 Antonio Camilo da Silva Filho1*
Antonio Camilo da Silva Filho1* Jeroniza Nunes Marchaukoski2
Jeroniza Nunes Marchaukoski2 Roberto Tadeu Raittz2
Roberto Tadeu Raittz2 Camilla Reginatto De Pierri3Diogo de Jesus Soares Machado2Cyntia Maria Telles Fadel-Picheth1Geraldo Picheth1
Camilla Reginatto De Pierri3Diogo de Jesus Soares Machado2Cyntia Maria Telles Fadel-Picheth1Geraldo Picheth1- 1Department of Clinical Analysis, Federal University of Parana, Curitiba, Brazil
- 2Department of Bioinformatics, Professional and Technical Education Sector, Federal University of Parana, Curitiba, Brazil
- 3Department of Biochemistry and Molecular Biology, Federal University of Parana, Curitiba, Brazil
Aeromonas are Gram-negative rods widely distributed in the environment. They can cause severe infections in fish related to financial losses in the fish industry, and are considered opportunistic pathogens of humans causing infections ranging from diarrhea to septicemia. The objective of this study was to determine in silico the contribution of genomic islands to A. hydrophila. The complete genomes of 17 A. hydrophila isolates, which were separated into two phylogenetic groups, were analyzed using a genomic island (GI) predictor. The number of predicted GIs and their characteristics varied among strains. Strains from group 1, which contains mainly fish pathogens, generally have a higher number of predicted GIs, and with larger size, than strains from group 2 constituted by strains recovered from distinct sources. Only a few predicted GIs were shared among them and contained mostly genes from the core genome. Features related to virulence, metabolism, and resistance were found in the predicted GIs, but strains varied in relation to their gene content. In strains from group 1, O Ag biosynthesis clusters OX1 and OX6 were identified, while strains from group 2 each had unique clusters. Metabolic pathways for myo-inositol, L-fucose, sialic acid, and a cluster encoding QueDEC, tgtA5, and proteins related to DNA metabolism were identified in strains of group 1, which share a high number of predicted GIs. No distinctive features of group 2 strains were identified in their predicted GIs, which are more diverse and possibly better represent GIs in this species. However, some strains have several resistance attributes encoded by their predicted GIs. Several predicted GIs encode hypothetical proteins and phage proteins whose functions have not been identified but may contribute to Aeromonas fitness. In summary, features with functions identified on predicted GIs may confer advantages to host colonization and competitiveness in the environment.
Introduction
The bacterial genome is composed of a core genome containing the genetic information required for essential functions, and a flexible gene pool, which encodes additional traits that can be beneficial under certain circumstances. The flexible gene pool represents variable chromosomal regions and includes mobile and accessory genetic elements, such as bacteriophages, plasmids, insertion sequences, and genomic islands (GIs) (Dobrindt et al., 2004).
Genomic islands are syntenic blocks of accessory genes acquired by horizontal gene transfer (HGT) which contribute to the diversification and adaptation of microorganisms and offer a selective advantage for host bacteria. According to their gene content, GIs are described as pathogenicity, resistance, symbiosis, metabolic or fitness islands (Weinstock et al., 2000; Juhas et al., 2009; Rao et al., 2020). Most GIs are relatively large segments of DNA, usually between 10 and 200 kb, detected by comparisons among closely related strains; however, regions with sizes < 10 kb have also been identified and named genomic islets (Hacker et al., 1990; Juhas et al., 2009; Li and Wang, 2021). GIs usually differ from chromosomes in terms of GC content, tetranucleotide frequency, and codon usage. They are often flanked by small directly repeated (DR) sequences and are often inserted into tRNA genes. They can carry functional or cryptic genes encoding integrases, factors related to plasmid conjugation, phages involved in GI transfer, and transposons or insertion sequence (IS) elements, which may be implicated in mobilizing genetic material onto or deleting DNA from GI (Buchrieser et al., 1998; Juhas et al., 2009; Partridge et al., 2018). Currently, bioinformatics tools developed for GI prediction (Vernikos and Parkhill, 2006; Pundhir et al., 2008; Che et al., 2014; Soares et al., 2016; Wei et al., 2016; Bertelli et al., 2017) are available for the study of these mobile elements.
Aeromonas are Gram-negative bacteria ubiquitous in aquatic environments and can be isolated from virtually every environmental niche where bacterial ecosystems exist (Janda and Abbott, 2010). There are 36 species recognized of Aeromonas (Fernández-Bravo and Figueras, 2020), of which A. hydrophila is the most studied and the first species of the genus to have a sequenced genome (Seshadri et al., 2006; Grim et al., 2014; Rasmussen-Ivey et al., 2016b). A. hydrophila has been isolated from fresh and marine waters, diseased fish, poikilothermic aquatic animals, and warm-blooded animals (Martin-Carnahan and Joseph, 2005). In fish, A. hydrophila can cause a variety of diseases, including septicemia, red sore disease, and ulcerative infections, and has been linked to fish death worldwide, resulting in great economic losses (Janda and Abbott, 2010; Hossain et al., 2013, 2014; Rasmussen-Ivey et al., 2016a). In humans, A. hydrophila is associated with both intestinal and extraintestinal diseases (Martin-Carnahan and Joseph, 2005), such as gastroenteritis, wound infection, septicemia, pneumonia, soft tissue infections, peritonitis, hepatobiliary tract infections, and necrotizing fasciitis (Janda and Abbott, 1998, 2010; Grim et al., 2014).
Genome sequencing and comparative genomic analysis have allowed major advances in the study of Aeromonas virulence. Highly virulent strains recovered from humans or fish have been identified, several genes have been associated with pathogenicity and some are related to GIs in some strains (Seshadri et al., 2006; Hossain et al., 2013; Grim et al., 2014; Pang et al., 2015; Rasmussen-Ivey et al., 2016a). The objective of this study was to analyze A. hydrophila genomes using a GI predictor, determine the distribution of predicted GIs, their characteristics, functions encoded, core genome content, and sharing among strains.
Materials and Methods
Aeromonas hydrophila Genomes
The complete genomes of A. hydrophila available at the National Center for Biotechnology Information database (Geer et al., 2010) as at 12/01/2019 were used in this study. The source and genome characteristics of the A. hydrophila strains are shown in Table 1.
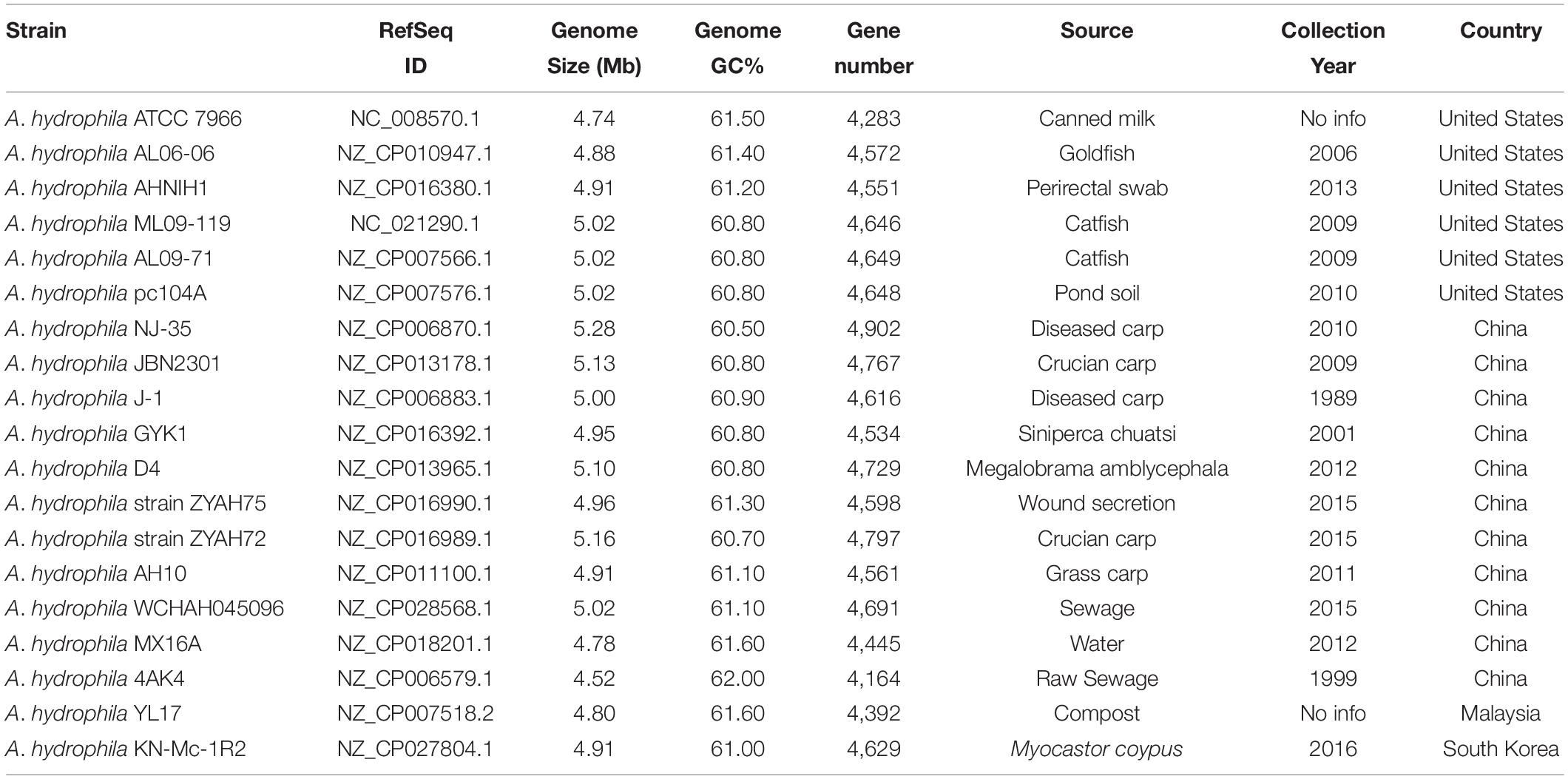
Table 1. General characteristics of Aeromonas hydrophila.
Phylogenetic Analysis
A phylogenetic tree was generated using the software Spaced Words Projection – SweeP (De Pierri et al., 2020) with the complete genomes of all Aeromonas species available from the NCBI database. Default SweeP criteria were used in the analysis. Data regarding the organisms used are shown in Supplementary Table 1.
Genomic Islands Prediction and Analysis
IslandViewer 4 (Bertelli et al., 2017) was used to predict GIs in A. hydrophila strains. GIs < 10 kb were considered genomic islands unless otherwise stated. Some GIs predicted by SIGI-HMM (Waack et al., 2006) and IslandPath-DIMOB (Hsiao et al., 2003) two components of IslandViewer 4, were partially superimposed, increasing the number of predictions. Then, a verification of these regions was conducted to determine the start and end of the GIs manually, avoiding misidentification in the number of predicted GIs. After the identification of each predicted GI by IslandViewer 4, the regions were selected and visualized using Artemis software (Rutherford et al., 2000) to generate .GBK and .FASTA files for each GI. These files contained the amino acid sequence, the name, and the protein ID of each product of each GI, and were used to create multi-fasta files for each microorganism. These files were submitted to RAST (Brettin et al., 2015) for standardization of gene nomenclature. The IslandViewer version 4.0 (Web Server) was used.
