 2020). There is therefore a paucity of data on the role that genomic surveillance plays in understanding the environmental dimensions of antibiotic resistance, particularly in Africa.
In this study, we investigated the antibiotic resistome, mobilome, virulome, and phylogenomic lineages of Enterococcus spp. obtained from a WWTP and its associated waters. Additionally, we assayed the role of the water environment in the dissemination of multi-drug resistant Enterococcus spp. which could be of clinical or veterinary importance.
Materials and Methods
Ethical Consideration
Ethical approval was received from the Biomedical Research Ethics Committee (Reference: BCA444/16) of the University of KwaZulu-Natal. Permission to collect water samples was sought and granted by uMgeni Water which owns and operates the investigated WWTP.
Study Site
Manual grab water samples were collected in sterile 500-mL containers from the influent (29°36′3.70″S 30°25′41.71″E), final effluent (29°35′49.97″S 30°26′19.74″E) of a major WWTP as well as upstream (29°36′10.73″S 30°25′29.97″E) and downstream (29°36′27.54″S 30°27′0.76″E) of its associated receiving water body in uMgungundlovu District, KwaZulu-Natal, South Africa.
The WWTP is the largest in Pietermaritzburg, the provincial capital of KwaZulu-Natal in South Africa. The WWTP discharges its final effluent into the uMsunduzi river, a key water source for domestic, agricultural, and recreational purposes to inhabitants of the several informal settlements along its banks (Moodley et al., 2016).
Bacterial Isolates
Enterococcus spp. were isolated from water samples collected fortnightly over 7 months (May 2018 to November 2018).
Putative identification was accomplished during enumeration using the Enterolert®/Quanti-Tray® 2000 system followed by phenotypic confirmation on Bile Aesculin Azide agar (Merck, Germany) or Slanetz and Bartley agar (Merck, Germany). Samples from, upstream and downstream river water as well as final effluent were diluted 1 mL in 100 mL (0.01 dilution) while the influent with its higher bacterial load was o diluted by 0.05 mL in 100 mL (0.005 dilution) using sterile water. A volume of 100 mL of each sample was analyzed using the Enterolert® Quanti-Tray® 2000 system (IDEXX Laboratories (Pty.) Ltd., Johannesburg, South Africa). Enterococcus spp. were obtained from positive quanti-trays, sub-cultured on Bile Aesculin Azide or Slanetz and Bartley agar and incubated at 41°C for 24–48 h. At least ten distinct colonies representing each sampling site were randomly selected from the Bile Aesculin Azide or Slanetz and Bartley agar and further sub-cultured onto the same media, respectively, to obtain pure colonies. Molecular confirmation of Enterococcus spp. was done using real-time polymerase chain reaction (rtPCR) of the tuf (Elongation factor tu) gene (Ke et al., 1999).
Antibiotic susceptibility determination was undertaken using the Kirby-Bauer method on a panel of sixteen commercial antibiotic discs which included: chloramphenicol (CHL, 30 μg), tetracycline (TET, 30 μg), ampicillin (AMP, 10 μg), nitrofurantoin (NIT, 300 μg), ciprofloxacin (CIP, 5 μg), levofloxacin (LVX, 5 μg), imipenem (IPM, 10 μg), linezolid (LZD, 30 μg), erythromycin (E, 15 μg), quinupristin–dalfopristin (Q–D, 15 μg) against E. faecium only, tigecycline (TGC, 15 μg), trimethoprim-sulfamethoxazole (SXT, 25 μg), vancomycin (VAN, 30 μg) and teicoplanin (TEC, 30 μg). Detection of high-level aminoglycoside resistance was ascertained using gentamicin (GEN, 120 μg) and streptomycin (STR, 300 μg) discs. Inhibition zones were measured, and the results were interpreted using the European Committee on Antimicrobial Testing (EUCAST) breakpoint tables (EUCAST, 2020). Breakpoints for chloramphenicol, tetracycline, erythromycin, linezolid, nitrofurantoin, vancomycin, and gentamicin were obtained from the Clinical and Laboratory Standards Institute (CLSI) interpretative charts (CLSI, 2020). Enterococcus faecalis ATCC 29212 was used for quality control.
Whole-Genome Sequencing and Analysis
Twelve MDR Enterococcus spp. isolates with similar antibiograms obtained from all four sampled sites were selected for WGS. Genomic DNA was extracted using the GenElute Bacterial Genomic DNA kit (Sigma Aldrich, St. Louis, United States) followed by quantification using the 260/280 nm wavelength on a Nanodrop 8000 (Thermo Scientific Waltham, MA, United States). Library preparation was done using the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA, United States). WGS was undertaken using an Illumina MiSeq machine (Illumina, San Diego, CA, United States). The raw reads were quality trimming using Sickle v1.331 and assembled spontaneously using the SPAdes v3.6.2 assembler. All contiguous sequences were subsequently submitted to GenBank and assigned accession numbers under Bio project PRJNA609064 (Supplementary Table 1).
The assembled genomes were analyzed for MLST sequence types on the MLST 1.8 database (Larsen et al., 2012) hosted by the Centre for Genomic Epidemiology (CGE)2. Acquired antimicrobial resistance genes and chromosomal point mutations including the DNA gyrase gyrA and parC genes (quinolone resistance) and the pbp5 gene (ampicillin resistance) were annotated using ResFinder3 set at default threshold ID (90%) and minimum length (60%) values. Plasmid replicons types were identified using PlasmidFinder 2.1 on the CGE website4. Virulence genes were determined using VirulenceFinder 2.0 on the CGE website5.
The assembled genomes were further analyzed for MGEs, including insertion sequences, using ISFinder6 (Siguier, 2006), and intact prophages using PHASTER7 (Zhou et al., 2011; Arndt et al., 2016). ICE and putative integrative and mobilisable elements (IME) were identified using the ICEberg database8. RAST SEEDVIEWER9 was also used to annotate and identify the genomes with integrons, and transposons. The synteny and genetic environment of ARGs and associated MGEs were investigated using the general feature format (GFF3) files from GenBank. The genetic environment of virulence genes detected in the study were also determined using a similar approach. The GFF files were imported into Geneious prime 2020.210 for analysis.
Phylogenetic Reconstruction
Whole-genome sequences of the E. faecalis and E. faecium isolates were compared with isolates curated from the PATRIC website11 from different African countries including South Africa. The genomes of E. hirae and E. durans isolates were compared to those of isolates belonging to the respective species curated from the PATRIC website from different countries across the world as there were no/few entries from Africa. Whole-genome sequences of all isolates were uploaded and analyzed on the CSI Phylogeny 1.4 pipeline12 that recognizes, screens, and validates the location of single nucleotide polymorphisms (SNPs) before deducing a phylogeny based on the concatenated alignment of the high-quality SNPs. SNPs were identified from the alignments using the mpileup module in SAMTools version 0.1.18 (Li et al., 2009). Selection of SNPs was based on default parameters in CSI Phylogeny (Kaas et al., 2014). The following reference genomes were used for each alignment; E. faecalis, (E. faecalis V583), E. faecium (E. faecium DO), E. hirae (E. hirae ATCC 9790), and E. durans (E. durans ATCC 6056). The phylogenetic tree was constructed using FastTree (Price et al., 2010). The generated phylogenetic trees were viewed, annotated, and edited using the Iterative Tree of Life (iTOL)13.
Results
Isolate Source and Antibiotic Susceptibility Patterns
A total of 579 Enterococcus spp. isolates were obtained from the different sampling points. Of these, 12 isolates were selected for WGS, distributed as follows: three isolates from the upstream site of the WWTP along the receiving river, four from the downstream site, three from the raw influent, and two were from the final effluent of the WWTP (Supplementary Table 1). Selected isolates consisted of E. faecalis (4 isolates), E. faecium (5), E. hirae (2), and E. durans (1) (Supplementary Table 1).
The resistance patterns displayed by the selected isolates from different time points and sampling sites are shown in Table 1. The resistance profile TET-SXT-STR was found in two isolates, one E. faecalis obtained from raw influent and one E. durans isolate from the influent. Two E. faecalis isolates from downstream and upstream sites had the same resistance profile ERY-TEC-TET-SXT, other resistance profiles shared by at least two isolates from different sites included E. faecium (QD-TET-SXT, QD-TET-SXT-STR), and E. hirae (NIT-SXT-STR) isolates. The resistance profiles TEC-QD-TET-SXT, and ERY-TEC-LZD-TET-SXT-GEN-STR were unique to individual isolates (Table 1).
