 Vojtěch Tláskal
Vojtěch Tláskal Victor Satler Pylro
Victor Satler Pylro Lucia Žifčáková
Lucia Žifčáková Petr Baldrian
Petr Baldrian- 1Laboratory of Environmental Microbiology, Institute of Microbiology of the Czech Academy of Sciences, Praha, Czechia
- 2Microbial Ecology and Bioinformatics Laboratory, Department of Biology, Federal University of Lavras (UFLA), Lavras, Brazil
The bacterial genus Sodalis is represented by insect endosymbionts as well as free-living species. While the former have been studied frequently, the distribution of the latter is not yet clear. Here, we present a description of a free-living strain, Sodalis ligni sp. nov., originating from decomposing deadwood. The favored occurrence of S. ligni in deadwood is confirmed by both 16S rRNA gene distribution and metagenome data. Pangenome analysis of available Sodalis genomes shows at least three groups within the Sodalis genus: deadwood-associated strains, tsetse fly endosymbionts and endosymbionts of other insects. This differentiation is consistent in terms of the gene frequency level, genome similarity and carbohydrate-active enzyme composition of the genomes. Deadwood-associated strains contain genes for active decomposition of biopolymers of plant and fungal origin and can utilize more diverse carbon sources than their symbiotic relatives. Deadwood-associated strains, but not other Sodalis strains, have the genetic potential to fix N2, and the corresponding genes are expressed in deadwood. Nitrogenase genes are located within the genomes of Sodalis, including S. ligni, at multiple loci represented by more gene variants. We show decomposing wood to be a previously undescribed habitat of the genus Sodalis that appears to show striking ecological divergence.
Introduction
Bacterial insect endosymbionts that occupy tissues of their hosts are widespread in multiple insect taxa (reviewed in Perlmutter and Bordenstein, 2020). The nature of such symbiosis may take the form of parasitism, mutualism or commensalism depending on the ecology of the bacterial symbiont. Parasitic bacteria may manipulate reproduction of their hosts, while mutualistic bacteria may provide essential vitamins or participate in nitrogen (N) cycling (Akman et al., 2002; Behar et al., 2005; Sabree et al., 2009). Commensal bacteria can use insects at some part of their life cycle as vectors for transmission to their target host organism, e.g., plants for which they are parasitic (Nadarasah and Stavrinides, 2011). The life strategy of the symbiont depends on the evolutionary history with the host and is driven by the length of its coexistence with the host (Toh et al., 2006). Different bacterial symbionts with distinct evolutionary histories have been described, e.g., in tsetse flies (Glossina sp.). Wigglesworthia glossinidia (Enterobacterales) provides vitamins to the tsetse fly and to other symbionts (Hall et al., 2019) and represents an obligate mutualist with a long coevolutionary history (Chen et al., 1999). Another tsetse fly enterobacterium, Sodalis glossinidius, shows a more recent association with its host as suggested by its relatively large genome that contains a high number of pseudogenes, which remain actively transcribed in a cell-free culture (Toh et al., 2006; Goodhead et al., 2020; Hall et al., 2020). Further hinting at the recent establishment of the Sodalis-tsetse symbiosis was the successful establishment and vertical transmission of the non-native Sodalis population in tsetse flies cleared from their native symbionts. It indicates lack of host-symbiont extensive coevolution (Weiss et al., 2006).
Sodalis endosymbionts are present in other insects with nutritionally restricted diets, such as bloodsucking Diptera (Nováková and Hypša, 2007; Chrudimský et al., 2012), Phthiraptera (Fukatsu et al., 2007; Boyd et al., 2016), sapsucking Hemiptera (Kaiwa et al., 2010) and Coleoptera (Heddi et al., 1999). Multiple Sodalis species have thus been identified within their hosts and shown to supply essential vitamins to the host (Heddi et al., 1999; Boyd et al., 2016). In contrast, only one free-living member of this genus has been described despite the apparent recent establishment of the host-dependent life strategy of the endosymbionts. Sodalis sp. strain HS, later described as S. praecaptivus was isolated from a hand wound of a human impaled with a tree branch (Clayton et al., 2012; Chari et al., 2015). Incidental isolation of Sodalis-related strain from such source was unexpected and as a consequence Sodalis became a textbook example of microbial specialization processes (Yong, 2016).
The genome-wide sequence comparison of Sodalis species shows S. glossinidius and the cereal weevil symbiont S. pierantonius strain SOPE to be closely related to S. praecaptivus. In particular, the S. pierantonius strain SOPE shares high genome similarity with free-living S. praecaptivus, and its symbiosis with the weevil shows evidence of recent origin (Clayton et al., 2012). Despite the presence of numerous pseudogenes, genes of both symbionts represent a subset of the genes in the S. praecaptivus genome, showing that the S. praecaptivus-like ancestor might be the ancestor for symbionts as well (Clayton et al., 2012). The genome characteristics of S. praecaptivus correspond to host-independent life and stabilizing gene selection pressure; in comparison with symbiotic relatives, it has a larger genome, fewer pseudogenes and fewer IS elements (Oakeson et al., 2014). Consequently, due to its wide spectrum of genes and high coding density, S. praecaptivus is able to utilize several carbon (C) sources, including chitin and plant sugars (cellobiose, xylose, rhamnose) and N sources, such as ammonia and nitrate (Chari et al., 2015). While S. praecaptivus is host-independent, it is able to colonize weevils and tsetse flies through quorum sensing suppression of its virulence, which suggests a mechanism of insect symbiosis development in the Sodalis group (Enomoto et al., 2017; Munoz et al., 2020). Despite the well-characterized phylogeny and genome properties of the Sodalis group, a systematic approach to assess the environmental distribution of free-living Sodalis strains has not been performed. It remains to be determined whether there are different levels of relatedness of non-symbiotic strains with symbionts in distinct insect taxa hosts.
Here, we present a Sodalis pangenome analysis that allowed us to characterize different guilds within this genus. Guilds differ in their gene composition as well as in their life strategy. Our results show that the Sodalis genus is functionally versatile with multiple ecological roles. We report novel free-living Sodalis species-level taxon associated in high abundances with decomposing wood, for which we propose the name Sodalis ligni sp. nov. This taxon is represented by the strain dw23T and can utilize C-rich labile wood-derived compounds and fix atmospheric nitrogen. Sodalis ligni is a globally distributed species that is directly related to symbiotic Sodalis members with characterized genomes while maintaining a non-symbiotic lifestyle and preferring deadwood habitats.
Materials and Methods
Sodalis Isolation
Wood samples for bacterial isolation were obtained in the Žofínský Prales National Nature Reserve, an unmanaged forest in the south of the Czech Republic (48°39′57″N, 14°42′24″E). The core zone of the forest reserve (42 ha) has never been managed, and human intervention stopped in 1838, when it was declared a reserve. This reserve thus represents a rare fragment of European temperate virgin forest with deadwood left to spontaneous decomposition. The reserve is situated at 730–830 m a.s.l., the bedrock is almost homogeneous and consists of fine to medium-grainy porphyritic and biotite granite. The annual average rainfall is 866 mm, and the annual average temperature is 6.2°C (Anderson-Teixeira et al., 2015). At present, the reserve is covered by a mixed forest in which Fagus sylvatica predominates, followed by Picea abies and Abies alba. The mean living tree volume is 690 m3 h–1, and the mean volume of coarse woody debris (logs, represented by tree trunks and their fragments) is 208 m3 h–1 (Král et al., 2010; Šamonil et al., 2013). Logs are repeatedly surveyed, and the approximate age of each log, the cause of death (e.g., stem breakage, windthrow, etc.) and the status before downing (fresh, decomposed) is known. The area has been subjected to the characterization of bacterial and fungal succession on deadwood (Baldrian et al., 2016; Tláskal et al., 2017; Odriozola et al., 2021), and the functional roles of the members of the deadwood microbiome were determined (Tláskal et al., 2021).
Wood samples were obtained as described previously in Tláskal et al. (2017). Briefly, in October 2013, four samples from selected logs were obtained by drilling with an electrical drill vertically along the whole decomposing stem. Part of the material was used for bacterial community characterization (Tláskal et al., 2017) and part was treated as follows: wood chips from each stem were pooled together and transported to the laboratory, where they were kept at 4°C until the next day. Wood material was shaken with 15 mL of Ringer solution for 2 h and diluted 104× to 106×. Dilutions were plated on nutrient-limited NB medium (0.26 g L–1 Nutrient Broth, 15 g L–1 agar, pH 5). The growth of bacterial colonies was recorded on Petri dishes using a marker for 8 weeks. Colony PCR was used to infer the taxonomy of the slow-growing strains that appeared in the later phase of cultivation. The PCR premix and cycling conditions were as follows: 2.5 μL 10 × buffer for DyNAzyme DNA Polymerase; 0.75 μL DyNAzyme II DNA polymerase (2 μL–1); 0.75 μL of BSA (20 mg mL–1); 0.5 μL of PCR Nucleotide Mix (10 mM); 1 μL of each primer eub530f (10 μM) and eub1100br (10 μM) (Lane, 1991) and sterile ddH2O up to 25 μL; amplification started at 94°C for 5 min, followed by 35 cycles of 94°C for 1 min, 62°C for 1 min, 72°C for 1 min and finished with a final setting of 72°C for 10 min. Sanger sequencing was performed using the reverse primer. The obtained sequences were compared by BLASTn with bacterial 16S rRNA gene-based community data from the same habitat (Tláskal et al., 2017). Bacterial strains with high similarity and coverage to the most abundant bacteria recovered by environmental DNA sequencing were selected for further cultivation and genome sequencing (Tláskal and Baldrian, 2021).