Tools and Databases for Traits Prediction
To determine the traits encoded by the predicted GIs, datasets containing the amino acid sequences of the GIs of each A. hydrophila strain were compared with the amino acid sequences of the databases indicated below using the online web server CD-HIT Suite: Biological Sequence Clustering and Comparison (CD-HIT-2D; version v4.8.1-2019-0228) (Huang et al., 2010). The minimum threshold for sequence identity cut-off was 70%. All other parameters of CD-HIT-2D are indicated in Supplementary Table 2.
The following databases were used to identify traits related to virulence, antibiotic resistance, or drug targets encoded on the predicted GIs:
Pathosystems Resource Integration Center – PATRIC (Wattam et al., 2017). The virulence factor database – VFDB (Liu et al., 2019). PHIDIAS: Pathogen-host interaction data integration and analysis system VICTORS (Sayers et al., 2019). The Comprehensive Antibiotic Resistance Database CARD (Jia et al., 2017). National Database of Antibiotic Resistant Organisms1. DrugBank (Wishart et al., 2018). Therapeutic Target Database (TTD) (Li et al., 2018). For the identification of metabolic traits, KEGG Mapper (Kanehisa and Sato, 2020) was used with family/genus 642 ID. The data obtained from the tests were manually curated.
Phage Prediction
Phage regions were analyzed using PHASTER Phage Search Tool Enhanced Release (Arndt et al., 2016). The PHASTER search phages regions using BLAST through file analysis in .GBK or .FASTA format. The sequences are compared with the NCBI Phage database and a phage database developed by Srividhya et al. (2006). Phage-like genes are then grouped into phage regions using DBSCAN (Ester et al., 1996). Regions corresponding to phages are assigned scores and colors depending on the level of identity and coverage of the region. Intact phage (> 90 score) are colored green; questionable phage (70-90 score) labeled blue; incomplete phage (< 70 score) are colored red. For this analysis, complete genomes of A. hydrophila were submitted to PHASTER to search for phage regions, and the results were compared with predicted GIs for identification of phage-containing GIs.
Shared Genomic Islands
To compare the predict GIs, genes of each GI were concatenated and used to create individual multi-fast files for each strain. After this step, BLASTp was used to infer homology, considering a minimum of 75% of identity and sequence coverage for comparisons between single GIs against all other predicted GIs in all strains. Power Bi, a software for generating interactive reports with data modeling tools, and chord diagrams were used to represent the proportion of GIs shared among the strains. An image was generated using the data obtained from the total number of shared GIs. The nodes were organized around a circle, with the proportion between points connected to each other, represented proportionally by the size of each arc2. Power Bi version 2.96.1061.0 (Windows 64-bit) was used in this study.
Core Genome and Related Genomic Islands
The core genome of A. hydrophila strains was determined using the web server EDGAR version 3.0 (Web Server) https://edgar3.computational.bio.uni-giessen.de/ (Dieckmann et al., 2021). This bioinformatics tool is based on ‘score ratio values’ (SRVs) methodology for orthology estimation. To calculate the SRVs, all BLAST scores are normalized against the maximum score, which is the self-hit of a query sequence. Then, the distribution of the SRVs is analyzed to derive a threshold that is tailored to the data, usually around an SRV of 0.3 (30%). Furthermore, an initial e-value cutoff of 1e-05 is used. This orthologous approach is such that only one-to-one pairs are found. For duplicated genes and paralogs, a single hit is found, and additional copies are missed. Pseudogenes are excluded from these analyses. Core genome calculation requires one reference genome, for which A. hydrophila ATCC 7966 was chosen. Results from the core genome (EDGAR) are presented with a locus tag for each gene and strain with the amino acid sequence. After identifying the genes from the core genome, they were compared with predicted GIs using CD-HIT, a tool developed for clustering very long microbial genomic sequences. In this analysis the comparison was performed gene-by-gene for all GIs of each strain using amino acid sequences. The cut-off rate used was 90% identity.
Cluster Alignments
Mauve, a tool for multiple genome alignments that can be used to view genomic rearrangements, inversions, and large insertions in related genomes, was used to analyze O antigen clusters. The alignment is shown as an image formed by colored blocks, each one of them representing genome region aligned and internally free of genomic rearrangements (Darling et al., 2004). Mauve version 2.4.0 (Windows 64-bit) was used in this study.
Statistical Methods
Variables with normal distributions (Kolmogorov-Smirnov) were reported as mean ± 1-standard deviation. Comparison among groups were tested with Student t-test (2-tailed) or by one-way ANOVA for multiple groups comparisons. A P-value < 0.05 was considered significant.
All calculations were performed using MedCalc version 17.6 (MedCalc Statistical Software bvba, Ostend, Belgium).
Results
Phylogenetic Analysis and General Characteristics of Aeromonas hydrophila Strains
A phylogenetic tree based on the complete genome of Aeromonas species was generated to verify the relationships among A. hydrophila isolates. A total of 96 genomes from 10 Aeromonas species and some strains identified only to the genus level were included in this analysis, the results of which are shown in Figure 1. Strains of A. schubertii, A. veronii, A. salmonicida, A. dhakensis, A. hydrophila, A. encheleia, A. media, A. rivipollensis, A. jandaei, and A. caviae were separated into distinct branches of the phylogenetic tree according to their species. However, there were some exceptions, with A. veronii WP2_S18_CRE03 grouped with A. jandaei strains 3299 and 3348; A. hydrophila YL17 grouped together A. dhakensis KN_Mc_6U21, and A. hydrophila 4AK4 with A. media T0-1-19 and A. rivipollensis KN_Mc_11N1, suggesting that these strains were misidentified at the species level. Thus, the genomes of strains YL17 and 4AK4 were removed from this study. Additionally, a small number of strains previously identified only at the genus level clustered together with certain species (Figure 1).
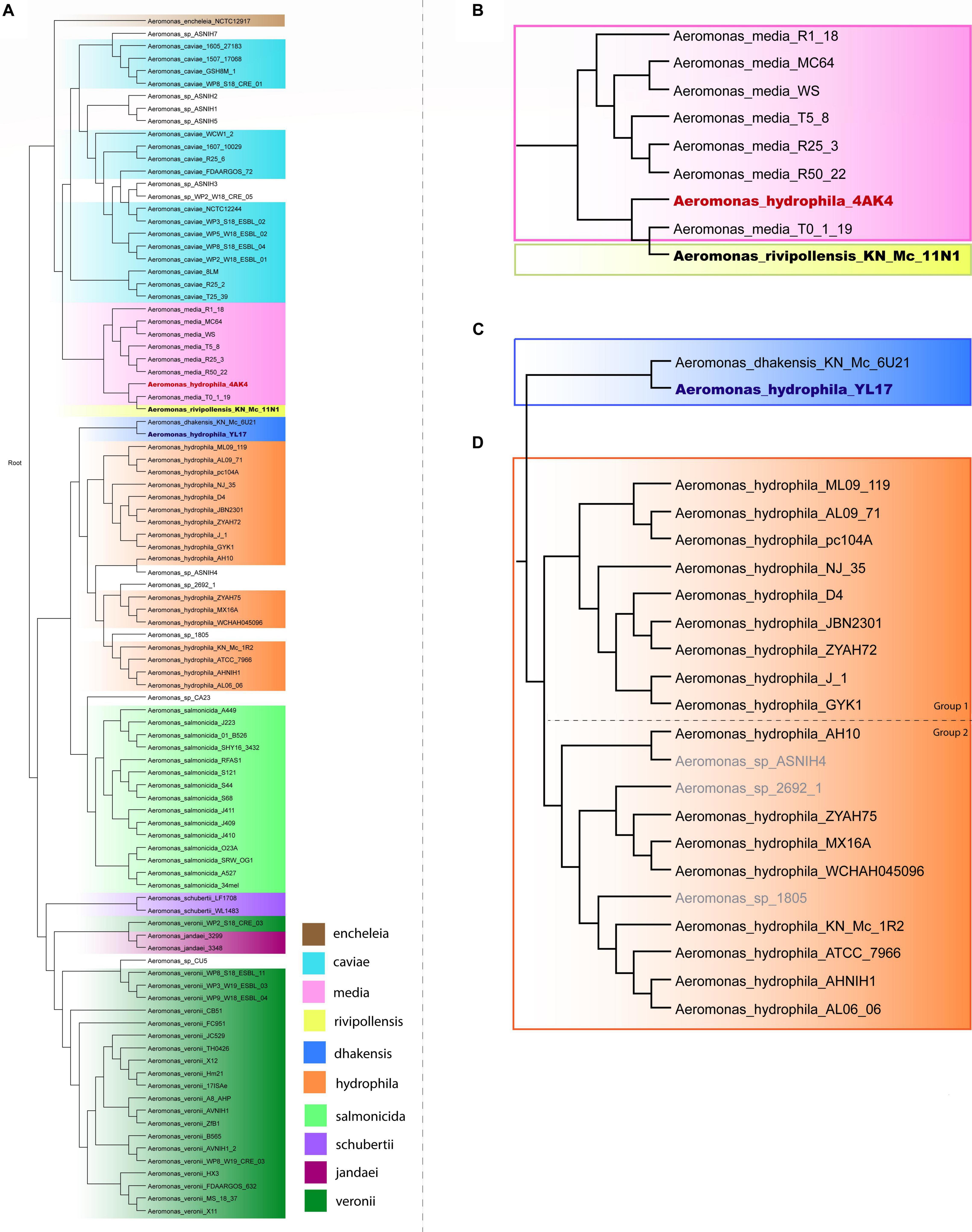
Figure 1. Phylogenetic tree of the Aeromonas species. (A) Phylogenetic tree generated from the whole genome of 96 Aeromonas strains. (B) Highlighted branch of “A. hydrophila 4AK4.” (C) Highlighted branch of “A. hydrophila YL17.” (D) Highlighted branch of A. hydrophila; strains analyzed are subgrouped into two main subclades, separated by dotted lines: ML09-119, AL09-71, pc104A, NJ-35, D4, JBN2301, ZYAH72, J-1, and GYK1 (group 1); AH10, ZYAH75, MX16A, WCHAH045096, KN-Mc-1R2, ATCC 7966, AHNIH1, and AL06-06 (group 2).
The 17 complete A. hydrophila genomes that were analyzed for predicted GIs differed in genome size, which varies by up to 0.54 Mb, as observed between strains ATCC 7966, which has the smallest genome (4.74 Mb) and NJ-35 (5.28 Mb) the largest; the GC content varied from 60.50% in strain NJ-35 to 61.60% in strain MX16A (Table 1).
Phylogenetic analysis classified the 17 A. hydrophila strains into two main groups (Figure 1). Group 1 contained nine strains, isolated in China or the United States, recovered from diseased fish, except for pc104A, which was recovered from the soil of a catfish pond (Table 1). The Chinese and American A. hydrophila strains were separated into different subclades of group 1. Group 2 contained strains recovered from different sources, including the environment (ATCC 7966, MX16A, and WCHAH045096), human clinical samples (ZYAH75, AHNIH1), Myocastor (KN-Mc-1R2), and diseased fish (AH10 and AL06-06). They were also separated into subclades according to their geographical origins.
Strains from groups 1 and 2 differ in relation to the mean genome size; 5,08 and 4,89 Mb, respectively. Interestingly, all strains of group 1 presented genomic GC contents below 61% (60.50% to 60.90%), while those of group 2 were 61% or higher (61.00 to 61.60%; Table 1), a significative difference (P < 0.001; t-test), however, stats were underpowered.
