 Yundan Liu1†Kaiyang Zheng1†Baohong Liu2†
Yundan Liu1†Kaiyang Zheng1†Baohong Liu2† Yantao Liang1,3*Siyuan You1Wenjing Zhang1Xinran Zhang1Yaqi Jie4Hongbing Shao1,3
Yantao Liang1,3*Siyuan You1Wenjing Zhang1Xinran Zhang1Yaqi Jie4Hongbing Shao1,3 Yong Jiang1,3
Yong Jiang1,3 Cui Guo1,3
Cui Guo1,3 Hui He1,3
Hui He1,3 Hualong Wang1,3
Hualong Wang1,3 Yeong Yik Sung3,5
Yeong Yik Sung3,5 Wen Jye Mok3,5
Wen Jye Mok3,5 Li Lian Wong3,5
Li Lian Wong3,5 Andrew McMinn1,6
Andrew McMinn1,6 Min Wang1,3,7*
Min Wang1,3,7*- 1College of Marine Life Sciences, Frontiers Science Center for Deep Ocean Multispheres and Earth System, Institute of Evolution and Marine Biodiversity, Ocean University of China, Qingdao, China
- 2Department of Hospital Infection Management, Qilu Hospital, Shandong University, Qingdao, China
- 3UMT-OUC Joint Centre for Marine Studies, Qingdao, China
- 4College of Letters and Science, University of Wisconsin–Madison, Madison, WI, United States
- 5Institute of Marine Biotechnology, Universiti Malaysia Terengganu, Kuala Terengganu, Malaysia
- 6Institute for Marine and Antarctic Studies, University of Tasmania, Hobart, TAS, Australia
- 7The Affiliated Hospital of Qingdao University, Qingdao, China
Marinobacter is the abundant and important algal-associated and hydrocarbon biodegradation bacteria in the ocean. However, little knowledge about their phages has been reported. Here, a novel siphovirus, vB_MalS-PS3, infecting Marinobacter algicola DG893(T), was isolated from the surface waters of the western Pacific Ocean. Transmission electron microscopy (TEM) indicated that vB_MalS-PS3 has the morphology of siphoviruses. VB_MalS-PS3 was stable from −20 to 55°C, and with the latent and rise periods of about 80 and 10 min, respectively. The genome sequence of VB_MalS-PS3 contains a linear, double-strand 42,168-bp DNA molecule with a G + C content of 56.23% and 54 putative open reading frames (ORFs). Nineteen conserved domains were predicted by BLASTp in NCBI. We found that vB_MalS-PS3 represent an understudied viral group with only one known isolate. The phylogenetic tree based on the amino acid sequences of whole genomes revealed that vB_MalS-PS3 has a distant evolutionary relationship with other siphoviruses, and can be grouped into a novel viral genus cluster with six uncultured assembled viral genomes from metagenomics, named here as Marinovirus. This study of the Marinobacter phage vB_MalS-PS3 genome enriched the genetic database of marine bacteriophages, in addition, will provide useful information for further research on the interaction between Marinobacter phages and their hosts, and their relationship with algal blooms and hydrocarbon biodegradation in the ocean.
Introduction
The Marinobacter genus is within the class Gammaproteobacteria, order Oceanospirillales and includes Gram-negative, aerobic, motile, halotolerant or halophilic, rod-shaped bacteria (Gauthier et al., 1992), and well known for algal-associated bacteria and its capacity of hydrocarbon degradation, particularly using aromatic and aliphatic hydrocarbons as its sole carbon source (Martin et al., 2003; Holtzapple and Schmidt-Dannert, 2007; Kim et al., 2017). Marinobacter genus is one of the important algal-associated bacterial groups in marine environments (Gonzalez et al., 2000; Buchan et al., 2014). Marinobacter algicola DG893(T) was isolated from paralytic shellfish toxin-producing dinoflagellates (Green et al., 2006), and had been proposed to generate photoactive siderophores that may offer a bioavailable form of iron to commensally associated phytoplankton, affecting algal growth and bloom dynamics (Yarimizu et al., 2018). In addition, petroleum pollution has become a serious environmental problem in the sea, along with the occasional and various release routes such as oil mining, accidental oil spills, shipping transportation, and industrial activities (Cui et al., 2008). Although many marine environments are seriously affected by hydrocarbon pollution, many microbes can utilize hydrocarbon as carbon sources and mediate the influences from hydrocarbon pollution (Antunes et al., 2007; Zenati et al., 2018). Most isolated Marinobacter strains from hydrocarbon-contaminated environments have been found to be able to efficiently degrade hydrocarbon and petroleum compounds (Martinez et al., 2003; Kim et al., 2006). Currently, the genus Marinobacter contains 81 species, and Marinobacter aquaeolei and its heterotypic synonym Pseudomonas nautica are the main hydrocarbon-degrading species (Hedlund et al., 2001; Gorshkova et al., 2003; Gu et al., 2007). Marinobacter squalenivorans and Marinobacter alkaliphilus also have the capacity to degrade hydrocarbon compounds (Shieh et al., 2003; Singer et al., 2011). These Marinobacter species have developed complex and diverse metabolic capacities to adapt to polluted and sometimes hypersaline environments. They can degrade hexadecane, fluoranthene, pristine, and octadecane and even metabolize the herbicide 1, 3-diphenylurea (Takai et al., 2005). The degradation of these hydrocarbon compounds can result in wax ester accumulation in the cytoplasm and influence their fatty acid composition (Hickford et al., 2004). The degradation products, however, can play key roles in marine systems, such as promoting the utilization of hydrocarbons and facilitating the absorption of nutrients (Gardner et al., 2004). This genus is potentially involved in complex ecological networks across trophic levels and could be ecologically and evolutionarily influenced by their bacteriophages (Amarillas et al., 2017).
Bacteriophages, i.e., bacterial viruses, are the most abundant and diverse biological entities on the Earth, with a total abundance of approximately 1030–1032 (Hammerl et al., 2016). The genomic and metagenomic analysis illustrated the tremendous genetic diversity and the huge unknown gene pool of bacteriophages (Li et al., 2016; Gong et al., 2018; Liang et al., 2019). By infecting and lysing their host cells, phages can control and regulate the abundance and structure of microbial communities and play an important role in global biogeochemical cycles (Pourcel et al., 2017). Viral lysing shunts microbial biomass into dissolved organic matter (DOM), mediating the recycling of macro- and micro-elements such as C, P, N, S, and Fe (De Leeuw et al., 2017). In addition, the expression of phage-encoded auxiliary metabolic genes (AMGs), such as dsrC and amoC genes, can manipulate the metabolic reprogramming of infected cells and enhance phage fitness (Zimmerman et al., 2020). However, more than 90% of the viral populations remain unknown (Gregory et al., 2019).
Despite the ecological importance and environmental remediation significance of the Marinobacter genus, our knowledge about their phages and potential interactions with host cells is still rare. To date, only two Marinobacter phages have been reported, including PS6 and AS1, which were isolated from the Bohai Sea of China and the Arabian Sea, respectively (as of May 2021; Zhu et al., 2018; Aparna et al., 2019). Both phages belong to the Siphoviridae family within the Caudovirales order, which typically has a long, flexible, and non-retractable tail (Amgarten et al., 2017; Ramphul et al., 2017). Compared with the booming metagenomic research, there are fewer studies describing the isolation and research of new marine bacteriophages. Therefore, there is a lack of culturable counterparts in the metagenomic viral sequences, which hinders the understanding of the ecological function and biological characteristics of marine viruses (Yang et al., 2021). In addition, isolation and genomic analysis of more Marinobacter phages could improve our understanding of their evolution, interaction with host cells, and potential roles in phytoplankton blooms and hydrocarbon-contaminated environments.
In this study, a new Marinobacter phage vB_MalS-PS3 was isolated from the surface water of the western Pacific Ocean and enriched the genetic database of Marinobacter bacteriophages. Comparative genomes of Marinobacter phages vB_MalS-PS3, AS1, and PS6 were also examined. Due to its novelty in the evolutionary relationship, vB_MalS-PS3 can be grouped into a novel viral cluster (VC) with six uncultured phage contigs from metagenomics, in addition, will provide useful information for further research on the interaction between Marinobacter and their phages. The genome sequence information reported here will provide an important basis for further study of the adaptive evolution and ecological role of Marinobacter phage and their hosts in the sea.
Materials and Methods
Phage Isolation, Purification, and Cross Infectivity Test
The 1 L sample water was collected from the western Pacific Ocean (10.6°N, 143.6167°E), during the spring of 2017, and filtered through 0.22 μm pore-size membranes (Millipore) and checked for the presence of phages by using the double-agar layer method (0.5% low-melt top agar) to obtain the plaques. Each plaque was cored, plaque-purified three times and resuspended in 500 μL of sterile SM buffer [100 mM NaCl, 81.2 mM MgSO4. 7H2O, 50 mM Tris–HCl (pH 7.5), 0.01% gelatin]. Purified bacteriophage was stored in SM buffer at 4°C for a few months for further processing (Liu et al., 2019). The host bacterial strain M. algicola DG893(T) was incubated in LB liquid medium at 28°C.
In order to establish the infection efficacy of each phage, host range tests were evaluated on a range of Marinobacter species by using a cross infectivity test (Table 1). 200 μl of bacteriophage suspension (108 PFU) was mixed with 200 μl of an overnight bacterial culture undergoing exponential growth (OD = 600) and with 4.5 ml of top agar (cooled to 45°C) and poured onto the LB agar, SM buffer alone was used as a blank control (Kajsík et al., 2019). After overnight cultivation at 28°C the plaques were counted by the double-layer agar method. The occurrence of plaques forming in the plate indicated the presence of phages.
