 Isis Gabriela Barbosa Carvalho1
Isis Gabriela Barbosa Carvalho1 Marcus Vinicius Merfa2
Marcus Vinicius Merfa2 Natália Sousa Teixeira-Silva1
Natália Sousa Teixeira-Silva1 Paula Maria Moreira Martins1
Paula Maria Moreira Martins1 Marco Aurélio Takita1
Marco Aurélio Takita1 Alessandra Alves de Souza1*
Alessandra Alves de Souza1*- 1Centro de Citricultura Sylvio Moreira, Instituto Agronômico, Cordeirópolis, Brazil
- 2Department of Entomology and Plant Pathology, Auburn University, Auburn, AL, United States
Copper-based compounds are widely used in agriculture as a chemical strategy to limit the spread of multiple plant diseases; however, the continuous use of this heavy metal has caused environmental damage as well as the development of copper-resistant strains. Thus, it is important to understand how the bacterial phytopathogens evolve to manage with this metal in the field. The MqsRA Toxin–Antitoxin system has been recently described for its function in biofilm formation and copper tolerance in Xylella fastidiosa, a plant-pathogen bacterium responsible for economic damage in several crops worldwide. Here we identified differentially regulated genes by X. fastidiosa MqsRA by assessing changes in global gene expression with and without copper. Results show that mqsR overexpression led to changes in the pattern of cell aggregation, culminating in a global phenotypic heterogeneity, indicative of persister cell formation. This phenotype was also observed in wild-type cells but only in the presence of copper. This suggests that MqsR regulates genes that alter cell behavior in order to prime them to respond to copper stress, which is supported by RNA-Seq analysis. To increase cellular tolerance, proteolysis and efflux pumps and regulator related to multidrug resistance are induced in the presence of copper, in an MqsR-independent response. In this study we show a network of genes modulated by MqsR that is associated with induction of persistence in X. fastidiosa. Persistence in plant-pathogenic bacteria is an important genetic tolerance mechanism still neglected for management of phytopathogens in agriculture, for which this work expands the current knowledge and opens new perspectives for studies aiming for a more efficient control in the field.
Introduction
Xylella fastidiosa is a phytopathogen with a broad host range that affects plants worldwide (Almeida et al., 2019). Plant diseases caused by this bacterium include citrus variegated chlorosis (CVC), Pierce’s disease (PD) in grapevines, and the olive quick decline syndrome (OQDS), which constitute important threats for these crops (Almeida et al., 2019; Saponari et al., 2019; Coletta-Filho et al., 2020). Copper-based compounds are widely used in agriculture as a chemical strategy to limit the spread of multiple plant diseases (Lamichhane et al., 2018). Although X. fastidiosa is not itself controlled by copper spraying, biocomplexes containing copper, zinc, and citric acid have been used to control X. fastidiosa in olive groves (Girelli et al., 2019). Copper has an important contribution in crop protection; however, there are many issues related to the use of this heavy metal such as phytotoxicity, soil accumulation, negative effects on soil biota, and development of copper-resistant strains (Lamichhane et al., 2018). Thus, regarding plant-pathogen interaction, it is important to understand how the bacterial phytopathogens evolve to deal with this metal in the field.
In X. fastidiosa, the mqsRA toxin–antitoxin (TA) system type II is a genetic mechanism that has been associated with tolerance to copper stress (Muranaka et al., 2012). There are six types of TA system, which are distinct according to the action, nature, and mechanisms used by the antitoxins to neutralize the activities of the toxins (Page and Peti, 2016). Typically, in these systems, the toxin gene product is a protein and the antitoxin gene is a non-coding RNA (in types I and III) or a protein (in types II, IV, V, and VI) (Page and Peti, 2016; Harms et al., 2018). Bacterial toxin-antitoxin (TA) systems encode a stable toxin that disrupts cellular function and its labile cognate antitoxin in the same operon. The antitoxin neutralizes toxin activity under normal conditions, while proteases degrade the antitoxin under stress, allowing the toxin activity (Wang and Wood, 2011; Fisher et al., 2017). Moreover, the antitoxin usually regulates the expression of its own TA operon by binding to a palindromic sequence in the promoter region and repressing its transcription (Wang et al., 2011). TA systems have been shown to play a role in persistence, biofilm formation, cell movement, pathogenicity, DNA maintenance, and phage-defense (Wang et al., 2011; Wen et al., 2014; Shidore and Triplett, 2017). In addition, they are highly expressed in persister cells and, thus, are generally responsible for the persistence phenotype (Wang and Wood, 2011; Fisher et al., 2017). A persister cell constitutes a tolerant cell (Lewis, 2010) originating from a population that displays antibiotic persistence, being a subpopulation phenomenon (sometimes referred to as heterotolerance) (Balaban et al., 2019), while, a tolerant cell is the capacity of an entire population of bacteria to survive a bactericidal antibiotic exposure (Balaban et al., 2019). Multidrug resistance in bacteria can occur by distinct ways like the accumulation of the resistance factors like plasmids or genes, each one encoding for resistance to a particular agent, and can or cannot occur along with the activity of multidrug efflux pumps (Nikaido, 2009).
The mqsRA TA system was originally described in Escherichia coli and shown to be involved in biofilm and persister cell formation (Wang and Wood, 2011). The toxin mqsR was the most induced gene in E. coli persisters and the first TA system to reduce persister formation upon deletion, while increasing this phenotype after overexpression (Kim and Wood, 2010). It has been demonstrated that X. fastidiosa may form persister cells under copper stress (Muranaka et al., 2012; Merfa et al., 2016), representing an important survival strategy still unexplored in plant pathogenic bacteria (Martins et al., 2018).
The MqsRA TA system is composed of the MqsR toxin, which is an endoribonuclease that degrades messenger RNA (mRNA) with GCU motifs and the MqsA antitoxin that binds and inactivates the toxin via its N-terminal domain (Brown et al., 2009; Yamaguchi et al., 2009; Lee et al., 2014). Due to its ability to selectively degrade mRNA, MqsR also acts as a global regulator (Wood et al., 2013). Thus, aiming to identify genes modulated by MqsR in X. fastidiosa, we overexpressed this toxin under the control of its native promoter and performed RNA-Seq when growing cells under normal and upon copper stress conditions.
Our results show that MqsR is a key gene regulator in the pathway tolerance of X. fastidiosa to copper stress, mediating several genes that prompt the cells to enter in a state that suggests the formation of persisters. In addition, copper induces MqsR-independent responses related to proteolysis and multidrug resistance through transcriptional regulator, transporters, and efflux pumps in order to increase the bacterial tolerance to this metal. This study presents unexplored mechanisms in phytopathogens that could have important impacts on how they can deal with agrochemicals and highlight the persistence phenomenon that could be occurring in the field.
Materials and Methods
Bacterial Strains and Transformation
The bacterial strains used in this study were the X. fastidiosa wild-type strain 11399 (Coletta-Filho et al., 2001; Niza et al., 2016) and 11399 overexpressing mqsR under the control of its native promoter (Xf-mqsR) (Merfa et al., 2016) (Supplementary Table 1). The increased amount of MqsR was previously confirmed by Western blot (Merfa et al., 2016). We transformed X. fastidiosa 11399 strain with the pXF20 empty vector (Lee et al., 2010), by electroporation (1.8 kV, 200 Ω, 25 μF) to serve as negative control (Xf-EV). The transformants were grown on selective medium PWG (phytone peptone; BD Biosciences, San Jose, CA, United States) 4.0 (g/L), trypticase peptone (BD) 1.0 (g/L), K2HPO4 (Sigma, St. Luis, MO, United States) 1.2 (g/L), hemin chloride stock (Sigma) 10 (mL/L), KH2PO4 1.0 (g/L), Gelzan (Sigma) 8.0 (g/L), MgSO4:7H2O, 0.4 (g/L), phenol red stock (Sigma; 0.2% (w/v) phenol red in distilled water) 10 (mL/L), glutamine (Sigma) 4 (g/L), and bovine serum albumin fraction-five (BSA) (Sigma) 3 (g/L); this medium was prepared according Davis et al. (1981) plates supplemented with 50 μg/mL kanamycin. The transformation was confirmed by PCR using a specific pair of primers to detect the pXF20 plasmid (Supplementary Figure 1). The primers used to confirm this transformation are oriV-pXF20-F 5’-GGTTTGTGAAAGCGCAGTG and trfA-pXF20-R 5’-ATTGCCAATTTGGACAGATG. The Xf-EV and Xf-mqsR strains were routinely grown on selective PWG plates supplemented with 50 μg/mL kanamycin at 28°C for 7 days.
Copper Sensitivity Assay
To evaluate the effects of copper on X. fastidiosa growth and formation of persisters, Xf-EV and Xf-mqsR cells were grown in PW broth (PWG without Gelzan) (Davis et al., 1981) and treated with 3 mM CuSO4.5H2O (Sigma). Control samples of both strains were grown in non-copper PW broth. Cells grown on solid PW were harvested from plates, resuspended in PBS buffer, and the optical density (OD600 nm) was adjusted to 0.3 and inoculated into PW broth to grow for another 7 days. The cells were then collected and the OD600 nm was adjusted to 0.1. From each of these bacterial suspensions, 10-mL aliquots were inoculated into 90 mL fresh PW broth and incubated at 28°C for 14 days at 150 rpm. Subsequently, Xf-EV and Xf-mqsR cells were exposed to 0 (“C-0” for Xf-EV, and “M-0” for Xf-mqsR) and 3 mM copper (“C-3” for Xf-EV and “M-3” for Xf-mqsR) for 24 h (Merfa et al., 2016). Each treatment was performed in duplicates for each strain. The cells of each culture were collected, rinsed with DEPC water, and resuspended in 11 mL of PBS buffer. An aliquot of 1 mL from each suspension was used to determine colony formation units (CFU/mL), and to perform electron microscopy analysis, as described below. The cells of the remaining 10 mL were collected under the same conditions and stored at −80°C for RNA extraction. Three independent biological replicates were performed.
Bacterial Growth Under Copper Stress
Aliquots of the entire experimental condition described above (Supplementary Figure 2) were collected to determine the CFU/mL of each biological experiment at the following time course: inoculation time (t0), 14 days after growth (stationary phase, Campanharo et al., 2003) in fresh PW broth when copper was added (t1) and 24 h after copper treatment (t2), completing 15 days of growth. From each sample, a 10-fold serial dilution was performed and plated in PWG to estimate CFU. Four replicates were used for each sample, which were grown at 28°C for 30 days. The measurements were performed in triplicates, and results were scored as the means ± standard deviation and compared using the Student’s t-test (p ≤ 0.05).