Characteristics of Predicted Genomic Islands of Aeromonas hydrophila Strains
The number of predicted GIs varied widely among the strains, from 13 in A. hydrophila ATCC 7966 and MX16A to 33 in strain NJ-35, and the average size ranged from 12402 bp in AL06-06 to 29317 in JBN2301. A significant difference was observed in terms of the ratio between the total GI sequence size and host genome size, which varied from 4% in strain ATCC 7966 to 12% in NJ-35. The variation in GC content within GIs ranged from 34% to 66%, both in A. hydrophila isolate AH10 (Table 2). In general, the genomes of group 1 strains presented a higher average number of predicted GIs, genes in GIs, larger mean GI size, and ratio of GIs relative to genome size than those of group 2 (Table 2).
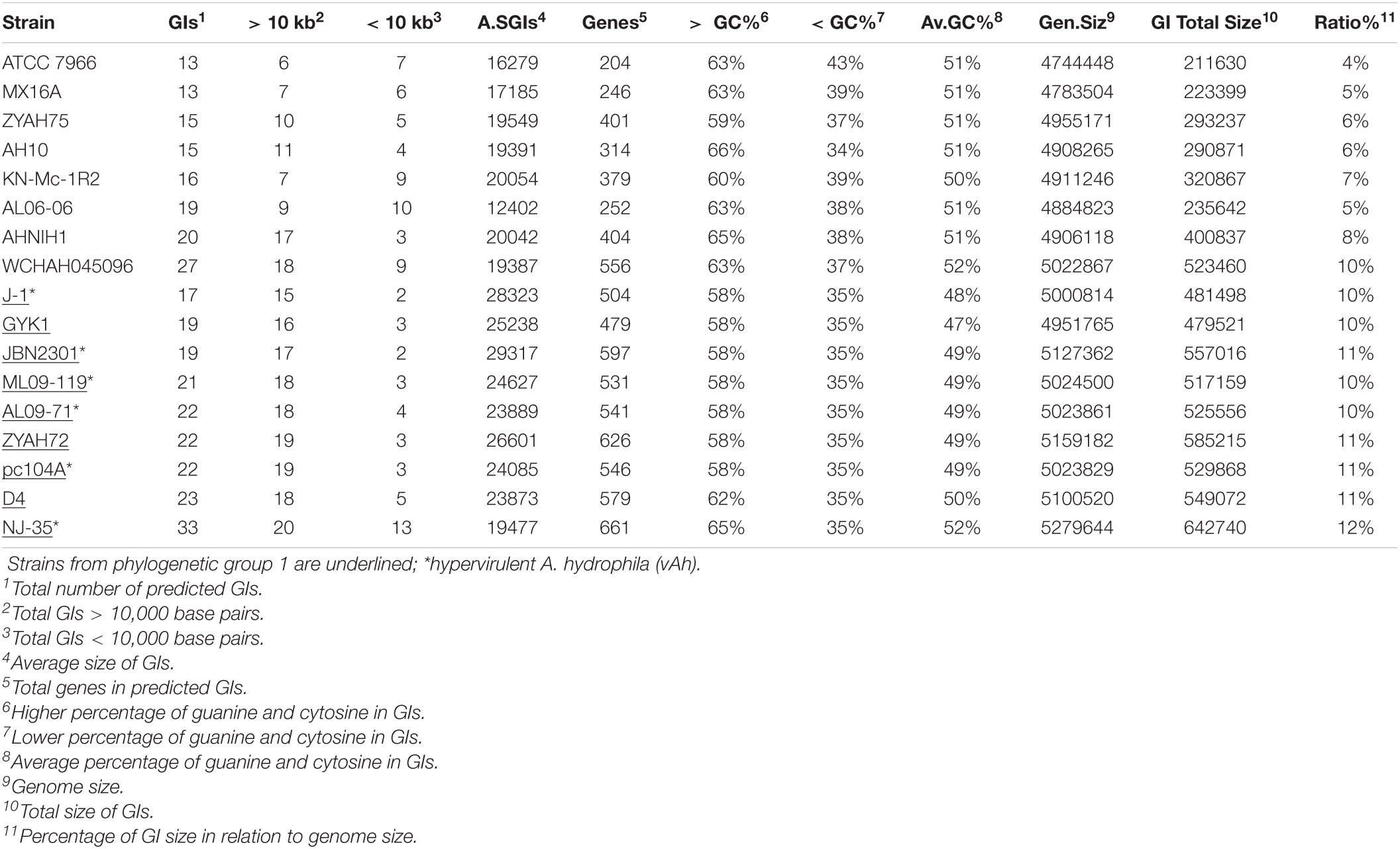
Table 2. General data of predicted genomic islands (GIs).
Altogether, the data indicated differences in relation to genome size and GC content, distribution, and characteristics of GIs of strains from groups 1 and 2. Genes identified on the predicted GIs are listed in Supplementary Table 3.
Functions Encoded by the Predicted Genomic Islands
Determination of the functions of genes in the predicted GIs was based on databases related to virulence, resistance, metabolism, drug targets, and RAST annotation. A complete list of genes with identified functions and the corresponding databases is shown in Supplementary Table 4. The main traits identified are described below.
Traits Related to Virulence
O antigen
Gene clusters encoding proteins associated with the biosynthesis of O antigen (O Ag) were identified in the GIs of all A. hydrophila strains. Ten distinct O Ag clusters were found among the 17 strains analyzed in this study, including serogroups O1, O25, and putative serogroups OX1, OX4, OX5, OX6, and OX7 (Figure 2). The O Ag cluster of strain ZYAHA75 presented identity and query coverage of 89.69% and 97%, respectively, with serogroup O18 of A. hydrophila strain PPD134/91 compared in Figure 2. The O Ag cluster of KN-Mc-1R2 contains cysN and cysD genes encoding enzymes related to sulfur metabolism, and that of strain MX16A is interrupted by genes encoding transposases, suggesting that HGT played a role in the formation of this cluster. The comparison of the OAg cluster sequence of strain MX16A showed higher homology with that of Aeromonas veroni A8-AHP, with 77% of coverage and 93% of identity. When compared to the same species, better results were observed with A. hydrophila G5380, with 50% of coverage and 94% of identity. For KN-MC-1R2, better results were obtained with Aeromonas hydrophila 3924, with 44% of coverage and 98% of identity. When compared with another species, 41% of cover and 89% of identity was found with the cluster of Aeromonas caviae KAM345.
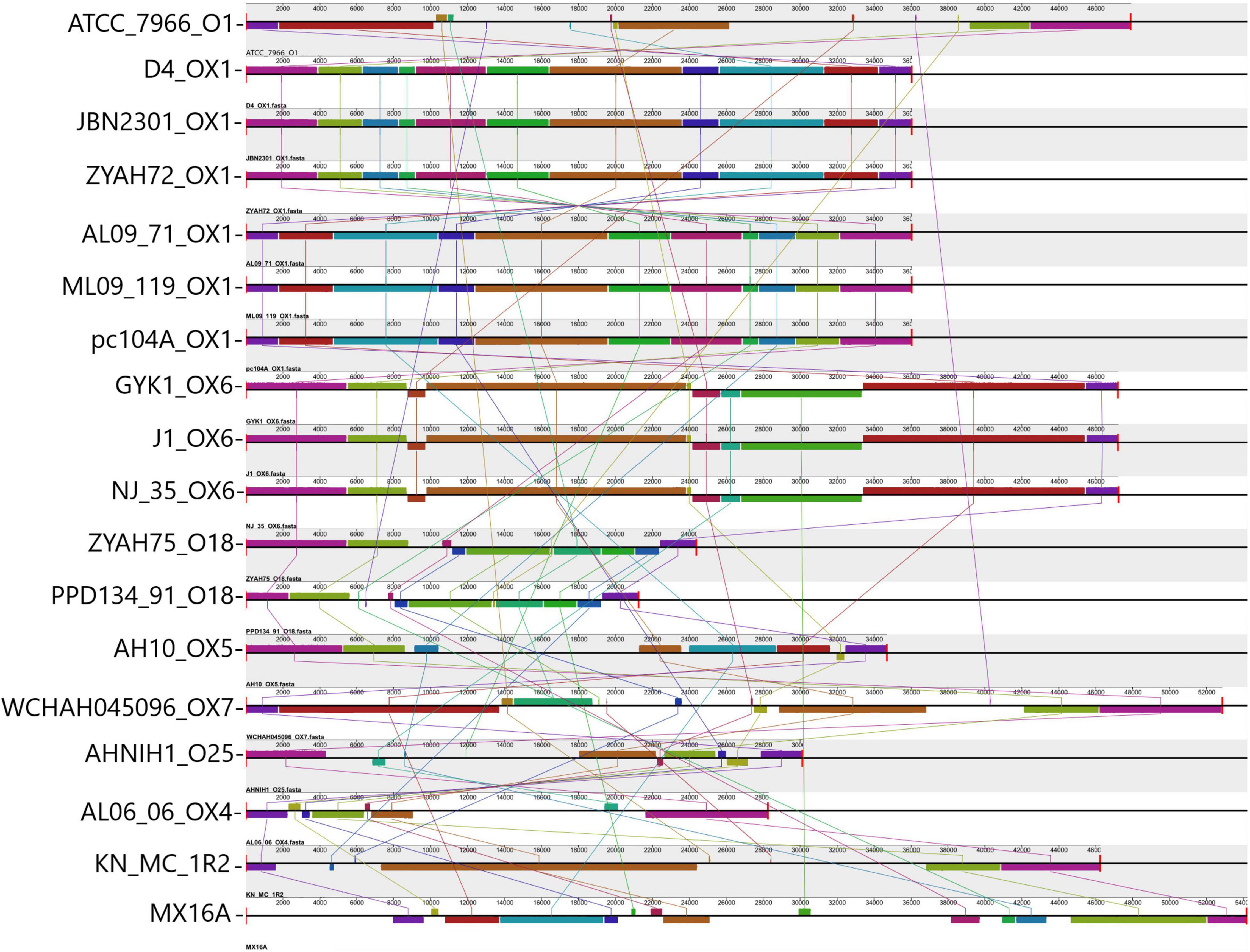
Figure 2. Comparison of O antigen biosynthesis clusters encoded in predicted genomic islands (GIs). Alignment of the O Ag cluster from 17 A. hydrophila strains analyzed and strain PPD134-91. Blocks with the same color indicate homologous regions. The scale indicates the size of the cluster. Strain designations and respective serogroups are indicated below the representation of the cluster. Serogroups identified OX1 (D4, JBN2301, ZYAH72, AL09-71, ML09-119, pc104A); OX6 (GYK1, J-1, NJ-35); O18 (comparison between PPD134/91 serogroup O18, and ZYAH75 cluster); OX5 (AH10); OX7 (WCHAH045096); O25 (AHNIN1); OX4 (AL06-06); O1 (ATCC 7966); not determined (KN-MC-1R2, MX16A). The O Ag cluster of strains pc104a, AL09-71, and ML09-119 are in the reverse orientation in the genome of the strains. The name of each strain and the respective serogroup are indicated in front of the blocks representing the O Ag cluster.
Flagella
Genes from polar flagella region 2 (flaA flaBGHJ maf-1) were identified in the predicted GIs of most strains, with additional genes found in several of them. Three types of predicted GI were observed according to the identity and number of genes encoded. One, approximately 7 kb encoding flaA flaBGHJ maf-1 and a hypothetical protein was observed in strains ATCC 7966, AL06-06, and AHNIH1. Predicted GIs of approximately 19 kb encoding the polar flagella region 2 together with other genes, including a flagellin-like, transposase, neuB, and flmD (related to polar flagella glycosylation), were found in all strains from group 1 and in AH-10. Finally, GIs of approximately 15 kb containing the polar flagella region 2 genes, neuB, flmD, and other genes distinct from those of the GI described above, were identified in strains WCHAH045096 and MX16A. In strains KN-Mc-1R2 and ZYAH75, no GIs containing flagellar region 2 genes were predicted, although these regions are present in their genomes.