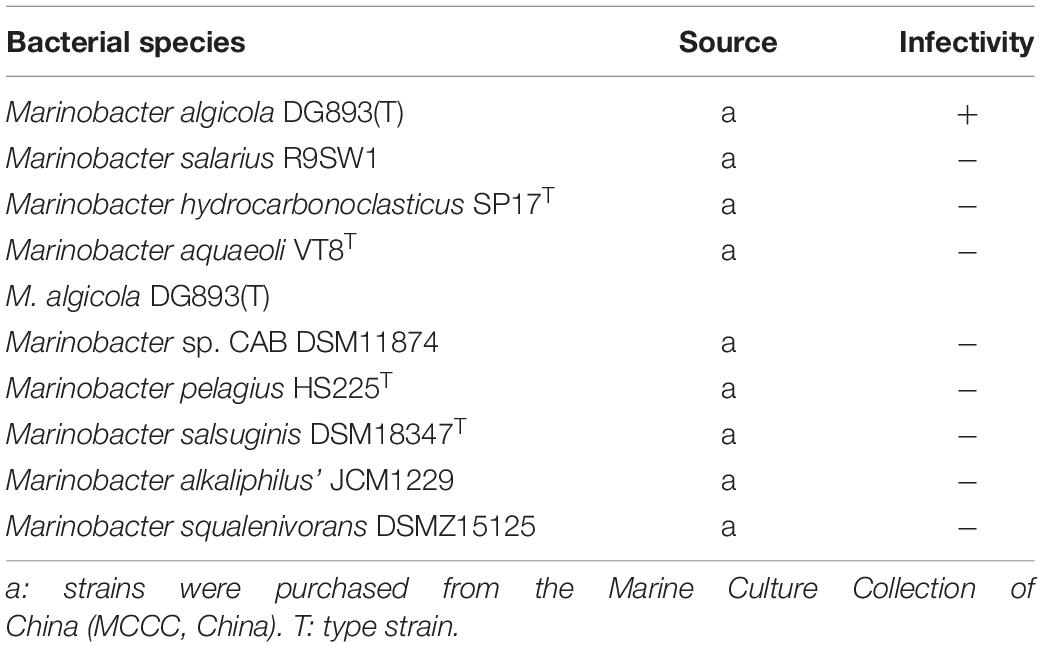
Table 1. Bacterial strains used in this study to determine phage host range.
Transmission Electron Microscopy
The morphology of the phage was examined by transmission electron microscopy (TEM). The purified phage particles were negatively stained with phosphotungstic acid (1% w/v, pH 7.0; Zhang et al., 2020). Stained particles were observed using a JEOL Model JEM-1200EX TEM at 100 kV. Phage dimensions were estimated based on the electron micrographs (Gao et al., 2017).
One-Step Growth Curve Assay and Biological Feature Assays
A modified soft-agar overlay method was used to test the one-step growth curve assay. The phage vB_MalS-PS3 was mixed with the host bacterium in the exponential growth phase (OD600) at a multiplicity of infection (MOI) of 0.1 and incubated at 37°C. To allow adsorption of the phages to the host bacteria the suspensions were incubated for 15 min before further centrifugation, then the infected culture was centrifuged at 8,000 × g for 1 min to remove the unabsorbed phages. Five ml of fresh LB liquid medium was used to suspend the cells. Samples were taken at 5-min intervals (up to 30 min) and 10-min intervals (up to 90 min) and the phage titer was then measured by plaque assays (Kwiatek et al., 2017).
To investigate phage stability at different temperatures, SM buffer was used to dilute phage vB_MalS-PS3. The mixtures were then incubated at −20, 4, 25, 45, 55, 65, 75, and 85°C for 2 h; the double-layer agar method was used to calculate the survival rate of each treated sample. To determine the phage capsid stability, the vB_MalS-PS3 phage was incubated over a pH range of 2–13 with 1 M NaOH or 1 M HCl for 2 h at adjusted optimum temperature; the double-layer agar method was used to evaluate the phage survival rate (Wang et al., 2017).
Extraction of Phage DNA, Sequencing, and Bioinformatic and Phylogenetic Analysis
Bacteriophage genomic DNA extraction was performed by Virus DNA Kit (OMEGA), utilizing DNase and RNase. Nucleic acids were detected by electrophoresis (Pires et al., 2011). Purified phage genomic DNA was sequenced using Illumina Miseq 2 × 300 paired-end sequence by the Novogene Company (Tianjin), the raw data were then filtered to get clean data and assembled using SOAP de novo v2.04 (Beilstein and Dreiseikelmann, 2006).
Open reading frames (ORFs) were predicted using a combination of Glimmer,1 RAST,2 and GeneMarkS3 (Schneiker et al., 2006). The initiation codon and terminate codon of each ORF were checked manually to avoid breakdown for long protein sequence. The sequences of ORFs were subjected to the BLASTp against by the non-redundant (nr) protein database of the GenBank,4 with E-values < 1E-5. Pfam5 with default parameters also were used to predict the function of the proteins (Mistry et al., 2020). tRNAs were predicted by the tRNAscan-SE program.6 The comparative genome figure was produced using Easyfig (Meng et al., 2017). Protein domains were predicted and analyzed using InterPro7 and CDD.8 Genome mapping was performed using CLC Main Workbench 20. The bacteriophage proteomic tree and genome alignments were conducted with Viptree9 (Nishimura et al., 2017). Viral genome sequences and taxonomic information of viruses and their hosts are based on the Virus-Host DB using tBLASTx (Mihara et al., 2016). Selected 1,265 viral genomes as reference sequences to build a phylogenetic tree, and 33 closely related genome sequences of vB_MalS-PS3 were selected to draw a rectangular proteomic tree. The heatmap of the shared genes between 32 closely related genome sequences of vB_MalS-PS3 was plotted using the ‘‘pheatmap’’ R package. The whole-genome phylogenetic tree based on amino acid sequences was constructed using the Virus Classification and Tree Building Online Resource (VICTOR10) and visualized with iTol (v.5; Meier-Kolthoff and Göker, 2017; Letunic and Bork, 2019). All pairwise comparisons of the nucleotide sequences were conducted using the genome-blast distance phylogeny (GBDP) method under settings recommended for prokaryotic viruses (Meier-Kolthoff et al., 2013; Meier-Kolthoff and Göker, 2017). Taxon boundaries at the species, genus and family level were estimated with the OPTSIL program, the recommended clustering thresholds and an F value (fraction of links required for cluster fusion) of 0.5 (Göker et al., 2009; Meier-Kolthoff et al., 2014; Meier-Kolthoff and Göker, 2017). The average nucleotide identity (ANI) was calculated using OrthANI software and JSpeciesWS11 to establish ANI phylogenetic trees (Lee et al., 2016; Richter et al., 2016).
Phage Homologs in Environmental Viral Sequences and Network Analysis
In order to know the accurate taxonomy information and search for homologs of vB_MalS-PS3, each coding sequence was queried against Integrated Microbial Genomes/Virus (IMG/VR) and Global Ocean Viromes 2.0 (GOV 2.0) database using all-versus-all BLASTp, with an E value cutoff of 10–5 (Paez-Espino et al., 2016; Gregory et al., 2019). VCs were identified using ClusterONE with default parameters which are defined in the vConTACT 2.0 (Nepusz et al., 2012; Jang et al., 2019). Network visualization and modular analysis were made by Gephi version 0.9.2 (Xue et al., 2018).
Genome Sequence Accession Number
The annotation results and related information were uploaded to GenBank to obtain the accession number. The complete genome sequence of bacteriophage vB_MalS-PS3 is available in the GenBank database under accession number MF959999.
Results and Discussion
Biological Features and Host Range of Phage vB_MalS-PS3
A novel Marionbacter phage vB_MalS-PS3 was isolated from the western Pacific Ocean, serving M. algicola DG893(T) as its host. Phytoplankton blooms can cause acute effects on marine ecosystems, one hypothesis is that bacteria producing photoactive iron carriers can provide a bioavailable form of iron for symbiotic phytoplankton, which in turn will affect the growth and bloom dynamics of algae (Yarimizu et al., 2018). M. algicola DG893(T) is a phytoplankton-associated bacterium that produces the photoactive siderophore vibrioferrin (Romano et al., 2013). Therefore, vB_MalS-PS3, the phage hosted by algae-associated bacteria, might influence the population dynamics of harmful algal blooms through influencing their host cells.
The bacteriophages were examined by TEM and classified on the basis of their morphological features to the order Caudovirales, tailed phages. Phage vB_MalS-PS3 has an icosahedron head (∼50 nm in diameter) and a long, non-contractile tail (∼100 nm), it was further classified into the Siphoviridae family based on siphoviral morphology (Figure 1A). The head is a protein capsid like icosahedron and the tail contains the tail tube, tail plate, and tail filament.
