Abstract
Plants live in association with microorganisms that positively influence plant development, vigor, and fitness in response to pathogens and abiotic stressors. The bulk of the plant microbiome is concentrated belowground at the plant root-soil interface. Plant roots secrete carbon-rich rhizodeposits containing primary and secondary low molecular weight metabolites, lysates, and mucilages. These exudates provide nutrients for soil microorganisms and modulate their affinity to host plants, but molecular details of this process are largely unresolved. We addressed this gap by focusing on the molecular dialog between eight well-characterized beneficial strains of the Pseudomonas fluorescens group and Brachypodium distachyon, a model for economically important food, feed, forage, and biomass crops of the grass family. We collected and analyzed root exudates of B. distachyon and demonstrated the presence of multiple carbohydrates, amino acids, organic acids, and phenolic compounds. The subsequent screening of bacteria by Biolog Phenotype MicroArrays revealed that many of these metabolites provide carbon and energy for the Pseudomonas strains. RNA-seq profiling of bacterial cultures amended with root exudates revealed changes in the expression of genes encoding numerous catabolic and anabolic enzymes, transporters, transcriptional regulators, stress response, and conserved hypothetical proteins. Almost half of the differentially expressed genes mapped to the variable part of the strains’ pangenome, reflecting the importance of the variable gene content in the adaptation of P. fluorescens to the rhizosphere lifestyle. Our results collectively reveal the diversity of cellular pathways and physiological responses underlying the establishment of mutualistic interactions between these beneficial rhizobacteria and their plant hosts.
Introduction
Plants are meta-organisms or holobionts that rely in part on their microbiome for specific functions and traits. The ability of the plant microbiome to influence plant development, vigor, health, and fitness in response to abiotic stressors associated with global climate change is documented by numerous studies (Lugtenberg and Kamilova, 2009). There is mounting evidence that plants actively recruit beneficial microbiomes, but many aspects of this process are still very much a black box (Reinhold-Hurek et al., 2015). The foundation for the differential affinity of rhizobacteria toward host plants is built upon complex chemical cross talk between microorganisms and plant roots. Up to 40% of photosynthetically fixed carbon is released by plant roots in the form of exudates and secretions, lysates, and mucilages (Curl and Truelove, 1986; Lynch, 1990; Whipps, 1990; Badri and Vivanco, 2009). The release of these compounds is actively controlled in response to environmental stimuli, and the composition of root exudates varies greatly according to plant species and physiological condition (Lynch, 1990; Nguyen, 2003; Phillips et al., 2004; De-la-Pena et al., 2008). The presence and composition of exudates strongly impact soil microorganisms, which is consistent with the idea that plants actively select and shape their root microbiota (Zolla et al., 2013).
Primary root exudates include simple and complex sugars, amino acids, polypeptides and proteins, organic, aliphatic and fatty acids, sterols, and phenolics (Nguyen, 2003; Badri and Vivanco, 2009; Badri et al., 2009). These compounds serve as carbon and energy sources for rhizobacteria, and the presence of the intact corresponding catabolic pathways is essential for competitive colonization of roots and disease suppression (Lugtenberg et al., 2001; Kamilova et al., 2005; Lugtenberg and Kamilova, 2009). Root exudates also contain numerous signal molecules and secondary metabolites, the significance of which is only now emerging (Walker et al., 2003; Bais et al., 2005, 2006). A handful of analyses of plant-induced gene expression by transcriptional profiling in vitro (Mark et al., 2005) or in the rhizosphere (Silby and Levy, 2004; Ramos-Gonzalez et al., 2005; Matilla et al., 2007; Barret et al., 2009) have identified multiple genes that are differentially regulated by exposure to roots or root exudates. Bacterial pathways expressed during rhizosphere colonization control utilization of plant-derived metabolites (Simons et al., 1996, 1997; Camacho-Carvajal, 2001; Lugtenberg and Kamilova, 2009), motility and chemotaxis (de Weert et al., 2002; Lugtenberg and Kamilova, 2009), phase variation (Dekkers et al., 1998; Sanchez-Contreras et al., 2002; van den Broek et al., 2005), outer membrane integrity (de Weert et al., 2006; Lugtenberg and Kamilova, 2009), and the ability to sequester limiting resources (Raaijmakers et al., 1995) and resist environmental stresses (Sarniguet et al., 1995; Miller and Wood, 1996; van Veen et al., 1997; Schnider-Keel et al., 2001). In its spatial and temporal properties, root colonization resembles biofilm formation, and biofilm-related pathways also have been implicated in adhesion to seeds and roots and rhizosphere colonization (Espinosa-Urgel et al., 2000; Hinsa et al., 2003; Yousef-Coronado et al., 2008; Fuqua, 2010; Martinez-Gil et al., 2010; Nielsen et al., 2011; Zboralski and Filion, 2020). Finally, root exudates strongly affect the expression of diverse plant growth promotion and biocontrol genes (Vacheron et al., 2013). Over the past decade, the genomes of numerous rhizosphere strains have been sequenced and analyzed, but functional genomics studies of rhizosphere competence lag behind the availability of sequence data.
This study explored the molecular dialog between the model host plant Brachypodium distachyon and several well-characterized rhizosphere strains of the Pseudomonas fluorescens group. Brachypodium is a small annual grass originating in semi-arid regions of the Middle East that has emerged as a prime model for economically important food, feed, forage, and biomass crops of the grass family (Bevan et al., 2010; Schwartz et al., 2010; Brkljacic et al., 2011; Hong et al., 2011; Tyler et al., 2014). The biology, extensive collection of resources, and research tools make B. distachyon an attractive model to investigate interactions between plants and root-associated microbes. Pseudomonads are ubiquitous Gram-negative γ-proteobacteria that colonize eukaryotic hosts and include both commensals and economically important pathogens of plants and animals (Moore et al., 2006; Schroth et al., 2006; Yahr and Parsek, 2006). The genus Pseudomonas currently comprises > 100 named species that have been separated based on multilocus sequence analysis into 14 species groups (Garrido-Sanz et al., 2016; Hesse et al., 2018). The P. fluorescens group is the most diverse regarding both the genetic distances within it, the number of species and the large pangenome that makes up > 50% of the pangenome of the genus as a whole (Loper et al., 2012). The group also encompasses an unusually high proportion of strains that inhabit the plant rhizosphere and possess plant growth promoting and biocontrol properties. Naylor et al. (2017) profiled bacterial communities associated with root tissues and rhizosphere of 18 different plant species of the Poaceae family. That study identified Pseudomonas among taxa constituting the core grass root microbiome and demonstrated that these bacteria were enriched in C3 plants, including wheat, rye, barley, oat, and Brachypodium. We confirmed the capacity of B. distachyon Bd21 to serve as a host for rhizobacteria of the P. fluorescens group in preliminary greenhouse assays with biocontrol strains P. synxantha 2-79, P. brassicacearum Q8r1-96, and P. protegens Pf-5. Results of these experiments revealed that all strains successfully established and colonized the roots of Brachypodium (Supplementary Table 1).
In this study, we focused on eight well-studied strains of the P. fluorescens complex that are supported by years of studies, numerous refereed publications, and high-quality genome sequences. By profiling transcriptomes of these strains during growth in root exudates of B. distachyon, we revealed the diversity of cellular pathways and physiological responses that underlie the establishment of mutualistic interactions between beneficial rhizobacteria and the host plant. Our results also confirmed that root exudates contain carbohydrates, amino acids, organic acids, and phenolics that serve as carbon and energy sources for rhizobacteria. The root exudates also contained osmoprotectants that may help microorganisms to persist in the rhizosphere of drought-stressed plants. The diversity of microbial genes perturbed by root exudates reflects the importance of the variable genome in adaptation of individual strains of Pseudomonas to the rhizosphere lifestyle.
Materials and Methods
Bacterial Strains Used in the Study
The eight Pseudomonas strains used for this study are P. synxantha 2-79 (Thomashow and Weller, 1988), P. fluorescens SBW25 (Silby et al., 2009), Pseudomonas sp. R1-43-08 (Parejko et al., 2012), P. brassicacearum Q8r1-96 (Raaijmakers and Weller, 1998), P. fluorescens Q2-87 (Bangera and Thomashow, 1996), P. chlororaphis 30-84 (Thomashow et al., 1990), P. fluorescens Pf0-1 (Silby et al., 2009), and P. protegens Pf-5 (Howell and Stipanovic, 1980). The selected organisms have been studied extensively for their role in biological control and plant growth promotion (Supplementary Table 2). The strains were maintained in the laboratory as frozen stocks (−80°C) and routinely cultured in King’s medium B (King et al., 1954) or 21C medium, which contained (per 1 L): 1.0 g of NH4Cl, 3.5 g of Na2HPO4⋅2H2O, 2.8 g of KH2PO4, 3.0 g of glucose, and 20 ml of a microelement solution (Smibert and Kreig, 1994; Halverson and Firestone, 2000).
Propagation of Plants and Collection of Root Exudates
B. distachyon Bd21 was established from seed material obtained from the USDA-ARS Plant Germplasm Introduction and Testing Research Unit (Pullman, WA, United States). Brachypodium seeds were imbibed for 3 days at 4°C and sown in 7 × 7 cm pots filled with Sunshine Potting Mix #4 (Sun Gro Horticulture, Agawam, MA, United States). Plants were grown in an IR-89X (Percival Scientific, Perry, IA, United States) controlled environment chamber retrofitted with 6500K and 3000K T5 54W grow lights (Spectralux) under a 20-h light, 24°C/4-h dark, 18°C cycle. Plants were watered and fertilized with Jack’s professional water-soluble fertilizer (20:20:20) (JR Peters, Allentown, PA, United States). After 12 weeks and plant senescence, seeds were collected, processed, and stored under desiccant and dark conditions at room temperature.
To collect root exudates, seeds of B. distachyon Bd21 were surface-sterilized, pregerminated, and placed in sterile 1 L wide-mouth glass jars containing 113 g of 6-mm glass beads and 25 ml distilled water. Jars were covered with vented caps and plants were grown hydroponically in an environmental controlled growth chamber under conditions described above. After 6 days, root exudates were extracted from individual jars and their sterility was confirmed by spotting on nutrient agar. Multiple batches of root exudates were collected, filtered (0.22 μm), aliquoted in Falcon tubes (10 ml), lyophilized, and stored at –80°C.
Metabolomic Profiling of Root Exudates
Exudates were analyzed for primary metabolites at the Murdock Metabolomics Laboratory at Washington State University (Pullman, WA, United States). Freeze-dried residues were suspended in 500 μl 50% aqueous acetonitrile and clarified by centrifugation for 20 min at 21,000 × g and 4°C. The liquid chromatography mass spectrometry analysis was conducted with a Synapt G2-S quadrupole-ion mobility spectrometry-time of flight mass spectrometer system equipped with an acquity ultra-performance liquid chromatograph (UPLC) and an acquity photodiode array detector (all from Waters, Milford, MA, United States). The exudate metabolites were separated on a SeQuant ZIC-pHILIC HPLC column (2.1 × 100 mm, 3 μm) (Milllipore Sigma, Burlington, MA, United States) using acetonitrile with 0.1% formic acid as solvent B and water with 0.1% formic acid as solvent A at a flow rate of 400 μl min–1 and the following linear gradient extending over 14 min: 0 min, 80% B; 4 min, 80% B, 6 min: 10% B; 7.5 min, 10% B; 10 min, 80% B; and 14 min, 80% B. Mass spectra were collected in positive ion mode over a range of m/z 50–1,200 with a scan time of 0.2 s. The Q-TOF-MS source was at 3.0 kV and 120°C; the sampling cone at 40 V, desolvation temperature was 250°C; cone gas and desolvation gas flow were at 0 and 850 L h–1, respectively. Leucine enkephalin was used for post-acquisition mass correction. Target compounds were visualized using selected ion chromatograms at 0.05 Da window width. The compound identification was based on comparison of chromatographic behavior and accurate masses to those of authentic standards.
