 Cinthia Alves-Barroco1
Cinthia Alves-Barroco1 João Caço1
João Caço1 Catarina Roma-Rodrigues1
Catarina Roma-Rodrigues1 Alexandra R. Fernandes1Ricardo Bexiga2
Alexandra R. Fernandes1Ricardo Bexiga2 Manuela Oliveira2Lélia Chambel3Rogério Tenreiro3
Manuela Oliveira2Lélia Chambel3Rogério Tenreiro3 Rosario Mato1*
Rosario Mato1* Ilda Santos-Sanches1†
Ilda Santos-Sanches1†- 1UCIBIO, Departamento de Ciências da Vida, NOVA School of Science and Technology/FCT NOVA, Universidade NOVA de Lisboa, Caparica, Portugal
- 2Centro de Investigação Interdisciplinar em Sanidade Animal, Faculdade de Medicina Veterinária, Universidade de Lisboa, Lisbon, Portugal
- 3Biosystems and Integrative Sciences Institute, Faculdade de Ciências, Universidade de Lisboa, Edifício TecLabs, Lisbon, Portugal
Streptococcus dysgalactiae subsp. dysgalactiae (SDSD) has been considered a strict animal pathogen. Nevertheless, the recent reports of human infections suggest a niche expansion for this subspecies, which may be a consequence of the virulence gene acquisition that increases its pathogenicity. Previous studies reported the presence of virulence genes of Streptococcus pyogenes phages among bovine SDSD (collected in 2002–2003); however, the identity of these mobile genetic elements remains to be clarified. Thus, this study aimed to characterize the SDSD isolates collected in 2011–2013 and compare them with SDSD isolates collected in 2002–2003 and pyogenic streptococcus genomes available at the National Center for Biotechnology Information (NCBI) database, including human SDSD and S. dysgalactiae subsp. equisimilis (SDSE) strains to track temporal shifts on bovine SDSD genotypes. The very close genetic relationships between humans SDSD and SDSE were evident from the analysis of housekeeping genes, while bovine SDSD isolates seem more divergent. The results showed that all bovine SDSD harbor Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas IIA system. The widespread presence of this system among bovine SDSD isolates, high conservation of repeat sequences, and the polymorphism observed in spacer can be considered indicators of the system activity. Overall, comparative analysis shows that bovine SDSD isolates carry speK, speC, speL, speM, spd1, and sdn virulence genes of S. pyogenes prophages. Our data suggest that these genes are maintained over time and seem to be exclusively a property of bovine SDSD strains. Although the bovine SDSD genomes characterized in the present study were not sequenced, the data set, including the high homology of superantigens (SAgs) genes between bovine SDSD and S. pyogenes strains, may indicate that events of horizontal genetic transfer occurred before habitat separation. All bovine SDSD isolates were negative for genes of operon encoding streptolysin S, except for sagA gene, while the presence of this operon was detected in all SDSE and human SDSD strains. The data set of this study suggests that the separation between the subspecies “dysgalactiae” and “equisimilis” should be reconsidered. However, a study including the most comprehensive collection of strains from different environments would be required for definitive conclusions regarding the two taxa.
Introduction
Streptococcus dysgalactiae (SD) includes two subspecies: S. dysgalactiae subsp. dysgalactiae (SDSD) and S. dysgalactiae subsp. equisimilis (SDSE) (Jensen and Kilian, 2012). While SDSE has been recognized as an increasingly important human pathogen, SDSD has been considered a strictly animal pathogen and is commonly associated with bovine mastitis (Zadoks et al., 2011; Rato et al., 2013). In the last years, although rare, human diseases by this subspecies have been reported (Koh et al., 2009; Park et al., 2012; Jordal et al., 2015; Chennapragada et al., 2018), and it is urgent to understand the role of this subspecies in human pathogenesis. Recently, we have reported bovine SDSD isolates’ ability to adhere to and internalize different human cell lines, including keratinocytes (Roma-Rodrigues et al., 2016; Alves-Barroco et al., 2018).
SDSD and SDSE, together with Streptococcus agalactiae, Streptococcus canis, and Streptococcus pyogenes, are the major pathogens of the pyogenic group. Previous studies reveal that these species are closely related. Phylogenetic analysis involving the Streptococcus core genome within the pyogenic clade displayed overall consensus. Though the issue is not entirely clear-cut, the most likely scenario is that the species tree has S. dysgalactiae and S. pyogenes as sister species and, together with S. canis, establishes a very closely related branch. These data suggest that they have recently descended from a common ancestor (Suzuki et al., 2011; Jensen and Kilian, 2012; Lefébure et al., 2012). The divergence among species has been mainly associated with mobile genetic elements (MGEs) (Lefébure et al., 2012). Indeed, about a quarter of streptococci’s genome consists of exogenous and mobile DNA, e.g., transposons, prophage, and plasmid. Thus, horizontal gene transfer (HGT) is considered the primary mechanism for generating diversity in this genus (Baiano and Barnes, 2009; Haenni et al., 2010; De la Casa-Esperón, 2012; Richards et al., 2012; Wong and Yuen, 2012; Rohde and Cleary, 2016). Overall, the MGEs are self-transmissible elements that are omnipresent in bacteria, harboring genes responsible for their maintenance, regulation, and mobility and genes encoding antimicrobial resistance, as well as other determinants, such as genes that encode virulence factors (Grohmann et al., 2003; McNeilly and McMillan, 2014).
Thus, many MGEs serve as shuttles for genes that are beneficial to bacteria during their proliferation in a host environment. Although several mechanisms can mediate genetic transfer in pyogenic streptococci, the transduction might be particularly relevant since bacteriophages have been detected in a considerable proportion (McShan and Nguyen, 2016). In S. pyogenes and SDSE, more than 20% of the accessory genomes consist of prophage DNA, highlighting the critical role of transduction in the evolution of these bacteria (Lier et al., 2015; Yamada et al., 2019).
The presence of phage-carrying S. pyogenes virulence genes among SDSD isolates from bovine mastitis was previously reported, namely, the streptococcal pyrogenic exotoxin genes (speK, speC, speL, and speM), the phage DNase (spd1), and determinants of antibiotic resistance located on MGEs (Rato et al., 2011; Abdelsalam et al., 2015). It was hypothesized that these virulence genes might contribute to the increased pathogenic potential; thus, SDSD should not be ignored as a potential human pathogen.
Despite the benefits of HGT, due to the vast diversity of the bacteriophages in the environment, the bacteria are under constant phage attack. To survive, the bacteria have devised various strategies that involve innate and adaptive immunity and interfere with the infection process (Barrangou and Marraffini, 2014). Among all of these strategies, the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas system is the one that provides the bacteria with the ability to “memorize” a piece of the invaders’ genetic material (Bhaya et al., 2011; Bondy-Denomy et al., 2013; Rath et al., 2015; Makarova et al., 2015; Makarova et al., 2018) by acquiring foreign nucleic acid fragments called spacers. Together with host Cas proteins, the spacers behave as an adaptive immune system in bacteria and archaea. Besides, the sequential order of the spacers in the array provides chronological information about the bacteria’s exposure to foreign nucleic acids (Lier et al., 2015), providing insights into host–phage interactions within a specific niche. These data show indicators of the evolution of the bacteria that carry them (Lier et al., 2015).
To date, studies on the presence of S. pyogenes phages among SDSD isolates are scarce. For example, it is not yet clear whether these phages (and other MGEs) contribute to more pathogenic SDSD phenotypes. The CRISPR/Cas system in the SDSD and its impact on HGT are examples of how little is known, so far, about this microorganism.
In the present study, SDSD isolates collected in two different periods (2002–2003, partially studied, and 2011–2013) were characterized and compared to obtain new information about molecular virulence profiles, antibiotypes, and population structure. We also investigated the CRISPR/Cas systems in SDSD strains to gain insights into defense systems and their impact on HGT. Additionally, pyogenic streptococcus genomes available at National Center for Biotechnology Information (NCBI) database, including human SDSD and SDSE strains, were included in this study to characterize CRISPR/Cas system, prophage regions, giving further insights on horizontal acquisitions of genetic elements particularly associated with resistance traits or virulence to track temporal shifts of SDSD genotypes.
Materials and Methods
Bacterial Isolates
Out of a total of 194 Gram-positive cocci, 37 SDSD were isolated and included in this study (collection II). The isolates were collected from 2011 to 2013, from 250 animals with mastitis of 12 dairy farms located in Portugal. The presumptive identification of the SDSD isolates was performed by traditional phenotypic tests such as colony morphology, type of hemolysis, Lancefield serologic groups using the SLIDEX Strepto Plus (Biomérieux, Marcy-l’Étoile, France), and absence of catalase.
For a comparative analysis, 18 strains of bovine SDSD (collection I), partially characterized (Rato et al., 2010, 2011, 2013) and collected between 2002 and 2003 from 248 animals with mastitis on eight farms located in Portugal, were included in the study.
Genomic DNA Extraction
Genomic DNA was extracted according to Alves-Barroco et al. (2019).
Identification by 16S rRNA Sequence
The identification of the SDSD isolates was confirmed by analysis of 16S rRNA gene sequencing. Amplification and sequencing were performed with the same primers (Takahashi et al., 1997). The sequencing of PCR products was carried out by STAB-Vida (Lisbon, Portugal). The sequences were analyzed and aligned using CLC Main Workbench 20.1 alignment program editor (QIAGEN, Venlo, Netherlands) and then compared with sequences from the NCBI database using the Basic Local Alignment Search Tool (BLAST)1. Subsequently, the nucleotide sequences were subjected to phylogenetic analysis using MEGA X (Kumar et al., 2018).
Pulsed-Field Gel Electrophoresis
The agarose plugs containing the genomic DNA from streptococcal strains were prepared as previously described (Rato et al., 2008). All pulsed-field gel electrophoresis (PFGE) patterns were analyzed visually and by computer-assisted cluster analysis using BioNumerics v. 6.6 (Applied Maths, Sint-Martens-Latem, Belgium). Levels of similarity were calculated using Dice coefficient, and unweighted pair group method with arithmetic mean (UPGMA) was used for clustering to produce band-based dendrograms (1.5% and no optimization). Groups of patterns with similarity of 100% were considered indistinguishable and were assigned to the same subtype of PFGE type.
Phylogenetic Analysis
For the phylogenetic analysis of bovine SDSD isolates, the Multilocus Sequence Analysis (MLSA) scheme was performed according to the method previously described for SDSE based on sequences of seven housekeeping genes (McMillan et al., 2010). Seven housekeeping genes, namely, glucose kinase gki, glutamine transport protein gtr, glutamate racemase murI, DNA mismatch repair protein mutS, transketolase recP, xanthine phosphoribosyl transferase xpt, and acetoacetyl-coathioloase atoB, were amplified; and all amplified DNA fragments were sequenced. The sequences were edited, aligned, and subjected to phylogenetic analysis using CLC Bio Main Workbench 20.1 alignment program editor (QIAGEN, Venlo, Netherlands). Housekeeping gene sequences were trimmed, concatenated, and subjected to phylogenetic analysis. For concatenated housekeeping alleles, phylogenetic relationships were inferred using the minimum evolution method, and the distances were computed using the maximum composite likelihood method.
For the phylogenetic analysis, bovine SDSD isolates representatives of collection I and II (a total of 15 strains) were selected based on their different resistance, virulence, and PFGE profiles. Additionally, the sequences of SDSD strains (n = 12), SDSE strains (n = 15), S. pyogenes strains (n = 11), and S. canis strains (n = 3) (Supplementary Table 1 in Supplementary File 1), available in the NCBI Nucleotide database, were included in this study for comparison.
Virulence Gene Screening
The following genetic determinants of S. pyogenes (i.e., speA, speB, speC, speF, speG, speH, speK, speL, speM, spegg, smeZ, spd1, and sdn) were screened in bovine SDSD isolates by PCR. Additionally, we investigated the presence of operon encoding streptolysin S (SLS), namely, sagA, sagB, sagE, and sagI genes. For each 25 μl PCR mixture were added 100 ng of bacterial DNA, 1 × reaction buffer for NZYTaq DNA polymerase, 2.5 mM of MgCl2, 0.4 mM of dNTPs NZYMix, 1 U of NZYTaq DNA polymerase (NZYTech, Lisbon, Portugal), and 1 μM of each primer (Thermo Fisher Scientific, Waltham, MA, United States). Gene expression assays by reverse-transcriptase PCR (RT-PCR) were performed according to Alves-Barroco et al. (2019). Negative results were confirmed in at least three independent assays. VSD13 and VSD7 were used as positive control. Primer sequences and amplicon expected size PCR conditions for amplifying all the genetic determinants are listed in Supplementary Table 2 (Supplementary File 2).