Scanning Electron Microscopy
Scanning electron microscopy was performed under the experimental conditions described above (Supplementary Figure 2). Briefly, an aliquot of the planktonic and biofilm cells was sampled 24 h after copper addition for each X. fastidiosa strain. Controls without copper were also collected for both Xf-EV and Xf-mqsR. Samples were centrifuged and resuspended in RNA Later solution (Thermo Fisher Scientific, Waltham, MA, United States), frozen in liquid nitrogen and stored at −80°C. For microscopy analysis, cells were thawed, centrifuged, fixed in 2.5% glutaraldehyde in 0.2 M sodium cacodylate buffer (v/v) and kept at 4°C until use. Preparation of samples for visualization was done according to Kozlowska et al. (2014). Electron micrographs were captured with a magnification of 4000 × using a Hitachi TM 3000 scanning electron microscope (Hitachi, Tokyo, Japan). The Xf-EV and Xf-mqsR cells were measured using the ImageJ software (ImageJ, 2018) to determine the length and proportion of elongated and small cells at 100 cells per treatment. Only cells longer than 4.0 μm were considered elongated (Liu et al., 2014; Merfa et al., 2016), while only cells with a length smaller than 2.0 μm were considered small. The length of each cell in each treatment was analyzed through comparison of means by one-way analysis of variance (ANOVA) followed by Holm–Sidak multiple comparison test or Tukey’s HSD test (p ≤ 0.05).
RNA Isolation and RNA-Seq
RNA-Seq reads were produced from 12 RNA samples: three from non-treated Xf-EV cells, three from non-treated Xf-mqsR, three from copper-treated cells of Xf-EV, and three from copper-treated Xf-mqsR cells. Total RNA was extracted using the hot phenol method (Khodursky et al., 2003), treated with DNase I RNase free (Qiagen, Hilden, Germany), purified using the RNeasy Plus Kit (Qiagen, Hilden, Germany) and eluted in 30 μL of RNase-free water. Concentrations were determined by spectrophotometry (NanoDrop 8000, Thermo Fisher Scientific). Ribo-Zero rRNATM Removal Kit (Illumina, San Diego, CA, United States) was used for rRNA removal. The depleted RNA was precipitated using ethanol according to the manufacturer’s instructions and resuspended in 10 μL of RNase-free water. Samples were quantified for the presence of rRNA using the 2100 Bioanalyzer system (Agilent Technologies, Santa Clara, CA, United States) at the Life Sciences Core Facility (LaCTAD). cDNA libraries were prepared using the Illumina TruSeq Stranded mRNA Library Prep Kit (Illumina). Sequencing was performed using the HiSeq High Output kit (Illumina) on a HiSeq 2500 system (Illumina), run with 2 × 100 bp paired-end reads.
RNA-Seq Data Analysis
The sequencing reads were analyzed in the FastQC program (Wingett and Andrews, 2018) and processed using Trimmomatic (Bolger et al., 2014) to remove adapters and extremities with poor quality. The reads were mapped to the genome of X. fastidiosa 9a5c (NCBI BioProject accession PRJNA271) using the STAR program (Dobin et al., 2013). From the mapped data, the gene-mapped reads were counted using the Subread package (Liao et al., 2019). Standardization and analysis of differential gene expression (p < 0.05) was performed using the EdgeR package (Robinson et al., 2010), computed using data from all three biological replicates. Differentially expressed genes obtained from EdgeR analyses were used for functional categorization by Blast2GO (Götz et al., 2008). Venny 2.1.0 (Oliveros, 2007) was used to show exclusive genes regulated in Xf-mqsR under copper stress.
Data Validation by Quantitative Real Time-PCR (RT-qPCR)
RNA samples were obtained from three other experiments using the same experimental condition as the RNA-Seq. A total of 250 ng of purified RNA from each condition was used as input for cDNA synthesis with the Reverse Transcription System kit (Promega, Madison, WI, United States). RT-qPCR was performed using the GoTaq qPCR Master Mix (Promega) in an ABI PRISM 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, United States). Relative expression values were normalized to the X. fastidiosa 16S ribosomal RNA endogenous control (Merfa et al., 2016). Cycling parameters were performed according to the manufacturer’s protocol. The relative expression quantification (RQ) was calculated as previously described (Livak and Schmittgen, 2001). The selected genes are based on the RNA-Seq analysis of M-0 and M-3 (Supplementary Table 2). Three independent biological replicates were used for data validation.
Palindrome Search
The motif 5’-ACC (N)7 GTT-3’ (Merfa et al., 2016), used as target sequence for DNA binding by the antitoxin MqsA, was searched in the genome of X. fastidiosa strain 9a5c using PATLOC (Mr̀azek and Xie, 2006), and also at the differentially expressed genes (DEG) data set herein generated.
Results
MqsR Overexpression Changes X. fastidiosa Phenotype
To evaluate the effects of copper on wild-type X. fastidiosa and the mqsR-overexpressing strain, bacterial growth with and without copper was evaluated. In a previous work we verified that overexpression of MqsR increased the formation of persister cells under 3 mM of copper stress (Merfa et al., 2016). Here, to access the phenotypic and genetic regulation mediated by MqsR, we used the same condition, where copper was added after 15 days of bacterial growth.
At the time of the inoculation (t0), and after 15 days of growth in fresh PW broth (t1), no significant difference in bacterial growth was observed between C-0 and M-0 (Figure 1). However, 24 h after addition of copper (t2), there was a significant reduction in population size of approximately 100-fold between copper-treated samples (C-3 and M-3) and their respective untreated controls (C-0 and M-0) (Figure 1). We observed an approximately 10% increase in cell survival after copper treatment in populations overexpressing mqsR (M-3) in comparison to the control (C-3). However, the difference in CFU counts between M-3 and C-3 was not significant (F = 0.06, p = 0.11) (Figure 1).
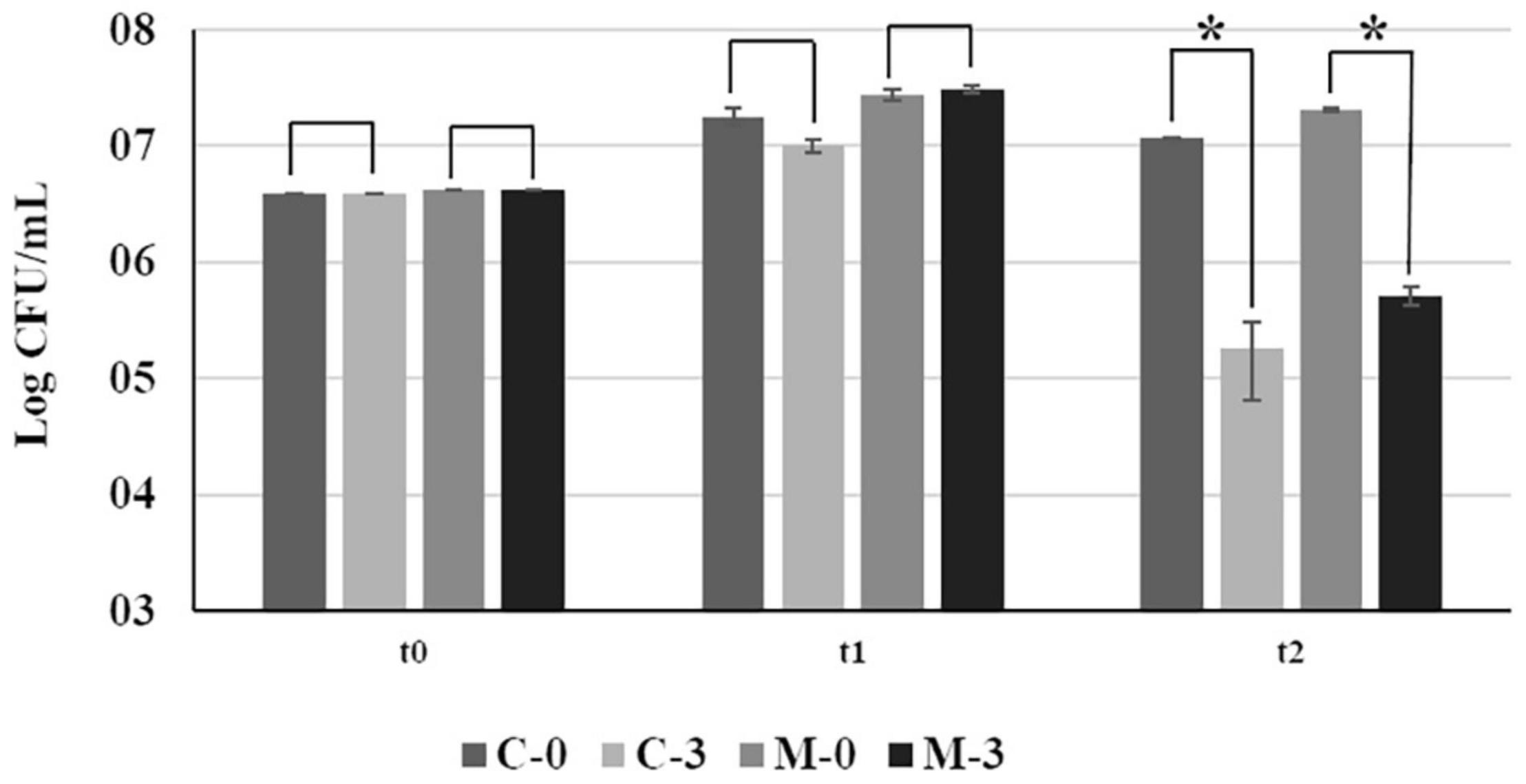
Figure 1. Bacterial growth of Xf-EV and Xf-mqsR under normal conditions and after copper stress at different time points. C-0: Xf-EV; C-3: Xf-EV + 3 mM CuSO4; M-0: Xf-mqsR; M-3: Xf-mqsR + 3 mM CuSO4. t0: initial inoculum; t1: 14 days of growth; t2: 24 h after copper treatment, at the end of 15 days. Three independent experiments were performed with similar results. * indicates statistical significance by Student’s t-test (p ≤ 0.05, n = 4).
To verify possible phenotypic changes in X. fastidiosa cells potentially caused by the overexpression of mqsR and copper treatment, samples from each experimental condition were used for scanning electron microscopy. Under normal growth condition, biofilm and planktonic cells in C-0 did not show any significant morphological change (Figures 2A,B). However, when copper was added (C-3) a reduction was observed in biofilm size (Figure 2C), and curiously, copper induced aggregation and elongated cells in the planktonic condition (Figure 2D, red arrows).