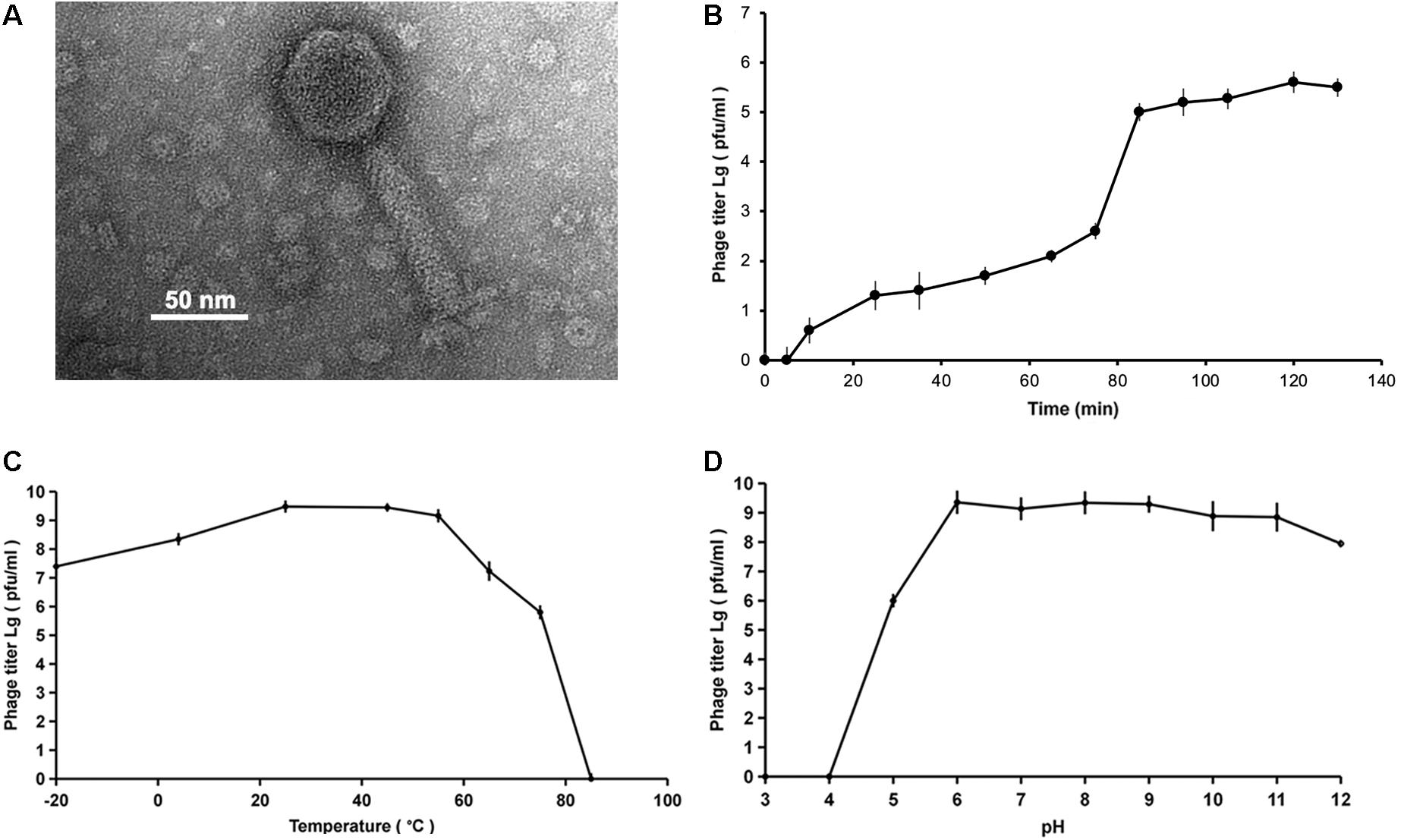
Figure 1. (A) Transmission electron microscopy (TEM) of bacteriophage vB_MalS-PS3. The scale bar is 50 nm. (B) One-step growth curve of vB_MalS-PS3. (C) The curve of thermal stability of vB_MalS-PS3. (D) The curve of capsid stability of vB_MalS-PS3. The Y-axis of the figure is a log scale.
The one-step growth curve showed that the latent period is approximately 80 min and the rise periods is about 10 min (Figure 1B). When the ambient temperature was between −20 and 55°C, the titer of vB_MalS-PS3 remained at a high level (Figure 1C). Optimum temperature of phage infection is 25°C. The phage titer dropped sharply when temperature up to 60°C and the maximum temperature that vB_MalS-PS3 maintains infectious activity is around 65°C. For capsid stability, no phage titer was detected when the pH was less than or equal to three. From pH 6 to 11, there is no significant loss of phage titer (Figure 1D). A significant decline of titer occurred when the pH was equal to or greater than 12.
From the result of cross-infection using double-layer medium, phage vB_MalS-PS3 had a narrow host range, which could not infect the other eight species tested in the Marinobacter genus expect M. algicola DG893(T) (Table 1).
Lambda-Like Genomic Elements Encoded by vB_MalS-PS3 With Two AMGs
The genome of vB_MalS-PS3 consists of a linear, double-stranded 42,168-bp DNA molecule with a GC content of 56.23% and no tRNA genes. The coding ratio of the phage genome is 89.27%.
The results of the prediction showed that the genome contains 54 ORFs (Figure 2 and Table 2), among them, in the positive strand, there were 20 ORFs, the other 34 ORFs were present in the negative strand. Most of the ORFs (45) are initiated by codon ATG, and nine are initiated by alternative codon GTG. From the result of ORFs annotations, it was found that 29 ORFs have high similarity with other published viral genes. Nineteen ORFs possess known functions. Other ORFs (n = 35) are ORFans for missing homologous proteins in current public databases.
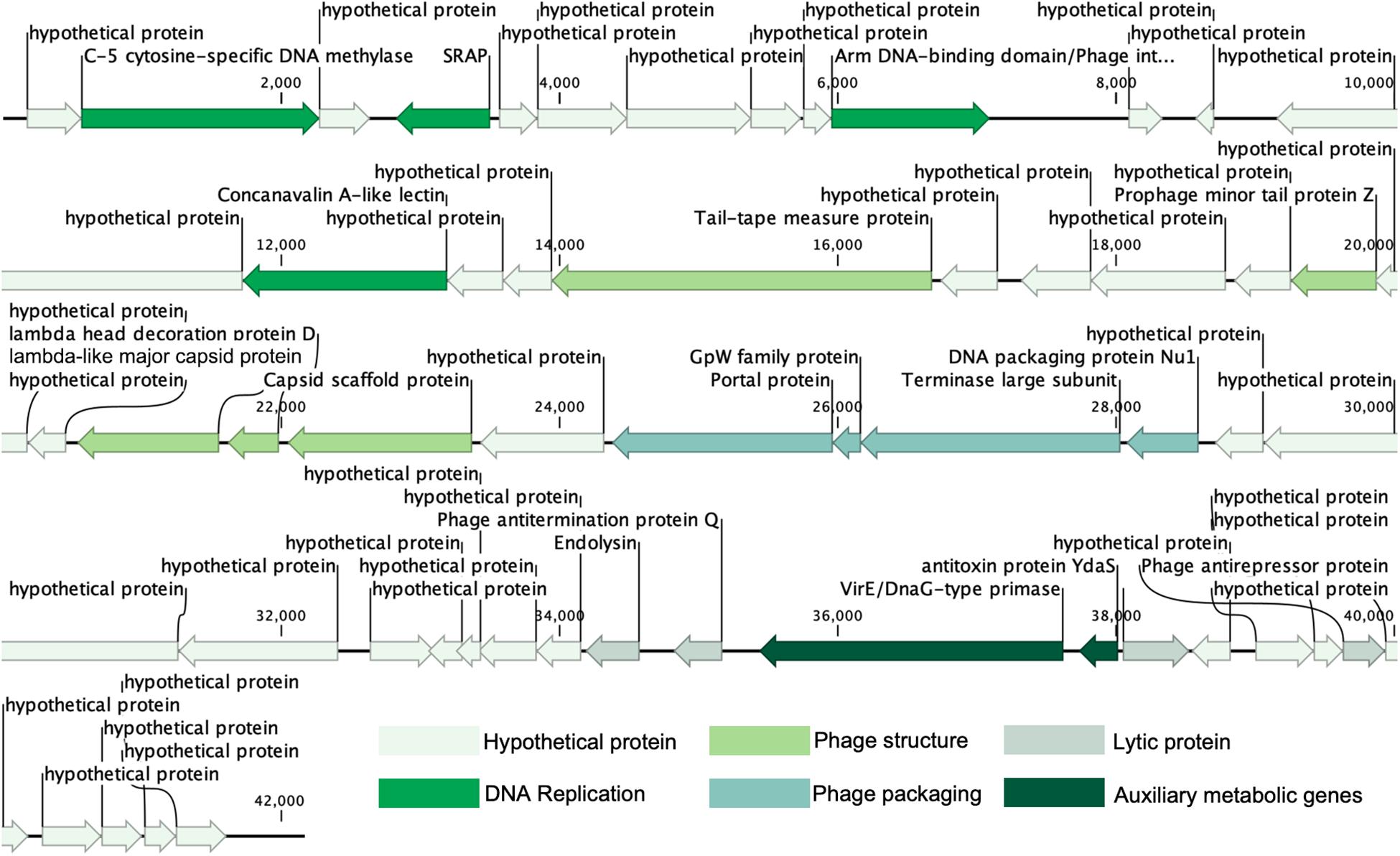
Figure 2. Genome map and functional annotation of the predicted proteins of vB_MalS-PS3.