By this approach, we were able to select two bacterial strains (labeled Sodalis sp. strain dw23T and strain dw96) with high similarity to the abundant cluster CL27 in community data (Tláskal et al., 2017), which is 100% similar to the 16S rRNA gene sequence with NCBI accession AJ011333.1, which is mislabeled as Yersinia sp. (Elo et al., 2000). The nearest relative with a complete genome sequence available was the Sodalis praecaptivus strain HS (CP006569.1, similarity 97.2%, Chari et al., 2015). Strains dw23T and dw96 originated from two distinct Fagus sylvatica stems 9 m and 12 m long, respectively, that had been decomposing for less than 5 years.
DNA Extraction, Sequencing, and Genome Assembly
The two Sodalis strains were cultivated in 50 mL of GY-VL55 liquid medium (Lladó et al., 2019) with shaking for 2 weeks at 23°C. After cultivation, the cells were pelleted and DNA was extracted using the ArchivePure DNA Yeast & Gram- + Kit (5 Prime, Germany) according to the manufacturer’s instructions. The DNA was quantified by a Qubit 2.0 Fluorometer (Life Technologies, United States), and sheared by Bioruptor Pico (Diagenode, Belgium) to an average length of 550 bp. Sequencing adapters were ligated by the TruSeq DNA PCR-Free Library Prep Kit (Illumina Inc., United States). The ligated library was sequenced on the Illumina MiSeq platform with 2 × 250 (strain dw23T) and 2 × 300 (strain dw96) paired-end runs.
Sodalis ligni dw23T was selected for further Nanopore MinION sequencing. DNA was extracted by mechanical cell lysis using vortex, followed by DNA binding and purification on AMPure XP magnetic beads (Beckman Coulter, United States) with 70% ethanol. The SQK-LSK108 ligation kit was used to prepare a long-read sequencing library according to the manufacturer’s instructions. The library was loaded onto a Nanopore flow-cell version FLO-MIN106 for a 48 h sequencing run. The obtained FAST5 reads were basecalled into FASTQ with local Albacore 2.1.7 (available via ONT community site1) with a minimal quality threshold of 7. Passed reads were scanned for remaining adapters, which were trimmed with Porechop 0.2.32.
The Sodalis sp. strain dw96 genome assembly used short reads only, and Sodalis ligni dw23T used a hybrid assembly of short and long reads. Assembly was performed with Unicycler 0.4.4 (Wick et al., 2017) in normal mode wrapping the following programs: SPAdes 3.11.1 (Bankevich et al., 2012), BLAST 2.2.28+ (Altschul et al., 1997), bowtie 2.2.4 (Langmead et al., 2009), samtools 1.6 (Li et al., 2009) and pilon 1.22 (Walker et al., 2014). Prokka 1.13 (Seemann, 2014) with RNAmmer (Lagesen et al., 2007) was used for gene calling, annotation and rRNA genes identification. Genome completeness and contamination were estimated using CheckM v1.1.3 (Parks et al., 2015). The genomes were compared with NCBI Prok database of complete prokaryotic genomes (release Dec-29-2020) using whole-genome based comparison in MiGA (Rodriguez-R et al., 2018).
Pangenome Analysis
The NCBI-genome-download 0.2.12 script3 was used to retrieve all sequenced genomes from the genus Sodalis (March, 2020). The two strains obtained from deadwood were added to the set of genomes. Sodalis-like endosymbionts and other related strains analyzed previously by Santos-Garcia et al. (2017) were included to estimate size of the core gene set of Sodalis-allied group which takes into account significant genome reduction reported for some of these symbionts. Further analysis was focused on Sodalis strains with large genomes while those symbionts which are in the advanced reductive evolution process were excluded from the analysis because of their smaller genomes with significantly distinct gene compositions (Santos-Garcia et al., 2017). Selected genomes are summarized in Supplementary Table 1 and consist of Sodalis sensu stricto strains from unpublished as well as published studies (Toh et al., 2006; Chrudimský et al., 2012; Clayton et al., 2012; Oakeson et al., 2014; Rosas-Pérez et al., 2017; Rubin et al., 2018). Genomes were subsequently analyzed within pangenome analysis in anvi’o 6.2 (Eren et al., 2015, see the “Code Availability” section) to identify deadwood-associated genes and genes shared by all the genomes. ANI was calculated using pyANI 0.2.10 (Pritchard et al., 2015). The resulting image was processed with Inkscape4. For CAZyme annotation, prokka 1.13 gene prediction was used to obtain sequences of genes from 11 Sodalis genomes. The amino acid sequences were compared with the dbCAN database version 07312018 using run-dbcan.py 2.0.11 script (Zhang et al., 2018) and hmmer 3.3 (Eddy, 2011). CAZymes with an e-value ≤ 1E−20 were considered for further annotation. Ward’s clustering was used to group genomes based on the Hellinger transformed counts of all the detected CAZymes and pheatmap 1.0.12 package was used to generate a CAZy heatmap (Kolde, 2019). Spearman correlation was used to calculate genome-related statistics.
nifH Phylogenetics
The curated database of nifH sequences classified to the phylum level or below was retrieved (Moynihan, 2020). Genes from the phylum Proteobacteria and a random nifH representative for each taxonomic group were selected. Two nifH genes from Sodalis sp. strain dw96 and three from Sodalis ligni were aligned with the retrieved nifH collection using the online MAFFT v7.475 (Katoh et al., 2018). Maximum likelihood tree was constructed using IQ-TREE v1.6.12 (-alrt 1000 -bb 1000) (Nguyen et al., 2014) using best-fit model GTR + F + R10 identified by ModelFinder (Kalyaanamoorthy et al., 2017) with UFBoot ultrafast bootstrap (Hoang et al., 2017). Tree was further edited using iTOL (Letunic and Bork, 2011).
Habitat Context—Soil and Deadwood
To confirm the presence of Sodalis in deadwood 16S rRNA gene datasets from previous studies published by Hoppe et al. (2015), Moll et al. (2018), Probst et al. (2018), and Šamonil et al. (2020) were screened for similarity with the 16S rRNA genes of strain dw23T and strain dw96 using BLAST 2.2.28+, and the relative abundance of the most abundant OTU within each study with >97% similarity to the cultivated strains was calculated.
Furthermore, three wood-associated Sodalis genomes together with genome of Sodalis praecapticus and genome of Sodalis pierantonius strain SOPE were used as a reference for the mapping of metagenomes of naturally decomposing wood obtained in the Žofínský prales Nature Reserve (Tláskal et al., 2021, unpublished). The metagenomic study included five age classes of differently decomposed deadwood of Fagus sylvatica, with the youngest age class decomposing for <5 years and the oldest age class decomposing for more than 41 years. Sodalis ligni dw23T was recovered from the same decomposing tree as one of the metagenomic and metatranscriptomic sample (SRA BioSample SAMN13762420). Mapping procedure used BWA-MEM 0.7.17 (Li, 2013) with the default settings, samclip 0.4.05 for the removal of mapped reads with hard- and soft-clipped flanking ends longer than 10 bp, samtools-1.9 (Li et al., 2009) for SAM/BAM format conversion and for filtering of mapped reads, and bedtools 2.29.2 (Quinlan and Hall, 2010) for the conversion of BAM into the BED format with the attached CIGAR string. Analysis was focused only on reads with mapping scores of 60, i.e., exact mapping to the unique position along the genome. The metagenomic sample SRR10968255 was omitted from mapping due to the low number of sequences.
The raw BWA-MEM 0.7.17 mapping files of the deadwood metagenomes to the S. ligni genome were used as inputs into the anvi’o 6.2 metagenomic pipeline to visualize the occurrence of S. ligni in individual deadwood age classes. No read filtering was applied to allow for single-nucleotide variants (SNVs) density calculation of S. ligni according to Reveillaud et al. (2019) (see “Code Availability” section). Similarly, metatranscriptomic reads from a previous study (Tláskal et al., 2021, unpublished) were mapped against the set of S. ligni genes to infer nitrogen fixation expression in decomposing wood. The data were summarized using R 4.0.0 (R Core Team, 2020) and tidyverse 1.3.0 package (Wickham et al., 2019). Significant differences were tested by the Kruskal-Wallis test using the agricolae package (de Mendiburu, 2017).
Insect Cell-Free Cultivation and Enzyme Activity
To assess growth on insect cell-free media containing wood and to exclude the possibility that the Sodalis isolates are obligatory symbionts of a deadwood-associated insect, wood pellets from fresh beech wood were milled using an Ultra Centrifugal Mill ZM 200 (Retsch, Germany); 0.3 g of fine wood dust was mixed with 30 mL of Ringer solution and autoclaved. Media were inoculated in duplicate with S. ligni colonies growing on plates. Inoculated liquid media containing wood as the only C source were left at 24°C on an orbital shaker for incubation. Aliquots of media were regularly sampled, and samples were immediately frozen. To quantify the increase in cell biomass, DNA from the aliquots was extracted using a DNeasy UltraClean Microbial Kit (Qiagen, Germany) according to the manufacturer’s protocol with an additional initial spin for wood dust removal. The bacterial rRNA gene copies in DNA were quantified by qPCR using the 1108f and 1132r primers (Wilmotte et al., 1993; Amann et al., 1995). Sanger sequencing from the primer 1492r (Lane, 1991) was used to confirm the presence of Sodalis-only DNA in the final aliquots.
The activity of the cell wall-associated fraction of enzymes was measured in cell suspension after 2 weeks of incubation in 50 mL of liquid GY-VL55 medium as described previously (Lasa et al., 2019). For the catalase and oxidase tests, bacterial cells from a GY-VL55 agar plate were transferred into 3% H2O2 solution and 1% N, N, N’, N’-tetramethyl-p-phenylenediamine dihydrochloride drops, respectively. The Biolog PM1 system was used according to the manufacturer’s instructions to identify growth on different C sources.