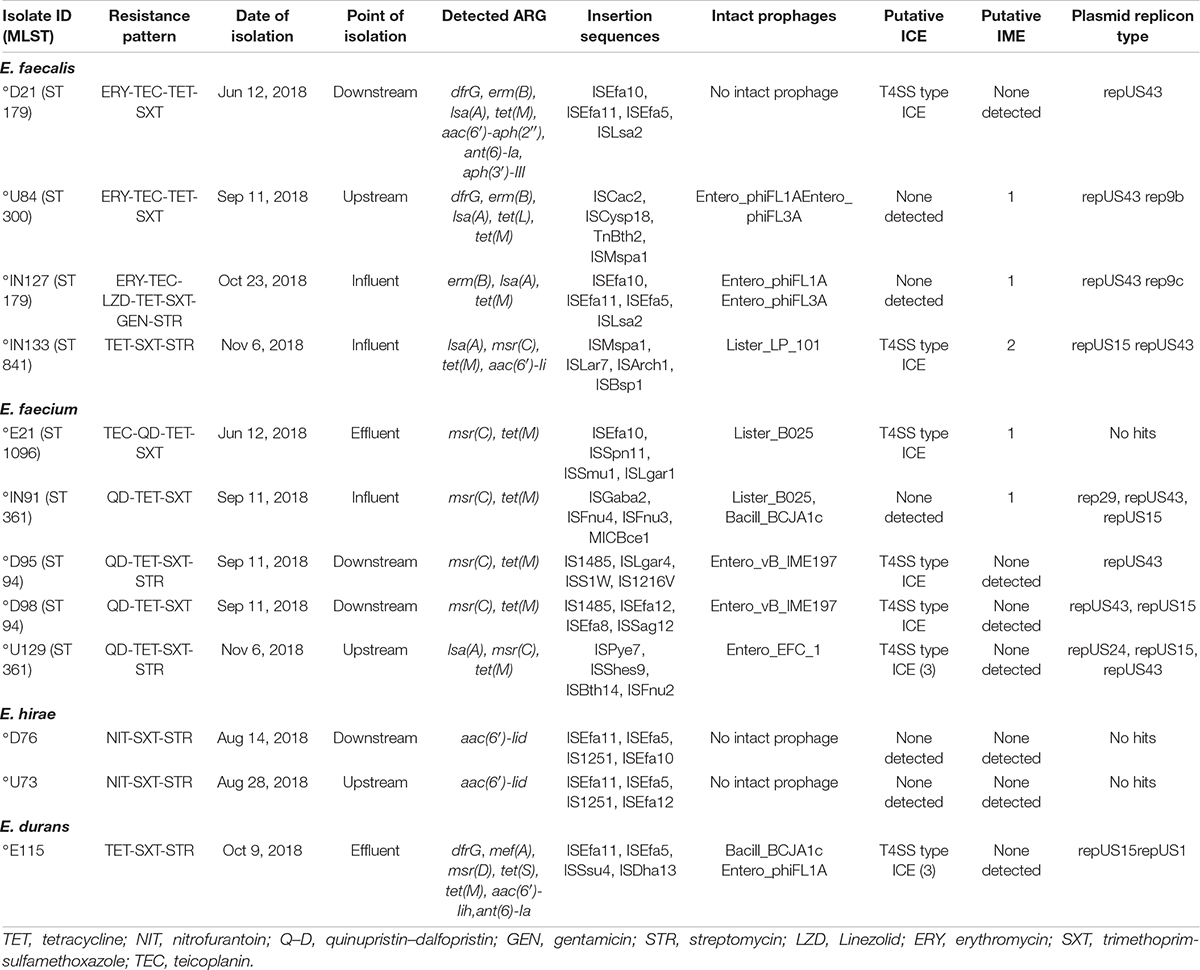
Table 1. Distribution of antibiotic resistance genes and mobile genetic elements in environmental Enterococcus spp.
Genome Characteristics
The genome and assembly characteristics of the Enterococcus spp. sequences are presented in Supplementary Table 1. The total assembled genome size ranged from 2.5–3.2 MB, the GC content ranged from 36.6–38.4, the N50, L50; the total number of contigs are also shown in Supplementary Table 1.
Antibiotic Resistance Genes
Several ARGs were present in the isolates, with each isolate harboring at least one ARG (Table 1). Most of the isolates belonging to all the sub-species harboured macrolides/streptogramins/lincosamides resistance genes lsa(A), msr(C), msr(D), erm(B), and mef(A). Other ARGs included the tetracycline resistance [tet(S), tet(M), and tet(L)], aminoglycoside resistance [aac(6′)-aph(2″), ant(6)-Ia, aph(3′)-III, aac(6′)-Iid, aac(6′)-Iih], and trimethoprim resistance (dfrG) gene (Table 1). In E. faecalis macrolide resistance was mediated by the erm(B) gene – two isolates from the influent (IN127, ST179), and downstream (D21, ST179) sites had the erm (B), isa(A), and tet(M) genes in common. Tetracycline resistance was mediated mainly by the tet(M) gene in all the TET resistant 10/12 (83.3%) isolates except for one E. faecalis isolate (U84 ST300) from the upstream site that had tet(M) and tet(L), as well as an E. durans, isolate (E115) from the effluent that had tet(M) and tet(S). In the E. faecium isolates resistance genes could not be linked to sequence type or source of isolation as all the isolates had the msr(C), and tet(M) genes in common (Table 1).
The quinolone resistance determinant regions (QRDRs) of the DNA gyrase (gyrA) and DNA topoisomerase IV genes (parC), were assayed for point mutations in all isolates. The gyrA (I259L∗, I306V∗, N708D∗, D759N∗, A811V∗, G819A∗, S820T∗, N708D∗), and parC (I699V∗, E707D∗, L773I∗) showed putatively novel mutations that were not linked to phenotypic resistance (Table 2). Only E. faecium isolates harboured mutations in all the assayed genes (Table 2). Point mutations in the pbp5 gene which encodes ampicillin resistance were mostly putatively novel mutations (Table 2). No mutations were found in E. faecalis, E. hirae, and E. durans.

Table 2. Point mutation in the gyrA, parC (quinolone resistance), and pbp5 (ampicillin resistance) genes in environmental Enterococcus spp.
Mobile Gene Elements (Plasmids, Insertion Sequences, Intact Prophages, and Integrons)
PlasmidFinder revealed a total of seven different plasmid associated replication genes (repUS15, repUS43, rep9c, rep9b, rep29, repUS24, repUS1). The repUS43 and repUS15 were the most common replicon types occurring in eight (66.7%) and five (41.6%) isolates, respectively (Table 1). A total of seven (58.3%) isolates had more than one plasmid replicon; however, no plasmid replicon types were detected in three isolates (one Enterococcus feacium and two E. hirae) (Table 1). There was no unique pattern concerning the replicon type, sequence type, and source of isolation. However, replicon type rep9b/c was only found in E. faecalis isolates with rep24 and rep29 being unique to E. faecium isolates.
Some ARGs were associated with insertion sequences (IS6, ISL3, and IS982), and transposons (Tn3 and Tn6000) with most of those associated with MGE being plasmid-borne (Table 3). However, the majority of ARGs were located on chromosomes and not associated with any MGEs (Supplementary Table 2). An E. faecium isolate (D95) from the downstream site harboured an efflux pump encoding macrolide resistance gene msr(A) that was associated with insertion sequence IS982. The contig carrying the msr(A) gene and associated MGE had very high similarity (99–100%) to a target sequence E. faecium HB-1 chromosome (CP040878.1) in GenBank (Table 3). An E. faecalis isolate (D21) from the downstream site had a plasmid-encoded trimethoprim resistance gene dfrG whose genetic environment had ISL3. The contig was highly similar to a target sequence in GenBank E. faecalis strain 133170041-3 plasmid pAD1 (CP046109.1) confirming carriage of the gene on a plasmid. Another isolate (U84) from the upstream site had a plasmid that co-carried the tetracycline resistance [tet(M) and tet(L)] and macrolide resistance erm(B) genes. The genetic environment of the resistance genes consisted of a recombinase and the Tn3 transposons and the contig was closely related to E. faecalis S7316 plasmid Ps7316optrA (LC499744.1) (Table 3). The E. durans isolate (E115) from the effluent site had an antibiotic resistance genetic island consisting of genes encoding resistance to aminoglycosides [ant(6)-Ia], chloramphenicol (catB), macrolides [msr(D) and mef(A)], and trimethoprim (dfrG). The resistance island had MGEs including several recombinases and the insertion sequence IS6 (Table 3). The resistance island was located on a contig that closely resembled a target sequence in GenBank E. faecalis strain transconjugant T4 plasmid pJH-T4 (KY290886.1) implying that it was located on a plasmid. The genetic environment of the tetracycline resistance gene tet(S) was associated with the insertion sequence IS6. Interestingly the contig carrying this tet(S) gene was highly similar to an E. faecalis strain C386 transposon Tn6000 (JN208881.1) (Table 3). The tet(M) resistance gene was consistently found associated with the tetracycline resistance leader peptide (tetrLpep) and a conjugative transposon/transfer protein (TcpC), genetic context tet(M): tetrLpep: TcpC (IN127, IN133, E21, E115, D21, U129) and the reverse context TcpC:tetrLpep:tet(M) in D95, D98, U84). The TcpC conjugative TcpC is required for efficient conjugative transfer and mediates tetracycline resistance. Notably, the genetic context was found on contigs with high similarity (98–100%) to Enterococcus spp. chromosomal sequences deposited in GenBank except in E. faecalis isolate U84 where the genes were co-carried on a plasmid with other ARGs (Table 3).