Components of Type VI Secretion System
Genes encoding VgrG and Hcp, components of the expelled puncturing structure of type VI secretion system (T6SS) were found in the predicted GIs of strains AH10, AL06-06, and KN-Mc-1R2. In the first two strains, VgrG-encoding genes are present in two predicted GIs.
Additionally, certain features that may potentially be associated with virulence are encoded in predicted GIs of some strains, including ankyrin proteins (AnkB), which are encoded together with catalase (KatE) and, in most of them, with super oxide dismutase; Fic (filamentation induced by cyclic AMP) proteins, of which two types PA0574 and KPN03553 were identified; pilin Flp; the zona occludens toxin and accessory cholera enterotoxin, were sequentially encoded (Table 3). Of the genes with identified functions, some were found in genomic islets, which contain low numbers of genes and are generally poorly explored or studied. Among the genes identified in genomic islets are those encoding Fic domain protein PA0574 (strain ZYAH72, GI-18); zone occludens toxin and accessory cholera enterotoxin (strains KN-Mc-1R2, GI-11, AHNIH1, GI-17), and VgrG (strains AH10, GI15; AL06-06, GI-16).
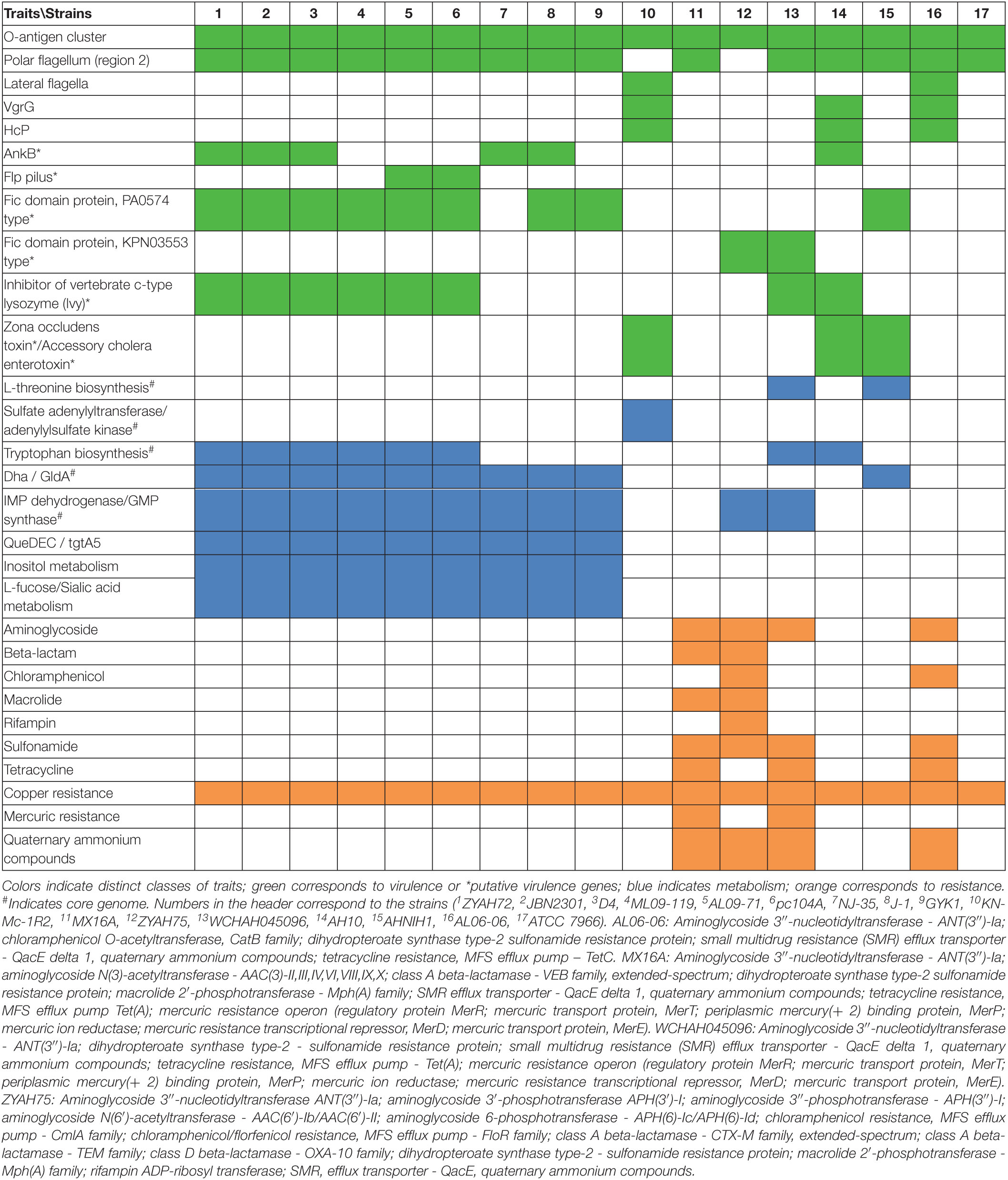
Table 3. Main traits encoded in predicted genomic islands (GIs).
Traits Related to Resistance
Genes associated with antibiotic resistance were restricted to the predicted GIs from strains AL06-06, ZYAH75, MX16A, and WCHAH045096. These genes were associated with resistance to sulfonamide, aminoglycoside, tetracycline, rifampin, chloramphenicol, and macrolides, in addition to TEM beta-lactamase, and distinct extended-spectrum beta-lactamases that are able to hydrolyze different beta-lactams.
Additionally, genes related to mercury, copper, and chromium compounds, and ammonium quaternary resistance were identified in the GIs of some strains (Table 3, Supplementary Table 4).
Traits Related to Metabolism
Genes or clusters of genes associated with metabolism were identified in the predicted GIs (Table 3).
Among them, the gene cluster dhaKLM, encoding subunits of dihydroxyacetone kinase (Dha), gldA, glycerol dehydrogenase (GldA), and dhaR, encoding the transcriptional regulator DhaR, are associated with glycerol metabolism.
Genes encoding the enzymes inosine-5′-monophosphate dehydrogenase (IMP dehydrogenase) and GMP synthase were also identified in the predicted GIs of several strains. They participate in purine metabolism in the synthesis of GMP from IMP.
Other predicted GIs contained genes related to amino acid biosynthesis. Enzymes that participate in the biosynthesis of tryptophan from chorismate are encoded by the predicted GIs from several strains. Additionally, genes encoding the bifunctional aspartate kinase/homoserine dehydrogenase I, homoserine kinase, and threonine synthase, enzymes that are part of the aspartate pathway of amino acid biosynthesis related to threonine biosynthesis, were identified in predicted GIs in a few strains.
The cysN and cysD genes encoding subunits of the enzyme sulfate adenylyltransferase, and cysC encoding adenylylsulfate kinase were found to be part of the predicted GIs of a certain strains. These enzymes participate in some steps in the process of assimilatory sulfate reduction, in which sulfate is reduced to hydrogen sulfide and then incorporated in cysteine and methionine biosynthesis.
Genes encoding 6-carboxytetrahydropterin synthase (QueD), 7-carboxy-7-deazaguanine synthase (QueE), and 7-cyano-7-deazaguanine synthase (QueC) were identified in several of the predicted GIs, and participate in the synthesis of queuosine.
Genes associated with carbohydrate metabolism, including sialic acid, fucose, and inositol metabolism, were also identified in the predicted GIs (Table 3).
Phages
PHASTER analysis indicated the presence of intact and questionable phages in strain genomes. For intact phages, the most common were Aeromonas phage phiO18P, Salmonella phage SEN8, Escherichia phage Lys12581Vzw, Shigella phage POCJ13, and for questionable phages Escherichia phage 520873 and Shigella phage POCJ13. Only strains ATCC 7966, GYK1, and MX16A did not have any phage-related regions with a score > 70. However, no intact phages were found on the predicted GIs, but several of them contained partial phage sequences (Supplementary Table 5).
Other Features Encoded in Predicted Genomic Islands
Several other features are encoded in predicted GIs, including proteins related to chemotaxis signal sensing/transduction, response regulators, and a two-component system; a type I restriction modification system, DNA repair proteins, transporters, toxin-antitoxin modules, cytochrome associated proteins, an inhibitor of vertebrate c-type lysozyme (Ivy), and flagella and pili biosynthesis-associated proteins. Finally, GIs encoding transposon-associated proteins, conjugation-related proteins, or almost exclusively hypothetical proteins, were also observed in A. hydrophila. The distribution of these features varied among strains (Supplementary Table 3).
Predicted Genomic Islands Containing Genes From Core Genome
The presence of genes from the core genome was detected in several of the predicted GIs from all strains (Supplementary Table 3), which reached 13% (strain AH10) to 27% (strain AL06-06), with an average of 18% among the A. hydrophila isolates.
However, in a few islands, a high number of genes from the core genome were detected. This was observed mainly in the predicted GIs containing the gene cluster dhaKLM gldA, which were identified as part of the core genome. In addition, genes encoding ribosomal proteins and transcription-related factors, rRNAs, RNA polymerase subunit ββ′, and some tRNAs, among others, are present in the predicted GIs. The presence of repeat regions and transposases between these genes was also observed, which may have contributed to its identification as a GI. The predicted GI containing the cluster scsABCD, associated with copper resistance, also contains a high proportion of genes from the core genome.
Other genes from the core genome in predicted GIs encode the enzyme IMP dehydrogenase, enzymes associated with biosynthesis of tryptophan or threonine, and the enzyme adenylylsulfate kinase. These and other genes related to the core genome are highlighted in blue in Supplementary Table 3.
Shared Genomic Islands
Several strains share predicted GIs, as indicated by comparative analysis performed considering at least 75% amino acid sequence identity and coverage as criteria (Figure 3; Supplementary Table 6). Some of the predicted GIs were shared among most of the strains, while others were shared only among those of phylogenetic group 1 or group 2. Strains from group 1 shared all or most of their predicted GIs, while those from group 2 shared only a few (Figure 3; Supplementary Table 6).