Screening of virulence genes and comparative analysis in silico among SDSD (from bovine, human, and fish strains), SDSE, S. canis, and S. pyogenes sequences (see Supplementary Table 1 in Supplementary File 1) were performed using the CLC Bio Main Workbench. Alignment of speC, speK, speL, speM, and spd1 sequence of bovine SDSD against the GenBank database using BLAST was performed to trace homolog genes among streptococcal species.
Antimicrobial Resistance Patterns
All SDSD strains were tested for antimicrobial resistance by disk diffusion method (Oxoid Ltd., Basingstoke, United Kingdom), and the determination of minimum inhibitory concentration (MIC) follows the guidelines from the Clinical and Laboratory Standards Institute (CLSI) for antimicrobial susceptibility tests for bacteria isolated from animals (CLSI M31-A3, 2008). The following antimicrobials were selected for testing, based on several criteria: (a) licensing for mastitis treatment in cattle—amoxicillin–clavulanic acid 30 μg (AMC), gentamicin 10 μg (CN), penicillin 10 units (P), pirlimycin 2 μg (PRL), and streptomycin 10 μg (S); and (b) use in human medicine—chloramphenicol 30 μg (CHL), erythromycin 15 μg (ERY), tetracycline 30 μg (TET), and vancomycin 30 μg (VA). The results were interpreted according to the CLSI guidelines (CLSI M31-A3, 2008). The antimicrobial manufacturers’ instructions were followed when CLSI guidelines were not available. The reference strain Staphylococcus aureus ATCC 29213 was used for quality control for both methods.
Macrolide Resistance Phenotypes
All the isolates were tested for resistance only to macrolides (M phenotype) or to macrolides, lincosamides, and streptogramin-B (MLSB phenotype). The MLSB resistance, inducible (iMLSB) or constitutive (cMLSB), was checked by a double-disk test with erythromycin and pirlimycin disks. Resistance only to lincosamides (L phenotype) and to lincosamides and streptogramin-A (LSA phenotype) was described previously (Malbruny et al., 2004; Rato et al., 2013).
Antimicrobial Resistance Gene Detection
All the isolates with M or MLSB phenotypes (Pires et al., 2005) were screened to detect ermA, ermB, and mefA macrolide resistance genes. Genes tetM, tetT, tetW, tetL, tetQ tetO, tetK, and tetS were screened among all tetracycline-resistant strains (Pires et al., 2005). All strains were tested for the presence of linB gene (lincosamide resistance) (Bozdogan et al., 1999). Primer sequences and amplicon expected sizes are listed in Supplementary Table 2 (Supplementary File 2). Negative results were confirmed in at least three independent assays. VSD13 and VSD19 strains were used as positive control.
CRISPR Array Sequencing and CRISPR-Associated (Cas) Gene Screening
In this study, the genome sequence of an SDSE strain ATCC 12394 (GenBank accession number: NC_017567) was used to design specific primers to amplify the CRISPR array and cas genes (Supplementary Table 2 in Supplementary File 2). PCRs were performed in a thermocycler (Biometra, Göttingen, Germany) in a final volume of 25 μl containing 100 ng of bacterial DNA, 1 × PCR buffer, 2.5 mM of MgCl2, 0.4 mM of dNTPs, 1 U of Taq polymerase (NZYTech, Lisbon, Portugal), and 1 μM of each primer (NZYTech, Lisbon, Portugal). PCR conditions consisted of an initial denaturation cycle (95°C for 5 min), followed by 35 cycles of denaturation (95°C for 30 s), annealing (52°C for 30 s), and extension (72°C for 80 s). A final extension at 72°C for 7 min was also performed. Milli-Q water was used as a negative control in each PCR. The PCR products of CRISPR array regions were purified using the Wizard PCR Preps DNA Purification System (Promega, Madison, WI, United States). Sequencing was performed by STAB-Vida (Lisbon, Portugal) using specific primers (CRISPR Sdys For and CRISPR Sdys Rev; Supplementary Table 2 in Supplementary File 2). The obtained DNA sequences were analyzed and edited using the software CLC Bio Main Workbench. For each CRISPR array sequence, the spacers, repeats, and flanking regions were determined using CRISPRfinder and CRISPRtionary2. All experiments were carried out in triplicate (Grissa et al., 2007).
The similarity of the spacers, repeats, and flanking regions was analyzed using software currently available for CRISPR miner to infer a large-scale microbe–phage infection network3. The spacers from each strain were selected and used to BLAST (blastn with parameters settings: e-value < 0.01, and up to three mismatches allowed) against both bacteria and archaea virus genomes of an integrated database from the NCBI viral Genome Resource. Matches with a query cover above 90% with 100% identity were considered.
Prophage Identification
Prophinder (Lima-Mendez et al., 2008) was used to screen for prophage-specifying regions among the genome of SDSD, SDSE, S. canis, and S. pyogenes strains (accession numbers of genomes are available in Supplementary Table 1 in Supplementary File 1). Prophage sequences were annotated using RAST (Aziz et al., 2008). Intact prophages were manually analyzed to confirm the presence of genes indispensable to produce functional phages: left attachment site (attL); lysogeny; DNA replication; transcriptional regulation; head; tail; lysis modules; and right attachment site (attR).
Statistical Analysis
GraphPad Prism version 7.0 was used for statistical analysis. Data analysis was performed using the chi-square method. Statistical significance was considered when p < 0.05.
Results
Bacterial Isolates and Identification
The SDSD isolates’ identification was confirmed using 16S rRNA gene sequencing. Out of 194 Gram-positive cocci, 37 isolates (19%) were identified as S. dysgalactiae species. Sequence analysis of the rRNA 16S from bovine SDSD isolates showed between 99.2% and 100% nucleotide identity to SDSD ATCC 2795, SDSD NCTC4670, SDSD NCTC13731, and SDSD NCTC4669 strains deposited in the NCBI Nucleotide database. UPGMA dendrogram shows the multiple alignments of rRNA 16S sequences of bovine SDSD (from this study), SDSD, SDSE, and S. pyogenes strains deposited in the NCBI database. See Supplementary Figure 1 (Supplementary File 3).
Pulsed-Field Gel Electrophoresis Profiles
The 55 SDSD isolates from subclinical mastitis isolated in Portugal during 2002–2003 (collection I) and 2011–2013 (collection II) were resolved into 45 PFGE types. Epidemiologically groups or clusters (at 70% similarity) revealed two major clonal groups, including close to 90% of all bovine isolates. The macro-restriction profiles are listed in Table 1. Two or more strains share six PFGE patterns, and 31 PFGE patterns were of single isolates. Results demonstrated that there are no distinct groups among isolates of collection I and collection II.
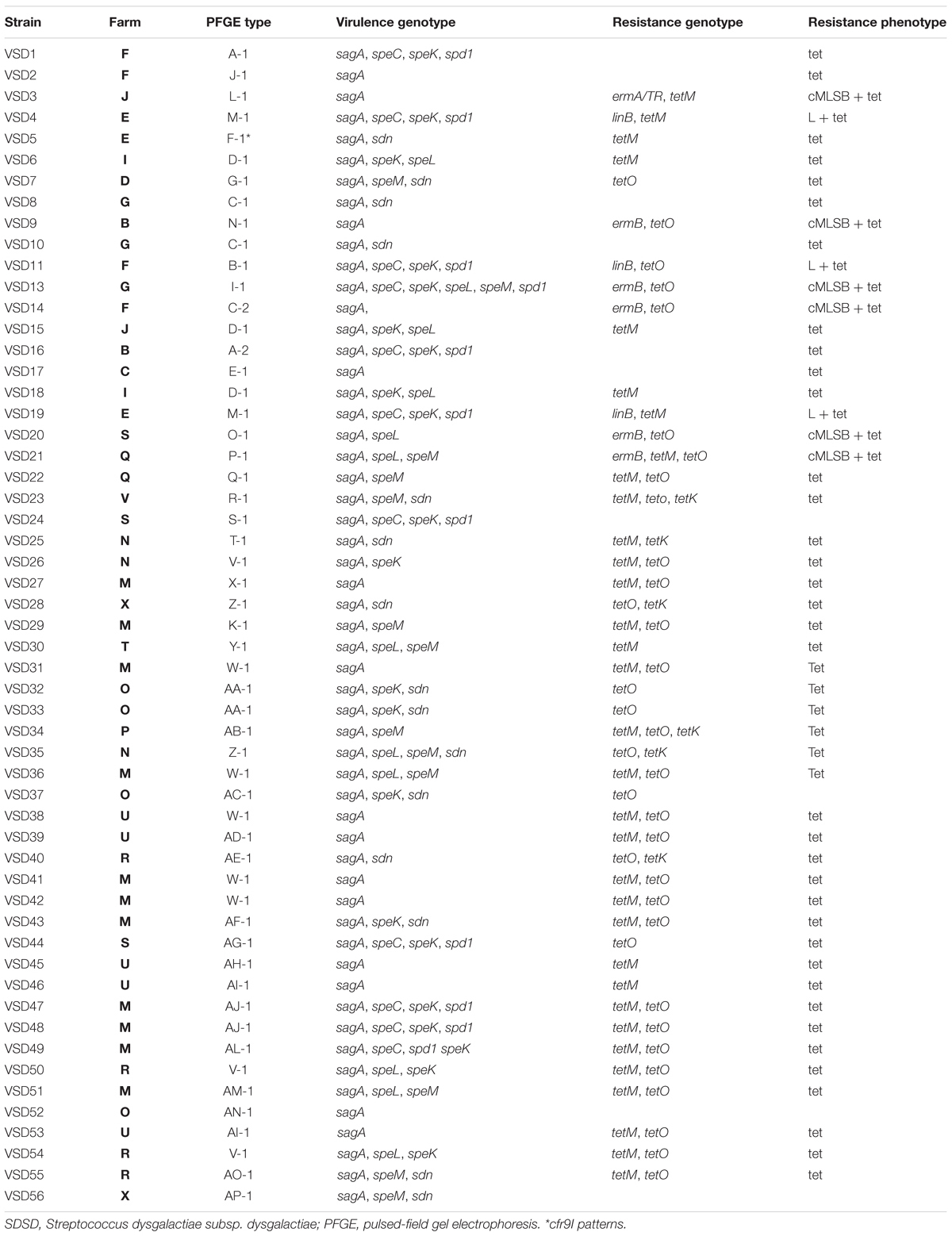
Table 1. Characterization of bovine SDSD isolates from collection I (VSD1 to VSD11 and VSD13 to VSD19) (2002–2003) and from collection II (VSD20 to VSD55) (2011–2013).
Phylogenetic Analysis
Alignment of concatenated sequences of seven housekeeping genes (gki, gtr, murI, mutS, recP, xpt, and atoB) was created using the standard parameters of ClustalW. Then a minimum evolution tree was obtained from sequences from bovine SDSD isolates (determined in this study) and sequences of a selection of SDSD, SDSE, S. pyogenes, and S. canis strains available in the NCBI database (Supplementary Table 1 in Supplementary File 1 and Figure 1).