Table 3. Mobile genetic elements associated with antibiotic resistance genes in Enterococcus spp.
A total of 32 IS families were detected in the genomes (Table 1). Ten IS families occurred more than once, with the ISEfa5 (5 isolates), ISEfa11 (5), and ISEfa10 (4) being predominant. ISEfa5 and ISEfa11 occurred in the same five isolates, covering all four species (Table 1). The IS did not follow source or sequence type, albeit two E. faecalis isolates (IN127, D21) belonging to ST179 had the same four ISs.
Intact prophages were found within 9/12 (75%) of the genomes. Three isolates comprising one E. faecalis and two E. hirae did not possess any intact prophages. A total of seven intact prophages were identified across all the investigated isolates, with Lister_LP_101 and Entero_EFC_1 being unique to individual isolates (Table 1). The Entero_phiFL1A was the most common prophage occurring in three different isolates from the upstream, influent, and effluent sites. The Entero_phiFL3A (n = 2) occurred in E. faecalis isolates from the upstream and influent site, Lister_B025 (n = 2) occurred in E. faecium isolates from the influent and effluent sites. The occurrence of intact prophages was not according to species, as several prophages occurred in different species including Entero_phiFL1A (E. faecalis and E. durans), Lister_B025 (E. faecium and E. hirae), and Bacill_BCJA1c (E. faecium and E. durans). E. faecalis and E. faecium isolates did not have any intact prophages in common. The intact prophages did not occur according to sequence type, although E. faecium isolates (D95, D98) belonging to ST179 had the same prophage Entero_vB_IME197.
Seven isolates had regions encoding the T4SS type ICE, with one E. faecium isolate (U129) from the upstream site and the effluent isolate E. durans (E115) having three regions each (Table 1). The IMEs were detected in five isolates (3 E. faecalis and 2 E. faecium). Two isolates (one E. faecalis and one E. faecium isolate) harboured both the ICE and IME. E. hirae isolates did not harbour any of the stated MGEs except for insertion sequences implying that these might be central in horizontal gene transfer, however, none of the ARGs in these isolates were associated with MGEs. The genome of environmental Enterococcus spp. consists of a rich diversity of MGEs including ISs, transposons, prophages, and plasmids that probably drive genetic exchange within and among these species.
Virulome of Enterococcus Isolates
A diversity of virulence genes was found in the E. faecium and E. faecalis isolates with none identified in E. hirae and E. durans (Table 4). For E. faecalis, a total of 20 different virulence genes were identified, including genes encoding sex pheromones, adhesion, cell invasion, aggregation, toxins, biofilm formation, cytolysin production, immunity, antiphagocytic activity, and proteases (Table 4). All the E. faecalis isolates had eleven of these genes (cCF10, cOB1, cad, camE, ace, SrtA, ebpA, ebpC, efaAfs, tpx, and gelE) in common. In E. faecium, only four virulence genes were identified and included adhesins (acm and efaAfm), a sex pheromone (cad), and an antiphagocytic factor (tpx) (Table 4).
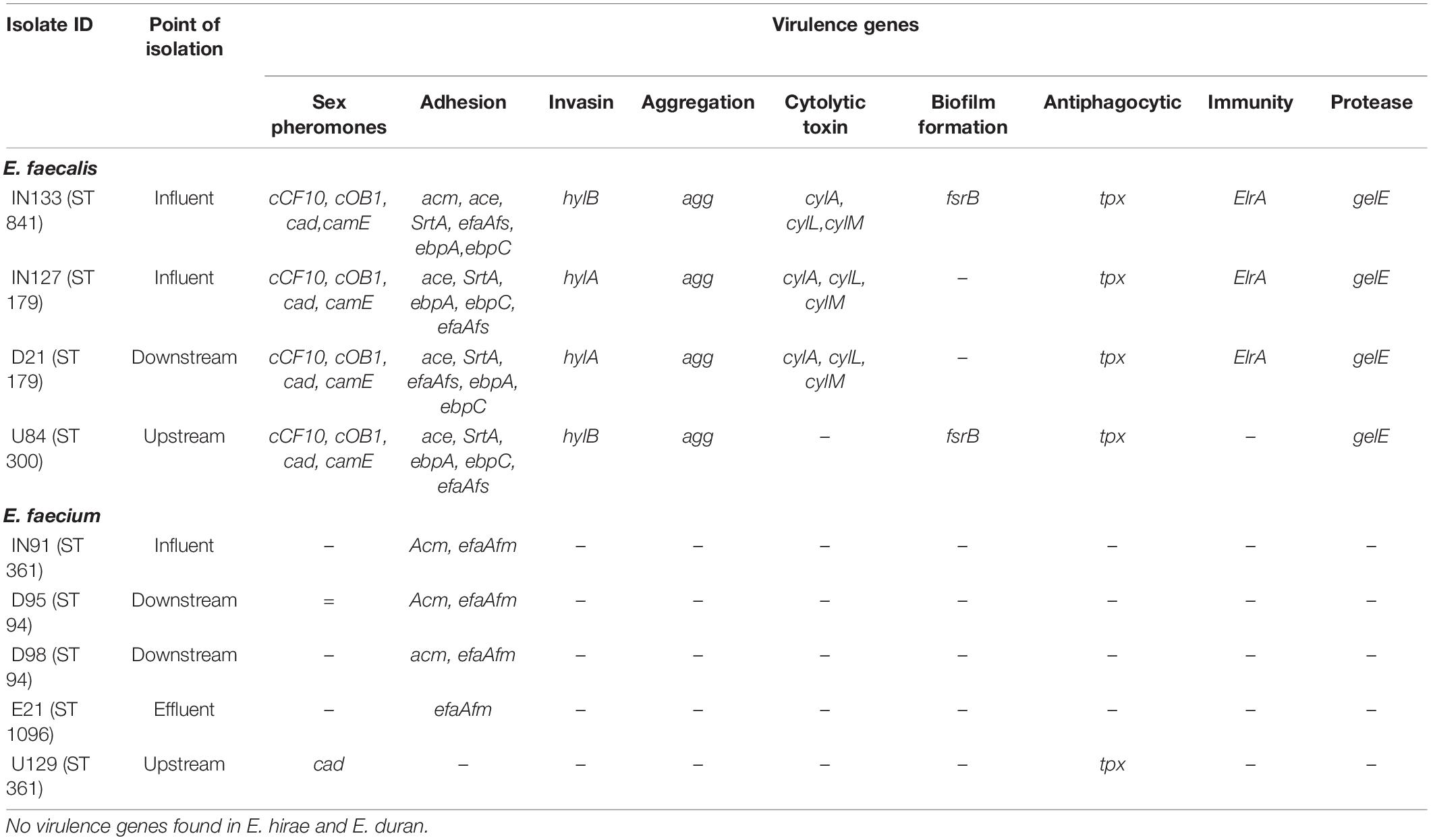
Table 4. Virulence gene profiles of environmental Enterococcus spp.
The virulence genes in E. faecium were mostly devoid of any association with MGEs. Among the E. faecalis isolates the gelE (protease) was co-carried with the fsrC (biofilm formation) virulence gene in a genetic environment that had an integrase and IS256. This occurred in two isolates from the influent (IN127) and downstream (D21) sites with genetic context gelE:fsrC::::integrase:::: IS256 (Table 5). The contigs bearing these virulence genes were highly similar (99 - 100%) to a chromosomal sequence in GenBank Enterococcus faecalis strain FDAARGOS_324 chromosome (CP028285.1) implying their carriage in the chromosome. Although, several virulence genes were found to occur together in other E. faecalis isolates their genetic environment did not contain any MGEs (Table 5). This implies that in addition to MGEs like ISs the transfer of virulence genes may be moderated by other processes that facilitate genetic exchange e.g., natural transformation.
