 Adolfo Isla1,2,3†
Adolfo Isla1,2,3† J. Eduardo Martinez-Hernandez4,5,6†
J. Eduardo Martinez-Hernandez4,5,6† Héctor A. Levipan7
Héctor A. Levipan7 Denise Haussmann3
Denise Haussmann3 Jaime Figueroa1,2
Jaime Figueroa1,2 Maria Cecilia Rauch1
Maria Cecilia Rauch1 Vinicius Maracaja-Coutinho4,8,9*
Vinicius Maracaja-Coutinho4,8,9* Alejandro Yañez2,10*
Alejandro Yañez2,10*- 1Instituto de Bioquímica y Microbiología, Universidad Austral de Chile, Valdivia, Chile
- 2Interdisciplinary Center for Aquaculture Research (INCAR), University of Concepcion, Concepción, Chile
- 3Departamento de Ciencias Básicas, Facultad de Ciencias, Universidad Santo Tomás, Santiago, Chile
- 4Centro de Modelamiento Molecular, Biofísica y Bioinformática – CM2B2, Facultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile, Santiago, Chile
- 5Programa de Doctorado en Genómica Integrativa, Vicerrectoría de Investigación, Universidad Mayor, Santiago, Chile
- 6Laboratorio de Biología de Redes, Centro de Genómica y Bioinformática, Facultad de Ciencias, Universidad Mayor, Santiago, Chile
- 7Laboratorio de Ecopatología y Nanobiomateriales, Departamento de Biología, Facultad de Ciencias Naturales y Exactas, Universidad de Playa Ancha, Valparaiso, Chile
- 8Instituto Vandique, João Pessoa, Brazil
- 9Beagle Bioinformatics, Santiago, Chile
- 10Facultad de Ciencias, Universidad Austral de Chile, Valdivia, Chile
Piscirickettsia salmonis is a bacterial pathogen that severely impact the aquaculture in several countries as Canada, Scotland, Ireland, Norway, and Chile. It provokes Piscirickettsiosis outbreaks in the marine phase of salmonid farming, resulting in economic losses. The monophyletic genogroup LF-89 and a divergent genogroup EM-90 are responsible for the most severe Piscirickettsiosis outbreaks in Chile. Therefore, the development of methods for quick genotyping of P. salmonis genogroups in field samples is vital for veterinary diagnoses and understanding the population structure of this pathogen. The present study reports the development of a multiplex PCR for genotyping LF-89 and EM-90 genogroups based on comparative genomics of 73 fully sequenced P. salmonis genomes. The results revealed 2,322 sequences shared between 35 LF-89 genomes, 2,280 sequences in the core-genome of 38 EM-90 genomes, and 331 and 534 accessory coding sequences each genogroup, respectively. A total of 1,801 clusters of coding sequences were shared among all tested genomes of P. salmonis (LF-89 and EM-90), with 253 and 291 unique sequences for LF-89 and EM-90 genogroups, respectively. The Multiplex-1 prototype was chosen for reliable genotyping because of differences in annealing temperatures and respective reaction efficiencies. This method also identified the pathogen in field samples infected with LF-89 or EM-90 strains, which is not possible with other methods currently available. Finally, the genome-based multiplex PCR protocol presented in this study is a rapid and affordable alternative to classical sequencing of PCR products and analyzing the length of restriction fragment polymorphisms.
Introduction
Piscirickettsia salmonis is a facultative intracellular Gram-negative bacterium (Fryer and Hedrick, 2003; Mauel et al., 2008) and the etiological agent of Piscirickettsiosis, a systemic infection that affects all farmed salmon species (Mauel et al., 2008). In Chile, the bacterium is widely distributed and costs the salmon farming industry up to US$700 million per year (Maisey et al., 2017). In the first half of 2019, P. salmonis was the leading cause of mortality in farmed Atlantic salmon and rainbow trout (Sernapesca, 2020b). Consequently, 93.85% of all antibiotics used between January and June, 2020 in Chilean seawater aquaculture (Sernapesca, 2020a) were used to treat Piscirickettsiosis outbreaks. P. salmonis is also present in Canada (Brocklebank et al., 1993), Scotland (Grant et al., 1996), Ireland (Rodger and Drinan, 1993), and Norway (Olsen et al., 1997). P. salmonis is also present in different non-salmonid fish species, such as Blackspot grouper (Epinephelus melanostigma) (Chen et al., 2000), White sea bass (Atractoscion nobilis) (Arkush et al., 2005), and European sea bass (Dicentrarchus labrax) (Athanassopoulou et al., 2004; McCarthy et al., 2005). It has also been identified in different fish species in Chile (Contreras-Lynch et al., 2015).
Initially, P. salmonis was misclassified as a member of the order Rickettsiales (Cvitanich et al., 1991). However, the bacterium was reclassified as a member of the Gammaproteobacteria class of the phylum Proteobacteria following 16S rRNA sequencing (Fryer et al., 1992). Geographically diverse P. salmonis isolates (e.g., from Chile, Norway, Canada, Ireland, among others) mainly belong to the monophyletic group denominated LF-89 (reference strain ATCC VR-1361) or an outlier group related to the Chilean EM-90 strain (Heath et al., 2000; Saavedra et al., 2017). Additionally, Saavedra et al. (2017), analyzed 500 field isolates of P. salmonis and showed that 50% of samples were phylogenetically related to EM-90 or LF-89 isolates. They also found that genetic groups (or genogroups) were widely distributed and responsible for outbreaks of Piscirickettsiosis in salmon farms.
The existence of two genetic groups was reinforced after a comparative genomic analysis of 19 fully sequenced P. salmonis isolates (Nourdin-Galindo et al., 2017). This notion has also been experimentally supported by studies based on Multilocus Sequence Typing (MLST) and PCR amplification of 16S rRNA genes followed by restriction fragment length polymorphism (PCR-RFLP) (Isla et al., 2019; Aravena et al., 2020). Significant differences in genomic and phenotypic traits between P. salmonis genogroups highlight the need to provide alternative methodologies for genotyping. For instance, comparative analyses of core-genomes have revealed diverse genes encoding molecular functions related to antioxidant activity, transcriptional factors, translational regulation, and genogroup-specific virulence factors (Nourdin-Galindo et al., 2017). Genomic differences between genogroups grown at different culture conditions can often be reflected at the phenotypic level. For instance, EM-90-like strains grown at 22°C have a mucoid phenotype in culture plates absent in LF-89 strains (Nourdin-Galindo et al., 2017; Saavedra et al., 2017). Additionally, challenge assays in fish result in macroscopic lesions in the intestines, brain, and skeletal muscle with an EM-90-like isolate, but not an LF-89-like isolate (Rozas-Serri et al., 2017). Furthermore, fish infected with an EM-90-like strain presented an exacerbated immune response and higher cumulative mortality in a shorter time than LF-89 infections (Rozas-Serri et al., 2017).
Genomic and phenotypic differences between EM-90 and LF-89 genogroups require new tools for fast discrimination of members belonging to both genogroups in field samples and laboratory cultures. Additionally, different farmed salmonids could be a source of diverse P. salmonis genogroups, especially of LF-89 and EM-90 genogroups. The present study outlines a multiplex PCR procedure for use with LF-89 and EM-90 genogroups based on a comparative genomics approach which is rapid, economical, and straightforward.
Methodology
Datasets
This study analyzed chromosomal whole-genome sequences belonging to 73 Chilean P. salmonis strains. Sequences were downloaded from the National Center for Biotechnology Information (NCBI) database (Sayers et al., 2020) in September 2020 (Supplementary Table 1). Excluding plasmid sequences and draft genomes (contig and/or scaffold level).
Prediction of Protein-Encoding Genes in P. salmonis Genomes
Prodigal was used to predict the complete repertoire of protein-encoding genes available in all P. salmonis genomes (Hyatt et al., 2010) with default parameters to obtain nucleotide and amino acid sequences for each genome. Predicted open reading frames (ORFs) were functionally annotated using an automated in-house pipeline using Eggnog-mapper version 2.0.1-14-gbf04860, based on EggNOG 4.5 using default parameters and Diamond for orthologous searches (Buchfink et al., 2015; Huerta-Cepas et al., 2017); COGs terms and KEGG numbers and pathways were transferred by EggNOG mapper annotation; and InterproScan 5.31-70.0 (Jones et al., 2014) to detect functional domains based on sequence similarity and hidden Markov models (HMMs) searches. A scheme of the used in-house pipeline is provided as Supplemental Material (Supplementalry Figure 1).