For gas chromatography, derivatization was carried out using a modification of the procedure of Lee and Fiehn (2008). The freeze-dried residues were suspended in 950 μl aqueous methanol (84%, v/v) and clarified by centrifugation for 15 min at 21,000 × g at 4°C. The supernatants were spiked with 1 μg of the internal standard salicylic acid-d6 (C/D/N Isotopes, Quebec, Canada) and dried in vacuo. The dry residues were suspended in 10 μl of O-methoxylamine hydrochloride (30 mg ml–1 in anhydrous pyridine, both from Millipore Sigma) and incubated while mixing (1,000 RPM) for 90 min at 30°C. Subsequently, samples were derivatized with 90 μl of MSTFA with 1% TMCS (Thermo Fisher Scientific, Waltham, MA, United States) for 30 min at 37°C. Gas chromatography-mass spectroscopy analysis was performed using a Pegasus 4D time-of-flight mass spectrometer (LECO, Saint Joseph MI) equipped with a MPS2 autosampler (Gerstel, Linthicum, MD) and a 7890A oven (Agilent Technologies, Santa Clara, CA, United States). The derivatization products were separated on a 30-m, 0.25 mm i.d., 0.25 μm df Rxi-5Sil column (Restek, Bellefonte, PA, United States) with an IntegraGuard precolumn using ultrapure He at a constant flow of 0.9 ml min–1 as carrier gas. The linear thermal gradient started with a 1-min hold at 70°C, followed by a ramp to 300°C at 10°C min–1. The final temperature was held for 5 min prior to returning to initial conditions. Mass spectra were collected at 17 spectra s–1. Peak identification was conducted using the Fiehn primary metabolite library (Kind et al., 2009) and an identity score cutoff of 700. Additionally, authentic standards for a number of primary metabolites were analyzed under identical conditions and the data used to compare the chromatographic behavior. Peak alignment and spectrum comparisons were carried out using the Statistical Compare feature of ChromaTOF software (LECO).
Isolation of RNA From Bacteria Cultured in Root Exudates and RNA-Seq
The strains were pregrown overnight at 25°C on 21C-glucose agar and then subcultured into 96-well microplates containing liquid 21C-glucose medium amended with Brachypodium exudates. The liquid medium was prepared by dissolving the lyophilized root exudate material in an appropriate volume of 21C-glucose medium to concentrate root exudates 20-fold. The growth medium was sterilized by passing it through a 0.22-μm membrane filter. The control cultures were grown under identical conditions in the absence of exudates. All treatments were inoculated at OD600 of 0.1 and incubated for 20 to 22 h until cultures entered late-exponential growth phase at 25°C in an atmosphere of 15% oxygen [created by a ProOx P110 oxygen controller (BioSpherix, Parish, NY, United States) with a hypoxia C-chamber]. The cells were stabilized by the addition RNAprotect reagent (QIAGEN, Germantown, MD, United States) and total RNA was purified using a RNeasy Protect Bacteria Mini Kit (QIAGEN) from three biological replicates of each strain cultured under control conditions and in exudates. The quality assessment of the extracted RNA samples was performed with a NanoDrop OneC Spectrophotometer (Thermo Fisher Scientific) and a 2100 Bioanalyzer (Agilent Technologies) and revealed A260/A280 and A260/A230 values of > 2.0 and a mean RNA integrity numbers (RIN) value of > 9.2.
Three biological replicates of RNA samples were shipped on dry ice to the DOE Joint Genome Institute (Walnut Creek, CA, United States), where rRNA was depleted and stranded RNA-Seq libraries were prepared, quantified by qPCR and sequenced using a HiSeq 2500 instrument (Illumina). The fastq file reads were filtered and processed with BBDuk1 to remove reads that contained 1 or more “N” bases, had an average quality score across the read less than 10 or had a minimum length < 51 bp or 33% of the full read length. Reads mapped with BBMap (see text footnote 2) to masked human, cat, dog, and mouse references at 93% identity were removed. Another category of removed sequences matched RNA spike-in, PhiX, common microbial contaminants, and ribosomal RNAs. The processed reads from each library were aligned to the reference genome using BBMap with only unique mappings allowed (BAMs/directory). If a read mapped to more than one location it was ignored. featureCounts (Liao et al., 2014) was used to generate raw gene counts, which were normalized to adjust for the length of each gene and total number of reads mapped for each library. The normalization formula used: n = [r/(l/1,000)]/(t/1,000,000), where n = normalized read count for gene (G) for library (L); r = raw read count for gene G for library L; l = gene G length; and t = total reads mapped for library L. Raw gene counts were used to evaluate the level of correlation between biological samples using Pearson’s correlation.
Bioinformatic Analysis
Count tables generated by the JGI RNA-Seq pipeline were input into DESeq2 (Love et al., 2014) to normalize and determine differential expression. Statistical significance was established through DESeq2 by using three biological replicates for control and root exudate conditions. Scatterplots were generated from the DESeq2 data table outputs using ggplot2. Genes differentially expressed between control and root exudate samples (log2 fold-changes –2 ≥ to ≤ 2, adjusted p value ≤ 0.05) were used in downstream analysis. The core genome and pangenome for the Pseudomonas strains used in this study were computed using the OthoMCL v.2.0, Species Tree Builder v.2.2.0, and Phylogenetic Pangenome Accumulation v1.4.0 apps implemented in the U.S. Department of Energy Systems Biology Knowledgebase (KBase) (Arkin et al., 2018). Additional comparisons were conducted with the PGAweb pangenome analysis pipeline (Chen et al., 2018). Differentially expressed genes were assigned to core, non-core, and singleton parts of each strain’s proteome by BLASTp with an E value cutoff of e-06, identity of 40% and coverage of 60%. Functional annotation of differentially expressed genes was carried out with the Blast2GO (Conesa and Gotz, 2008) and visualized in WEGO 2.0 (Ye et al., 2018). Additional manual curation was performed using tools implemented in the Integrated Microbial Genomes (IMG) database (Markowitz et al., 2012), Pseudomonas Genome Database (Winsor et al., 2009), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2008), and Geneious 10.2.3 (Biomatters, Auckland, New Zealand). Metabolic functions encoded by the differentially expressed genes were mapped using iPath 3.0 (Darzi et al., 2018). Phylogenetic analyses were carried out by building multiple sequence alignments with MAFFT v7.222 (Katoh and Standley, 2013) and inferring neighbor-joining (NJ) phylogenies with Geneious Tree Builder. The resultant phylogenetic trees were visualized with iTOL (Letunic and Bork, 2016). Reproducibility of clades within the inferred NJ trees was assessed by bootstrap resampling with 1,000 replicates.
Characterization of Carbon Source Utilization With Biolog Phenotype Microarrays
The utilization of carbon sources was analyzed using Phenotype MicroArrays (Biolog, Hayward, CA, United States) as follows. The bacteria were cultured overnight on Luria-Bertani agar at 25°C, after which cells were harvested and suspended in inoculating fluid (IF-0). The transmittance of the suspension was adjusted to 42% using a Biolog turbidimeter. The cell suspension was mixed with IF−0 containing Dye Mix A (Biolog) to achieve a final transmittance of 85%. One hundred microliter aliquots of the adjusted cell suspension were inoculated into PM01 and PM02A plates, which were then incubated in an OmniLog Phenotype MicroArray System (Biolog) at 25°C for 48 h. The formation of formazan was recorded at 15 min intervals, and data were analyzed using OmniLog Parametric Analysis software v1.20.02 (Biolog). Relative growth of the studied strains was normalized to growth on D-glucose and visualized using Heatmapper (Babicki et al., 2016).
Data Availability
Sequences generated in this project were deposited under NCBI BioProject accession numbers PRJNA439743 through PRJNA439790.
Results
Metabolomic Profiling of Root Exudates of B. distachyon
Metabolomics analysis of lyophilized root exudates revealed the presence of numerous plant metabolites, 86 of which were identified by matching their spectra to the LECO/Fiehn Metabolomics library (Supplementary Table 3). These metabolites included (i) carbohydrates and their derivatives (glucose, fructose, xylose, sucrose, trehalose, maltose, galactose, and others); (ii) sugar alcohols (β-mannosylglycerate, myo-inositol, galactinol, 2-deoxyerythritol, ribitol, threitol and cellobitol); (iii) amino acids and derivatives (glutamine, tyrosine, glutamic acid, asparagine, aspartic acid, valine, phenylalanine, isoleucine, glycine, serine, proline, leucine, tryptophan, cysteine, methionine, citrulline, and others); (iv) organic acids (aconitic, allantoic, γ-aminobutyric, azelaic, citric, fumaric, 2-furoic, D-glyceric, 3-hydroxypropionic, α-ketoadipic, malic, methylmalonic, nicotinic, quinic, succinic, threonic); and (v) assorted metabolites including heterocyclic compounds, phenolics, and biogenic amines, etc (3-hydroxypyridine, maleimide, noradrenaline, 4-hydroxy-3-methoxybenzoate, 5-methoxytryptamine, uracil, aminomalonic acid, palmitic acid, and urea). Results of the analysis also revealed that root exudates of B. distachyon contain hydroxyectoine and the quaternary amine (QA) glycine betaine (Supplementary Figure 1).
Phylogenetic and Pangenome Analyses of Pseudomonas Strains Used in the Study
We used a set of phylogenetic markers suggested by Mulet et al. (2010) to investigate the relatedness of the eight strains used in this study to distinct lineages recognized within the P. fluorescens species complex. The multilocus sequence analysis based on the concatenated sequences of the housekeeping genes rrs (16S rRNA), gyrB, rpoB, and rpoD identified R1-43-08 (along with strains 2-79 and SBW25) as a member of the P. fluorescens subgroup (Figure 1). The rest of the strains clustered closely with four additional subgroups of the P. fluorescens complex, namely P. corrugata (strains Q2-87 and Q8r1-96), P. koreensis (Pf0-1), P. protegens (Pf-5), and P. chlororaphis (30–84). The genomes of the eight rhizosphere Pseudomonas strains varied in size by 1.43 megabase (ranging from 5.65 to 7.07 Mb) and contained between 5,166 and 6,363 protein-coding genes (Figure 2A). The shared gene content was characterized with OrthoMCL, which uses all-against-all BLASTp followed by the Markov Cluster algorithm to identify protein groups shared between the compared genomes, as well as groups representing species-specific gene expansion families (Li et al., 2003). The pangenome analysis revealed a core comprised of approximately 3,179 orthologs that were shared among all strains and represented 50.0% to 61.5% of each predicted proteome (Figures 2A,B). The non-core pangenome contained genes shared by two or more (but not all) strains and contained between 1,482 and 2,080 orthologs, which corresponded to 28.7–36.3% of individual proteomes. The rest of the predicted protein-coding genes were strain-specific singletons that comprised 7.5% to 15.1% of the strain’s predicted proteomes. In respect to divergence from the core genome, strain Pf-5 was found to possess the highest proportion of unique genes (n = 949) followed by 2-79 (n = 887). The entire pangenome of the Pseudomonas strains encompassed over 12,000 homolog and singleton gene families.
FIGURE 1

Neighbor joining phylogeny showing the relationship of the eight strains used in this study (indicated by red triangles) to different species of the P. fluorescens complex. The phylogeny was established based on the concatenated sequences of the housekeeping genes rrs (16S rRNA), gyrB (subunit B of DNA gyrase), rpoB (β subunit of RNA polymerase), and rpoD (sigma 70 factor subunit of RNA polymerase). Distance matrices were calculated by the Jukes-Cantor method. Colored circles on tree nodes indicate bootstrap values (1,000 replicates) that vary between 60% (smallest circle) and 100% (largest circles).
FIGURE 2

Pangenome analysis of the studied Pseudomonas strains. (A) The innermost circle shows the number of orthologous protein families shared among all eight strains used in this study. The second circle shows orthologs present in two or more (but not all) strains, whereas the outermost circle represents strain-specific singletons. Values in brackets under strain names correspond to the total number of protein-coding genes predicted in each genome. (B) The gradual expansion of the pangenome (blue color) and contraction of the core genome (green color) following the sequential addition of genomes from the dataset. Box plots indicate the 25th and 75th percentiles and medians (horizontal lines) with whiskers corresponding to the 10th and 90th percentiles. The input order was randomized to avoid any bias due to the sequential addition of new genomes. The pangenome size increases steadily without reaching a plateau even after the addition of 11,939 non-redundant gene families. At the same time, the core genome converged to 3,179 genes. (C) The pangenome-based phylogenomic analysis of the studied strains. Here, the pangenome was defined with OrthoMCL, and orthologous gene sets were then partitioned into the core, singleton, and non-core (the remaining ortholog sets) categories. These categories were calculated for each node in the Maximum Likelihood species tree, using the set of genomes for which that node represents the ancestor. The results of the ortholog partitioning are shown in pie charts placed at tree nodes. Numbers indicate bootstrap support values. The analysis was conducted in KBase (Arkin et al., 2018).
Further homolog family-based comparisons identified Q8r1-96 and R1-43-08 as the most distantly related strains, with 3349 shared homologs (Supplementary Table 4A). Q8r1-96 and Q2-87, which shared 4,489 homologs, were the most closely related strains. The partitioning of homolog gene families into the core, non-core, and singleton parts of the pangenome agreed with phylogenetic relationships of the strains deduced from the analysis of a selected subset of COGs (Clusters of Orthologous Groups) (Figure 2C and Supplementary Tables 4B,C). The COG-based phylogeny supported the multilocus sequence analysis and revealed that the eight Pseudomonas strains form three distinct clusters, the first of which contained 2-79, R1-30-84, and SBW25. The second cluster included Q8r1-96 and Q2-87, whereas the third encompassed strains 30-84, Pf-5, and Pf0-1.