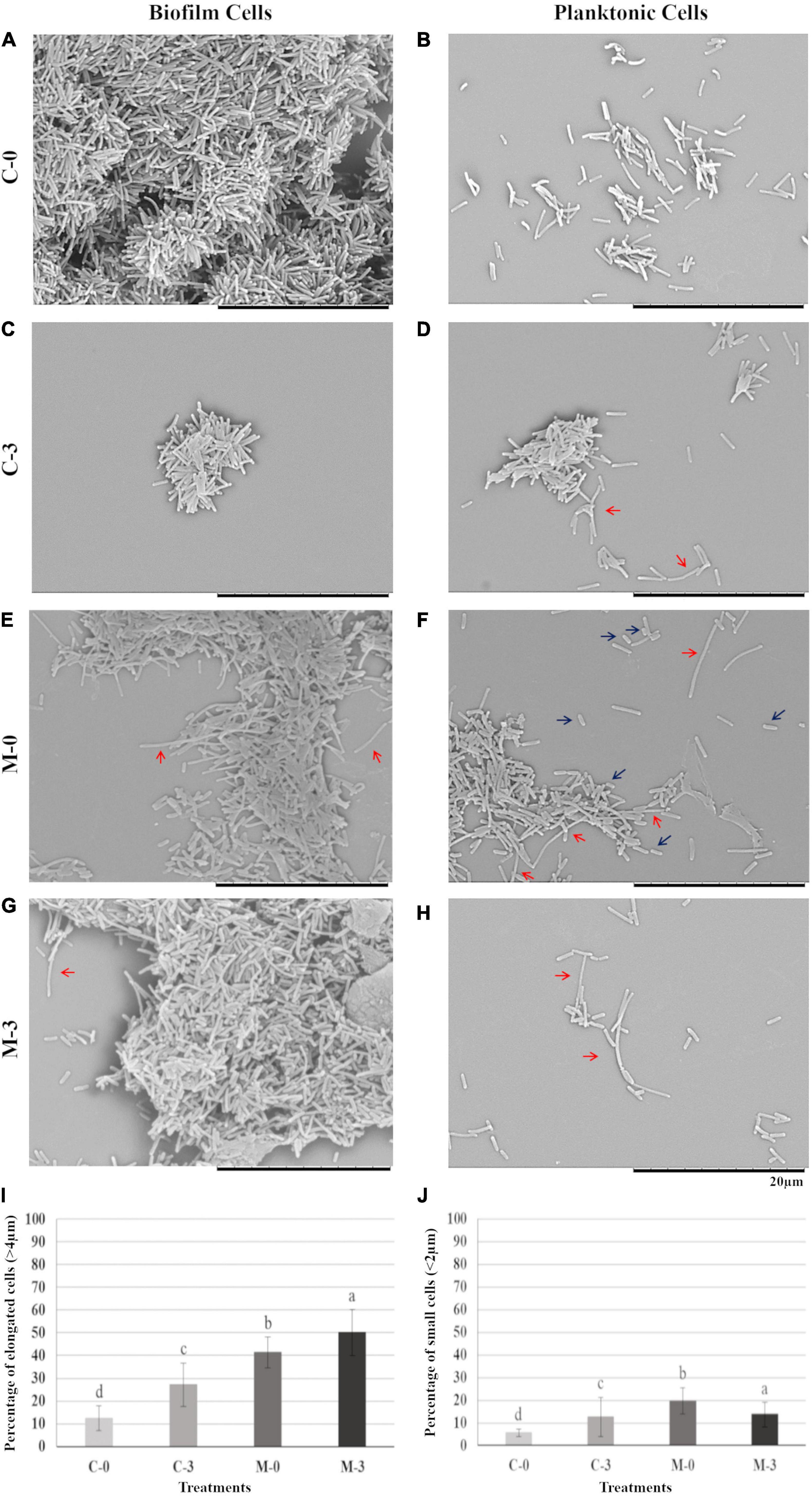
Figure 2. Biofilm and planktonic behavior of Xf-EV and Xf-mqsR cells under copper stress. Left column: cells in biofilm. Right column: planktonic cells. (A,B) Representative pictures of C-0: Xf-EV cells without copper treatment. (C,D) Representative pictures of C-3: Xf-EV cells treated with 3 mM CuSO4. (E,F) Representative pictures of M-0: Xf-mqsR cells without copper treatment. (G,H) Representative pictures of M-3: Xf-mqsR cells treated with 3 mM CuSO4. Red arrows show elongated cells and blue arrows show short cells. Scale bar: 20 μm. (I) Percentage of planktonic cells longer than 4.0 μm. (J) Percentage of planktonic cells shorter than 2.0 μm in the different treatments. Different letters on top of column bars indicate significant difference as analyzed by one-way ANOVA in SigmaPlot followed by Tukey’s HSD test (p ≤ 0.05; n = 3 biological replicates, with 100 internal replicates each). C-0: Xf-EV cells without copper treatment; C-3: Xf-EV cells treated with 3 mM of CuSO4; M-0: Xf-mqsR cells without copper treatment; M-3: Xf-mqsR cells treated with 3 mM of CuSO4.
On the other hand, X. fastidiosa overexpressing mqsR (M-0) presented more elongated cells even without copper stress (Figures 2E,F, red arrows), and at an even greater extent than C-3 (Figure 2D, red arrows). In addition, X. fastidiosa overexpressing mqsR (M-0) displayed a phenotypic heterogeneity that can be demonstrated by the presence of a higher population of shorter cells when compared to the other treatments (Figure 2F, blue arrows). Elongated cells were also observed in X. fastidiosa overexpressing mqsR in presence of copper (M-3) in both biofilm and planktonic conditions (Figures 2G,H, red arrows).
Microscopy images for each condition (n = 100) were used for counting elongated and short cells in the planktonic fraction (Figures 2I,J). The results showed a good agreement with the visual observation, with a higher population of elongated cells in X. fastidiosa overexpressing mqsR (M-0) compared to C-0. Copper induced an increase of elongated cells in both populations (C-3 and M-3). Interestingly the number of elongated cells in M-0 is naturally even higher than C-3 (Figure 2I). Similarly, higher percentages of short cells were observed in presence of copper (C-3 and M-3) or X. fastidiosa overexpressing mqsR (M-0) (Figure 2J).
Overall, these results show that besides copper treatment, mqsR overexpression also led to changes in X. fastidiosa morphology and pattern of aggregation, culminating in a global phenotypic heterogeneity. Interestingly, heterogeneous phenotypes in single bacterial populations have been described as indicative of persister cells (Michiels et al., 2016; Fisher et al., 2017).
RNA-Seq Data
RNA-Seq reads were produced for C-0, C-3, M-0, and M-3 (Supplementary Figure 3). Raw sequencing reads were deposited under the NCBI Bio-Project ID PRJNA718853. Average post-trim read length ranged from 36 to 105 bp, the reads aligned to the genome of X. fastidiosa 9a5c. Variable rRNA and small RNA depletion efficiencies between samples resulted in 0–7.3% in library preparation. Mapped reads were used to determine transcript boundaries and normalized expression for all protein-coding genes by EdgeR (Supplementary Material 1, Data Sets 1–5). Pearson’s correlation coefficient for protein-coding gene expression between experimental replicates ranged from 0.89 to 0.93. Highlighted DEGs of libraries were characterized according to the biological process by Blast2GO (Supplementary Material 1, Data Sets 6–11).
mqsR Differentially Modulates Global Gene Expression of X. fastidiosa
To investigate global expression changes likely to be associated with the phenotypes described above, we performed RNA sequencing analysis. To identify which genes were modulated by MqsR under normal growth conditions, we assessed the pairwise comparison between M-0/C-0 libraries (without copper treatment). Amongst the DEGs, 189 genes showed upregulation by the overexpression of mqsR alone, while 164 genes were downregulated (Supplementary Material 1, Data Set 1; p < 0.05). RNA-Seq expression values (log2 fold-change) were confirmed by RT-qPCR for 10 selected genes based on Table 1, with a Pearson correlation coefficient of 0.89 (Supplementary Figure 4). According to the data obtained through RNA-Seq (Supplementary Material 1, Data Set 1), the selected genes that are possibly modulated by MqsR are listed in Table 1.
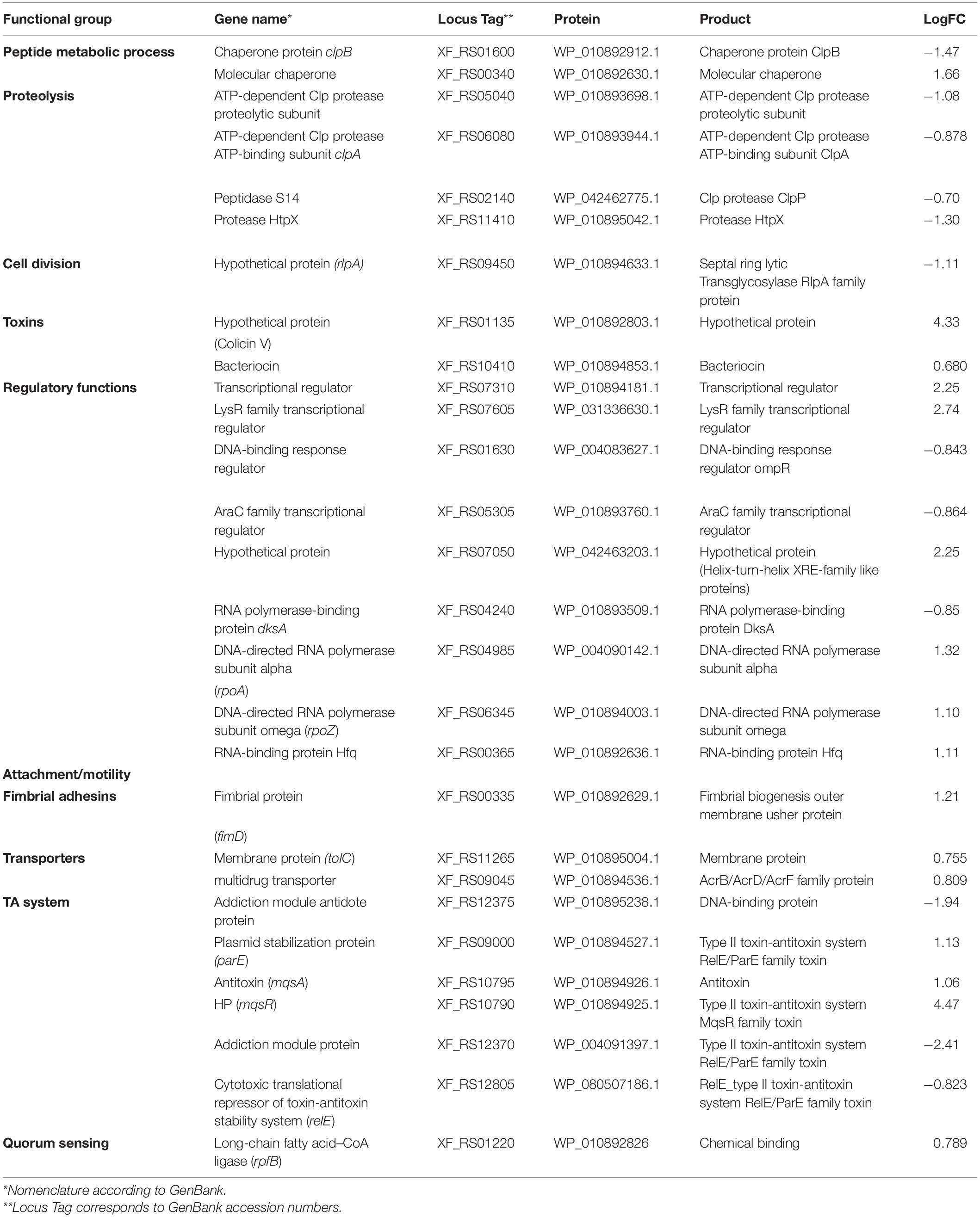
Table 1. Genes modulated by MqsR in X. fastidiosa.