Comparative Analysis and Selection of Core Sequences
The conservation of all predicted genes was analyzed, and the gene repertoires of P. salmonis LF-89 and EM-90 genogroups were compared. The amino acid sequences were recovered in FASTA format for all genes predicted in the prediction step. These sequences were then grouped using CD-HIT stand-alone, version 4.8.1 (Fu et al., 2012) with 75% of sequence similarity and 50% coverage of both the largest and shortest sequences as a cutoff value to consider a given protein a member of a given cluster. A binary presence-absence matrix was constructed considering each gene cluster for each genogroup and obtained the core genes for each genogroup. These subsets were compared with the same parameters described previously (only the unique core genes from each genogroup were recovered). Finally, to corroborate the resulting comparisons, each gene was aligned against the opposite genogroup genome with GMAP version 2017-01-14 (Wu et al., 2016).
Development and Optimization of a Multiplex PCR
Genes unique to each genogroup (LF-89 and EM-90) were annotated using the database of Clusters of Orthologous Groups of proteins (COGs) and EggNOG 4.5 (Huerta-Cepas et al., 2017). Evaluated sequences were annotated as “Non-Identified,” “Poorly Characterized” and removed those classified as “Information Storage and Processing.” Each sequence was manually curated using the BlastP tool against the P. salmonis database using 60% identity, 50% coverage, and E-value <10–5 (Ye et al., 2012). Primer sets were designed with the Primer-BLAST tool (Ye et al., 2012) using default parameters and the top gene/protein with functional annotations representative of each genogroup. Final amplicon sizes ranged between 100 and 200 nt. The annealing temperatures and efficiency of the reactions for each primer set were evaluated via conventional and quantitative PCR (qPCR), using chromosomal DNA from P. salmonis LF-89 EM-90 as templates. The primer sets with better experimental results in conventional PCRs for each genogroup were used for multiplex PCR. The reaction efficiency of the chosen primer sets was then evaluated by qPCR.
Bacterial Cultures and Identification
All P. salmonis isolates analyzed in this study were grown using the SHK-1 cell line (a macrophage-like cell line from S. salar head kidney) for 10–15 days. The culture medium was centrifuged at 1,000 × g for 5 min at 18°C following the cytopathic effects. The supernatant was removed, the pellet was recovered, and grown up on Austral TS-HEM solid and in Austral SRS-broth culture media (Yañez et al., 2013; Yáñez et al., 2014). Culture purity was verified using Gram staining, IFAT, and nested PCRs (Mauel et al., 1996; Yañez et al., 2013).
Bacterial DNA Extraction
Chromosomal DNA of the P. salmonis strains was extracted using the NucleoSpin Tissue Genomic DNA Purification Kit (Macherey-Nagel) as per the manufacturer’s recommendations. DNA quality was evaluated visually by agarose gel electrophoresis (0.8% wv–1 in TAE 1X buffer, SyberSafe, Invitrogen). DNA concentration was adjusted between 10 and 20 ng μL–1 with a ScanDrop analyzer (Analytik Jena) and stored samples at 4°C until further PCR amplification.
PCRs and Visualization
DNA was amplified in a 25 μL reaction using the GoTaq Green Master Mix (Promega), following the manufacturer’s instructions. DNA fragments were amplified using a MaxyGene II Thermal Cycler (Axygen Scientific), and amplified products were detected on a 1.8% wv–1 agarose gel in TAE 1X buffer with SyberSafe using standard procedures. Real-time PCRs were conducted with GoTaq qPCR Master Mix (Promega) and a CFX96 Real-Time System (BIO-RAD).
DNA Sequencing of 16S rRNA Genes
16S rRNA gene sequences were amplified using the 16SF and 16SR primer pair (Table 1) and a MaxyGene II Thermal Cycler (Axygen Scientific). The thermocycling protocol was as follows: initial denaturation at 95°C for 3 min, 35 cycles of amplification (denaturation at 95°C for 1 min, annealing at 45°C for 1 min and extension at 72°C for 1 min), and a final extension at 72°C for 5 min. The amplicons were visualized using agarose gel electrophoresis (1.8% wv–1 in TAE 1X buffer) and SyberSafe following standard procedures and used the E.Z.N.A Gel Extraction Kit (Omega-Biotek) to recover amplicons from agarose gels (according to the manufacturer’s instructions). DNA was stored at −20°C until further use. Macrogen (Korea) was used to sequence DNA samples using the dideoxynucleotide method (Sanger et al., 1977) and the above primer pair. All chromatograms were manually verified to ensure high sequencing quality results.
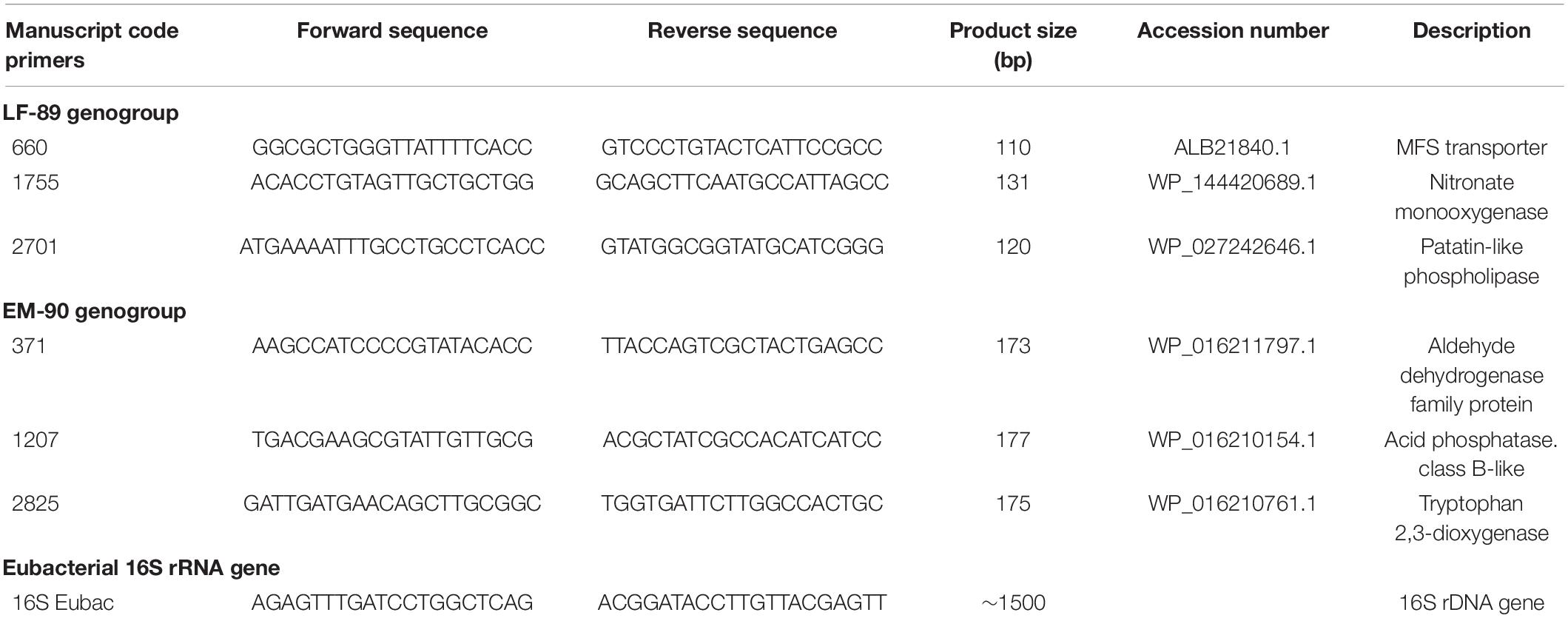
Table 1. List of primers used for genotyping P. salmonis isolates.
Phylogenetic Analysis
The genotyping methodology developed in the present study was verified using a phylogenetic tree constructed from 16S rRNA gene sequences of randomly selected P. salmonis isolates and by visualizing amplicons from the multiplex PCR on an agarose gel. The MEGA version X (Kumar et al., 2018) software was used to perform phylogenetic and molecular evolutionary analyses based on 16S rRNA gene sequences aligned with the DNA ClustalW algorithm using default parameters. An evolutionary molecular model was constructed using the option “Best D.N.A./Protein Model” from the “Model Selection” tool and phylogenetic reconstruction with the neighbor-joining tree algorithm with 1,000 bootstrap replicates. For this analysis, the 16S rRNA gene sequence was used of Francisella noatunensis as an outgroup.