Correlating the Composition of Root Exudates With Metabolic Profiles of Pseudomonas Strains
We used the Phenotype MicroArray PM1 and PM2 plates to profile the eight Pseudomonas strains for the utilization of 190 different carbon sources. Results of the analysis identified 90 compounds that supported growth and clustered by their intensities of utilization into three distinct groups (Figure 3). Group I was comprised of 30 highly metabolized carbon sources, which included several amino acids and intermediates of glycolysis, pyruvate metabolism, and citrate cycle. Approximately half of these compounds were catabolized by all eight strains, and included several organic acids (fumaric, citric, gluconic, malic, and pyroglutamic), amino acids (Glu, Asn, Gln, Asp, Pro, Ala, and γ-aminobutyric acid), carbohydrates (glucose, mannose, and mannitol), and the purine nucleoside inosine. Group II was composed of 44 chemically diverse carbon sources that were variably utilized by the strains. These compounds were carbohydrates, organic acids, amino acids, phenolics, and polyols, and included known compatible solutes and intermediates of metabolism of pentoses, galactose, starch, and sucrose. Group III encompassed the rest of the Phenotype MicroArray test panel and contained compounds that were not catabolized by the tested strains. Among several notable exceptions were α-hydroxyglutamic acid- γ-lactone, putrescine, and itaconic, citramalic, and succinamic acids, which supported the growth of strains 2-79, 30-84, Pf-5, and SBW25. We further matched the carbon metabolic profiles of the Pseudomonas strains against the list of plant-derived metabolites from the root exudates of B. distachyon Bd21. Interestingly, many carbon sources from the Phenotype MicroArray panel were also present in the root exudates of B. distachyon Bd21, and some of these compounds (glucose, mannose, galactose, fructose, γ-aminobutyric acid, aspartic acid, citric acid, malic acid, fumaric acid, quinic acid, alanine, glutamine, and glutamic acid) were catabolized by all strains used in this study, while others (e.g., xylose, trehalose, m-inositol) were actively utilized only by certain organisms (Figure 3). The comparison of catabolic profiles across the eight studied Pseudomonas strains revealed the presence of three distinct clusters. The first cluster contained strains Q8r1-96 and Q2-87, which consumed very similar sets of carbon sources, as well as strain Pf0-1. The second cluster was composed of 2-79, R1-43-08, SBW25, and 30-84, whereas the third cluster was represented by a single strain, Pf-5. The overall similarity of the catabolic profiles partially agreed with the separation of the strains into different subgroups of the P. fluorescens complex (see above).
FIGURE 3
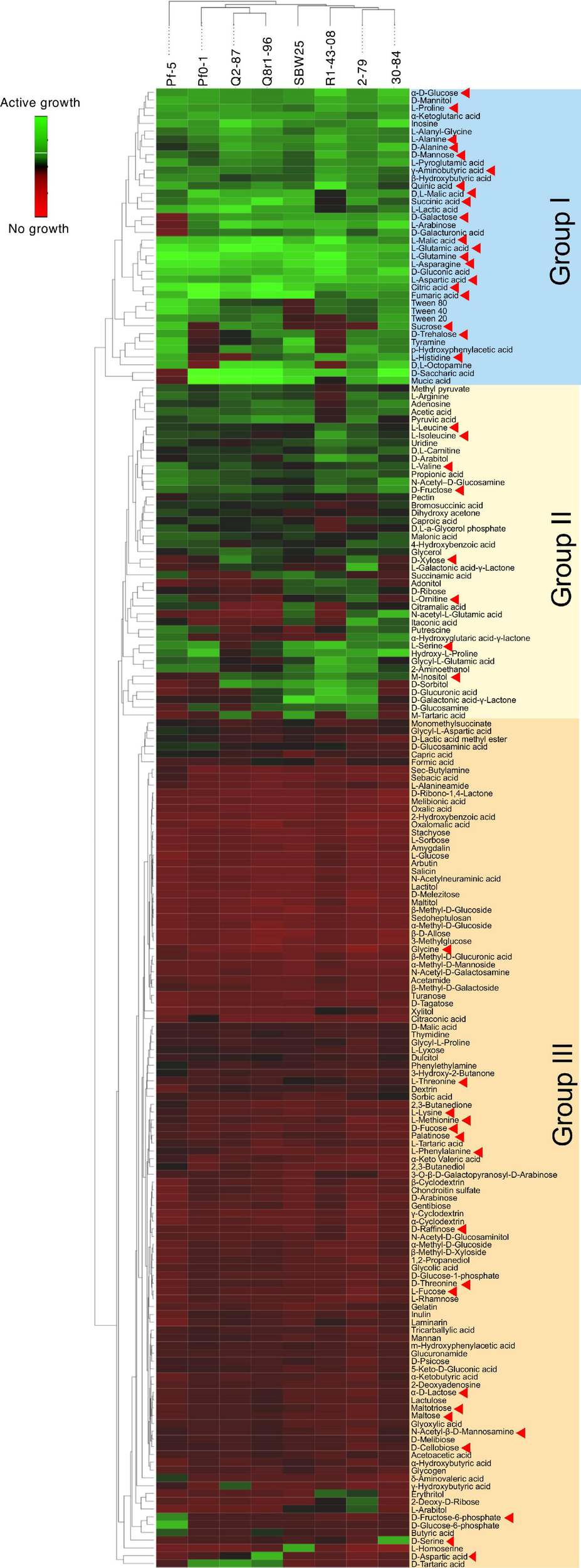
Biolog Phenotype MicroArray profiling the eight rhizosphere Pseudomonas strains used in the study. The hierarchical clustering analysis was carried out using the average linkage method with Euclidean distances. Carbon sources identified by red arrowheads were also detected in the sterile root exudates of B. distachyon Bd21.
Analysis of the RNA-seq Results
In order to understand the cellular responses of rhizosphere Pseudomonas to plant exometabolites, we analyzed the transcriptome changes in cultures grown in the presence of root exudates. Under field conditions, rhizobacteria colonize plant roots in the form of surface-attached microaerobic biofilms (Hojberg et al., 1999). To mimic these conditions, the eight Pseudomonas strains were grown statically at 72% air saturation in 21C-glucose medium amended with root exudates and then processed to extract total RNA (Supplementary Figure 2). A total of 995 million raw sequencing reads were generated from the RNA samples by using the Illumina HiSeq-2500 platform, averaging 20.7 million reads per sample. The removal of low-quality and rRNA sequences resulted in a total of 793 million filtered reads that were mapped onto the eight Pseudomonas genomes with a mean of 7.48 million mapped fragments per genome. The differentially abundant transcripts were identified by setting a p value of 0.05 (adjusted for multiple testing) and the log2 fold-change (FC) threshold ≥± 2.0 (Figure 4 and Supplementary Tables 5–12). When compared with the control conditions, an average of 204 genes per strain were differentially expressed in the presence of root exudates, with the highest (n = 425) and lowest (n = 112) numbers observed, respectively, in SBW25 and Q2-87 (Figure 4). Overall, more genes were induced than repressed in response to exudates, but the actual numbers in each category varied substantially depending on the identity of the Pseudomonas strain. In most strains, the bulk of the differentially expressed genes was almost equally distributed between the core (mean, 48.2%) and non-core (mean, 45.8%) parts of the genome, whereas the strain-specific singleton genes constituted on average only 5.9% (Figure 4B). One notable exception was observed in Q8r1-96, where all differentially expressed genes belonged to the core (73.8%) and non-core (26.2%) parts of the genome. Another notable pattern was observed in R1-43-08, where the majority of genes affected by the presence of root exudate fell into the non-core category (56.3%). The highest proportion of differentially expressed singletons (11.3 and 10.4%, respectively) was identified in strains SBW25 and Pf-5.
FIGURE 4
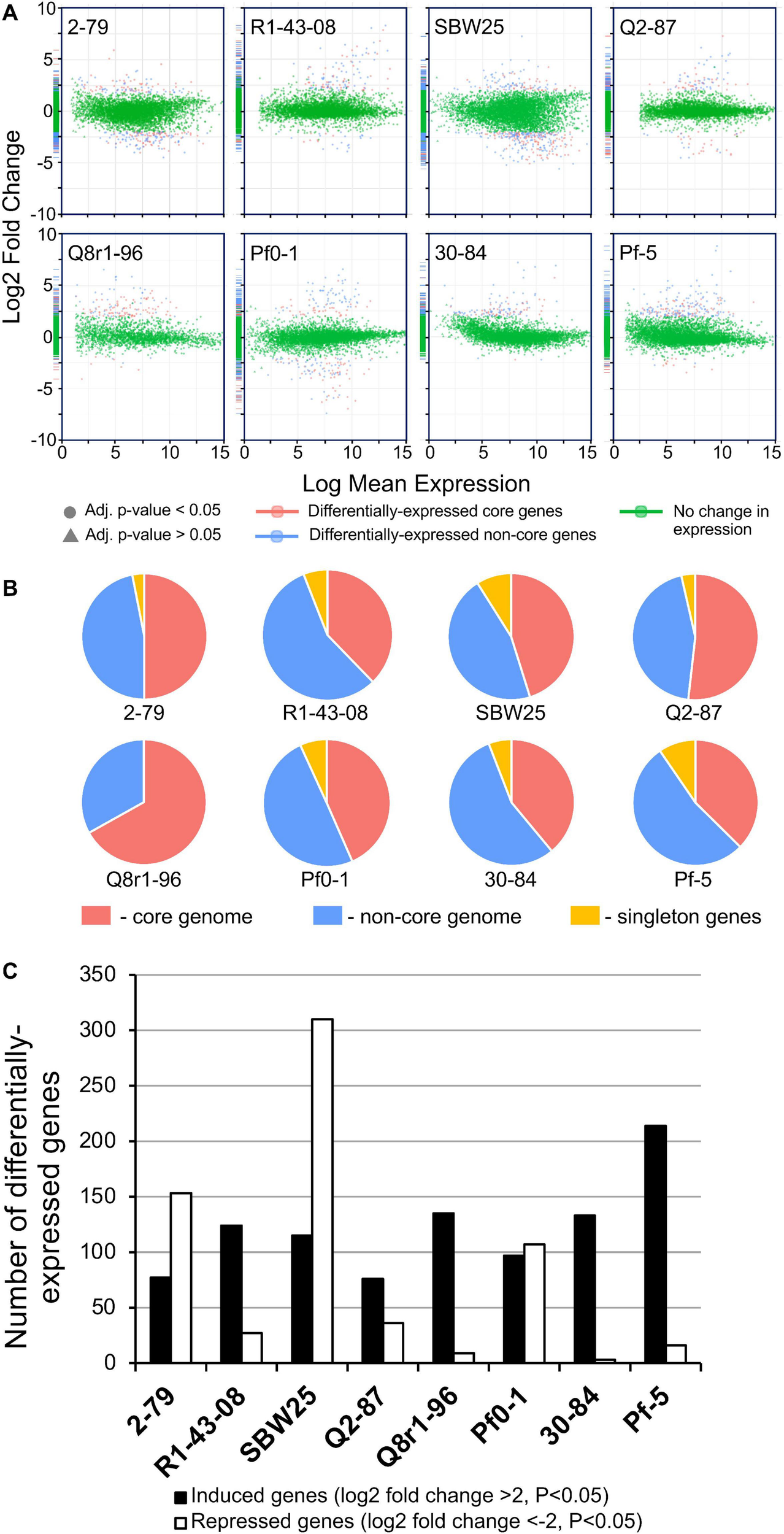
(A) Log ratio versus abundance plots (MA-plots) showing the changes in gene expression in response to root exudates. The differentially expressed core and non-core genes are shown in red and blue, respectively. Green color indicates genes with a log2 fold-change and/or adjusted p values below the established threshold. (B) Circular diagrams depicting the distribution of differentially expressed genes among the core, non-core, and singleton proteomes of individual Pseudomonas strains. (C) The number of genes per genome that were induced and repressed by B. distachyon root exudates.
We further explored how the identified differentially expressed genes were distributed across genomes of the eight studied rhizosphere strains. The pairwise BLASTp comparisons identified 2-79 and SBW25 as two strains that shared the highest number of genes (n = 101) induced or repressed in response to root exudates (Table 1). The second pair of strains with a significant number of similar differentially expressed genes (n = 86) was Q8r1-96 and Pf-5, which was followed by Pf0-1 and 30-84, which shared 56 differentially expressed genes. These patterns of shared genes were also observed when the results of the pairwise BLASTp comparisons were converted into a binary gene presence/absence matrix, which was then subjected to cluster analysis using a UPGMA algorithm based on Sorensen’s dissimilarity index or examined by non-metric multidimensional scaling (NMDS) (Figure 5).