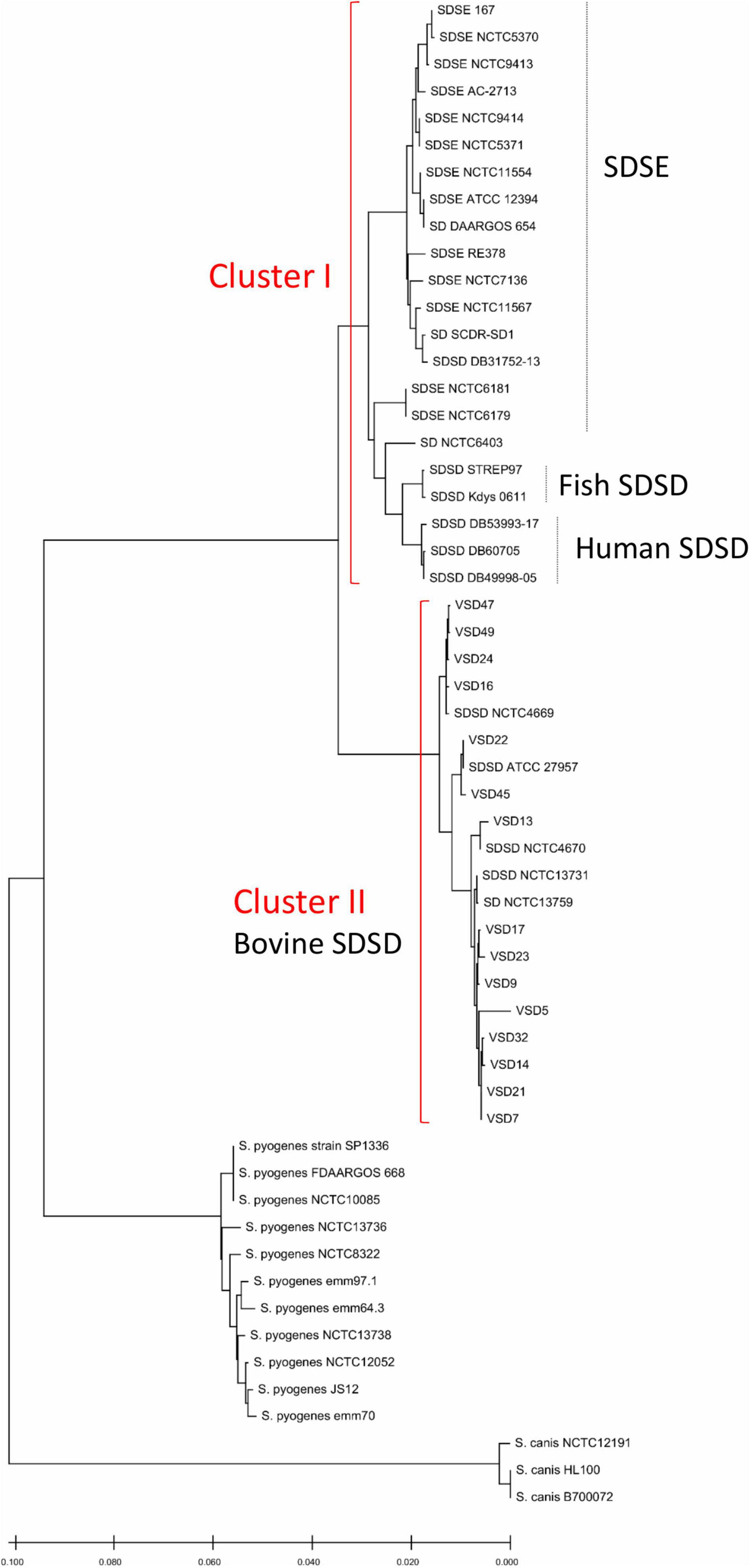
Figure 1. Phylogenetic analysis based on concatenated sequences of seven housekeeping genes (gki, gtr, murI, mutS, recP, xpt, and atoB) showing the position of the 15 bovine SDSD isolates and sequences of strains available in the National Center for Biotechnology Information (NCBI) database, including SDSD strains (n = 12), SDSE strains (n = 15), Streptococcus pyogenes strains (n = 11), Streptococcus canis strains (n = 3). VSD, Vet S. dysgalactiae subsp. dysgalactiae. Bovine SDSD isolates: ATCC 27957; NCTC13731; NCTC4669; NCTC4670 and VSDs. Human SDSD strains: DB31752-13; DB49998-05; DB60705-15, and DB53993-17. Fish SDSD strain: SDSD Kdys0611. SDSE, S. dysgalactiae subsp. equisimilis. The phylogenetic relationships were inferred using the minimum evolution method, and phylogenetic distances were estimated using the maximum composite likelihood method using the MEGA (Kumar et al., 2018). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree.
Streptococcus canis and S. pyogenes constitute monophyletic clusters. The S. dysgalactiae cluster was divided into two groups: cluster 1—human and fish SDSD strains, and SDSE; cluster 2—bovine SDSD isolates. Data show that human SDSD strains are closer to SDSE than bovine SDSD isolates. Interestingly, this phylogenetic relationship between SDSD and SDSE obtained by MLSA does not reflect the relationship based on the 16S rRNA sequences (Figure 1 and Supplementary Figure 1 in Supplementary File 3). The accession numbers of gki, gtr, murI, mutS, recP, xpt, and atoB genes are present in Supplementary Table 3 (Supplementary File 5).
Virulence Genes
All the 37 bovine SDSD isolates (collection II), screened by PCR, carry at least one of the following superantigens (SAgs) genes (speC, speK, speL, and speM), DNase (spd1), streptodornase (sdn), or SLS (sagA). None of the strains carry speA, speB, speF, speG, spegg, and speH SAgs genes; smeZ mitogenic exotoxin gene; and sagB, sagE, and sagI genes.
The results showed that the SAgs genes were unevenly distributed among all the 37 bovine SDSD isolates (see Table 1). The screening revealed the following distribution: speC, 20%; speK, 40%; speL, 20%; speM, 23.6%; spd1, 20%; and sdn, 27.2% positive isolates. Transcriptional analysis showed that all virulence genes were transcribed in all SDSD isolates that carried them.
Similar virulence gene patterns were found among bovine SDSD from collection I (Rato et al., 2010, 2011) and II. No relationships were found between virulence profiles and the farm or macro-restriction patterns (see Table 1).
The comparison of the distribution of virulence genes between SDSD isolates from collection I and II is shown in Figure 2. Chi-square statistical analysis revealed that each independent virulence gene proportion was not statistically different between collections.
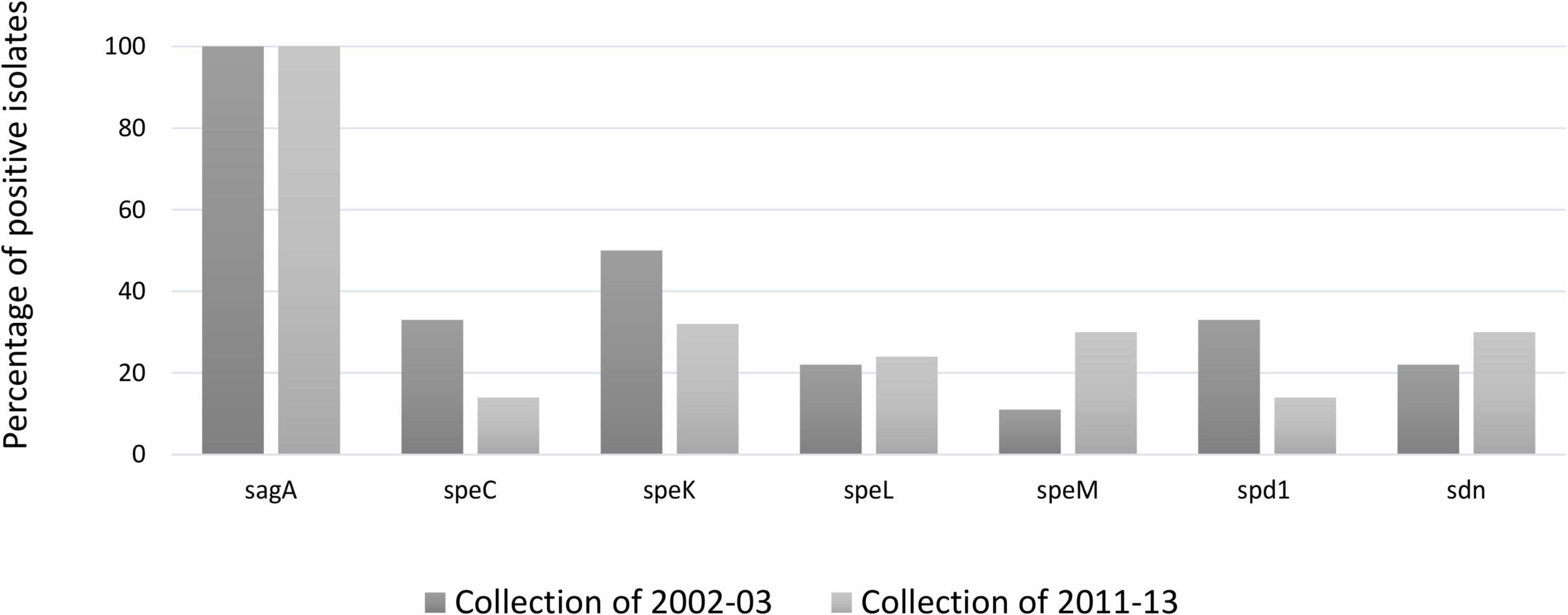
Figure 2. Distribution (%) of group A Streptococcus pyogenes virulence gene among Streptococcus dysgalactiae subsp. dysgalactiae collections isolate in 2002–2003 and 2011–2013. speC, speK, speL, speM: encode streptococcal pyrogenic exotoxins; spd1: DNase; sdn: streptodornase (DNase). The statistical significance of the data was determined by a chi-square test (χ2), where a probability value (p) ≤ 0.05 was considered as statistically significant.
The comparison of the homologous sequences of bovine SDSD SAgs genes with S. pyogenes pyrogenic exotoxin gene from the NCBI databases, showed high identity: >98% identity for speC; >97% for speK; >98% for speM; and >97% for speL. In addition, a high identity was also found for DNAses spd1 (>98%) (Supplementary Tables 4–7 in Supplementary File 4).
For a comparative analysis, the presence of virulence genes in the genomes of the streptococci listed in Supplementary Table 1 (Supplementary File 1) was analyzed. None of the speB, speC, speF, speG, speH, speK, speL, speM, and smeZ virulence genes were detected among human SDSD or SDSE strains. Streptodornase (sdn) was found among the human SDSE strains (3/15), S. pyogenes strains (2/15), bovine SDSD isolates (5/20), but it was not detected in human SDSD or S. canis strains. spegg gene was found in SDSE, S. pyogenes, and human SDSD, but not in bovine SDSD; and the smeZ was found only among S. pyogenes strains (Supplementary Figure 2 in Supplementary File 3). The nucleotide sequences of spegg gene in human and fish SDSD strains revealed >99% identity with its homolog in S. pyogenes from the NCBI database.
In addition, all the bovine SDSD isolates from collection I and the genomes listed in Supplementary Table 1 (Supplementary File 1) were tracked for the presence of the SLS operon. The comparative analysis showed that of the nine genes that are part of the SLS operon, the bovine SDSD isolates only had sagA gene (Table 1), while the SDSE, human and fish SDSD, S. pyogenes, and S. canis strains harbored the complete operon (Figures 3A,B and Supplementary Figure 2 in Supplementary File 3). Figure 3C shows the amino acid sequence of SagA protein of the bovine SDSD isolates deduced through in silico analysis, compared with SDSE, SDSD, S. canis, and S. pyogenes sequences. The alignment of SagA peptide demonstrated that the leader-peptide region is highly conserved, while the pro-peptide region is more variable. In this last region, the bovine SDSD isolates present a deletion of two cysteine residues.
