 Paula Gómez1
Paula Gómez1 Laura Ruiz-Ripa1Rosa Fernández-Fernández1Haythem Gharsa2Karim Ben Slama2
Laura Ruiz-Ripa1Rosa Fernández-Fernández1Haythem Gharsa2Karim Ben Slama2 Ursula Höfle3Myriam Zarazaga1
Ursula Höfle3Myriam Zarazaga1 Mark A. Holmes4
Mark A. Holmes4 Carmen Torres1*
Carmen Torres1*- 1Area of Biochemistry and Molecular Biology, OneHealth-UR Research Group, University of La Rioja, Logroño, Spain
- 2Laboratoire des Microorganismes et Biomolécules Actives, Faculté des Sciences de Tunis, Université de Tunis El Manar, Tunis, Tunisia
- 3Health and Biotechnology SaBio Research Group, Instituto de Investigación en Recursos Cinegéticos IREC (CSIC-UCLM-JCCM), Ciudad Real, Spain
- 4Department of Veterinary Medicine, University of Cambridge, Cambridge, United Kingdom
Most methicillin resistant Staphylococcus aureus (MRSA) isolates harboring mecC gene belong to clonal complex CC130. This lineage has traditionally been regarded as animal-associated as it lacks the human specific immune evasion cluster (IEC), and has been recovered from a broad range of animal hosts. Nevertheless, sporadic mecC-MRSA human infections have been reported, with evidence of zoonotic transmission in some cases. The objective of this study was to investigate the whole-genome sequences of 18 S. aureus CC130 isolates [13 methicillin-resistant (mecC-MRSA) and five methicillin-susceptible (MSSA)] from different sequences types, obtained from a variety of host species and origins (human, livestock, wild birds and mammals, and water), and from different geographic locations, in order to identify characteristic markers and genomic features. Antibiotic resistance genes found among MRSA-CC130 were those associated with the SSCmecXI element. Most MRSA-CC130 strains carried a similar virulence gene profile. Additionally, six MRSA-CC130 possessed scn-sak and one MSSA-ST130 had lukMF’. The MSSA-ST700 strains were most divergent in their resistance and virulence genes. The pan-genome analysis showed that 29 genes were present solely in MRSA-CC130 (associated with SCCmecXI) and 21 among MSSA-CC130 isolates (associated with phages). The SCCmecXI, PBP3, GdpP, and AcrB were identical at the amino acid level in all strains, but some differences were found in PBP1, PBP2, PBP4, and YjbH proteins. An examination of the host markers showed that the 3’ region of the bacteriophage φ3 was nearly identical to the reference sequence. Truncated hlb gene was also found in scn-negative strains (two of them carrying sak-type gene). The dtlB gene of wild rabbit isolates included novel mutations. The vwbp gene was found in the three MSSA-ST700 strains from small ruminants and in one MSSA-ST130 from a red deer; these strains also carried a scn-type gene, different from the human and equine variants. Finally, a phylogenetic analysis showed that the three MSSA-ST700 strains and the two MSSA-ST130 strains cluster separately from the remaining MRSA-CC130 strains with the etD2 gene as marker for the main lineage. The presence of the human IEC cluster in some mecC-MRSA-CC130 strains suggests that these isolates may have had a human origin.
Introduction
Staphylococcus aureus is a common colonizer of the nasopharynx and skin of animals and humans; however, it is also a versatile opportunistic pathogen causing a wide variety of diseases from mild skin problems to life-threatening bacteraemias. The situation may be complicated when infections are caused by methicillin-resistant S. aureus (MRSA) isolates. Currently, the expression of mecA gene, as well as of other mec homolog genes, mecC and mecB, have been described in S. aureus conferring methicillin resistance (Becker et al., 2018).
The mecC-gene has been found in several MRSA lineages, mainly associated with animals, such as CC130, CC49, ST425, CC599, and CC1943. The ruminant associated CC130 is the most commonly found mecC lineage (Paterson et al., 2014a; Zarazaga et al., 2018). mecC-MRSA-CC130 was first described in cattle and in humans in the United Kingdom, Denmark, and Ireland (García-Álvarez et al., 2011; Shore et al., 2011). Since then, this lineage has been detected in diverse hosts in many European countries, with cattle and wildlife (including free grazing domesticated animals) being the most common hosts (Zarazaga et al., 2018). The prevalence of mecC-MRSA in people seems to be low (Paterson et al., 2014a, b; Lozano et al., 2020), however, the zoonotic transmission from livestock to people has been reported (Harrison et al., 2013), as well as its ability to cause disease (Petersen et al., 2013). This mecC-MRSA-CC130 lineage seems to be susceptible to many non-β-lactam agents and lacks major human virulence factors (Cuny et al., 2011; Monecke et al., 2013; Paterson et al., 2014a). However, they are carriers of a novel allele of exfoliative toxin gene (named etd2), which could explain the wide variety of hosts (Monecke et al., 2013). Adaptation of S. aureus to particular host species can be associated with mobile genetic elements (MGEs) or chromosomal mutations. In particular, the genes of the human specific immune evasion cluster (IEC) are considered to be a marker indicating some degree of human host adaptation. This IEC system is found in seven different configurations (types A–G) depending on the combination of five genes (scn, chp, sak, sea/sep); the scn gene (encodes a staphylococcal complement inhibitor) is included in all IEC types, and is often used as a marker of IEC-positive isolates, and is functionally essential (van Wamel et al., 2006). None of the mecC-MRSA reported strains harbored the scn gene (essential for the IEC system) (Lozano et al., 2020), with the exception of a few isolates belonging to ST1945, ST1581, and ST1583 previously described by our group from wildlife and extensively farmed domestic animals (Gómez et al., 2014, 2015; Ruiz-Ripa et al., 2019) and one ST1945 MRSA strain from a human sample (Harrison et al., 2017); it is worth noting that all these IEC-positive isolates were of type-E (carrying the scn and sak genes).
The methicillin-susceptible S. aureus (MSSA) isolates of the CC130 clonal complex are commonly found in cattle and are an important cause of disease (Monecke et al., 2016). The mecA gene has never been found in isolates belonging to the CC130 clonal complex and S. aureus-CC130 was initially described as a MSSA of animals from Europe and Africa (Smith et al., 2014). The ST700 lineage is part of CC130 by definition, as a single locus variant of ST130 (tsi allele different between them). MSSA-ST700 isolates are frequently found in Italian sheep populations (Azara et al., 2017; Vitale et al., 2018) and ST700 and some of its single locus variants (CC700) may be considered as a distinct, or separate, lineage due to its independent evolution and different epidemiology (Smith et al., 2014).
Studies of the intrinsic Penicillin-Binding-Proteins (PBPs) of S. aureus have shown that PBPs may contain mutations that affect β-lactam resistance, as highlighted by the case of a PBP4 capable of conferring high-level and broad-spectrum resistance to β-lactams, comparable to that provided by PBP2a (Chan et al., 2016).
In order to better understand the genetic characteristics of S. aureus CC130, the objective of this study was to analyze data from whole genome sequencing (WGS) of a collection of CC130 S. aureus strains (MRSA and MSSA) belonging to different sequences types, obtained from various host species, and from different geographic locations, in order to identify distinctive markers and genomic features of public health relevance.