Results
Comparative Genomics Identifies Unique Gene Candidates as Markers for EM and LF Genogroups
In this study, 73 fully sequenced P. salmonis genomes were retrieved from the NCBI database (Supplementary Table 1) and separated into LF-89, and EM-90 genogroups, based on hierarchical clustering of all ORFs predicted with the CD-Hit tool (Supplementary Figure 2). A total of 5,467 coding sequences were predicted, of which 2,653 and 2,814 were identified in LF-89 and EM-90 genomes, respectively.
Each genogroup’s sequences were separated into the core, and accessory coding sequences based on ORFs predicted for LF-89 and EM-90 genomes. In silico analysis of the LF-89 genogroup revealed 2,322 sequences shared between 35 genomes and showed that LF-89 genomes have 331 accessory coding sequences (Figure 1A) and the 38 EM-90 genomes shared 2,280 sequences in the core genome and had 534 accessory coding sequences (Figure 1B). A total of 1,801 clusters of shared gene sequences were found between LF-89 and EM-90 genogroups. LF-89 and EM-90 genogroups had 253 and 291 unique genes, respectively (Figure 1C).
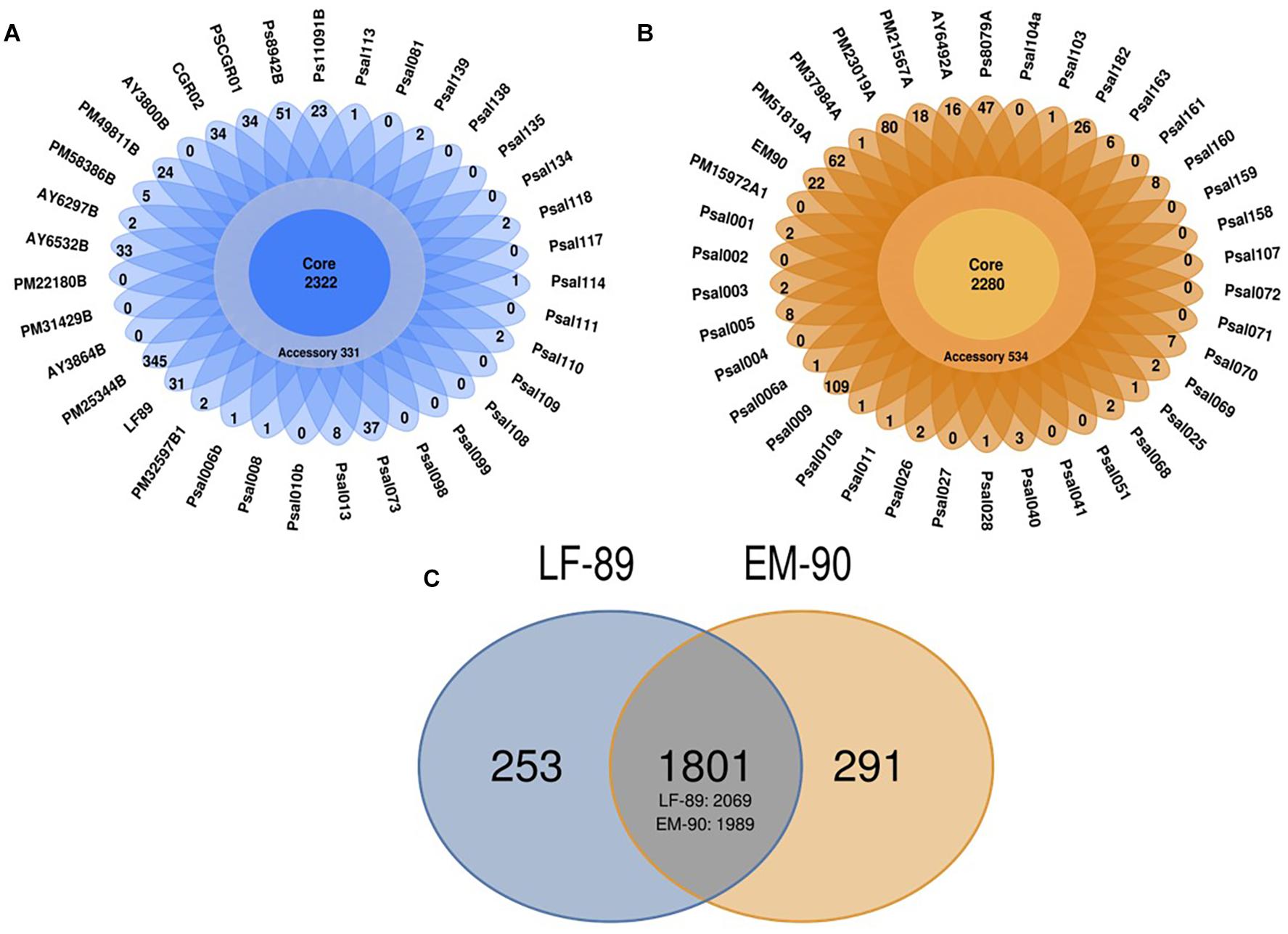
Figure 1. In silico identification of core-genome in 73 genomes of P. salmonis. (A, B) Core and Accessory genes in LF-89 and EM-90 genomes, respectively. (C) Ven-diagram of unique and shared sequences between LF-89 and EM-90 genomes. The intersection corresponds to shared gene clusters, and the numbers under the intersection correspond to the total number of genes counted.
Subsequently, 5,467 predicted coding sequences were functionally annotated using the COGs database from EggNOG 4.5 tool, and 158 and 250 sequences were annotated in LF-89 and EM-90 genogroups, respectively, after selecting the unique sequences in each genogroup (E-value annotation < 0.001).
Sequences were classified into three categories; Cellular Process & Signaling, Metabolism, and Poorly Characterized (Figure 2), and annotated the Poorly Characterized sequences in each dataset. Sequences annotated as Non-Identified, and Information storage and Processing were mobile elements (transposases and integrases), and therefore was removed from further analysis. Each selected sequence annotated by COG categories was manually curated against the P. salmonis database on NCBI using the BlastP tool to exclude sequences whose descriptions included “Hypothetical Protein” and “Unknown Function.” A total of 22 and 18 unique sequences were identified in the LF-89 and EM-90 genogroups, respectively (Supplementary Table 2). The characterization of each sequence group also indicated that 11 and 14 sequences were associated with “Metabolism” in LF-89 and EM-90 genetic groups, respectively. Moreover, three gene sequences of the LF-89 genogroup were related to “Cellular Processing and Signaling,” while only a sequence associated with this category was found in the EM-90 genogroup. The rest of the sequences were classified as “Function unknown” according to COG annotation in both genogroups.
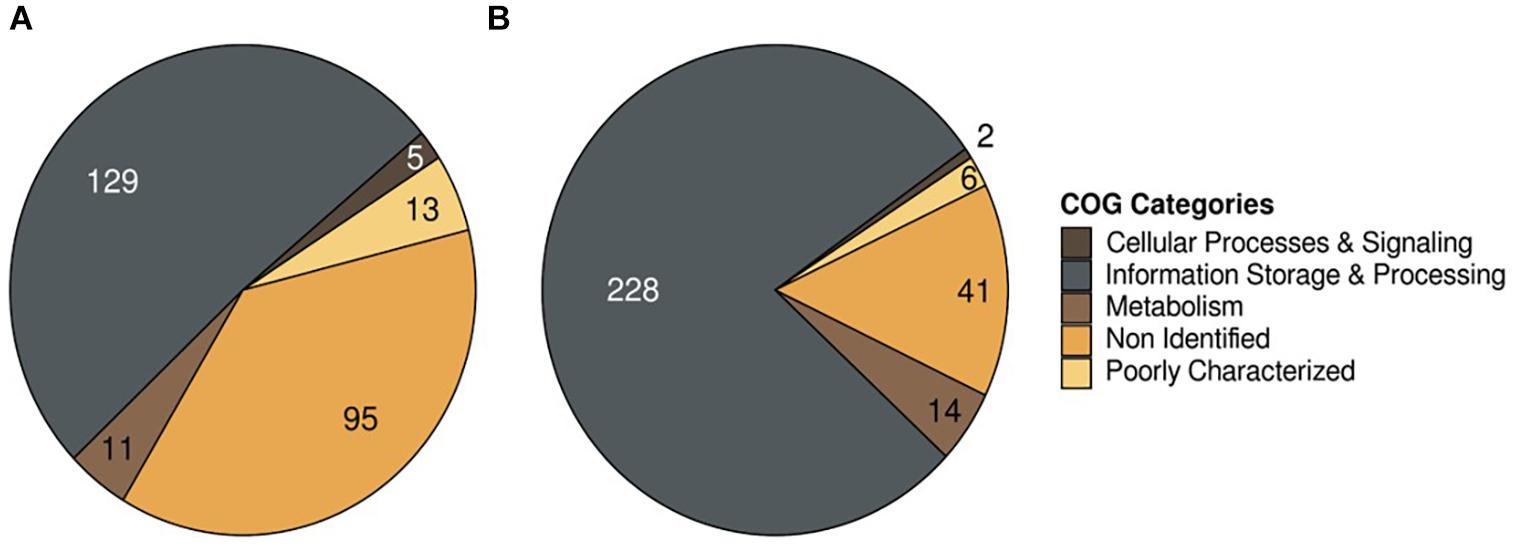
Figure 2. COG annotation of unique genes in P. salmonis genogroups. (A) LF-89 genogroup; (B) EM-90 genogroup.