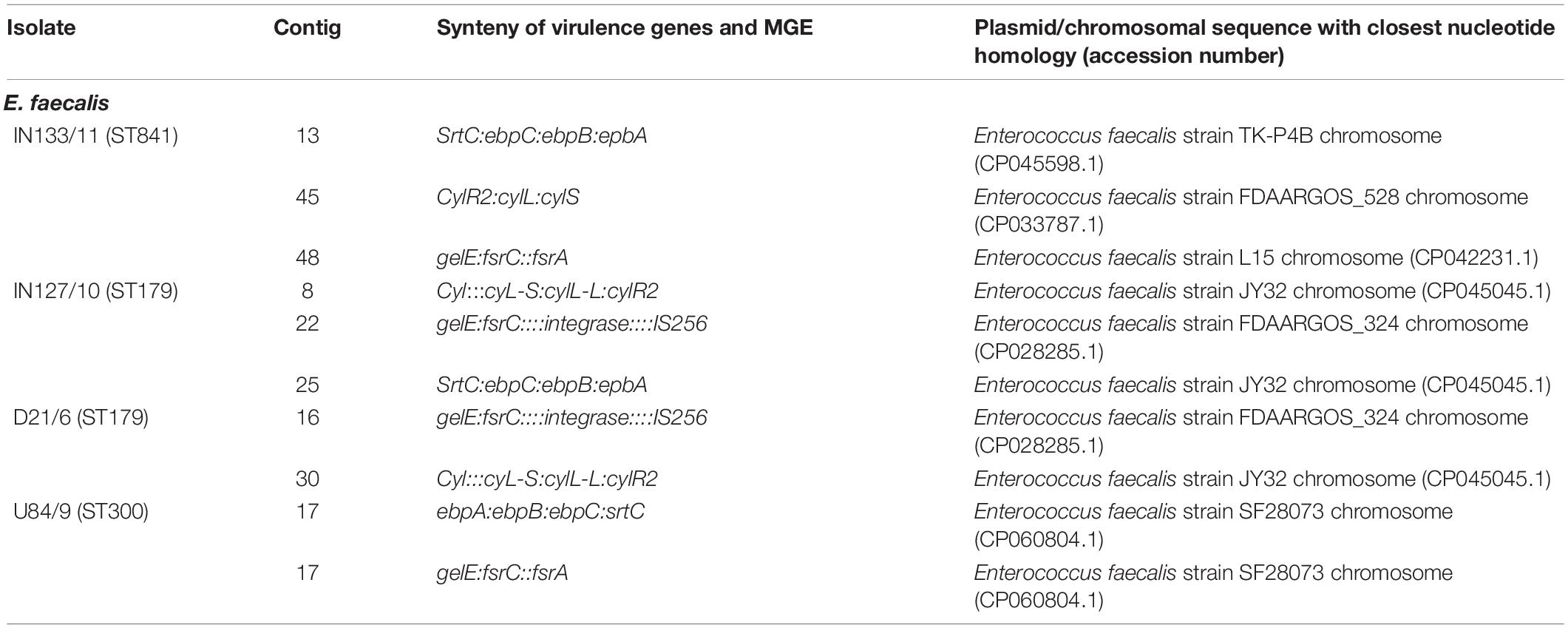
Table 5. Mobile genetic elements associated with virulence genes in E. faecalis isolates.
MLST and Phylogenomics
MLST analysis revealed that two E. faecalis isolates had a common sequence type ST179; the rest of the isolates had unique sequence types, ST841, and ST300 (Table 1). The E. faecium genomes belonged to three sequence types, ST94 (n = 2), ST361 (n = 2), and ST1096 (n = 1).
Phylogenetic analysis of the E. faecalis genomes from this study and those from other studies in Africa showed that the isolates were more closely related to animal and environmental isolates than to clinical isolates (Figure 1). An isolate obtained from the influent (IN133 and ST841) was more closely related to a Tunisian isolate (1351.1813, ST859) from chicken meat. An isolate (U84, ST300) from the upstream site was closely related to an isolate (1351.4175, ST271) from agricultural soil obtained in the same district of uMgungundlovu in KZN, South Africa. However, the other two isolates (D21, IN127), both ST179, clustered together and were in the same node as environmental isolates obtained from the soil and chicken litter of a sugarcane farm in KZN, South Africa (Figure 1).
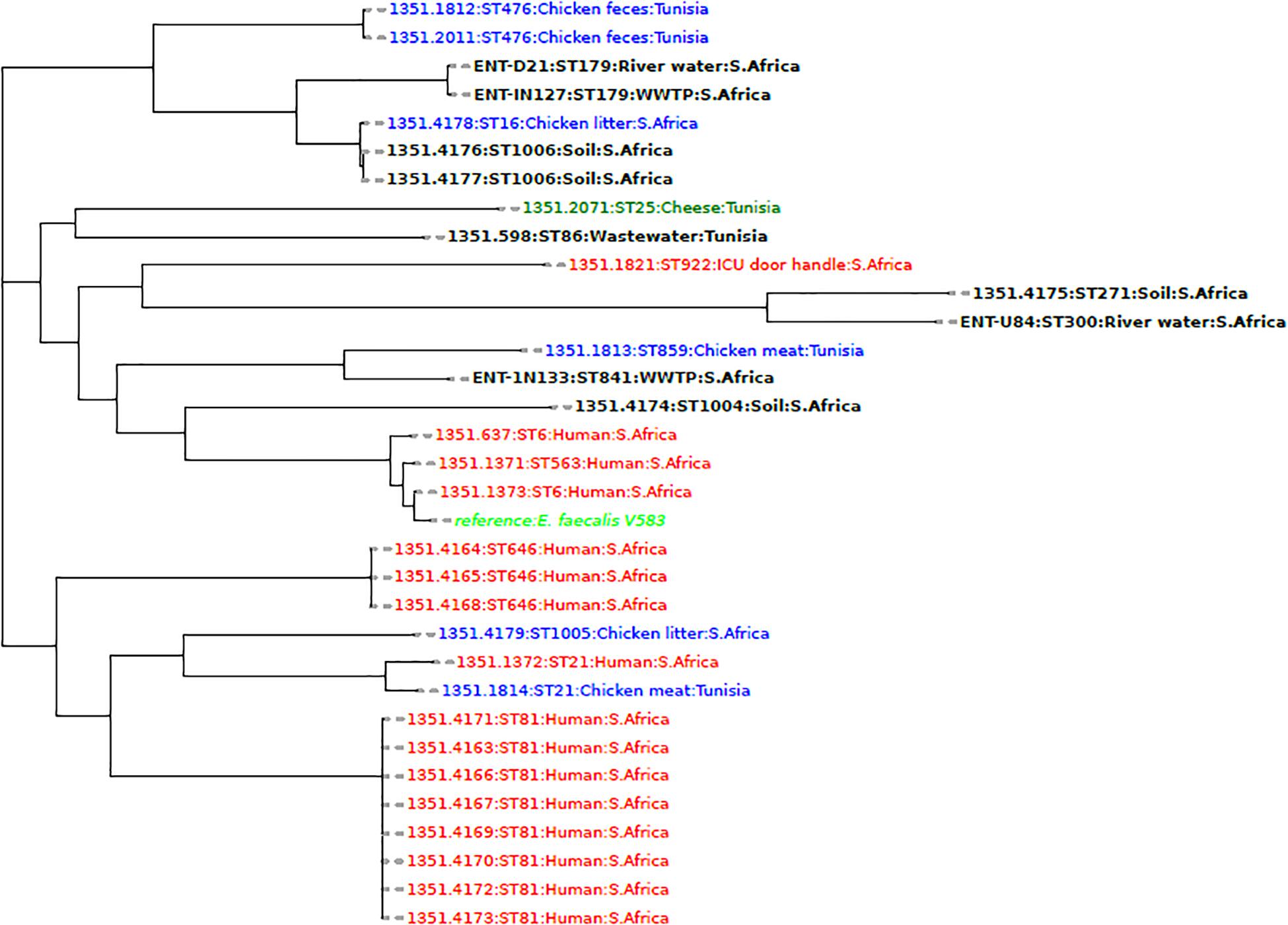
Figure 1. A phylogenomic tree showing the relationship of E. faecalis isolates from this study with African isolates from humans (red), animals (blue), and the environment (black) obtained from the PATRIC database (https://www.patricbrc.org/).
Comparison of E. faecium genomes with other WGS isolates from Africa revealed that the isolates from this study D95, D98 (ST94), and IN91, U129 (ST361) clustered together according to sequence type, but formed a separate clade with isolates obtained from the chicken litter at a sugarcane farm in KZN, South Africa. An isolate E21 (ST1096), was found in a different clade and clustered closely with a South African soil isolate from the same farm in KZN (Figure 2).