Functional categorization of these 353 DEGs comprised genes associated with peptide metabolic process, transport, proteolysis, transcriptional regulation, and RNA metabolic processes (Figure 3, Supplementary Material 1, and Data Sets 6, 7). Genes associated with proteolysis were exclusively downregulated, including the proteases clpA and clpP. On the other hand, genes related to peptide metabolism were exclusively upregulated, including those related to ribosomal subunit scaffolding of RNA polymerase (RNAP), such as rpoA and rpoZ. These genes are also listed in the regulatory function category, together with mqsR, lysR, and the post-transcriptional regulator hfq, which were induced. The regulators genes mqsR, rpoZ, and lysR are related to bacterial survival, stress responses, and pathogenicity (Maddocks and Oyston, 2008; Santiago et al., 2015; Merfa et al., 2016; Weiss et al., 2017). rpoZ mutants of Mycobacterium smegmatis were deficient in motility and biofilm formation, consequently affecting the formation of extracellular matrix (Mathew and Chatterji, 2006). Besides, the overexpression of transcriptional regulator type LysR from X. fastidiosa in E. coli was described to a play role in maturation of biofilm during its development (Santiago et al., 2015). In X. fastidiosa, it is important to emphasize that the formation of biofilm is characterized as the main pathogenicity mechanism (Coletta-Filho et al., 2020). The transport category included the upregulation of tolC and acrB, both related to efflux pumps (Weston et al., 2018) and bacterial persistence (Pu et al., 2016). Efflux pumps are important for broad cellular homeostasis during stress responses. They export a wide variety of compounds, such as signaling molecules and antimicrobial compounds (Langevin and Dunlop, 2018). rlpA, a gene involved in cell division (Jorgenson et al., 2014; Berezuk et al., 2018), was downregulated, and fimD, which is involved with type I fimbrial adhesin (Meng et al., 2005), was upregulated. These genes are involved in bacterial physiology and biofilm formation, respectively. Furthermore, our results showed TA-related genes. The relE (XF_RS12805) toxin was downregulated by MqsR; this gene is associated with inhibition translation by cleavage of mRNA in the ribosome (Fiebig et al., 2010). Another toxin, parE gene, was upregulated and it is responsible for inhibiting gyrase and thereby blocks chromosome replication (Jiang et al., 2002). Modulation of relE and parE suggests that these bacterial cells maintain basal activities with reduced metabolism as shown in persister cells (Lewis, 2007). In addition, the repression of rlpA, inhibiting cell division and induction of the toxin encoding parE (Yuan et al., 2011), which inhibits bacterial division, could contribute with the observed elongated phenotype. Taken together, these observations suggest that overexpression of mqsR contributes to bacterial survival during stress response by activating pathogenicity regulators and inhibiting proteolysis and cell division.
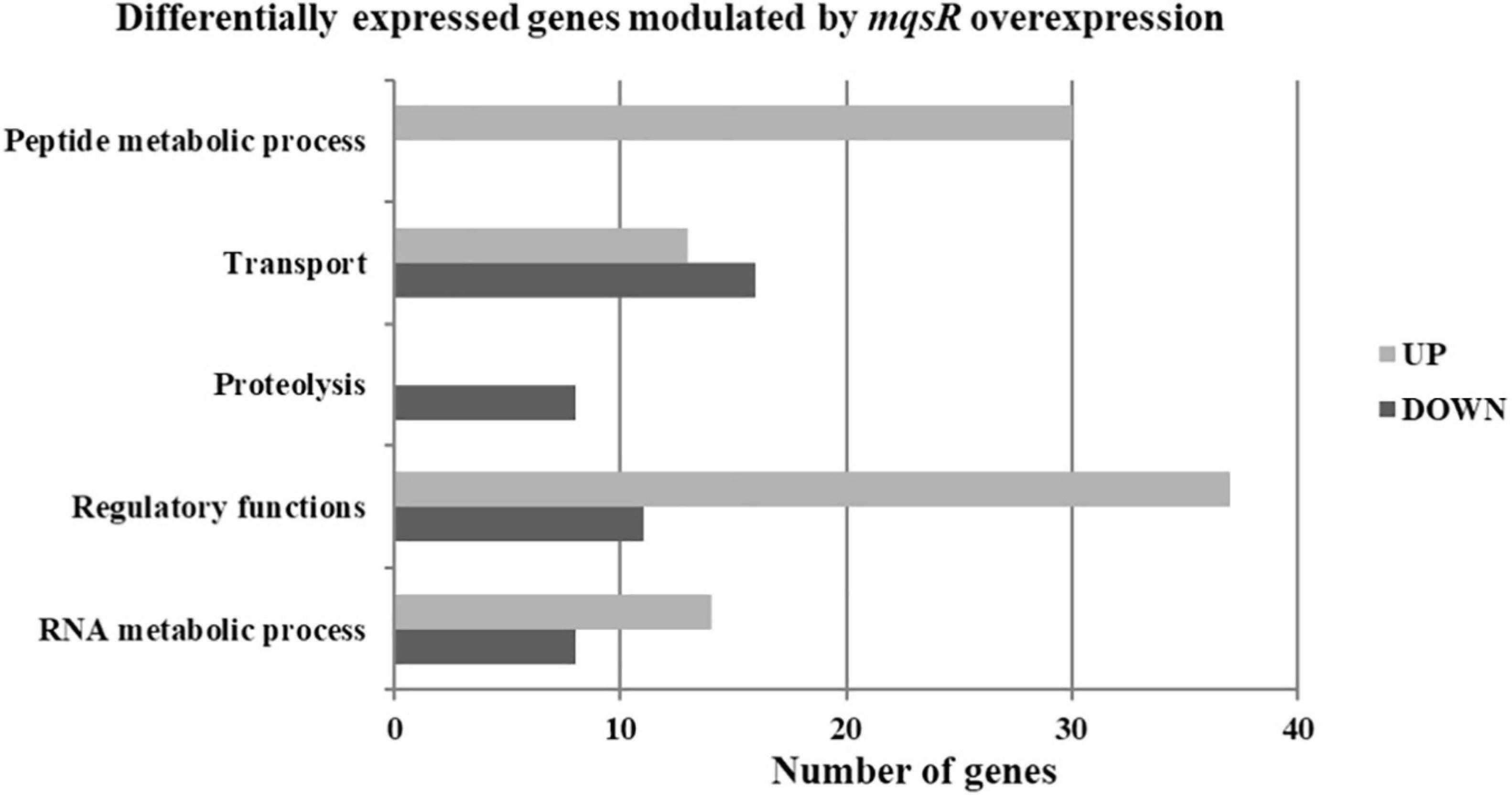
Figure 3. Gene ontology categorization of differentially expressed genes in X. fastidiosa in response to overexpression of mqsR. Genes were classified by functional category of biological processes using Blast2GO. UP: upregulated genes; DOWN: downregulated genes.
Overexpression of mqsR Modulates Translation in X. fastidiosa Under Copper Stress
To identify the influence of copper on the gene expression, the following pairwise comparisons of the sequencing libraries were performed: i. M-3/M-0, and ii. C-3/C-0. Each pairwise comparison generated 417 and 662 DEGs, respectively. The M-3/M-0 comparison resulted in 238 upregulated and 179 downregulated genes (Supplementary Material 1, Data Set 2), while the C-3/C-0 analysis resulted in 335 upregulated and 327 downregulated genes (Supplementary Material 1, Data Set 3).
To verify X. fastidiosa genes modulated by MqsR in response to copper stress, a Venn diagram was used to compare the up- and downregulated genes in the M-3/M-0 and C-3/C-0 libraries (Figure 4). This comparison provided genes modulated only by mqsR-overexpressing cells under copper stress (M-3/M-0), resulting exclusively in 111 upregulated and 84 downregulated genes (Figure 4 and Supplementary Material 1, Data Set 4).
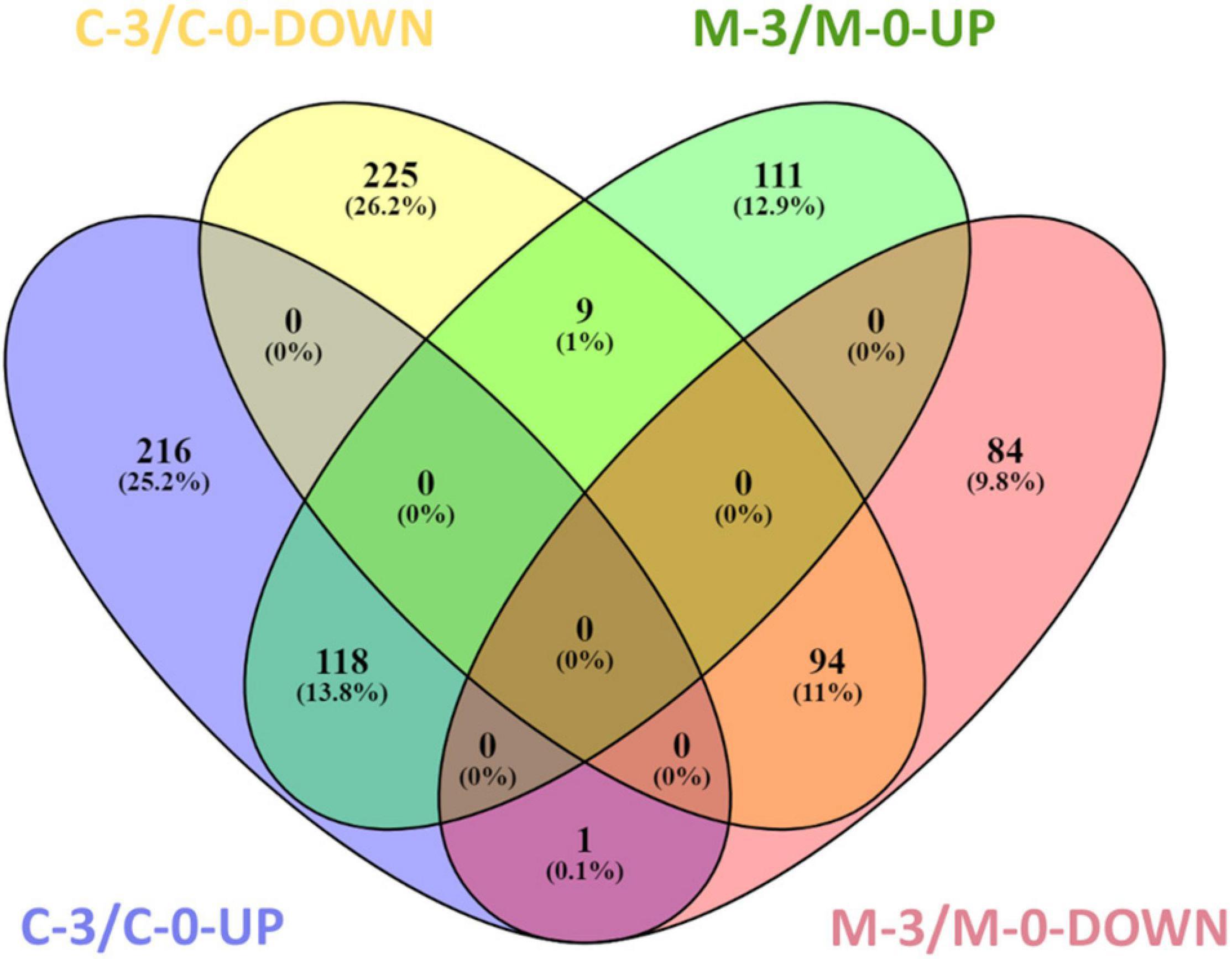
Figure 4. Venn diagram representing the number of up and downregulated genes in X. fastidiosa under copper stress. M-3/M-0: pairwise comparison between the M-3 (Xf-mqsR + CuSO4) and M-0 (Xf-mqsR) libraries. C-3/C-0: pairwise comparison between the C-3 (Xf-EV + CuSO4) and C-0 (Xf-EV) libraries. UP: upregulated genes; DOWN: downregulated genes.