Finally, six sequences were randomly selected (three for each genogroup) to design PCR primers with the Primer-Blast tool. Primers were designed such that amplicons were close to 130 bp in the LF-89 genogroup and approximately 170 bp in the EM-90 genogroup. The sequence was then manually analyzed using the BlastN tool to select the primer sets that specifically amplified the LF-89 or EM90 genogroup in silico (Table 1).
Optimization of Genotyping PCR
The primer sets’ annealing temperatures for each genogroup were optimized by gradient PCR in separate reactions using genomic DNA from LF-89 (ATCC VR-1361) and EM-90 isolates as templates. Each set produced single amplicons at all temperatures in a range between 55 and 68°C (Table 1). The primers’ specificities were tested using DNA templates from their target and non-target genogroups using sequenced DNA samples, phylogenetic analysis of 16S rRNA gene sequences (unpublished data), and MLST for P. salmonis. Each primer set only amplified its target genogroup, and amplicons were ∼130 and ∼170 bp in the LF-89 and EM-90 samples. No other bands were observed on agarose gels (Figure 3).
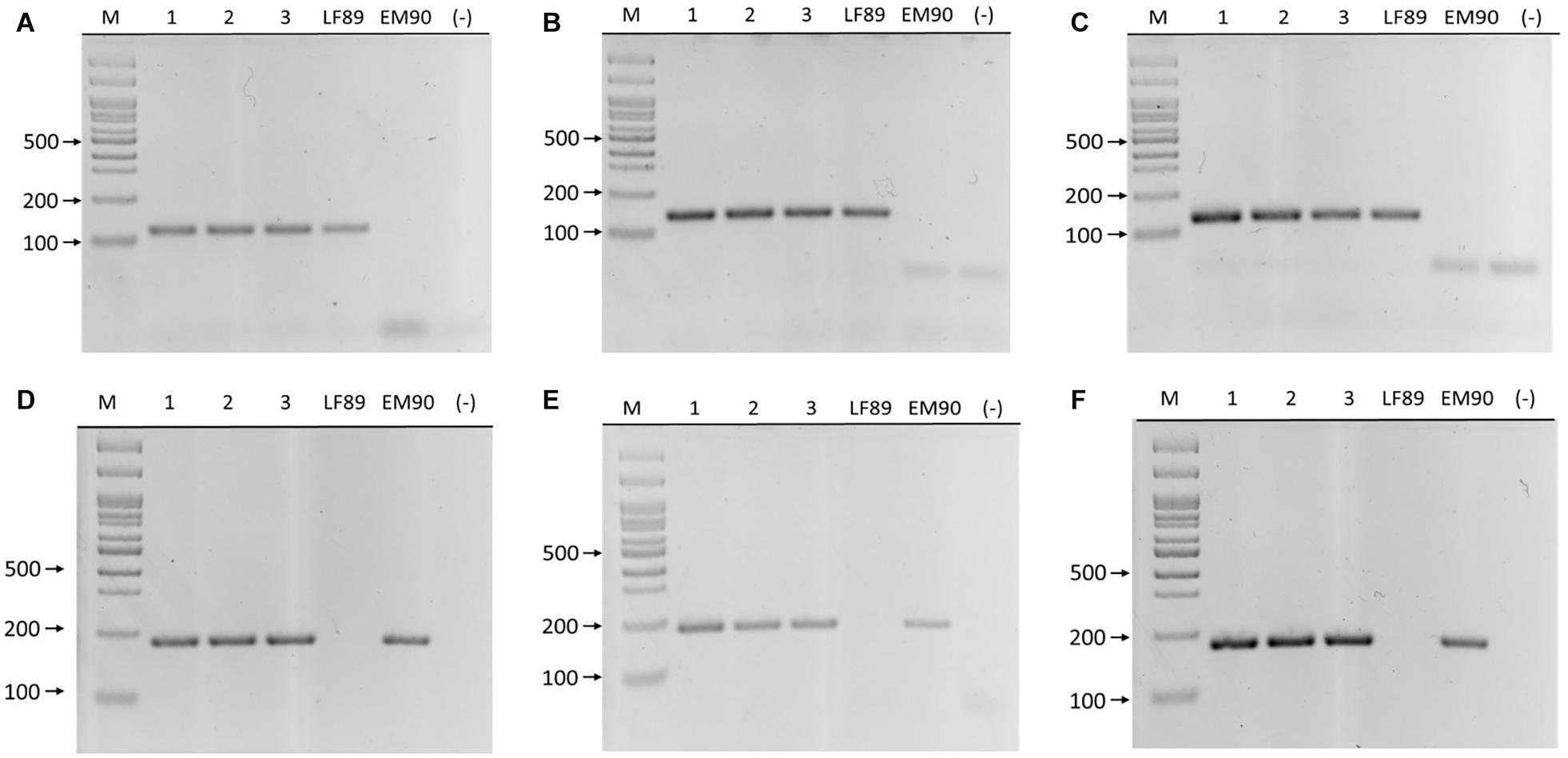
Figure 3. PCR amplification of LF-89 and EM-90 unique genes in P. salmonis. Upper panels (A–C) primer pairs 660, 1755, and 2701, respectively. Samples (1) AUS005, (2) AUS008, (3) AUS025, LF-89 strain, EM-90 like strain, and (–) negative control. Bottom panels (D–F) primer pairs 1207, 371, and 2825, respectively. Samples (1) AUS045, (2) AUS067, (3) AUS100, LF-89 strain, EM-90 like strain, and (–) negative control. (M) Molecular weight marker (100 bp Ladder).
The six primer sets were combined in nine Multiplex PCR prototypes (Multiplex-1 to Multiplex-9), evaluated their specificity with DNA from each genogroup, and discarded the prototypes with non-specific amplification by conventional PCR. The Multiplex-1 (1755/1207 primer mix), Multiplex-2 (2701/371 primer mix), and Multiplex-3 (660/1207 primer mix) prototypes were selected because they showed specific amplification of each genogroup (Figure 4) when PCR products were visualized via agarose gel electrophoresis. Three Multiplex PCR prototypes were used to identify both genogroups in a sample with a 1:1 ratio of DNA from both genogroups and observed two amplicons of the expected sizes (Figure 4, sample 3).