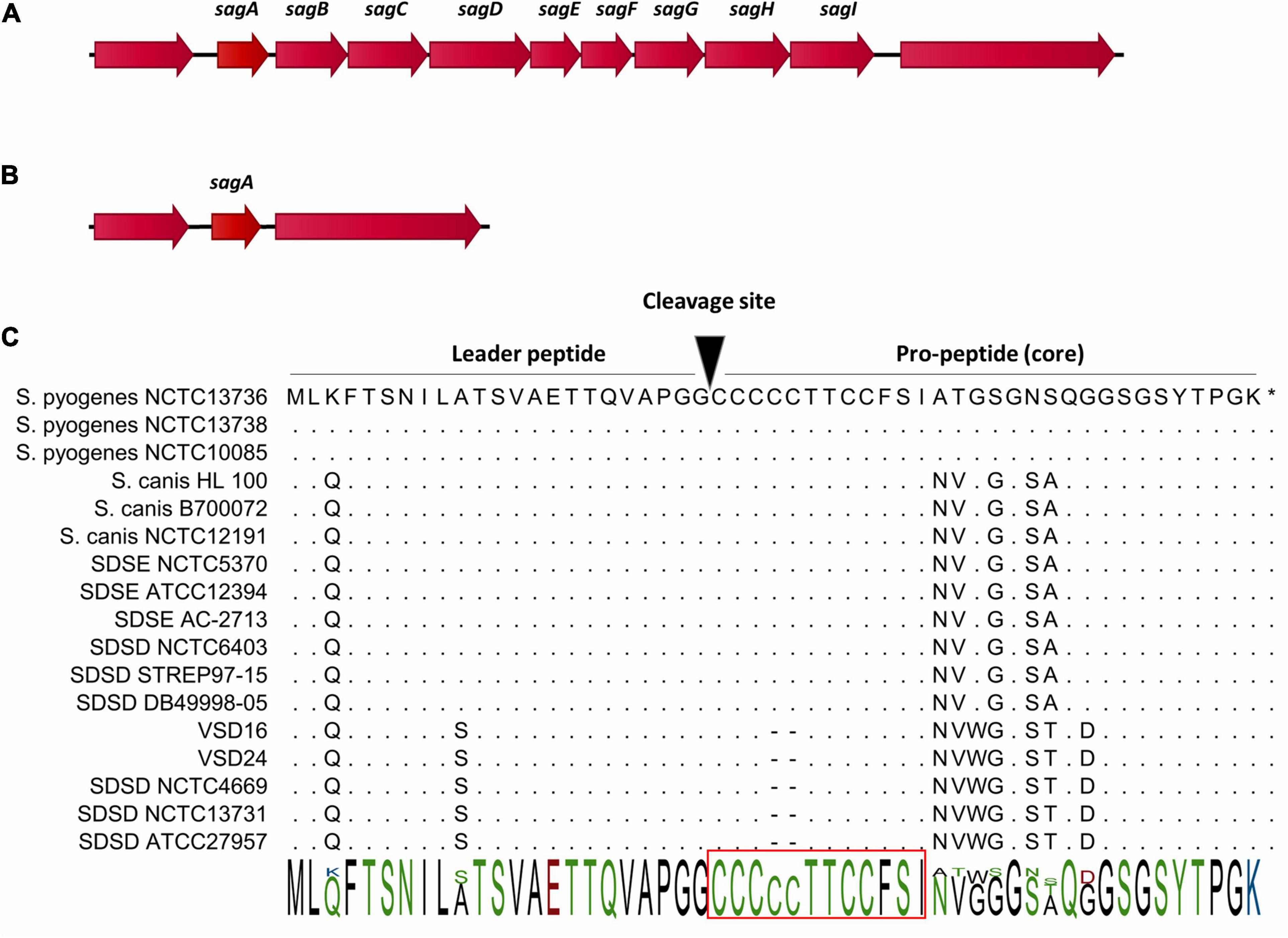
Figure 3. Gene cluster organization of streptolysin S (SLS) operon and precursor peptide sequences of SLS cytolysins (SagA). (A) Organization of the SLS operon in Streptococcus dysgalactiae subsp. equisimilis (SDSE), human and fish S. dysgalactiae subsp. dysgalactiae (SDSD), Streptococcus pyogenes, and Streptococcus canis. The operon encoding SLS includes the prepropeptide structural gene (sagA), followed by eight genes responsible for the conversion of SagA into SLS (sagBCD), transport across the membrane (sagFGHI), and accountable for leader cleavage (sagE) (Datta et al., 2005). (B) Organization of the genomic region flanking sagA in bovine SDSD isolates. (C) Alignment of SagA peptide, the precursor of SLS. SLS core region possesses a highly conserved N-terminus, while the C-terminus is more variable. The putative leader peptide cleavage site is shown. Red rectangle, minimal core region required for hemolytic activity of SLS in S. pyogenes. STAB-Vida performed sequencing of sagA. Deduced amino acid sequences from this bovine allele were compared with sequences from the National Center for Biotechnology Information (NCBI) database and were analyzed with the CLC-bio Main Workbench sequence alignment tool (QIAGEN, Netherlands).
Antimicrobial Resistance Patterns
Among the 37 bovine isolates from collection II, the highest antimicrobial resistance patterns were observed for tetracycline (90%), followed by streptomycin (69%) and gentamicin (60%). None of the streptococcal isolates tested showed resistance to amoxicillin/clavulanic acid, cefazolin, cefoperazone, chloramphenicol, penicillin, rifaximin, and vancomycin (Table 1 and Figure 4). Two isolates resistant to pirlimycin and erythromycin presented the cMLSB phenotype (Table 1), while the phenotype M (erythromycin-resistant only) was not detected. The two strains showed the cMLSB phenotype (constitutive MLSB). Strains of the cMLSB phenotype harbored ermB gene, while ermA and mefA (macrolide resistance genes) were not detected (Table 1). The resistance to tetracycline was associated with tetM, tetK, and tetO (tetS, tetT, tetW, tetL, and tetQ negative). Figure 4 compares the resistance phenotypes distribution and genotypes among the 55 bovine SDSD isolates from collection I (Rato et al., 2013) and collection II. Chi-square statistical analysis revealed that each independent resistance phenotype proportion was not statistically different between collections. However, statistically significant differences were observed between tetM, tetK, and tetO.
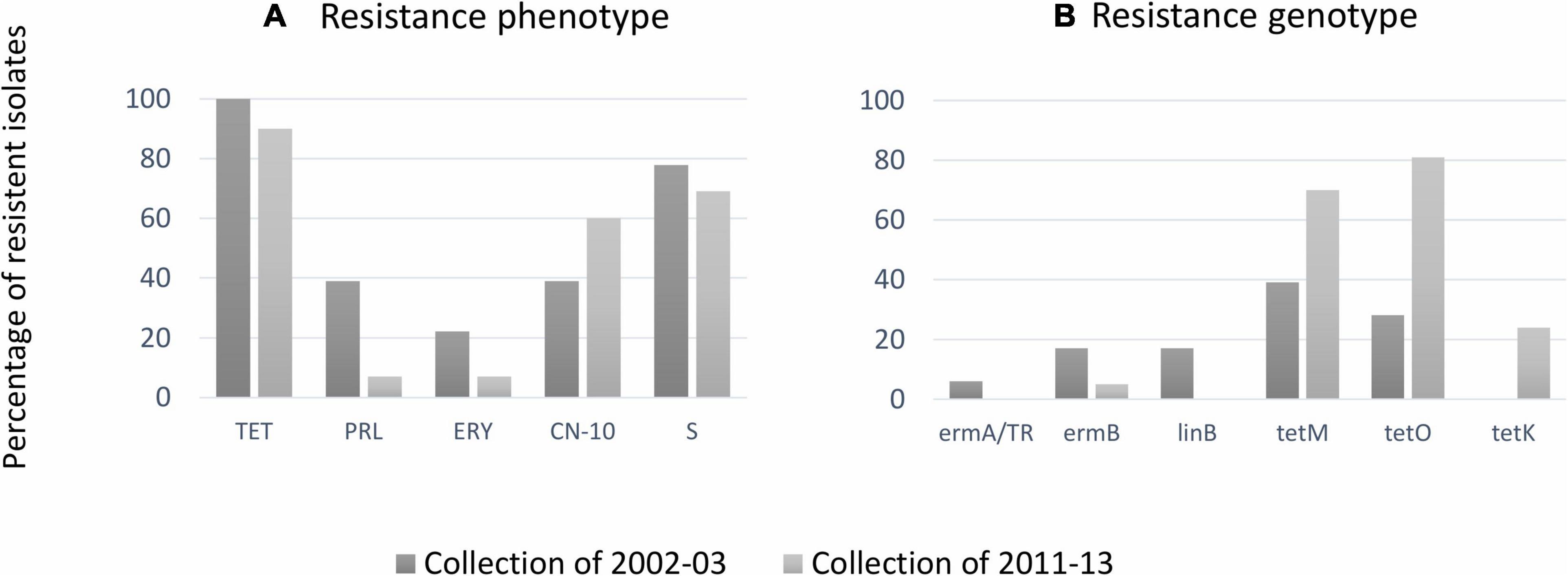
Figure 4. (A) Percentages of antimicrobial resistance among bovine Streptococcus dysgalactiae subsp. dysgalactiae (SDSD) isolates from the present study against erythromycin (ERY), gentamicin (CN), pirlimycin (PRL), streptomycin (S), and tetracycline (TET). Resistance was not observed against amoxicillin–clavulanic acid (AMC), cefazolin (KZ), chloramphenicol (CHL), penicillin (P), rifaximin (RAX), and vancomycin (VA). (B) Distribution resistance genotypes among bovine SDSD: ermA and ermB: macrolide resistance genes; linB: lincosamide resistance gene; tetK, tetM, and tetO: resistance to tetracycline genes. The statistical significance of the data was determined by a chi-square test (χ2), where a probability value (p) ≤ 0.05 was considered as statistically significant.
CRISPR System Analysis and Prophage Identification
SDSD isolates from collection I of 2002–2003 (N = 18) and collection II of 2011–2013 (N = 37) were analyzed by sequencing the CRISPR loci. Overall, the analysis of CRISPR array discloses a high intrinsic diversity among bovine SDSD understudy, demonstrating the distinguish groups among isolates from both collections (Figure 5).
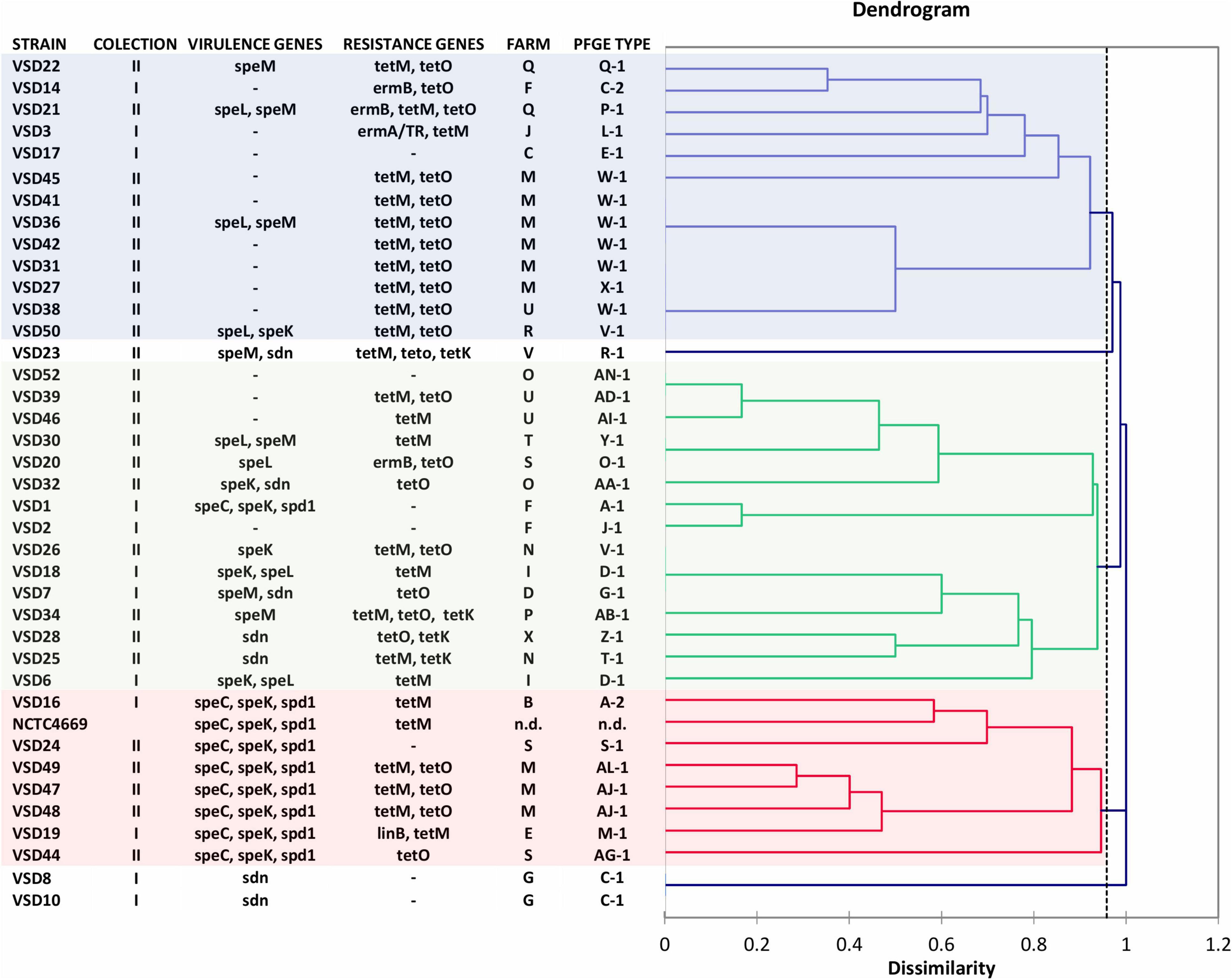
Figure 5. Dendrogram based on spacer CRISPR IIA cluster of Streptococcus dysgalactiae subsp. dysgalactiae isolates of mastitis in cattle collected in 2002–2003 (Rato et al., 2010, 2011, 2013) (VSD1 to VSD19—collection I) and 2011–2013 (VSD20 to VSD56—collection II). Streptococcus pyogenes phages encode genes speC, speK, speL, speM, spd1, and sdn. The dendrogram was obtained by means of Dice coefficient and the agglomerative clustering of the unweighted pair group method with arithmetic mean (UPGMA).