In the search for genes oppositely modulated between Xf-EV (C-3/C-0) and Xf-mqsR (M-3/M-0), a set of nine genes were found to be downregulated in C-3/C-0 and upregulated in M-3/M-0. Of those, there is the yeiP elongation factor (XF_RS09585), three ribosomal subunits (XF_RS00715, XF_RS00285, XF_RS09575), the msrB (XF_RS03590) and yuxK (XF_RS04035), the aminotransferase astC (XF_RS06015, also known as argM or cstC), and two hypothetical proteins (XF_RS03860, XF_RS05645). Interestingly though, the ribosomal protein (XF_RS12125) was the only one showing an opposite behavior, being induced in C-3/C-0 but suppressed in M-3/M-0. This protein is involved in translation, and accordingly, this category was downregulated in M-3/M-0. In E. coli, it is known that persister cells have very low metabolism, with non-growing cells as a result of a depletion in translation and, thus, in protein production capacity, cessation of transcription and reduction in ATP production (Kwan et al., 2013; Kim et al., 2018). Moreover, the ability to wake up from this persister state was related to ribosome content (Kim et al., 2018). Our results show categories such as peptide metabolic processes and translation downregulated, suggesting low-metabolism and depletion of protein production in X. fastidiosa in such condition.
Finally, 94 genes remained downregulated, and 118 genes were upregulated in both conditions. We believe that these sets of genes are modulated due to treatment with copper itself and are likely to be independent of MqsR functions (Supplementary Material 1, Data Set 5).
Next, the functional characterization of the M-3/M-0 data set was performed to identify which genes are differentially modulated by the overexpression of MqsR under copper stress. RNA-Seq log2 fold-change values were confirmed by RT-qPCR for 10 selected genes selected from Table 2 with a Pearson correlation coefficient of 0.93 (Supplementary Figure 4). Genes related to translation and peptide metabolic processes were exclusively repressed in the M-3/M-0 libraries, whereas proteolysis and drug metabolic processes were induced (Figure 5A and Supplementary Material 1, Data Sets 8, 9). Other categories identified in this analysis included transport, regulatory functions, and RNA metabolic processes. Therefore, besides lowering the metabolism, the cells activate these specific salvage mechanisms allowing copper tolerance.
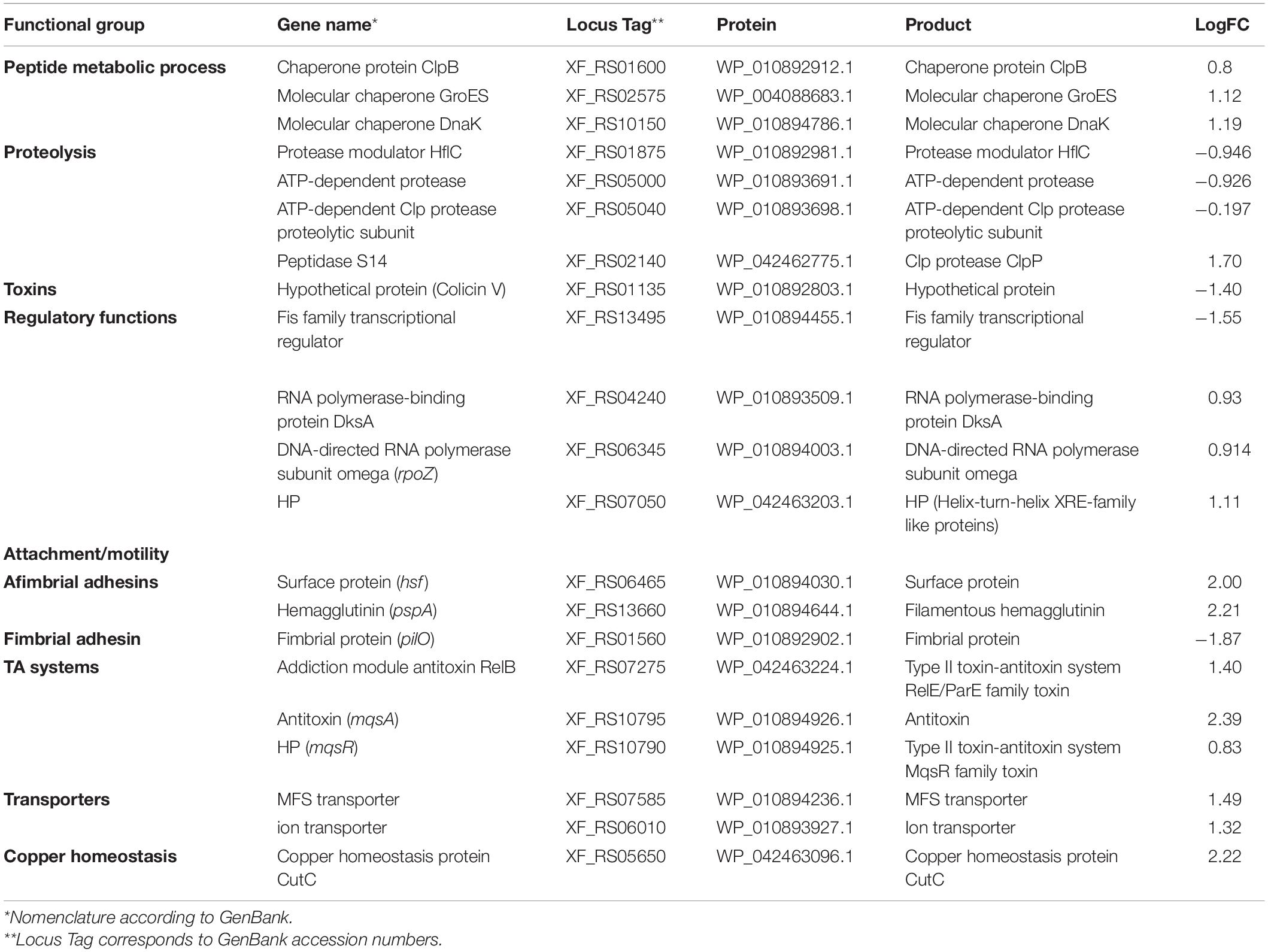
Table 2. Genes modulated by MqsR in X. fastidiosa under copper stress.
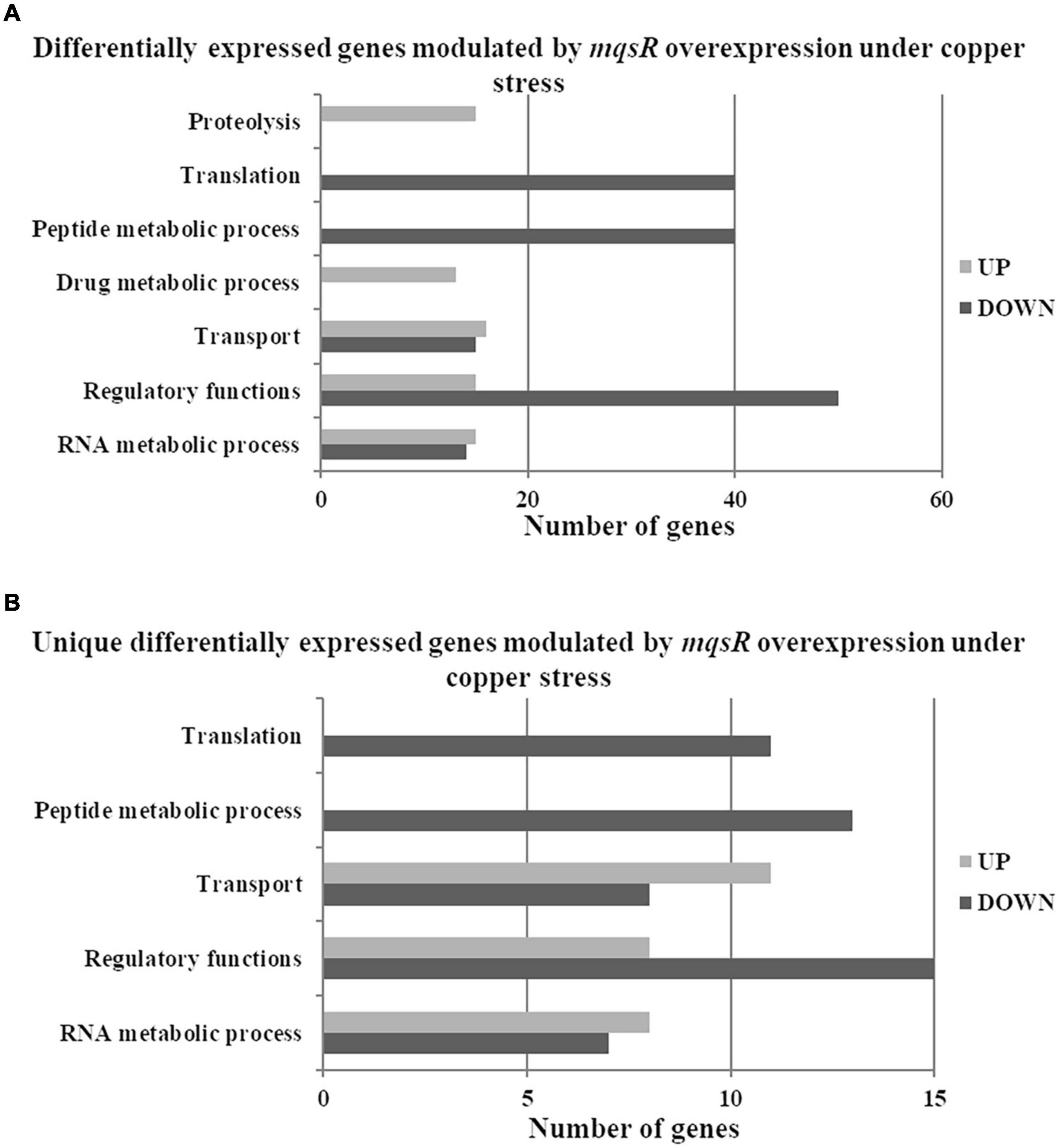
Figure 5. Gene Ontology categorization of differentially expressed genes in X. fastidiosa overexpressing mqsR in response to copper. (A) Genes were classified by functional category of biological processes using Blast2GO. (B) Categorization of differentially expressed genes unique to Xf-mqsR in response to copper. UP: upregulated genes; DOWN: downregulated genes.