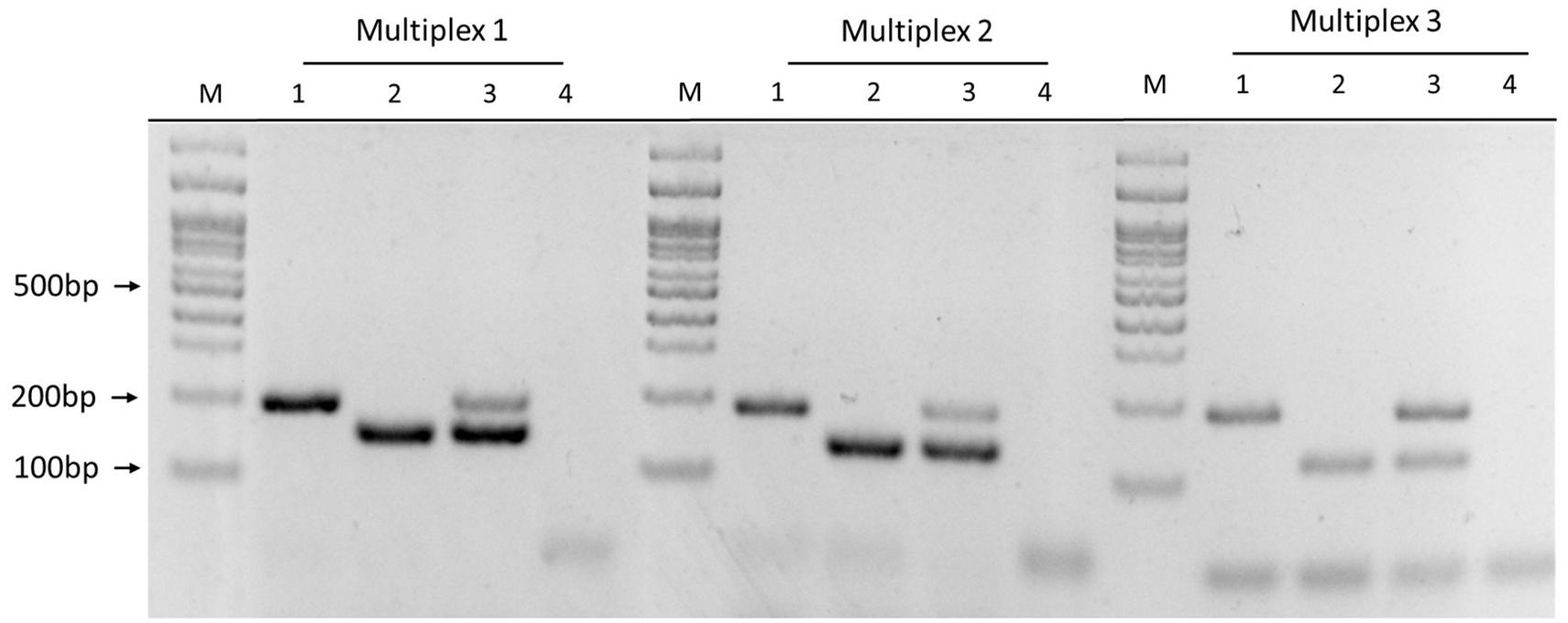
Figure 4. Multiplex PCR prototype using different primer mixes. Multiplex-1: 1755/1207 primer mix; Multiplex-2: 2701/371 primer mix; Multiplex-3: 660/1207 primer mix. Samples: (1) EM-90 strain, (2) LF-89 strain, (3) LF-89 and EM-90 like mix (1:1 ratio), (4) negative control, and (M) Molecular weight marker (100 bp Ladder).
The efficiency of each Multiplex PCR prototype was evaluated by absolute qPCR using the standard curve method. The Multiplex-1 prototype was 100.44 and 96.76% efficient with DNA of LF-89 and EM-90 isolates, respectively (Figure 5A); the Multiplex-2 prototype was 116.5 and 140.9% efficient, respectively, and the Multiplex-3 prototype was 118.4 and 93.2% efficient, respectively (Supplementary Figures 3A,B). Additionally, the melting temperatures of LF-89 and EM-90 PCR products from the Multiplex-1 prototype had a difference of 1.5°C (Figure 5B); however, they were the same for the Multiplex 2 and 3 prototypes (77.5 and 80°C, respectively) (Supplementary Figures 3A,B). The Multiplex-1 prototype was selected to validate our methodology using field samples because of the differences in melting temperatures between resulting amplicons from LF-89 and EM-90 DNA templates and the efficiencies of PCR reactions.
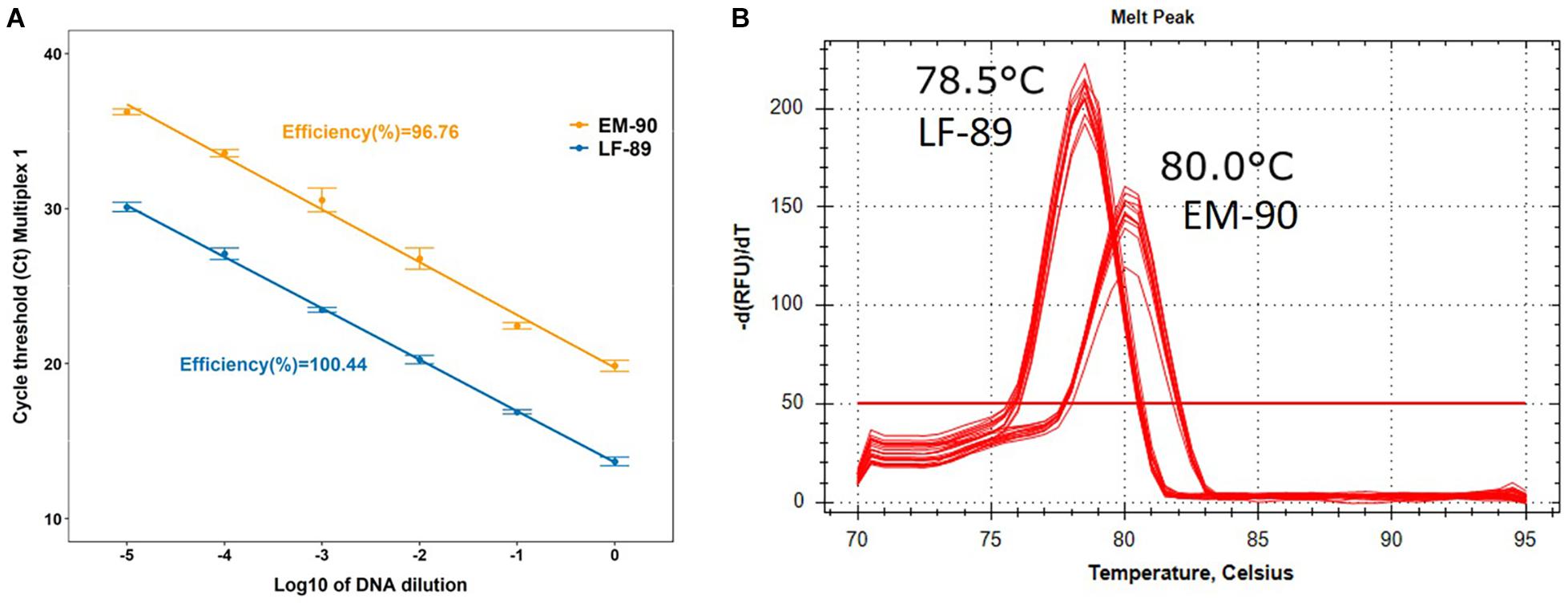
Figure 5. Characterization of Multiplex-1 prototype using qPCR. (A) Standard curve and efficiency of reaction using LF-89 and EM-90-like isolates as DNA templates. (B) Melting temperature of the amplicon from LF-89 and EM-90-like isolates.
Validation of Multiplex PCR
DNA was extracted from 48 P. salmonis isolates (kept in pure culture in our laboratory) with a NucleoSpin Tissue Genomic DNA Purification Kit and analyzed them using spectrophotometry. The samples were obtained from different hosts, years, and locations in Southern Chile (Table 2). Gram staining, IFAT, nested PCRs (data not shown) confirmed the purity of each culture.