TABLE 1
| Strain | 2-79 | SBW25 | R1-43-08 | Q8r1-96 | Q2-87 | 30-84 | Pf0-1 | Pf-5 |
| 2–79 | 260 | |||||||
| SBW25 | 101 | 425 | ||||||
| R1-43-08 | 30 | 25 | 151 | |||||
| Q8r1-96 | 32 | 39 | 21 | 145 | ||||
| Q2-87 | 27 | 28 | 25 | 31 | 112 | |||
| 30-84 | 27 | 23 | 24 | 32 | 28 | 136 | ||
| Pf0-1 | 38 | 50 | 29 | 29 | 50 | 56 | 205 | |
| Pf-5 | 36 | 41 | 52 | 86 | 29 | 55 | 40 | 230 |
The number of differentially expressed genes shared among the eight studied strains of rhizosphere Pseudomonas.
The pairwise comparisons were conducted by BLASTp with the following cutoff parameters: E-value < 1e-06, minimum percent identity > 40%, and minimum percent coverage > 65%. The black diagonal cells show the number of differentially expressed genes per strain. In other words, these are self comparison values.
FIGURE 5

Comparison of the eight Pseudomonas strains based on the content (presence/absence) of genes differentially expressed in the presence of root exudates. (A) UPGMA clustering based on the Sorensen’s dissimilarity index. (B) non-metric multidimensional scaling (NMDS) analysis.
The differentially expressed Pseudomonas genes were subjected to Blast2Go analysis and Gene Ontology (GO) annotation (Figure 6). Metabolic process, catalytic activity, and membrane were the most common annotation terms across the three primary GO term categories (i.e., biological process, molecular function, and cellular component). A total of 1,694 GO terms was assigned to 805 upregulated genes, with the majority of the GO terms related to molecular function (682, 40.3%), followed by biological process (669, 39.5%), and cellular component (343, 20.2%). In the 539 downregulated gene category, 1,101 GO terms were assigned to biological process (420, 38.1%), molecular function (417, 37.9%), and cellular component (264, 24.0%). Within biological process, metabolic process, cellular process, localization, response to stimulus, and regulation were over-represented. Within molecular function, the largest proportion was assigned to catalytic activity, binding, and transporter activity categories. Within cellular component, the majority were assigned to membrane, membrane part, cell, and cell part categories. Across the eight strains, 37–42% of differentially expressed genes had no Gene Ontology IDs and encoded various conserved hypothetical proteins.
FIGURE 6

Gene Ontology (GO) classification of Pseudomonas genes that were induced (red bars) or repressed (gray bars) in response to root exudates of B. distachyon Bd21. The terms were derived from 93 different functional groups (GO subcategories level 4). The GO terms were assigned with Blast2GO (Conesa and Gotz, 2008) and visualized in WEGO 2.0 (Ye et al., 2018). On a WEGO histogram, the percentage of 100 is defined as the total number of genes assigned a GO term. However, the subcategories do not add up to 100% because many genes fall into more than one functional class and are therefore annotated by multiple GO terms.
Functional Classification of Shared Differentially Expressed Genes
The interrogation of RNA-seq data revealed multiple cellular pathways that were differentially regulated in bacterial cultures incubated with root exudates (Supplementary Figures 3, 4). Although none of these differentially regulated pathways were shared by all eight strains, the cross-strain comparisons revealed several types of common and specific transcriptomic responses that were elicited by the presence of plant exometabolites (Table 2). The visual representation of core gene expression patterns is provided in Supplementary Figure 5, which shows heatmaps of expression profiles and p-adj values for core genes shared by the studied strains. The figure is accompanied by Supplementary Table 13 that lists predicted functions of genes constituting the four distinct clusters observed after hierarchical clustering of gene expression values. The first category of shared differentially expressed pathways functioned in the uptake and catabolism of selected carbohydrates, quaternary ammonium compounds (QAs), and phenolics. All strains except for R1-43-08, responded to root exudates by inducing the fructose-specific phosphoenolpyruvate (PEP)-dependent phosphotransferase system (PTSFru). The components of this system are encoded by a conserved operon and include the cytoplasmic polyprotein EI/HPr/EIIAFru (FruB), the 1-phosphofructokinase FruK, and the fructose-specific permease EIIBC (FruA) (Chavarria et al., 2016). The PTSFru system functions by acquiring high-energy phosphates from PEP and sequentially passing them, via the EI/HPr/EIIAFru domains of FruB, to the EIIB component of FruA. The phosphates are ultimately transferred by the EIIC transporter to fructose yielding fructose 1-phosphate, which is channeled into the central metabolic pathways through the action of the phosphofructokinase FruK.
TABLE 2
| Predicted function | Strainb | |||||||
| 2–79 | SBW25 | R1-43-08 | Q8r1-96 | Q2-87 | 30–84 | Pf0-1 | Pf-5 | |
| Uptake and catabolism of fructose | ||||||||
| D-fructose PTS system, IIC component | 2756598827 (2.7) | 649634314 (2.3) | 2597873629 (3.6) | 2597850083 (2.7) | 2597856046 (3.6) | 637740645 (2.9) | 637318202 (2.8) | |
| 1-phosphofructokinase | 2756598828 (2.9) | 649634313 (3.1) | 2597873628 (3.9) | 2597850082 (2.7) | 2597856045 (3.4) | 637740644 (3.1) | 637318201 (2.7) | |
| D-fructose PTS system, IIA component | 2756598829 (2.6) | 649634312 (3.0) | 2597873627 (3.7) | 2597850081 (2.6) | 2597856044 (3.6) | 637740643 (2.9) | 637318200 (3.2) | |
| Uptake and catabolism of arabinose | ||||||||
| MFS superfamily transporter | 2756599521 (2.2) | 649635836 (3.6) | 2756590067 (4.9) | 2597851595 (3.1) | 2597859759 (4.2) | 637743102 (2.9) | ||
| L-arabinonate dehydratase | 2756599520 (3.1) | 649635835 (4.3) | 2756590066 (5.5) | 2597851594 (5.4) | 2597859760 (4.0) | 637743103 (3.5) | ||
| Interconversion of alpha- and beta-anomers of aldoses | ||||||||
| Aldose epimerase superfamily protein | 2756599919 (2.7) | 2597878613 (4.2) | 2597849545 (3.1) | 2597860977 (4.2) | 637742166 (3.4) | 637323358 (3.5) | ||
| Uptake and catabolism of quaternary ammonium compounds | ||||||||
| Choline dehydrogenase BetA | 2597874908 (2.3) | 2597851450 (2.1) | ||||||
| Transcriptional regulator GbdR | 2756597125 (−3.7) | 649639087 (−3.9) | ||||||
| Membrane dipeptidase, dgc operon | 2756597136 (1.9) | 2756592046 (2.0) | 2597878321 (3.8) | 2597849833 (2.9) | 2597860696 (2.6) | 637323077 (3.3) | ||
| Hypothetical protein, dgcAB operon | 2756597137 (2.4) | 2756592045 (2.3) | 2597878320 (3.4) | 2597849834 (2.7) | 2597860695 (2.3) | 637323076 (3.3) | ||
| Dimethyl Gly demethylase DgcA | 2756597138 (2.7) | 2756592044 (2.0) | 2597878317 (3.3) | 2597849835 (2.7) | 2597860694 (2.2) | 637323075 (3.2) | ||
| Dimethyl Gly demethylase DgcB | 2756597139 (2.3) | 2597878318 (3.6) | 2597849836 (2.6) | 2597860693 (2.4) | 637323074 (3.0) | |||
| Betaine demethylase, GbcA subunit | 2756597143 (2.1) | 2756592039 (2.3) | 2597878312 (4.3) | 637323070 (3.7) | ||||
| Betaine demethylase, GbcB subunit | 2597878311 (3.9) | 2597860689 (2.0) | 637323069 (3.2) | |||||
| Ser hydroxymethyltransferase, sox operon | 2756597149 (2.1) | 2756592033 (2.0) | 2597878308 (2.9) | 2597849846 (2.7) | 637323064 (3.1) | |||
| Sarcosine oxidase, γ subunit, SoxG | 2597878304 (2.9) | |||||||
| Sarcosine oxidase, α subunit, SoxA | 2756592028 (2.1) | 2597878305 (3.0) | 637323061 (2.4) | |||||
| Sarcosine oxidase, δ subunit, SoxD | 2597878306 (2.5) | 637323062 (2.2) | ||||||
| Sarcosine oxidase, β subunit, SoxB | 2756597150 (2.4) | 2756592032 (1.9) | 2597878307 (2.6) | 637323063 (2.3) | ||||
| Betaine substrate-binding protein CbcX | 2756590368 (2.3) | 2597878336 (2.3) | 2597850794 (3.1) | 637742655 (-3.5) | ||||
| ABC transporter, ATP-binding protein CbcV | 2597878338 (2.3) | 637742656 (-3.5) | ||||||
| Uptake and catabolism of myo-inositol | ||||||||
| 5-dehydro-2-deoxygluconokinase, IolC | 2756592881 (2.5) | 2597876275 (4.3) | 2597857598 (2.4) | 637319925 (4.2) | ||||
| 2-keto-myo-inositol dehydratase, IolE | 2756592884 (2.5) | 2597876273 (4.6) | 2597857602 (2.4) | 637319928 (4.1) | ||||
| 5-deoxy-glucuronate isomerase, IolB | 2756592883 (2.3) | 2597876272 (4.3) | 2597857600 (2.4) | 637319927 (4.3) | ||||
| 2-keto-myo-inositol isomerase, IolL | 2756592882 (2.7) | 2597857599 (2.4) | 637319926 (4.4) | |||||
| 3D-(3,5/4)-trihydroxycyclohexane-1,2-dione acylhydrolase, IolD | 2756592885 (2.4) | 2597876269 (4.8) | 2597857603 (2.5) | 637319929 (4.3) | ||||
| Myo-inositol 2-dehydrogenase, IolG | 2756595203 (1.9) | 2756592886 (2.2) | 2597876268 (4.8) | 2597857604 (2.0) | 637319930 (3.9) | |||
| Inositol transport substrate-binding protein | 2756592888 (2.0) | 2597876265 (3.8) | 2597851513 (2.8) | 637319932 (3.8) | ||||
| Inositol transport permease protein | 2756592890 (1.9) | 2597876263 (3.6) | 2597851515 (2.2) | 637319934 (3.5) | ||||
| Inositol transport ATP-binding protein | 2756592889 (2.2) | 2597876264 (3.8) | 2597851514 (2.7) | 637319933 (3.6) | ||||
| Catabolism of phenolics | ||||||||
| Muconate cycloisomerase | 2597859089 (3.1) | 637742838 (3.4) | 637321199 (4.8) | |||||
| Muconolactone delta-isomerase | 2597859088 (2.6) | 637742837 (3.5) | 637321198 (4.4) | |||||
| Catechol 1,2-dioxygenase | 2597859087 (2.0) | 637742836 (2.9) | 637321197 (3.5) | |||||
| AraC-type DNA-binding protein | 2597859086 (2.0) | 637321196 (2.2) | ||||||
| Benzoate 1,2-dioxygenase, α subunit | 2756599329 (2.7) | 2597859085 (3.4) | 637742843 (3.9) | 637321195 (3.8) | ||||
| Benzoate 1,2-dioxygenase, β subunit | 2756599330 (2.2) | 2597859084 (3.3) | 637742842 (4.2) | 637321194 (4.8) | ||||
| Benzoate 1,2-dioxygenase, reductase subunit | 2756599331 (2.1) | 2597859083 (3.4) | 637742841 (3.9) | 637321193 (4.3) | ||||
| Copper homeostasis | ||||||||
| pre-Q0 reductase/7-cyano-7-deazaguanine reductase CinQ | 2756597439 (2.7) | 649635068 (1.8) | 2597874689 (3.4) | 637319306 (2.5) | ||||
| Cupredoxin-like copper-binding protein CinA | 2756597440 (2.8) | 649635067 (4.3) | 2756590986 (2.03) | 2597853017 (7.3) | 2597857153 (5.7) | 637743734 (3.5) | 637319305 (3.4) | |
| Heavy metal response regulator CinR | 649635066 (1.8) | 2597874687 (2.1) | 2597853018 (3.2) | 2597857152 (2.4) | 637743735 (2.1) | 637319304 (2.5) | ||
| Heavy metal sensor histidine kinase CinS | 649635065 (2.0) | 2597857151 (2.2) | 637743736 (2.0) | 637319303 (2.0) | ||||
| Copper resistance protein CopA | 2597877412 (5.9) | 2597850492 (5.6) | 2597857966 (6.9) | 637743691 (5.2) | 637320232 (6.4) | |||
| Copper resistance protein CopB | 2597850491 (5.3) | 2597857965 (6.8) | 637743692 (5.5) | 637320231 (6.0) | ||||
| Copper resistance protein CopC | 2597850490 (4.6) | 2597857964 (7.2) | 637743693 (5.4) | |||||
| Copper resistance protein CopD | 2597850489 (4.9) | 2597857963 (6.9) | 637743694 (5.2) | |||||
| Conserved hypothetical protein | ||||||||
| Aldose epimerase superfamily protein | 2756599919 (2.7) | 2597878613 (4.2) | 2597849545 (3.1) | 2597860977 (4.2) | 637742166 (3.4) | 637323358 (3.5) | ||
| Uptake and catabolism of sulfonates | ||||||||
| FMN-dependent monooxygenase SsuE | 2756592254 (−1.9) | 637745334 (−3.5) | ||||||
| Sulfonate substrate-binding protein SsuA | 649639261 (−2.0) | 2756592253 (−2.2) | 2597878518 (−2.6) | 2597849636 (−3.9) | 637745333 (−7.0) | 637323272 (−2.7) | ||
| FMN-dependent monooxygenase SsuD | 649639260 (−1.9) | 2756592252 (−2.3) | 2597849637 (−2.8) | 637745332 (−5.8) | 637323271 (−2.3) | |||
| Sulfonate permease protein SsuC | 649639259 (−2.4) | 2756592251 (−2.1) | 2597878516 (−1.9) | 2597849638 (−3.2) | 637745331 (−5.8) | 637323270 (−2.5) | ||
| Sulfonate transport ATP-binding protein SsuB | 649639258 (−2.5) | 2756592250 (−2.3) | 2597849639 (−2.5) | 637745330 (−5.6) | 637323269 (−2.7) | |||
| Molybdopterin binding protein SsuF | 649639257 (−3.1) | 2756592249 (−2.7) | 2597878514 (−2.9) | 2597849641 (−3.3) | 637745339 (−6.2) | 637323268 (−2.7) | ||
| Uptake and catabolism of taurine | ||||||||
| Taurine substrate-binding protein TauA | 2597854917 (−2.0) | 637740095 (−4.1) | ||||||
| Taurine transport ATP-binding protein TauB | 2756592398 (−1.9) | 637740094 (−3.7) | 637317614 (−1.9) | |||||
| Taurine permease protein TauC | 2756592398 (−1.9) | 637740093 (−3.9) | 637317613 (−2.0) | |||||
| Taurine dioxygenase TauD | 2597854920 (−1.8) | 637740092 (−3.6) | ||||||
The distribution and predicted functions of selected differentially expressed genesa.