The proteolysis category included another peptidase S4 clpP (XF_RS02140) and tldD (XF_RS04775) metalloprotease, important regulators of bacterial metabolism. These genes are related to protein degradation. Although yet unclear, tldD was described as a putative regulator of chromosome-encoded TA system activities (Hu et al., 2012). Among the upregulated genes involved in transport, there were genes that encode ion transporters and sulfate transporters belonging to the ABC transporter family. ABC transporters are known to be involved in the influx or efflux of a wide diversity of molecules, and also with antimicrobial peptide resistance (Orelle et al., 2019). The categories associated with translation and peptide metabolic process showed downregulated genes encoding ribosomal subunits and the elongation factors EF-Tu and EF-G. Interestingly EF-Tu is described as the most enriched protein in X. fastidiosa outer membrane vesicles (OMVs) important for pathogen systemic dissemination throughout the host xylem vessels (Feitosa-Junior et al., 2019). The category linked to regulatory functions showed various downregulated genes, such as the global regulator fis, which is involved in virulence and pathogenicity. The rice pathogen Dickeya zeae showed remarkably decreased virulence capacity after fis deletion (Lv et al., 2018). This global virulence regulator is involved in exopolysaccharide production, motility, biofilm formation, and cellular aggregation in Dickeya zeae. All these processes are of utmost importance for X. fastidiosa pathogenicity, being associated with host colonization. The rpoA and a DNA-binding regulator hypothetical protein (XF_RS07050) were also repressed in the overexpressing strain under copper treatment.
To identify genes exclusively modulated by MqsR under copper stress, we analyzed the gene ontology of the 111 upregulated genes and 84 downregulated genes presented in Figures 4, 5B (Supplementary Material 1, Data Sets 4, 10, 11). The exclusively downregulated categories included translation and the peptide metabolic process (Figure 5B and Supplementary Material 1, Data Sets 10, 11). Other categories identified were transport, regulatory functions, and RNA metabolic process. The genes dksA and rpoZ from the regulatory functions group are transcriptional regulators associated with stress responses (Mathew and Chatterji, 2006; Wang et al., 2018) and were induced under copper stress. The highlighted genes that are modulated by MqsR under copper stress are listed in Table 2.
Considering all the above-mentioned results, we built a hypothetical model for the mqsR overexpression and its influence on the X. fastidiosa regulatory mechanisms under normal and copper-induced stress conditions (Figure 6).
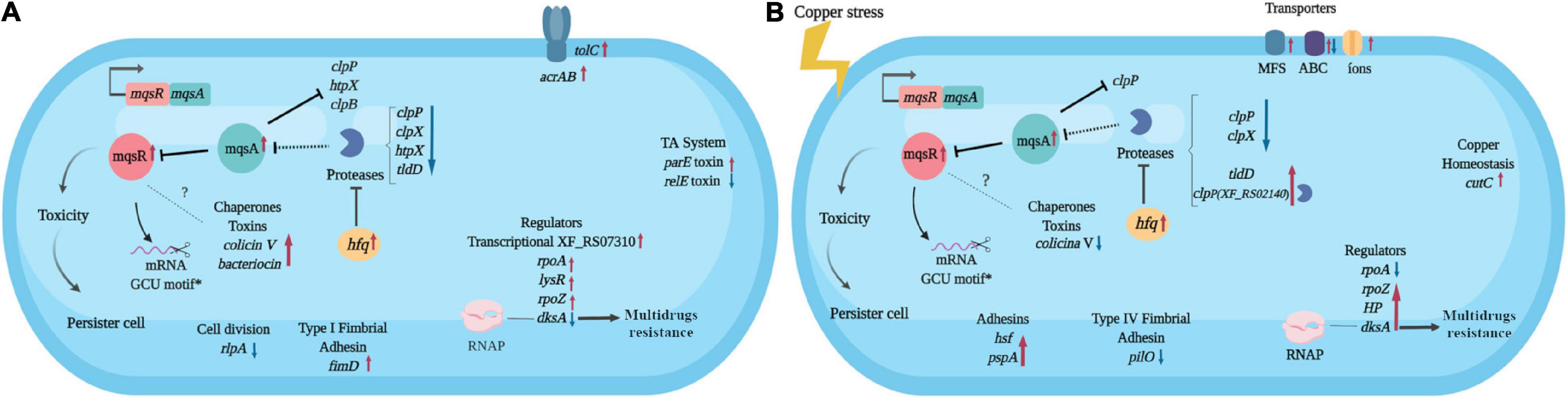
Figure 6. Hypothetical model of the X. fastidiosa MqsR-dependent regulon. Response to overexpression of mqsR in X. fastidiosa (A) and under copper stress (B). Arrows pointing to the side (→) indicate direct regulation and up (↑) red arrows indicate upregulation, while ⊥ indicates inhibition and down (↓) blue arrows indicate downregulation. Dashes indicate direct binding and dotted lines indicate hypothetical regulations. *Demonstrated by Lee et al. (2014). The figure was created in BioRender.com.
Discussion
Copper-containing compounds are among the most used chemicals in agriculture (Lamichhane et al., 2018). The antimicrobial effects of copper were previously attributed to stress-induced responses in many bacterial plant pathogens (Wright et al., 2019; Martins et al., 2021), including X. fastidiosa (Rodrigues et al., 2008; Muranaka et al., 2012; Merfa et al., 2016; Ge et al., 2021). In this bacterium, the MqsRA TA system was reported to play a key role when the pathogen is under copper stress. MqsRA is likely to function as an indicator for exogenous stressors through the induction of cell elongation, formation of structured biofilm aggregations, and reduction in cell movement (Merfa et al., 2016). To better understand the roles the toxin MqsR may be playing over stress-induced responses in X. fastidiosa, we assessed the major phenotypic outcomes and the global transcriptional profile of the mqsR-overexpressing strain under copper-stress conditions through microscopy and RNA-Seq analysis.
The mqsR overexpression triggers genetic response where cells activate genes associated to stress adaptation (Figure 6A), from which many are conserved in the presence of copper (Figure 6B). These characteristics suggest that increasing the amount of MqsR leads to a priming effect of cells to stresses that normally induce expression of mqsR, like copper. We observed an approximately 10% increase in cell survival after copper treatment in the population overexpressing mqsR and considering that, in stationary phase, only up to ∼1% of cells are persisters (Keren et al., 2004; Lewis, 2007), we can infer that a higher number of persisters were present under this condition. Therefore, our results demonstrate that the presence of the stressor is not needed to induce the genes and consequent cell morphology changes when mqsR is overexpressed. These morphologies include the elongated cell formation and population heterogeneity, indicative of persister cell activation (Michiels et al., 2016; Fisher et al., 2017), which is supported by tolC induction (Figure 6A), that was associated with E. coli persistence (Pu et al., 2016). Indeed, the presented results fit perfectly in the mathematical model in which systems that do not present bistability produce the hysteretic switch to the persistent state (Fasani and Savageau, 2013), represented in our condition by the overexpression of mqsR.
Besides, the overexpression of mqsR in E. coli exhibited cellular toxicity, resulting in increased persister cell formation (Kim et al., 2010). Taken together and with previous results (Muranaka et al., 2012; Merfa et al., 2016), the role of mqsR in X. fastidiosa seems to be similar to E. coli which involves the induction of persister cells.
It has been demonstrated that the MqsRA TA system in X. fastidiosa likely autoregulates its own expression to balance the toxin and antitoxin in the most beneficial ratio for the cells to oppose the stress (Merfa et al., 2016). The mqsR overexpression itself presents a stress condition to the cell, thus to inactivate the toxin, the antitoxin MqsA should be produced to reach a T:A balance (Brown et al., 2013). Indeed, we observed an induction of mqsA under both conditions (Figures 6A,B). An upregulation of hfq in Xf-mqsR was observed in both conditions with and without copper stress. The hfq gene encodes an RNA chaperone that, among other regulatory functions, is related to the downregulation of proteases (Kim and Wood, 2010). It suggests that hfq is a key gene in the autoregulation of the MqsRA TA system, and we propose it could be one of the factors responsible for keeping the ideal T:A ratio in the cell by controlling the expression of proteases and consequently the cell morphologies observed in this work.
The genes modulated clpP, hfq, and clpB by MqsR in X. fastidiosa resemble those modulated by the same regulon in E. coli (Kim et al., 2010). These genes are involved in stress responses and contribute to toxicity and, consequently, to persister cell formation in E. coli (Kim and Wood, 2010; Kim et al., 2010). Differences in the global transcriptional profile were also observed, suggesting a potential X. fastidiosa-exclusive mechanism. Among the exclusive genes modulated by MqsR only in X. fastidiosa are two gene regulators (XF_RS07050 and XF_RS07310). The regulator XF_RS07310 has the same type of HTH domain as the MqsA antitoxin, suggesting that it could also bind to promoter regions of target genes and modulate their expression. Some regulators related to bacterial survival and stress responses previously described in several bacteria were also modulated (Maddocks and Oyston, 2008; Santiago et al., 2015; Merfa et al., 2016; Weiss et al., 2017).
The MqsA antitoxin regulates the expression of mqsRA and other genes in E. coli by binding to palindromic sequences in their promoter regions and repressing their expression (Brown et al., 2009; Wang and Wood, 2011; Soo and Wood, 2013). The MqsA antitoxin encoded by X. fastidiosa has the same amino acid residues in its HTH domain responsible for DNA binding (Merfa et al., 2016). Therefore, we searched for the MqsA-like palindromic sequence 5’-AAC (N)7 GTT in the genome of X. fastidiosa (Supplementary Table 3), seeking to identify those genes that were specifically differentially expressed in our RNA-Seq analyses. We investigated gene regulations in conditions where mqsRA expression is increased, such as under copper stress and mqsR overexpression. We verified 526 palindromic regions throughout the X. fastidiosa genome, with 77 corresponding to intergenic regions (Supplementary Table 3). Among the DEGs, a few showed the searched palindromic sequence in their intergenic regions (Supplementary Table 4). These genes included clpP, htpX, clpB, and rpfB, besides mqsR itself. According to data RNA-Seq, clpP and clpB genes remained downregulated, while mqsA expression remained upregulated, suggesting that MqsA may be regulating proteolysis under stress conditions in mqsR overexpression.
In our model, copper stress induces responses independent of MqsR involving protein degradation and multidrug resistance. When mqsR is overexpressed under copper stress, other clpP (XF_RS02140) and tldD encoding proteases were induced (Figure 6B). Thus, the observed upregulation of proteases could contribute to the consequent upregulation of mqsRA. The regulator dksA, which plays an important role in the multidrug resistance in E. coli (Wang et al., 2018), shifted from downregulation in normal growth conditions to upregulation under copper stress, supporting its role of multidrug resistance. Copper also induces the expression of transporter genes associated with multidrug efflux pumps including cutC, which is specific for copper efflux (Rodrigues et al., 2008; Li et al., 2009). It has been shown that multidrug efflux pumps induce persistence, and persister cells combine active efflux with passive numbness to survive antibiotic attacks (Pu et al., 2016). This demonstrates the interplay between resistance and tolerance mechanisms, which are complementary and redundant bacterial strategies to survive under stress conditions (Lewis, 2007).