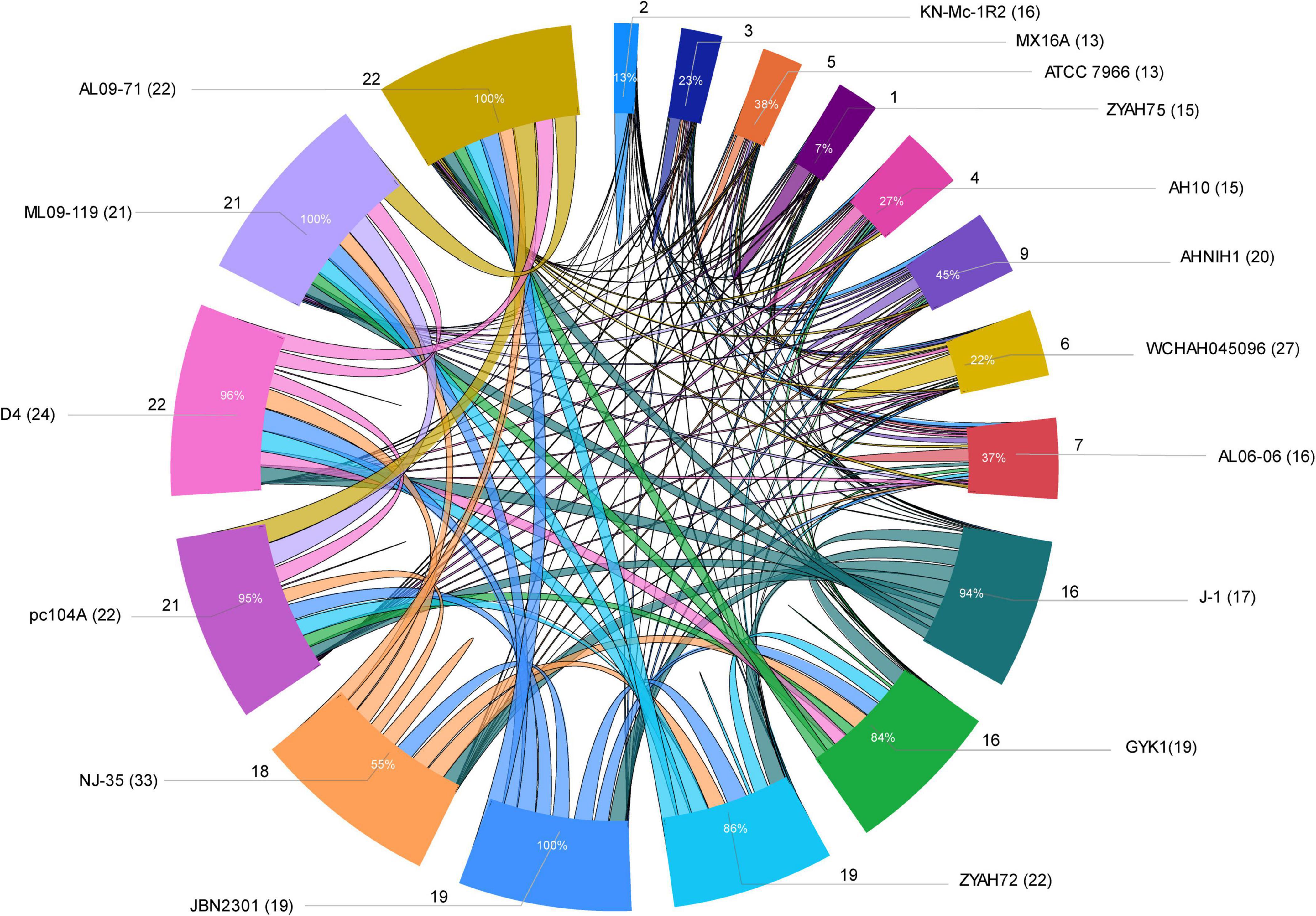
Figure 3. Sharing of predicted genomic islands (GIs) among A. hydrophila strains. Ratio of GIs shared among strains. Distribution of the data is presented as the total number of predicted GIs, the total number of shared GIs, and their related percentages. The circle indicates the predicted GIs shared proportionally by the size of each arc; the names of the strains are indicated outside the circle with the number of predicted GIs in parentheses; the number of GIs shared is indicated by the lines and the proportion inside the arc; lines connecting strains indicate sharing of predicted GIs.
The main predicted GIs with identified functions shared among two or more strains from both groups included those encoding ScsABCD, DhaKLM/GldA, and enzymes associated with tryptophan biosynthesis/Ivy. Of these, the first is the most widespread and is present in all strains except KN-Mc-1R2. This strain also contains the cluster ScsABCD; however, it is encoded in a larger GI that did not meet the criteria used in this study to define a shared island with other strains. The predicted GIs with characterized functions shared only among strains from group 1 included those encoding inositol and sialic acid/L-fucose metabolism; QueDEC, the O antigen cluster for putative OX1 or OX6 serogroups, and Fic PA0574 (Supplementary Tables 3,6). Additionally, predicted GIs encoding toxin/antitoxin systems, phages or hypothetical proteins are shared among them. In group 2, predicted GIs encoding the zona occludens toxin/accessory cholera enterotoxin and flagellar regions 1 and 2 were the most common shared GIs (Supplementary Tables 3,6).
Discussion
In this study, complete genomes of A. hydrophila strains were analyzed using the Island Viewer 4 bioinformatics tool for GI prediction in order to determine the distribution of GIs and their contributions in such strains, however, while some of the features encoded have functions defined, others are putative and have to be experimentally demonstrated in Aeromonas. A phylogenetic analysis performed with SweeP software showed that two strains identified as A. hydrophila (4AK4 and YL17) were misidentified at species level. These findings are in agreement with the studies by Beaz-Hidalgo et al. (2015) and Moriel et al. (2021) which indicated several problems with the taxonomic affiliation of Aeromonas genomes deposited at the NCBI database. These findings show the relevance of using complete genomes for taxonomy of Aeromonas. The A. hydrophila strains were classified into two distinct phylogenetic groups (Figure 1). Group 1 contained strains isolated from diseased fish, except pc104A, which was recovered from the soil of a catfish pond that experienced an epidemic outbreak of septicemia caused by Aeromonas (Pang et al., 2015). Most of the strains were related to epidemic outbreaks of motile Aeromonas septicemia in catfish that occurred in the United States (ML09-119, AL09-71, and pc104A) or carp in China (NJ-35, JBN2301, and J1), and share a recent common ancestor, as shown by comparative genomic analyses (Hossain et al., 2014). These strains were previously classified as hypervirulent A. hydrophila (vAh) based on phenotypic and genotypic tests or in silico analysis for pathotype-specific PCR of vAh isolates (Pang et al., 2015; Rasmussen-Ivey et al., 2016a).
Strains ZYAH72, D4 and GYK1, isolated from diseased fish in China, are also part of phylogenetic group 1. These United States and Chinese strains were separated into distinct subclades of group 1 (Figure 1), what is in agreement with the data of Pang et al. (2015).
Strains from phylogenetic group 2 were recovered from different sources and were also separated into two subclades based on their geographical origins (Figure 1), and the South Korean strain grouped together with the United States strains. Three of these strains (AH10, ATCC 7966, and AL06-06) were previously studied and considered to be non-virulent A. hydrophila (non-vAh) in relation to the hypervirulent vAh strains recovered from fish disease outbreaks (Pang et al., 2015; Rasmussen-Ivey et al., 2016a). Our data confirmed the results of other phylogenetic studies indicating the separation of vAh from non-epidemical strains (Pang et al., 2015; Rasmussen-Ivey et al., 2016a).
Strains from groups 1 and 2 differed in several characteristics, including genome size, number of predicted GIs, GI average size, and ratio of GIs relative to genome size (Table 2), in which strains from group 1 generally present greater numbers. Genomic GC content also varied among these strains, from 60.50% to 60.90% in group 1 and from 61% to 61.60% in group 2. Strains from groups 1 and 2 also differed in terms of the functions encoded on the predicted GIs, although some features were common among them (Table 3).
The distribution of predicted GIs containing the main functions identified, and some features whose function have yet to be demonstrated in A. hydrophila, varied among the strains (Table 3), while some were common and other were encoded in a few strains. Among the identified characteristics are clusters for the synthesis of O Ag. O Ag, the most surface-exposed part of the lipopolysaccharide, a component of the outer membrane of Gram-negative bacteria, mediates pathogenicity by protecting infecting bacteria from serum complement killing and phagocytosis. It consists of repeating oligosaccharide subunits whose variability confers immunological specificity (Tomás, 2012). Several O Ag gene cluster types have been described in A. hydrophila (Hossain et al., 2013; Pang et al., 2015; Cao et al., 2018) and previously associated with GIs (Hossain et al., 2013; Pang et al., 2015). Gene clusters for O Ag were found in the predicted GIs of all strains; however, they differed in their gene content and were related to 10 distinct serogroups (Figure 2).
Thirteen of the strains analyzed in this study had their O Ag gene clusters previously determined (Hossain et al., 2013; Pang et al., 2015; Cao et al., 2018).
The putative serogroup OX1 was proposed for strains ML09-119, AL09-71, pc104a, JBN2301, and D4 strains (Cao et al., 2018). Strain ZYAH72, which had no previously analyzed O Ag cluster, shared the same cluster with the strains above (Figure 2). Thus, it should also be included in the putative OX1 serogroup. Strains NJ-35 and GYK1 were classified as putative serogroups OX6 (Cao et al., 2018). According to our data, strain J-1 should also be included in this serogroup because it shares the same O Ag biosynthesis cluster. This is in agreement with Pang et al. (2015), who showed that J-1 and NJ-35 have identical O Ag gene clusters.
All other strains had unique O Ag clusters. In contrast to the vAh fish pathogens that presented putative OX1 or OX6 O Ag clusters, strains AL06-06 and AH10 were also recovered from diseased fish but were considered as non-vAh (Rasmussen-Ivey et al., 2016a) and presented clusters OX4 and OX5, respectively (Cao et al., 2018). Strain ATCC 7966 belongs to the O1 serogroup (Seshadri et al., 2006), AHNIH1 to O25, WCHAH045096 to OX7 (Cao et al., 2018). The ZYAHA75 O cluster is similar to O18, and those of MX16A and KN-MC-1R2 were not determined (Figure 2).
Thus, no O Ag gene cluster was shared among strains from phylogenetic groups 1 and 2. All strains from group 2 exhibited unique O Ag clusters. Of the 10 distinct serogroups detected, only OX1 and OX6 were shared, and the identification of these clusters might be an alert for potential virulent strains related to outbreaks of epidemic diseases in fish.
Several genes associated with polar flagella biogenesis and chemotaxis were identified in the predicted GIs of most strains (Table 3; Supplementary Table 3). Mesophilic Aeromonas have a polar flagellum for swimming in liquid, and some strains can also express multiple inducible lateral flagella for swarming over viscous environments or surfaces (Janda and Abbott, 2010). The polar flagellum genes are clustered in five distinct regions, whereas lateral flagella genes are organized in only one cluster (Tomás, 2012). Beyond their role in motility, in Aeromonas flagella act as adhesins for human intestinal enterocytes and in biofilm formation (Kirov et al., 2004), and are essential for adhesion and the ability to invade fish cell lines (Beaz-Hidalgo and Figueras, 2013). In the current study, GIs containing gene clusters for polar flagella region 2 (flaA flaBGHJ maf-1) (Canals et al., 2006) were predicted. However, the data indicated that gene content downstream of polar flagella region 2 is heterogeneous in different strains (Supplementary Table 3) and that genes associated with flagella glycosylation, such as flmD and neuB (Tabei et al., 2009; Wilhelms et al., 2012; Fulton et al., 2015) are adjacent to region 2 in certain A. hydrophila isolates. These data are in agreement with a recent study (Forn-Cuní et al., 2021) that showed that pse/flm genes (flmA flmB neuA flmD neuB) related to polar flagellin glycosylation are clustered in highly polymorphic GIs with three main genomic glycosylation islands identified.
Groups I and III contain almost exclusively the pse genes (flmA flmB neuA flmD neuB). Aeromonas belonging to these groups have flagellins modified with a single monosaccharide pseudaminic acid derivative (Forn-Cuní et al., 2021). Strains from phylogenetic group 1 and strain AH-10 belong to the glycosylation genomic island group I, while MX16A and WCHA045096 were included in group III according to Forn-Cuní et al. (2021).
The glycosylation GI group II contains other genes between the pse/flm genes, and are present in strains that modify polar flagellins with heterogeneous glycan moieties. Strains ATCC 7966, AL06-06, and AHNIHI are part of this group together with KN-Mc-1R2 and ZYAHA75 (Forn-Cuní et al., 2021). Flagellin glycosylation is essential for flagellar function (Tabei et al., 2009) and polar flagellar glycan composition can differ between A. hydrophila strains (Wilhelms et al., 2012; Fulton et al., 2015; Forn-Cuní et al., 2021). Because Aeromonas flagella have several roles in pathogenesis (Kirov et al., 2004; Beaz-Hidalgo and Figueras, 2013), based on data showing that strains ML09-119 and AL09-71 (pathogens of catfish), AH10, J-1, JBN2301, and NJ 35 (pathogen from carp) share the same predicted GIs related to flagellar region 2, and results from Forn-Cuní et al. (2021), showing that they belong to the glycosylation GI group I, we speculate that flagellin glycosylation patterns may favor interactions with determined hosts.