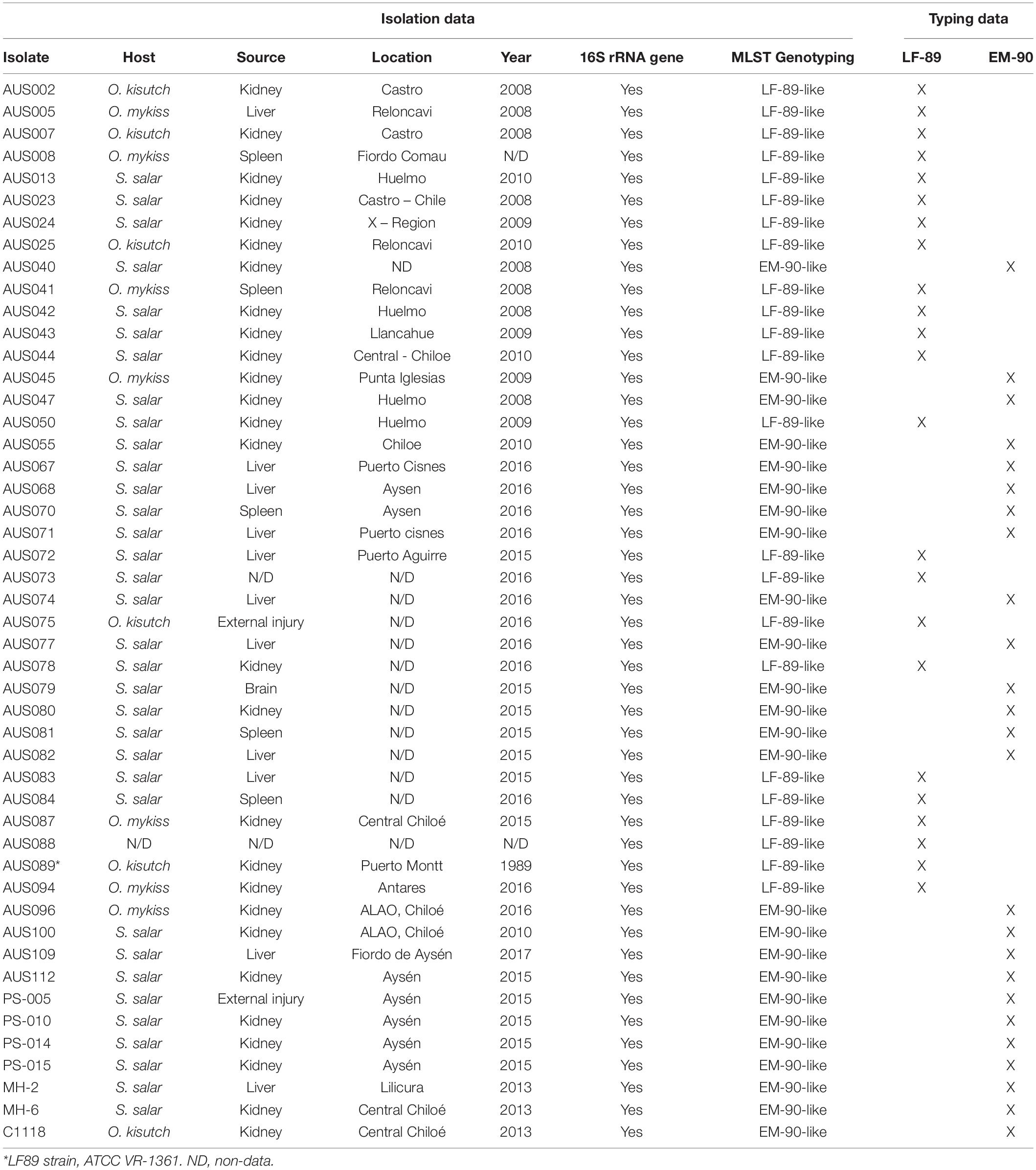
Table 2. Genotyping results using multiplex PCR for 48 P. salmonis isolates obtained from different hosts (Salmo salar, Oncorhynchus mykiss, and Oncorhynchus kisutch), sources, years, and locations in Chile.
The samples were analyzed using the Multiplex-1 prototype by conventional PCR and visualized amplicons via agarose gel electrophoresis. These results were compared with genotyping using the MLST assay developed for P. salmonis, amplification, sequencing, and phylogenetic analysis of 16S rRNA genes. The same results were obtained with the three genotyping methodologies (Table 2 and Figure 6); 48% of isolates (n = 23) corresponded to the LF-89 genogroup, and the rest (n = 25) corresponded to the EM-90 genogroup. Genomic DNA was extracted from ten bacterial fish pathogens (donated by Ruben Avendaño-Herrera, Universidad Andres Bello) to evaluate the Multiplex-1 prototype specificity. The identification of each microorganism via amplification and sequencing of 16S rRNA genes was used as negative controls. No non-specific bands were observed with the Multiplex-1 prototype by conventional PCR (Supplementary Table 3) using DNA templates from non-target bacteria.
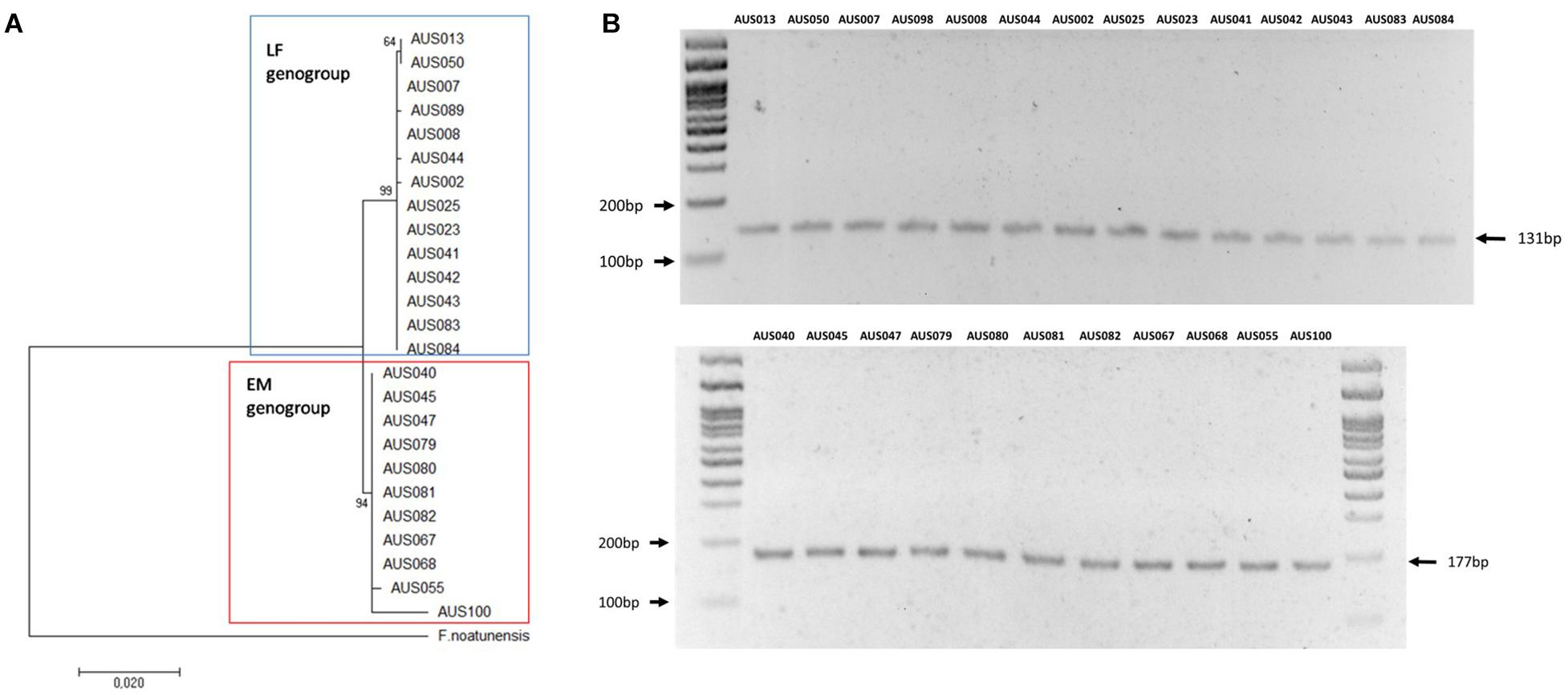
Figure 6. Comparative result of Multiplex-1 prototype and phylogenetic analysis of 16s rRNA gene sequences. (A) Molecular phylogenetic analysis using the maximum likelihood method. The Kimura 2-parameter model was used and Neighbor-Joining with 1,000 bootstraps. The 16S rRNA gene sequence of F. noatunensis was used as an outgroup. (B) Multiplex-1 prototype; upper and bottom panels depict isolates of LF-89 and EM-90 genogroups, respectively.
Discussion
Piscirickettsia salmonis is the bacterium responsible for Piscirickettsiosis that affects seriously aquaculture worldwide, including the Chilean salmonid industry (Maisey et al., 2017). The disease has important economic impacts which has encouraged to many researchers to develop diverse strategies for the molecular characterization of the major clusters of strains associated to field disease outbreaks. In this sense, diverse studies based on the 16S rRNA gene analysis (sequencing, PCR-RFLP) (Mauel et al., 1999; Mandakovic et al., 2016; Aravena et al., 2020), phenotypic characterization (Otterlei et al., 2016), MLST (Isla et al., 2019) and comparative genomics (Bohle et al., 2014; Bravo and Martinez, 2016; Nourdin-Galindo et al., 2017) have indicated the existence of LF-89 and EM-90 genogroups containing epidemiologically relevant P. salmonis stains. Both genogroup isolates show differential susceptibility to florfenicol, oxytetracycline, and quinolones (Saavedra et al., 2017). Also, they have different culture temperatures, whereas the optimal growth temperature of LF-89 genogroups is 16 to18°C, EM-90 isolates grow best between 18 to 22°C (Saavedra et al., 2017). These genetic groups also cause different levels of morbidity during challenge assays; EM-90 isolates cause higher mortality over shorter periods than the LF-89 strain (Rozas-Serri et al., 2017, 2018). Additionally, EM-90 isolates are more pathogenic than LF-89 isolates and exhibit increased intracellular replication and decreased host survival rate (Rozas-Serri et al., 2017). These differences highlight the need to develop methodologies for rapid genotyping of P. salmonis in field samples.