Results
S. ligni was able to grow on laboratory media in which beech wood was the only source of C, reaching a cell density up to 1.3 × 108 mL–1 within 4 weeks of incubation. This growth excluded the possibility that it is an obligate symbiont of deadwood-associated insects. Hybrid assembly of the Sodalis ligni dw23T provided 32 contigs (>200 bp, N50: 3.8 Mbp), a total genome size of 6.4 Mbp, GC content 54.96% and a mean coverage of 99×. Assembly of the Sodalis sp. strain dw96 provided 85 contigs (>200 bp, max 0.72 Mbp, N50: 0.25 Mbp), a total genome size of 5.9 Mbp, GC content 54.04% and a mean coverage of 52×. Genome completeness was 98.8 and 99.9% for the Sodalis ligni dw23T and strain dw96, respectively. Contamination was not detected in the assembled genomes. Sodalis ligni dw23T was placed within the genus Sodalis (P = 0.211) and belongs to a species not represented in the NCBI database of prokaryotic genomes (P = 0.0025).
We retrieved 11 Sodalis genomes from NCBI together with the strain metadata (Supplementary Table 1). Pangenome analysis divided the available Sodalis genomes into three distinct guilds based on gene frequency (Figure 1). One group consisted of two genomes of Sodalis glossinidius morsitans (strains Sg and ASM), which are tsetse fly endosymbionts. The two Sodalis ligni dw23T and strain dw96 isolated in this study from deadwood formed the second group together with the free-living Sodalis sp. strain 159R retrieved from NCBI as an anaerobic lignin degrader. The third group consisted of endosymbionts of hymenopteran, hemipteran, dipteran and coleopteran insects. The gene frequency, gene count and genome length of S. praecaptivus have high similarity with the third group of endosymbionts, thus placing this free-living strain distant from deadwood-associated Sodalis sp. strains dw23T and dw96 despite their relatively high 16S rRNA gene similarity (97.2 and 97.8%, respectively). The ANI between strain dw23T or strain dw96 and the rest of the analyzed genomes ranged from 76 to 78%, except for strain 159R (98 and 88%, respectively, Figure 1 and Supplementary Table 2). The ANI between strains dw23T and dw96 was 88%. The genome size of all Sodalis strains and their gene count were closely correlated (ρ = 0.82, P = 0.004), while the genome size and gene counts per Kbp were negatively correlated (ρ = -0.63, P = 0.04). Free-living strains exhibited the lowest coding density (0.87–0.90 genes per kbp) and longer genes than symbionts (947 ± 7 bp and 732 ± 39 bp, respectively, P < 0.01, Supplementary Table 1).
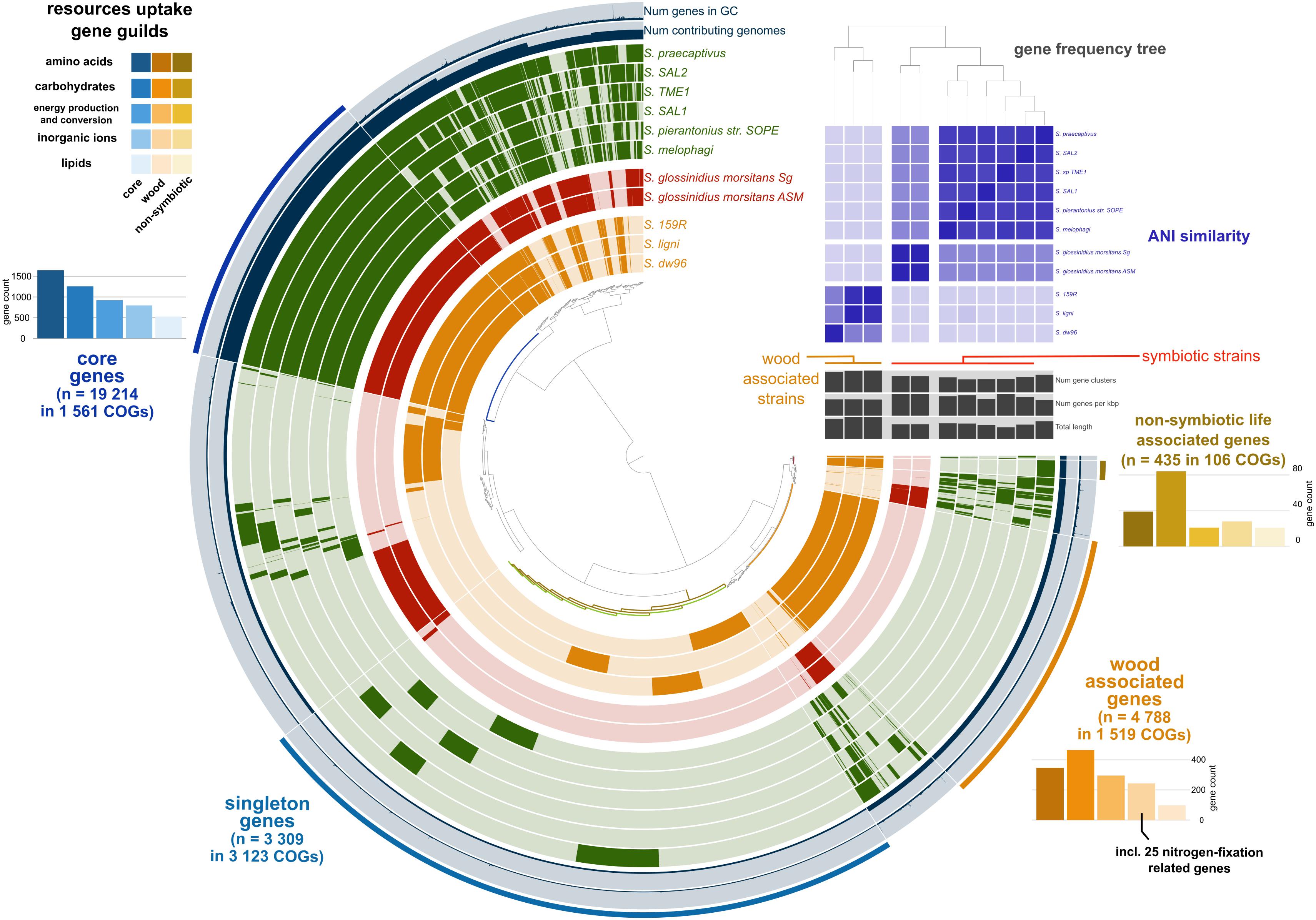
Figure 1. Pangenome analysis of the available Sodalis genomes. Genes (indicated by dark colors within the circles) were clustered based on orthologous similarity (inner tree), and genome layers were sorted based on a gene frequency tree (right upper tree). Sodalis guilds are color coded; deadwood-associated strains in orange, tsetse fly symbionts in red and insect symbionts with S. praecaptivus in green. ANI is indicated as the upper right heatmap. Pangenome analysis identified core genes shared by all genomes (bar plots in hues of blue), genes associated with non-symbiotic life (bar plots in hues of yellow) and genes associated with wood habitat (bar plots in hues of orange). Individual bar plots show counts of genes divided according to their function.
Pangenome analysis allowed us to identify clusters of orthologous genes (COGs) that were shared among strain guilds. Genes shared by all the genomes represent the core Sodalis pangenome (in average 1,747 genes per genome from 1,561 core gene clusters, 35.6% of genes from the average Sodalis genome, Figure 1). The core gene set shared with the Sodalis-like symbionts with extremely reduced genomes is represented by in average 130 genes from only 125 gene clusters. The COG categories with the highest share of classified core genes were translation, amino acid and carbohydrate metabolism, cell wall/membrane biosynthesis and replication categories (Figures 1, 2). Genes shared only among free-living strains were less frequent (in average 109 genes from 106 gene clusters; 2.0% per average free-living Sodalis genome) and were dominated by genes related to carbohydrate metabolism. Gene sharing among wood-associated strains was relatively common (in average 1,596 genes from 1,519 gene clusters, 28.4% per average wood-associated Sodalis genome), these genes were mostly classified in carbohydrate and amino acid utilization and transcription regulation COG categories. The nitrogenase gene nifH and genes encoding an ABC-type molybdate transport system were significantly enriched in the accessory genes of the wood-associated guild (adj. q-value = 0.01). Sodalis ligni dw23T, Sodalis sp. strain dw96 and Sodalis sp. strain 159R each contained nifHDK operons, while nitrogen fixation genes were absent in other Sodalis genomes. Similar to symbionts, wood-associated strains contained type III secretion system components.