VgrG and Hcp play roles in Aeromonas virulence, influencing bacterial motility, protease production, and biofilm formation, inducing host cell toxicity, apoptosis, and activation of macrophages (Suarez et al., 2008, 2010a,b; Sha et al., 2013). They are components of the T6SS which in addition to virulence plays roles in inter-bacterial competition. T6SS is encoded within gene clusters containing the core genes of the secretion machinery, however, additional copies of hcp and vgrG can be found outside the main cluster (Cianfanelli et al., 2016; Moriel et al., 2021), which explains the presence of these genes in two distinct predicted GIs in certain strains (Supplementary Table 3). However, a search in the genome showed that strain AL06-06 does not have the complete T6SS cluster, differently from strains AH10 and KN-Mc-1R2. This is in agreement with Tekedar et al. (2019) which showed that this strain have only three components of T6SS, hcp, tssH and vgrG. In their study they also showed that virulent strains including ML09-119, AL09-71 and pc104A also have only these T6SS components, and that hcp1 and vgrG1 contribute for virulence of the ML09-119 in catfish.
Some other genes with functions identified in predicted GIs (Supplementary Table 3) are associated with virulence in other bacteria, but their roles in Aeromonas have yet to be determined. Among them are the zonula occludens toxin and the accessory cholera enterotoxin, which have been described in Vibrio cholerae and have the ability to affect intestinal function in rabbits (Trucksis et al., 1993; Fasano et al., 1997). Another is the ankyrin protein, AnkB. In plant-pathogens Xanthomonas oryzae pv. oryzae and X. oryzae pv. oryzicola, AnkB participates in biofilm formation, swimming ability, exopolysaccharide production, and defense against oxidative stress. In these bacteria, the ankB gene is located 58 bp downstream of catB, which encodes the enzyme catalase. AnkB affects catB gene expression, catalase activity, and sensitivity to H2O2 (Pan et al., 2018). In A. hydrophila, the ankB gene is in tandem with katE, which encodes catalase, and in most strains with a gene encoding a superoxide dismutase precursor. Although A. hydrophila is an opportunistic pathogen in animals, this led us to speculate regarding its roles in defense against oxidative stress, as shown in X. oryzae.
Fic proteins are widespread in bacteria and are often found encoded in GIs. They may play a role as toxins secreted from pathogens, or as the toxin component of toxin/antitoxin modules. However, the great majority of Fic proteins have unknown functions (Veyron et al., 2018).
Several genes from the core genome were found in the predicted GIs (Supplementary Table 3), which should be further considered. Except for the scsABCD locus associated with copper resistance, which was present in the predicted GIs of all strains analyzed, the distribution of GIs encoding resistance-associated features was limited to four strains in phylogenetic group 2 (Table 3). The full scs operon appears to be limited to some Enterobacteriaceae (Salmonella enterica, Serratia proteamaculans, Citrobacter koseri, and Klebsiella pneumoniae), Aeromonas hydrophila, and Photobacterium profundum (Subedi et al., 2019). The scsABCD locus was originally identified in S. enterica Typhimurium, and is required to deal with copper and H2O2 stress, helping to increase Salmonella survival under severe copper and oxidative stress, hostile conditions encountered by the pathogen during its intracellular survival (López et al., 2018) thus, contributing to virulence. However, copper may also be found in water (Domek et al., 1984). Considering that Aeromonas are ubiquitous in aquatic habitats and are considered as opportunistic pathogens (Janda and Abbott, 2010), potential roles of scsABCD may contribute to fitness in environment and virulence in the host.
In A. hydrophila, scsSBCD is located in the predicted GIs distinct from those encoding features associated with resistance (Supplementary Table 3).
Aeromonas have intrinsic resistance to some β-lactams due to the production of one to three inducible β-lactamases. The resistance phenotypes vary in relation to the species according the production of one to three inducible β-lactamases, with five basic phenotypes described (Fosse, 2010). In A. hydrophila three classes of β-lactamases, a class B metallo-β-lactamase, a class C cephalosporinase, and a class D oxacillinase were described (Fosse, 2010; Janda and Abbott, 2010), conferring to the species the penicillinase-cephalosporinase-carbapenemase phenotype exhibiting resistance to ampicillin, amoxicillin, ticarcillin, cephalothin, and profile susceptible or intermediate to imipenem (Fosse, 2010). We looked for genes encoding these beta lactamases and subclass B2 metallo-β-lactamase (CphA family, or ImiH or ImiS), cephalosporin-hydrolyzing class C β-lactamase (CepS or CepH) and OXA-12 family class D β-lactamase (AmpH/OXA-724 or AmpS/OXA-725 or OXA-726) were identified in the chromosome of all 17 strains. Additionally, a tetracycline efflux MFS transporter TetE was identified in the chromosome of 2 strains.
Aeromonas are usually susceptible to aminoglycosides, tetracyclines, chloramphenicol, sulfonamides, trimethoprim, nitrofurans, nalidixic acid, fluoroquinolones (Fosse, 2010), carbapenems, third- and fourth-generation cephalosporins, macrolides, monobactams and extended spectrum penicillins (Janda and Abbott, 2010; Fernández-Bravo and Figueras, 2020). However, strains presenting resistance to one or more of these antibiotics are common in clinical and environmental strains (Aravena-Román et al., 2012; Esteve et al., 2015). Studies on molecular antibiotic resistance have shown that class 1 integrons, involved in intraspecific and interspecific dissemination of resistance, and large plasmids are associated to resistance to several antibiotics in Aeromonas (Nguyen et al., 2014; Hughes et al., 2016).
Aeromonas hydrophila multidrug-resistant strains are frequent, mainly among clinical isolates (Esteve et al., 2015; Hughes et al., 2016). Among the strains analyzed, the following contains plasmids, as informed at the NCBI database: JBN2301 (3 plasmids), D4 (4 plasmids), AHNIH1 (1 plasmid), AL06-06 (3 plasmids) and WCHAH045096 (6 plasmids). Plasmids from strains JBN2301and AL06-06 do not contain resistance genes. In strain D4, only one of the plasmids contain a gene encoding a quinolone resistance pentapeptide repeat protein QnrS2. The plasmid of strain AHNIH1, contains several genes encoding for resistance, as described (Hughes et al., 2016). Includes three class A beta-lactamases (KPC-2, SHV-12 and CARB-12) and genes encoding for resistance to aminoglycosides, chloramphenicol, fluoroquinolones, macrolides, sulfonamide and trimethoprim. Three of the plasmids of WCHAH045096 carry resistance genes. They are associated to the resistance to several classes of antibiotics including aminoglycosides, sulfonamide, trimethoprim, quinolone, tetracycline and carbapenem.
Predicted GIs containing antibiotic resistance associated genes had heterogeneous distribution among the A. hydrophila (Supplementary Table 4), possibly reflecting horizontal gene transfer and selection with the exposure to antibiotics in their environments. These predicted GIs were identified in four strains, recovered from water, sewage, fish or human, indicating that antibiotic resistance associated with GIs is spread in clinical and environmental strains. Most of these predicted GIs also encode transposases, integrases, and mobile elements, and one of them also includes proteins related to conjugation (Supplementary Table 3). In the strain AL06-06, a predicted GI encodes for aminoglycoside, sulfonamide, chloramphenicol and tetracycline resistance. The predicted GI of WCHAH045096 encodes for resistance to the same antibiotics described above, and contains genes for several proteins associated with mercury resistance. The predicted GI of strain MX16A encodes antibiotic resistance for tetracycline, aminoglycoside, chloramphenicol, sulfonamide, macrolide and beta-lactams e proteins related to mercury resistance. Strain ZYAH75 has three predicted GIs containing resistance genes. One of them contain genes for resistance to aminoglycoside and sulfonamide together with proteins for mercury resistance. This predicted GI is a mosaic of proteins associated with mobile elements, it encodes conjugative transfer proteins TrbJ, TrbK and TrbK, IncP-type oriT binding proteins TraJ and TraK, together with integrase, transposase, IS and mobile element proteins. Genes for chloramphenicol and aminoglycoside resistance are encoded in two other islands, which contains additionally genes encoding class A extended-spectrum beta-lactamase CTM-3, class A broad-spectrum beta-lactamases TEM-1, and gene for sulfonamide resistance; or genes for resistance to macrolides and quinolone, oxacillin-hydrolyzing class D beta-lactamase OXA-10, and also proteins associated with mercury resistance. Integron integrase Intl1 are encoded in two of the predicted GI of ZYAH75, and those from strains WCHAH045096 and MX16A. In several of these predicted GIs there is more than one gene encoding for aminoglycoside modifying enzymes, and for sulfonamide resistance. Therefore, in addition to the intrinsic resistance characteristic of the species, some strains analyzed contain determinants for resistance to tetracycline in the chromosome (2 strains); plasmids carrying genes for resistance to one and up to six distinct classes of antibiotics, and even more than one plasmid related to resistance, and predicted GIs in which several genes of resistance are encoded, the most common are those related to the aminoglycoside and sulfonamide resistance. Aeromonas are ubiquitous in aquatic environments and widely distributed, being found in every environmental niche where bacterial ecosystems exist, facilitating the contact with humans (Janda and Abbott, 2010). Thus, the presence of GIs carrying resistance genes in Aeromonas may impact the fitness of the bacteria, facilitate the resistance dissemination, and spread of resistant strains in several environments.
Some of the predicted GIs shared among strains from phylogenetic group 1 encode metabolic pathways for utilization of myo-inositol and L-fucose/sialic acid, which were previously described in GIs of A. hydrophila strains NJ-35, J-1, ML09-119, AL09-71, and pc104A (Pang et al., 2015). In the current study, these features were also found in the predicted GIs of other strains of group 1. These pathways may be linked to the full virulence of fish disease epidemics A. hydrophila (Pang et al., 2015), suggesting that these GIs may be related to fitness or enhancing adaptation to hosts and competitiveness.
Other predicted GIs common to vAh contain genes encoding QueD, QueE, and QueC (Supplementary Table 3), which participate in the synthesis of PreQ0, a precursor of queuosine- (Q) tRNA modification in bacteria. PreQ0 is reduced to PreQ1 and inserted in the G residue at position 34 of tRNAs with GUN anticodons by tRNA guanosine transglycosylase (bTGT); however, the enzyme can also use PreQ0 when PreQ1 is absent (Thiaville et al., 2016). However, predicted A. hydrophila GIs encoding QueDEC also encode TgtA5, a variant of TGT that has divergent features from bTGT, helicases, and proteins involved in DNA repair (Supplementary Table 3). Similar clusters have been described in a GI of Salmonella enterica serovar Montevideo and by comparative genomic analysis in several other bacteria, including A. hydrophila ML09-119. In S. enterica serovar Montevideo, Kineococcus radiotolerans, Comamonas testosteroni, and Sphingopyxis alaskensis, clustering of tgtA5 and preQ0 synthesis genes is involved in inserting 7-deazapurine derivatives into DNA, suggesting a new and complex system of DNA modification (Thiaville et al., 2016). The SAM-dependent methyltransferase HI0095 (UbiE paralog), which is associated with virulence in vAh (Rasmussen-Ivey et al., 2016a) is also encoded in this predicted GI. Thus, the roles of these genes in A. hydrophila DNA modification and/or virulence have to be determined.