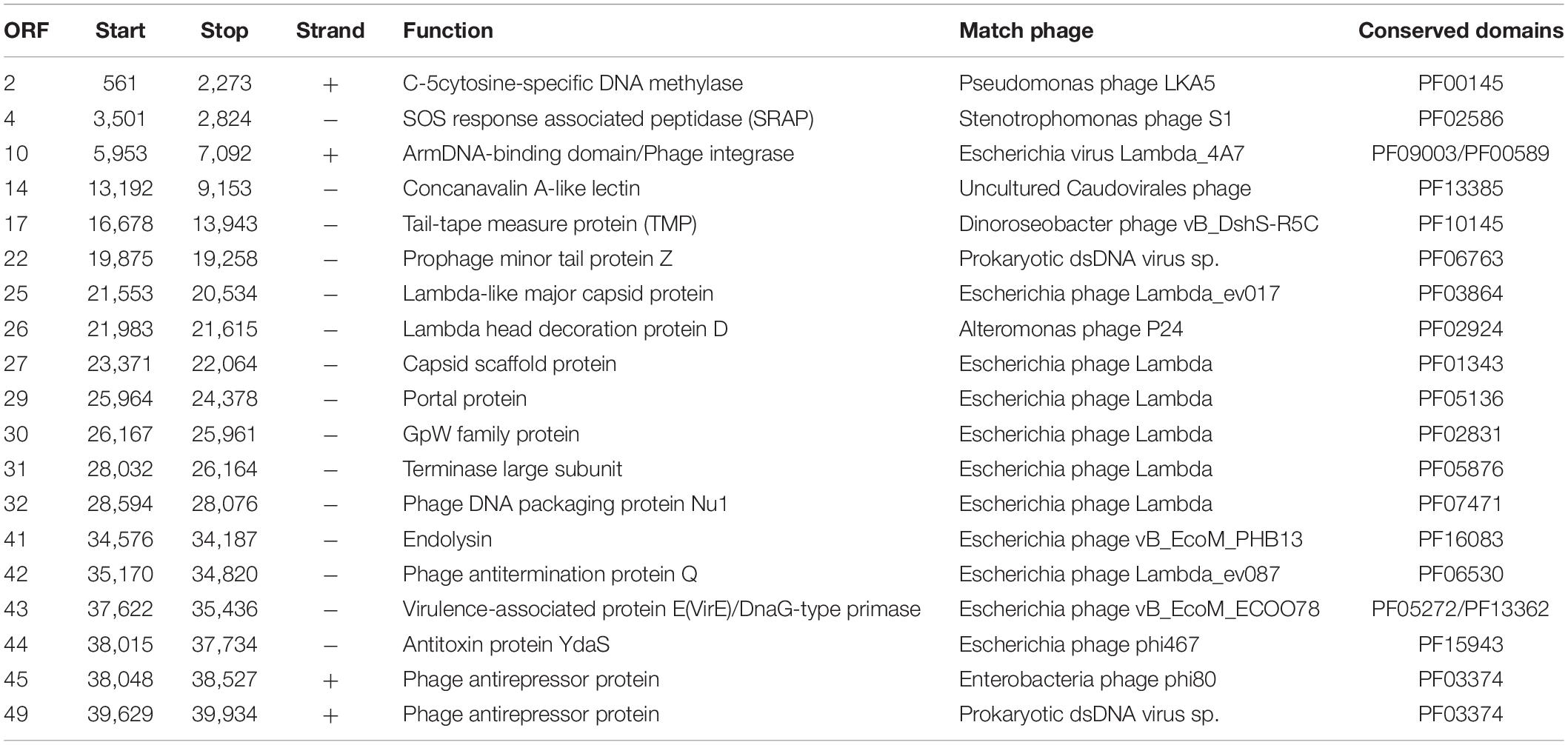
Table 2. The conserved domains detected of vB_MalS-PS3.
Nineteen conserved protein domains were detected (Table 2). Three viral genetic functional modules based on these protein domains were classified, including DNA replication and nucleotide metabolism, phage structure and additional functions (phage packaging, lysis).
ORF10 is the integrase of vB_MalS-PS3, indicating its temperate propagation property in natural habitats. It can catalyze site-specific integration and excision of temperate bacteriophages and other mobile genetic elements from the bacterial host chromosome and shows similarity to Escherichia virus Lambda_4A7 (Tay et al., 2010). Two functional domains were detected in this ORF, including the Arm DNA-binding domain (PF09003) and the Phage integrase family (PF00589). PF09003 and PF00589 locate at the beginning of ORF10 (amino acids sites: from 1 to 63) and another segment (from 188 to 357), respectively. PF09003 is a part of lambda-like integrase, which is responsible for high-affinity binding to each of the five DNA arm-type sites and is also a context-sensitive modulator of DNA cleavage (Wojciak et al., 2002). These two domains are prevalent in Siphoviridae based on the information provided by Pfam, especially for those temperate siphoviruses. Similar integration and recombination progress with Escherichia phage Lambda might be applied by phage vB_MalS-PS3. ORF2, coding the enzyme of C-5 cytosine-specific DNA methylase, modifies the phage DNA to avoid the degradation or restriction of enzymes from the host cell, or methylation marks the phage DNA to protect it from its own nucleases which degrade the host DNA during the early stages of infection (Aravind et al., 2013). From current information of this protein in Pfam (PF00145), most viral-encoded C-5 cytosine-specific DNA methylase is from Siphoviridae (n = 264), occupying 44.5% number of related sequences in viruses. This indicates this enzyme might be much more prevalent in Siphoviridae than other viruses. ORF2 shows homology to Pseudomonas phage LKA5, which might play an important role in the lysogenic life cycle of vB_MalS-PS3. ORF4 coding proteins, which have a similarity to known virus proteins in the database, contained a conservative field, which is related to the peptide enzyme (SRAP) in the SOS response. It can thus be concluded that ORF4-encoded proteins may be associated with DNA repair. Abasic (AP) sites are one of the most common DNA lesions that block replicative polymerases. SRAP proteins shield the AP site from endonucleases and error-prone polymerases (Mohni et al., 2019). Thus, the expression of SRAP encoded by vB_MalS-PS3 might protect the replication of viral genomes from occasional interruption inside cells.
Two protein domains were included in ORF43, including Toprim domain (PF13362; from 203 to 307) and Virulence-associated protein E (PF05272; from 429 to 619). PF13362 is a DnaG-type primase (Aravind and Landsman, 1998), while PF05272 is a typical AMG. ORF43 is homologous with Escherichia phage vB_EcoM_ECOO78 and contains a P-loop motif. Protein with the domain of PF05272 can aggravate the pathogenicity of some pathogenic bacteria (Ji et al., 2016) so that ORF43 might promote interspecific or intraspecific competition ability for those vB_MalS-PS3-infected Marinobacter. ORF 44 encodes antitoxin protein YdaS (PF15943) and shows homologous with Escherichia phage phi467. YdaS is a putative bacterial antitoxin that can neutralize the toxin YdaT, while YdaS is an AMG. This entry also includes some bacteriophage proteins, such as cro from Pseudomonas phage D3 and gene 30 protein from Enterobacteria phage phi80 (Yamaguchi and Inouye, 2011).
ORF29, ORF 30, ORF31, and ORF32 encode four packaging-associated genes, including portal protein, GpW family protein, terminase large subunit and Phage DNA packaging protein Nu1, respectively. All ORFs show homology to the Escherichia phage Lambda, which contains Lambda family phage portal protein (PF05136), gpW (PF02831), Terminase_GpA domain (PF05876), and Phage_Nu1domain (PF07471), respectively. Despite the Lambda-like terminase homolog was found in the genome of vB_MalS-PS3, it is still unknown whether the Lambda-like-terminase-mediated process could participate in the DNA packaging of vB_MalS-PS3. Hence, the functions of Lambda-like terminase in DNA packaging of vB_MalS-PS3 is still unclear. We hypothesized that it might introduce staggered nick to generate the cohesive ends (similar to cosN) from other specific site recognition to implement the function of DNA shearing. This also confirmed the potential highly hybridized genotype of PS3 to some extent. Different viral gene possesses a divergent origin, suggesting the special phylogenic status and evolutionary history of PS3. As a series of lambda-homologous proteins found in the genome of PS3, which might be the gene signature of viral evolutionary history of PS3, we speculate that this phage might be originated from Lambdavirus in the past, but acquired or loss some other genes. Hence, the PS3 was not clustered with Lambdavirus in the phylogenetic tree.
Among the proteins directly involved in phage structure, ORF25 is a Lambada-like major capsid protein with Phage major capsid protein E domain (PF03864). ORF 22 is a Prophage minor tail protein Z (PF06763), and this family consists of several prophage minor tail protein Z like sequences from Escherichia coli, Salmonella typhimurium and Lambda-like bacteriophages. ORF17 encodes the tail-tape measure protein (TMP) and shows homology to Dinoroseobacter phage vB_DshS-R5C. Forty-four isolated viruses with homologs of ORF17 were reported in GenBank, and the genome size of all related phages infecting Proteobacteria was higher than 70,000-bp. To the best of our knowledge, vB_MalS-PS3 is the only small-genome Proteobacteria phages encoding vB_DshS-R5C-like TMP. ORF27 is a capsid scaffold protein with Peptidase family S49 domain (PF01343), which is homologous with Escherichia phage Lambda and other several lambda-like phages. Several genes associated with genome replication that are commonly found in phages, such as helicase, DNA-binding protein and ligase, are missing from the genome of vB_MalS-PS3 based on current annotation information. Potentially, other unknown viral replication genes may be encoded by the vB_MalS-PS3 genome, or its replication process depends on the host enzyme system, which is unclear currently.
From the view of the genomic signature of vB_MalS-PS3, it is a Lambda-like temperate siphoviruses with a hybrid viral genome. Both replication and structure functional modules contained Lambda-like genetic elements. The genes of phage vB_MalS-PS3 are from diverse siphoviral lineages, such as Lambdavirus, Nanhaivirus, and Chivirus.
Phage vB_MalS-PS3 Represented a New Siphoviral Genus
To identify the exact taxonomic position of the phage vB_MalS-PS3, the phylogenetic tree based on the viral proteome of whole genomes was established by Viptree (Figure 3A), and the 32 closest phages in the proteome tree were selected together with Marinobacter phage vB_MalS-PS3 to establish a rectangular tree for subsequent comparison and analysis (Figure 3B). The results of phylogenetic analysis and gene-shared heatmap revealed that vB_MalS-PS3 represents a separate cluster and is far from the other isolated phages. To confirm this finding, we chose two phages complete genome sequences (Marinobacter phage AS1 and Marinobacter phage PS3), carried out the comparative genomic analysis with vB_MalS-PS3 (Figure 3C). Similar to the results of the phylogenetic tree analysis, vB_MalS-PS3 showed low similarity in either Marinobacter phage AS1 or Marinobacter phage PS6. The results indicated the novelty of vB_MalS-PS3 and had a relatively distant evolutionary relationship which represents a novel VC.