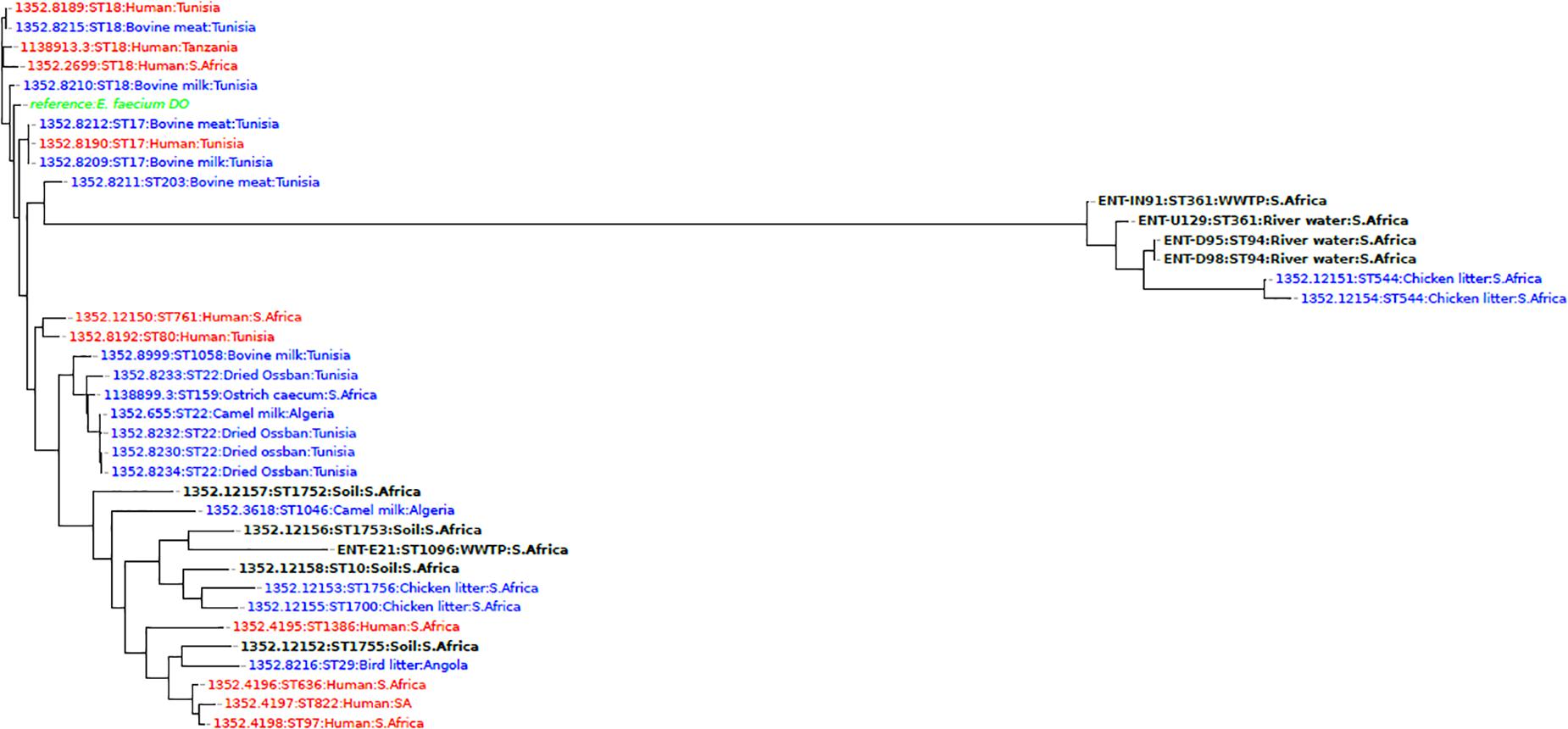
Figure 2. A phylogenomic tree showing the relationship of E. faecium isolates from this study with African isolates from humans (red), animals (blue), and the environment (black) obtained from the PATRIC database (https://www.patricbrc.org/).
Phylogenetic analysis of E. hirae isolates revealed that the E. hirae isolates were more closely related to livestock and environmental isolates. The upstream isolate U73 was closely related and clustered closely with isolates from Goa (Tibetan antelope) fecal matter obtained in China, suggesting that the isolate could be of animal origin. The other isolate (D76) was also closely related and clustered together with isolates from fermented vegetables from Malaysia, signifying that the isolate may be from an agricultural source (Figure 3). The E. durans isolate clustered closely with a bovine isolate (53345.56) obtained from South Africa and an isolate frim chicken (53345.33) from the United States implying that it is an animal-associated isolate (Figure 4).
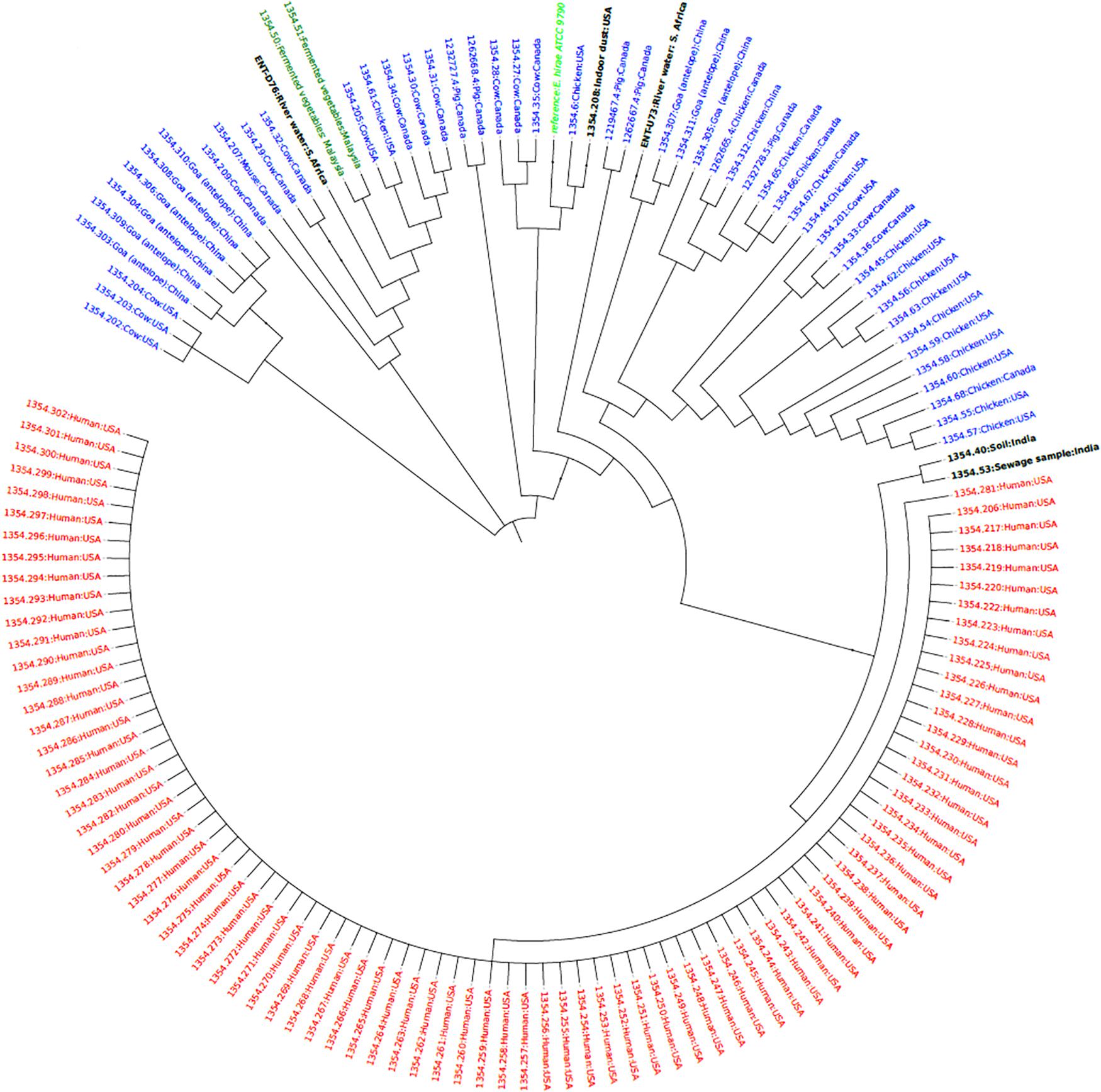
Figure 3. Phylogenetic tree based on SNP differences in the core genomes of E. hirae isolates from this study (ENT D76 and ENT U73) and other isolates from humans (red), animals (blue), environment (black) obtained from the PATRIC database (https://www.patricbrc.org/).