Among the 55 bovine SDSD isolates analyzed for the CRISPR array, 49 were PCR positive (but only 41 strains were CRISPR spacer positive; see Supplementary File 6), and four were PCR negative. For two strains, the amplified product showed high homology (>99%) with ISSdy transposase (ORF A, GenBank accession: VDZ39966.1 and ORF B, GenBank accession: VDZ39965.1).
A high degree of direct repeat (DR) sequence (5′GTTTTAGAGCTGTGCTGTTTCGAATGGTTCCAAAAC′3) conservation was observed among bovine SDSD isolates. The DR showed sequence similarity to typical repeat sequences of other streptococci species, such as S. agalactiae with 100% of identity (Lier et al., 2015), Streptococcus thermophilus with an average identity of about 85% (Hu et al., 2020), and S. pyogenes with >95% of identity (Nozawa et al., 2011).
A total of 158 spacers were identified among the 41 CRISPR spacer positive strains, of which 80 (50%) were found to be unique. The number of spacers in each CRISPR array was eight on average, ranging from a maximum of 26 to a minimum of 2 (Supplementary Tables 8, 9 in Supplementary File 6). Most strains (39 of 41) shared at least one spacer. Only VSD4 and VSD13 did not share spacers; these strains harbored six and three unique spacers, respectively. The SDSD VSD5, VSD9, VSD11, VSD29, VSD33, VSD37, and VSD40 isolates were CRISPR spacer negative (Supplementary File 6).
Three major groups were observed based on the common spacer sequences or absence of spacers (Figure 5 and Supplementary Table 9 in Supplementary File 6). Some old spacers were detected in bovine SDSD that harbored the same combinations of SAgs. For example, the 41, 42, 62, 63, 64, 65, 66, 118, 119, 120, and 121 spacers were detected among isolates from collection I (VSD16 and VSD19) and collection II (VSD24, VSD47, VSD48, and VSD49) harboring speC, speK, and spd1 genes (Figure 5 and Supplementary File 6). Some of these spacers were also detected in SDSD NCTC4669 (isolated in 1935).
About 72% (114/158) spacers matched with sequences present in the genome of S. pyogenes, S. agalactiae, S. canis, and S. dysgalactiae with a similarity >95. A BLAST was carried out to determine if the spacers had an origin in bacteriophage. Sequences above 95% of identity, in both query coverage and percent identity, were considered. Of the 158 spacer sequence, 29 (18.4%) showed high identity (bit scores ≥ 50) with prophage regions. These regions correspond to phage isolated from S. pyogenes and phages belonging to others species, namely, S. agalactiae str. ILRI005; Listeria phage B054; Flavobacterium phage 11b; Streptococcus phage SpSL1; Synechococcus phage S-CAM7; Bacillus virus IEBH; and Bacillus virus 250 (Table 2). About 28% of the sequence spacers of SDSD were not identified in the NCBI database.
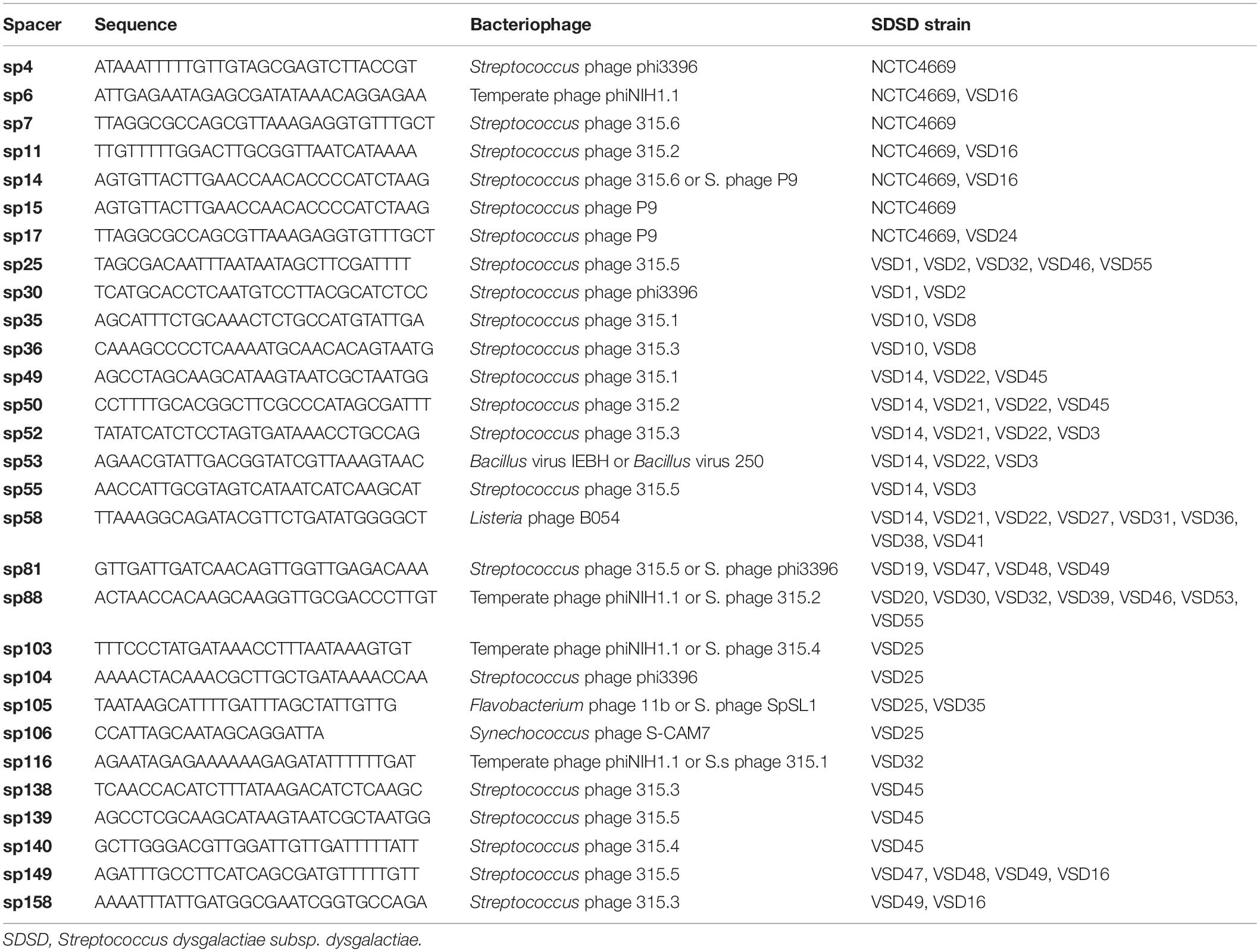
Table 2. Identification and nucleotide sequence of the bacteriophage spacers in a CRISPR array of SDSD.
Due to the similarity of some spacers between contemporary bovine SDSD isolates and SDSD NCTC4669 (isolated in 1935, the oldest documented with sequenced genome) (Chanter et al., 1997), we analyze the sequences adjacent to a CRISPR array of this oldest strain. The genetic map of this region consisted of only four cas genes (cas9, cas1, cas2, and csn2), corresponding to the CRISPR type IIA system. Additionally, the ISSdys transposase gene was found in type IIA system.
The genes associated with the type IIA CRISPR system were searched among the 55 bovine SDSD isolates (collection I and II). All bovine SDSD isolates were PCR positive to cas9, cas1, cas2, and csn2 genes. Additionally, two of the 55 strains were PCR positive for ISSdys transposase.
To elucidate the relationship between the prophages and CRISPR systems among SDSD strains, we analyzed CRISPR array, CRISPR-associated (Cas) proteins, and prophage structure using complete genome sequences of SDSD listed in Supplementary Table 1 (Supplementary File 1). For a comparative analysis, the complete genome of SDSE, S. canis, and S. pyogenes was also analyzed.
The CRISPR/Cas loci of the SDSD and SDSE strains analyzed were classified as type I-C and/or type II-A (Table 3). SDSD and SDSE genomes showed a high number of spacers when compared with S. pyogenes. While CRISPR arrays of S. pyogenes have between 0 and 10 spacers, SDSD and SDSE can have more than 40 spacers (Table 3). No CRISPR system was observed in 15% and 42% of the SDSE and S. pyogenes genomes, respectively, while in all SDSD strains, CRISPR/Cas system is present.

Table 3. Identification of CRISPR/Cas systems and prophage or prophage-like regions.
The total number of prophage regions obtained through in silico analysis was 228 in 42 genomes, with an average of 5.45 prophages per genome (Table 3). S. pyogenes had the highest number of complete prophages per genome, i.e., 2.1, followed by SDSD with two complete prophages per genome. SDSE and S. canis had 1.15 and 1 complete prophages per genome.
The SDSD STREP97-15 strain presented the highest number of prophage regions (20); however, only three prophage regions appear to be complete. Among human SDSD strains, only SDSD DB60705-15 harbored complete prophages (3). SDSD DB31752-13, DB49998-05, and DB53993-17 did not harbor any complete prophages. On the other hand, all bovine and fish SDSD strains harbored complete prophages. The complete prophage genomes of the SDSD strains are listed in Table 4. Overall, there was no statistical difference between the total number of prophage regions in type IC SDSD strains and type IIA SDSD strains.