No distinctive characteristics of strains from group 2 were identified, and most isolates shared only a small number of predicted GIs (Supplementary Table 6), suggesting a high degree of strain diversity. Of note, several of these strains have multiple determinants of antimicrobial resistance encoded by their predicted GIs. Group 2 contains only eight strains; however, they may better reflect the diversity of GIs in A. hydrophila because previous studies have shown that most strains from group 1 are highly related and constitute a clonal group (Hossain et al., 2014; Pang et al., 2015; Rasmussen-Ivey et al., 2016a).
Addendum
At the end of this study, new complete genomes of A. hydrophila were available in the NCBI database reaching a total of 40, of which 19 were previously analyzed. Information on the new 21 strains is showed in Supplementary Table NST1. A complementary phylogenetic analysis including all 40 genomes indicated that four of the strains classified as A. hydrophila were misidentified at species level; two are the new strains NEB724 and B11, and YL17 and 4AK4 which were previously excluded from our study (Supplementary Figure 1). The 36 A. hydrophila were separated in two main branches (Supplementary Figure 1). However, one branch contains only new strains isolated from chicken (4 strains). The other branch includes subgroups containing the strains previously classified in group 1 (Vah) plus new isolates from snakes (2), fish (1) and without info (1); and the strains from previous group 2, in which new isolates from water (4), fish (4), human (1), and no source info (2) were included (Supplementary Figure 1). The raw data of Island Viewer4 analysis performed with the new genomes are showed in Supplementary Table NST2. Among the 19 new genomes of A. hydrophila, 735 GIs were predicted, however, these GIs were not manually curated. Gene clusters for O Ag biosynthesis were identified in predicted GIs. The relationship among the O antigen gene clusters of the 36 A. hydrophila was analyzed through nucleotide sequences alignment performed using the Clustal Omega Web Server3, with “default” parameters and the software IQ-TREE - Efficient Tree Reconstruction (version 1.6.12 – Windows 64; Nguyen et al., 2015, Mol. Biol. Evol., 32:268–274, 2015), which was used to build the phylogenetic tree of the clusters. The parameters used were “default” for ModelFinder, with bootstrap and maximum likelihood values. Results are indicated in the (Supplementary Figure 2), which indicates that two of the new strains present O Ag cluster already identified among the strains previously analyzed. Strain LHW39 presents the OX1cluster which was observed among some of the VAh, and strain HX-3 the OX-5 cluster, identified in strain AH10. Fourteen O Ag cluster, apparently distinct of those identified among strains previously analyzed, were found in GIs of 17 new genomes (Supplementary Figure 2); similar O Ag clusters were found in GIs of strains 23-C-23 and WCX23-1; Brac6 and ONP3-1; GSH8-2 and WP8-S18.ESBL02. Predicted GIs containing resistance genes were found in 5 strains (Supplementary Table NST3), and are related with resistance to tetracycline (4 strains), beta-lactam (class A extended-spectrum beta-lactamase CTX-M-14; 1 strain), macrolide (2 strains), aminoglycosides and sulfonamide (1 strain). The number of resistance features found in the predicted GIs of the new genomes varies from 1 to 3, in contrast with those observed in the previous analyzes which range from 3 to 9. These preliminary results confirm the previous analyzes indicating that some features, such as O Ag clusters, are associated to GIs in all strains. Features encoded on predicted GIs of the new genomes confirms the contribution of these elements for the diversity and antimicrobial resistance of A. hydrophila.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
AS designed the study, collected and data analysis, and manuscript writing. JM did the funding acquisition, data evaluation, and original draft writing. RR did the conception and design of the manuscript. CD did the phylogeny analysis. DJ developed scripts and contributed to data analysis. CF-P did the data analysis and manuscript writing. GP reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The funding was provided by Coordination of Improvement of Higher Level Personnel (CAPES/Brazil); Computational Biology Project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the Graduate Programs in Clinical Analysis; Bioinformatics; Biochemistry and Molecular Biology of the Federal University of Paraná for the support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.769380/full#supplementary-material
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance
- ^ https://github.com/Microsoft/PowerBI-visuals-chord
- ^ https://www.ebi.ac.uk/Tools/msa/clustalo/
References
Aravena-Román, M., Inglis, T. J. J., Henderson, B., Riley, T. V., and Changa, B. J. (2012). Antimicrobial susceptibilities of Aeromonas strains isolated from clinical and environmental sources to 26 antimicrobial agents. Antimicrob. Agents Chemother. 56, 1110–1112. doi: 10.1128/AAC.05387-11
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Beaz-Hidalgo, R., and Figueras, M. J. (2013). Aeromonas spp. whole genomes and virulence factors implicated in fish disease. J. Fish Dis. 36, 371–388. doi: 10.1111/jfd.12025
Beaz-Hidalgo, R., Hossain, M. J., Liles, M. R., and Figueras, M.-J. (2015). Strategies to avoid wrongly labelled genomes using as example the detected wrong taxonomic affiliation for Aeromonas genomes in the GenBank database. PLoS One 10:e0115813. doi: 10.1371/journal.pone.0115813
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group, Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 10:8365. doi: 10.1038/srep08365
Buchrieser, C., Prentice, M., and Carniel, E. (1998). The 102-kilobase unstable region of Yersinia pestis comprises a high-pathogenicity island linked to a pigmentation segment which undergoes internal rearrangement. J. Bacteriol. 180, 2321–2329.
Canals, R., Ramirez, S., Vilches, S., Horsburgh, G., Shaw, J. G., Tomás, J. M., et al. (2006). Polar flagellum biogenesis in Aeromonas hydrophila. J. Bacteriol. 188, 542–555. doi: 10.1128/JB.188.2.542-555.2006
Cao, H., Wang, M., Wang, Q., Xu, T., Du, Y., Li, H., et al. (2018). Identifying genetic diversity of O antigens in Aeromonas hydrophila for molecular serotype detection. PLoS One 13:e0203445. doi: 10.1371/journal.pone.0203445
Che, D., Wang, H., Fazekas, J., and Chen, B. (2014). An accurate genomic island prediction method for sequenced bacterial and archaeal genomes. J. Proteomics Bioinform. 7, 214–221. doi: 10.4172/jpb.1000322
Cianfanelli, F. R., Monlezun, L., and Coulthurst, S. J. (2016). Aim, load, fire: the type VI secretion system, a bacterial Nanoweapon. Trends Microbiol. 24, 51–62.
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
De Pierri, C. R., Voyceik, R., Santos de Mattos, L. G. C., Kulik, M. G., Camargo, J. O., Repula de Oliveira, A. M., et al. (2020). SWeeP: representing large biological sequences datasets in compact vectors. Sci. Rep. 10:91.
Dieckmann, M. A., Beyvers, S., Nkouamedjo-Fankep, R. C., Hanel, P. H. G., Jelonek, L., Blom, J., et al. (2021). EDGAR3.0: comparative genomics and phylogenomics on a scalable infrastructure. Nucleic Acids Res. 49, W185–W192.
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Domek, M. J., LeChevallier, M. W., Cameron, S. C., and McFeters, G. A. (1984). Evidence for the role of copper in the injury process of coliform bacteria in drinking water. Appl. Environ. Microbiol. 48, 289–293. doi: 10.1128/aem.48.2.289-293.1984
Ester, M., Kriegel, H., Sander, J., and Xu, X. (1996). “A density-based algorithm for discovering clusters in large spatial databases with noise,” in Proceedings of the KDD-1996 Second International Conference on Knowledge Discovery and Data Mining, (Menlo Park, CA: AAAI Press), 226–231.
Esteve, C., Alcaide, E., and Giménez, M. J. (2015). Multidrug-resistant (MDR) Aeromonas recovered from the metropolitan area of Valencia (Spain): diseases spectrum and prevalence in the environment. Eur. J. Clin. Microbiol. Infect. Dis. 34, 137–145. doi: 10.1007/s10096-014-2210-z
Fasano, A., Uzzau, S., Fiore, C., and Margaretten, K. (1997). The enterotoxic effect of Zonula occludens Toxin on rabbit small intestine involves the paracellular pathway. Gastroenterology 112, 839–846.
Fernández-Bravo, A., and Figueras, M. J. (2020). An update on the genus Aeromonas: taxonomy, epidemiology, and pathogenicity. Microorganisms 8:129. doi: 10.3390/microorganisms8010129
Forn-Cuní, G., Fulton, K. M., Smith, J. C., Twine, S. M., Mendoza-Barberà, E., Tomás, J. M., et al. (2021). Polar flagella glycosylation in Aeromonas: genomic characterization and involvement of a specific glycosyltransferase (Fgi-1) in heterogeneous flagella glycosylation. Front. Microbiol. 11:595697. doi: 10.3389/fmicb.2020.595697
Fosse, T. (2010). “Chapter 42. Aeromonas, vibrio and plesiomonas,” in Antibiogram, eds P. Courvalin, R. Leclercq, and L. B. Rice (Paris: ESKA Publishing, ASM Press), 509–518.
Fulton, K. M., Mendoza-Barberá, E., Twine, S. M., Tomás, J. M., and Merino, S. (2015). Polar glycosylated and lateral non-glycosylated flagella from Aeromonas hydrophila strain AH-1 (Serotype O11). Int. J. Mol. Sci. 16, 28255–28269. doi: 10.3390/ijms161226097
Geer, L. Y., Marchler-Bauer, A., Geer, R. C., Han, L., He, J., He, S., et al. (2010). The NCBI BioSystems database. Nucleic Acids Res. 38, D492–D496. doi: 10.1093/nar/gkp858
Grim, C. J., Kozlova, E. V., Ponnusamy, D., Fitts, E. C., Sha, J., Kirtley, M. L., et al. (2014). Functional genomic characterization of virulence factors from necrotizing fasciitis-causing strains of Aeromonas hydrophila. Appl. Environ. Microbiol. 80, 4162–4183. doi: 10.1128/AEM.00486-14
Hacker, J., Bender, L., Ott, M., Wingender, J., Lund, B., Marre, R., et al. (1990). Deletions of chromosomal regions coding for fimbriae and hemolysins occur in vitro and in vivo in various extra intestinal Escherichia coli isolates. Microb. Pathog. 8, 213–225. doi: 10.1016/0882-4010(90)90048-U
Hossain, M. J., Sun, D., McGarey, D. J., Wrenn, S., Alexander, L. M., Martino, M. E., et al. (2014). An Asian origin of virulent Aeromonas hydrophila responsible for disease epidemics in United States-farmed catfish. mBio 5:e00848-14. doi: 10.1128/mBio.00848-14
Hossain, M. J., Waldbieser, G. C., Sun, D., Capps, N. K., Hemstreet, W. B., et al. (2013). Implication of lateral genetic transfer in the emergence of Aeromonas hydrophila isolates of epidemic outbreaks in channel catfish. PLoS One 8:e80943. doi: 10.1371/journal.pone.0080943
Hsiao, W., Wan, I., Jones, S. J., and Brinkman, F. S. L. (2003). IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19, 418–420. doi: 10.1093/bioinformatics/btg004
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics (Oxf. Engl.) 26, 680–682. doi: 10.1093/bioinformatics/btq003
Hughes, H. Y., Conlan, S. P., Lau, A. F., Dekker, J. P., Michelin, A. V., Youn, J. H., et al. (2016). Detection and whole-genome sequencing of carbapenemase-producing Aeromonas hydrophila isolates from routine perirectal surveillance culture. J. Clin. Microbiol. 54, 1167–1170. doi: 10.1128/JCM.03229-15
Janda, J. M., and Abbott, S. L. (1998). Evolving concepts regarding the genus Aeromonas: an expanding panorama of species, disease presentations, and unanswered questions. Clin. Infect. Dis. 27, 332–344.