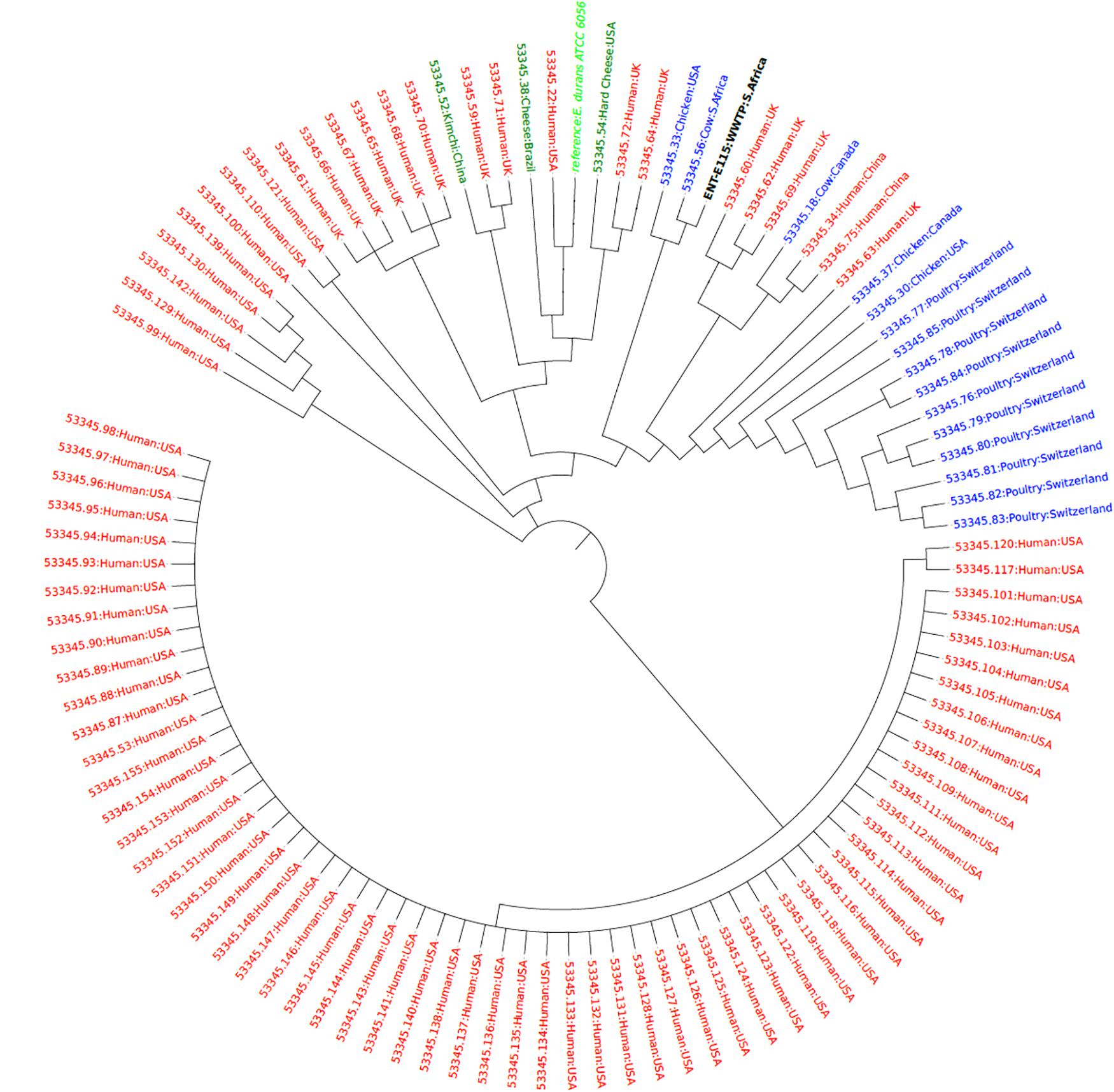
Figure 4. Phylogenetic tree showing the relationship of the E. durans isolate (black label) with other isolates obtained from humans (red), animals (blue), and food sources (green) from the PATRIC database (https://www.patricbrc.org/).
Discussion
Bioinformatics tools were used to analyze the whole genomes of MDR Enterococcus isolates (n = 12) with similar antibiograms obtained from a WWTP and the receiving water bodies at different sampled points and at different timelines. While many ARGs were carried on plasmids, transposable elements, and insertion sequences, most were, carried on chromosomes with no association with MGEs. A few virulence genes were associated with ISs, with most occurring on chromosomes. The abundance of MGEs observed in the Enterococci genomes, however, signifies their importance in gene rearrangements and horizontal gene transfer in these environmental isolates. This study is one of the first studies to explore the resistome, virulome, mobilome, clonality, and phylogenomics of Enterococcus spp. obtained from the water environment in Africa.
Tetracycline resistance genes identified in this study included the tet(M), tet(S), and tet(L). The tet(M) gene was present in the E. durans isolate and in all the E. faecium and E. faecalis isolates (Table 1). The tet(M) and tet(S) encode for ribosome protection proteins and the tet(L) encodes an efflux pump (Tao et al., 2010). There was a high concordance between the phenotypic AST and genotypic data with regards to TET resistance. The genetic context tet(M):tetrLpep:TcpC (or its reverse) was found in 9/10 (90%) of isolates that harboured the tet(M) gene (Table 3). The TcpC gene encodes a conjugative TcpC which is essential for efficient conjugative transfer and has previously been associated with conjugative tetracycline resistance plasmids in Clostridium perfringens (Bannam et al., 2006). Most of the tet(M) genes were located on chromosomes except for E. faecalis isolate U84 where the genetic context was associated with a plasmid. The genes involved in ribosome protection including tet(M) are typically found on both plasmids and self-conjugative transposons in chromosomes (Roberts, 1996) as evidenced in this study. Transfer of tet(M) in environmental Enterococci is possibly mediated by the conjugative TcpC.
Resistance to macrolides was associated mainly with the erm(B) and msr(C) genes. The erm(B) encodes a ribosomal methylase and is considered to be the most common gene responsible for resistance to erythromycin in enterococci; the methylase can also result in resistance to lincosamides, and streptogramin B (Miller et al., 2015). The rRNA methylases, erm(A), erm(B), and erm(C) modify specific nucleotides in the 23S rRNA and block macrolide binding (Chancey et al., 2015). Resistance to macrolides may also be caused by mutations in the 23S ribosomal RNA gene or be mediated by efflux pumps (Miller et al., 2015). All isolates that were phenotypically resistant to erythromycin had the erm(B) gene (Table 1). In Enterococcus spp. the erm(B) gene is considered the most widespread erythromycin resistance gene (Zaheer et al., 2020). The msr(C) gene which encodes an efflux pump was identified in all the E. faecium isolates which is consistent with earlier studies that stated that the gene seems to be specific for this species (Zaheer et al., 2020). The genome of the E. durans isolate had the efflux pump encoding genes msr(D) and mef(A) which were unique to this isolate. The macrolide efflux (mef) genes were initially identified in Streptococcus pyogenes (Sutcliffe et al., 1996) and S. pneumoniae (Gay and Stephens, 2001) and have been noted to always occur upstream and to be co-transcribed with an ATP-binding subunit ABC-transporter gene msr(D), functioning as a dual efflux pump (Ambrose et al., 2005). The genes were located on a resistance island consisting of MGEs (recombinase, IS6) together with chloramphenicol, aminoglycoside, and trimethoprim resistance genes (Table 3). There is a possibility that this resistance island is transmissible within and across sub-species, although its transferability was not experimentally investigated. Although E. durans strains rarely cause infection, the occurrence of these resistance genes implies the importance of these organisms as environmental reservoirs which could potentially mediate the transfer of these genes to pathogens of clinical or veterinary importance.
Enterococci are inherently resistant (low-level) to aminoglycosides, mostly due to the presence of the aac(6′)-Ii gene. Some isolates, however, exhibit high-level resistance to gentamicin and streptomycin and are clinically important (Sanderson et al., 2020). The presence of other acquired aminoglycoside resistance genes including aac(6′)-Ie–aph(2″)-Ia, aph(3′)-IIIa, and ant(6)-Ia confers high-level resistance to various aminoglycosides (Said et al., 2015). Except for E. durans isolate E115 none of the aminoglycoside resistance genes were associated with MGEs and most were borne on the chromosome (Table 3). Isolate E115 had the ant(6)-Ia gene which formed part of a resistance island that was on a plasmid. The isolate exhibited high-level resistance to streptomycin (Table 1).