aThe shared differentially expressed genes were identified by BLASTp with the cutoff parameters of E-value < 1e-06, minimum percent identity > 40%, and minimum percent coverage > 65%.
bValues in columns indicate JGI IMG gene IDs followed by the corresponding fold-change (FC) values (shown in brackets).
In all strains except for Q8r1-96 and Pf-5, the exposure to root exudates resulted in the induction of two genes adjacent to the fru cluster that encoded a Major Facilitator Superfamily (MFS) transporter and an L-arabinonate dehydratase (Table 2). These genes are predicted to participate in the uptake and catabolism of L-arabinose, where L-arabinonate dehydratase plays an important role by converting L-arabinonate to 2-dehydro-3-deoxy-L-arabinonate (Rahman et al., 2017). In SBW25, R1-43-08, and Q2-87, we also observed the induction of genes encoding components of the AraFGH complex, an ATP-Binding Cassette (ABC) superfamily transporter involved in the import of arabinose into the cell (Supplementary Tables 6, 7, 9). Finally, all strains except SBW25 and R1-43-08 responded to the presence of exudates by upregulating a conserved gene encoding an aldose epimerase superfamily protein. Such enzymes equilibrate alpha- and beta-anomers of aldoses and ensure that stereospecific enzymes involved in the metabolism of free sugars do not act as metabolic bottlenecks (Abayakoon et al., 2018). Although some aldose epimerases have been linked to specific pathways, the Pseudomonas gene identified in this study could not be assigned to a particular metabolic process based on sequence analysis and genomic location.
Several Pseudomonas strains responded to the presence of root exudates by upregulating genes involved in the uptake and catabolism of myo-inositol and possibly other stereoisomers of inositol (Table 2). The upregulated catabolic genes encode the dehydrogenase IolG, which oxidizes myo-inositol to its corresponding ketone, as well as IolE, IolD, IolB, and IolC that collectively convert the 2-keto-myo-inositol to acetyl-CoA and the glycolysis intermediate dihydroxyacetone phosphate (Yoshida et al., 2008; Kohler et al., 2011). In R1-43-08, Q8r1-96, Q2-87, and Pf-5, the upregulated functions also involved components of the putative inositol-specific ABC transporter. The cross-genome comparisons revealed that in all studied strains except for Pf0-1, components of the myo-inositol utilization pathway were encoded within a well-conserved gene cluster which, in addition to catabolic and transport functions, also encodes a dedicated transcriptional repressor.
All studied strains of Pseudomonas carry multiple genes involved in scavenging the quaternary ammonium compounds choline, glycine betaine (GB), carnitine, choline-O-sulfate, and sarcosine from the environment. Many of these genes were differentially expressed, including those encoding parts of the ABC transporter CbcXWV, which is predicted to function in the uptake of choline under water-replete conditions (Table 2). Among enzymes induced in the presence of root exudates were the choline dehydrogenase BetA, which converts choline to glycine betaine and a network of enzymes (i.e., the Rieske family oxygenase GbcAB, the dimethyglycine demethylase DgcAB, and the sarcosine oxidase SoxBDAG) that sequentially convert GB to glycine. In 2-79 and SBW25, this group of differentially regulated genes also included an AraC-family transcriptional activator GbdR, which perceives intercellular levels of GB and induces genes involved in the transport and catabolism of glycine betaine and detoxification of the catabolic byproducts (Hampel et al., 2014).
The last category of activated catabolic pathways included the catechol branch of the β-ketoadipate pathway for the degradation of aromatic compounds. In strains 30-84, Pf0-1, and Pf-5, growth on root exudates resulted in upregulation of catechol-1,2-dioxygenase, muconate cycloisomerase, and muconolactone isomerase, which collectively cleave the catechol ring and convert it to β-ketoadipate enol-lactone (Harwood and Parales, 1996). Finally, analysis of the P. synxantha 2-79 transcriptome identified an induction of benABC genes encoding subunits of benzoate 1,2-dioxygenase, an oxidoreductase that generates catechol from benzoate.
In addition to various catabolic pathways, the exposure to root exudates also induced several genes involved in the homeostasis of copper (Table 2). Four of these genes form a conserved cluster in genomes of the strains and encode the periplasmic copper-sensing two-component system CinRS, the plastocyanin/azurin-like protein CinA, and the NADPH-dependent pre-Q0 reductase CinQ. Also, in strains Q2-87, 30-84, Pf0-1, and Pf-5, we observed upregulation of a conserved operon encoding the multicopper oxidase CopA, the periplasmic copper-binding protein CopC, the inner membrane protein CopD, and outer membrane protein CopB. In several Gram-negative bacteria, these Cop proteins are thought to have dual functions and participate both in the uptake of essential copper as well as in the sequestration of excess copper in the periplasm and outer membrane.
The analysis of shared downregulated pathways revealed that most of the strains respond to the presence of root exudates by repressing genes involved in the uptake and catabolism of sulfur compounds (Table 2). In strains SBW25, R1-43-08, Q8r1-96, Q2-87, Pf0-1, and Pf-5, this response involved the ssuEADCB operon responsible for the utilization of alkanesulfonates as sulfur sources. The ssu operon is highly conserved in fluorescent pseudomonads and encodes the FMNH2-dependent monooxygenase SsuD and the NAD (P)H-dependent FMN reductase SsuE, which together catalyze the desulfonation of alkanesulfonates. Also, the ssu locus contains genes for the molybdopterin-binding protein SsuF and the alkanesulfonate-specific ABC-type transporter consisting of the sulfonate substrate-binding protein SsuA, sulfonate permease protein SsuC, and sulfonate transport ATP-binding protein SsuB. Finally, in R1-43-08, Q2-87, Pf0-1, and Pf-5, growth on root exudates coincided with repression of the tauABCD operon, which allows these strains to utilize taurine (2-aminoethanesulfonate) as a sulfur source. The repressed tau genes encoded the 2-oxoglutarate-dependent taurine dioxygenase TauD and substrate-binding, ATP-binding, and permease components of the taurine-specific ABC transporter TauABC.
Other Differentially Expressed Pathways
In addition to their effect on several shared cellular pathways, growth on root exudates resulted in the induction or repression of numerous strain-specific genes. In closely related P. synxantha 2-79 and P. fluorescens SBW25, we observed differential expression of genes involved in energy metabolism, transport of amino acids, and surface attachment (Supplementary Tables 5, 6). Other notable differentially expressed pathways included 2–79 gene clusters that encode enzymes for the catabolism of trehalose, a prophage, and toxin/antitoxin system, as well as the SBW25 operon predicted to control the synthesis of the capsular exopolysaccharide colonic acid. The response of Pseudomonas sp. R1-43-08 to root exudates also involved differential expression of different energy metabolism pathways. In addition, we observed the upregulation of genes involved in the uptake and catabolism of xylose (also upregulated in 2–79) and repression of enzymes for the biosynthesis of phenazine-1-carboxylic acid and assimilation of inorganic sulfur and L-cysteine biosynthesis (Supplementary Table 7).
The analysis of the Q8r1-96 transcriptome revealed perturbation of different metabolic pathways including genes encoding components of cytochrome C oxidase, transport and catabolism of sorbitol/mannitol, metabolism of butanoic acid, and biosynthesis of exopolysaccharides alginate and poly-β-1-6-N-acetylglucosamine (Supplementary Table 8). In P. fluorescens Q2-87, we identified differential expression of genes involved in metabolism of galactose, tryptophan, tyrosine, glycine, serine, and threonine (Supplementary Table 9), while in P. chlororaphis 30-84, growth on exudates activated the biosynthesis of molybdopterin cofactor, catabolism of galactonate and acetoin, and uptake and catabolism of putrescine (Supplementary Table 10). The response of P. protegens Pf-5 to root exudates involved upregulation of acetoin dehydrogenase, which converts acetoin to acetaldehyde and acetyl-CoA, as well as pathways for the utilization of glycolate and putrescine (Supplementary Table 11). Also induced were genes for the production of pyrrolnitrin and PhlG hydrolase, which modulate the metabolic loads attributed to the synthesis of 2,4-diacetylphloroglucinol. The differentially expressed genes of P. fluorescens Pf0-1 included, among others, operons encoding cytochrome C oxidase and enzymes for catabolism of malonic acid (Supplementary Table 12). Yet another interesting finding involved the induction of assorted genes acting in the homeostasis of iron and defense against reactive oxygen species (ROS). We observed activation of iron dicitrate transporters (SBW25 and 30-84), genes for the biosynthesis of siderophores ornicorrugatin (SBW25) and pyochelin (Pf-5), heme-degrading enzymes (2–79, 30–84), TonB siderophore receptors, and components of the energy-transducing inner membrane complex TonB-ExbB-ExbD (2–79 and Pf-5). The differentially expressed ROS defense pathways were represented by different catalases in strains 2–79, R1-43-08, Q8r1-96, Q2-87, Pf0-1, and Pf-5 and organic hydroperoxide resistance proteins in strains SBW25 and R1-43-08. Finally, in SBW25, Q2-87, 30–84, and Pf0-1, the addition of exudates resulted in the upregulation of peroxiredoxins that detoxify H2O2, peroxynitrite, and aliphatic and aromatic hydroperoxides.
Discussion
Our analysis of B. distachyon root exudates revealed a complex mix of primary and secondary metabolites, thus supporting the view of the plant rhizosphere as a carbon-rich niche for soil microorganisms. Our results were in agreement with a recent report of 27 different sugars, amino acids, and organic acids in Brachypodium exudates (Kawasaki et al., 2016). We confirmed the presence of exometabolites identified in that study, along with dozens of additional analytes that were identified by matching their mass-spectra and retention indices to the LECO/Fiehn Metabolomics library (Supplementary Table 3). The complementation of the metabolomic analysis with profiling of the bacteria by Biolog Phenotype MicroArrays revealed that a substantial proportion of the characterized exudate constituents were catabolized by a collection of eight Pseudomonas strains from across the P. fluorescens group that is known to form associations with plant roots. The amendment of Pseudomonas cultures with root exudates caused changes in the expression of multiple genes encoding catabolic and anabolic enzymes, predicted transporters, transcriptional regulators, stress response, and conserved hypothetical proteins. In most strains, these differentially expressed genes were almost equally split between the core and variable genome regions, mirroring the substantial strain-to-strain variation in the genome size and gene content within the P. fluorescens species complex (Loper et al., 2012).