Janda, J. M., and Abbott, S. L. (2010). The genus Aeromonas : taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 23, 35–73. doi: 10.1128/CMR.00039-09
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Juhas, M., van der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136
Kanehisa, M., and Sato, Y. (2020). KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 29, 28–35. doi: 10.1002/pro.3711
Kirov, S. M., Castrisios, M., and Shaw, J. G. (2004). Aeromonas flagella (Polar and Lateral) are enterocyte Adhesins that contribute to biofilm formation on surfaces. Infect. Immun. 72, 1939–1945.
Li, W., and Wang, A. (2021). Genomic islands mediate environmental adaptation and the spread of antibiotic resistance in multiresistant Enterococci – evidence from genomic sequences. BMC Microbiol. 21:55. doi: 10.1186/s12866-021-02114-4
Li, Y. H., Yu, C. Y., Li, X. X., Zhang, P., Tang, J., Yang, Q., et al. (2018). Therapeutic target database update 2018: enriched resource for facilitating bench-to-clinic research of targeted therapeutics. Nucleic Acids Res. 46, D1121–D1127. doi: 10.1093/nar/gkx1076
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
López, C., Checa, S. K., and Soncini, F. C. (2018). CpxR/CpxA controls scsABCD transcription to counteract copper and oxidative stress in Salmonella enterica serovar typhimurium. J. Bacteriol. 200:e00126-18. doi: 10.1128/JB.00126-18
Martin-Carnahan, M., and Joseph, S. W. (2005). “Aeromonadales ord. nov,” in Bergey’s Manual of Systematic Bacteriology: The Proteobacteria, Part B: The gammaproteobacteria, Vol. 2, eds D. J. Brenner, N. R. Krieg, J. T. Staley, G. M. Garrity, D. R. Boone, P. de Vos, et al. (New York, NY: Springer), 556–587. doi: 10.1007/0-387-28022-712
Moriel, B., Prediger, K. C., Souza, E. M., Pedrosa, F. O., Fadel-Picheth, C. M. T., and Cruz, L. M. (2021). In silico comparative analysis of Aeromonas type VI secretion system. Braz. J. Microbiol. 52, 229–243. doi: 10.1007/s42770-020-00405-y
Nguyen, H. N. K., Van, T. T. H., Nguyen, H. T., Smooker, P. M., Shimeta, J., and Coloe, P. J. (2014). Molecular characterization of antibiotic resistance in Pseudomonas and Aeromonas isolates from catfish of the Mekong Delta, Vietnam. Vet. Microbiol. 171, 397–405. doi: 10.1016/j.vetmic.2014.01.028
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Pan, X., Xu, S., Wu, J., Duan, Y., Zheng, Z., Wang, J., et al. (2018). Ankyrin-like protein AnkB interacts with CatB, affects catalase activity, and enhances resistance of Xanthomonas oryzae pv. oryzae and Xanthomonas oryzae pv. oryzicola to phenazine-1-carboxylic acid. Appl. Environ. Microbiol. 84:e02145-17. doi: 10.1128/AEM.02145-17
Pang, M., Jiang, J., Xie, X., Wu, Y., Dong, Y., Kwok, A. H. Y., et al. (2015). Novel insights into the pathogenicity of epidemic Aeromonas hydrophila ST251 clones from comparative genomics. Sci. Rep. 5:09833. doi: 10.1038/srep09833
Partridge, S. R., Kwong, S. M., Firth, N., and Jensen, S. O. (2018). Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31:e00088-17. doi: 10.1128/CMR.00088-17
Pundhir, S., Vijayvargiya, H., and Kumar, A. (2008). PredictBias: a server for the identification of genomic and pathogenicity islands in prokaryotes. In Silico Biol. 8, 223–234.
Rao, R. T., Sharma, S., Sivakumar, N., and Jayakumar, K. (2020). Genomic islands and the evolution of livestock-associated Staphylococcus aureus genomes. Biosci. Rep. 40:BSR20202287. doi: 10.1042/BSR20202287
Rasmussen-Ivey, C. R., Hossain, M. J., Odom, S. E., Terhune, J. S., Hemstreet, W. G., Shoemaker, C. A., et al. (2016b). Classification of a hypervirulent Aeromonas hydrophila pathotype responsible for epidemic outbreaks in warm-water fishes. Front. Microbiol. 7:1615. doi: 10.3389/fmicb.2016.01615
Rasmussen-Ivey, C. R., Figueras, M. J., McGarey, D., and Liles, M. R. (2016a). Virulence factors of Aeromonas hydrophila: in the wake of reclassification. Front. Microbiol. 7:1337. doi: 10.3389/fmicb.2016.01337
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Sayers, S., Li, L., Ong, E., Deng, S., Fu, G., Lin, Y., et al. (2019). Victors: a web- based knowledge base of virulence factors in human and animal pathogens. Nucleic Acids Res. 47, D693–D700. doi: 10.1093/nar/gky999
Seshadri, R., Joseph, S. W., Chopra, A. K., Sha, J., Shaw, J., Graf, J., et al. (2006). Genome sequence of Aeromonas hydrophila ATCC 7966T: Jack of all trades. J. Bacteriol. 188, 8272–8282. doi: 10.1128/JB.00621-06
Sha, J., Rosenzweig, J. A., Kozlova, E. V., Wang, S., Erova, T. E., Kirtley, M. L., et al. (2013). Evaluation of the roles played by Hcp and VgrG type 6 secretion system effectors in Aeromonas hydrophila SSU pathogenesis. Microbiology 159, 1120–1135. doi: 10.1099/mic.0.063495-0
Soares, S. C., Geyik, H., Ramos, R. T., de Sá, P. H., Barbosa, E. G., Baumbach, J., et al. (2016). GIPSy: genomic island prediction software. J. Biotechnol. 232, 2–11. doi: 10.1016/j.jbiotec.2015.09.008
Srividhya, K. V., Rao, G. V., Raghavenderan, L., Mehta, P., Prilusky, J., Manicka, S., et al. (2006). “Database and comparative identification of prophages,” in Intelligent Control and Automation, Lecture Notes in Control and Information Sciences, Vol. 344, eds D.-S. Huang, K. Li, and G. W. Irwin (Berlin: Springer), 863–868.
Suarez, G., Sierra, J. C., Erova, T. E., Sha, J., Horneman, A. J., and Chopra, A. K. (2010a). A type VI secretion system effector protein, VgrG1, from Aeromonas hydrophila that induces host cell toxicity by ADP ribosylation of actin. J. Bacteriol. 192, 155–168. doi: 10.1128/JB.01260-09
Suarez, G., Sierra, J. C., Kirtley, M. L., and Chopra, A. K. (2010b). Role of Hcp, a type 6 secretion system effector, of Aeromonas hydrophila in modulating activation of host immune cells. Microbiology 156, 3678–3688. doi: 10.1099/mic.0.041277-0
Suarez, G., Sierra, J. C., Sha, J., Wang, S., Erova, T. E., Fadl, A. A., et al. (2008). Molecular characterization of a functional type VI secretion system from a clinical isolate of Aeromonas hydrophila. Microb. Pathog. 44, 344–361. doi: 10.1016/j.micpath.2007.10.005
Subedi, P., Paxman, J. J., Wang, G., Ukuwela, A. A., Xiao, Z., and Heras, B. (2019). The Scs disulfide reductase system cooperates with the metallochaperone CueP in Salmonella copper resistance. J. Biol. Chem. 294, 15876–15888. doi: 10.1074/jbc.RA119.010164
Tabei, S. M. B., Hitchen, P. G., Day-Williams, M. J., Merino, S., Vart, R., Pang, P. C., et al. (2009). An Aeromonas caviae genomic Island is required for both O-antigen lipopolysaccharide biosynthesis and flagellin glycosylation. J. Bacteriol. 191, 2851–2863. doi: 10.1128/JB.01406-08
Tekedar, H. C., Abdelhamed, H., Kumru, S., Blom, J., Karsi, A., and Lawrence, M. L. (2019). Comparative genomics of Aeromonas hydrophila secretion systems and mutational analysis of hcp1 and vgrG1 genes from T6SS. Front. Microbiol. 9:3216. doi: 10.3389/fmicb.2018.03216
Thiaville, J. J., Kellner, S. M., Yuan, Y., Hutinet, G., Thiaville, P. C., Jumpathong, W., et al. (2016). Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc. Natl. Acad. Sci. U.S.A. 113, E1452–E1459. doi: 10.1073/pnas.1518570113
Tomás, J. M. (2012). The main Aeromonas pathogenic factors. Int. Sch. Res. Netw. ISRN Microbiol. 2012:256261. doi: 10.5402/2012/256261
Trucksis, M., Galen, J. E., Michalski, J., Fasano, A., and Kaper, J. B. (1993). Accessory cholera enterotoxin (Ace), the third toxin of a Vibrio cholerae virulence cassette. Proc. Natl. Acad. Sci. U.S.A. 90, 5267–5271.
Vernikos, G. S., and Parkhill, J. (2006). Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22, 2196–2203. doi: 10.1093/bioinformatics/btl369
Veyron, S., Peyroche, G., and Cherfils, J. (2018). FIC proteins: from bacteria to humans and back again. Pathog. Dis. 76:fty012. doi: 10.1093/femspd/fty012
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142. doi: 10.1186/1471-2105-7-142
Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., et al. (2017). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542. doi: 10.1093/nar/gkw1017
Wei, W., Gao, F., Du, M. Z., Hua, H. L., Wang, J., Guo, F. B., et al. (2016). Zisland Explorer: detect genomic Islands by combining homogeneity and heterogeneity properties. Brief. Bioinform. 18, 357–366. doi: 10.1093/bib/bbw019
Weinstock, G. M., Sodergren, E. J., Smajs, D., and Norris, S. J. (2000). “Identification of virulence genes in silico: infectious disease genomics,” in Virulence Mechanisms of Bacterial Pathogens, 3rd Edn, eds K. A. Brogden, J. A. Roth, T. B. Stanton, C. A. Bolin, F. C. Minion, and M. J. Wannemuehler (Washington, DC: American Society of Microbiology), 251–261.
Wilhelms, M., Fulton, K. M., Twine, S. M., Tomás, J. M., and Merino, S. (2012). Differential glycosylation of polar and lateral flagellins in Aeromonas hydrophila AH-3. J. Biol. Chem. 287, 27851–27862. doi: 10.1074/jbc.M112.376525
Keywords: Aeromonas hydrophila, genomic island, virulence, metabolism, antibiotic resistance
Citation: da Silva Filho AC, Marchaukoski JN, Raittz RT, De Pierri CR, de Jesus Soares Machado D, Fadel-Picheth CMT and Picheth G (2021) Prediction and Analysis in silico of Genomic Islands in Aeromonas hydrophila. Front. Microbiol. 12:769380. doi: 10.3389/fmicb.2021.769380
Received: 01 September 2021; Accepted: 01 November 2021;
Published: 29 November 2021.
Edited by:
Daniel Yero, Universidad Autónoma de Barcelona, SpainReviewed by:
Gabriel Forn-Cuní, Leiden University, NetherlandsAntony T. Vincent, Laval University, Canada
Copyright © 2021 da Silva Filho, Marchaukoski, Raittz, De Pierri, de Jesus Soares Machado, Fadel-Picheth and Picheth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Camilo da Silva Filho, YW50b25pby5jYW1pbG9maWxob0BnbWFpbC5jb20=