A diversity of virulence genes was identified in the genomes of the sequenced E. faecalis and E. faecium isolates (Table 4). The gelE and fsr genes have been shown to occur together in E. faecalis isolates from healthy and sick animals (Šeputiene et al., 2012). The fsrABDC operon has been shown to regulate the expression of the gelE gene and other virulence genes (Hancock and Perego, 2004). The gelE encodes an extracellular zinc endopeptidase that cleaves a broad range of substrates including collagen and gelatin. It accentuates the pathogenesis of endocarditis caused by E. faecalis (Thurlow et al., 2010). The gelE and fsr genes occurred together in several E. faecalis isolates including IN133 and U84 that had genetic context gelE:fsrC::fsrA (Table 5). Isolates D21 and IN127 both had genetic context gelE:fsrC::::integrase:::: IS256 suggesting that IS256 plays a role in the transmission of these virulence genes. The IS256 is prevalent in the genomes of MDR enterococci and staphylococci where it occurs either independently or is associated with ARGs or virulence genes involved in biofilm formation (Hennig and Ziebuhr, 2010; Kim et al., 2019). Other virulence genes were not associated with MGEs suggesting that processes like natural transformation may be important in the transfer of these genes. Generally, the repertoire of virulence genes revealed in this study point to the presence of potentially pathogenic Enterococcus spp. in the investigated water environment.
MLST revealed distinct sequence types that are associated with clinical, animal, and agricultural sources. For E. faecalis isolates, ST179 was the most common sequence type (n = 2) a finding similar to other studies (Zaheer et al., 2020). For the E. faecium isolates, ST94 (n = 2) and ST361 (n = 2) were the most prevalent sequence types. In the study by Zaheer et al. (2020) the ST94 was the most abundant ST in the cattle feedlot catch basin and was found in other sources namely, surface water, urban wastewater, and from the clinic. This possibly points to the ubiquitous nature of this sequence type. The ST361 is not one of the notable E. faecium STs as it has not been implicated in clinical cases which are mostly attributed to the ST17, ST18 and, ST78 lineages (Palmer et al., 2014). A study from the United Kingdom used WGS to investigate the prevalence of vancomycin-resistant E. faecium in 20 WWTPs and reported an E. faecium ST361 from a WWTP (Gouliouris et al., 2018).
The phylogenomics of E. faecalis and E. faecium isolates revealed that all isolates were closely related to environmental or animal isolates, and not clinical isolates (Figures 1, 2). However, the E. faecalis influent isolate (IN133, ST841) harbored the cytolysin genes that have been attributed to clinical E. faecalis isolates intimating pathogenic potential (Zaheer et al., 2020). Phylogenetic analysis of the E. hirae isolates in this study revealed a close association with other animal and environmental isolates (Figure 3). E. hirae is known to inhabit a variety of animals and plants (Byappanahalli et al., 2012) and has been widely associated with cattle feces, chicken broilers, and associated production systems (Rehman et al., 2018; Zaheer et al., 2020). The E. durans isolate was closely related to animal isolates (Figure 4), indicating that it may be of animal origin. E. durans isolates are known to inhabit humans, animals, and insects and occasionally cause human infections (Byappanahalli et al., 2012). The isolate investigated in this study lacked virulence determinants and is most likely a potential reservoir of ARGs. Although a small subset of Enterococcus spp. isolates were used, this study adds to the limited knowledge of the resistome, virulome, mobilome, and phylogenies in environmental Enterococci in Africa. Future studies should look to use a larger sample size and greater diversity of Enterococcus spp. from diverse geographical locations.
Conclusion
This is the first report of genomic diversity of Enterococcus spp. found in wastewater and associated river water in KwaZulu-Natal, South Africa. Enterococcus spp. showed a rich repertoire of ARGs and virulence factors implying that the water environment is a substantive reservoir of MDR microbes which are potential pathogens. Genomic analysis of the Enterococci isolates allowed for the description of the resistome, virulome, and mobilome as well as the determination of phylogenetic relationships with animal, agricultural and environmental isolates. Such work allows a deeper understanding of the potential transmission dynamics related to the spread of antibiotic resistance in the water environment.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author Contributions
SE, JM, AA, and DA: co-conceptualized the study. JM: performed the experiments and wrote the manuscript. JM, AA, and DA: analyzed the data. SE, AA, and DA: supervision. SE: funding acquisition. All authors undertook critical revision of the manuscript and reviewed, edited, and approved the final manuscript.
Funding
This work was based on the research supported by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation of South Africa (Grant No. 98342), the National Research Foundation Competitive Grant for Rated Researchers (Grant No. 106063), jointly funded by the South African Medical Research Council (SAMRC), and UK Medical Research Council Newton Fund and the SAMRC Self-Initiated Research Grant. The funding source was not involved in study design, sample collection, data analysis, and interpretation.
Disclaimer
Any opinion, finding, and conclusion or recommendation expressed in this material is that of the authors, and neither the NRF nor the other funding bodies accept any liability in this regard.
Conflict of Interest
SE is chairperson of the Global Respiratory Infection Partnership and a member of the Global Hygiene Council, funded by unrestricted educational grants from Reckitt and Benckiser (Pty.), United Kingdom.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank all members of the Antimicrobial Research Unit (ARU) at UKZN for their assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.648454/full#supplementary-material
Supplementary Table 1 | Genome and assembly characteristics of sequenced Enterococcus spp. isolates from a wastewater treatment plant and its associated waters.
Supplementary Table 2 | Antibiotic resistance genes not associated with any mobile genetic elements in enterococci.
Footnotes
- ^ https://github.com/najoshi/sickle
- ^ http://cge.cbs.dtu.dk/services/MLST/
- ^ https://cge.cbs.dtu.dk/services/ResFinder/
- ^ https://cge.cbs.dtu.dk/services/PlasmidFinder/
- ^ https://cge.cbs.dtu.dk/services/VirulenceFinder/
- ^ https://isfinder.biotoul.fr/
- ^ https://phaster.ca/
- ^ http://db-mml.sjtu.edu.cn/ICEberg/
- ^ http://rast.nmpdr.org/seedviewer.cgi
- ^ https://www.geneious.com
- ^ https://www.patricbrc.org/
- ^ https://cge.cbs.dtu.dk/services/CSIPhylogeny/
- ^ https://itol.embl.de/
References
Alexander, J., Hembach, N., and Schwartz, T. (2020). Evaluation of antibiotic resistance dissemination by wastewater treatment plant effluents with different catchment areas in Germany. Sci. Rep. 10, 1–9. doi: 10.1038/s41598-020-65635-4
Ambrose, K. D., Nisbet, R., and Stephens, D. S. (2005). Macrolide efflux in Streptococcus pneumoniae is mediated by a dual efflux pump (mel and mef) and is erythromycin inducible. Antimicrob. Agents Chemother. 49, 4203–4209. doi: 10.1128/AAC.49.10.4203-4209.2005
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bannam, T. L., Teng, W. L., Bulach, D., Lyras, D., and Rood, J. I. (2006). Functional identification of conjugation and replication regions of the tetracycline resistance plasmid pCW3 from Clostridium perfringens. J. Bacteriol. 188, 4942–4951. doi: 10.1128/JB.00298-06
Bengtsson-Palme, J., Kristiansson, E., and Larsson, D. G. J. (2018). Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol. Rev. 42, 68–80. doi: 10.1093/femsre/fux053
Berendonk, T. U., Manaia, C. M., Merlin, C., Kassinos, D. F., Cytryn, E., Walsh, F., et al. (2013). Tackling antibiotic resistance: the environmental framework. Nat. Rev. Microbiol. 13, 310–317. doi: 10.1038/nrmicro3439
Bourafa, N., Loucif, L., Boutefnouchet, N., and Rolain, J. M. (2015). Enterococcus hirae, an unusual pathogen in humans causing urinary tract infection in a patient with benign prostatic hyperplasia: First case report in Algeria. N. Microbes N. Infect. 8, 7–9. doi: 10.1016/j.nmni.2015.08.003
Byappanahalli, M. N., Nevers, M. B., Korajkic, A., Staley, Z. R., and Harwood, V. J. (2012). Enterococci in the Environment. Microbiol. Mol. Biol. Rev. 76, 685–706. doi: 10.1128/mmbr.00023-12
Chancey, S. T., Bai, X., Kumar, N., Drabek, E. F., Daugherty, S. C., Colon, T., et al. (2015). Transcriptional attenuation controls macrolide inducible efflux and resistance in Streptococcus pneumoniae and in other gram-positive bacteria containing mef/mel (msr(D)) elements. PLoS One 10:1–23. doi: 10.1371/journal.pone.0116254
CLSI (2020). CLSI M100-ED30:2020 Performance Standards for Antimicrobial Susceptibility Testing, 30th Edition. Available online at: http://em100.edaptivedocs.net/GetDoc.aspx?doc=CLSI M100 ED30:2020 doi: 10.1016/j.drup.2016.04.003 (accessed May 11, 2020).
Conte, D., Kasuko, J., Nogueira, S., Marenda, T., Lima, R., De, et al. (2017). Characterization of CTX-M enzymes, quinolone resistance determinants, and antimicrobial residues from hospital sewage, wastewater treatment plant, and river water. Ecotoxicol. Environ. Saf. 136, 62–69. doi: 10.1016/j.ecoenv.2016.10.031
EUCAST (2020). Testing Breakpoint tables for interpretation of MICs and zone diameters. Available online at: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_10.0_Breakpoint_Tables.pdf (accessed May 11, 2020).