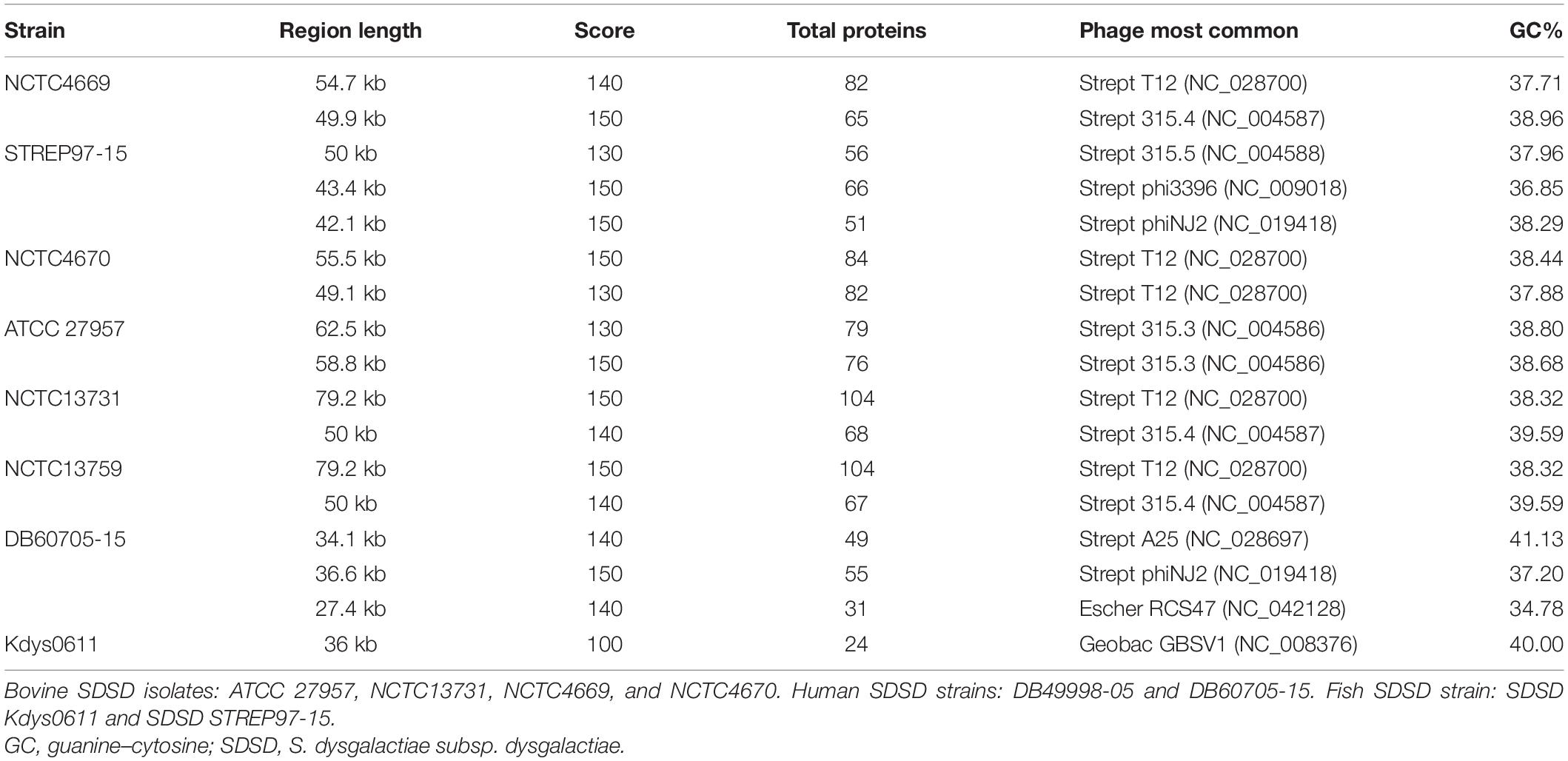
Table 4. Prophage regions identified by computational tools for the genome of Streptococcus dysgalactiae subsp. dysgalactiae strains.
Discussion
In the present study, the MLSA was performed according to the method previously described (McMillan et al., 2010). In a previous study, the phylogeny of S. canis, S. dysgalactiae, Streptococcus equi, and S. pyogenes species based on MLSA (map, pfl, ppaC, pyk, rpoB, sodA, and tuf genes) was reconstructed according to this analysis; S. dysgalactiae includes two separate clusters corresponding to the “dysgalactiae” and “equisimilis” subspecies (Jensen and Kilian, 2012). Surprisingly, our results show the separation of bovine SDSD from human SDSD strains in different clusters, the latter being closer to the SDSE (Figure 1).
In this study, we searched for the presence of different S. pyogenes virulence genes (encoded by MGE or either chromosomal) among bovine SDSD isolates, namely, genes encoding DNAses, SAgs, and SLS.
Some of the SAgs genes are phage- and transposon-associated among S. pyogenes strains (Proft and Fraser, 2016). Previous studies have related some of these genes to an increase in the pathogenic potential of SDSD isolates (Rato et al., 2010, 2011). Our results show that the SAgs genes (speC, speK, speL, and speM) and spd1 are unevenly distributed among bovine SDSD isolates (from collection I and collection II) but not among human and fish SDSD strains (Table 1 and Supplementary Figure 2 in Supplementary File 3).
The speC and spd1 (initially identified in the M1 phage) genes were always detected together, and it was also found to be associated with speK (initially identified in the M3 phage) among bovine SDSD isolates, indicating the probable poly-lysogeny, as described for S. pyogenes (Giovanetti et al., 2015). We also found the speC–spd1–speK genes in the Strept T12 NC_028700 prophage of the bovine SDSD NCTC4669 strain.
Alignment of speC, speK, speL, speM, and spd1 sequence of bovine SDSD against the GenBank database, using BLAST, revealed a nucleotide identity of 97% to >99% with the homologous S. pyogenes virulence (Supplementary File 5). Although the genomes of the bovine SDSD isolates from collection I and II had not been sequenced, the high homology of SAgs and the presence of these genes in prophage regions among S. pyogenes could indicate that similar phages (or even the same) are shared among the bovine SDSD and S. pyogenes. The high identity of the SAgs and the different habitats of SDSD (animals) and S. pyogenes (human pathogen) may suggest that the HGT events occurred before habitat separation.
We analyzed the presence of virulence genes among streptococcal genomes listed in Supplementary Table 1 (Supplementary File 1; Supplementary Figure 2 in Supplementary File 3). We found the streptococcal pyrogenic exotoxin G gene (spegg) in SDSE, S. pyogenes, and human SDSD, but not in bovine SDSD. The presence of spegg has been documented in fish SDSD strains (Abdelsalam et al., 2010); however, to our knowledge, the presence of this gene has not been reported so far among human SDSD strains. Nucleotide sequences from the human and fish SDSD strains revealed >99% identity with the homologous spegg of S. pyogenes from the NCBI database. To date, there is no evidence of HGT of the spegg; so its absence may mean the loss of this in bovine SDSD isolates common ancestor.
SDSD (α-hemolytic) and SDSE (β-hemolytic) have been distinguished based on their hemolytic properties on blood agar in routine laboratory practice. The hemolytic activity is mainly attributed to SLS production among the pyogenic group of streptococci.
In this study, the results showed the loss of sagB-I genes among bovine SDSD isolates. At the same time, the presence of the SLS operon was observed in human and fish SDSD, SDSE, S. pyogenes, and S. canis (β-hemolytic strains). Our data are the first to suggest that hemolytic differences between human SDSD strains (β-hemolytic) and bovine SDSD isolates (α-hemolytic) may be related to the loss of SagB-I and mutation of sagA gene observed in bovine SDSD isolates (Figure 3). Likely, the exact incidence of SDSD human infections on a global basis can be underestimated, mainly because of the failure to distinguish SDSD from SDSE strains in clinical laboratories.
Even though SDSD remains susceptible to most prescribed antibiotics, a significant number of treatment failures have been reported (Trieu-Cuot et al., 1993; Arthur et al., 1995; González Pedraza Avilés and Ortiz Zaragoza, 2000; Schwarz et al., 2004; Woodford, 2005; Pletz et al., 2006; Kimura et al., 2013; Cattoir, 2016; Doumith et al., 2017; Lai et al., 2017). Although tetracycline is not indicated for bovine mastitis-associated streptococci treatment in Portugal nowadays, high resistance to this class of antibiotics has been observed among bovine SDSD isolates. We observed a significant increase in the frequency of tetM, tetK, and tetO tetracycline resistance genes among bovine SDSD of collection II compared with bovine strains of the collection I. This frequency is likely associated with the fact that tetracycline has been extensively used to treat infections in bovine for many decades; tetracycline resistance determinants are linked to other resistance genes. For example, tetracycline resistance genes can be acquired via MGEs (e.g., plasmids and/or transposons), which also harbored erythromycin resistance genes (Brenciani et al., 2007, 2010; Emaneini et al., 2014; Santoro et al., 2014; Cattoir, 2016), contributing to a multiresistant phenotype. In our SDSD isolates, we identified the presence of ermB/tetO/tetK and ermB/tetO macrolide/tetracycline resistance genes, suggesting a possible horizontal gene co-transference of these genes.
Since the sequential order of the spacers in the CRISPR array provides chronological information about the bacteria’s exposure to foreign nucleic acids (Lier et al., 2015), providing insights into host–phage interactions within a specific niche, herein, we investigated the occurrence of CRISPR/Cas systems and CRISPR spacer content in bovine SDSD isolates to gain insight into the population diversity and the impact of this system on HGT.
Our results showed that all bovine SDSD strains characterized in the present study carried the CRISPR/Cas IIA system. In addition, we also observed a high degree of polymorphism in the CRISPR spacers providing a high ability to discriminate between strains. About 18.4% of spacers match known prophages with over 96% identity indicating a cross-species exchange of genetic material. The most frequent bacteriophages are listed in Table 2. Some studies have suggested that some common phages act as spacer donators (Le Rhun et al., 2019; Hu et al., 2020); thus, different spacers in different strains but identified as the same phage may be correlated with phage’s evolution (Sorek et al., 2013; Landsberger et al., 2018; Watson et al., 2018). About 28% of the sequence spacers of SDSD were not identified in the database, probably because of the rapid evolution of phage sequences, and sequences of new phages are not available.
The widespread presence of this CRISPR/Cas system among SDSD bovine strains, the high conservation of repeated sequences, and the polymorphism observed among the CRISPR IIA array spacers of these strains may lead us to consider them as indicators of the activity of the system (Figure 5 and Supplementary Table 9 in Supplementary File 6).
To elucidate the relationship between the prophages and CRISPR systems among SDSD strains, we analyzed CRISPR array, CRISPR-associated (Cas) proteins, and prophage structure using complete genome sequences of SDSD listed in Supplementary Table 1 (Supplementary File 1). For a comparative analysis, complete genome of SDSE, S. canis, and S. pyogenes were also analyzed.
We observed that the CRISPR II-A system had increased the number of spacers relative to CRISPR I-C. Indeed, CRISPR II-A locus is the most widespread type among streptococci strains and has the most significant number of spacers. Several studies concluded that type II-A CRISPR might be the primary phage prevention or elimination system in the streptococcus genome (Serbanescu et al., 2015; Yamada et al., 2019; Hu et al., 2020).
The genomes of SDSD and SDSE show a high number of spacers compared with those of S. pyogenes (Table 3). Some studies have speculated on the loss of the CRISPR array and/or the CRISPR/Cas locus in some clinical isolates. In this regard, it was proposed that the lack of CRISPR/Cas could indicate an adaptation to acquire new virulence genes. It has also been hypothesized that the low number of spacers acquired by S. pyogenes could be a strategy to acquire virulence genes and thus increase its pathogenic capacity (Le Rhun et al., 2019). Among our SDSD strains, there was no statistically significant difference in the total number of prophages in the strains with type IC and type IIA systems. A more significant number of prophage regions would be expected in the absence of the CRISPR/Cas system; however, we observe SDSD strains with an active CRISPR/Cas system harboring several prophage regions per genome.
Despite the different origins of SDSD strains, most prophages are related and were identified initially in S. pyogenes (Table 4). The phages’ specificity can explain the high frequency of S. pyogenes prophages among SDSD isolates; that is, the interaction between phage particles and bacteria generally involves specific receptors located in the bacterial cell membranes (Stone et al., 2019). The fact that SDSD and S. pyogenes do not share the same host makes it seem likely that the transfer of phages harboring streptococcal pyrogenic exotoxins has occurred before the divergence of ecological niches between the two species.
In conclusion, our results showed a significant increase in the frequencies of the tetM, tetO, and tetK genes over time, although tetracycline is not indicated for bovine mastitis-associated streptococcus treatment in Portugal nowadays. Our data suggest that carriage of S. pyogenes prophage virulence genes (speC, speK, speL, speM, and spd1) is maintained over time and seems to be a property of bovine SDSD, but not of human and fish SDSD and SDSE strains. The high homology of SAgs genes between bovine SDSD and S. pyogenes strains may suggest that the HGT events occurred before habitat separation.
This study documented for the first time the widespread presence of the CRISPR/Cas IIA system among bovine SDSD isolates, and the polymorphism observed in spacer content can be considered indicators of the system activity. However, no correlation was observed between the number of spacers CRISPR IIA and the number of prophages in the SDSD genomes. Further studies are necessary to understand better the CRISPR/Cas systems among SDSD strains, such as other CRISPR systems, and its impact on the evolution of SDSD virulence.