Materials and Methods
Strains Included in the Study
Eighteen S. aureus strains of the clonal complex CC130 were included in this study for genomic comparison. These strains were as follows: (1) 13 MRSA, carrying the mecC gene, and belonging to the sequence types ST130, ST1945, ST3061, ST1571, ST1581, and ST1583; (2) two MSSA-ST130; and (3) three MSSA-ST700 (as a possible divergent CC130 lineage). These 18 MRSA-CC130, MSSA-ST130, and MSSA-ST700 strains were studied by WGS, having been collected during previous studies from different host samples: animals from extensive farms [four red deer (Cervus elaphus), two sheep (Ovis sp.), and one goat (Capra sp.)] (Gharsa et al., 2015; Gómez et al., 2015), wildlife [four magpies (Pica pica), two wild rabbits (Oryctolagus cuniculus), one wood mouse (Apodemus sylvaticus), one white stork (Ciconia ciconia), and one cinereous vulture (Aegypius monachus)] (Gómez et al., 2014, 2016; Ruiz-Ripa et al., 2019), the environment (one river water) (Gómez et al., 2017), and humans (one skin lesion of a cattle farmer)] (Benito et al., 2016). The characteristics of the included strains are indicated in Table 1.
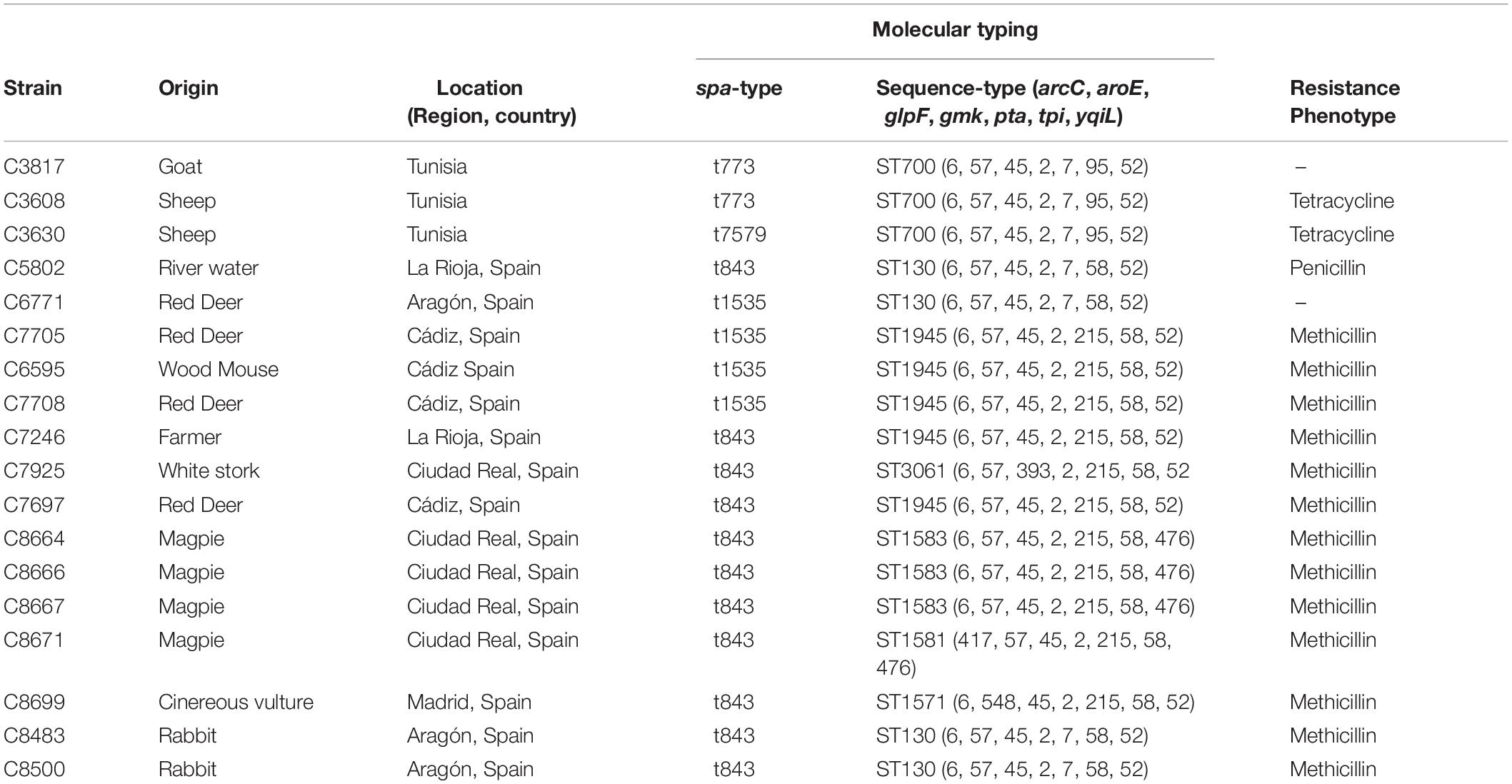
Table 1. Characteristics of the 18 S. aureus CC130 strains (13 MRSA and 5 MSSA).
Whole Genome Sequencing and Analysis of Sequences
Genomic DNA from each isolate was extracted with MasterPureTM DNA Purification Gram Positive (Cambio, United Kingdom). WGS was performed on an Illumina HiSeq 2000 using paired-end mode (100 bp). De novo assembly and initial annotation was carried out using bioinformatic tools at the Wellcome Trust Sanger Institute. Reordering of the contigs was performed by alignment against S. aureus LGA251 genome (GenBank accession number: NC_017349) using Mauve (Rissman et al., 2009). Predicted coding sequences were identified and annotated automatically using RAST (Aziz et al., 2008) and manually with Genious Prime (Biomatters, Auckland, New Zealand). The resistance and virulence genotypes as well as the presence of rep genes were studied using ResFinder, VirulenceFinder and PlasmidFinder, respectively1. In silico analysis of the presence of antimicrobial substances related genes was performed using some genome-mining tools as antiSMASH and BAGEL (de Jong et al., 2006; Blin et al., 2019). PHASTHER Search Tool was used to determine the presence of prophage sequences (Arndt et al., 2016). When the study required it, the sequences were compared using Clustal Omega2.
The pan-genome was analyzed to estimate the core genome and the accessory or variable genome using Roary (Page et al., 2015) and BLAST-Ring-Image-Generator (BRIG) was employed to obtain a visual comparison with S. aureus LGA251 genome as reference (GenBank accession number: NC_017349) (Alikhan et al., 2011). Phylogenetic trees were generated using Geneious Prime with default settings.
Results and Discussion
Whole Genome Sequencing Results
The genome data of the 13 MRSA-CC130, two MSSA-ST130 and three MSSA-ST700 strains analyzed in this study have been placed in the European nucleotide archive3, and general sequence data, with the accession numbers are shown in Supplementary Table 1.
Antimicrobial and Heavy Metal Resistance and Virulence Genotype
The resistance genotype analysis showed that all MRSA-CC130 strains contained the mecC as well as the blaZ-SCCmecXI (β-lactamase), arsB (arsenite efflux pump), and arsC (arsenate reductase) genes, which are described as being part of SCCmecXI element (Shore et al., 2011). No other resistance genes were detected among MRSA-CC130 strains, which agrees with the fully susceptible phenotype for non-β-lactams previously found in these mecC-positive strains. Among MSSA strains, three out of the five showed resistance to at least one of the antimicrobial agents tested, one MSSA-ST130 strain for penicillin (with blaZ gene) and two MSSA-ST700 strains for tetracycline [with tet(K) gene] (Table 2).