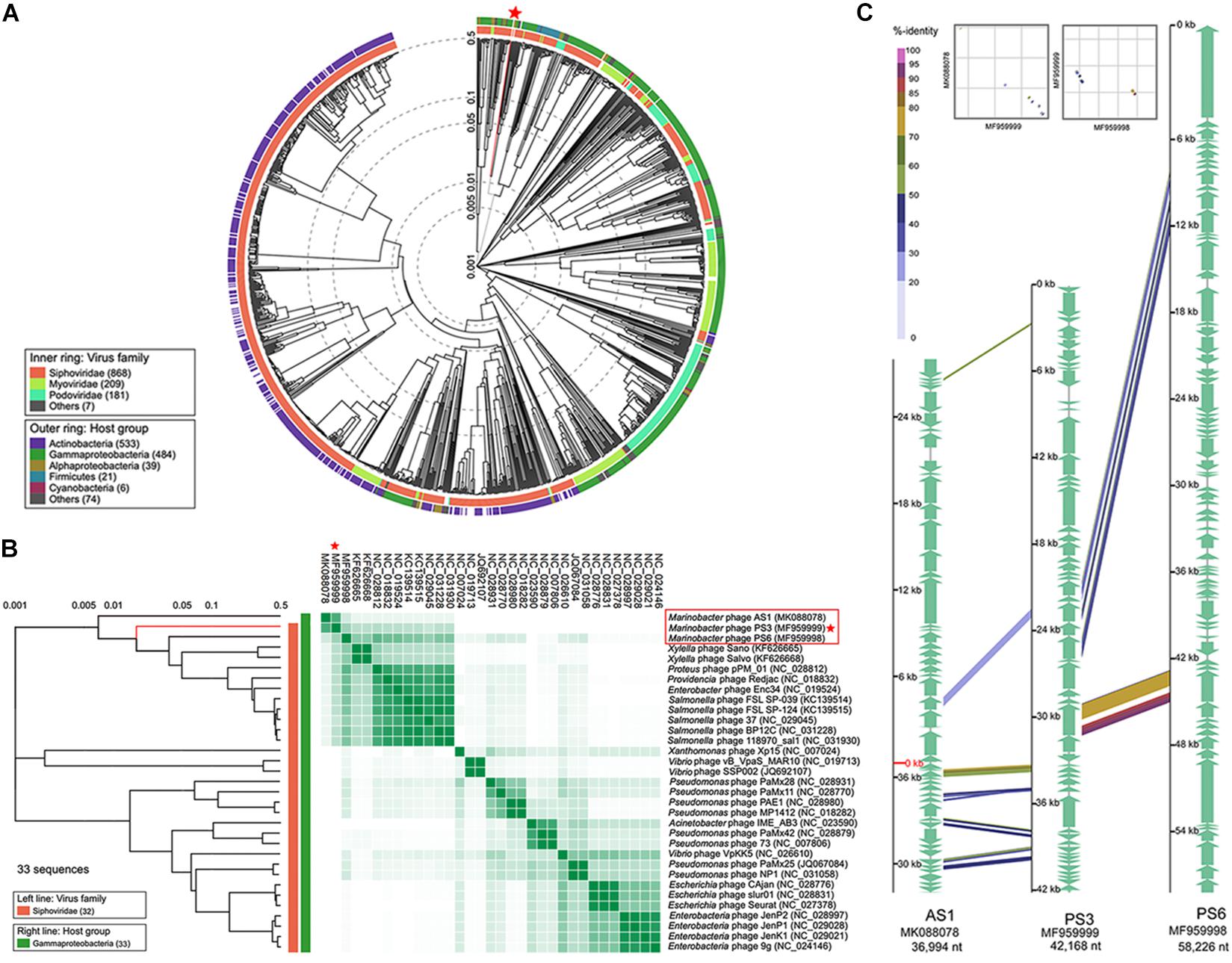
Figure 3. Phylogenetic and comparative genomic analysis of vB_MalS-PS3. (A) Viral phylogenetic tree of whole phage vB_MalS-PS3 genomes represented in the circular view. (B) Viral phylogenetic tree represented in the rectangular view with 32 closely related genome sequences of vB_MalS-PS3, and heatmap showing the percentage of shared genes among 33 genome sequences. The ratio of shared genes was based on all-to-all BLASTP analysis (E-value < 1E-10). (C) Genomic organization of the Marinobacter bacteriophage vB_MalS-PS3 compared to Marinobacter bacteriophage AS1 and Marinobacter bacteriophage PS6.
Although vB_MalS-PS3 exhibits no significant genomic synteny with other known phage isolates, high similarity with some metagenomic assembled genome virals (MAGs) from IMG/VR and GOV 2.0 database using all-versus-all BLASTp. Therefore, six MAGs with high similarity (shared genes > 50%) to vB_MalS-PS3 were retrieved from IMG/VR datasets. Then, 168 viruses (28 RefSeq from NCBI, 139 IMG/VR and GOV 2.0 metagenomic viral sequences and vB_MalS-PS3) were classified into different VCs using the vConTACT 2.0 (Figure 4A). Six phages and vB_MalS-PS3 were grouped as a VC (VC 364_0) from the result of genome-content-based analysis (Supplementary Table 1). In further comparative genomic analysis, all six phages clustered with vB_MalS-PS3 revealed considerably conserved in whole-genome synteny. Most of the homologous ORFs throughout these phage genomes located in the packaging module (portal protein, terminase large subunit and phage DNA packaging) and structural module (TMP, major capsid protein, head decoration protein D and capsid scaffold protein; Figure 4C).
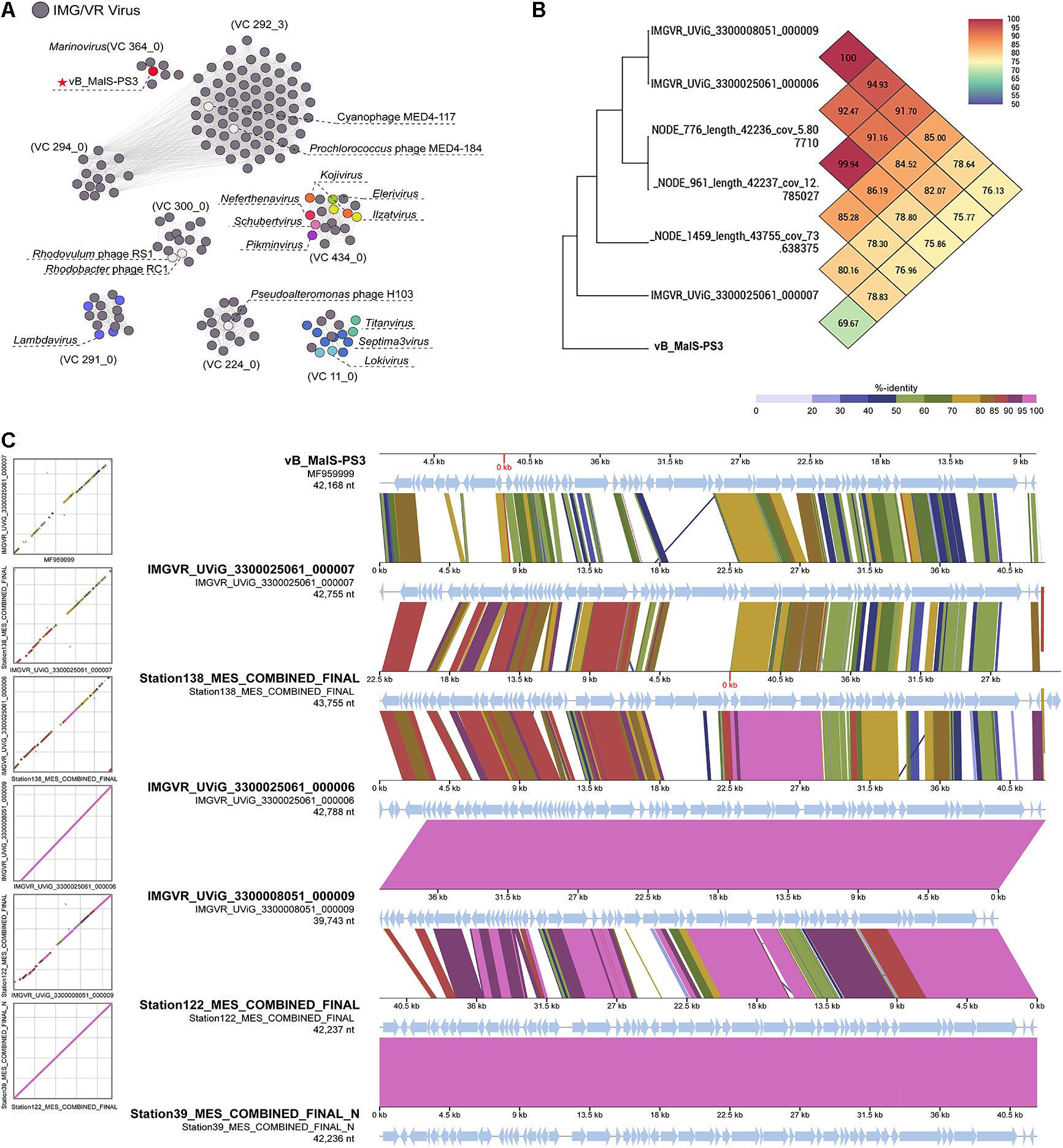
Figure 4. (A) Gene-content-based viral network of vB_MalS-PS3, Siphoviridae virus from NCBI RefSeq database, and related environmental viral sequences from Integrated Microbial Genomes/Virus (IMG/VR). The nodes represent the viral genomic sequences. The edges represent the similarity score between genomes based on shared gene content. The gray circles indicate the related environmental viral sequences from IMG/VR. In each viral cluster (VC), viral genomes that belong to different genus clusters classified and ratified by the International Committee on Taxonomy of Viruses (ICTV) are indicated by different colors. VCs generated using vConTACT2, the network was visualized using Gephi version 0.9.2. (B) Genome comparison between the vB_MalS-PS3 and the environmental viral sequences in IMG/VR. (C) Heat map based on OrthoANI values calculated using OAT software.