Overall, with the results herein presented, we were able to expand the knowledge on the genes and mechanisms associated with MqsR, as well as the function of the MqsRA TA system in X. fastidiosa. MqsR regulates genes that alter cell behavior in order to prime them to respond to environmental stress, which is related to induction of persistence. The persistence in plant-pathogenic bacteria is an important tolerance mechanism to this agrochemical which is still neglected in the management of agricultural diseases.
Data Availability Statement
The datasets generated and analyzed for this study can be found under the NCBI Bio-Project ID PRJNA718853. Other data used in this study are available on request from the corresponding author.
Author Contributions
AS and MT conceived and designed this research, provided reagents, analytical tools, and revised the manuscript. IC and PM conducted the experiments and analyzed the data. IC, MM, NT-S, MT, and AS wrote the manuscript. IC, MM, PM, MT, and AS contributed to the interpretation of the data and provided intellectual input. All authors read and approved the final manuscript.
Funding
This work was supported by a research grant from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP-2013/10957-0) and also from INCT Citrus (Proc. CNPQ465440/2014-2 and FAPESP 2014/50880-0). IC was an MSc. student from the Graduate Program in Tropical and Subtropical Agriculture (IAC), supported by a fellowship from FAPESP (2016/15741-4) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, grant 001). NT-S and PM are post-doctoral fellows supported by FAPESP (2019/01447-5 and 2018/18550, respectively). AS and MT are recipients of research fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the staff of the Life Sciences Core Facility (LaCTAD) from the State University of Campinas (UNICAMP) for the RNA-Seq (Genomics) analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.712564/full#supplementary-material
Supplementary Figure 1 | X. fastidiosa transformed with the pXF20 empty vector.
Supplementary Figure 2 | Experimental design.
Supplementary Figure 3 | Reads of RNA-Seq.
Supplementary Figure 4 | Validation RNA-Seq data.
Supplementary Table 1 | Bacterial strains and plasmids.
Supplementary Table 2 | Primers used for real-time quantitative PCR.
Supplementary Table 3 | MqsA-like palindromic sequences (5’-AAC (N)7 GTT-3’) found in the genome of X. fastidiosa 9a5c.
Supplementary Table 4 | Genes with the MqsA-like palindromic sequence identified in the RNA-Seq.
Supplementary Data Set 1 | Differential gene expression analysis between the M-0/C-0.
Supplementary Data Set 2 | Differential gene expression analysis between the M-3/M-0.
Supplementary Data Set 3 | Differential gene expression analysis between the C-3/C-0.
Supplementary Data Set 4 | Differential gene expression analysis between the downregulated and upregulated genes from the C-3/C-0 and M-3/M-0.
Supplementary Data Set 5 | Venn diagram of differentially expressed genes in M-3/M-0 and C-3/C-0.
Supplementary Data Set 6 | Functional characterization of proteins encoded by genes downregulated by MqsR in X. fastidiosa from Figure 3.
Supplementary Data Set 7 | Functional characterization of proteins encoded by genes upregulated by MqsR in X. fastidiosa from Figure 3.
Supplementary Data Set 8 | Functional characterization of proteins encoded by genes upregulated by MqsR in X. fastidiosa under copper stress from Figure 5A.
Supplementary Data Set 9 | Functional characterization of proteins encoded by genes downregulated by MqsR in X. fastidiosa under copper stress from Figure 5A.
Supplementary Data Set 10 | Functional characterization of proteins encoded by unique genes downregulated by MqsR in X. fastidiosa under copper stress from Figure 5B.
Supplementary Data Set 11 | Functional characterization of proteins encoded by unique genes upregulated by MqsR in X. fastidiosa under copper stress from Figure 5B.
References
Almeida, R. P. P., De La Fuente, L., Koebnik, R., Lopes, J. R. S., Parnell, S., and Scherm, H. (2019). Addressing the new global threat of Xylella fastidiosa. Phytopathology 109, 172–174. doi: 10.1094/PHYTO-12-18-0488-FI
Balaban, N. Q., Helaine, S., Lewis, K., Ackermann, M., Aldridge, B., Andersson, D. I., et al. (2019). Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 17, 441–448. doi: 10.1038/s41579-019-0196-3
Berezuk, A. M., Glavota, S., Roach, E. J., Goodyear, M. C., Krieger, J. R., and Khursigara, C. M. (2018). Outer membrane lipoprotein RLPA is a novel periplasmic interaction partner of the cell division protein FTSK in Escherichia coli. Sci. Rep. 81:12933. doi: 10.1038/s41598-018-30979-5
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brown, B. L., Grigoriu, S., Kim, Y., Arruda, J. M., Davenport, A., Wood, T. K., et al. (2009). Three dimensional structure of the MQSR:MqsA complex: a novel TA pair comprised of a toxin homologous to RELE and an antitoxin with unique properties. PLoS Pathog. 5:e1000706. doi: 10.1371/journal.ppat.1000706
Brown, B. L., Lord, D. M., Grigorius, S., Peti, W., and Pages, R. (2013). The Escherichia coli toxin MqsR destabilizes the transcriptional repression complex formed between the antitoxin MqsA and the mqsRA operon promoter. J. Biol. Chem. 288, 1286–1294. doi: 10.1074/jbc.M112.421008
Campanharo, J. C., Lemos, M. V. F., Lemos, E. G., and de, M. (2003). Growth optimization procedures for the phytopathogen Xylella fastidiosa. Curr. Microbiol. 462, 0099–0102. doi: 10.1007/S00284-002-3829-Z
Coletta-Filho, H., Della, Takita, M. A., De Souza, A. A., Aguilar-Vildoso, C. I., and Machado, M. A. (2001). Differentiation of strains of Xylella fastidiosa by a variable number of tandem repeat analysis. Appl. Environ. Microbiol. 67, 4091–4095. doi: 10.1128/AEM.67.9.4091-4095.2001
Coletta-Filho, H. D., Castillo, A. I., Laranjeira, F. F., de Andrade, E. C., Silva, N. T., de Souza, A. A., et al. (2020). Citrus variegated chlorosis: an overview of 30 years of research and disease management. Trop. Plant Pathol. 45, 175–191. doi: 10.1007/s40858-020-00358-5
Davis, M. J., French, W. J., and Schaad, N. W. (1981). Axenic culture of the bacteria associated with phony disease of peach and plum leaf scald. Curr. Microbiol. 6, 309–314. doi: 10.1007/BF01566883
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Fasani, R. A., and Savageau, M. A. (2013). Molecular mechanisms of multiple toxin-antitoxin systems are coordinated to govern the persister phenotype. Proc. Natl. Acad. Sci. U.S.A. 110, E2528–E2537. doi: 10.1073/PNAS.1301023110
Feitosa-Junior, O. R., Stefanello, E., Zaini, P. A., Nascimento, R., Pierry, P. M., Dandekar, A. M., et al. (2019). Proteomic and metabolomic analyses of Xylella fastidiosa OMV-enriched fractions reveal association with virulence factors and signaling molecules of the DSF family. Phytopathology 109, 1344–1353. doi: 10.1094/PHYTO-03-19-0083-R
Fiebig, A., Castro Rojas, C. M., Siegal-Gaskins, D., and Crosson, S. (2010). Interaction specificity, toxicity and regulation of a paralogous set of ParE/RelE-family toxin-antitoxin systems. Mol. Microbiol. 77, 236–251. doi: 10.1111/j.1365-2958.2010.07207.x
Fisher, R. A., Gollan, B., and Helaine, S. (2017). Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 15, 453–464. doi: 10.1038/nrmicro.2017.42
Ge, Q., Liu, R., Cobine, P. A., Potnis, N., and De La Fuente, L. (2021). Phenotypic and phylogenetic characterization of cu homeostasis among Xylella fastidiosa Strains. Pathogens 10:495. doi: 10.3390/pathogens10040495
Girelli, C. R., Angilè, F., Coco, L. D., Migoni, D., Zampella, L., Marcelletti, S., et al. (2019). 1H-NMR metabolite fingerprinting analysis reveals a disease biomarker and a field treatment response in xylella fastidiosa subsp. Pauca-infected olive trees. Plants 8:115. doi: 10.3390/plants8050115
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Harms, A., Brodersen, D. E., Mitarai, N., and Gerdes, K. (2018). Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell 70, 768–784. doi: 10.1016/j.molcel.2018.01.003
Hu, Y., Peng, N., Han, W., Mei, Y., Chen, Z., Feng, X., et al. (2012). An archaeal protein evolutionarily conserved in prokaryotes is a zinc-dependent metalloprotease. Biosci. Rep. 32, 609–618. doi: 10.1042/BSR20120074
ImageJ. (2018). Available online at: https://imagej.nih.gov/ij/all-notes.html (accessed March 25, 2021).