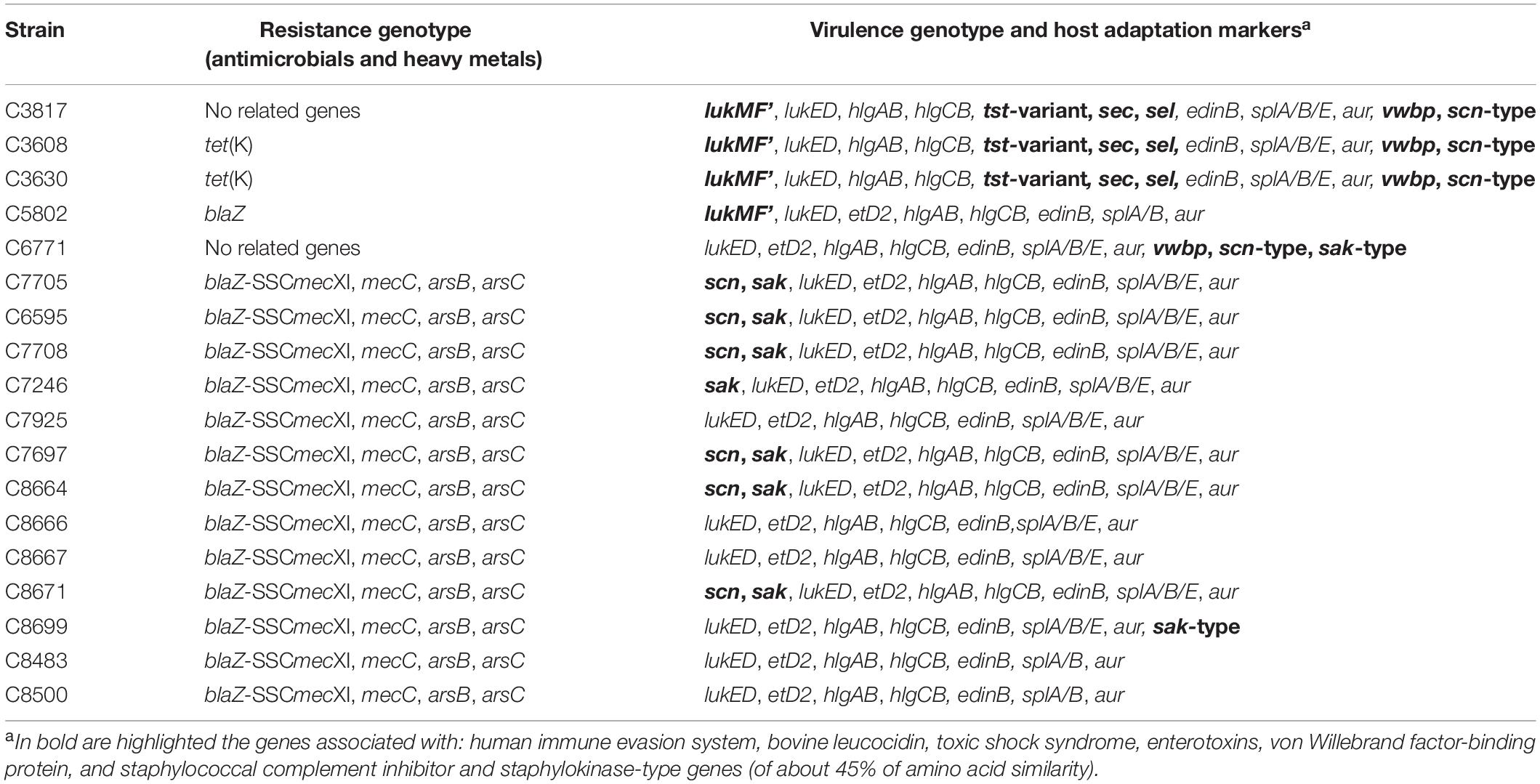
Table 2. Antimicrobial resistance and virulence genes detected in the 18 S. aureus CC130 strains included in this study.
A list of selected virulence and/or fitness genes are shown in Table 2. All the strains carried the genes: lukED, hlgAB, hlgCB, edinB, splA/B, and aur. Nevertheless, some differences were detected with respect to genes belonging to the IEC system, leucocidins, exfoliative toxins, allele variant of toxic shock syndrome toxin, enterotoxins, and immune evasion proteases. The three MSSA strains of lineage ST700 carried sec and sel genes, and also a variant of tst with an amino acid sequence closer to the tst gene found associated with bovine origin than with the one of human origin (Monecke et al., 2007); this combination of pyrogenic toxin superantigens is associated with the pathogenicity island SaPIbov (Fitzgerald et al., 2001), and has been previously described in strains from ruminants with the same ST (Luzzago et al., 2014). The ST700 strains were obtained from apparently healthy animals although a subclinical mastitis cannot be ruled out. All of them presented the tst-variant, sec, and sel virulence genes, as well as the lukMF’ gene, previously found in isolates from cases of mastitis (Schlotter et al., 2012). All our CC130 strains, except those belonging to ST700, carried the etD2 gene. The lukMF’ genes, encoding a leucocidin strongly associated with ruminants (Monecke et al., 2007), were only detected in four MSSA strains obtained from sheep and goats (MSSA-ST700) and from river water (MSSA-ST130); these data support the association of this leucocidin with a ruminant origin, and also may suggests that the strain from river water could have a bovine origin.
On the other hand, the lukED, hlgAB, hlgCB, edinB, splA/B, and aur genes were present in the 18 strains. Usually, S. aureus has up to 6 types of toxin genes in the core genome (HlgAB, HlgCB, and LukAB) (Alonzo and Torres, 2014). The combination of LukED with splA/splB genes has been detected previously among other clonal complexes (Jamrozy et al., 2012), generally being found on the genomic island νSaβ, highly conserved in some lineages (McCarthy and Lindsay, 2013). Other genes, such as aur (immune evasion proteases), edinB (exfoliative toxin) or splA/B/E (immune evasion proteases), are found highly conserved in S. aureus (Sabat et al., 2008; Munro et al., 2010; Paharik et al., 2016). Nevertheless, the splE gene was absent in three of our strains, and some authors suggested the implication of this nuclease in clinical manifestations (Stach et al., 2018). The analysis of genes encoding bacteriocins revealed the presence of the gene encoding the bacteriocin lactococcin 972 (GenBank accession number: NC_004955) in all the analyzed strains; furthermore, this gene showed in all isolates an identical genetic environment, which corresponds to the one found in the reference sequence of S. aureus LGA251.
Comparison Between the Strains
The pan-genome study showed that a total of 2,318 genes were included in all strains, 539 were in two or more strains, and 345 were unique genes of specific strains. Circular genome comparison of MSSA and MRSA strains (LGA251 as reference) showed some differences between MRSA and MSSA strains (Supplementary Figure 1). It was determined that 29 genes were present in all 13 MRSA and in none of the MSSA strains (mainly associated with SCCmecXI mobile genetic element), and 21 genes in all 5 MSSA strains and not in the MRSA (mostly associated with phages) (Supplementary Table 2). It has been reported that the core genome is largely preserved within the same lineage (McCarthy et al., 2011). In addition, we analyzed the presence of unique genes in scn-positive (n = 6) and scn-negative strains (n = 12). The scn-negative strains did not carry unique genes, however, scn-positive strains presented different genes encoding proteins associated with bacteriophages (including the human scn-IEC gene), that were not present among scn-negative strains (Supplementary Table 3).