The analysis of transcriptome responses to root exudates revealed several types of cellular pathways present in the strains used in this study. The first category of such pathways was involved in the catabolism of carbohydrates such as fructose, arabinose, myo-inositol, xylose, trehalose, and galactose. Among these catabolic traits, the ability to utilize fructose as a carbon source is highly conserved among fluorescent pseudomonads. In contrast, growth on arabinose, myo-inositol, xylose, and trehalose is variably present and was traditionally used to differentiate species and biovars within the P. fluorescens group (Barrett et al., 1986). We speculate that such variably distributed pathways contribute to the differential affinity of pseudomonads toward host plants and/or to determine which strains flourish in response to growing roots and changing environments. Several independent studies have confirmed the importance of carbohydrate catabolism pathways for the biology of rhizosphere pseudomonads. For example, in vivo expression technology (IVET) profiling of P. fluorescens SBW25 identified xylose isomerase among genome regions essential for the colonization of sugar beet seedlings (Liu et al., 2015), whereas a genome-wide Tn-Seq screen of Pseudomonas simiae identified genes for the catabolism of myo-inositol among traits essential for the colonization of Arabidopsis thaliana roots (Cole et al., 2017).
The response of rhizosphere Pseudomonas to Brachypodium root exudates also involved pathways for the uptake and metabolism of amino acids. We observed differential expression of genes encoding the hydrophobic (HAAT) and polar (PAAT) amino acid uptake transporters in strains 2-79, SBW25, Q2-87, Pf0-1, and Pf-5. Other related genes encoded enzymes for the catabolism of valine and glutamic acid (2-79); metabolism of tryptophan, glycine, serine, and threonine (Q2-87); and biosynthesis of methionine (Q8r1-96). It is plausible that the abundance of amino acids in root exudates is also linked to the repression of pathways involved in the catabolism of sulfonates and taurine that was observed in several strains (Table 2). Although the preferred source of sulfur for P. fluorescens is unknown, in the closely related P. aeruginosa, the sulfur starvation response is triggered by the growth on any sulfur compound other than sulfate, thiocyanate, and cysteine (Hummerjohann et al., 1998). This fact, together with the presence of cysteine and cystine in the root exudates, suggest that root exudates of Brachypodium may serve as an important source of sulfur for rhizosphere Pseudomonas. These findings also agree well with the reported scarcity of inorganic sulfate in the soil, and the presence of sulfur mostly in the form of organic compounds, including amino acids, proteins, sulfate esters, and sulfonates (Autry and Fitzgerald, 1990).
Another interesting result of this study was the concerted activation of copper and iron homeostasis pathways observed in all of the Pseudomonas strains used in this work. In bacteria, an excess of copper is toxic and triggers oxidative stress due to the formation of free radicals, as well as disruption of protein metalation and stability of iron-sulfur clusters (Bondarczuk and Piotrowska-Seget, 2013). On the other hand, copper is an essential trace element used as a cofactor in different enzymes. Similarly, although elevated levels of iron cause redox stress, this element is also found in active energy metabolism enzymes and is crucial for bacterial growth (Andrews et al., 2003). The analysis of metal homeostasis genes identified in this study suggests that their induction was likely triggered by the deficiency of copper and iron in bacterial cultures grown in the presence of root exudates. We attribute this effect to the ability of some components of root exudates to chelate soil metals.
Despite the abundance of iron in the soil, its bioavailability is limited due to the low solubility of Fe (III) oxyhydrates at neutral pH. The non-graminaceous plants circumvent this problem by acidifying the rhizosphere and secreting flavins, phenolics, and organic acids that chelate iron. The reduction of these ferric chelates releases soluble ferrous iron taken up by root cells (Kobayashi and Nishizawa, 2012). Graminaceous plants, like Brachypodium, acquire iron by secreting into the soil non-protein amino acids of the mugineic acid (MA) group, which act as Fe (III)-chelating phytosiderophores. In addition to iron, low-molecular-weight organic acids and phytosiderophores bind other divalent and trivalent metals (including copper) and contribute to heavy-metal tolerance in plants (Chen et al., 2017). It is plausible that the presence of these plant exometabolites is responsible for the deficit of iron and copper observed in Pseudomonas cultures grown in the presence of root exudates. These results further underscore the importance of diverse and redundant metal homeostasis pathways found in genomes of the P. fluorescens group for the ability of these organisms to colonize and persist in the plant rhizosphere.
Recently, Klonowska et al. (2018) examined transcriptomic responses of symbiotic nitrogen-fixing bacteria to root exudates of the legume plant Mimosa pundica, which has an unusual ability to support both alpha- (Rhizobium) and beta-rhizobia (Cupriavidus and Burkholderia). Using RNA-seq, the authors characterized genes involved in the perception of root exudates in the nodulating bacteria Burkholderia phymatum STM815, Cupriavidus taiwanensis LMG19424, and Rhizobium mesoamericanum STM3625. Interestingly, the analysis of differentially expressed genes revealed induction of pathways involved in the catabolism of fructose, xylose, myo-inositol, and protocatechuate/catechol. Also upregulated were some copper homeostasis, siderophore biosynthesis, and oxidative stress genes. Finally, the analytical profiling of M. pundica exudates revealed an overlap with Brachypodium in the types of carbohydrates, amino acids, and organic acids present. These findings suggest that differentially expressed genes shared by multiple strains of the group P. fluorescens are not unique to the Brachypodium-Pseudomonas system but represent a set of conserved cellular pathways involved in the perception of plant exometabolites by different clades of rhizosphere-dwelling Proteobacteria.
Most strains included in this study were originally selected based on the ability to colonize the rhizosphere and produce secondary metabolites that alleviate the plant stress response and/or inhibit soilborne pathogens. It has been suggested that plant metabolites released into the rhizosphere affect the biocontrol activity of plant-beneficial pseudomonads (de Werra et al., 2011). We provide further support to this hypothesis by demonstrating that in some strains, root exudates modulate the expression of genes for the catabolism of the plant growth-promoting metabolites acetoin and 2,3-butanediol. The exposure to exudates also affected the expression of genes for the synthesis of well-characterized antifungal compounds pyrrolnitrin, phenazine-1-carboxylic acid, and 2,4-diacetylphloroglucinol. The modulatory effects were strain-specific, suggesting significant differences in the regulatory networks involved in the perception of plant signals and regulation of the production of antibiotics and growth-promoting metabolites.
The final significant finding of this study was the induction of catabolism of quaternary amines (QAs) observed in multiple strains of the P. fluorescens group during growth on root exudates. This observation was supported by the detection of glycine betaine in the root secretions of B. distachyon. The presence of QAs in plant tissues and the capacity of these metabolites to provide stress protection and nutrients to plant pathogens and symbionts were reported before (Boncompagni et al., 1999; Chen et al., 2013; Kabbadj et al., 2017), but our study is among the first to highlight the potential importance of these metabolites for rhizosphere interactions. Pseudomonads do not synthesize QAs de novo but have evolved many pathways to scavenge them from eukaryotic hosts, where these metabolites are abundant due to the prominence of phosphatidylcholine in cellular membranes. Strains of P. fluorescens carry genes for the conversion of choline, carnitine, and glycine betaine to glycine, as well as quaternary amine transporters of the BCCT and ABC families that are also conserved in the opportunistic human pathogen P. aeruginosa and the plant pathogen P. syringae (Galvao et al., 2006; Chen et al., 2013; Wargo, 2013b).
In P. aeruginosa, choline catabolism genes are essential for the ability of this pathogen to persist during lung infection (Wargo, 2013a). Similarly, a P. syringae mutant deficient in BetT, OpuC, and CbcXWV quaternary amine transporters had reduced fitness during colonization of bean and soybean leaves under greenhouse and field conditions (Chen et al., 2013). Depending on water availability, P. aeruginosa and P. syringae catabolize exogenously supplied QAs as carbon and nitrogen sources or accumulate them as osmoprotectants (Chen et al., 2013; Wargo, 2013b). Our ongoing work in P. synxantha 2–79 unraveled similar physiological responses and demonstrated that QA transporters function differentially and redundantly in the uptake of quaternary amines as nutrients (Pablo and Mavrodi, unpublished). In contrast, under water stress, the QAs choline, betaine, and carnitine are accumulated preferentially for osmoprotection. Under drought stress, a 2–79 mutant devoid of all known QA transporters was less competitive in the colonization of the Brachypodium rhizosphere than its wild-type parental strain. Interestingly, our metabolomic profiling of root exudates also revealed proline, glutamine, and hydroxyectoine. These metabolites act as compatible solutes in different groups of microorganisms (Yancey et al., 1982; Empadinhas and da Costa, 2008), suggesting an important role of root exudates in the ability of Pseudomonas to persist in the rhizosphere of drought-stressed plants.
Statements
Data availability statement
Sequences generated in this project were deposited under NCBI BioProject accession numbers PRJNA439743 through PRJNA439790.
Author contributions
DM, OM, and LT conceived the research project. OM and JM collected root exudates. OM and DM cultured strains and extracted total RNA. AB and DG performed metabolomic analysis of root exudates. DM, JP, and AF analyzed RNA-seq data. LE, KH, and IP conducted Biolog analyses. DM, AF, OM, DW, and LT wrote the manuscript. All authors contributed to the manuscript revision.
Funding
This study was funded by NSF grant IOS-1656872 and by an award from the DOE Joint Genome Institute’s Community Science Program. The authors also acknowledge support from Australian Research Council Discovery grant (DP160103746) and Mississippi INBRE, funded by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant P20GM103476.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.651282/full#supplementary-material
Footnotes
References
1
AbayakoonP.LingfordJ. P.JinY.BengtC.DaviesG. J.YaoS.et al (2018). Discovery and characterization of a sulfoquinovose mutarotase using kinetic analysis at equilibrium by exchange spectroscopy.Biochem. J.4751371–1383. 10.1042/bcj20170947
2
AndrewsS. C.RobinsonA. K.Rodriguez-QuinonesF. (2003). Bacterial iron homeostasis.FEMS Microbiol. Rev.27215–237. 10.1016/s0168-6445(03)00055-x
3
ArkinA. P.CottinghamR. W.HenryC. S.HarrisN. L.StevensR. L.MaslovS.et al (2018). KBase: the United States department of energy systems biology knowledgebase.Nat. Biotechnol.36566–569.
4
AutryA. R.FitzgeraldJ. W. (1990). Sulfonate S: a major form of forest soil organic sulfur.Biol. Fertil. Soils1050–56.
5
BabickiS.ArndtD.MarcuA.LiangY.GrantJ. R.MaciejewskiA.et al (2016). Heatmapper: web-enabled heat mapping for all.Nucleic Acids Res.44W147–W153.
6
BadriD. V.VivancoJ. M. (2009). Regulation and function of root exudates.Plant Cell Environ.32666–681. 10.1111/j.1365-3040.2009.01926.x
7
BadriD. V.WeirT. L.Van Der LelieD.VivancoJ. M. (2009). Rhizosphere chemical dialogues: plant-microbe interactions.Curr. Opin. Biotechnol.20642–650. 10.1016/j.copbio.2009.09.014
8
BaisH. P.PrithivirajB.JhaA. K.AusubelF. M.VivancoJ. M. (2005). Mediation of pathogen resistance by exudation of antimicrobials from roots.Nature434217–221. 10.1038/nature03356
9
BaisH. P.WeirT. L.PerryL. G.GilroyS.VivancoJ. M. (2006). The role of root exudates in rhizosphere interations with plants and other organisms.Annu. Rev. Plant Biol.57233–266. 10.1146/annurev.arplant.57.032905.105159
10
BangeraM. G.ThomashowL. S. (1996). Characterization of a genomic locus required for synthesis of the antibiotic 2,4-diacetylphloroglucinol by the biological control agent Pseudomonas fluorescens Q2-87.Mol. Plant Microbe Interact.983–90.