Phages are regarded as members of the same genus by the International Committee on Taxonomy of Viruses (ICTV) when their nucleotide sequence identities are greater than 70% (Adriaenssens and Brister, 2017). The ANI value between vB_MalS-PS3 and the other six environmental viral sequences ranged from 69.97 to 100% (Figure 4B), confirming these phages (VC 364_0) might be identified as a new genus. But the application of ANI to taxonomy is limited to sequences with high coverage at the whole genome level (Feng et al., 2021). Therefore, whole-genome-based phylogenic analysis was conducted for vB_MalS-PS3 and the other six MAGs through VICTOR (Figure 5). The whole-genome phylogeny and OPTSIL taxon results are implied that vB_MalS-PS3 and the other six environmental viral sequences are clustering together (shown in red), comparing with the identified siphoviral genera in ICTV. In conclusion, all results confirmed that vB_MalS-PS3 and related MAGs are a novel unassigned viral genus, while vB_MalS-PS3 is the only isolates in this putative genus and named here as Marinovirus.
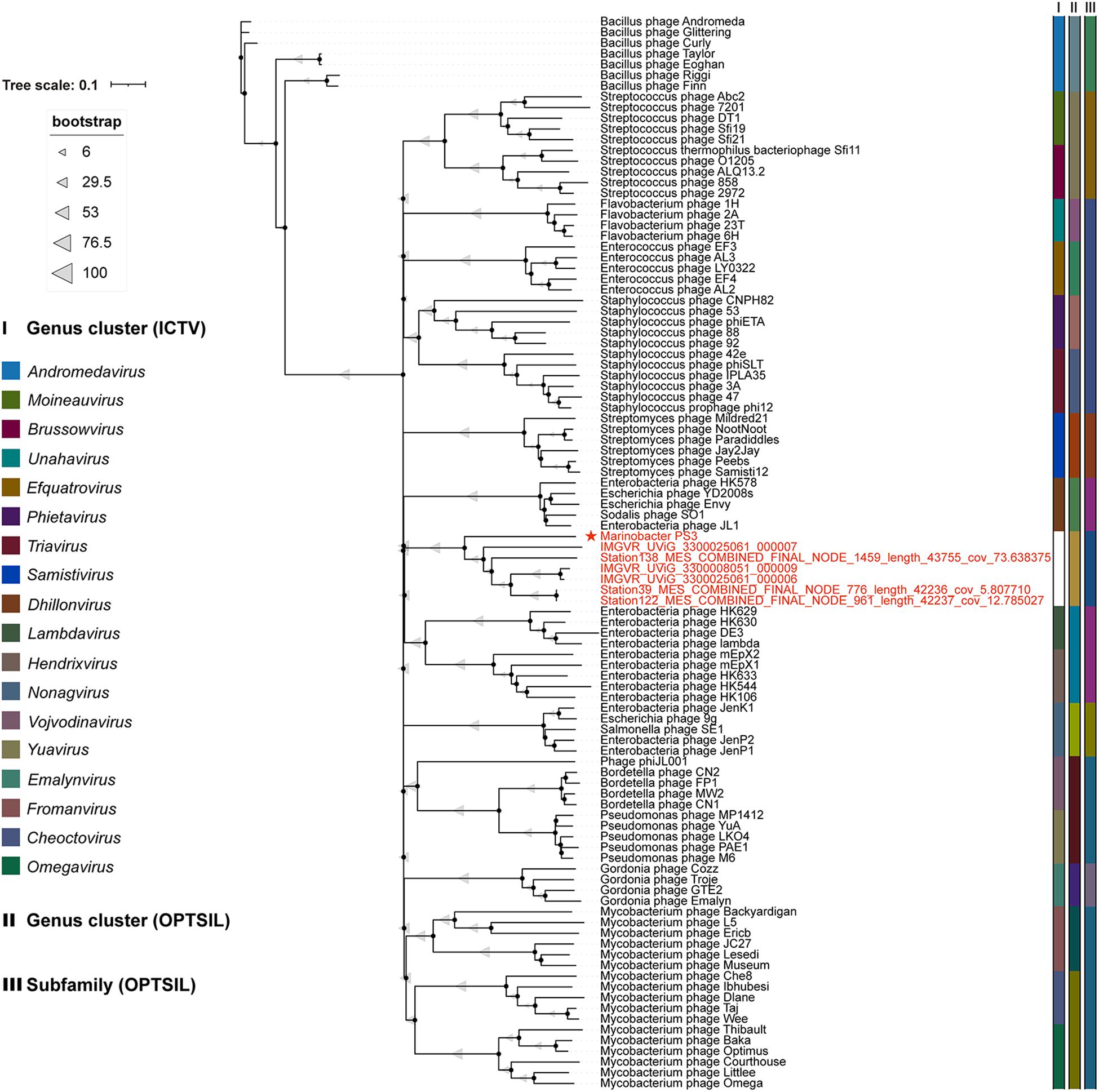
Figure 5. Genome BLAST distance phylogeny (GBDP) tree of 100 siphovirus genomes. vB_MalS-PS3 shows the phylogenomic GBDP trees inferred using the formula D6 and yielding average support of 57%. The numbers above branches are GBDP pseudo-bootstrap support values from 100 replications. The branch lengths of the resulting Virus Classification and Tree Building Online Resource (VICTOR) trees are scaled in terms of the respective distance formula used. The ICTV taxonomy and predicted OPTSIL taxon are shown. Each genus is indicated by a unique color. The members in VC 364_0 (Figure 4A) are shown in red.
Conclusion
Bacteriophages play a significant role in marine biogeochemical cycles by affecting their host’s physiology and ecology. In this study, we describe a novel Lambda-like temperate phage vB_MalS-PS3, which infects M. algicola DG893(T). In addition to the comparatively special position in phylogeny, the genomic signature of vB_MalS-PS3 shows a mosaic pattern as a novel temperate phage, which might make it a viral isolate, representing a genus of uncultured viruses, namely Marinovirus. Investigation of phage functional genes and other less well-studied genes will no doubt enable phage genome analysis to be more explicit and detailed. In subsequent studies on the phages-host system, some functional genes will need to be analyzed in more detail, including putative integration function, structural and morphogenesis genes, tape measure protein, other structural proteins, early regulation region, host lysis, DNA modification, small ORFs and other noteworthy features. Its evolutionary linage is novel and represents a cluster together with some uncultured virus sequences.
The study will contribute to the understanding of the role of lysogenetic viruses in marine ecosystems, particularly Marinobacter bacteriophages, including their host specificity, adaptation to host defense systems and propagation dynamics in natural systems. In the future, isolation, genome sequencing and interaction analysis of phages-hosts systems of Marinobacter can improve our understanding about their host defense systems and co-evolution relationships. Currently, databases of genes and proteins are extremely limited, and more phages need to be isolated and identified from different environments. Marinobacter phages have important effects, not only on lysing the host but also on the manipulating host to enroll the global biogeochemical circle through viral chronic infections. This study will contribute to the understanding of the physiological, genetic diversity and genomic characteristics of phages and their hosts in different aquatic environments. This will enable the exploration of the ecological characteristics of the system of phages-host under different conditions and the characteristics of their interactions.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
YTL and MW: conceptualization and project administration. YDL: methodology, writing, and original draft preparation. KZ: software. WZ, XZ, and HS: validation. YJ and YQJ: formal analysis. CG: investigation. HW: resources. SY: data curation. AM and BL: writing, review, and editing. YS, WM, and LW: visualization. HH: supervision. YTL, MW, and AM: funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This research was financially supported by the Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao; No. 2018SDKJ0406-6), the National Key Research and Development Program of China (2018YFC1406704), the Fundamental Research Funds for the Central Universities (202072002 and 201812002), the National Natural Science Foundation of China (Nos. 41976117 and 41606153), and 973 Program (No. 2013CB429704).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
These sequence data (IMGVR_UViG_3300008051_000009, IMGVR_UViG_3300025061_000007, and IMGVR_UViG_33000 25061_000006) were produced by the US Department of Energy Joint Genome Institute http://www.jgi.doe.gov/in collaboration with the user community.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.726074/full#supplementary-material
Footnotes
- ^ http://ccb.jhu.edu/software/glimmer/index.shtml
- ^ http://rast.nmpdr.org
- ^ http://topaz.gatech.edu/GeneMark/
- ^ https://www.ncbi.nlm.nih.gov
- ^ https://pfam.xfam.org/search/sequence
- ^ http://lowelab.ucsc.edu/tRNAscan-SE/
- ^ https://www.ebi.ac.uk/interpro/
- ^ https://www.ncbi.nlm.nih.gov/cdd
- ^ https://www.genome.jp/viptree/
- ^ https://ggdc.dsmz.de/victor.php
- ^ http://jspecies.ribohost.com/jspeciesws/#analyse
References
Adriaenssens, E., and Brister, J. R. (2017). How to name and classify your phage: an informal guide. Viruses 9:70. doi: 10.3390/v9040070
Amarillas, L., Rubí-Rangel, L., Chaidez, C., González-Robles, A., Lightbourn-Rojas, L., and León-Félix, J. (2017). Isolation and characterization of phiLLS, a novel phage with potential biocontrol agent against multidrug-resistant Escherichia coli. Front. Microbiol. 8:1355. doi: 10.3389/fmicb.2017.01355
Amgarten, D., Martins, L. F., Lombardi, K. C., Antunes, L. P., de Souza, A. P. S., Nicastro, G. G., et al. (2017). Three novel Pseudomonas phages isolated from composting provide insights into the evolution and diversity of tailed phages. BMC Genomics 18:346. doi: 10.1186/s12864-017-3729-z
Antunes, A., Franca, L., Rainey, F. A., Huber, R., Nobre, M. F., Edwards, K. J., et al. (2007). Marinobacter salsuginis sp. nov., isolated from the brine–seawater interface of the shaban deep. red sea. Int. J. Syst. Evol. Microbiol. 57, 1035–1040. doi: 10.1046/j.1462-2920.2002.00351.x
Aparna, S., Parvathi, A., Ram, A. P., and Sime-Ngando, T. (2019). Genome analysis of Marinobacter phage AS1 suggests its close interactions with host Marinobacter sp. Aquat. Microbial. Ecol. 83, 119–129. doi: 10.3354/ame01912
Aravind, L., Anand, S., and Iyer, L. M. (2013). Novel autoproteolytic and DNA-damage sensing components in the bacterial SOS response and oxidized methylcytosine-induced eukaryotic DNA demethylation systems. Biol. Dir. 8, 1–11.