SCCmecXI Element and Penicillin Binding Proteins (PBPs)
The structure of the SCCmecXI element in the 13 MRSA CC130 strains was compared with the one of MRSA strain M10/0061 (GenBank accession number: FR823292), used as reference. This structure seems to be highly conserved among the 13 MRSA-CC130 strains, pointing out the potential of this type of SCCmec to be transferred among S. aureus, due to the relatively small size of this mobile genetic element, approximately 30 Kb (Shore et al., 2011). In fact, it has been suggested that SCCmecXI could have originated in another species or genus, being distantly related to the other SCCmec elements and possibly SCCmecXI represent an ancestral form (Shore et al., 2011).
The results of the study of amino acid changes in PBPs and in other three proteins previously associated with β-lactam resistance (Ba et al., 2014, 2019) are shown in Table 3. Amino acid changes in our strains were included, as well as those of MRSA LGA251 using the corresponding sequences of MSSA ATCC 25923 (GenBank accession number: CP009361) as reference for all sequences, except for PBP2c. In the case of PBP2c, the sequence of MRSA LGA251 was used as reference. The PBP2c protein, encoded by the mecC gene, presented a 100% of amino acid similarity to PBP2c of MRSA LGA251. As regards the other PBPs, some amino acid changes were found, especially in PBP3 and PBP4. These amino acid changes seem to be repeated in the 18 strains, including MSSA ones, with some few exceptions (PBP1: T371I in a MSSA-ST130 water strain; PBP2: T439M in a MRSA-ST1583 magpie strain; PBP4: D28N in MSSA-ST700 strains, and A288T in two MRSA-ST130 rabbit strains). Nevertheless, differences were greater in the case of MRSA LGA251. It should be noted that previous studies indicate that mutations in PBP4 are related to increased MICs for β-lactams (Alexander et al., 2018), and a modified PBP1 had been previously associated with a reduced susceptibility in S. lugdunensis, but not in S. aureus (Kotsakis et al., 2012). Only one of the changes detected in this study, T371I in PBP1, was previously reported, combined in that case with other PBP mutations in a clinical MRSA ST1 strain lacking mec gene (Ba et al., 2014); in our case, the strain which harbored the T371I change was MSSA. In addition to PBPs, the study of GdpP, YjbH, and AcrB proteins, which could be implicated in β-Lactam resistance (Banerjee et al., 2010; Göhring et al., 2011; Ba et al., 2019), showed the same amino acid changes in all analyzed strains, with the exception of YjbH in which two changes (L95V, A83P) were detected in all strains, but in MSSA-ST700, MRSA-ST130 and MSSA-CC130 where only one change was found (L95V).
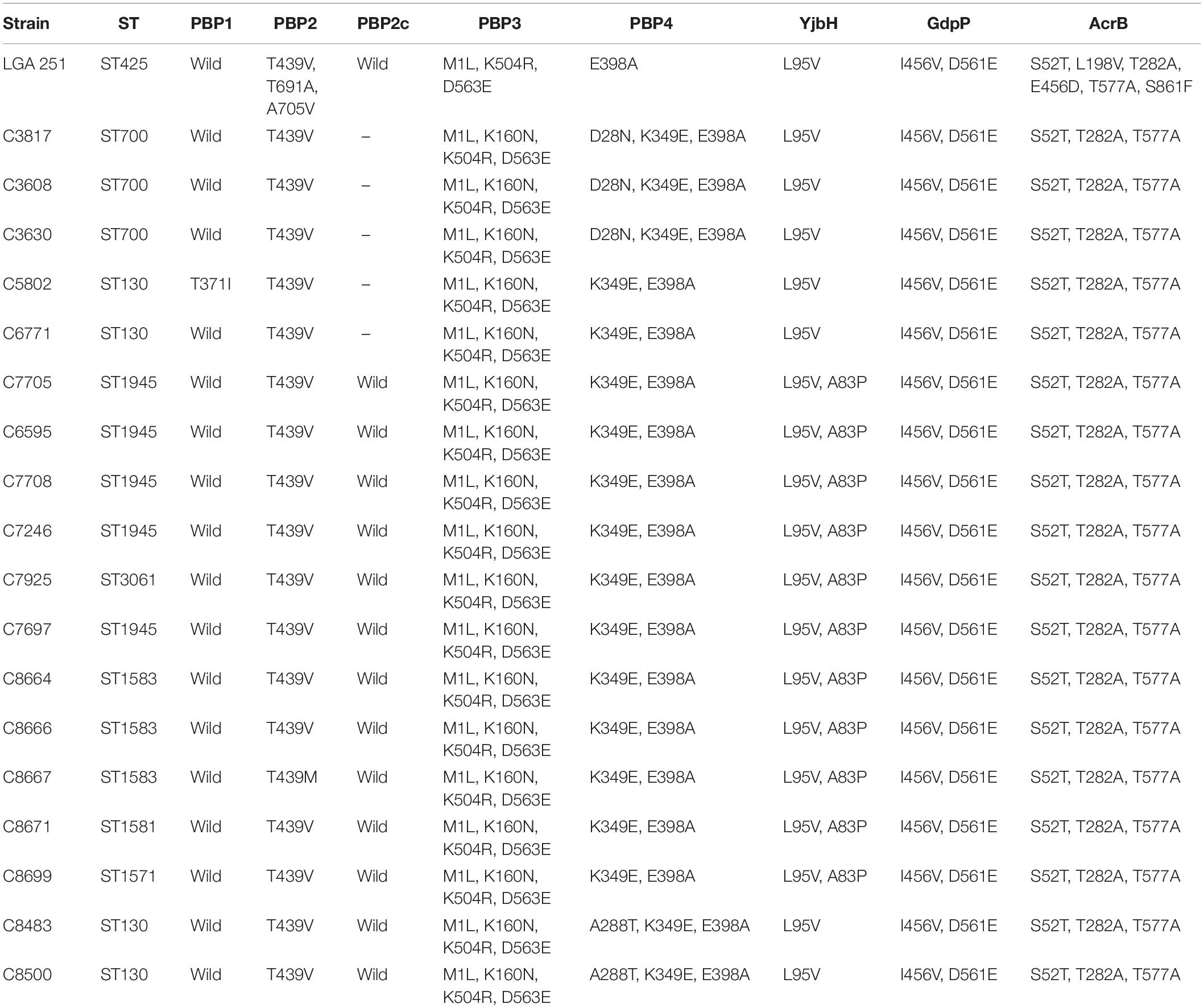
Table 3. Identified amino acid changes in PBPs 1, 2, 2c, 3, 4, YjbH, GdpP, and AcrB proteins of the 18 S. aureus strains included in this study and also of MRSA LGA251 strain (MRSA LGA251 as used as reference strain for PBP2c and MSSA ATCC 25923 as reference strain for PBPs 1, 2, 3, 4, YjbH, GdpP, and AcrB protein analysis).
Host Adaptation, Prophages, and Other Mobile Genetic Elements
The presence of the scn gene in some of our mecC-positive MRSA-CC130 strains is a remarkable feature since mecC-MRSA, as well as CC130 strains in general, are considered animal-associated, and IEC system is considered a human adaptation marker. The 3’ conserved region of the β-hemolytic bacteriophage φ3 (approximately 8,000 pb) of the mecC-positive strain C6595 (IEC type E, isolated of a wood mice) was compared with the IEC of the reference strain MRSA252 (GenBank accession number: BX571856, type A) (Figure 1), and no differences were observed apart from the different content in genes that give rise to the type of IEC. It can be assumed that these strains have an advantage in colonizing and/or infecting humans, as already was described in unusual IEC-positive MRSA livestock associated CC398 strains (Cuny et al., 2015; Pérez-Moreno et al., 2017; Ceballos et al., 2019).
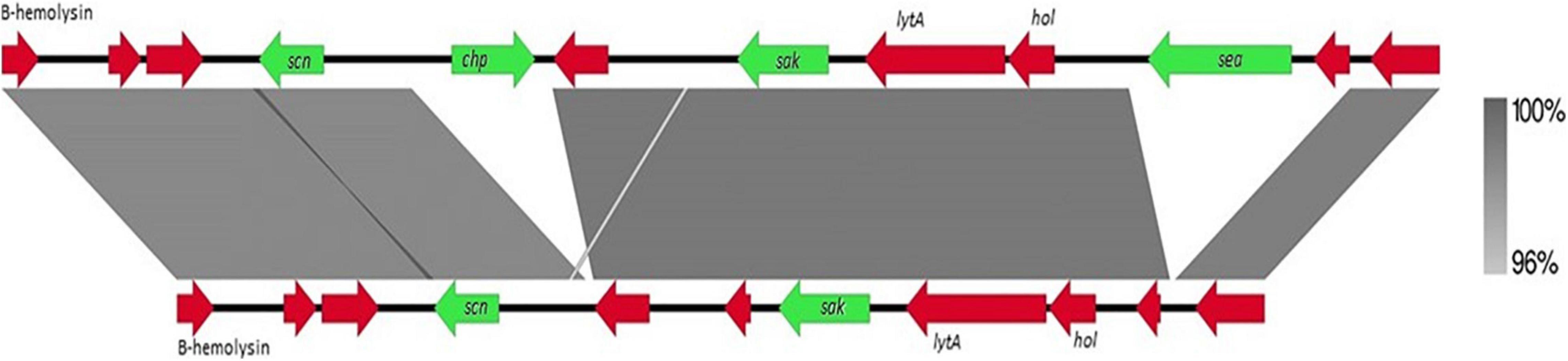
Figure 1. Comparison of 3′ conserved region of β-hemolytic bacteriophage (Φ3) between reference strain MRSA-252 (above) and C6595 (below). The percentage of similarity is indicated (right). Arrows in green corresponding to the IEC genes, and in red other coding sequences.
The hlb gene was also analyzed, showing that it was truncated in all the strains that presented the IEC system. The truncated hlb gene was also found in other three scn-negative strains (C6771, C7246, and C8699), which showed an integrated phage of about 45 kb, that only contained phage-related genes; it should be highlighted that C6771 and C8699 isolates contained a sak-related gene with a 45% of similarity respect to the sak IEC virulence gene (GenBank accession number: NC_026016). The dtlB gene, present in the two MRSA-ST130 mecC-positive strains isolated from healthy wild rabbits (C8483 and C8500), showed the following amino acid changes with respect to the reference MRSA252 strain (GenBank accession number: BX571856): (a) C8483 (I227T, A382S, and ∗405Q); (b) C8500 (A382S, G401D, and K402R). None of the strains presented the mutations T113K, Y250H, or ∗405Y previously described (Viana et al., 2015), and only the A382S change present in both strains has not been previously reported (Viana et al., 2015; Holmes et al., 2016). The vwbp gene (SaPIbov5, GenBank accession number: JP5338 used as reference) was found in the three MSSA-ST700 strains from small ruminants and in the MSSA-ST130 strain from a red deer, indicating in this case an adaptation to the host (Viana et al., 2010).
Phage analysis showed 12 different intact prophages in genome with an average of 2 ± 2 prophage regions per genome. The strains C3608, C3630, C3817, and C6771 (3 MSSA-ST700 and one MSSA-ST130, that also carried the vwbp gene), showed an identical coding sequence contained in a phage described as a protein related to the expression of fibrinogen (scn-type gene), but different from human variant (47% similarity of amino acid sequence with WP_000702262 as reference) and from the new variant described and associated with the evasion of the equine immune system (45% similarity of amino acid sequence with WP_106096712 as reference) (Supplementary Figure 2).
Only three strains (C3608, C3630 and C5802, corresponding with two MSSA-ST700 and one MSSA-ST130), presented rep genes: rep7, rep24, repUS23, rep5. In addition, only rep7 was detected showing a 100% nucleotide identity in two of the strains (C3608 and C3630). The gene rep7 has been previously described widely distributed in other CCs within the species S. aureus (Lozano et al., 2012).
Phylogenetic Analysis
The phylogenetic tree (Figure 2) showed that the three MSSA strains belonging to ST700, clearly constituted a separate clade from the remaining CC130 strains included in this study. Our ST700 MSSA strains were very different from the other 15 strains of the studied collection based on the results from all the analyses performed, supporting the consideration of ST700 as a lineage distinct from CC130 (Smith et al., 2014). The other 2 MSSA-ST130 strains also form a distinct clade in this collection.
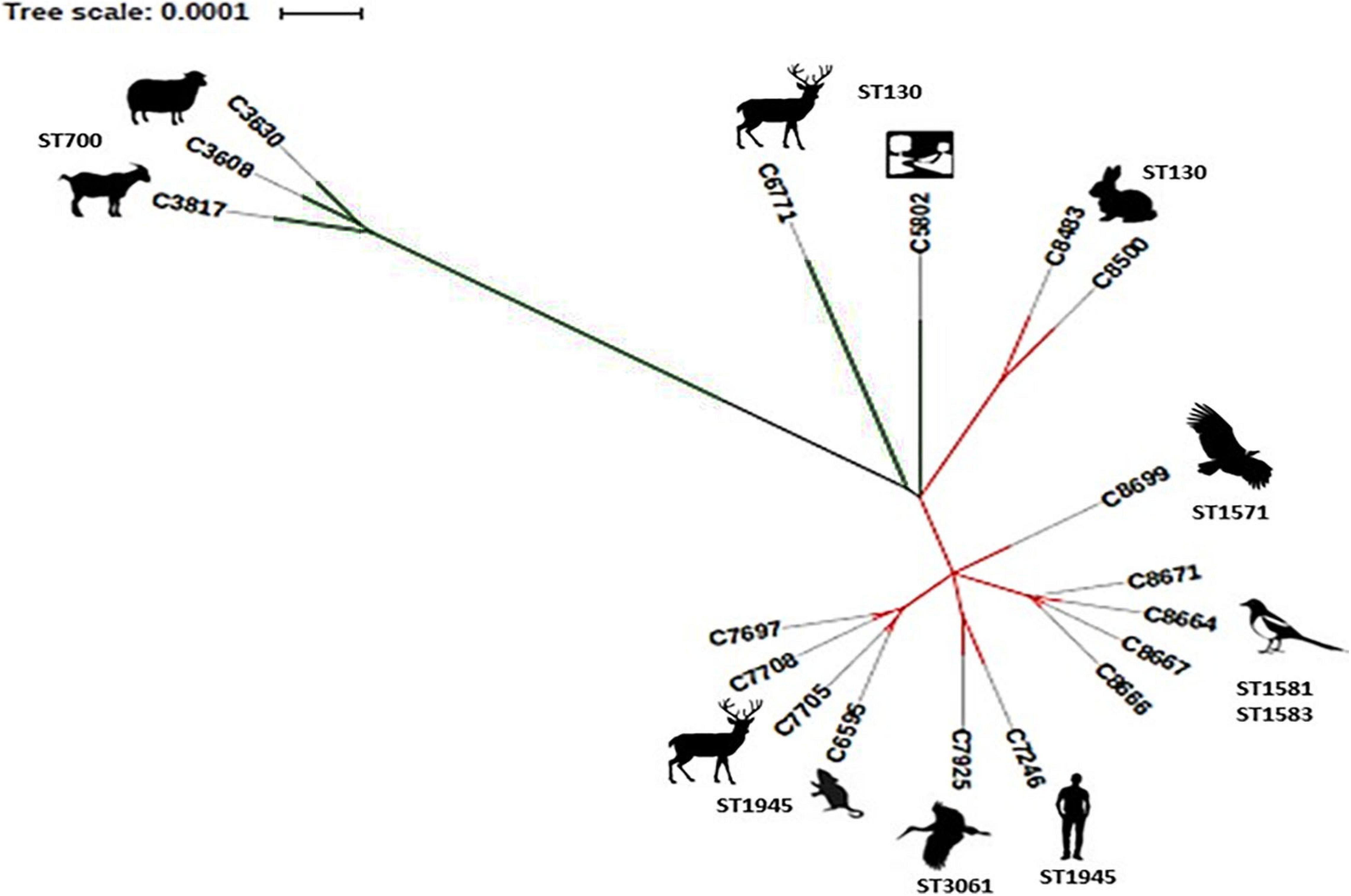
Figure 2. Phylogenetic tree showing the phylogenetic distance between 18 strains CC130 included in this study. Green lines corresponding to MSSA and red lines to MRSA strains.
Finally, the 13 MRSA-CC130 strains are grouped and the following associations can be seen: (1) C8483 and C8500 from rabbits from Aragon are clustered together indicating the possible animal-animal transfer; (2) C6595, C7697, C7705, and C7708, from red deer and small mammals from the same geographical area, highlighting C7705 and C6595 MRSA strains that were indistinguishable (by this analysis), which might suggest an interspecies transmission event; (3) C8666, C8667, C8664, and C8671, all isolated from magpies in the same location; (4) C7925 and C7246 that were isolated in a different geographical area, of different origins (stork and human, respectively), and with different STs; and (5) C8699 (from vulture) that is grouped individually.
Conclusion
Taking into account the relatively small number of strains included in this study, the comparison of fifteen strains CC130 from different animal origins, geographical locations and STs, demonstrated clear differences between isolates depending if they were mecC-positive or mecC-negative and between sequence types. Markedly divergent results from the three MSSA-ST700 isolates reinforce the idea of considering this lineage as distinctly separate from CC130. The etD2 gene appears to be a genetic marker of CC130 lineage (MSSA and MRSA), which is missing from ST700 strains although further studies are required to confirm this. The presence of IEC system in some of the MRSA-mecC from animals opens questions about the origins and evolution of mecC-MRSA.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
CT and MH conceived and designed the study. PG, LR-R, RF-F, HG, KB, and UH performed the initial sampling procedure and the initial characterization of isolates. PG performed laboratory work. PG, MZ, CT, and MH interpreted the results and contributed to producing the first draft of the manuscript. All authors have revised and agreed to the final version of the manuscript.
Funding
The work performed in the University of La Rioja was financed by projects SAF2016-76571-R and PID2019-106158RB-I00 of the Agencia Estatal de Investigación (AEI) of Spain and the Fondo Europeo de Desarrollo Regional (FEDER) of EU. Work in Cambridge was supported by a UK-China AMR Partnership Initiative (MR/P007201/1) held by the Department of Veterinary Medicine, University of Cambridge.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.655994/full#supplementary-material
Supplementary Figure 1 | Circular comparison of MSSA in Green-blue rings and MRSA in warm colors using LGA251 as reference.% GC content and GC Skew are represented in innermost circles (colors indicated in the coded legend).
Supplementary Figure 2 | (A) Amino acid comparative of scn-like found in 4 MSSA isolates of this study. (B) Amino acid comparative between one scn-like found in this study, one scn-equine (WP_106096712 as reference) and one scn-human (WP_000702262 as reference).
Supplementary Table 1 | Results of whole genome sequence and accession number of the strains included in this study.
Supplementary Table 2 | Result of unique genes in MRSA and in MSSA strains analyzed by Roary pipeline.
Supplementary Table 3 | Result of unique genes in scn positive and scn negative strains analyzed by Roary pipeline.
Footnotes
References
Alexander, J. A. N., Chatterjee, S. S., Hamilton, S. M., Eltis, L. D., Chambers, H. F., and Strynadka, N. C. J. (2018). Structural and kinetic analysis of penicillin-binding protein 4 (PBP4)-mediated antibiotic resistance in Staphylococcus aureus. J. Biol. Chem. 293, 19854–19865. doi: 10.1074/jbc.RA118.004952
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Alonzo, F., and Torres, V. J. (2014). The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol. Mol. Biol. Rev. 78, 199–230. doi: 10.1128/MMBR.00055-13
Arndt, D., Grant, J., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Azara, E., Piras, M. G., Parisi, A., and Tola, S. (2017). Antimicrobial susceptibility and genotyping of Staphylococcus aureus isolates collected between 1986 and 2015 from ovine mastitis. Vet. Microbiol. 205, 53–56. doi: 10.1016/j.vetmic.2017.05.006
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Ba, X., Harrison, E. M., Edwards, G. F., Holden, M. T., Larsen, A. R., Petersen, A., et al. (2014). Novel mutations in penicillin-binding protein genes in clinical Staphylococcus aureus isolates that are methicillin resistant on susceptibility testing, but lack the mec gene. J. Antimicrob. Chemother. 69, 594–597. doi: 10.1093/jac/dkt418
Ba, X., Kalmar, L., Hadjirin, N. F., Kerschner, H., Apfalter, P., Morgan, F. J., et al. (2019). Truncation of GdpP mediates β-lactam resistance in clinical isolates of Staphylococcus aureus. J. Antimicrob. Chemother. 74, 1182–1191. doi: 10.1093/jac/dkz013
Banerjee, R., Gretes, M., Harlem, C., Basuino, L., and Chambers, H. F. (2010). A mecA-negative strain of methicillin-resistant Staphylococcus aureus with high-level β-lactam resistance contains mutations in three genes. Antimicrob. Agents Chemother. 54, 4900–4902. doi: 10.1128/AAC.00594-10
Becker, K., van Alen, S., Idelevich, E. A., Schleimer, N., Seggewiß, J., Mellmann, A., et al. (2018). Plasmid-encoded transferable mecB-mediated methicillin resistance in Staphylococcus aureus. Emerg. Infect. Dis. 24, 242–248. doi: 10.3201/eid2402.171074
Benito, D., Gómez, P., Aspiroz, C., Zarazaga, M., Lozano, C., and Torres, C. (2016). Molecular characterization of Staphylococcus aureus isolated from humans related to a livestock farm in Spain, with detection of MRSA-CC130 carrying mecC gene: a zoonotic case? Enferm. Infecc. Microbiol. Clin. 34, 280–285. doi: 10.1016/j.eimc.2015.03.008
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Ceballos, S., Aspiroz, C., Ruiz-Ripa, L., Reynaga, E., Azcona-Gutiérrez, J. M., Rezusta, A., et al. (2019). Epidemiology of MRSA CC398 in hospitals located in Spanish regions with different pig-farming densities: a multicentre study. J. Antimicrob. Chemother. 74, 2157–2161. doi: 10.1093/jac/dkz180
Chan, L. C., Gilbert, A., Basuino, L., da Costa, T. M., Hamilton, S. M., Dos Santos, K. R., et al. (2016). PBP 4 mediates high-level resistance to new-generation cephalosporins in Staphylococcus aureus. Antimicrob. Agents Chemother. 60, 3934–3941. doi: 10.1128/AAC.00358-16
Cuny, C., Abdelbary, M., Layer, F., Werner, G., and Witte, W. (2015). Prevalence of the immune evasion gene cluster in Staphylococcus aureus CC398. Vet. Microbiol. 177, 219–223. doi: 10.1016/j.vetmic.2015.02.031
Cuny, C., Layer, F., Strommenger, B., and Witte, W. (2011). Rare occurrence of methicillin-resistant Staphylococcus aureus CC130 with a novel mecA homologue in humans in Germany. PLoS One 6:e24360. doi: 10.1371/journal.pone.0024360
de Jong, A., van Hijum, S. A. F. T., Bijlsma, J. J. E., Kok, J., and Kuipers, O. P. (2006). BAGEL: a web-based bacteriocin genome mining tool. Nucleic Acids Res. 34, W273–W279. doi: 10.1093/nar/gkl237
Fitzgerald, J. R., Monday, S. R., Foster, T. J., Bohach, G. A., Hartigan, P. J., Meaney, W. J., et al. (2001). Characterization of a putative pathogenicity island from bovine Staphylococcus aureus encoding multiple superantigens. J. Bacteriol. 183, 63–70. doi: 10.1128/JB.183.1.63-70.2001
García-Álvarez, L., Holden, M. T. G., Lindsay, H., Webb, C. R., Brown, D. F. J., Curran, M. D., et al. (2011). Methicillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect. Dis. 11, 595–603. doi: 10.1016/S1473-3099(11)70126-8
Gharsa, H., Ben Slama, K., Gómez-Sanz, E., Lozano, C., Zarazaga, M., Messadi, L., et al. (2015). Molecular characterization of Staphylococcus aureus from nasal samples of healthy farm animals and pets in Tunisia. Vector Borne Zoonotic Dis. 15, 109–115. doi: 10.1089/vbz.2014.1655
Göhring, N., Fedtke, I., Xia, G., Jorge, A. M., Pinho, M. G., Bertsche, U., et al. (2011). New role of the disulfide stress effector YjbH in β-lactam susceptibility of Staphylococcus aureus. Antimicrob. Agents Chemother. 55, 5452–5458. doi: 10.1128/AAC.00286-11
Gómez, P., Casado, C., Sáenz, Y., Ruiz-Ripa, L., Estepa, V., Zarazaga, M., et al. (2017). Diversity of species and antimicrobial resistance determinants of Staphylococci in superficial waters in Spain. FEMS Microbiol. Ecol. 93:fiw208. doi: 10.1093/femsec/fiw208
Gómez, P., González-Barrio, D., Benito, D., García, J. T., Viñuela, J., Zarazaga, M., et al. (2014). Detection of methicillin-resistant Staphylococcus aureus (MRSA) carrying the mecC gene in wild small mammals in Spain. J. Antimicrob. Chemother. 69, 2061–2064. doi: 10.1093/jac/dku100
Gómez, P., Lozano, C., Camacho, M. C., Lima-Barbero, J. F., Hernández, J. M., Zarazaga, M., et al. (2016). Detection of MRSA ST3061-t843-mecC and ST398-t011-mecA in white stork nestlings exposed to human residues. J. Antimicrob. Chemother. 71, 53–57. doi: 10.1093/jac/dkv314
Gómez, P., Lozano, C., González-Barrio, D., Zarazaga, M., Ruiz-Fons, F., and Torres, C. (2015). High prevalence of methicillin-resistant Staphylococcus aureus (MRSA) carrying the mecC gene in a semi-extensive red deer (Cervus elaphus hispanicus) farm in Southern Spain. Vet. Microbiol. 177, 326–331. doi: 10.1016/j.vetmic.2015.03.029
Harrison, E. M., Coll, F., Toleman, M. S., Blane, B., Brown, N. M., Török, M. E., et al. (2017). Genomic surveillance reveals low prevalence of livestock- associated methicillin-resistant Staphylococcus aureus in the East of England. Sci. Rep. 7:7406. doi: 10.1038/s41598-017-07662-2
Harrison, E. M., Paterson, G. K., Holden, M. T., Larsen, J., Stegger, M., Larsen, A. R., et al. (2013). Whole genome sequencing identifies zoonotic transmission of MRSA isolates with the novel mecA homologue mecC. EMBO Mol. Med. 5, 509–515. doi: 10.1002/emmm.201202413
Holmes, M. A., Harrison, E. M., Fisher, E. A., Graham, E. M., Parkhill, J., Foster, G., et al. (2016). Genomic analysis of companion rabbit Staphylococcus aureus. PLoS One 11:e0151458. doi: 10.1371/journal.pone.0151458
Jamrozy, D. M., Fielder, M. D., Butaye, P., and Coldham, N. G. (2012). Comparative genotypic and phenotypic characterisation of methicillin-resistant Staphylococcus aureus ST398 isolated from animals and humans. PLoS One 7:e40458. doi: 10.1371/journal.pone.0040458
Kotsakis, S. D., Tzouvelekis, L. S., and Zerva, L. (2012). Staphylococcus lugdunensis strain with a modified PBP1A/1B expressing resistance to β-lactams. Eur. J. Clin. Microbiol. Infect. Dis. 31, 169–172. doi: 10.1007/s10096-011-1289-8
Lozano, C., Fernández-Fernández, R., Ruiz-Ripa, L., Gómez, P., Zarazaga, M., and Torres, C. (2020). Human mecC-carrying MRSA: clinical implications and risk factors. Microorganisms 28:1615. doi: 10.3390/microorganisms8101615
Lozano, C., García-Migura, L., Aspiroz, C., Zarazaga, M., Torres, C., and Aarestrup, F. M. (2012). Expansion of a plasmid classification system for Gram-positive bacteria and determination of the diversity of plasmids in Staphylococcus aureus strains of human, animal, and food origins. Appl. Environ. Microbiol. 78, 5948–5955. doi: 10.1128/AEM.00870-12
Luzzago, C., Locatelli, C., Franco, A., Scaccabarozzi, L., Gualdi, V., Viganò, R., et al. (2014). Clonal diversity, virulence-associated genes and antimicrobial resistance profile of Staphylococcus aureus isolates from nasal cavities and soft tissue infections in wild ruminants in Italian Alps. Vet. Microbiol. 170, 157–161. doi: 10.1016/j.vetmic.2014.01.016
McCarthy, A. J., and Lindsay, J. A. (2013). Staphylococcus aureus innate immune evasion is lineage-specific: a bioinfomatics study. Infect. Genet. Evol. 19, 7–14. doi: 10.1016/j.meegid.2013.06.012
McCarthy, A. J., Witney, A. A., Gould, K. A., Moodley, A., Guardabassi, L., Voss, A., et al. (2011). The distribution of mobile genetic elements (MGEs) in MRSA CC398 is associated with both host and country. Genome Biol. Evol. 3, 1164–1174. doi: 10.1093/gbe/evr092
Monecke, S., Gavier-Widén, D., Hotzel, H., Peters, M., Guenther, S., Lazaris, A., et al. (2016). Diversity of Staphylococcus aureus isolates in european wildlife. PLoS One 11:e0168433. doi: 10.1371/journal.pone.0168433