11
BarretM.Frey-KlettP.Guillerm-ErckelboudtA. Y.BoutinM.GuernecG.SarniguetA. (2009). Effect of wheat roots infected with the pathogenic fungus Gaeumannomyces graminis var. tritici on gene expression of the biocontrol bacterium Pseudomonas fluorescens Pf29Arp.Mol. Plant-Microbe Interact.221611–1623. 10.1094/mpmi-22-12-1611
12
BarrettE. L.SolanesR. E.TangJ. S.PalleroniN. J. (1986). Pseudomonas fluorescens biovar V: its resolution into distinct component groups and the relationship of these groups to other P. fluorescens biovars, to P. putida, and to psychrotrophic pseudomonads associated with food spoilage.J. Gen. Microbiol.1322709–2721. 10.1099/00221287-132-10-2709
13
BevanM. W.GarvinD. F.VogelJ. P. (2010). Brachypodium distachyon genomics for sustainable food and fuel production.Curr. Opin. Biotechnol.21211–217. 10.1016/j.copbio.2010.03.006
14
BoncompagniE.OsterasM.PoggiM. C.Le RudulierD. (1999). Occurrence of choline and glycine betaine uptake and metabolism in the family Rhizobiaceae and their roles in osmoprotection.Appl. Environ. Microbiol.652072–2077. 10.1128/aem.65.5.2072-2077.1999
15
BondarczukK.Piotrowska-SegetZ. (2013). Molecular basis of active copper resistance mechanisms in Gram-negative bacteria.Cell Biol. Toxicol.29397–405. 10.1007/s10565-013-9262-1
16
BrkljacicJ.GrotewoldE.SchollR.MocklerT.GarvinD. F.VainP.et al (2011). Brachypodium as a model for the grasses: today and the future.Plant Physiol.1573–13.
17
Camacho-CarvajalM. M. (2001). Molecular Characterization of the Roles of Type 4 pili, NDH-I and PyrR in Rhizosphere Colonization of Pseudomonas fluorescens WCS365. Dissertation, University of Leiden, Leiden.
18
ChavarriaM.Goni-MorenoA.De LorenzoV.NikelP. I. (2016). A metabolic widget adjusts the phosphoenolpyruvate-dependent fructose influx in Pseudomonas putida.mSystems1:e00154-16.
19
ChenC.LiS.McKeeverD. R.BeattieG. A. (2013). The widespread plant-colonizing bacterial species Pseudomonas syringae detects and exploits an extracellular pool of choline in hosts.Plant J.75891–902. 10.1111/tpj.12262
20
ChenX.ZhangY.ZhangZ.ZhaoY.SunC.YangM.et al (2018). PGAweb: a web server for bacterial pan-genome analysis.Front. Microbiol.9:1910. 10.3389/fmicb.2018.01910
21
ChenY. T.WangY.YehK. C. (2017). Role of root exudates in metal acquisition and tolerance.Curr. Opin. Plant Biol.3966–72. 10.1016/j.pbi.2017.06.004
22
ColeB. J.FeltcherM. E.WatersR. J.WetmoreK. M.MucynT. S.RyanE. M.et al (2017). Genome-wide identification of bacterial plant colonization genes.PLoS Biol.15:e2002860. 10.1371/journal.pbio.2002860
23
ConesaA.GotzS. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics.Int. J. Plant Genomics2008:619832.
24
CurlE. A.TrueloveB. (1986). The Rhizosphere.Berlin: Springer-Verlag.
25
DarziY.LetunicI.BorkP.YamadaT. (2018). iPath3.0: interactive pathways explorer v3.Nucleic Acids Res.46W510–W513.
26
de WeertS.DekkersL.BitterW.TuinmanS.WijfjesA.Van BoxtelR.et al (2006). The two-component colR/S system of Pseudomonas fluorescens WCS365 plays a role in rhizosphere competence through maintaining the structure and function of the outer membrane.FEMS Microbiol. Ecol.58205–213. 10.1111/j.1574-6941.2006.00158.x
27
de WeertS.VermeirenH.MuldersI. H.KuiperI.HendrickxN.BloembergG. V.et al (2002). Flagella-driven chemotaxis towards exudate components is an important trait for tomato root colonization by Pseudomonas fluorescens.Mol. Plant Microbe Interact.151173–1180. 10.1094/mpmi.2002.15.11.1173
28
de WerraP.HuserA.TabacchiR.KeelC.MaurhoferM. (2011). Plant- and microbe-derived compounds affect the expression of genes encoding antifungal compounds in a pseudomonad with biocontrol activity.Appl. Environ. Microbiol.772807–2812. 10.1128/aem.01760-10
29
DekkersL. C.PhoelichC. C.Van Der FitsL.LugtenbergB. J. (1998). A site-specific recombinase is required for competitive root colonization by Pseudomonas fluorescens WCS365.Proc. Natl. Acad. Sci. U.S.A.957051–7056. 10.1073/pnas.95.12.7051
30
De-la-PenaC.LeiZ.WatsonB. S.SumnerL. W.VivancoJ. M. (2008). Root-microbe communication through protein secretion.J. Biol. Chem.28325247–25255. 10.1074/jbc.m801967200
31
EmpadinhasN.da CostaM. S. (2008). Osmoadaptation mechanisms in prokaryotes: distribution of compatible solutes.Int. Microbiol.11151–161.
32
Espinosa-UrgelM.SalidoA.RamosJ. L. (2000). Genetic analysis of functions involved in adhesion of Pseudomonas putida to seeds.J. Bacteriol.1822363–2369. 10.1128/jb.182.9.2363-2369.2000
33
FuquaC. (2010). Passing the baton between laps: adhesion and cohesion in Pseudomonas putida biofilms.Mol. Microbiol.77533–536. 10.1111/j.1365-2958.2010.07250.x
34
GalvaoT. C.De LorenzoV.CanovasD. (2006). Uncoupling of choline-O-sulphate utilization from osmoprotection in Pseudomonas putida.Mol. Microbiol.621643–1654. 10.1111/j.1365-2958.2006.05488.x
35
Garrido-SanzD.Meier-KolthoffJ. P.GökerM.MartínM.RivillaR.Redondo-NietoM. (2016). Genomic and genetic diversity within the Pseudomonas fluorescens complex.PLoS One11:e0150183. 10.1371/journal.pone.0150183
36
HalversonL. J.FirestoneM. K. (2000). Differential effects of permeating and nonpermeating solutes on the fatty acid composition of Pseudomonas putida.Appl. Environ. Microbiol.662414–2421. 10.1128/aem.66.6.2414-2421.2000
37
HampelK. J.LabauveA. E.MeadowsJ. A.FitzsimmonsL. F.NockA. M.WargoM. J. (2014). Characterization of the GbdR regulon in Pseudomonas aeruginosa.J. Bacteriol.1967–15. 10.1128/jb.01055-13
38
HarwoodC. S.ParalesR. E. (1996). The beta-ketoadipate pathway and the biology of self-identity.Annu. Rev. Microbiol.50553–590. 10.1146/annurev.micro.50.1.553
39
HesseC.SchulzF.BullC. T.ShafferB. T.YanQ.ShapiroN.et al (2018). Genome-based evolutionary history of Pseudomonas spp.Environ. Microbiol.202142–2159.
40
HinsaS. M.Espinosa-UrgelM.RamosJ. L.O’TooleG. A. (2003). Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein.Mol. Microbiol.49905–918. 10.1046/j.1365-2958.2003.03615.x
41
HojbergO.SchniderU.WintelerH. V.SorensenJ.HaasD. (1999). Oxygen-sensing reporter strain of Pseudomonas fluorescens for monitoring the distribution of low-oxygen habitats in soil.Appl. Environ. Microbiol.654085–4093. 10.1128/aem.65.9.4085-4093.1999
42
HongS. Y.ParkJ. H.ChoS. H.YangM. S.ParkC. M. (2011). Phenological growth stages of Brachypodium distachyon: codification and description.Weed Res.51612–620. 10.1111/j.1365-3180.2011.00877.x
43
HowellC. R.StipanovicR. D. (1980). Suppression of Pythium ultimum induced damping-off of cotton seedlings by Pseudomonas fluorescens and its antibiotic pyoluteorin.Phytopathology70712–715. 10.1094/phyto-70-712
44
HummerjohannJ.KuttelE.QuadroniM.RagallerJ.LeisingerT.KerteszM. A. (1998). Regulation of the sulfate starvation response in Pseudomonas aeruginosa: role of cysteine biosynthetic intermediates.Microbiology1441375–1386. 10.1099/00221287-144-5-1375
45
KabbadjA.MakoudiB.MouradiM.PaulyN.FrendoP.GhoulamC. (2017). Physiological and biochemical responses involved in water deficit tolerance of nitrogen-fixing Vicia faba.PLoS One12:e0190284. 10.1371/journal.pone.0190284
46
KamilovaF.ValidovS.AzarovaT.MuldersI.LugtenbergB. (2005). Enrichment for enhanced competitive plant root tip colonizers selects for a new class of biocontrol bacteria.Environ. Microbiol.71809–1817. 10.1111/j.1462-2920.2005.00889.x
47
KanehisaM.ArakiM.GotoS.HattoriM.HirakawaM.ItohM.et al (2008). KEGG for linking genomes to life and the environment.Nucleic Acids Res.36D480–D484.
48
KatohK.StandleyD. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability.Mol. Biol. Evol.30772–780. 10.1093/molbev/mst010
49
KawasakiA.DonnS.RyanP. R.MathesiusU.DevillaR.JonesA.et al (2016). Microbiome and exudates of the root and rhizosphere of Brachypodium distachyon, a model for wheat.PLoS One11:e0164533. 10.1371/journal.pone.0164533
50
KindT.WohlgemuthG.LeeD. Y.LuY.PalazogluM.ShahbazS.et al (2009). FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry.Anal. Chem.8110038–10048. 10.1021/ac9019522
51
KingE. O.WardM. K.RaneyD. E. (1954). Two simple media for the demonstration of pyocyanin and fluorescein.J. Lab. Clin. Med.44301–307.
52
KlonowskaA.MelkonianR.MicheL.TisseyreP.MoulinL. (2018). Transcriptomic profiling of Burkholderia phymatum STM815, Cupriavidus taiwanensis LMG19424 and Rhizobium mesoamericanum STM3625 in response to Mimosa pudica root exudates illuminates the molecular basis of their nodulation competitiveness and symbiotic evolutionary history.BMC Genomics19:105. 10.1186/s12864-018-4487-2
53
KobayashiT.NishizawaN. K. (2012). Iron uptake, translocation, and regulation in higher plants.Annu. Rev. Plant Biol.63131–152. 10.1146/annurev-arplant-042811-105522
54
KohlerP. R.ChoongE. L.RossbachS. (2011). The RpiR-like repressor IolR regulates inositol catabolism in Sinorhizobium meliloti.J. Bacteriol.1935155–5163. 10.1128/jb.05371-11
55
LeeD.FiehnO. (2008). High quality metabolomic data for Chlamydomonas reinhardtii.Plant Meth.4:7. 10.1186/1746-4811-4-7
56
LetunicI.BorkP. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees.Nucleic Acids Res.44W242–W245.
57
LiL.StoeckertC. J.Jr.RoosD. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes.Genome Res.132178–2189. 10.1101/gr.1224503
58
LiaoY.SmythG. K.ShiW. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features.Bioinformatics30923–930. 10.1093/bioinformatics/btt656
59
LiuY.RaineyP. B.ZhangX. X. (2015). Molecular mechanisms of xylose utilization by Pseudomonas fluorescens: overlapping genetic responses to xylose, xylulose, ribose and mannitol.Mol. Microbiol.98553–570. 10.1111/mmi.13142
60
LoperJ. E.HassanK. A.MavrodiD. V.DavisE. W.LimC. K.ShafferB. T.et al (2012). Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions.PLoS Genet.8:e1002784. 10.1371/journal.pgen.1002784
61
LoveM. I.HuberW.AndersS. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.Genome Biol.15:550.
62
LugtenbergB.KamilovaF. (2009). Plant-growth-promoting rhizobacteria.Annu. Rev. Microbiol.63541–556.
63
LugtenbergB. J.DekkersL.BloembergG. V. (2001). Molecular determinants of rhizosphere colonization by Pseudomonas.Annu. Rev. Phytopathol.39461–490.
64
LynchJ. M. (1990). “Microbial metabolites,” in The Rhizosphere, ed.LynchJ. M. (Chichester: John Wiley & Sons), 177–206.
65
MarkG. L.DowJ. M.KielyP. D.HigginsH.HaynesJ.BaysseC.et al (2005). Transcriptome profiling of bacterial responses to root exudates identifies genes involved in microbe-plant interactions.Proc. Natl. Acad. Sci. U.S.A.10217454–17459. 10.1073/pnas.0506407102
66
MarkowitzV. M.ChenI. M.PalaniappanK.ChuK.SzetoE.GrechkinY.et al (2012). IMG: the integrated microbial genomes database and comparative analysis system.Nucleic Acids Res.40D115–D122.