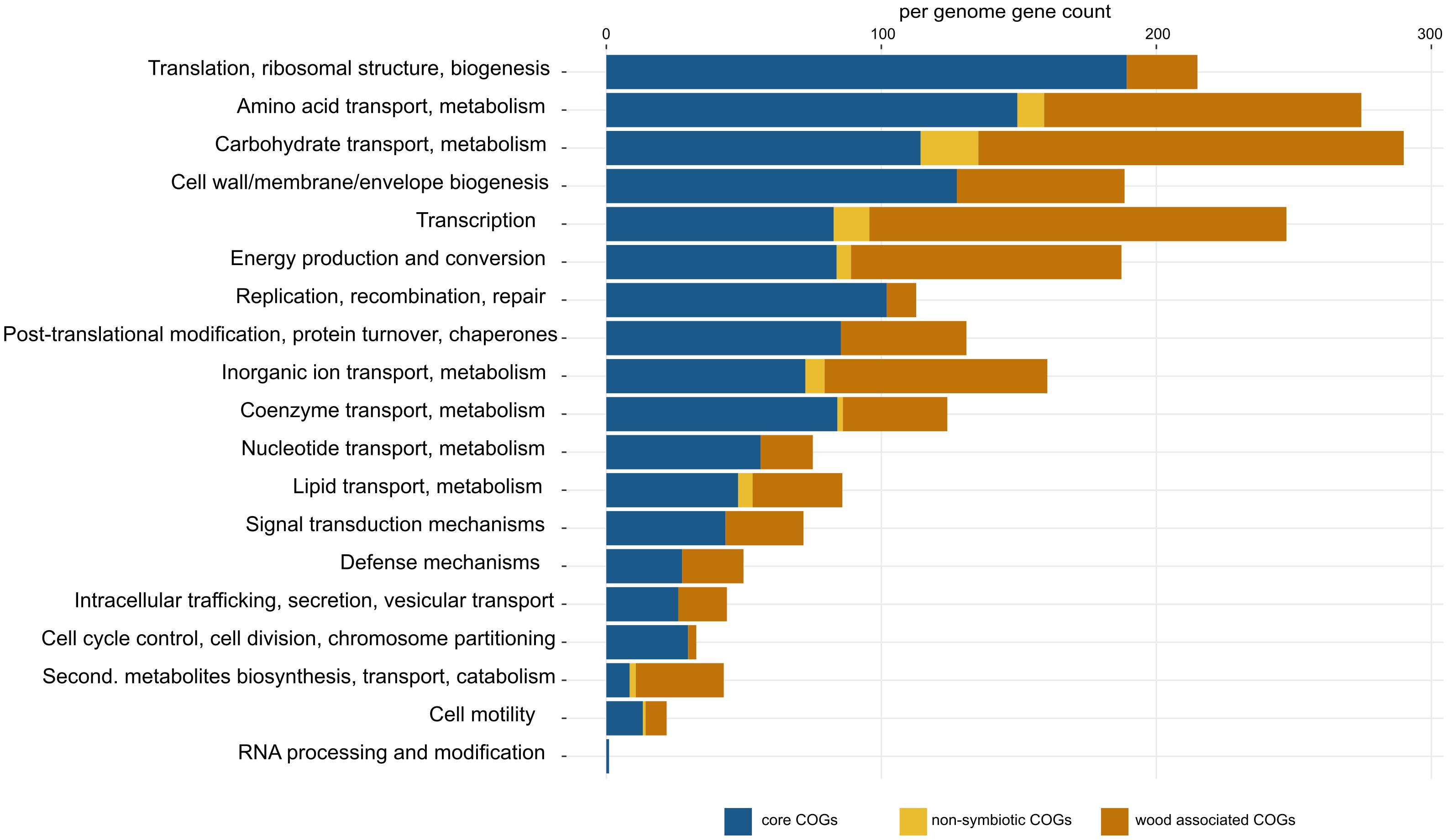
Figure 2. Detailed list of COG functions in Sodalis core genes (blue), non-symbiotic life-associated accessory genes (yellow) and wood-associated accessory genes (orange). Gene counts were corrected for the number of genomes in each group (core = 11, free-living = 4, wood-associated = 3). Per-genome numbers of additional genes with unknown function are not displayed, and their counts are: core = 403, free-living = 42, wood-associated = 651.
In total, 833 genes encoding carbohydrate-active enzymes from 66 CAZy families were identified across all the genomes. Free-living strains together with the symbiotic Sodalis sp. strain SAL2 contained the highest per-genome share of CAZy, with >1.62% of the total genes being identified as CAZymes (Supplementary Tables 3, 4). Wood-associated strains showed the highest diversity of CAZy, possessing >80% of the detected CAZy families each. The lowest diversity of CAZy (43.9% of detected families) was recorded in tsetse fly-associated strains. CAZy richness in individual genomes was correlated with CAZy abundance (ρ = 0.7, P = 0.02). The distribution of detected CAZymes followed Sodalis guild differentiation, with distinct CAZy patterns for wood-associated strains, tsetse fly symbionts and other symbionts (Figure 3A). Additionally, the functional annotation of CAZy families showed to some extent their conservation within Sodalis guilds (Figure 3B and Supplementary Table 4). In comparison to symbionts, wood-associated strains had more genes for α-glucan, cellobiose, xylobiose and chitin utilization. Genes for cellulose, pectin and hemicelluloses degradation were also detected; however, the counts of these CAZymes were comparable with those in the genomes of symbionts. Glycoconjugate utilization CAZymes were enriched in wood dwellers as well as free-living S. praecaptivus. Lytic polysaccharide monooxygenases (LPMOs) were not detected in the genomes of wood-associated strains.
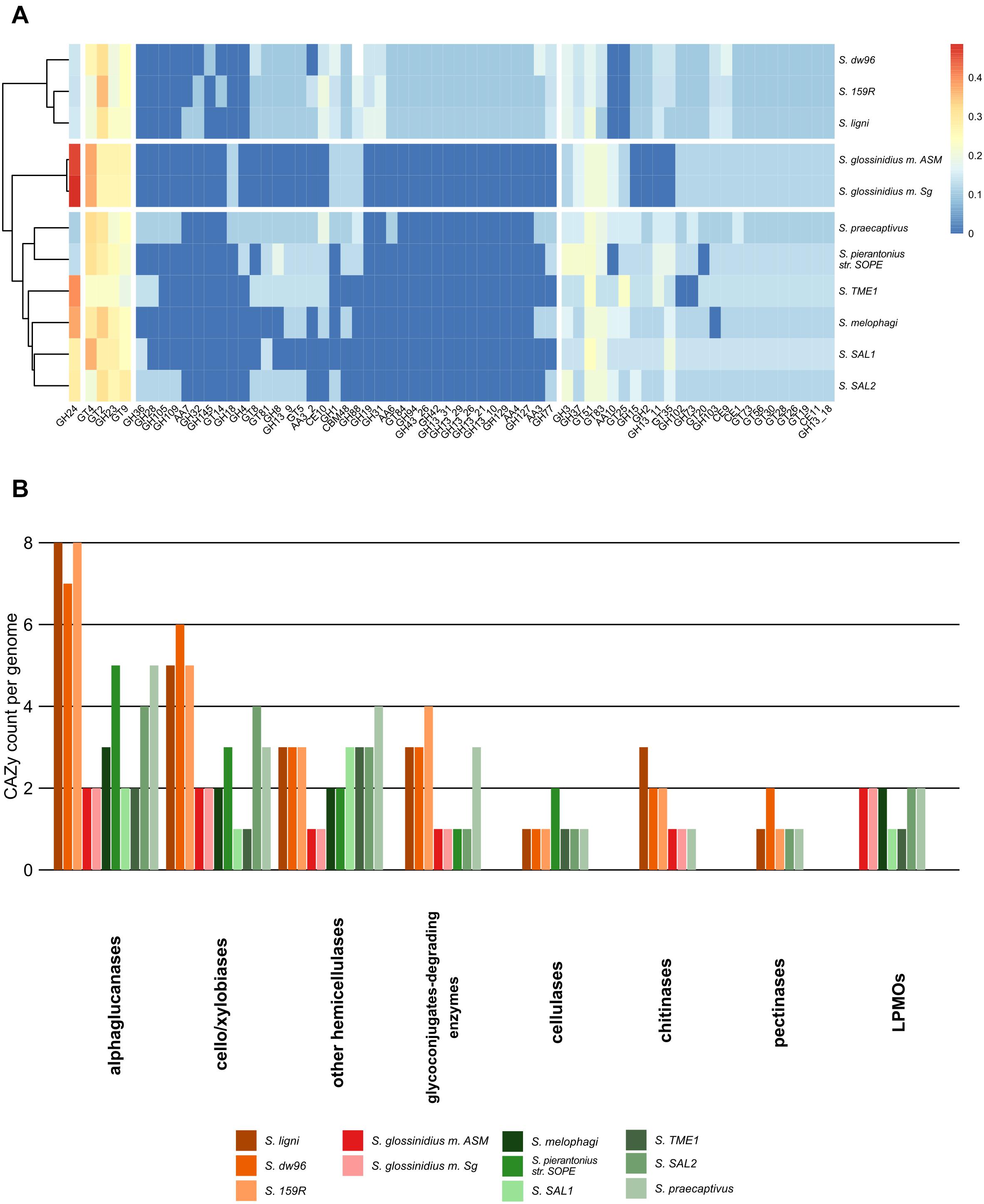
Figure 3. CAZy presence in the Sodalis genomes. (A) CAZy heatmap with clustering analysis shows the CAZyme abundance pattern, which follows Sodalis guild differentiation into wood-associated strains, tsetse symbionts and other symbiotic strains. Legend shows transformed counts. (B) Functional annotation of the CAZymes found in the genomes of Sodalis genomes. Genes for labile substrate utilization, such as cello/xylobiose and α-glucan, are increased in deadwood-associated strains.
16S rRNA genes assigned to Sodalis were present in other published community studies from deadwood (see the “Data Availability” section) and typically exhibited high relative abundance. The mean abundance of one Sodalis OTU related to strains dw23T and strain dw96 at 98.8% similarity was 2.79 ± 0.86% (n = 155, spruce deadwood in Finland, Schigel et al., unpublished data). Another Sodalis OTU (98% similarity to the presented genomes) was found on beech and spruce wood at 0.57–0.70% (Hoppe et al., 2015). When comparing deadwood of 13 tree species after 6 years of decomposition, the broadleaved species Betula sp., Populus spp., Carpinus sp. showed the highest abundance of Sodalis OTU (Supplementary Table 5; Moll et al., 2018). The abundance of other Sodalis OTU (99.6% similarity to the presented genomes) also differed in samples of spruce wood cubes (0.23 ± 0.16%, n = 26) and surrounding soil (0.004 ± 0.002%, n = 30, P < 0.001, Probst et al., 2018). Similarly, sequences assigned to Sodalis (100% similarity) were enriched in decomposing beech trees in Žofínský prales Nature Reserve (0.93 ± 0.48%, n = 24, P < 0.001), when compared to underlying soil (0.0004 ± 0.0002%, n = 48, Šamonil et al., 2020).