Jiang, Y., Pogliano, J., Helinski, D. R., and Konieczny, I. (2002). PARE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 44, 971–979. doi: 10.1046/j.1365-2958.2002.02921.x
Jorgenson, M. A., Chen, Y., Yahashiri, A., Popham, D. L., and Weiss, D. S. (2014). The bacterial septal ring protein RlpA is a lytic transglycosylase that contributes to rod shape and daughter cell separation in Pseudomonas aeruginosa. Mol. Microbiol. 93, 113–128. doi: 10.1111/mmi.12643
Keren, I., Kaldalu, N., Spoering, A., Wang, Y., and Lewis, K. (2004). Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230, 13–18. doi: 10.1016/S0378-1097(03)00856-5
Khodursky, A. B., Bernstein, J. A., Peter, B. J., Rhodius, V., Wendisch, V. F., and Zimmer, D. P. (2003). “Escherichia coli spotted double-strand dna microarrays: rna extraction, labeling, hybridization, quality control, and data management,” in Functional Genomics, eds M. J. Brownstein and A. Khodursky (New Jersey, NJ: Humana Press), 61–78. doi: 10.1385/1-59259-364-x:61
Kim, J. S., Yamasaki, R., Song, S., Zhang, W., and Wood, T. K. (2018). Single cell observations show persister cells wake based on ribosome content. Environ. Microbiol. 20, 2085–2098. doi: 10.1111/1462-2920.14093
Kim, Y., Wang, X., Zhang, X. S., Grigoriu, S., Page, R., Peti, W., et al. (2010). Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin CSPD. Environ. Microbiol. 12, 1105–1121. doi: 10.1111/j.1462-2920.2009.02147.x
Kim, Y., and Wood, T. K. (2010). Toxins HHA and CSPD and small RNA regulator HFQ are involved in persister cell formation through MQSR in Escherichia coli. Biochem. Biophys. Res. Commun. 391, 209–213. doi: 10.1016/j.bbrc.2009.11.033
Kozlowska, J., Vermeer, L. S., Rogers, G. B., Rehnnuma, N., Amos, S.-B. T. A., Koller, G., et al. (2014). Combined systems approaches reveal highly plastic responses to antimicrobial peptide challenge in Escherichia coli. PLoS Pathog. 10:e1004104. doi: 10.1371/journal.ppat.1004104
Kwan, B. W., Valenta, J. A., Benedik, M. J., and Wood, T. K. (2013). Arrested protein synthesis increases persister-like cell formation. Antimicrob. Agents Chemother. 57, 1468–1473. doi: 10.1128/AAC.02135-12
Lamichhane, J. R., Osdaghi, E., Behlau, F., Köhl, J., Jones, J. B., and Aubertot, J. N. (2018). Thirteen decades of antimicrobial copper compounds applied in agriculture. Review. Agron. Sustain. Dev. 38:28. doi: 10.1007/s13593-018-0503-9
Langevin, A. M., and Dunlop, M. J. (2018). Stress introduction rate alters the benefit of AcrABTolC efflux pumps. J. Bacteriol. 200:e00525. doi: 10.1128/JB.00525-17
Lee, M. W., Rogers, E. E., and Stenger, D. C. (2010). Functional characterization of replication and stability factors of an incompatibility group P-1 plasmid from Xylella fastidiosa. Appl. Environ. Microbiol. 76, 7734–7740. doi: 10.1128/AEM.01921-10
Lee, M. W., Tan, C. C., Rogers, E. E., and Stenger, D. C. (2014). Toxin-antitoxin systems Mqsr/YGIT and DINJ/RELE of Xylella fastidiosa. Physiol. Mol. Plant Pathol. 87, 59–68. doi: 10.1016/j.pmpp.2014.07.001
Lewis, K. (2007). Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5, 48–56. doi: 10.1038/nrmicro1557
Lewis, K. (2010). Persister cells. Annu. Rev. Microbiol. 64, 357–372. doi: 10.1146/annurev.micro.112408.134306
Li, Y., Du, J., Zhang, P., and Ding, J. (2009). Crystal structure of human copper homeostasis protein CutC reveals a potential copper-binding site. J. Struct. Biol. 169, 399–405. doi: 10.1016/j.jsb.2009.10.012
Liao, Y., Smyth, G. K., and Shi, W. (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47:e47. doi: 10.1093/nar/gkz114
Liu, D., Almeida, R., Coletta-Filho, H., and Lopes, J. (2014). “Xylella fastidiosa,” in Manual of Security Sensitive Microbes and Toxins, ed. D. Liu (Boca Raton, FL: CRC Press), 841–850.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lv, M., Chen, Y., Liao, L., Liang, Z., Shi, Z., Tang, Y., et al. (2018). Fis is a global regulator critical for modulation of virulence factor production and pathogenicity of Dickeya zeae. Sci. Rep. 8:341. doi: 10.1038/s41598-017-18578-2
Maddocks, S. E., and Oyston, P. C. F. (2008). Structure and function of the LYSR-type transcriptional regulator (LTTR) family proteins. Microbiology 154, 3609–3623. doi: 10.1099/mic.0.2008/022772-0
Martins, P. M. M., Merfa, M. V., Takita, M. A., and De Souza, A. A. (2018). Persistence in phytopathogenic bacteria: Do we know enough? Front. Microbiol. 9:1099. doi: 10.3389/fmicb.2018.01099
Martins, P. M. M., Wood, T. K., and de Souza, A. A. (2021). Persister cells form in the plant pathogen xanthomonas citri subsp. citri under different stress conditions. Microorganisms 9:384. doi: 10.3390/microorganisms9020384
Mathew, R., and Chatterji, D. (2006). The evolving story of the omega subunit of bacterial RNA polymerase. Trends Microbiol. 14, 450–455. doi: 10.1016/j.tim.2006.08.002
Meng, Y., Li, Y., Galvani, C. D., Hao, G., Turner, J. N., Burr, T. J., et al. (2005). Upstream migration of Xylella fastidiosa via pilus-driven twitching motility. J. Bacteriol. 187, 5560–5567. doi: 10.1128/JB.187.16.5560-5567.2005
Merfa, M. V., Niza, B., Takita, M. A., and De Souza, A. A. (2016). The MqsRA toxin-antitoxin system from Xylella fastidiosa plays a key role in bacterial fitness, pathogenicity, and persister cell formation. Front. Microbiol. 7:904. doi: 10.3389/fmicb.2016.00904
Michiels, J. E., Van den Bergh, B., Verstraeten, N., and Michiels, J. (2016). Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist. Updat. 29, 76–89. doi: 10.1016/j.drup.2016.10.002
Mrázek, J., and Xie, S. (2006). Pattern locator: a new tool for finding local sequence patterns in genomic DNA sequences. Bioinformatics 22, 3099–3100. doi: 10.1093/bioinformatics/btl551
Muranaka, L. S., Takita, M. A., Olivato, J. C., Kishi, L. T., and de Souza, A. A. (2012). Global expression profile of biofilm resistance to antimicrobial compounds in the plant-pathogenic bacterium Xylella fastidiosa reveals evidence of persister cells. J. Bacteriol. 194, 4561–4569. doi: 10.1128/JB.00436-12
Nikaido, H. (2009). Multidrug resistance in bacteria. Annu. Rev. Biochem. 78, 119–146. doi: 10.1146/annurev.biochem.78.082907.145923
Niza, B., Merfa, M. V., Alencar, V. C., Menegidio, F. B., Nunes, L. R., Machado, M. A., et al. (2016). Draft genome sequence of 11399, a transformable citrus-pathogenic strain of Xylella fastidiosa. Genome Announc. 4:e01124. doi: 10.1128/genomeA.01124-16
Oliveros, J. C. (2007). Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online at: https://bioinfogp.cnb.csic.es/tools/venny/ (accessed February 2, 2021).
Orelle, C., Mathieu, K., and Jault, J. M. (2019). Multidrug ABC transporters in bacteria. Res. Microbiol. 170, 381–391. doi: 10.1016/j.resmic.2019.06.001
Page, R., and Peti, W. (2016). Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 12, 208–214. doi: 10.1038/nchembio.2044
Pu, Y., Zhao, Z., Li, Y., Zou, J., Ma, Q., Zhao, Y., et al. (2016). Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol. Cell 62, 284–294. doi: 10.1016/j.molcel.2016.03.035
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Rodrigues, C. M., Takita, M. A., Coletta-Filho, H. D., Olivato, J. C., Caserta, R., Machado, M. A., et al. (2008). Copper resistance of biofilm cells of the plant pathogen Xylella fastidiosa. Appl. Microbiol. Biotechnol. 77, 1145–1157. doi: 10.1007/s00253-007-1232-1
Santiago, A. S., Santos, C. A., Mendes, J. S., Toledo, M. A. S., Beloti, L. L., Souza, A. A., et al. (2015). Characterization of the LysR-type transcriptional regulator YcjZ-like from Xylella fastidiosa overexpressed in Escherichia coli. Protein Expr. Purif. 113, 72–78. doi: 10.1016/j.pep.2015.05.003
Saponari, M., Giampetruzzi, A., Loconsole, G., Boscia, D., and Saldarelli, P. (2019). Xylella fastidiosa in olive in apulia: where we stand. Phytopathology 109, 175–186. doi: 10.1094/PHYTO-08-18-0319-FI
Shidore, T., and Triplett, L. R. (2017). Toxin-antitoxin systems: implications for plant disease. Annu. Rev. Phytopathol. 55, 161–179. doi: 10.1146/annurev-phyto-080516-035559
Soo, V. W. C., and Wood, T. K. (2013). Antitoxin MqsA represses curli formation through the master biofilm regulator CsgD. Sci. Rep. 3:3186. doi: 10.1038/srep03186
Wang, J., Cao, L., Yang, X., Wu, Q., Lu, L., and Wang, Z. (2018). Transcriptional analysis reveals the critical role of RNA polymerase-binding transcription factor, DksA, in regulating multi-drug resistance of Escherichia coli. Int. J. Antimicrob. Agents 52, 63–69. doi: 10.1016/j.ijantimicag.2018.05.002
Wang, X., Kim, Y., Hong, S. H., Ma, Q., Brown, B. L., Pu, M., et al. (2011). Antitoxin MQSA helps mediate the bacterial general stress response. Nat. Chem. Biol. 7, 359–366. doi: 10.1038/nchembio.560
Wang, X., and Wood, T. K. (2011). Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 77, 5577–5583. doi: 10.1128/AEM.05068-11
Weiss, A., Moore, B. D., Tremblay, M. H. J., Chaput, D., Kremer, A., and Shaw, L. N. (2017). The ω subunit governs RNA polymerase stability and transcriptional specificity in Staphylococcus aureus. J. Bacteriol. 199:e00459. doi: 10.1128/JB.00459-16
Wen, Y., Behiels, E., and Devreese, B. (2014). Toxin-antitoxin systems: their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 70, 240–249. doi: 10.1111/2049-632X.12145
Weston, N., Sharma, P., Ricci, V., and Piddock, L. J. V. (2018). Regulation of the AcrAB-TolC efflux pump in Enterobacteriaceae. Res. Microbiol. 169, 425–431. doi: 10.1016/j.resmic.2017.10.005
Wingett, S. W., and Andrews, S. (2018). FastQ screen: a tool for multi-genome mapping and quality control. F1000Res. 7:1338. doi: 10.12688/f1000research.15931.2
Wood, T. K., Knabel, S. J., and Kwan, B. W. (2013). Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 79, 7116–7121. doi: 10.1128/AEM.02636-13
Wright, B. W., Kamath, K. S., Krisp, C., and Molloy, M. P. (2019). Proteome profiling of Pseudomonas aeruginosa PAO1 identifies novel responders to copper stress. BMC Microbiol. 19:69. doi: 10.1186/s12866-019-1441-7
Yamaguchi, Y., Park, J. H., and Inouye, M. (2009). MqsR, a crucial regulator for quorum sensing and biofilm formation, is a GCU-specific mRNA interferase in Escherichia coli. J. Biol. Chem. 284, 28746–28753. doi: 10.1074/jbc.M109.032904
Keywords: persister cells, toxin-antitoxin (TA), phytopathogenic bacteria, copper tolerance system, stress adaptation
Citation: Carvalho IGB, Merfa MV, Teixeira-Silva NS, Martins PMM, Takita MA and de Souza AA (2021) Overexpression of mqsR in Xylella fastidiosa Leads to a Priming Effect of Cells to Copper Stress Tolerance. Front. Microbiol. 12:712564. doi: 10.3389/fmicb.2021.712564
Received: 20 May 2021; Accepted: 09 August 2021;
Published: 20 September 2021.
Edited by:
Raymond J. Turner, University of Calgary, CanadaReviewed by:
Bork Ansgar Berghoff, Justus-Liebig-Universität Gießen, GermanyXiuli Hao, Huazhong Agricultural University, China
Copyright © 2021 Carvalho, Merfa, Teixeira-Silva, Martins, Takita and de Souza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Alves de Souza, ZGVzb3V6YUBjY3NtLmJy; YWxlc3NhbmRyYS5jb2xldHRhQHNwLmdvdi5icg==