Analysis of 73 genomes showed that P. salmonis could be segregated into two genogroups with 2,322 and 2,280 core sequences in LF-89 and EM-90 genogroups. The number of sequences was similar to that reported by Nourdin-Galindo et al. (2017), who reported core-genomes composed of 2,170 and 2,228 sequences for LF-89 and EM-90 genogroups respectively. Interestingly, different unique genes were identified than Nourdin-Galindo et al., especially in the case of the LF-89 genogroup. 253 and 291 unique sequences were identified in LF-89 and EM-90, whereas Nourdin-Galindo et al., identified 437 and 282 unique sequences in LF-89 and EM-90 clusters. These variations could be due to differences in the pipeline used to identify pan/core-genomes and unique genes. However, the most important factor in the decrease in the number of unique sequences identified in each genogroup may be the higher number of genomes analyzed. Thirty-five genomes of the LF-89 genogroup and 38 genomes of the EM-90 genogroup were analyzed in the present study compared to the 13 and 6 genomes analyzed by Nourdin-Galindo et al. (2017). Therefore, variability could be caused by the total number of genomes analyzed, so we highly recommend using at least five genomes for robust characterization of pan-genomes (Vernikos et al., 2015). Additionally, the core genome sizes in both genogroups were estimated to be similar to previous estimations using comparative genomics, where ∼8% of the bacterial genome was composed of core genes (Lapierre and Gogarten, 2009).
The description of the core/pan-genomes and unique genes or strain-specific sequences allowed us to identify molecular markers for phylogenetic analysis (Caputo et al., 2019). Three primer sets were designed to identify samples belonging to the LF-89 or EM-90 genogroup by conventional PCR by analyzing 73 complete P. salmonis genome sequences. A multiplex PCR for this classification was then validated by conventional and qPCR. Our taxonomic methodology for genotyping differs from classical approximations based on polymorphisms of 16S rRNA gene sequences, where primer sets are designed for small variable regions of these genes (Mauel et al., 1996). We recommend using the 16S rRNA gene to establish phylogenetic affiliations; however, this gene is highly conserved; therefore, it is not ideal for distinguishing closely related microorganisms (Rosselló-Mora and Amann, 2001).
Regarding the characterization of the 16S rDNA gene of P. salmonis, initial research showed that the sequences have high similarity (98.5%) with a difference of 22 nucleotides between the LF-89 and EM-90 genogroups (Mauel et al., 1999). The 16S rDNA analysis requires DNA sequencing, which is costly and time-consuming. Therefore, there is a need for new genotyping strategies for microorganisms. Others have proposed methods based on PCR-RLFP (Aravena et al., 2020) and MLST (Isla et al., 2019); however, these methodologies are qualitative and only indicate the presence or absence of the pathogen in a sample. Previous reports have used relative qPCR to measure the growth of P. salmonis in different tissues by amplification of 16S rRNA genes, Rozas-Serri et al. (2018) showed the importance of quantifying the load and growth of pathogens in post-smolt salmon infected with LF-89 or EM-90 strains (Rozas-Serri et al., 2018). They were able to quantify the growth of each strain by identifying and amplifying genes encoding conserved cellular functions in the core genome of each genogroup.
Our multiplex method generates amplicons with different melting temperatures and allows fast identification of the sample’s genogroup. The strain-mixed described by Isla et al., 2019 was omitted because there were very few whole-genome sequences in the database, so the core-genome of those strains could not be determined.
The Multiplex-1 prototype was used for analyzing DNA from other bacterial fish pathogens and showed that our primer sets specifically amplify genes unique to P. salmonis LF-89 and EM-90 genogroups. Additionally, our genotyping results of P. salmonis samples clustered into the two expected genogroups. The distribution of samples was also compared with the previous characterization of the same samples using the MLST developed for P. salmonis (Isla et al., 2019) or phylogenetic analysis of 16S rRNA gene sequences. The same dichotomous distribution of samples was observed using both methodologies and found both genogroups are widely distributed among the different fish hosts. This finding differs from previous reports reporting that EM-90 genogroups are responsible for most Piscirickettsiosis cases (Saavedra et al., 2017; Aravena et al., 2020). Therefore, we recommend using a higher number of field isolates and epidemiological studies to take different hosts into account.
In the present study, Bioinformatic analysis of fully sequenced P. salmonis genomes was used to design specific PCR primers against LF-89 and EM-90 genogroups to construct a novel diagnostic multiplex PCR tool. The multiplex-1 prototype is rapid, precise, and reliable and will reduce diagnosis times and costs. It will be helpful in routine veterinary diagnoses when using fish samples directly or pure and mixed bacterial cultures.
Data Availability Statement
The dataset analyzed for this study can be found in Genbank (https://www.ncbi.nlm.nih.gov/genbank/), and accession numbers can be found in the article/Supplementary Material (Supplementary Table 1).
Author Contributions
AY and VM-C: conceptualization. JF, AY, and AI: funding acquisition. AY and MR: project administration. AI, JM-H, and DH: methodology, sampling, and analysis. AI, VM-C, and JM-H: writing—original draft. AY and HL: writing—review and editing. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, DT, declared a shared affiliation, with the authors, JM-H and VM-C, to the handling editor at the time of the review.
Funding
This work was supported by grants FONDAP INCAR N°15110027, Vicerrectoría de Investigación, Desarrollo y Creación Artística (VIDCA) from Universidad Austral de Chile and CONICYT-PFCHA Doctorado Nacional 2015-21151459.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.673216/full#supplementary-material
References
Aravena, P., Pulgar, R., Ortiz-Severín, J., Maza, F., Gaete, A., Martínez, S., et al. (2020). PCR-RFLP detection and genogroup identification of Piscirickettsia salmonis in field samples. Pathogens 9:358. doi: 10.3390/pathogens9050358
Arkush, K. D., McBride, A. M., Mendonca, H. L., Okihiro, M. S., Andree, K. B., Marshall, S., et al. (2005). Genetic characterization and experimental pathogenesis of Piscirickettsia salmonis isolated from white seabass Atractoscion nobilis. Dis. Aquat. Organ. 63, 139–149. doi: 10.3354/dao063139
Athanassopoulou, F., Groman, D., Prapas, T., and Sabatakou, O. (2004). Pathological and epidemiological observations on rickettsiosis in cultured sea bass (Dicentrarchus labrax L.) from Greece. J. Appl. Ichthyol. 20, 525–529. doi: 10.1111/j.1439-0426.2004.00571.x
Bohle, H., Henríquez, P., Grothusen, H., Navas, E., Sandoval, A., Bustamante, F., et al. (2014). Comparative genome analysis of two isolates of the fish pathogen Piscirickettsia salmonis from different hosts reveals major differences in virulence-associated secretion systems. Genome Announc. 2:e01219-14. doi: 10.1128/genomeA.01219-14
Bravo, C., and Martinez, V. (2016). Whole-genome comparative analysis of the pathogen Piscirickettsia salmonis. Vet. Microbiol. 30, 36–43. doi: 10.1016/j.vetmic.2016.10.015
Brocklebank, J. R., Evelyn, T. P. T., Speare, D. J., and Armstrong, R. D. (1993). Rickettsial septicemia in farmed Atlantic and chinook salmon in British columbia: clinical presentation and experimental transmission. Can. Vet. J. 34, 745–748.
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Caputo, A., Fournier, P. E., and Raoult, D. (2019). Genome and pan-genome analysis to classify emerging bacteria. Biol. Direct. 14:5. doi: 10.1186/s13062-019-0234-0
Chen, S. C., Wang, P. C., Tung, M. C., Thompson, K. D., and Adams, A. (2000). A Piscirickettsia salmonis-like organism in grouper, Epinephelus melanostigma, in Taiwan. J. Fish Dis. 23, 415–418. doi: 10.1046/j.1365-2761.2000.00250.x
Contreras-Lynch, S., Olmos, P., Vargas, A., Figueroa, J., González-Stegmaier, R., Enríquez, R., et al. (2015). Identification and genetic characterization of Piscirickettsia salmonis in native fish from southern Chile. Dis. Aquat. Org. 115, 233–244. doi: 10.3354/dao02892
Cvitanich, J. D., Gárate, O., and Smith, C. E. (1991). The isolation of a rickettsia-like organism causing disease and mortality in Chilean salmonids and its confirmation by Koch’s postulate. J. Fish Dis. 14, 121–145. doi: 10.1111/j.1365-2761.1991.tb00584.x
Fryer, J. L., and Hedrick, R. P. (2003). Piscirickettsia salmonis: a gram-negative intracellular bacterial pathogen of fish. J. Fish Dis. 26, 251–262. doi: 10.1046/j.1365-2761.2003.00460.x
Fryer, J. L., Lannan, C. N., Giovannoni, S. J., and Wood, N. D. (1992). Piscirickettsia salmonis gen. nov., sp. nov., the causative agent of an epizootic disease in salmonid fishes. Int. J. Syst. Bacteriol. 42, 120–126. doi: 10.1099/00207713-42-1-120
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Grant, A. N., Brown, A. G., Cox, D. I., Birkbeck, T. H., and Griffen, A. A. (1996). Rickettsia-like organism in farmed salmon. Vet. Rec. 138:423.