The exact mapping of the deadwood metagenomic data to the Sodalis genomes confirmed the higher abundances of the S. ligni, Sodalis sp. strain dw96 and Sodalis sp. strain 159R guild in a deadwood habitat than S. praecaptivus and S. pierantonius strain SOPE (P < 0.001, Figure 4A). For the three wood-associated strains, the average number of exactly mapped reads was 808 ± 220 reads per million reads (812 ± 397 reads per million for S. ligni only). S. ligni showed an even distribution throughout the gradient of deadwood decomposition stages when considering the proportion of mapped reads from the total reads available (Figure 4B). The rate of SNVs per one mapped read was 0.03 ± 0.01 (n = 23), and the density of SNVs against the S. ligni genome showed homogenous Sodalis populations in the deadwood metagenomes, with mean SNVs of 0.17 ± 0.07% except for the outlier population in one young deadwood metagenome (1.7% SNVs, Figure 4B). Most SNVs occurred in the third codon wobbling position (Supplementary Figure 1). Three distinct nifHDK operons were identified in the genome of Sodalis ligni. Two of these operons successfully called reads from two deadwood metatranscriptome samples (SRA accessions SRR10968245, SRR10968250). Notably, the same decomposing tree (BioSample SAMN13762420) served as the source of RNA for the latter metatranscriptome, as well as for the cultivation of S. ligni. Phylogenetic analysis of the nifH gene across the phylum Proteobacteria showed placement of Sodalis nitrogenase genes into distinct clades common to other Enterobacterales, with the exception of one Sodalis ligni nifH placed on a distant branch (Figure 5).
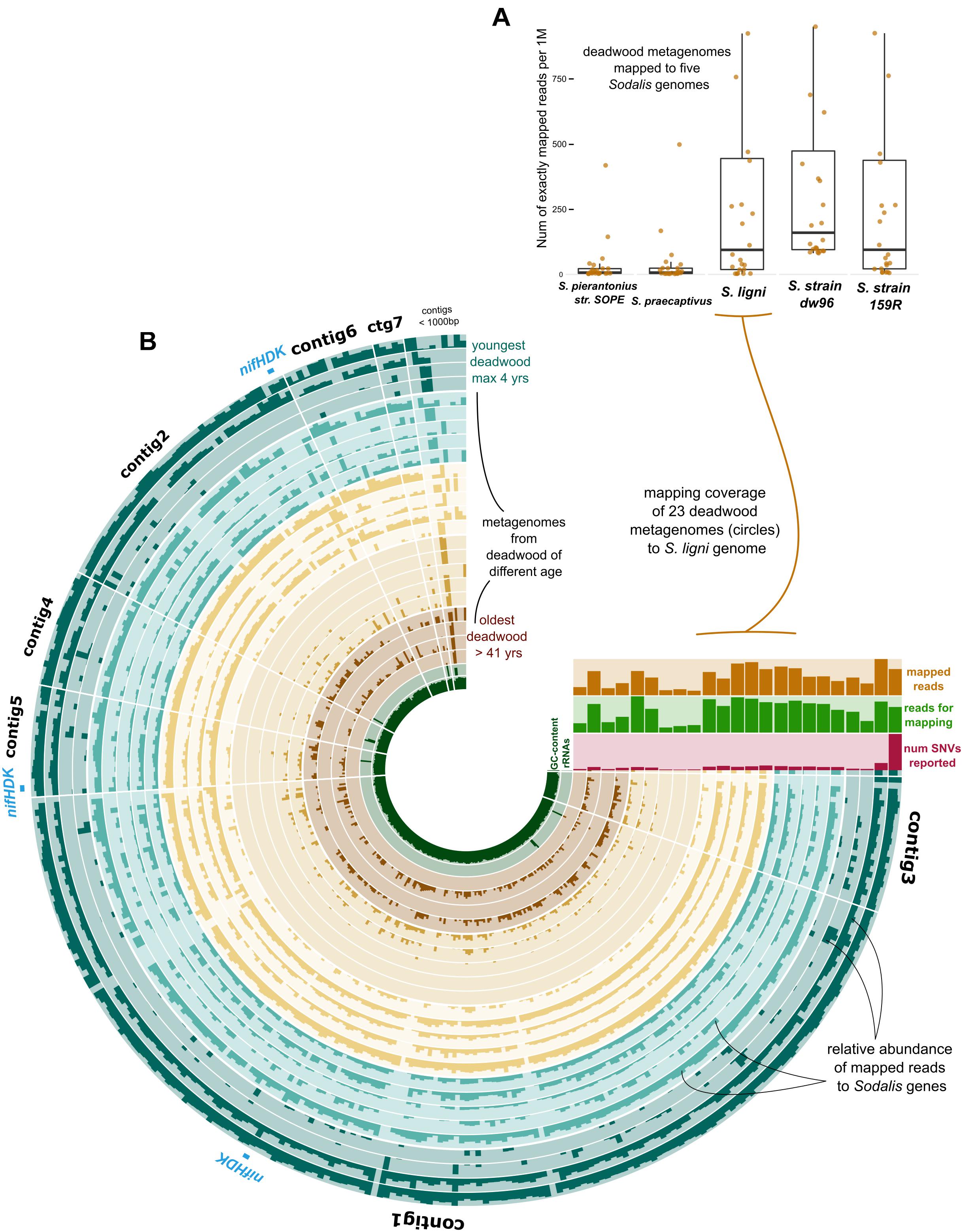
Figure 4. Deadwood metagenome mapping against the genome of S. ligni. (A) Boxplots show deadwood mapping count for wood-associated strains and S. pierantonius strain SOPE and S. praecaptivus. (B) The anvi’o plot with circles representing metagenome samples, the color coding of circles follows grouping of samples based on the length of deadwood decomposition. Dark bar plots within circles represent the relative abundance of reads mapped to S. ligni genes. Contigs were ordered based on the Euclidean distance and Ward linkage.
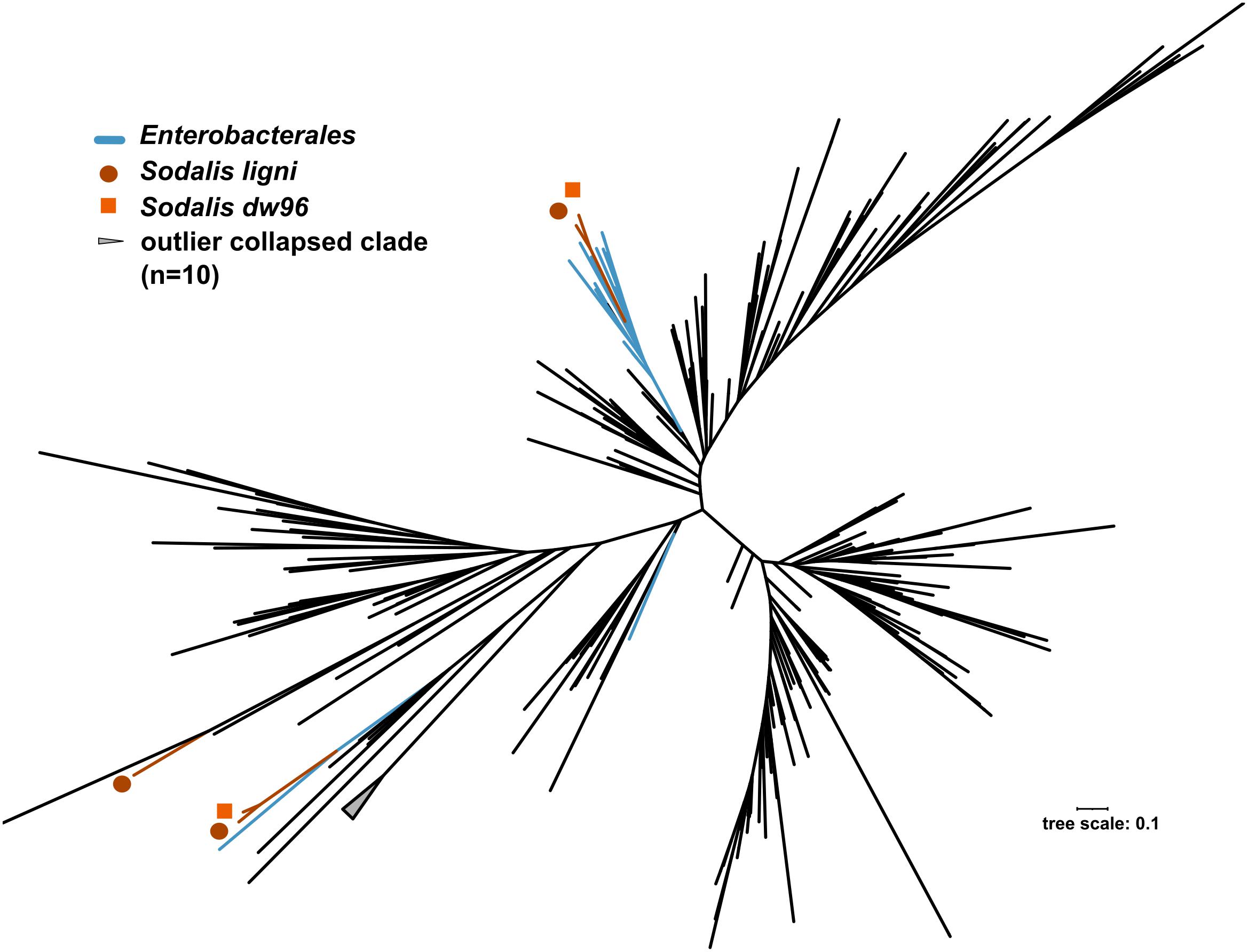
Figure 5. Phylogenetic tree based on nifH genes described from members of the phylum Proteobacteria. Blue branches denote Enterobacterales. Positions of the three nifH sequences from Sodalis ligni are labeled with dark brown circles, two of which are placed together with other enterobacteria. Two nifH sequences from Sodalis dw96 are labeled with bright brown squares, and both are placed together with other nifH from enterobacteria.
Description of Sodalis ligni sp. nov.
Sodalis ligni (lig’ni. L. gen. n. ligni of wood, referring to the isolation of the type strain from wood material).