Gay, K., and Stephens, D. S. (2001). Structure and dissemination of a chromosomal insertion element encoding macrolide efflux in Streptococcus pneumoniae. J. Infect. Dis. 184, 56–65. doi: 10.1086/321001
Gouliouris, T., Raven, K. E., Ludden, C., Blane, B., Corander, J., Horner, C. S., et al. (2018). Genomic surveillance of enterococcus faecium reveals limited sharing of strains and resistance genes between livestock and humans in the United Kingdom. MBio 9, 1–15. doi: 10.1128/mBio.01780-18
Hancock, L. E., and Perego, M. (2004). The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J. Bacteriol. 186, 5629–5639. doi: 10.1128/JB.186.17.5629-5639.2004
Hendriksen, R. S., Bortolaia, V., Tate, H., Tyson, G. H., Aarestrup, F. M., and McDermott, P. F. (2019). Using Genomics to Track Global Antimicrobial Resistance. Front. Public Heal. 7:00242. doi: 10.3389/fpubh.2019.00242
Hennig, S., and Ziebuhr, W. (2010). Characterization of the transposase encoded by IS256, the prototype of a major family of bacterial insertion sequence elements. J. Bacteriol. 192, 4153–4163. doi: 10.1128/JB.00226-10
Huijbers, P. M. C., Flach, C. F., and Larsson, D. G. J. (2019). A conceptual framework for the environmental surveillance of antibiotics and antibiotic resistance. Environ. Int. 130:104880. doi: 10.1016/j.envint.2019.05.074
Kaas, R. S., Leekitcharoenphon, P., Aarestrup, F. M., and Lund, O. (2014). Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9, 1–8. doi: 10.1371/journal.pone.0104984
Karkman, A., Do, T. T., Walsh, F., and Virta, M. P. (2018). Antibiotic-resistance genes in waste water. Trends Microbiol. 26, 220–228. doi: 10.1016/j.tim.2017.09.005
Karkman, A., Do, T. T., Walsh, F., and Virta, M. P. J. (2017). Antibiotic-resistance genes in wastewater. Trends Microbiol. 26, 220–228. doi: 10.1016/j.tim.2017.09.005
Ke, D., Picard, O. I. S. J., Martineau, F., Me, C., Roy, P. H., Ouellette, M., et al. (1999). Development of a PCR Assay for Rapid Detection of Enterococci. J. Clin. Microbiol. 37, 3497–3503. doi: 10.1128/jcm.37.11.3497-3503.1999
Kim, Y. Bin, Seo, K. W., Shim, J. B., Son, S., Hyun, N. E. B., and Lee, Y. J. (2019). Molecular characterization of antimicrobial-resistant Enterococcus faecalis and Enterococcus faecium isolated from layer parent stock. Poult. Sci. 98, 5892–5899. doi: 10.3382/ps/pez288
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, L., et al. (2012). Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 1355–1361. doi: 10.1128/JCM.06094-11
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Medeiros, A. W., Pereira, R. I., Oliveira, D. V., Martins, P. D., d’Azevedo, P. A., Van der Sand, S., et al. (2014). Molecular detection of virulence factors among food and clinical Enterococcus faecalis strains in South Brazil. Brazilian J. Microbiol. 45, 327–332. doi: 10.1590/S1517-83822014005000031
Miller, W. R., Munita, J. M., Arias, C. A., Santiago, A., De, and Unit, A. R. (2015). Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti Infect. Ther. 12, 1221–1236. doi: 10.1586/14787210.2014.956092
Moodley, B., Birungi, G., and Ndungu, P. (2016). Detection and Quantification of Emerging Organic Pollutants in the Umgeni and Msunduzi Rivers. WRC Report No.2215/1/16, South Africa: Commission Pretoria.
Palmer, K. L., van Schaik, W., Willems, R. J., and Gilmore, M. S. (2014). “Enterococcal genomics,” in Enterococci: From Commensals to Leading Causes of Drug Resistant Infection [Internet].
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS One 5:9490. doi: 10.1371/journal.pone.0009490
Rehman, M. A., Yin, X., Zaheer, R., Goji, N., Amoako, K. K., McAllister, T., et al. (2018). Genotypes and Phenotypes of Enterococci Isolated From Broiler Chickens. Front. Sustain. Food Syst. 2:83. doi: 10.3389/fsufs.2018.00083
Roberts, M. C. (1996). Tetracycline resistance determinants: Mechanisms of action, regulation of expression, genetic mobility, and distribution. FEMS Microbiol. Rev. 19, 1–24. doi: 10.1016/0168-6445(96)00021-6
Ryu, B. H., Hong, J., Jung, J., Kim, M. J., Sung, H., Kim, M. N., et al. (2019). Clinical characteristics and treatment outcomes of Enterococcus durans bacteremia: a 20-year experience in a tertiary care hospital. Eur. J. Clin. Microbiol. Infect. Dis. 38, 1743–1751. doi: 10.1007/s10096-019-03605-z
Said, B. L., Klibi, N., Lozano, C., Dziri, R., Slama, B. K., Boudabous, A., et al. (2015). Diversity of enterococcal species and characterization of high-level aminoglycoside resistant enterococci of samples of wastewater and surface water in Tunisia. Sci. Total Environ. 53, 11–17. doi: 10.1016/j.scitotenv.2015.05.091
Sanderson, H., Ortega-Polo, R., Zaheer, R., Goji, N., Amoako, K. K., Brown, R. S., et al. (2020). Comparative genomics of multidrug-resistant Enterococcus spp. isolated from wastewater treatment plants. BMC Microbiol. 20:1–17. doi: 10.1186/s12866-019-1683-4
Šeputiene, V., Bogdaite, A., Ružauskas, M., and Sužiedeliene, E. (2012). Antibiotic resistance genes and virulence factors in Enterococcus faecium and Enterococcus faecalis from diseased farm animals: Pigs, cattle and poultry. Pol. J. Vet. Sci. 15, 431–438. doi: 10.2478/v10181-012-0067-6
Siguier, P. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Su, M., Satola, S. W., and Read, T. D. (2019). Genome-based prediction of bacterial antibiotic resistance. J. Clin. Microbiol. 57, 1–15. doi: 10.1128/JCM.01405-18
Sutcliffe, J., Tait-Kamradt, A., and Wondrack, L. (1996). Streptococcus pneumoniae and Streptococcus pyogenes resistant to macrolides but sensitive to clindamycin: A common resistance pattern mediated by an efflux system. Antimicrob. Agents Chemother. 40, 1817–1824. doi: 10.1128/aac.40.8.1817
Tao, R., Ying, G., Su, H., Zhou, H., and Sidhu, J. P. S. (2010). Detection of antibiotic resistance and tetracycline resistance genes in Enterobacteriaceae isolated from the Pearl rivers in South China. Environ. Pollut. 158, 2101–2109. doi: 10.1016/j.envpol.2010.03.004
Thurlow, L. R., Thomas, V. C., Narayanan, S., Olson, S., Fleming, S. D., and Hancock, L. E. (2010). Gelatinase contributes to the pathogenesis of endocarditis caused by Enterococcus faecalis. Infect. Immun. 78, 4936–4943. doi: 10.1128/IAI.01118-09
Zaheer, R., Cook, S. R., Barbieri, R., Goji, N., Cameron, A., Petkau, A., et al. (2020). Surveillance of Enterococcus spp. reveals distinct species and antimicrobial resistance diversity across a One-Health continuum. Sci. Rep. 10, 1–16. doi: 10.1038/s41598-020-61002-5
Keywords: Enterococcus spp., whole-genome sequencing, wastewater treatment plant, antibiotic resistance, South Africa
Citation: Mbanga J, Amoako DG, Abia ALK, Allam M, Ismail A and Essack SY (2021) Genomic Analysis of Enterococcus spp. Isolated From a Wastewater Treatment Plant and Its Associated Waters in Umgungundlovu District, South Africa. Front. Microbiol. 12:648454. doi: 10.3389/fmicb.2021.648454
Received: 31 December 2020; Accepted: 19 May 2021;
Published: 14 June 2021.
Edited by:
Jorge Andres Olivares Pacheco, Pontificia Universidad Católica de Valparaíso, ChileReviewed by:
Ana P. Tedim, Institute of Health Sciences Studies of Castilla y León (IECSCYL), SpainAna Paula Guedes Frazzon, Federal University of Rio Grande do Sul, Brazil
Copyright © 2021 Mbanga, Amoako, Abia, Allam, Ismail and Essack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joshua Mbanga, am9zaG1iYW5nYUBnbWFpbC5jb20=; am9zaHVhLm1iYW5nYUBudXN0LmFjLnp3