Heath, S., Pak, S., Marshall, S., Prager, E. M., and Orrego, C. (2000). Monitoring Piscirickettsia salmonis by denaturant gel electrophoresis and competitive PCR. Dis. Aquat. Organ. 41, 19–29. doi: 10.3354/dao041019
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 8:119. doi: 10.1186/1471-2105-11-119
Isla, A., Saldariaga-Cordoba, M., Fuentes, D. E., Albornoz, R., Haussmann, D., Mancilla-Schulz, J., et al. (2019). Multilocus sequence typing detects new Piscirickettsia salmonis hybrid genogroup in Chilean fish farms: evidence for genetic diversity and population structure. J. Fish Dis. 42, 721–737. doi: 10.1111/jfd.12976
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lapierre, P., and Gogarten, J. P. (2009). Estimating the size of the bacterial pan-genome. Trends Genet. 25, 107–110. doi: 10.1016/j.tig.2008.12.004
Maisey, K., Montero, R., and Christodoulides, M. (2017). Vaccines for piscirickettsiosis (salmonid rickettsial septicaemia, SRS): the Chile perspective. Expert. Rev. Vaccines 16, 215–228. doi: 10.1080/14760584.2017.1244483
Mandakovic, D., Glasner, B., Maldonado, J., Aravena, P., González, M., and Cambiazo, V. (2016). Genomic-based restriction enzyme selection for specific detection of Piscirickettsia salmonis by 16S rDNA PCR-RFLP. Front. Microbiol. 7:643. doi: 10.3389/fmicb.2016.00643
Mauel, M. J., Giovannoni, S. J., and Fryer, J. L. (1996). Development of polymerase chain reaction assays for detection, identification, and differentiation of Piscirickettsia salmonis. Dis. Aquat. Organ. 26, 189–195. doi: 10.3354/dao026189
Mauel, M. J., Giovannoni, S. J., and Fryer, J. L. (1999). Phylogenetic analysis of Piscirickettsia salmonis by 16S, internal transcribed spacer (ITS) and 23S ribosomal DNA sequencing. Dis. Aquat. Organ. 35, 115–123. doi: 10.3354/dao035115
Mauel, M. J., Ware, C., and Smith, P. A. (2008). Culture of Piscirickettsia salmonis on enriched blood agar. J. Vet. Diagnostic. Investig. 20, 213–214. doi: 10.1177/104063870802000211
McCarthy, Ú, Steiropoulos, N. A., Thompson, K. D., Adams, A., Ellis, A. E., and Ferguson, H. W. (2005). Confirmation of Piscirickettsia salmonis as a pathogen in European sea bass Dicentrarchus labrax and phylogenetic comparison with salmonid strains. Dis. Aquat. Org. 64, 107–119. doi: 10.3354/dao064107
Nourdin-Galindo, G., Sánchez, P., Molina, C. F., Espinoza-Rojas, D. A., Oliver, C., Ruiz, P., et al. (2017). Comparative pan-genome analysis of Piscirickettsia salmonis reveals genomic divergences within genogroups. Front. Cell. Infect. Microbiol. 7:459. doi: 10.3389/fcimb.2017.00459
Olsen, A. B., Melby, H. P., Speilberg, L., Evensen, O., and Hastein, T. (1997). Piscirickettsia salmonis infection in atlantic salmon Salmo salar in norway-epidemiological, pathological and microbiological findings. Dis. Aquat. Org. 31, 35–48. doi: 10.3354/dao031035
Otterlei, A., Brevik, ØJ., Jensen, D., Duesund, H., Sommerset, I., Frost, P., et al. (2016). Phenotypic and genetic characterization of Piscirickettsia salmonis from Chilean and Canadian salmonids. BMC Vet. Res. 12:55. doi: 10.1186/s12917-016-0681-0
Rodger, H. D., and Drinan, E. M. (1993). Observation of a rickettsia-like organism in Atlantic salmon. Salmo salar L., in Ireland. J. Fish Dis. 16, 361–369. doi: 10.1111/j.1365-2761.1993.tb00869.x
Rosselló-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev. 25, 39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x
Rozas-Serri, M., Ildefonso, R., Peña, A., Enriquez, E., Barrientos, S., and Maldonado, L. (2017). Comparative pathogenesis of piscirickettsiosis in Atlantic salmon (Salmo salar L.) post-smolt experimentally challenged with LF-89-like and EM-90-like Piscirickettsia salmonis isolates. J. Fish Dis. 40, 1451–1472. doi: 10.1111/jfd.12671
Rozas-Serri, M., Peña, A., Arriagada, G., Enríquez, R., and Maldonado, L. (2018). Comparison of gene expression in post-smolt Atlantic salmon challenged by LF-89-like and EM-90-like Piscirickettsia salmonis isolates reveals differences in the immune response associated with pathogenicity. J. Fish Dis. 41, 539–552. doi: 10.1111/jfd.12756
Saavedra, J., Hernandez, N., Osses, A., Castillo, A., Cancino, A., Grothusen, H., et al. (2017). Prevalence, geographic distribution and phenotypic differences of Piscirickettsia salmonis EM-90-like isolates. J. Fish Dis. 40, 1055–1063. doi: 10.1111/jfd.12581
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74, 5463–5467. doi: 10.1073/pnas.74.12.5463
Sayers, E. W., Cavanaugh, M., Clark, K., Ostell, J., Pruitt, K. D., and Karsch-Mizrachi, I. (2020). GenBank. Nucleic Acids Res. 48, D84–D86. doi: 10.1093/nar/gkz956
Sernapesca (2020a). Report on Antimicrobial Use in the National Salmoniculture Industry. Valparaiso: National Service of Fisheries.
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Wu, T. D., Reeder, J., Lawrence, M., Becker, G., and Brauer, M. J. (2016). GMAP and GSNAP for genomic sequence alignment: enhancements to speed. Accuracy, and Functionality. Methods Mol. Biol. 1418, 283–334. doi: 10.1007/978-1-4939-3578-9_15
Yañez, A. J., Silva, H., Valenzuela, K., Pontigo, J. P., Godoy, M., Troncoso, J., et al. (2013). Two novel blood-free solid media for the culture of the salmonid pathogen Piscirickettsia salmonis. J. Fish Dis. 36, 587–591. doi: 10.1111/jfd.12034
Yáñez, A. J., Valenzuela, K., Matzner, C., Olavarría, V., Figueroa, J., and Avendaño-Herrera, R. (2014). Broth microdilution protocol for minimum inhibitory concentration (MIC) determinations of the intracellular salmonid pathogen Piscirickettsia salmonis to florfenicol and oxytetracycline. J. Fish Dis. 37, 505–509. doi: 10.1111/jfd.12144
Keywords: Chile, genotyping, Piscirickettsia salmonis detection, salmonid fish, comparative genomic
Citation: Isla A, Martinez-Hernandez JE, Levipan HA, Haussmann D, Figueroa J, Rauch MC, Maracaja-Coutinho V and Yañez A (2021) Development of a Multiplex PCR Assay for Genotyping the Fish Pathogen Piscirickettsia salmonis Through Comparative Genomics. Front. Microbiol. 12:673216. doi: 10.3389/fmicb.2021.673216
Received: 27 February 2021; Accepted: 17 May 2021;
Published: 11 June 2021.
Edited by:
Daniel Yero, Autonomous University of Barcelona, SpainReviewed by:
Mudit Chandra, Guru Angad Dev Veterinary and Animal Sciences University, IndiaDante Travisany, University of Chile, Chile
Hasan C. Tekedar, Mississippi State University, United States
Copyright © 2021 Isla, Martinez-Hernandez, Levipan, Haussmann, Figueroa, Rauch, Maracaja-Coutinho and Yañez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinicius Maracaja-Coutinho, dmluaWNpdXMubWFyYWNhamFAdWNoaWxlLmNs; Alejandro Yañez, YXlhbmV6QHVhY2guY2w=
†These authors have contributed equally to this work and share first authorship