Cells are aerobic, Gram-negative, cocci (0.6 μm) or short rods (1.2 × 0.6 μm). They grow separately without forming groups. Colonies on solid medium are circular, regularly edged, smooth, mucoid and white. Catalase positive and oxidase negative. The enzymatic in vitro assays were positive for β-glucosidase, β-xylosidase, N-acetyl-glucosaminidase, phosphodiesterase, acid phosphatase and lipase. L-serine, D-saccharic acid, N-acetyl-beta-D-mannosamine, D-cellobiose, D-galactose, L-fucose, fumaric acid, L-lactic acid, β-methyl-D-glucoside, adonitol, dulcitol, glycerol, L-galactonic acid-γ-lactone, and monomethyl succinate could be used as C sources. Optimal growth occurs on GY-VL55 medium (Lladó et al., 2019) and GY agar at pH 4.5–5.5 at 24°C.
The type strain is dw23T (=CECT 30299T = BCCM/LMG 32208T), isolated from decomposing wood of Fagus sylvatica L. in a temperate mixed forest, Central Europe. The genomic G + C content of the type strain is 54.96%.
Discussion
The genus Sodalis represents the model for the development of insect-associated lifestyle and host-symbiont interactions due to its evolutionarily young transition from a free-living to a symbiotic bacterium (Hall et al., 2020; Maire et al., 2020). Moreover, the tsetse fly symbiont Sodalis glossinidius is able to grow independently on its host under laboratory conditions, further pointing to the recent development of its non-facultative symbiosis (Goodhead et al., 2020). Until now, S. praecaptivus was the only free-living species described, which hints that there is a more diversified spectrum of life strategies than only association with insects (Clayton et al., 2012; Chari et al., 2015). Here, we describe two other free-living isolates from the Sodalis genus, one of which, strain dw23T, is suggested to represent the type strain for Sodalis ligni sp. nov. within the family Enterobacterales. The genome-to-genome alignment of the available Sodalis genomes shows ∼77% ANI with our strain, thus supporting the establishment of a new species within the Sodalis genus (Konstantinidis et al., 2006; Richter and Rosselló-Móra, 2009; Barco et al., 2020). This is supported also by comparison with NCBI database of prokaryotic genomes (Rodriguez-R et al., 2018). In contrast to Sodalis insect endosymbionts, S. ligni occupies deadwood and possesses genome characteristics connected with non-symbiotic life: a larger genome, a higher number of longer genes and a lower coding density (Toh et al., 2006; Oakeson et al., 2014). It refers to independent nutrient provisioning from more complex resources, the necessity for more sophisticated regulatory systems in fluctuating environments and a large effective population of free-living taxa in contrast to their symbiotic relatives (Giovannoni et al., 2014; Bobay and Ochman, 2018). We identified three ecological guilds within the genus Sodalis: tsetse endosymbionts, endosymbionts of other insect taxa and the wood-associated guild presented here. This differentiation is consistent based on ANI and on the functional genome content when considering either all genes or their subset CAZymes.
S. ligni appears in multiple 16S rRNA-based deadwood studies and is usually ranked among the most abundant taxa. Due to the multi-copy character of the 16S rRNA gene (Větrovský and Baldrian, 2013), amplicon studies might provide imperfect abundance estimates. This is true especially for the family Enterobacterales, as the mean number of 16S rRNA genes is 7 ± 0.5 (n = 3,130, Stoddard et al., 2015), which overestimates their abundance when comparing relative counts of 16S rRNA. Seven copies of 16S rRNA were also confirmed in the genome of S. ligni. Hybrid assembly using short and long reads helped to identify their individual copies, which are otherwise usually merged into one consensus copy by the assembler when assembling short reads only (Waters et al., 2018).
Despite the limitations of abundance estimates, amplicon studies showed clear habitat selection of S. ligni, which preferentially colonizes deadwood rather than the underlying soil (Probst et al., 2018; Šamonil et al., 2020). Moreover, Sodalis-related metagenome-assembled genomes are also absent in the recent large-scale cultivation-independent genome catalog, suggesting a low abundance of S. ligni in global soils (Nayfach et al., 2020). We therefore propose that S. ligni is a member of a wood-associated Sodalis guild. The independence of S. ligni from insect hosts is supported by its ability to grow solely on wood. The wood-associated guild further contains the other two isolates, Sodalis strain dw96 and Sodalis strain 159R, the former of which was identified in the present study and the latter was identified in an unpublished study. Other related Sodalis members, the previously described free-living S. praecaptivus and symbiont Sodalis pierantonius strain SOPE, do not show a deadwood preference (Oakeson et al., 2014). While for the symbiont this is not surprising, the main habitat and ecological functions of S. praecaptivus remain to be identified, considering its genome composition (which is closer to symbionts than to wood-associated strains), wood cultivation source and ability to grow in human and insect tissues (Clayton et al., 2012; Enomoto et al., 2017; Munoz et al., 2020). Based on the presence of virulence factors, S. praecaptivus may represent a plant pathogen transmitted by insects (Clayton et al., 2012) and thus may prefer living plants rather than decomposing wood.
Preferential colonization of wood of particular tree species by S. ligni might occur, as was observed in the data from conifers and broadleaved trees (Hoppe et al., 2015; Moll et al., 2018). However, further research across more deadwood types is needed to disentangle potential tree species selection. The abundance of S. ligni in decomposing wood does not seem to vary over tens of years, which is a typical lifetime for decomposing trees in temperate forests (Přívětivý et al., 2016), and the S. ligni population is homogenous with regard to SNVs density.
The core genes shared by all Sodalis strains contain genes for crucial cell functions, mainly translation, replication, transcription, amino acid and carbohydrate metabolism and cell wall/membrane biosynthesis. Furthermore, pangenome analysis unveiled a relatively low number of genes specific for non-symbiotic life, while accessory genes specific for wood-associated guild represent a significant portion of the total identified genes and are involved in, e.g., amino acid and carbohydrate metabolism. Such gene-specific enrichment is connected with a more diverse set of nutrient resources in deadwood habitats. The presence of type III secretion system genes in wood-associated strains might serve as an evolutionary predisposition for host infection by related symbiotic Sodalis members (Maire et al., 2020). The composition of CAZymes of wood-associated strains differentiates them from symbionts as well. Substrates targeted by CAZymes show that wood-associated Sodalis members utilize rather labile plant polymers or chitin to obtain C and do not serve as important cellulose degraders. The broader degradation capabilities were described for other deadwood-associated bacterial taxa such as those from the phyla Acidobacteria and Bacteroidetes (Tláskal and Baldrian, 2021). The ecological function of free-living Sodalis strains might be resolved based on the multiplied presence of nifHDK operons expressing nitrogenase, a key enzyme for nitrogen fixation which is dependent on the metal cofactors (e.g., molybdenum). The sequence difference between nitrogenase variants shows the importance of these genes for the bacterium. To reduce atmospheric nitrogen is energetically costly, and thus, there is probably a selection pressure in the Sodalis deadwood guild to not combine nitrogen fixation with resource-intensive degradation of recalcitrant plant polymers. Nitrogen fixation within the family Enterobacterales is known from taxa living in association with plants (Rosenblueth et al., 2004), with leaf-cutter ant fungus gardens (Pinto-Tomás et al., 2009) and with Diptera (Behar et al., 2005). Deadwood was previously described as a hotspot of nitrogen fixation by which bacteria enrich material with a low nitrogen content and relieve the nitrogen limitation on microbial colonization (Tláskal et al., 2021). Deadwood-associated S. ligni thus represents a candidate for nitrogen fixation and one of the rarely described free-living nitrogen fixing Enterobacterales members.
To conclude, we identified S. ligni as a member of the wood-associated Sodalis guild, and we provide a comprehensive analysis of its genome potential, substrate utilization and ecological function in the context of other known isolated Sodalis sensu stricto strains. Members of the Sodalis genus show broad life strategies with distinct specialization, indicating all-rounder character of this genus, including free-living deadwood inhabitants, non-symbiotic strains with the ability to colonize internal insect and human tissues and recently established symbionts in several insect orders. Such an extensive niche differentiation is known from another enterobacterial species, Escherichia coli (Tenaillon et al., 2010; Luo et al., 2011), but has not been described for Sodalis-related taxa. There is not enough evidence and more isolates from related sources are needed to determine whether deadwood served as a reservoir pool for symbiotic bacteria diversification via genome reduction or whether there was another common ancestor for both wood-associated and insect-associated guilds.
Code Availability
The above methods indicate the programs used for analysis within the relevant sections. The code for reproducing all sequence processing is provided at https://github.com/TlaskalV/Deadwood-Sodalis.
Data Availability Statement
Data described in this manuscript, including raw sequences from short-read sequencing and genome assembly files, have been deposited in the NCBI under BioProject accession numbers PRJNA599932 (Sodalis ligni dw23T) and PRJNA613885 (Sodalis sp. strain dw96). We further deposited MinION long-read sequencing data in ENA nucleotide archive (PRJEB43208), 16S rRNA sequences of Sodalis clustered in other deadwood studies (10.6084/m9.figshare.13227464), anvi’o pangenome databases (10.6084/m9.figshare.13221065), anvi’o deadwood metagenome mapping databases (10.6084/m9.figshare.13221074) and nifH tree in Newick format (10.6084/m9.figshare.14562279).
Author Contributions
VT conceived the study with input from PB. VT performed the strain isolation, pangenome analysis, metagenomic mapping, and strain characterization. LŽ and VP performed the library preparation and sequencing, genome assembly, and annotation. VT and PB wrote the original version of the manuscript. All authors contributed to the final version of the manuscript.
Funding
This work was supported by the Czech Science Foundation (21-09334J).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.668644/full#supplementary-material
Supplementary Table 1 | List of available Sodalis genomes together with genome, life strategy metadata and description of referencing studies.
Supplementary Table 2 | ANI statistics calculated for 11 available Sodalis genomes.
Supplementary Table 3 | Total count, gene share and richness of CAZymes identified in Sodalis genomes. Deadwood-associated strains followed by S. praecaptivus contain the highest number of diverse CAZy.
Supplementary Table 4 | Per family counts of CAZymes and their assigned activities in individual genomes.