Our data are the first to suggest that hemolytic differences between human SDSD strains (β-hemolytic) and bovine SDSD isolates (α-hemolytic) may be related to the loss of SagB-I and mutation of sagA gene observed in bovine SDSD isolates. The SagA alignment reveals higher homology between human and fish SDSD and SDSE strains when compared with the bovine SDSD isolates. The close genetic relationship between non-bovine SDSD and SDSE was also clear from phylogenetic analysis based on MLSA, while bovine SDSD isolates seem more divergent.
The data set of this study suggests that the separation between the subspecies “dysgalactiae” and “equisimilis” should be reconsidered. However, a study including the most comprehensive collection of strains from different environments would be required for definitive conclusions regarding the two taxa.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
IS-S, CA-B, AF, and RM contributed to the idea or design of the research. RB and MO collaborated with the collection of bovine mastitis samples. CA-B contributed to the experimental work, analysis of sequences, and performed complete genomes. JC and CR-R contributed to the research of virulence genes. LC and RT contributed with the support in the BioNumerics software and assistance in the analysis. CA-B wrote down the first draft and the subsequent revisions of the manuscript. RM and AF performed the revision and editing of the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Unidade de Ciências Biomoleculares Aplicadas-UCIBIO, which is financed by national funds from FCT/MEC (UIDP/04378/2020 and UIDB/04378/2020) and also by projects PTDC/CVT-EPI/4651/2012 and PTDC/CVT-EPI/6685/2014. FCT-MEC is also acknowledged for grant SFRH/BD/118350/2016 to CA-B.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.686413/full#supplementary-material
Footnotes
- ^ www.ncbi.nlm.nih.gov/BLAST
- ^ https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index
- ^ http://www.microbiome-bigdata.com/CRISPRminer/index.php/Home/Index/inde
References
Abdelsalam, M., Chen, S. C., and Yoshida, T. (2010). Dissemination of streptococcal pyrogenic exotoxin G (spegg) with an IS-like element in fish isolates of Streptococcus dysgalactiae. FEMS Microbiol. Lett. 309, 105–113. doi: 10.1111/j.1574-6968.2010.02024.x
Abdelsalam, M., Fujino, M., Eissa, A. E., Chen, S. C., and Warda, M. (2015). Expression, genetic localization and phylogenic analysis of NAPlr in piscine Streptococcus dysgalactiae subspecies dysgalactiae isolates and their patterns of adherence. J. Adv. Res. 6, 747–755. doi: 10.1016/j.jare.2014.05.005
Alves-Barroco, C., Roma-Rodrigues, C., Balasubramanian, N., Guimarães, M. A., Ferreira-Carvalho, B. T., Muthukumaran, J., et al. (2019). Biofilm development and computational screening for new putative inhibitors of a homolog of the regulatory protein BrpA in Streptococcus dysgalactiae subsp. dysgalactiae. Int. J. Med. Microbiol. 309, 169–181. doi: 10.1016/j.ijmm.2019.02.001
Alves-Barroco, C., Roma-Rodrigues, C., Raposo, L. R., Brás, C., Diniz, M., Caço, J., et al. (2018). Streptococcus dysgalactiae subsp. dysgalactiae isolated from milk of the bovine udder as emerging pathogens: In vitro and in vivo infection of human cells and zebrafish as biological models. Microbiol. Open 8:e00623. doi: 10.1002/mbo3.623
Aminov, R. I., Garrigues-Jeanjean, N., and Mackie, R. I. (2001). Molecular ecology of tetracycline resistance: development and validation of primers for detection of tetracycline resistance genes encoding ribosomal protection proteins. Appl. Environ. Microbiol. 67, 22–32. doi: 10.1128/AEM.67.1.22-32.2001
Arthur, M., Depardieu, F., Molinas, C., Reynolds, P., and Courvalin, P. (1995). The vanZ gene of Tn1546 from Enterococcus faecium BM4147 confers resistance to teicoplanin. Gene 154, 87–92. doi: 10.1016/0378-1119(94)00851-i
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genom. 9:75. doi: 10.1186/1471-2164-9-75
Baiano, J. C., and Barnes, A. C. (2009). Towards control of Streptococcus iniae. Emerg Infect Dis. 15, 1891–1896. doi: 10.3201/eid1512.090232
Barrangou, R., and Marraffini, L. A. (2014). CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol. Cell. 54, 234–244. doi: 10.1016/j.molcel.2014.03.011
Bhaya, D., Davison, M., and Barrangou, R. (2011). CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45, 273–297. doi: 10.1146/annurev-genet-110410-132430
Bondy-Denomy, J., Pawluk, A., Maxwell, K. L., and Davidson, A. R. (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432. doi: 10.1038/nature11723
Bozdogan, B., Berrezouga, L., Kuo, M. S., Yurek, D. A., Farley, K. A., Stockman, B. J., et al. (1999). A new resistance gene, linB, conferring resistance to lincosamides by nucleotidylation in Enterococcus faecium HM1025. Antimicrob. Agents Chemother. 43, 925–929. doi: 10.1128/AAC.43.4.925
Brenciani, A., Bacciaglia, A., Vecchi, M., Vitali, L. A., Varaldo, P. E., and Giovanetti, E. (2007). Genetic elements carrying erm(B) in Streptococcus pyogenes and association with tet(M) tetracycline resistance gene. Antimicrob. Agents Chemother. 51, 1209–1216. doi: 10.1128/AAC.01484-06
Brenciani, A., Bacciaglia, A., Vignaroli, C., Pugnaloni, A., Varaldo, P. E., and Giovanetti, E. (2010). Ψ m46.1, the main Streptococcus pyogenes element carrying mef(A) and tet(O) genes. Antimicrob. Agents Chemother. 54, 221–229. doi: 10.1128/AAC.00499-09
Cattoir, V. (2016). “Mechanisms of Antibiotic Resistance,” in Streptococcus pyogenes: Basic Biology to Clinical Manifestations, ed. J. J. Ferretti (University of Oklahoma Health Sciences Center).
Chanter, N., Collin, N., Holmes, N., Binns, M., and Mumford, J. (1997). Characterization of the Lancefield group C streptococcus 16S-23S RNA gene intergenic spacer and its potential for identification and sub-specific typing. Epidemiol. Infect. 118, 125–135. doi: 10.1017/s0950268896007285
Chennapragada, S. S., Ramphul, K., Barnett, B. J., Mejias, S. G., and Lohana, P. (2018). A rare case of Streptococcus dysgalactiae subsp. dysgalactiae human zoonotic infection. Cureus 10:e2901. doi: 10.7759/cureus.2901
Darenberg, J., Luca-Harari, B., Jasir, A., Sandgren, A., Pettersson, H., Schalén, C., et al. (2007). Molecular and clinical characteristics of invasive group A streptococcal infection in Sweden. Clin Infect Dis. 45, 450–458. doi: 10.1086/519936
Datta, V., Myskowski, S. M., Kwinn, L. A., Chiem, D. N., Varki, N., Kansal, R. G., et al. (2005). Mutational analysis of the group A streptococcal operon encoding streptolysin S and its virulence role in invasive infection. Mol. Microbiol. 56, 681–695. doi: 10.1111/j.1365-2958.2005.04583.x
De la Casa-Esperón, E. (2012). Horizontal transfer and the evolution of host-pathogen interactions. Int. J. Evol. Biol. 2012:679045. doi: 10.1155/2012/679045
Doumith, M., Mushtaq, S., Martin, V., Chaudhry, A., Adkin, R., Coelho, J., et al. (2017). Genomic sequences of Streptococcus agalactiae with high-level gentamicin resistance, collected in the BSAC bacteraemia surveillance. J. Antimicrob. Chemother. 72, 2704–2707. doi: 10.1093/jac/dkx207
Emaneini, M., Mirsalehian, A., Beigvierdi, R., Fooladi, A. A., Asadi, F., Jabalameli, F., et al. (2014). High incidence of macrolide and tetracycline resistance among Streptococcus agalactiae strains isolated from clinical samples in Tehran, Iran. Maedica 9, 157–161.
Giovanetti, E., Brenciani, A., Morroni, G., Tiberi, E., Pasquaroli, S., Mingoia, M., et al. (2015). Transduction of the Streptococcus pyogenes bacteriophage Φm46.1, carrying resistance genes mef(A) and tet(O), to other Streptococcus species. Front. Microbiol. 5:746. doi: 10.3389/fmicb.2014.00746
González Pedraza Avilés, A., and Ortiz Zaragoza, M. C. (2000). Streptococcus pyogenes: in vitro susceptibility and role of beta-lactamase producing bacteria in the persistence of streptococcal pharyngotonsillitis. Aten. Primaria 25, 542–545. doi: 10.1016/s0212-6567(00)78564-7
Green, N. M., Beres, S. B., Graviss, E. A., Allison, J. E., McGeer, A. J., Vuopio-Varkila, J., et al. (2005). Genetic diversity among type emm28 group A Streptococcus strains causing invasive infections and pharyngitis. J. Clin. Microbiol. 43, 4083–4091. doi: 10.1128/JCM.43.8.4083-4091.2005
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Grohmann, E., Muth, G., and Espinosa, M. (2003). Conjugative plasmid transfer in gram-positive bacteria. Microbiol. Mol. Biol. Rev. 67, 277–301. doi: 10.1128/mmbr.67.2.277-301.2003
Haenni, M., Saras, E., Bertin, S., Leblond, P., Madec, J. Y., and Payot, S. (2010). Diversity and mobility of integrative and conjugative elements in bovine isolates of Streptococcus agalactiae, S. dysgalactiae subsp. dysgalactiae, and S. uberis. Appl. Environ. Microbiol. 76, 7957–7965. doi: 10.1128/AEM.00805-10
Hu, T., Cui, Y., and Qu, X. (2020). Characterization and comparison of CRISPR Loci in Streptococcus thermophilus. Arch. Microbiol. 202, 695–710. doi: 10.1007/s00203-019-01780-3
Jasir, A., Tanna, A., Efstratiou, A., and Schalén, C. (2001). Unusual occurrence of M type 77, antibiotic-resistant group A streptococci in southern Sweden. J. Clin. Microbiol. 39, 586–590. doi: 10.1128/JCM.39.2.586-590.2001
Jensen, A., and Kilian, M. (2012). Delineation of Streptococcus dysgalactiae, its subspecies, and its clinical and phylogenetic relationship to Streptococcus pyogenes. J. Clin. Microbiol. 50, 113–126. doi: 10.1128/JCM.05900-11
Jordal, S., Glambek, M., Oppegaard, O., and Kittang, B. R. (2015). New tricks from an old cow: infective endocarditis caused by Streptococcus dysgalactiae subsp. dysgalactiae. J. Clin. Microbiol. 53, 731–734. doi: 10.1128/JCM.02437-14
Kimura, K., Nagano, N., Nagano, Y., Suzuki, S., Wachino, J., Shibayama, K., et al. (2013). High frequency of fluoroquinolone- and macrolide-resistant streptococci among clinically isolated group B streptococci with reduced penicillin susceptibility. J Antimicrob Chemother. 68, 539–542. doi: 10.1093/jac/dks423
Koh, T. H., Sng, L. H., Yuen, S. M., Thomas, C. K., Tan, P. L., Tan, S. H., et al. (2009). Streptococcal cellulitis following preparation of fresh raw seafood. Zoonoses Public Health 56, 206–208. doi: 10.1111/j.1863-2378.2008.01213.x
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lai, L., Dai, J., Tang, H., Zhang, S., Wu, C., Qiu, W., et al. (2017). Streptococcus suis serotype 9 strain GZ0565 contains a type VII secretion system putative substrate EsxA that contributes to bacterial virulence and a vanZ-like gene that confers resistance to teicoplanin and dalbavancin in Streptococcus agalactiae. Vet. Microbiol. 205, 26–33. doi: 10.1016/j.vetmic.2017.04.030
Landsberger, M., Gandon, S., Meaden, S., Rollie, C., Chevallereau, A., Chabas, H., et al. (2018). Anti-CRISPR Phages cooperate to overcome CRISPR-Cas immunity. Cell 174, 908–916.e28. doi: 10.1016/j.cell.2018.05.058
Le Rhun, A., Escalera-Maurer, A., Bratovič, M., and Charpentier, E. (2019). CRISPR-Cas in Streptococcus pyogenes. RNA Biol. 16, 380–389. doi: 10.1080/15476286.2019.1582974
Lefébure, T., Richards, V. P., Lang, P., Pavinski-Bitar, P., and Stanhope, M. J. (2012). Gene repertoire evolution of Streptococcus pyogenes inferred from phylogenomic analysis with Streptococcus canis and Streptococcus dysgalactiae. PLoS One 7:e37607. doi: 10.1371/journal.pone.0037607
Lier, C., Baticle, E., Horvath, P., Haguenoer, E., Valentin, A. S., Glaser, P., et al. (2015). Analysis of the type II-A CRISPR-Cas system of Streptococcus agalactiae reveals distinctive features according to genetic lineages. Front. Genet. 6:214. doi: 10.3389/fgene.2015.00214
Lima-Mendez, G., Van Helden, J., Toussaint, A., and Leplae, R. (2008). Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 24, 863–865. doi: 10.1093/bioinformatics/btn043
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev.Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2018). Classification and nomenclature of CRISPR-Cas systems: Where from here? CRISPR J. 1, 325–336. doi: 10.1089/crispr.2018.0033
Malbruny, B., Werno, A. M., Anderson, T. P., Murdoch, D. R., and Leclercq, R. (2004). A new phenotype of resistance to lincosamide and streptogramin A-type antibiotics in Streptococcus agalactiae in New Zealand. J. Antimicrob. Chemother. 54, 1040–1044. doi: 10.1093/jac/dkh493
Matsumoto, M., Hoe, N. P., Liu, M., Beres, S. B., Sylva, G. L., Brandt, C. M., et al. (2003). Intrahost sequence variation in the streptococcal inhibitor of complement gene in patients with human pharyngitis. J. Infect Dis. 187, 604–612. doi: 10.1086/367993
McMillan, D. J., Bessen, D. E., Pinho, M., Ford, C., Hall, G. S., Melo-Cristino, J., et al. (2010). Population genetics of Streptococcus dysgalactiae subspecies equisimilis reveals widely dispersed clones and extensive recombination. PLoS One 5:e11741. doi: 10.1371/journal.pone.0011741
McNeilly, C. L., and McMillan, D. J. (2014). Horizontal gene transfer and recombination in Streptococcus dysgalactiae subsp. equisimilis. Front. Microbiol. 5:676. doi: 10.3389/fmicb.2014.00676
McShan, W. M., and Nguyen, S. V. (2016). “The Bacteriophages of Streptococcus pyogenes. 2016 Feb 10 [Updated 2016 Mar 25],” in Streptococcus pyogenes : Basic Biology to Clinical Manifestations [Internet], eds J. J. Ferretti, D. L. Stevens, and V. A. Fischetti (Oklahoma (OK): University of Oklahoma Health Sciences Center).