67
Martinez-GilM.Yousef-CoronadoF.Espinosa-UrgelM. (2010). LapF, the second largest Pseudomonas putida protein, contributes to plant root colonization and determines biofilm architecture.Mol. Microbiol.77549–561. 10.1111/j.1365-2958.2010.07249.x
68
MatillaM. A.Espinosa-UrgelM.Rodriguez-HervaJ. J.RamosJ. L.Ramos-GonzalezM. I. (2007). Genomic analysis reveals the major driving forces of bacterial life in the rhizosphere.Genome Biol.8:R179.
69
MillerK. J.WoodJ. M. (1996). Osmoadaptation by rhizosphere bacteria.Annu. Rev. Microbiol.50101–136. 10.1146/annurev.micro.50.1.101
70
MooreE. R. B.TindallB. J.Martins Dos SantosV. A. P.PieperD. H.RamosJ.-L.PalleroniN. J. (2006). “Nonmedical Pseudomonas,” in The Prokaryotes, edsDworkinM.FalkowS.RosenbergE.SchleiferK.-H.StackebrandtE. (New York, NY: Springer), 646–703.
71
MuletM.LalucatJ.Garcia-ValdesE. (2010). DNA sequence-based analysis of the Pseudomonas species.Environ. Microbiol.121513–1530.
72
NaylorD.DeGraafS.PurdomE.Coleman-DerrD. (2017). Drought and host selection influence bacterial community dynamics in the grass root microbiome.ISME J.112691–2704. 10.1038/ismej.2017.118
73
NguyenC. (2003). Rhizodeposition of organic C by plants: mechanisms and controls.Agronomie23375–396. 10.1051/agro:2003011
74
NielsenL.LiX.HalversonL. J. (2011). Cell-cell and cell-surface interactions mediated by cellulose and a novel exopolysaccharide contribute to Pseudomonas putida biofilm formation and fitness under water-limiting conditions.Environ. Microbiol.131342–1356. 10.1111/j.1462-2920.2011.02432.x
75
ParejkoJ. A.MavrodiD. V.MavrodiO. V.WellerD. M.ThomashowL. S. (2012). Population structure and diversity of phenazine-1-carboxylic acid producing fluorescent Pseudomonas spp. from dryland cereal fields of central Washington state (USA)Microb. Ecol.63226–241. 10.1007/s00248-012-0015-0
76
PhillipsD. A.FoxT. C.KingM. D.BhuvaneswariT. V.TeuberL. R. (2004). Microbial products trigger amino acid exudation from plant roots.Plant Physiol.1362887–2894. 10.1104/pp.104.044222
77
RaaijmakersJ. M.VandersluisI.KosterM.BakkerP. A. H. M.WeisbeekP. J.SchippersB. (1995). Utilization of heterologous siderophores and rhizosphere competence of fluorescent Pseudomonas spp.Can. J. Microbiol.41126–135. 10.1139/m95-017
78
RaaijmakersJ. M.WellerD. M. (1998). Natural plant protection by 2,4-diacetylphloroglucinol-producing Pseudomonas spp. in take-all decline soils.Mol. Plant Microbe Interact.11144–152. 10.1094/mpmi.1998.11.2.144
79
RahmanM. M.AndbergM.ThangarajS. K.ParkkinenT.PenttilaM.JanisJ.et al (2017). The crystal structure of a bacterial L-arabinonate dehydratase contains a [2Fe-2S] cluster.ACS Chem. Biol.121919–1927. 10.1021/acschembio.7b00304
80
Ramos-GonzalezM. I.CamposM. J.RamosJ. L. (2005). Analysis of Pseudomonas putida KT2440 gene expression in the maize rhizosphere: in vivo expression technology capture and identification of root-activated promoters.J. Bacteriol.1874033–4041. 10.1128/jb.187.12.4033-4041.2005
81
Reinhold-HurekB.BungerW.BurbanoC. S.SabaleM.HurekT. (2015). Roots shaping their microbiome: global hotspots for microbial activity.Annu. Rev. Phytopathol.53403–424. 10.1146/annurev-phyto-082712-102342
82
Sanchez-ContrerasM.MartinM.VillacierosM.O’GaraF.BonillaI.RivillaR. (2002). Phenotypic selection and phase variation occur during alfalfa root colonization by Pseudomonas fluorescens F113.J. Bacteriol.1841587–1596. 10.1128/jb.184.6.1587-1596.2002
83
SarniguetA.KrausJ.HenkelsM. D.MuehlchenA. M.LoperJ. E. (1995). The sigma factor σs affects antibiotic production and biological control activity of Pseudomonas fluorescens Pf-5.Proc. Natl. Acad. Sci. U.S.A.9212255–12259. 10.1073/pnas.92.26.12255
84
Schnider-KeelU.LejbolleK. B.BaehlerE.HaasD.KeelC. (2001). The sigma factor AlgU (AlgT) controls exopolysaccharide production and tolerance towards desiccation and osmotic stress in the biocontrol agent Pseudomonas fluorescens CHA0.Appl. Environ. Microbiol.675683–5693. 10.1128/aem.67.12.5683-5693.2001
85
SchrothM. N.HildebrandD. C.PanopoulosN. J. (2006). “Phytopathogenic pseudomonads and related plant-associated pseudomonads,” in The Prokaryotes, edsDworkinM.FalkowS.RosenbergE.SchleiferK.-H.StackebrandtE. (New York, NY: Springer), 714–740. 10.1007/0-387-30746-x_23
86
SchwartzC. J.DoyleM. R.ManzanedaA. J.ReyP. J.Mitchell-OldsT.AmasinoR. M. (2010). Natural variation of flowering time and vernalization responsiveness in Brachypodium distachyon.Bioenergy Res.338–46. 10.1007/s12155-009-9069-3
87
SilbyM. W.Cerdeno-TarragaA. M.VernikosG. S.GiddensS. R.JacksonR. W.PrestonG. M.et al (2009). Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens.Genome Biol.10:R51.
88
SilbyM. W.LevyS. B. (2004). Use of in vivo expression technology to identify genes important in growth and survival of Pseudomonas fluorescens Pf0-1 in soil: Discovery of expressed sequences with novel genetic organization.J. Bacteriol.1867411–7419. 10.1128/jb.186.21.7411-7419.2004
89
SimonsM.Van Der BijA. J.BrandI.De WegerL. A.WijffelmanC. A.LugtenbergB. J. (1996). Gnotobiotic system for studying rhizosphere colonization by plant growth-promoting Pseudomonas bacteria.Mol. Plant Microbe Interact.9600–607. 10.1094/mpmi-9-0600
90
SimonsM.Van Der BijA. J.BrandJ.De WegerL. A.WijffelmanD. A.LugtenbergB. J. J. (1997). Amino acid synthesis is necessary for tomato root colonization by Pseudomonas fluorescens strain WCS365.Mol. Plant Microbe Interact.10102–106. 10.1094/mpmi.1997.10.1.102
91
SmibertR. M.KreigN. R. (1994). “Phenotypic characterization,” in Methods for General and Molecular Bacteriology, edsGerhardtP.MurrayR. G. E.WoodW. A.KreigN. R. (Washington, DC: American Society of Microbiology), 607–654.
92
ThomashowL. S.WellerD. M. (1988). Role of a phenazine antibiotic from Pseudomonas fluorescens in biological control of Gaeumannomyces graminis var. tritici.J. Bacteriol.1703499–3508. 10.1128/jb.170.8.3499-3508.1988
93
ThomashowL. S.WellerD. M.BonsallR. F.PiersonL. S. (1990). Production of the antibiotic phenazine-1-carboxylic acid by fluorescent Pseudomonas species in the rhizosphere of wheat.Appl. Environ. Microbiol.56908–912. 10.1128/aem.56.4.908-912.1990
94
TylerL.FangelJ. U.FagerstromA. D.SteinwandM. A.RaabT. K.WillatsW. G.et al (2014). Selection and phenotypic characterization of a core collection of Brachypodium distachyon inbred lines.BMC Plant Biol.14:25. 10.1186/1471-2229-14-25
95
VacheronJ.DesbrossesG.BouffaudM. L.TouraineB.Moenne-LoccozY.MullerD.et al (2013). Plant growth-promoting rhizobacteria and root system functioning.Front. Plant Sci.4:356. 10.3389/fpls.2013.00356
96
van den BroekD.BloembergG. V.LugtenbergB. (2005). The role of phenotypic variation in rhizosphere Pseudomonas bacteria.Environ. Microbiol.71686–1697. 10.1111/j.1462-2920.2005.00912.x
97
van VeenJ. A.Van OverbeekL. S.Van ElsasJ. D. (1997). Fate and activity of microorganisms introduced into soil.Microbiol. Mol. Biol. Rev.61121–135. 10.1128/.61.2.121-135.1997
98
WalkerT. S.BaisH. P.HalliganK. M.StermitzF. R.VivancoJ. M. (2003). Metabolic profiling of root exudates of Arabidopsis thaliana.J. Agric. Food Chem.512548–2554.
99
WargoM. J. (2013a). Choline catabolism to glycine betaine contributes to Pseudomonas aeruginosa survival during murine lung infection.PLoS One8:e56850. 10.1371/journal.pone.0056850
100
WargoM. J. (2013b). Homeostasis and catabolism of choline and glycine betaine: lessons from Pseudomonas aeruginosa.Appl. Environ. Microbiol.792112–2120.
101
WhippsJ. M. (1990). “Carbon economy,” in The Rhizosphere, ed.LynchJ. M. (Chichester: John Wiley & Sons), 59–97.
102
WinsorG. L.Van RossumT.LoR.KhairaB.WhitesideM. D.HancockR. E.et al (2009). Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes.Nucleic Acids Res.37D483–D488.
103
YahrT. L.ParsekM. R. (2006). “Pseudomonas aeruginosa,” in The Prokaryotes, edsDworkinM.FalkowS.RosenbergE.SchleiferK.-H.StackebrandtE. (New York, NY: Springer), 704–713.
104
YanceyP. H.ClarkM. E.HandS. C.BowlusR. D.SomeroG. N. (1982). Living with water stress: evolution of osmolyte systems.Science2171214–1222.
105
YeJ.ZhangY.CuiH.LiuJ.WuY.ChengY.et al (2018). WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update.Nucleic Acids Res.46W71–W75.
106
YoshidaK.YamaguchiM.MorinagaT.KineharaM.IkeuchiM.AshidaH.et al (2008). myo-Inositol catabolism in Bacillus subtilis.J. Biol. Chem.28310415–10424.
107
Yousef-CoronadoF.TraviesoM. L.Espinosa-UrgelM. (2008). Different, overlapping mechanisms for colonization of abiotic and plant surfaces by Pseudomonas putida.FEMS Microbiol. Lett.288118–124.
108
ZboralskiA.FilionM. (2020). Genetic factors involved in rhizosphere colonization by phytobeneficial Pseudomonas spp.Comput. Struct. Biotechnol. J.183539–3554.
109
ZollaG.BakkerM. G.BadriD. V.ChaparroJ. M.SheflinA. M.ManterD. K.et al (2013). “Understanding root-microbiome interactions,” in Molecular Microbial Ecology of the Rhizosphere, ed.De BruijnF. J. (Hoboken, NJ: John Wiley & Sons), 745–754.
Summary
Keywords
Pseudomonas, Brachypodium, rhizosphere, root exudates, transcriptome
Citation
Mavrodi OV, McWilliams JR, Peter JO, Berim A, Hassan KA, Elbourne LDH, LeTourneau MK, Gang DR, Paulsen IT, Weller DM, Thomashow LS, Flynt AS and Mavrodi DV (2021) Root Exudates Alter the Expression of Diverse Metabolic, Transport, Regulatory, and Stress Response Genes in Rhizosphere Pseudomonas. Front. Microbiol. 12:651282. doi: 10.3389/fmicb.2021.651282
Received
09 January 2021
Accepted
08 March 2021
Published
14 April 2021
Volume
12 - 2021
Edited by
Barbara Pivato, Institut National de Recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), France
Reviewed by
Cara Helene Haney, The University of British Columbia, Canada; Xingang Zhou, Northeast Agricultural University, China
Updates
Copyright
© 2021 Mavrodi, McWilliams, Peter, Berim, Hassan, Elbourne, LeTourneau, Gang, Paulsen, Weller, Thomashow, Flynt and Mavrodi.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dmitri Mavrodi, dmitri.mavrodi@usm.eduAlex Flynt, alex.flynt@usm.edu
†These authors have contributed equally to this work
This article was submitted to Microbe and Virus Interactions with Plants, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.