Supplementary Table 5 | Relative abundances of a Sodalis OTU with 99.6% similarity to S. ligni among deadwood of 13 tree species after 6 years of decomposition from the study Moll et al. (2018). Data represent means and standard errors.
Supplementary Figure 1 | Single-nucleotide variants (SNVs) detected after mapping deadwood metagenome reads to the S. ligni genome shown as boxplots of the occurrence of SNVs for each codon position. Each dot represents one metagenome sample.
Footnotes
- ^ https://community.nanoporetech.com
- ^ https://github.com/rrwick/Porechop
- ^ https://github.com/kblin/ncbi-genome-download
- ^ https://inkscape.org/
- ^ https://github.com/tseemann/samclip
References
Akman, L., Yamashita, A., Watanabe, H., Oshima, K., Shiba, T., Hattori, M., et al. (2002). Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat. Genet. 32, 402–407. doi: 10.1038/ng986
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Amann, R. I., Ludwig, W., and Schleifer, K.-H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169. doi: 10.1128/mr.59.1.143-169.1995
Anderson-Teixeira, K. J., Davies, S. J., Bennett, A. C., Muller-landau, H. C., and Wright, S. J. (2015). CTFS-ForestGEO: a worldwide network monitoring forests in an era of global change. Glob. Chang. Biol. 21, 528–549. doi: 10.1111/gcb.12712
Baldrian, P., Zrůstová, P., Tláskal, V., Davidová, A., Merhautová, V., and Vrška, T. (2016). Fungi associated with decomposing deadwood in a natural beech-dominated forest. Fungal Ecol. 23, 109–122. doi: 10.1016/j.funeco.2016.07.001
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barco, R. A., Garrity, G. M., Scott, J. J., Amend, J. P., Nealson, K. H., and Emerson, D. (2020). A genus definition for Bacteria and Archaea based on a standard genome relatedness index. mBio 11:e002475-19. doi: 10.1128/mBio.02475-19
Behar, A., Yuval, B., and Jurkevitch, E. (2005). Enterobacteria-mediated nitrogen fixation in natural populations of the fruit fly Ceratitis capitata. Mol. Ecol. 14, 2637–2643. doi: 10.1111/j.1365-294X.2005.02615.x
Bobay, L.-M., and Ochman, H. (2018). Factors driving effective population size and pan-genome evolution in bacteria. BMC Evol. Biol. 18:153. doi: 10.1186/s12862-018-1272-4
Boyd, B. M., Allen, J. M., Koga, R., Fukatsu, T., Sweet, A. D., Johnson, K. P., et al. (2016). Two bacterial genera, Sodalis and Rickettsia, associated with the Seal Louse Proechinophthirus fluctus (Phthiraptera: Anoplura). Appl. Environ. Microbiol. 82, 3185–3197. doi: 10.1128/AEM.00282-16
Chari, A., Oakeson, K. F., Enomoto, S., Grant Jackson, D., Fisher, M. A., and Dale, C. (2015). Phenotypic characterization of Sodalis praecaptivus sp. nov., a close non-insect-associated member of the Sodalis-allied lineage of insect endosymbionts. Int. J. Syst. Evol. Microbiol. 65, 1400–1405. doi: 10.1099/ijs.0.000091
Chen, X., Li, S., and Aksoy, S. (1999). Concordant evolution of a symbiont with its host insect species: molecular phylogeny of genus Glossina and its bacteriome-associated endosymbiont, Wigglesworthia glossinidia. J. Mol. Evol. 48, 49–58. doi: 10.1007/PL00006444
Chrudimský, T., Husník, F., Nováková, E., and Hypša, V. (2012). Candidatus Sodalis melophagi sp. nov.: phylogenetically independent comparative model to the tsetse fly symbiont Sodalis glossinidius. PLoS One 7:e40354. doi: 10.1371/journal.pone.0040354
Clayton, A. L., Oakeson, K. F., Gutin, M., Pontes, A., Dunn, D. M., von Niederhausern, A. C., et al. (2012). A novel human-infection-derived bacterium provides insights into the evolutionary origins of mutualistic insect-bacterial symbioses. PLoS Genet. 8:e1002990. doi: 10.1371/journal.pgen.1002990
de Mendiburu, F. (2017). Agricolae: Statistical Procedures for Agricultural Research. R Package Version 1.2-4.
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Elo, S., Maunuksela, L., Salkinoja-Salonen, M., Smolander, A., and Haahtela, K. (2000). Humus bacteria of Norway spruce stands: plant growth promoting properties and birch, red fescue and alder colonizing capacity. FEMS Microbiol. Ecol. 31, 143–152. doi: 10.1111/j.1574-6941.2000.tb00679.x
Enomoto, S., Chari, A., Clayton, A. L., and Dale, C. (2017). Quorum sensing attenuates virulence in Sodalis praecaptivus. Cell Host Microb. 21, 629–636. doi: 10.1016/j.chom.2017.04.003
Eren, A. M., Esen, ÖC., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platform for ‘Omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319
Fukatsu, T., Koga, R., Smith, W. A., Tanaka, K., Nikoh, N., Sasaki-Fukatsu, K., et al. (2007). Bacterial endosymbiont of the slender pigeon louse, Columbicola columbae, allied to endosymbionts of grain weevils and tsetse flies. Appl. Environ. Microbiol. 73, 6660–6668. doi: 10.1128/AEM.01131-07
Giovannoni, S. J., Cameron Thrash, J., and Temperton, B. (2014). Implications of streamlining theory for microbial ecology. ISME J. 8, 1553–1565. doi: 10.1038/ismej.2014.60
Goodhead, I., Blow, F., Brownridge, P., Hughes, M., Kenny, J., Krishna, R., et al. (2020). Large-scale and significant expression from pseudogenes in Sodalis glossinidius - A facultative bacterial endosymbiont. Microb. Genom. 6:e000285. doi: 10.1099/mgen.0.000285
Hall, R. J., Flanagan, L. A., Wood, A. J., Thomas, H., Springthorpe, V., Thorpe, S., et al. (2019). A tale of three species: adaptation of Sodalis glossinidius to tsetse biology, Wigglesworthia metabolism, and host diet. mBio 10:e002106-18. doi: 10.1128/mBio.02106-18
Hall, R. J., Thorpe, S., Thomas, G. H., and Wood, A. J. (2020). Simulating the evolutionary trajectories of metabolic pathways for insect symbionts in the genus Sodalis. Microb. Genomics 6:mgen000378. doi: 10.1099/mgen.0.000378
Heddi, A., Grenier, A. M., Khatchadourian, C., Charles, H., and Nardon, P. (1999). Four intracellular genomes direct weevil biology: nuclear, mitochondrial, principal endosymbiont, and Wolbachia. Proc. Natl. Acad. Sci. U.S.A. 96, 6814–6819. doi: 10.1073/pnas.96.12.6814
Hoang, D. T., Chernomor, O., Haeseler, A., Von Minh, B. Q., and Vinh, L. S. (2017). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hoppe, B., Krüger, D., Kahl, T., Arnstadt, T., Buscot, F., Bauhus, J., et al. (2015). A pyrosequencing insight into sprawling bacterial diversity and community dynamics in decaying deadwood logs of Fagus sylvatica and Picea abies. Sci. Rep. 5:9456. doi: 10.1038/srep09456
Kaiwa, N., Hosokawa, T., Kikuchi, Y., Nikoh, N., Meng, X. Y., Kimura, N., et al. (2010). Primary gut symbiont and secondary, Sodalis-allied symbiont of the scutellerid stinkbug Cantao ocellatus. Appl. Environ. Microbiol. 76, 3486–3494. doi: 10.1128/AEM.00421-10
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., Rozewicki, J., and Yamada, K. D. (2018). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Konstantinidis, K. T., Ramette, A., and Tiedje, J. M. (2006). The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 361, 1929–1940. doi: 10.1098/rstb.2006.1920
Král, K., Janík, D., Vrška, T., Adam, D., Hort, L., Unar, P., et al. (2010). Local variability of stand structural features in beech dominated natural forests of Central Europe: implications for sampling. For. Ecol. Manag. 260, 2196–2203. doi: 10.1016/j.foreco.2010.09.020
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY: Wiley).
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Lasa, A. V., Mašínová, T., Baldrian, P., and Fernández-López, M. (2019). Bacteria from the endosphere and rhizosphere of Quercus spp. use mainly cell wall-associated enzymes to decompose organic matter. PLoS One 14:e0214422. doi: 10.1371/journal.pone.0214422
Letunic, I., and Bork, P. (2011). Interactive Tree of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478. doi: 10.1093/nar/gkr201
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint]. Available online at: http://arxiv.org/abs/1303.3997
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352 (accessed May 11, 2019).
Lladó, S. F., Větrovský, T., and Baldrian, P. (2019). Tracking of the activity of individual bacteria in temperate forest soils shows guild-specific responses to seasonality. Soil Biol. Biochem. 135, 275–282. doi: 10.1016/j.soilbio.2019.05.010
Luo, C., Walk, S. T., Gordon, D. M., Feldgarden, M., Tiedje, J. M., and Konstantinidis, K. T. (2011). Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc. Natl. Acad. Sci. U.S.A. 108, 7200–7205. doi: 10.1073/pnas.1015622108
Maire, J., Parisot, N., Ferrarini, M. G., Vallier, A., Gillet, B., Hughes, S., et al. (2020). Spatial and morphological reorganization of endosymbiosis during metamorphosis accommodates adult metabolic requirements in a weevil. Proc. Natl. Acad. Sci. U.S.A. 117, 19347–19358. doi: 10.1073/pnas.2007151117
Moll, J., Kellner, H., Leonhardt, S., Stengel, E., Dahl, A., Buscot, F., et al. (2018). Bacteria inhabiting deadwood of 13 tree species reveal great heterogeneous distribution between sapwood and heartwood. Environ. Microbiol. 20, 3744–3756. doi: 10.1111/1462-2920.14376
Moynihan, M. A. (2020). nifHdada2 GitHub Repository. Zenodo. Available online at: https://zenodo.org/record/3958405/export/xm#.YKU5vbczbIU (accessed January 20, 2021).