Ng, L. K., Martin, I., Alfa, M., and Mulvey, M. (2001). Multiplex PCR for the detection of tetracycline resistant genes. Mol. Cell. Probes. 15, 209–215. doi: 10.1006/mcpr.2001.0363
Nozawa, T., Furukawa, N., Aikawa, C., Watanabe, T., Haobam, B., Kurokawa, K., et al. (2011). CRISPR inhibition of prophage acquisition in Streptococcus pyogenes. PLoS One 6:e19543. doi: 10.1371/journal.pone.0019543
Park, M. J., Eun, I. S., Jung, C. Y., Ko, Y. C., Kim, Y. J., Kim, C. K., et al. (2012). Streptococcus dysgalactiae subspecies dysgalactiae infection after total knee arthroplasty: a case report. Knee Surg. Relat. Res. 24, 120–123. doi: 10.5792/ksrr.2012.24.2.120
Pires, R., Rolo, D., Gama-Norton, L., Morais, A., Lito, L., Salgado, M. J., et al. (2005). Group A Streptococci from carriage and disease in Portugal: evolution of antimicrobial resistance and T antigenic types during 2000-2002. Microb. Drug Resist. 11, 360–370. doi: 10.1089/mdr.2005.11.360
Pletz, M. W., McGee, L., Van Beneden, C. A., Petit, S., Bardsley, M., Barlow, M., et al. (2006). Fluoroquinolone resistance in invasive Streptococcus pyogenes isolates due to spontaneous mutation and horizontal gene transfer. Antimicrob. Agents Chemother. 50, 943–948. doi: 10.1128/AAC.50.3.943-948.2006
Proft, T., and Fraser, J. D. (2016). “Streptococcal Superantigens: Biological properties and potential role in disease. Feb 10,” in Streptococcus pyogenes : Basic Biology to Clinical Manifestations [Internet], eds J. J. Ferretti, D. L. Stevens, and V. A. Fischetti (Oklahoma City (OK): University of Oklahoma Health Sciences Center).
Rath, D., Amlinger, L., Rath, A., and Lundgren, M. (2015). The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie 117, 119–128. doi: 10.1016/j.biochi.2015.03.025
Rato, M. G., Bexiga, R., Florindo, C., Cavaco, L. M., Vilela, C. L., and Santos-Sanches, I. (2013). Antimicrobial resistance and molecular epidemiology of streptococci from bovine mastitis. Vet. Microbiol. 161, 286–294. doi: 10.1016/j.vetmic.2012.07.043
Rato, M. G., Bexiga, R., Nunes, S. F., Cavaco, L. M., Vilela, C. L., and Santos-Sanches, I. (2008). Molecular epidemiology and population structure of bovine Streptococcus uberis. J. Dairy Sci. 91, 4542–4551. doi: 10.3168/jds.2007-0907
Rato, M. G., Bexiga, R., Nunes, S. F., Vilela, C. L., and Santos-Sanches, I. (2010). Human group A streptococci virulence genes in bovine group C streptococci. Emerg Infect. Dis. 16, 116–119. doi: 10.3201/eid1601.090632
Rato, M. G., Nerlich, A., Bergmann, R., Bexiga, R., Nunes, S. F., Vilela, C. L., et al. (2011). Virulence gene pool detected in bovine group C Streptococcus dysgalactiae subsp. dysgalactiae isolates by use of a group A S. pyogenes virulence microarray. J. Clin. Microbiol. 49, 2470–2479. doi: 10.1128/JCM.00008-11
Richards, V. P., Zadoks, R. N., Pavinski Bitar, P. D., Lefébure, T., Lang, P., Werner, B., et al. (2012). Genome characterization and population genetic structure of the zoonotic pathogen Streptococcus canis. BMC Microbiol. 12:293. doi: 10.1186/1471-2180-12-293
Rohde, M., and Cleary, P. P. (2016). “Adhesion and invasion of Streptococcus pyogenes into host cells and clinical relevance of intracellular streptococci. 10,” in Streptococcus pyogenes: Basic Biology to Clinical Manifestations [Internet], eds J. J. Ferretti, D. L. Stevens, and V. A. Fischetti (Oklahoma City (OK): University of Oklahoma Health Sciences Center).
Roma-Rodrigues, C., Alves-Barroco, C., Raposo, L. R., Costa, M. N., Fortunato, E., Baptista, P. V., et al. (2016). Infection of human keratinocytes by Streptococcus dysgalactiae subspecies dysgalactiae isolated from milk of the bovine udder. Microbes Infect. 18, 290–293. doi: 10.1016/j.micinf.2015.11.005
Santoro, F., Vianna, M. E., and Roberts, A. P. (2014). Variation on a theme; an overview of the Tn916/Tn1545 family of mobile genetic elements in the oral and nasopharyngeal streptococci. Front. Microbiol. 5:535. doi: 10.3389/fmicb.2014.00535
Schmitz, F. J., Beyer, A., Charpentier, E., Normark, B. H., Schade, M., Fluit, A. C., et al. (2003). Toxin-gene profile heterogeneity among endemic invasive European group A streptococcal isolates. J. Infect. Dis. 188, 1578–1586. doi: 10.1086/379230
Schwarz, S., Kehrenberg, C., Doublet, B., and Cloeckaert, A. (2004). Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev. 28, 519–542. doi: 10.1016/j.femsre.2004.04.001
Serbanescu, M. A., Cordova, M., Krastel, K., Flick, R., Beloglazova, N., Latos, A., et al. (2015). Role of the Streptococcus mutans CRISPR-Cas systems in immunity and cell physiology. J. Bacteriol. 197, 749–761. doi: 10.1128/JB.02333-14
Sorek, R., Lawrence, C. M., and Wiedenheft, B. (2013). CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 82, 237–266. doi: 10.1146/annurev-biochem-072911-172315
Stone, E., Campbell, K., Grant, I., and McAuliffe, O. (2019). Understanding and Exploiting Phage-Host Interactions. Viruses 11:567. doi: 10.3390/v11060567
Suzuki, H., Lefébure, T., Hubisz, M. J., Pavinski Bitar, P., Lang, P., Siepel, A., et al. (2011). Comparative genomic analysis of the Streptococcus dysgalactiae species group: gene content, molecular adaptation, and promoter evolution. Genome Biol. Evol. 3, 168–185. doi: 10.1093/gbe/evr006
Takahashi, T., Kaneko, M., Mori, Y., Tsuji, M., Kikuchi, N., and Hiramune, T. (1997). Phylogenetic analysis of Staphylococcus based on the 16S rDNA sequence and assignment of clinical isolates from animals. J. Vet. Med. Sci. 59, 775–783. doi: 10.1292/jvms.59.775
Trieu-Cuot, P., de Cespédès, G., Bentorcha, F., Delbos, F., Gaspar, E., and Horaud, T. (1993). Study of heterogeneity of chloramphenicol acetyltransferase (CAT) genes in streptococci and enterococci by polymerase chain reaction: characterization of a new CAT determinant. Antimicrob. Agents Chemother. 37, 2593–2598. doi: 10.1128/aac.37.12.2593
Watson, B., Staals, R., and Fineran, P. C. (2018). CRISPR-Cas-mediated phage resistance enhances horizontal gene transfer by transduction. mBio 9, e02406–17. doi: 10.1128/mBio.02406-17
Wong, S. S., and Yuen, K. Y. (2012). Streptococcus pyogenes and re-emergence of scarlet fever as a public health problem. Emerg. Microbes Infect. 1:e2. doi: 10.1038/emi.2012.9
Woodford, N. (2005). Biological counterstrike: antibiotic resistance mechanisms of Gram-positive cocci. Clin. Microbiol. Infect. 11, 2–21. doi: 10.1111/j.1469-0691.2005.01140.x
Yamada, S., Shibasaki, M., Murase, K., Watanabe, T., Aikawa, C., Nozawa, T., et al. (2019). Phylogenetic relationship of prophages is affected by CRISPR selection in Group A Streptococcus. BMC Microbiol. 19:24. doi: 10.1186/s12866-019-1393-y
Keywords: Streptococcus dysgalactiae subsp. dysgalactiae, CRISPR typing, phylogenetic relationships, virulence genes, phage genes
Citation: Alves-Barroco C, Caço J, Roma-Rodrigues C, Fernandes AR, Bexiga R, Oliveira M, Chambel L, Tenreiro R, Mato R and Santos-Sanches I (2021) New Insights on Streptococcus dysgalactiae subsp. dysgalactiae Isolates. Front. Microbiol. 12:686413. doi: 10.3389/fmicb.2021.686413
Received: 30 March 2021; Accepted: 15 June 2021;
Published: 15 July 2021.
Edited by:
Eric Altermann, AgResearch Ltd., New ZealandReviewed by:
Franklin Nobrega, University of Southampton, United KingdomEligia Maria Szewczyk, Medical University of Łódź, Poland
Copyright © 2021 Alves-Barroco, Caço, Roma-Rodrigues, Fernandes, Bexiga, Oliveira, Chambel, Tenreiro, Mato and Santos-Sanches. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosario Mato, ci5sYWJham9zQGZjdC51bmwucHQ=
†In memory of Professor Ilda S. Sanches – For her work and contributions to Streptococcus spp. research