Aravind, L., and Landsman, D. (1998). AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 26, 4413–4421. doi: 10.1093/nar/26.19.4413
Beilstein, F., and Dreiseikelmann, B. (2006). Bacteriophages of freshwater brevundimonas vesicularis isolates. Res. Microbiol. 157, 213–219. doi: 10.1016/j.resmic.2005.07.005
Buchan, A., Le Cleir, G. R., Gulvik, C. A., and Gonzalez, J. M. (2014). Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 12, 686–698. doi: 10.1038/nrmicro3326
Cui, Z., Lai, Q., Dong, C., and Shao, Z. (2008). Biodiversity of polycyclic aromatic hydrocarbon-degrading bacteria from deep sea sediments of the Middle Atlantic Ridge. Environ. Microbiol. 10, 2138–2149. doi: 10.1111/j.1462-2920.2008.01637.x
De Leeuw, M., Baron, M., Brenner, A., and Kushmaro, A. (2017). Genome analysis of a novel broad host range Proteobacteria phage isolated from a bioreactor treating industrial wastewater. Genes 8:40. doi: 10.3390/genes8010040
Feng, X., Yan, W., Wang, A., Ma, R., Chen, X., Lin, T.-H., et al. (2021). A novel broad host range phage infecting alteromonas. Viruses 13:987. doi: 10.3390/v13060987
Gao, Y., Liu, Q., Wang, M., Zhao, G., Jiang, Y., Malin, G., et al. (2017). Characterization and genome sequence of marine Alteromonas gracilis phage PB15 isolated from the Yellow Sea. China. Curr. Microbiol. 74, 821–826.
Gardner, R. A., Kinkade, R., Wang, C., and Phanstiel, O. (2004). Total synthesis of petrobactin and its homologues as potential growth stimuli for marinobacter h ydrocarbonoclasticus, an oil-degrading bacteria. J. Org. Chem. 69, 3530–3537. doi: 10.1002/chin.200438207
Gauthier, M. J., Lafay, B., Christen, R., Fernandez, L., Acquaviva, M., Bonin, P., et al. (1992). Marinobacter hydrocarbonoclasticus gen. nov., sp. nov., a new, extremely halotolerant, hydrocarbon-degrading marine bacterium. Int. J. Syst. Bacteriol. 42, 568–576. doi: 10.1099/00207713-42-4-568
Göker, M., García-Blázquez, G., Voglmayr, H., Tellería, M. T., and Martín, M. P. (2009). Molecular taxonomy of phytopathogenic fungi: a case study in Peronospora. PLoS One 4:e6319. doi: 10.1371/journal.pone.0006319
Gong, Z., Liang, Y., Wang, M., Jiang, Y., Yang, Q., Xia, J., et al. (2018). Viral diversity and its relationship with environmental factors at the surface and deep sea of Prydz Bay. Antarct. Front. Microbiol. 9:2981. doi: 10.3389/fmicb.2018.02981
Gonzalez, J. M., Simo, R., Massana, R., Covert, J. S., Casamayor, E. O., Pedros-Alio, C., et al. (2000). Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl. Environ. Microbiol. 66, 4237–4246. doi: 10.1128/aem.66.10.4237-4246.2000
Gorshkova, N. M., Ivanova, E. P., Sergeev, A. F., Zhukova, N. V., Alexeeva, Y., Wright, J. P., et al. (2003). Marinobacter excellens sp. nov., isolated from sediments of the Sea of Japan. Int. J. Syst. Evol. Microbiol. 53, 2073–2078. doi: 10.1099/ijs.0.02693-0
Green, D. H., Bowman, J. P., Smith, E. A., Gutierrez, T., and Bolch, C. J. (2006). Marinobacter algicola sp. nov., isolated from laboratory cultures of paralytic shellfish toxin-producing dinoflagellates. Int. J. Syst. Evol. Microbiol. 56, 523–527. doi: 10.1099/ijs.0.63447-0
Gregory, A. C., Zayed, A. A., Conceição-Neto, N., Temperton, B., Bolduc, B., Alberti, A., et al. (2019). Marine DNA viral macro-and microdiversity from pole to pole. Cell 177, 1109–1123. doi: 10.1038/s41579-019-0304-4
Gu, J., Cai, H., Yu, S.-L., Qu, R., Yin, B., Guo, Y.-F., et al. (2007). Marinobacter gudaonensis sp. nov., isolated from an oil-polluted saline soil in a Chinese oilfield. Int. J. Syst. Evol. Microbiol. 57, 250–254. doi: 10.1099/ijs.0.64522-0
Hammerl, J. A., Göllner, C., Al Dahouk, S., Nöckler, K., Reetz, J., and Hertwig, S. (2016). Analysis of the first temperate broad host range brucellaphage (BiPBO1) isolated from B. inopinata. Front. Microbiol. 7:24. doi: 10.3389/fmicb.2016.00024
Hedlund, B. P., Geiselbrecht, A. D., and Staley, J. T. (2001). Marinobacter strain NCE312 has a Pseudomonas-like naphthalene dioxygenase. FEMS Microbiol. Lett. 201, 47–51. doi: 10.1111/j.1574-6968.2001.tb10731.x
Hickford, S. J., Küpper, F. C., Zhang, G., Carrano, C. J., Blunt, J. W., and Butler, A. (2004). Petrobactin sulfonate, a new siderophore produced by the marine bacterium marinobacter hydrocarbonoclasticus. J. Nat. Prod. 67, 1897–1899. doi: 10.1021/np049823i
Holtzapple, E., and Schmidt-Dannert, C. (2007). Biosynthesis of isoprenoid wax ester in Marinobacter hydrocarbonoclasticus DSM 8798: identification and characterization of isoprenoid coenzyme A synthetase and wax ester synthases. J. Bacteriol. 189:3804. doi: 10.1128/jb.01932-06
Jang, H. B., Bolduc, B., Zablocki, O., Kuhn, J. H., Roux, S., Adriaenssens, E. M., et al. (2019). Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 37, 632–639. doi: 10.1038/s41587-019-0100-8
Ji, X., Sun, Y., Liu, J., Zhu, L., Guo, X., Lang, X., et al. (2016). A novel virulence-associated protein, vapE, in Streptococcus suis serotype 2. Mol. Med. Rep. 13, 2871–2877. doi: 10.3892/mmr.2016.4818
Kajsík, M., Bugala, J., Kadlièeková, V., Szemes, T., Turòa, J., and Drahovská, H. (2019). Characterization of Dev-CD-23823 and Dev-CT57, new Autographivirinae bacteriophages infecting Cronobacter spp. Arch. Virol. 164, 1383–1391. doi: 10.1007/s00705-019-04202-3
Kim, B.-Y., Weon, H.-Y., Yoo, S.-H., Kim, J.-S., Kwon, S.-W., Stackebrandt, E., et al. (2006). Marinobacter koreensis sp. nov., isolated from sea sand in Korea. Int. J. Syst. Evol. Microbiol. 56, 2653–2656. doi: 10.1099/ijs.0.64231-0
Kim, J.-O., Lee, H.-J., Han, S.-I., and Whang, K.-S. (2017). Marinobacter halotolerans sp. nov., a halophilic bacterium isolated from a saltern crystallizing pond. Int. J. Syst. Evol. Microbiol. 67, 460–465. doi: 10.1099/ijsem.0.001653
Kwiatek, M., Parasion, S., Rutyna, P., Mizak, L., Gryko, R., Niemcewicz, M., et al. (2017). Isolation of bacteriophages and their application to control Pseudomonas aeruginosa in planktonic and biofilm models. Res. Microbiol. 168, 194–207. doi: 10.1016/j.resmic.2016.10.009
Lee, I., Kim, Y. O., Park, S.-C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, Y., Wang, M., Liu, Q., Song, X., Wang, D., Ma, Y., et al. (2016). Complete genomic sequence of bacteriophage H188: a novel Vibrio kanaloae phage isolated from Yellow Sea. Curr. Microbiol. 72, 628–633. doi: 10.1007/s00284-015-0984-6
Liang, Y., Wang, L., Wang, Z., Zhao, J., Yang, Q., Wang, M., et al. (2019). Metagenomic analysis of the diversity of DNA viruses in the surface and deep sea of the South China sea. Front. Microbiol. 10:1951. doi: 10.3389/fmicb.2019.01951
Liu, Y., Zhao, L., Wang, M., Wang, Q., Zhang, X., Han, Y., et al. (2019). Complete genomic sequence of bacteriophage P23: a novel Vibrio phage isolated from the Yellow Sea. China Virus Genes 55, 834–842. doi: 10.1007/s11262-019-01699-3
Martin, S., Márquez, M., Sánchez-Porro, C., Mellado, E., Arahal, D., and Ventosa, A. (2003). Marinobacter lipolyticus sp. nov., a novel moderate halophile with lipolytic activity. J. Med. Microbiol. 53, 1383–1387. doi: 10.1099/ijs.0.02528-0
Martinez, J. S., Carter-Franklin, J. N., Mann, E. L., Martin, J. D., Haygood, M. G., and Butler, A. (2003). Structure and membrane affinity of a suite of amphiphilic siderophores produced by a marine bacterium. Proc. Nat. Acad. Sci. 100, 3754–3759. doi: 10.1073/pnas.0637444100