Monecke, S., Gavier-Widen, D., Mattsson, R., Rangstrup-Christensen, L., Lazaris, A., Coleman, D. C., et al. (2013). Detection of mecC-positive Staphylococcus aureus (CC130-MRSA-XI) in diseased European hedgehogs (Erinaceus europaeus) in Sweden. PLoS One 8:e66166. doi: 10.1371/journal.pone.0066166
Monecke, S., Kuhnert, P., Hotzel, H., Slickers, P., and Ehricht, R. (2007). Microarray based study on virulence-associated genes and resistance determinants of Staphylococcus aureus isolates from cattle. Vet. Microbiol. 125, 128–140. doi: 10.1016/j.vetmic.2007.05.016
Munro, P., Benchetrit, M., Nahori, M. A., Stefani, C., Clément, R., Michiels, J. F., et al. (2010). The Staphylococcus aureus epidermal cell differentiation inhibitor toxin promotes formation of infection foci in a mouse model of bacteremia. Infect. Immun. 78, 3404–3411. doi: 10.1128/IAI.00319-10
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Paharik, A. E., Salgado-Pabon, W., Meyerholz, D. K., White, M. J., Schlievert, P. M., and Horswill, A. R. (2016). The Spl serine proteases modulate Staphylococcus aureus protein production and virulence in a rabbit model of pneumonia. mSphere 1:e00208-16. doi: 10.1128/mSphere.00208-16
Paterson, G. K., Harrison, E. M., and Holmes, M. A. (2014a). The emergence of mecC methicillin-resistant Staphylococcus aureus. Trends Microbiol. 22, 42–47. doi: 10.1016/j.tim.2013.11.003
Paterson, G. K., Morgan, F. J., Harrison, E. M., Cartwright, E. J., Török, M. E., Zadoks, R. N., et al. (2014b). Prevalence and characterisation of human mecC methicillin-resistant Staphylococcus aureus isolates in England. J. Antimicrob. Chemother. 69, 907–910. doi: 10.1093/jac/dkt462
Pérez-Moreno, M. O., Centelles-Serrano, M. J., Nogales-López, J., Domenech-Spanedda, M. F., Lozano, C., and Torres, C. (2017). Unusual presence of the immune evasion gene cluster in livestock-associated MRSA of lineage CC398 causing peridural and psoas abscesses in a poultry farmer. Enferm. Infecc. Microbiol. Clin. 35, 651–654. doi: 10.1016/j.eimc.2016.07.008
Petersen, A., Stegger, M., Heltberg, O., Christensen, J., Zeuthen, A., Knudsen, L. K., et al. (2013). Epidemiology of methicillin-resistant Staphylococcus aureus carrying the novel mecC gene in Denmark corroborates a zoonotic reservoir with transmission to humans. Clin. Microbiol. Infect. 19, E16–E22. doi: 10.1111/1469-0691.12036
Rissman, A. I., Mau, B., Biehl, B. S., Darling, A. E., Glasner, J. D., and Perna, N. T. (2009). Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics 25, 2071–2073. doi: 10.1093/bioinformatics/btp356
Ruiz-Ripa, L., Gómez, P., Alonso, C. A., Camacho, M. C., de la Puente, J., Fernández-Fernández, R., et al. (2019). Detection of MRSA of Lineages CC130-mecC and CC398-mecA and Staphylococcus delphini-lnu(A) in Magpies and Cinereous Vultures in Spain. Microb. Ecol. 78, 409–415. doi: 10.1007/s00248-019-01328-4
Sabat, A. J., Wladyka, B., Kosowska-Shick, K., Grundmann, H., van Dijl, J. M., Kowal, J., et al. (2008). Polymorphism, genetic exchange and intragenic recombination of the aureolysin gene among Staphylococcus aureus strains. BMC Microbiol. 8:129. doi: 10.1186/1471-2180-8-129
Schlotter, K., Ehricht, R., Hotzel, H., Monecke, S., Pfeffer, M., and Donat, K. (2012). Leukocidin genes lukF-P83 and lukM are associated with Staphylococcus aureus clonal complexes 151, 479, 226 and 133 isolated from bovine udder infections in Thuringia. Germany. Vet. Res. 43:42. doi: 10.1186/1297-9716-43-42
Shore, A. C., Deasy, E. C., Slickers, P., Brennan, G., O’Connell, B., Monecke, S., et al. (2011). Detection of staphylococcal cassette chromosome mec type XI carrying highly divergent mecA, mecI, mecR1, blaZ, and ccr genes in human clinical isolates of clonal complex 130 methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 55, 3765–3773. doi: 10.1128/AAC.00187-11
Smith, E. M., Needs, P. F., Manley, G., and Green, L. E. (2014). Global distribution and diversity of ovine associated Staphylococcus aureus. Infect. Genet. Evol. 22, 208–215. doi: 10.1016/j.meegid.2013.09.008
Stach, N., Kalinska, M., Zdzalik, M., Kitel, R., Karim, A., Serwin, k., et al. (2018). Unique substrate specificity of SplE serine protease from Staphylococcus aureus. Structure 26, 572–579. doi: 10.1016/j.str.2018.02.008
van Wamel, W. J. B., Rooijakkers, S. H. M., Ruyken, M., van Kessel, K. P. M., and van Strijp, J. A. G. (2006). The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 188, 1310–1315. doi: 10.1128/JB.188.4.1310-1315.2006
Viana, D., Blanco, J., Tormo-Más, M. A., Selva, L., Guinane, C. M., Baselga, R., et al. (2010). Adaptation of Staphylococcus aureus to ruminant and equine hosts involves SaPI−carried variants of von Willebrand factor−binding protein. Mol. Microbiol. 77, 1583–1594. doi: 10.1111/j.1365-2958.2010.07312.x
Viana, D., Comos, M., McAdam, P. R., Ward, M. J., Selva, L., Guinane, C. M., et al. (2015). A single natural nucleotide mutation alters bacterial pathogen host-tropism. Nat. Genet. 47, 361–366. doi: 10.1038/ng.3219
Vitale, M., Gaglio, S., Galluzzo, P., Cascone, G., Piraino, C., Di Marco Lo Presti, V., et al. (2018). Antibiotic resistance profiling, analysis of virulence aspects and molecular genotyping of Staphylococcus aureus isolated in Sicily, Italy. Foodborne Pathog. Dis. 15, 177–185. doi: 10.1089/fpd.2017.2338
Keywords: MRSA, MSSA, whole genome sequencing, CC130, ST700, IEC, etD2
Citation: Gómez P, Ruiz-Ripa L, Fernández-Fernández R, Gharsa H, Ben Slama K, Höfle U, Zarazaga M, Holmes MA and Torres C (2021) Genomic Analysis of Staphylococcus aureus of the Lineage CC130, Including mecC-Carrying MRSA and MSSA Isolates Recovered of Animal, Human, and Environmental Origins. Front. Microbiol. 12:655994. doi: 10.3389/fmicb.2021.655994
Received: 19 January 2021; Accepted: 03 March 2021;
Published: 25 March 2021.
Edited by:
Magdalena Rzewuska, Warsaw University of Life Sciences, PolandReviewed by:
Darren Trott, University of Adelaide, AustraliaPeter Kinnevey, Dublin Dental University Hospital, Ireland
Copyright © 2021 Gómez, Ruiz-Ripa, Fernández-Fernández, Gharsa, Ben Slama, Höfle, Zarazaga, Holmes and Torres. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen Torres, Y2FybWVuLnRvcnJlc0B1bmlyaW9qYS5lcw==