Munoz, M. M., Spencer, N., Enomoto, S., Dale, C., and Rio, R. V. M. (2020). Quorum sensing sets the stage for the establishment and vertical transmission of Sodalis praecaptivus in tsetse flies. PLoS Genet. 16:e1008992. doi: 10.1371/journal.pgen.1008992
Nadarasah, G., and Stavrinides, J. (2011). Insects as alternative hosts for phytopathogenic bacteria. FEMS Microbiol. Rev. 35, 555–575. doi: 10.1111/j.1574-6976.2011.00264.x
Nayfach, S., Roux, S., Seshadri, R., Udwary, D., Varghese, N., Schulz, F., et al. (2020). A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 39, 499–509. doi: 10.1038/s41587-020-0718-6
Nguyen, L., Schmidt, H. A., Haeseler, A., and Von Minh, B. Q. (2014). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nováková, E., and Hypša, V. (2007). A new Sodalis lineage from bloodsucking fly Craterina melbae (Diptera, Hippoboscoidea) originated independently of the tsetse flies symbiont Sodalis glossinidius. FEMS Microbiol. Lett. 269, 131–135. doi: 10.1111/j.1574-6968.2006.00620.x
Oakeson, K. F., Gil, R., Clayton, A. L., Dunn, D. M., Von Niederhausern, A. C., Hamil, C., et al. (2014). Genome degeneration and adaptation in a nascent stage of symbiosis. Genome Biol. Evol. 6, 76–93. doi: 10.1093/gbe/evt210
Odriozola, I., Abrego, N., Tláskal, V., Zrůstová, P., Morais, D., Větrovský, T., et al. (2021). Fungal communities are important determinants of bacterial community composition in deadwood. mSystems 6:e01017-20. doi: 10.1128/mSystems.01017-20
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Perlmutter, J. I., and Bordenstein, S. R. (2020). Microorganisms in the reproductive tissues of arthropods. Nat. Rev. Microbiol. 18, 97–111. doi: 10.1038/s41579-019-0309-z
Pinto-Tomás, A. A., Anderson, M. A., Suen, G., Stevenson, D. M., Chu, F. S. T., Wallace Cleland, W., et al. (2009). Symbiotic nitrogen fixation in the fungus gardens of leaf-cutter ants. Science 326, 1120–1123. doi: 10.1126/science.1173036
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2015). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Přívětivý, T., Janík, D., Unar, P., Adam, D., Král, K., and Vrška, T. (2016). How do environmental conditions affect the deadwood decomposition of European beech (Fagus sylvatica L.)? For. Ecol. Manag. 381, 177–187. doi: 10.1016/j.foreco.2016.09.033
Probst, M., Gómez-Brandón, M., Bardelli, T., Egli, M., Insam, H., and Ascher-Jenull, J. (2018). Bacterial communities of decaying Norway spruce follow distinct slope exposure and time-dependent trajectories. Environ. Microbiol. 20, 3657–3670. doi: 10.1111/1462-2920.14359
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Reveillaud, J., Bordenstein, S. R., Cruaud, C., Shaiber, A., Esen, ÖC., Weill, M., et al. (2019). The Wolbachia mobilome in Culex pipiens includes a putative plasmid. Nat. Commun. 10:1051. doi: 10.1038/s41467-019-08973-w
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rodriguez-R, L. M., Gunturu, S., Harvey, W. T., Rosselló-Mora, R., Tiedje, J. M., Cole, J. R., et al. (2018). The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 46, W282–W288. doi: 10.1093/nar/gky467
Rosas-Pérez, T., Vera-Ponce de León, A., Rosenblueth, M., Ramírez-Puebla, S. T., Rincón-Rosales, R., Martínez-Romero, J., et al. (2017). “The symbiome of Llaveia cochineals (Hemiptera: Coccoidea: Monophlebidae) includes a gammaproteobacterial cosymbiont Sodalis TME1 and the known Candidatus Walczuchella monophlebidarum,” in Insect Physiology and Ecology, ed. V. D. C. Shields (London: IntechOpen), 115–134. doi: 10.5772/66442
Rosenblueth, M., Martínez, L., Silva, J., and Martínez-Romero, E. (2004). Klebsiella variicola, a novel species with clinical and plant-associated isolates. Syst. Appl. Microbiol. 27, 27–35. doi: 10.1078/0723-2020-00261
Rubin, B. E. R., Sanders, J. G., Turner, K. M., Pierce, N. E., and Kocher, S. D. (2018). Social behaviour in bees influences the abundance of Sodalis (Enterobacteriaceae) symbionts. R. Soc. Open Sci. 5:180369. doi: 10.1098/rsos.180369
Sabree, Z. L., Kambhampati, S., and Moran, N. A. (2009). Nitrogen recycling and nutritional provisioning by Blattabacterium, the cockroach endosymbiont. Proc. Natl. Acad. Sci. U.S.A. 106, 19521–19526. doi: 10.1073/pnas.0907504106
Šamonil, P., Daněk, P., Baldrian, P., Tláskal, V., Tejnecký, V., and Drábek, O. (2020). Convergence, divergence or chaos? Consequences of tree trunk decay for pedogenesis and the soil microbiome in a temperate natural forest. Geoderma 376:114499. doi: 10.1016/j.geoderma.2020.114499
Šamonil, P., Schaetzl, R. J., Valtera, M., Goliáš, V., Baldrian, P., Vašíčková, I., et al. (2013). Crossdating of disturbances by tree uprooting: can treethrow microtopography persist for 6000 years? For. Ecol. Manag. 307, 123–135. doi: 10.1016/j.foreco.2013.06.045
Santos-Garcia, D., Silva, F. J., Morin, S., Dettner, K., and Kuechler, S. M. (2017). The all-rounder Sodalis: a new bacteriome-associated endosymbiont of the lygaeoid bug Henestaris halophilus (Heteroptera: Henestarinae) and a critical examination of its evolution. Genome Biol. Evol. 9, 2893–2910. doi: 10.1093/gbe/evx202
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Stoddard, S. F., Smith, B. J., Hein, R., Roller, B. R. K., and Schmidt, T. M. (2015). rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 43, D593–D598. doi: 10.1093/nar/gku1201
Tenaillon, O., Skurnik, D., Picard, B., and Denamur, E. (2010). The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 8, 207–217. doi: 10.1038/nrmicro2298
Tláskal, V., and Baldrian, P. (2021). Deadwood-inhabiting bacteria show adaptations to changing carbon and nitrogen availability during decomposition. Front. Microbiol. 12, 1353. doi: 10.3389/fmicb.2021.685303
Tláskal, V., Brabcová, V., Větrovský, T., Jomura, M., López-Mondéjar, R., Monteiro, M. O. L., et al. (2021). Complementary roles of wood-inhabiting fungi and bacteria facilitate deadwood decomposition. mSystems 6:e001078-20. doi: 10.1128/mSystems.01078-20
Tláskal, V., Zrůstová, P., Vrška, T., and Baldrian, P. (2017). Bacteria associated with decomposing dead wood in a natural temperate forest. FEMS Microbiol. Ecol. 93:fix157. doi: 10.1093/femsec/fix157
Toh, H., Weiss, B. L., Perkin, S. A. H., Yamashita, A., Oshima, K., Hattori, M., et al. (2006). Massive genome erosion and functional adaptations provide insights into the symbiotic lifestyle of Sodalis glossinidius in the tsetse host. Genome Res. 16, 149–156. doi: 10.1101/gr.4106106
Větrovský, T., and Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly omprovement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Waters, N. R., Abram, F., Brennan, F., Holmes, A., and Pritchard, L. (2018). riboSeed: leveraging prokaryotic genomic architecture to assemble across ribosomal regions. Nucleic Acids Res. 46:e68. doi: 10.1093/nar/gky212
Weiss, B. L., Mouchotte, R., Rio, R. V. M., Wu, Y. N., Wu, Z., Heddi, A., et al. (2006). Interspecific transfer of bacterial endosymbionts between tsetse fly species: infection establishment and effect on host fitness. Appl. Environ. Microbiol. 72, 7013–7021. doi: 10.1128/AEM.01507-06
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wickham, H., Averick, M., Bryan, J., Chang, W., Mcgowan, L. D. A., François, R., et al. (2019). Welcome to the Tidyverse. J. Open Source Softw. 4:1686. doi: 10.21105/joss.01686
Wilmotte, A., Van der Auwera, G., and De Wachter, R. (1993). Structure of the 16S ribosomal RNA of the thermophilic cyanobacterium Chlorogloeopsis HTF (‘Mastigocladus laminosus HTF’) strain PCC7518, and phylogenetic analysis. FEBS Lett. 317, 96–100. doi: 10.1016/0014-5793(93)81499-P
Yong, E. (2016). I Contain Multitudes: The Microbes Within us and a Grander View of Life. London: HarperCollins.
Keywords: insect symbionts, Sodalis, deadwood, nitrogen fixation, free-living, non-symbiotic
Citation: Tláskal V, Pylro VS, Žifčáková L and Baldrian P (2021) Ecological Divergence Within the Enterobacterial Genus Sodalis: From Insect Symbionts to Inhabitants of Decomposing Deadwood. Front. Microbiol. 12:668644. doi: 10.3389/fmicb.2021.668644
Received: 16 February 2021; Accepted: 17 May 2021;
Published: 11 June 2021.
Edited by:
Paolina Garbeva, Netherlands Institute of Ecology (NIOO-KNAW), NetherlandsReviewed by:
Eva Novakova, University of South Bohemia in České Budějovice, CzechiaRosario Gil, University of Valencia, Spain
Copyright © 2021 Tláskal, Pylro, Žifčáková and Baldrian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vojtěch Tláskal, dGxhc2thbEBiaW9tZWQuY2FzLmN6