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., and Göker, M. (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33, 3396–3404. doi: 10.1093/bioinformatics/btx440
Meier-Kolthoff, J. P., Hahnke, R. L., Petersen, J., Scheuner, C., Michael, V., Fiebig, A., et al. (2014). Complete genome sequence of DSM 30083 T, the type strain (U5/41 T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genomic Sci. 9, 1–19. doi: 10.1186/1944-3277-9-2
Meng, X., Wang, M., You, S., Wang, D., Li, Y., Liu, Z., et al. (2017). Characterization and complete genome sequence of a novel Siphoviridae bacteriophage BS5. Curr. Microbiol. 74, 815–820. doi: 10.1007/s00284-017-1221-2
Mihara, T., Nishimura, Y., Shimizu, Y., Nishiyama, H., Yoshikawa, G., Uehara, H., et al. (2016). Linking virus genomes with host taxonomy. Viruses 8:66. doi: 10.3390/v8030066
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar Gustavo, A., Sonnhammer, E. L. L., et al. (2020). Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi: 10.1093/nar/gkaa913
Mohni, K. N., Wessel, S. R., Zhao, R., Wojciechowski, A. C., Luzwick, J. W., Layden, H., et al. (2019). HMCES maintains genome integrity by shielding abasic sites in single-strand DNA. Cell 176, 144–153. doi: 10.3410/f.734636888.793557375
Nepusz, T., Yu, H., and Paccanaro, A. (2012). Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 9:471. doi: 10.1038/nmeth.1938
Nishimura, Y., Yoshida, T., Kuronishi, M., Uehara, H., Ogata, H., and Goto, S. (2017). ViPTree: the viral proteomic tree server. Bioinformatics 33, 2379–2380. doi: 10.1093/bioinformatics/btx157
Paez-Espino, D., Chen, I.-M. A., Palaniappan, K., Ratner, A., Chu, K., Szeto, E., et al. (2016). IMG/VR: a database of cultured and uncultured DNA Viruses and retroviruses. Nucleic Acids Res. 45:gkw1030. doi: 10.1093/nar/gkw1030
Pires, D., Sillankorva, S., Faustino, A., and Azeredo, J. (2011). Use of newly isolated phages for control of Pseudomonas aeruginosa PAO1 and ATCC 10145 biofilms. Res. Microbiol. 162, 798–806. doi: 10.1016/j.resmic.2011.06.010
Pourcel, C., Midoux, C., Vergnaud, G., and Latino, L. (2017). A carrier state is established in Pseudomonas aeruginosa by phage LeviOr01, a newly isolated ssRNA levivirus. J. Gen. Virol. 98, 2181–2189. doi: 10.1099/jgv.0.000883
Ramphul, C., Casareto, B. E., Dohra, H., Suzuki, T., Yoshimatsu, K., Yoshinaga, K., et al. (2017). Genome analysis of three novel lytic Vibrio coralliilyticus phages isolated from seawater. Okinawa, Japan. Mar. Genomics 35, 69–75. doi: 10.1016/j.margen.2017.06.005
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Romano, A., Trimble, L., Hobusch, A. R., Schroeder, K. J., Amin, S. A., Hartnett, A. D., et al. (2013). Regulation of iron transport related genes by boron in the marine bacterium Marinobacter algicola DG893. Metallomics 5, 1025–1030. doi: 10.1039/c3mt00068k
Schneiker, S., Dos Santos, V. A. M., Bartels, D., Bekel, T., Brecht, M., Buhrmester, J., et al. (2006). Genome sequence of the ubiquitous hydrocarbon-degrading marine bacterium Alcanivorax borkumensis. Nat. Biotechnol. 24, 997–1004. doi: 10.1038/nbt1232
Shieh, W. Y., Jean, W. D., Lin, Y.-T., and Tseng, M. (2003). Marinobacter lutaoensis sp. nov., a thermotolerant marine bacterium isolated from a coastal hot spring in Lutao. Taiwan. Can. J. Microbiol. 49, 244–252. doi: 10.1007/s00284-017-1251-9
Singer, E., Webb, E. A., Nelson, W. C., Heidelberg, J. F., Ivanova, N., Pati, A., et al. (2011). Genomic potential of Marinobacter aquaeolei, a biogeochemical “opportunitroph”. Appl. Environ. Microbiol. 77:2763. doi: 10.1128/aem.01866-10
Takai, K., Moyer, C. L., Miyazaki, M., Nogi, Y., Hirayama, H., Nealson, K. H., et al. (2005). Marinobacter alkaliphilus sp. nov., a novel alkaliphilic bacterium isolated from subseafloor alkaline serpentine mud from ocean drilling program site 1200 at south chamorro seamount. Mariana Forearc. Extremophiles 9, 17–27. doi: 10.1007/s00792-004-0416-1
Tay, Y., Ho, C., Droge, P., and Ghadessy, F. J. (2010). Selection of bacteriophage lambda integrases with altered recombination specificity by in vitro compartmentalization. Nucleic Acids Res. 38:e25. doi: 10.1093/nar/gkp1089
Wang, C., Chen, Q., Zhang, C., Yang, J., Lu, Z., Lu, F., et al. (2017). Characterization of a broad host-spectrum virulent Salmonella bacteriophage fmb-p1 and its application onduck meat. Virus Res. 236, 14–23. doi: 10.1016/j.envint.2019.05.062
Wojciak, J. M., Sarkar, D., Landy, A., and Clubb, R. T. (2002). Arm-site binding by λ-integrase: solution structure and functional characterization of its amino-terminal domain. Proc. Nat. Acad. Sci. 99, 3434–3439. doi: 10.1073/pnas.052017999
Xue, Y., Chen, H., Yang, J. R., Liu, M., Huang, B., and Yang, J. (2018). Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 12, 2263–2277. doi: 10.1038/s41396-018-0159-0
Yamaguchi, Y., and Inouye, M. (2011). Regulation of growth and death in Escherichia coli by toxin–antitoxin systems. Nat. Rev. Microbiol. 9, 779–790. doi: 10.1038/nrmicro2651
Yang, M., Xia, Q., Du, S., Zhang, Z., Qin, F., and Zhao, Y. (2021). Genomic characterization and distribution pattern of a novel marine OM43 Phage. Front. Microbiol. 12:651326. doi: 10.3389/fmicb.2021.651326
Yarimizu, K., Cruz-López, R., and Carrano, C. J. (2018). Iron and harmful algae blooms: Potential algal-bacterial mutualism between Lingulodinium polyedrum and Marinobacter algicola. Front. Mar. Sci. 5:180. doi: 10.3389/fmars.2018.00180
Zenati, B., Chebbi, A., Badis, A., Eddouaouda, K., Boutoumi, H., El Hattab, M., et al. (2018). A non-toxic microbial surfactant from Marinobacter hydrocarbonoclasticus SdK644 for crude oil solubilization enhancement. Ecotoxicol. Environ. Saf. 154, 100–107. doi: 10.1016/j.ecoenv.2018.02.032
Zhang, X., Liu, Y., Wang, M., Wang, M., Jiang, T., Sun, J., et al. (2020). Characterization and genome analysis of a novel marine Alteromonas phage P24. Curr. Microbiol. 77, 2813–2820. doi: 10.1007/s00284-020-02077-1
Zhu, M., Wang, M., Jiang, Y., You, S., Zhao, G., Liu, Y., et al. (2018). Isolation and complete genome sequence of a novel Marinobacter phage B23. Curr. Microbiol. 75, 1619–1625. doi: 10.1007/s00284-018-1568-z
Keywords: bacteriophage, Marinobacter, hydrocarbon biodegradation, genome and comparative genome analysis, phylogenetic analysis, algal-associated bacteria
Citation: Liu Y, Zheng K, Liu B, Liang Y, You S, Zhang W, Zhang X, Jie Y, Shao H, Jiang Y, Guo C, He H, Wang H, Sung YY, Mok WJ, Wong LL, McMinn A and Wang M (2021) Characterization and Genomic Analysis of Marinobacter Phage vB_MalS-PS3, Representing a New Lambda-Like Temperate Siphoviral Genus Infecting Algae-Associated Bacteria. Front. Microbiol. 12:726074. doi: 10.3389/fmicb.2021.726074
Received: 06 July 2021; Accepted: 03 August 2021;
Published: 25 August 2021.
Edited by:
Heejoon Myung, Hankuk University of Foreign Studies, South KoreaReviewed by:
Ricardo Cruz-López, Universidad Autónoma de Baja California, MexicoPaul Hyman, Ashland University, United States
Copyright © 2021 Liu, Zheng, Liu, Liang, You, Zhang, Zhang, Jie, Shao, Jiang, Guo, He, Wang, Sung, Mok, Wong, McMinn and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yantao Liang, bGlhbmd5YW50YW9Ab3VjLmVkdS5jbg==; Min Wang, bWluZ3dhbmdAb3VjLmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship