Abstract
Quorum sensing (QS) is a bacterial cell-cell communication system with genetically regulated mechanisms dependent on cell density. Canonical QS systems in gram-negative bacteria possess an autoinducer synthase (LuxI family) and a transcriptional regulator (LuxR family) that respond to an autoinducer molecule. In Gram-positive bacteria, the LuxR transcriptional regulators “solo” (not associated with a LuxI homolog) may play key roles in intracellular communication. Arthrobacter sp. UMCV2 is an actinobacterium that promotes plant growth by emitting the volatile organic compound N, N-dimethylhexadecylamine (DMHDA). This compound induces iron deficiency, defense responses in plants, and swarming motility in Arthrobacter sp. UMCV2. In this study, the draft genome of this bacterium was assembled and compared with the genomes of type strains of the Arthrobacter genus, finding that it does not belong to any previously described species. Genome explorations also revealed the presence of 16 luxR-related genes, but no luxI homologs were discovered. Eleven of these sequences possess the LuxR characteristic DNA-binding domain with a helix-turn-helix motif and were designated as auto-inducer-related regulators (AirR). Four sequences possessed LuxR analogous domains and were designated as auto-inducer analogous regulators (AiaR). When swarming motility was induced with DMHDA, eight airR genes and two aiaR genes were upregulated. These results indicate that the expression of multiple luxR-related genes is induced in actinobacteria, such as Arthrobacter sp. UMCV2, by the action of the bacterial biocompound DMHDA when QS behavior is produced.
Introduction
Quorum sensing (QS) is a bacterial cell-cell communication system based on the production, release, and detection of signal molecules called autoinducers (AI). QS regulates gene expression in response to changes in bacterial population density and produces coordinated behavior, based on environmental conditions (Fuqua et al., 1994; Waters and Bassler, 2005; Papenfort and Bassler, 2016). Cellular functions regulated by QS include bioluminescence, antibiotic synthesis, virulence factor production, biofilm induction, sporulation, and swarming motility (Polkade et al., 2016; Bramhachari et al., 2018).
The lux operon, responsible for bioluminescence in the model organism Vibrio fischeri (Aliivibrio fischeri), is composed of six structural genes (luxA-E and luxG) and the regulatory genes luxI and luxR that encode homonym proteins (Fuqua et al., 1996; Miller and Bassler, 2001). LuxI is an acyl-synthase of approximately 190 amino acids that uses the acyl carrier protein as a substrate to add the N-acyl group to L-homoserine lactone derived from S-adenosylmethionine and produce acyl-homoserine lactones (AHLs)., the main autoinducers in Gram-negative bacteria. In other bacteria, LuxI homologs add different N-acyl groups to l-homoserine lactones to produce different AHLs (Fuqua et al., 1994; Zavilgelsky and Manukhov, 2001). LuxR is a transcriptional regulator of approximately 250 amino acids with characteristic modular architecture consisting of an autoinducer binding domain in the N-terminal region (Shadel et al., 1990; Slock et al., 1990) and a DNA-binding domain with a helix-turn-helix (HTH) motif in the C-terminal region (Choi and Greenberg, 1991; Fuqua et al., 1994).
Quorum sensing systems were identified in both Gram-negative and Gram-positive bacteria (Hawver et al., 2016). In Gram-negative bacteria, the more studied QS systems are regulated by members of the AHL family (Swift et al., 1994) which act as diffusible signal molecules, whose synthesis is performed by members of the LuxI family. Above a threshold concentration, these molecules bind to members of the transcriptional regulator LuxR family (Fuqua et al., 1996). The stable LuxR-AHL complex subsequently binds to the lux box, a specific regulatory sequence in the promoter region, to activate or suppress the transcription of its target genes (Fuqua et al., 2001).
The analysis of different proteobacterial genomes has revealed the wide presence of homologs of the transcriptional regulator luxR without the presence of their corresponding homolog LuxI, thus referred to as luxR orphans or “solos” (Fuqua, 2006; Case et al., 2008; Patankar and González, 2009). These transcriptional regulators possess the same structural organization as canonical LuxR (Bez et al., 2021). Stenotrophomonas maltophilia encodes the transcriptional regulator SmoR, a non-LuxI-associated LuxR homolog that binds to oxo-C8-homoserine lactone produced by Pseudomonas aeruginosa. SmoR regulates its operon transcription and promotes swarming motility in S. matophilia (Martínez et al., 2015). LuxR solo proteins are involved in inter-kingdom signaling, responding to the signals produced by eukaryotes (Venturi and Fuqua, 2013; Kan et al., 2017). A subfamily of luxR is present exclusively in plant-associated bacterial (PAB) responses to plant low-molecular weight compounds (González et al., 2013; González and Venturi, 2013; Venturi and Fuqua, 2013). Structurally, PAB-LuxR is very similar to canonical LuxR but differs in one or two of the aromatic or hydrophobic amino acids normally conserved in the auto-inducer binding domain of canonical LuxR proteins (González and Venturi, 2013). PAB-LuxR proteins are present in phytopathogenic bacteria as XccR of Xanthomonas campestris (Zhang et al., 2007), or OryR of Xanthomonas oryzae (Ferluga and Venturi, 2009) and in plant beneficial bacteria, such as Pseudomonas sp. GM79 (Coutinho et al., 2018) and PsrR of Kosakonia sp. KO348 (Mosquito et al., 2020). Other LuxR proteins respond to endogenous molecules other than AHLs and are therefore not associated with LuxI synthases. PluR, present in Photorhabdus luminescens, responds to α-pyrones produced by the ketosynthase PpyS, whereas PauR of the human pathogen Photorhabdus asymbiotica senses dialkylresorcinols and cyclohexanediones produced by the bacteria (Brachmann et al., 2013; Brameyer et al., 2014, 2015).
Other molecules, such as volatile organic compounds (VOCs) produced by bacteria, act as signaling molecules regulating processes that are frequently regulated by QS (Schulz-Bohm et al., 2017; Xie et al., 2018). Resistance against antibiotics, virulence, and motility are examples of behaviors regulated by bacterial VOCs. The mechanisms involved in the communication mediated by VOCs and the regulation of these behaviors are primarily unknown (Kim et al., 2013; Enkataraman et al., 2014; Mansurova et al., 2018). The participation of luxR transcriptional regulators is not discarded.
In gram-positive bacteria, QS systems are usually regulated by auto-inducing peptides (Monnet and Gardan, 2015; Hawver et al., 2016; Aframian and Eldar, 2020), released by specific transporters into the extracellular media, where they are detected by a transmembrane receptor. Autoinducer detection triggers a signaling pathway through the successive phosphorylation/dephosphorylation of a sensing transmembrane histidine kinase and a transcriptional regulator that interacts with DNA and controls the QS response (Lyon and Novick, 2004; Dufour and Lévesque, 2013; Monnet et al., 2014). Although LuxR-LuxI systems are marginally present in Gram-positive bacteria, luxR solo genes are frequently found in that group of bacteria (Rajput and Kumar, 2017).
Arthrobacter sp. UMCV2 is an actinobacterium (Gram-positive) isolated from the Zea mays rhizosphere (Valencia-Cantero et al., 2007). This Arthrobacter strain promotes plant growth through the emission of VOCs, predominantly N, N-dimethylhexadecylamine (dimethylhexadecylamine, DMHDA) (Velázquez-Becerra et al., 2011). DMHDA emitted by Arthrobacter sp. UMCV2 accumulates in the extracellular medium and modulates the growth and swarming motility of Arthrobacter sp. UMCV2 and other bacteria (Velázquez-Becerra et al., 2013; Martínez-Cámara et al., 2020). In this study, we analyzed the Arthrobacter sp. UMCV2 genome, identified and classified the Arthrobacter sp. UMCV2 luxR-related genes, and demonstrated the upregulation of luxR-related genes by DMHDA when motility-associated genes are induced.
Materials and methods
Biological material
The strain used in this study was Arthrobacter sp. UMCV2 (Valencia-Cantero et al., 2007), deposited at the Microorganism Collection of the National Center of Genetic Resources (Boulevard de la Biodiversidad 400, Rancho las Cruces, 47600 Tepatitlán de Morelos, Jalisco, México) having accession number CM-CNRG-691. The bacterial strain was routinely cultured on nutrient agar (NA) or nutrient broth (NB) at 22°C.
Chemicals
The chemical compound N, N-dimethylhexadecylamine (DMHDA) was purchased from Sigma Aldrich (St Louis MO USA, catalog 40460) and dissolved in ethanol. Equal volumes of solvent were used in all the treatments.
Genomic DNA extraction
From a single colony culture, Arthrobacter sp. UMCV2 was inoculated into 50 ml of NB at 22°C with agitation at 180 rpm for 2 days. Genomic DNA was extracted from 20 ml of liquid culture using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA, catalog A1120), according to the manufacturer’s instructions. The quality and quantity of genomic DNA were assessed by agarose gel electrophoresis using a NanoDrop 1000 (Thermo Scientific, Rockford, IL, USA).
Genome sequencing, assembly, annotation, and phylogenetic analysis
The genome of Arthrobacter sp. UMCV2 was sequenced at the genomic sequencing facilities of LANGEBIO, Cinvestav-IPN (Irapuato-México), using an Illumina MiSeq platform generating three paired-end libraries, with coverage of 57x, the original data were assembled into 461 contigs or scaffolds using Newbler v2.9 software. To order the contigs, A. chlorophenolicus A6 was used as the reference genome. A summary of the project information is presented in Supplementary Table 1. The genome sequence was submitted to the RAST web service (MG-RAST 4.0.3)1 for automated annotation (Aziz et al., 2008). The sequence was deposited in the GenBank database of the National Center for Technology Information (NCBI)2 with accession number (CP024915.1).
A genomic-based phylogenetic tree, a 16S-based phylogenetic tree and digital DNA–DNA hybridization (dDDH) were performed with the assembled genome using the Type Strain Genome Server (TYGS) pipeline (Meier-Kolthoff and Göker, 2019; Meier-Kolthoff et al., 2022).3 Trees were inferred using FastME 2.1.6.1 from Genome BLAST Distance Phylogeny (GBDP), distance formula d5 (Farris, 1972; Meier-Kolthoff et al., 2013; Lefort et al., 2015), comparing the genomes of Arthrobacter sp. UMCV2, with type strains deposited in the DSMZ database.
In silico analysis of Arthrobacter sp. UMCV2 luxR-related genes
In addition to NCBI gene notation, a complementary search for luxR and corresponding LuxI homologous genes was performed with RAST MG-RAST 4.0.3 (see above) and PATRIC 3.6.12 (Brettin et al., 2015).4 Putative luxR homologs were analyzed using InterPro v62.0 (Blum et al., 2021).5
Phylogenetic and molecular evolutionary analyses were conducted using MEGA version X (Kumar et al., 2018) using the maximum likelihood method and the Jones-Taylor-Thornton (JTT) matrix-based model (Jones et al., 1992). The bootstrap consensus tree inferred from 1,000 replicates was used to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). Initial trees for the heuristic search were obtained automatically by applying the Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model followed by selecting the topology with a superior log-likelihood value. The analysis involved 44 amino acid sequences. There were 1,098 positions in the final dataset.
Bacterial motility assay
The effect of DMHDA on bacterial motility was tested on NA plates with DMHDA at concentrations of 0, 0.1, 0.2, 3.0, 4.0, 5.0, and 6.0 μM. Subsequently, 5 μl of bacterial suspension (OD595 = 1) was added to each plate center. The swarming motility was assessed following the protocols described by Martínez-Cámara et al. (2020) with a few modifications, such as petri dishes prepared with NB plus 0.5% agar and incubated for 120 h at 23°C. The motility diameter was measured using a digital Vernier caliper (Mitutoyo Corporation, Tokyo Japan, catalog 500-196-30) and the results were reported in centimeters of bacterial extension on the media. Three independent assays, each with four replicates, were performed.
Gene expression analysis
RNA extraction was performed for three biological replicates of Arthrobacter sp. UMCV2 from swarming motility, as previously described (Martínez-Cámara et al., 2020). RNA was extracted using TRI reagent (Sigma Aldrich, St Louis, MO, USA, catalog T9424) and treated with DNase I to remove the remaining genomic DNA. The integrity of the RNA was assessed by visualization on 1.2% agarose gel. The final concentration was estimated using NanoDrop (Thermo Scientific, Rockford, IL, USA). cDNA synthesis was performed using 500 ng RNA template and SuperScript First-Strand Synthesis System (Life Technologies/Gibco-BRL CA, USA). Reverse transcription quantitative real-time PCRs (RT-qPCR) were performed on an ABI StepOneTM System thermocycler (Applied Biosystems; Foster City, CA, USA). Oligonucleotides were designed (using the NCBI for Biotechnology Information primer design tool) to amplify airR1-12 and aiaR1-4 (Supplementary Table 2). Oligonucleotides to amplify fliC, flgL, fliM, motA, and recA have been previously reported (Martínez-Cámara et al., 2020), recA was used as the normalizer gene. RT-qPCR analysis was performed using the SYBR-Green kit (Applied Biosystems, Foster City, CA, USA, catalog 4385612) and the following protocol: 5 μl SYBR Green, 1 μl direct and inverse oligo, 2 μl cDNA, and 1 μl water. Samples were run using the following protocol: 95°C for 5 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min. To prepare the melting curve, samples were run at 95°C for 15 s and 60°C for 1 min, and the temperature was subsequently raised to 95°C at a rate of 0.3°C/s. Gene expression was evaluated using the comparative ΔΔ Ct method according to Livak and Schmittgen (2001).
Statistical analysis
The results were analyzed using Student’s t-test or analysis of variance and Duncan’s multiple range test for multiple (p ≤ 0.05).
Results
Genome properties
The Arthrobacter sp. UMCV2 assembled genome sequence consists of a single circular chromosome of 3,435,243 bp with a 69.3% of GC content (Figure 1). Automatic gene functional annotation predicted 3,114 genes, consisting of 3,067 protein-coding and 47 RNA genes (Table 1). The gene distribution had representatives of 23 “Clusters of Orthologous Groups” (COGs) functional categories (Supplementary Table 3). The genome size of Arthrobacter sp. UMCV2 is similar to the genome size of closed-related type strains of Arthrobacter species deposited in the DSMZ database (Supplementary Table 4) that range between 3.17 Gb of A. echini and 4.42 Gb of A. sedimenti. Genomic-based and 16S-based phylogenetic trees were generated using the complete genome sequences. 16S genes of the closed-related bacterial type strain deposited in the DSMZ database were detected using the pipeline of the SYGS server (Meier-Kolthoff and Göker, 2019). Both phylogenetic trees presented a clear affiliation of the UMCV2 strain with the Arthrobacter genus. However, further genomic (Figure 2A) or 16S (Figure 2B) phylogenetic trees did not conclusively supported a taxonomic position at the species level. The 16S-based phylogenetic tree showed clustering of the Arthrobacter sp. UMCV2 strain with the bacterial species A. echini, A. bussei, A. agilis, A. cheniae, A. ruber, and A. sedimenti. The genomic-based phylogenetic tree retains the same type of clustering and clearly shows a divergence of the UMCV2 branch from the other Arthrobacter species.
FIGURE 1
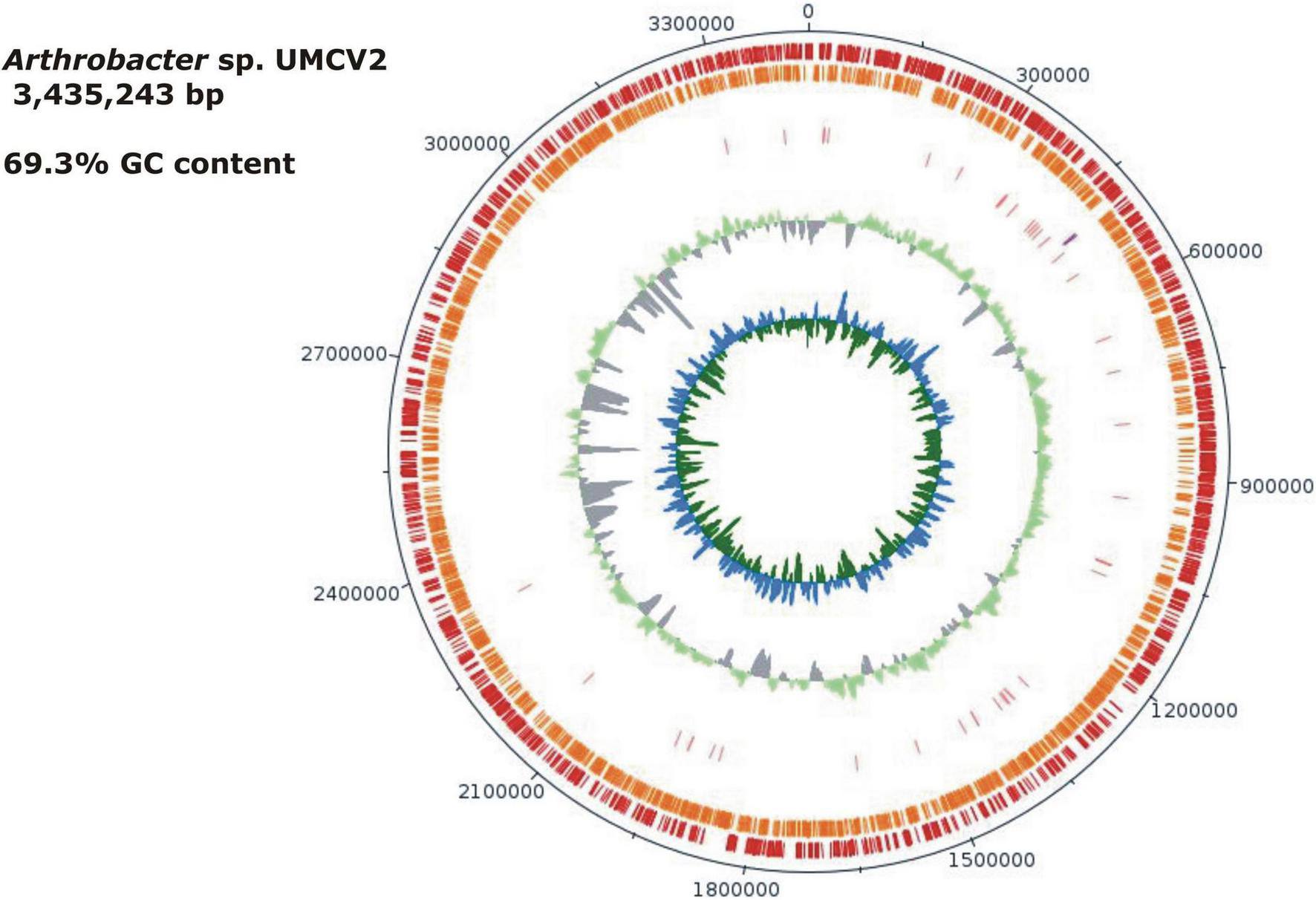
Graphical map of the Arthrobacter sp. UMCV2. From outside to the center: genes on the forward strand (red), genes on the reverse strand (orange), RNA genes (pink tRNAs, purple rRNAs), the guanine-cytosine (GC) ratio is in light green and gray. The inner circle shows the GC skew in blue and green. Gaps between individual contigs are not presented. The order of the contigs was assumed to correspond to a reference genome. Contigs not matching the reference genome were ordered from the largest to the smallest. Ordered contigs were joined with 50 bp of “N.” This figure was drawn with the DNA plotter software.
TABLE 1
| Attribute | Value | % of total |
| Genome size (bp) | 3,435,243 | 100 |
| DNA coding region (bp) | 2,735,154 | 79.62 |
| DNA G + C content (bp) | 1,902,572 | 69.3 |
| DNA scaffolds | 461 | 100 |
| Total genes | 3,114 | 100 |
| Protein coding genes | 3,067 | 98.49 |
| RNA genes | 47 | 1.51 |
| Genes assigned to COGs | 1,884 | 60.50 |
| Genes with Pfam domains | 1,930 | 61.95 |
Arthrobacter sp. UMCV2 genome statistics.
FIGURE 2
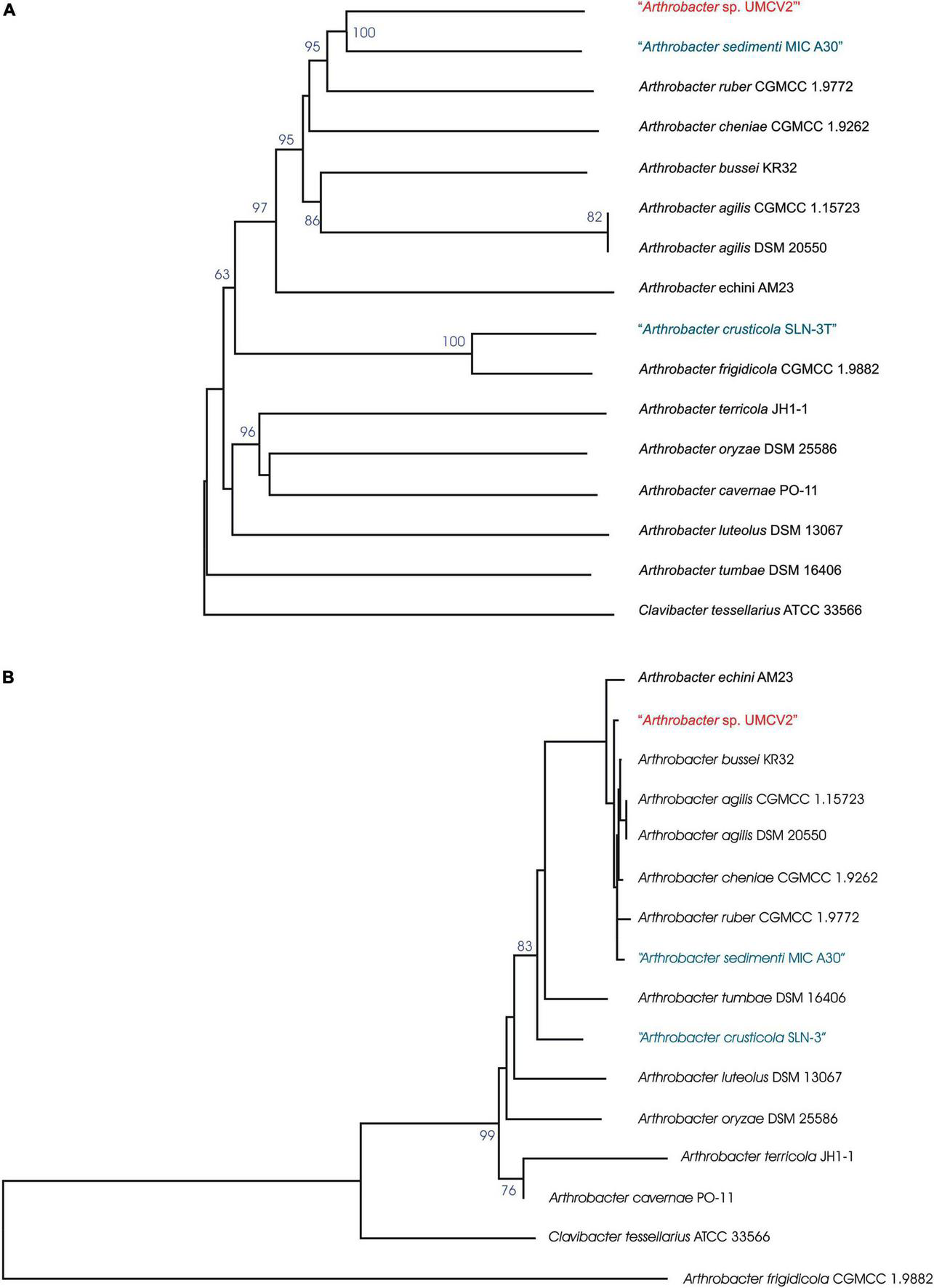
Arthrobacter sp. UMCV2 phylogenetic tree based on the genomic (A) and (B) 16S rRNA gene sequences. The trees were inferred from genome BLAST distance phylogeny (GBDP) distances. The branch lengths were scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values >60% from 100 replications, with average branch support of 77.4% for genomic tree and 57.2% for 16S rRNA gene sequences tree. The trees were rooted at the midpoint. Species in blue were mentioned as not validly published by the DSMZ server https://lpsn.dsmz.de/ at the time of writing.
Relatednesses between the genomes of Arthrobacter sp. UMCV2 and closed-related type strains of Arthrobacter species were calculated as dDDH percentages. Concordantly with the phylogenetic tree topologies, the highest dDDH values of Arthrobacter sp. UMCV2 were produced with cluster members of type strains such as A. bussei, A. agilis, A. cheniae, A. ruber, and A. sedimenti, ranging from 40.3% (with A. ruber) to 36.2% (with A. bussei). Similar dDDH values were produced among type strains of the same cluster ranging from 47% (A. agilis to A. bussei) to 31.4% (A. agilis to A. sedimenti) (Table 2). These results strongly suggest that Arthrobacter sp. UMCV2 belongs to a species not yet described within the genus Arthrobacter.
TABLE 2
| Bacterial strainb | Arthrobacter sp. UMCV2 |
Arthrobacter sedimenti
MIC A30 T |
Arthrobacter ruber CGMCC 1.9772T | Arthrobacter cheniae CGMCC 1.9262T | Arthrobacter bussei KR32T | Arthrobacter agilis CGMCC 1.15723 | Arthrobacter agilis DSM20550T | Arthrobacter echini AM23T | Arthrobacter frigidicola CGMCC 1.9882T | Arthrobacter crusticola SLN-3TT | Arthrobacter cavernae PO-11T | Arthrobacter oryzae DSM 25586T |
| Arthrobacter sp. UMCV2 | − | |||||||||||
| Arthrobacter sedimenti MIC A30T | 36.7 | − | ||||||||||
| Arthrobacter ruber CGMCC 1.9772T | 40.3 | 35.8 | − | |||||||||
| Arthrobacter cheniae CGMCC 1.9262T | 34.5 | 34.9 | 38.6 | − | ||||||||
| Arthrobacter bussei KR32T | 36.2 | 32.4 | 38.1 | 37.6 | − | |||||||
| Arthrobacter agilis CGMCC 1.15723 | 37.3 | 31.4 | 38.7 | 36.5 | 46.9 | − | ||||||
| Arthrobacter agilis DSM20550T | 37.3 | 31.4 | 38.6 | 36.4 | 47.0 | 100 | − | |||||
| Arthrobacter echini AM23T | 23.7 | 23.3 | 26.9 | 22.0 | 24.7 | 25.2 | 25.2 | − | ||||
| Arthrobacter frigidicola CGMCC 1.9882T | 18.4 | 18.6 | 20.2 | 18.4 | 18.7 | 19.0 | 19.0 | 17.0 | − | |||
| Arthrobacter crusticola SLN-3TT | 19.1 | 19.2 | 20.6 | 18.7 | 19.2 | 19.2 | 19.2 | 17.4 | 70.8 | − | ||
| Arthrobacter cavernae PO-11T | 14.4 | 14.6 | 14.7 | 14 | 14.3 | 14.1 | 14.5 | 13.9 | 14.5 | 14.5 | − | |
| Arthrobacter oryzae DSM 25586T | 14.3 | 14.6 | 14.6 | 13.9 | 14.4 | 14.3 | 14.3 | 13.8 | 14.5 | 14.4 | 20.8 | − |
| Arthrobacter luteolus DSM 13067T | 14.2 | 14.2 | 14.4 | 13.7 | 14.3 | 14.2 | 14.2 | 14.1 | 14.5 | 14.8 | 14.5 | 14.3 |
Genomic relatedness matrix of Arthrobacter sp. UMCV2 and closed-related type strains calculated as digital DNA–DNA hybridization (dDDH) percentagesa.
adDDH vales were calculated according to the formula one of the type strain genome server (TYGs) pipeline as described by Meier-Kolthoff and Göker (2019).
bType bacterial strains are marked withT.
Identification of characteristic LuxR domains
Analysis of the Arthrobacter sp. UMCV2 genome was performed with the MG-RAST and PATRIC servers and 18 putative luxR homolog sequences, but no LuxI putative homologs were identified. Protein sequences corresponding to the putative luxR homologs were examined to identify LuxR-characteristic domains using the InterPro server. Aliivibrio fischeri LuxR (Fuqua et al., 1994) and Pseudomonas sp. GM79 PipR (Coutinho et al., 2018) were included in the analysis as references for canonical experimentally characterized LuxR proteins, Streptomyces purpurogeneiscleroticus NRRL B-2952 LuxR (Rajput and Kumar, 2017) as a canonical in silico characterized LuxR protein from actinobacteria, and Streptomyces SN-593 RevU (Panthee et al., 2020) as an experimentally characterized LuxR-related protein from actinobacteria. All four reference proteins presented a characteristic DNA-binding domain at the C-terminal end (IPR000792). The canonical LuxR proteins displayed an autoinducer binding domain (IPR005143) at the N-terminal end, whereas RevU (LAL-LuxR protein) Streptomyces SN-593 exhibited an alternative ATPase domain (IPR041664) (Figure 3). Interestingly, the length of the RevU protein was significantly longer (923 a.a.) than the canonical LuxR (250 a.a.).
FIGURE 3

Conserved domains found in the model LuxR proteins and LuxR-related sequences from Arthrobacter sp. UMCV2. LuxR-related sequences were localized to the genome of Arthrobacter sp. UMCV2 employs the informatic tools MG-RAST 4.0.3 and PATRIC 3.6.12. Domains were localized in the protein sequences employing the informatics tool InterPro v62.0. Proteins are represented with a blue line and their lengths are compared with the upper scale. Conserved domains are localized in the proteins with colored boxes. Protein names, the strain of origin, and national center for technology information (NCBI) locus are indicated in the left column.
Further, we analyzed 18 Arthrobacter sp. UMCV2 putative LuxR proteins for conserved domains (Figure 3). Eleven sequences showed the characteristic DNA-binding domain IPR000792 at the C-terminal end (designated as airR sequences), four showed the IPR001867 domain described as a DNA-binding domain type OmpR/PhoB (designated as aiaR sequences), one sequence contained the IPR005561 domain described as transcription antitermination regulator, and two sequences did not show conserved C-terminal domains (Figure 2). At the N-terminal end, 10 sequences matched the IPR001789 domain, described as a signal transduction response regulator receiver domain, three sequences matched the ATPase domains (IPR041664 or IPR003593), and three sequences did not show conserved domains at the N-terminal end. The putative LuxR homologs in Arthrobacter sp. UMCV2 did not contain the canonical LuxR autoinducer-binding domain IPR005143. Only one sequence earlier recognized as a putative LuxR homolog sequence showed the unrelated domain IPR011051 (RmlC-like cupin domain superfamily). Similar to RevU from Streptomyces SN-593, five sequences were significantly longer (489–926 a.a.) than canonical LuxR sequences. Sequences CVO76_16125 and CVO76_05180 with the domains IPR011051 and IPR005561, respectively, were discarded as LuxR-related since their domains are discordant with LuxR proteins (Figure 3).
Evolutionary relationships of LuxR-related sequences
LuxR-related sequences were used to construct a phylogenetic tree to observe their association with previously experimentally or in silico characterized LuxR-related sequences (Figure 4 and Supplementary Table 6). All Actinobacteria LuxR-related proteins clustered together with the 16 putative LuxR homologs from Arthrobacter sp. UMCV2 (AirR and AiaR sequences), three LuxR or LuxR-related sequences from Streptomyces and Streptosporangium, and three luxR or luxR-related sequences from A. terricola or Arthrobacter species. Whereas all the other analyzed luxR-related sequences from Proteobacteria clustered together with LuxR of A. fischeri (Figure 4). Among the LuxR-related sequences of Arthrobacter sp. UMCV2, three principal clusters were observed. Cluster I grouped proteins of approximately 218–439 a.a. with the concurrence of the domains IPR001789 and IPR000792 (six proteins) or the IPR000792 domain alone (two proteins). LuxR from Arthrobacter terricola (previously characterized in silico) belonged to this cluster (Figure 4). Cluster II grouped proteins with the domain IPR001867 (the four AiaR proteins) and Cluster III grouped larger proteins (507–916 a.a.) with the LuxR-related proteins of Streptomyces, Streptosporangium, and Arthrobacter multispecies (Figures 3, 4). We included sequences of LuxR associated with LuxI and LuxR solos, although no preferential clustering between luxR solo genes was observed. These results indicate that the 16 analyzed sequences from Arthrobacter sp. UMCV2 belongs to the Actinobacteria group of LuxR-related sequences separate from the canonical LuxR from Proteobacteria, suggesting early divergence between these transcriptional regulators in both phyla.
FIGURE 4
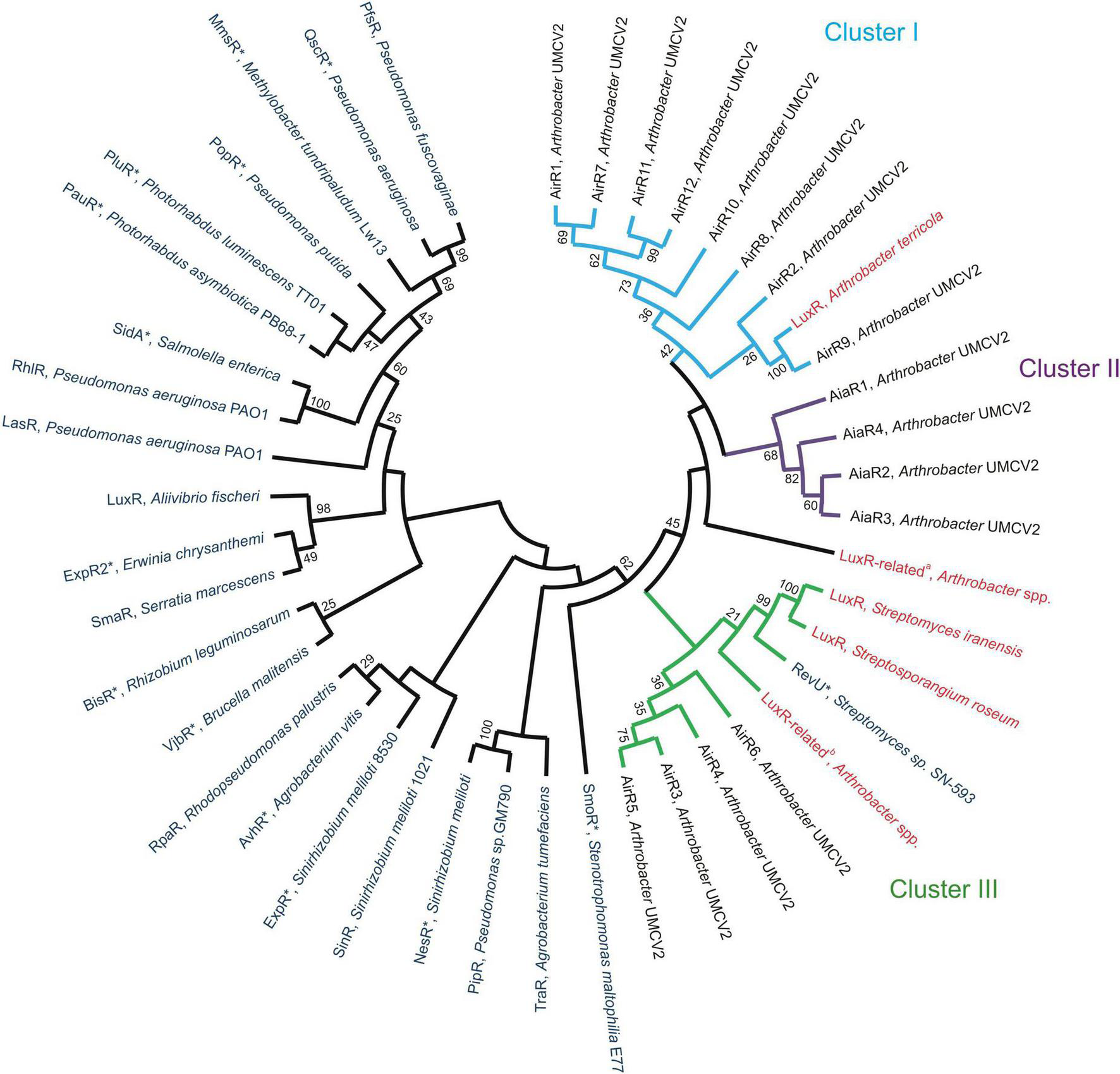
Evolutionary relationships among Arthrobacter LuxR-related sequences. The evolutionary relationship was inferred using the Maximum Likelihood method and jones-taylor-thornton (JTT) matrix-based model. The bootstrap consensus tree was inferred from 1,000 replicates. Blue sequences have been experimentally characterized, red denotes sequences informatically characterized and black is Arthrobacter sp. UMCV2 sequences established in this work. Cluster I (light blue, sequences with domains IPR001789, and IPR000792 or IPR000792 alone), Cluster II (purple, sequences with domains IPR001789, and IPR001867), and Cluster III (green, larger proteins) are emphasized. Accession numbers of employed sequences are provided in Supplementary Table 5. LuxR solos are marked with *.
N, N,-dimethylhexadecylamine modulates Arthrobacter sp. UMCV2 swarming motility
Arthrobacter sp. UMCV2 was placed in NB plates supplemented with DMHDA 1.0–6.0 μM to observe the bacterial swarming motility. After 72 h, Arthrobacter sp. UMCV2 showed an enhancement of 66, 61, and 59% in swarming motility on plates at 1.0, 2.0, and 3.0 μM DMHDA, respectively, compared with controls although, a drastic motility decrease was observed on plates with 4.0 and 5.0 μM DMHDA (Figure 5). These results show that DMHDA modulates swarming motility, a behavior usually associated with quorum sensing signaling that involves the bacterial flagellar apparatus.
FIGURE 5
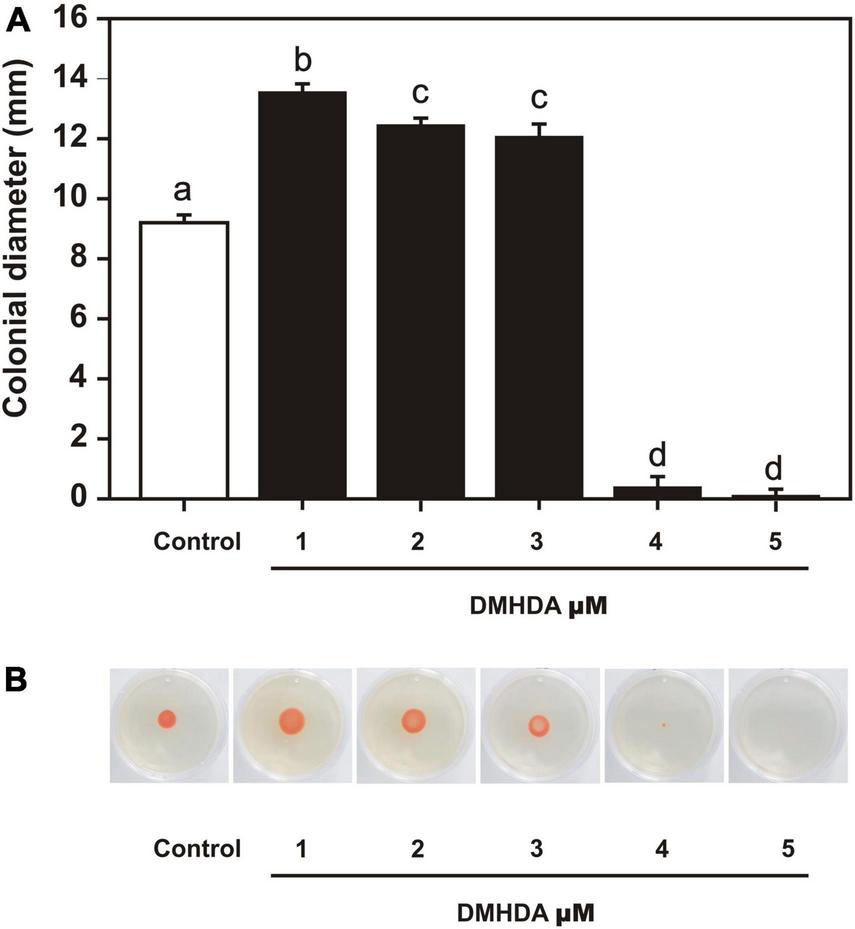
The effect of dimethylhexadecylamine (DMHDA) on Arthrobacter sp. UMCV2 swarming motility. Nutrient agar plates were prepared with various concentrations of DMHDA and inoculated with 5 μl bacterial suspension. After 72 h of culturing, colony diameters were measured. Bars, and error bars on panel (A), represent the mean ± standard error values, respectively, for four biological replicates. Letters indicate means that differ significantly, following Duncan’s multiple range test (p < 0.05). Panel (B) shows representative images of the treatments.
N, N,-dimethylhexadecylamine modulates the expression of auto-inducer related regulators and auto-inducer analog regulator genes
The expression of airR and aiaR genes, together with the flagellar genes flgL, fliC, and motA, were quantified in bacterial growth with 1.0 μM DMHDA and compared with the respective controls (0.0 μM). Eigth of the airR and two of the aiaR genes exhibited statistically significant induction. The overexpression of airR5 was 55-fold, followed by airR3 and airR7, over expressing by 25-fold. The overexpression of airR1, airR2, airR6, and aiaR3 ranged between 14- and 6-fold. The overexpression of airR11, aiaR2, and aiaR3 ranged between 4- and 1.6-fold (Figure 6A). With the increase in swarming motility, the expression of flagellar genes flgL, fliC, and motA also increased, showing an overexpression ranging from 8- to 4-fold (Figure 6B). These results showed that DMHDA modulates the expression of different airR and aiaR genes at different magnitudes, simultaneous with flagellar genes.
FIGURE 6
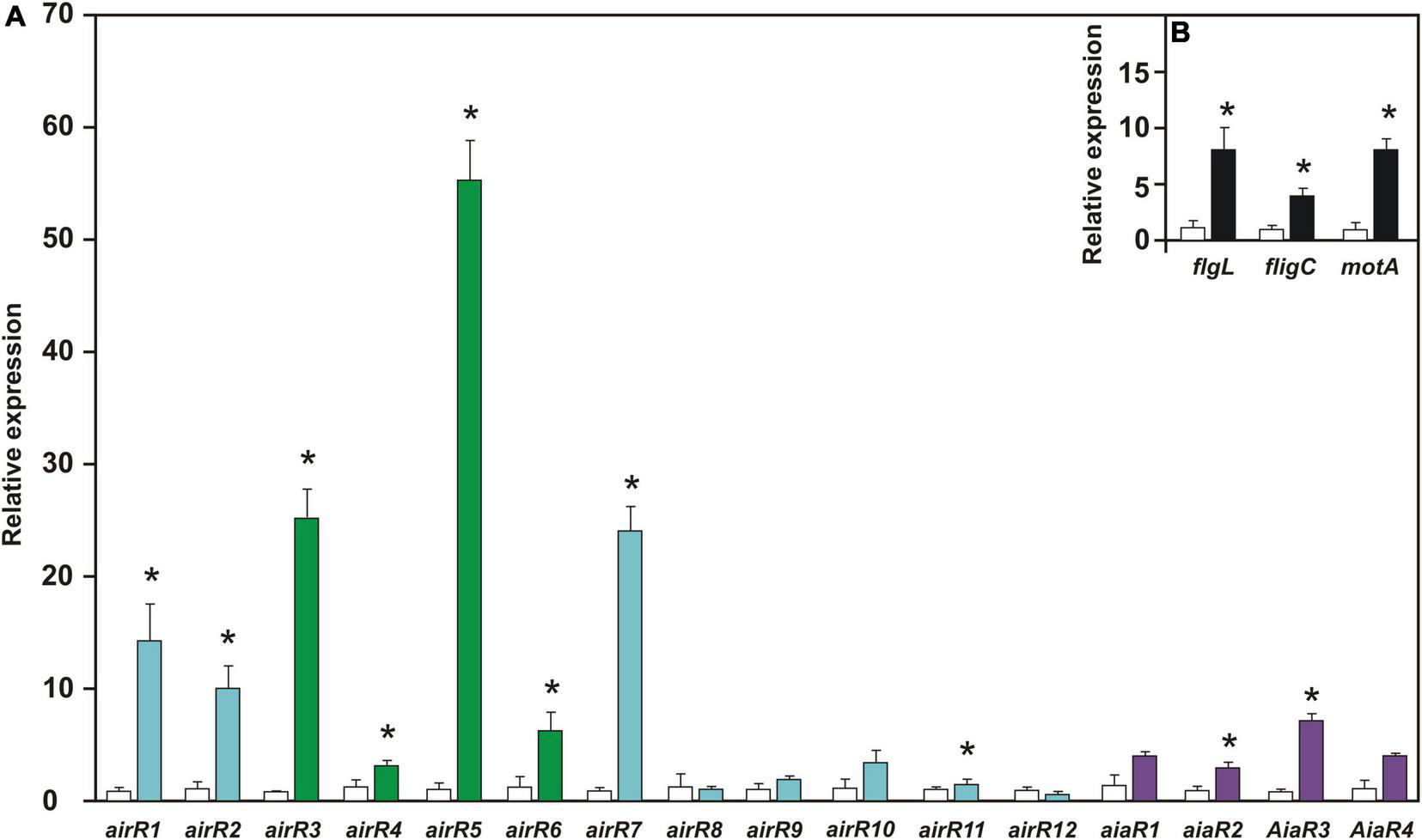
The relative expression of auto-inducer-related regulators (airR), auto-inducer analog regulator (aiaR), and motility-involved genes in Arthrobacter sp. UMCV2 induced with DMHDA. Arthrobacter sp. UMCV2 was induced with dimethylhexadecylamine (DMHDA). After 72 h, relative expression of airR, aiaR genes panel (A), and motility-involved genes panel (B) were determined. Values represent mean ± standard errors of relative expression of three biological replicates in reference to control (with bars). Asterisks indicate significant differences between treatments calculated using Student’s t-test (p < 0.05). Genes belonging to Cluster I, II, and III of Figure 4 are colored in light blue, purple, and green, respectively.
Discussion
Arthrobacter sp. UMCV2 is a plant growth-promoting rhizobacterium that was isolated while screening for iron-reducing bacteria, capable of supplying ferrous iron to plants in alkaline soils where this metal is limited to plant growth and development (Valencia-Cantero et al., 2007). This beneficial bacterium also promotes plant growth through the emission of the volatile organic compound DMHDA (Velázquez-Becerra et al., 2011). DMHDA acts as an inter-kingdom signaling molecule interacting with the plant cytokinin signaling pathway involving the AHK2 receptor (Vázquez-Chimalhua et al., 2021a) and producing in this way, a cross-talk with the jasmonic-acid pathway (Vázquez-Chimalhua et al., 2019). Plant organogenesis and growth are modulated by DMHDA through modifying the balance between stem cell niche and Jasmonic acid-dependent gene expression (Vázquez-Chimalhua et al., 2021b). DMHDA also induces expression of genes involved in plant systemic defense mechanisms and Fe-acquisition strategies, which are suggested to enhance photosynthesis and biomass production (Orozco-Mosqueda et al., 2013; Castulo-Rubio et al., 2015; Montejano-Ramírez et al., 2020). Previous results showed that Arthrobacter sp. UMCV2 adopts an endophytic lifecycle and colonizes plant tissues, providing benefits to host plants (Montejano-Ramírez et al., 2018; Hernández-Soberano et al., 2020).
Arthrobacter sp. UMCV2 genome and taxonomy
Arthrobacter sp. UMCV2 was initially identified as Arthrobacter agilis since its 16S rRNA sequences showed a percent identity above 99.5% (Valencia-Cantero et al., 2007). However, the 16S rRNA sequences of A. echini (Lee et al., 2016), A. ruber (Liu et al., 2018), A. bussei (Flegler et al., 2020), A. sedimenti (Lin et al., 2020), and A. cheniae (Yang et al., 2021) also share a percent identity above 99.5% with the 16S rRNA gene of Arthrobacter sp. UMCV2. In this study, the TYGS pipeline was employed to produce phylogenetic trees of the type strains of the species most closely related to Arthrobacter sp. UMCV2 on 16S rRNA and genomic terms (Figure 2), to determine the specific taxonomic position of Arthrobacter sp. UMCV2. Although our results place UMCV2 in the Arthrobacter genus, Arthrobacter sp. UMCV2 is not included in the A. agilis branch or in branches together with other type strains. Then, we used a dDDH approach to calculate the relatedness between the genomes of Arthrobacter sp. UMCV2 and type strains of closely related species. dDDH is a recognized standard procedure for genome sequence-based species delimitation (Meier-Kolthoff et al., 2013). Thus, we found that the higher dDDH values were shared between Arthrobacter sp. UMCV2 and a cluster of type strains including A. bussei, A. agilis, A. cheniae, A. ruber, and A. sedimenti, ranging from 40.3 to 36.2%, which are clearly below the 70%, which is generally accepted as the species boundary (Chun et al., 2018); concordantly, dDDH values detected among type strains were similarly low (Table 2). Therefore, we conclude that Arthrobacter sp. UMCV2 might be a new species that can eventually be described.
The Arthrobacter sp. UMCV2 genome has a typical size for the Arthrobacter genus. The genes involved in survival and colonization, such as SOD, showed aptitudes for colonization of roots (Kim et al., 2000; Alquéres et al., 2013), in addition to the operon ars, which confers resistance to arsenic and arsenate, a toxic metalloid (Wang et al., 2009). The presence of fhuDCB genes can be important in siderophore piracy (Koster, 1991; Galet et al., 2015), which is important for competition in soil.
Arthrobacter sp. UMCV2 possesses luxR-related genes
The presence of luxR annotated genes was particularly interesting since luxR genes are involved in the QS process. Previously, we found that DMHDA induces swarming motility in Arthrobacter sp. UMCV2, frequently recognized as a behavior regulated by QS (Martínez-Cámara et al., 2020). The canonical luxI-luxR system is typically the central component of QS in Gram-negative bacteria (Whiteley et al., 2017), but is exceptionally present in Gram-positive bacteria (Rajput and Kumar, 2017). Additionally, luxI homologs in the Arthrobacter sp. UMCV2 genome were not found. In contrast, 18 predicted protein sequences homologous to luxR were localized.
We used a domain-based approach to analyze putative luxR-related proteins and compared them with four well-characterized LuxR proteins. We included the first protein described as LuxR, originally found in A. fischeri (Fuqua et al., 1994), as a reference protein. PipR from Pseudomonas sp. GM79 was included as a reference LuxR since it is a well-experimentally characterized protein belonging to the LuxR protein family that responds to the plant derivative compound N-(2-hydroxyethyl)-2-(2-hydroxyethylamino) acetamide through a periplasmic binding protein. PipR has been included in the subfamily PAB that integrates LuxR proteins that respond to low-weight plant molecules (Mosquito et al., 2020; Xu, 2020). The LuxR protein from Streptomyces purpurogeneiscleroticus NRRL B-2952 (Rajput and Kumar, 2017) was also included as a reference since it is part of a canonical QS LuxI-LuxR system present in actinobacteria characterized in silico. Additionally, RevU was included as a non-canonical LuxR protein from actinobacteria (Panthee et al., 2020) since this protein binds the biomediator BR-1, a β-carboline compound, to regulate the synthesis of reveromycin in Streptomyces SN-593. RevU belongs to the LAL-LuxR subfamily (large ATP-binding regulators of the LuxR family), which is characterized by its large size (over 900 a.a. residues) and an ATP-binding domain in the N-terminus. Although it is present in gram-negative bacteria, it is more frequently found in actinobacteria (De Schrijver and De Mot, 1999; Panthee et al., 2020). Canonical LuxR from A. fischeri and S. purpurogeneiscleroticus, as well as PipR, displayed the same architecture composed of the autoinducer binding domain IPR005143 at the N-terminal end and the DNA-binding domain IPR000792 at the C-terminal. Unsurprisingly, in the LAL-LuxR subfamily member RevU protein, the autoinducer binding domain was replaced by an ATPase domain in addition to the large size of the protein (Figure 3).
None of the Arthrobacter sp. UMCV2 sequences showed architectures present in the model luxR sequences, although 11 sequences exhibited the DNA-binding domain IPR000792 (Figure 3). This is a HTH domain that was previously used as a criterion to include proteins in the luxR protein family (De Schrijver and De Mot, 1999; Lopes-Santos et al., 2012; Rajput and Kumar, 2017). We conclude that these 11 proteins belong to the LuxR transcription regulator family and are designated as Auto-inducer-related Regulators (AirR). Among these proteins, eight were grouped in cluster I (Figure 4) and six of them possessed a signal transduction response regulator receiver domain (IPR001789) that substitutes the autoinducer binding domain of canonical LuxR. In other proteins with HTH domains, the autoinducer binding domain is substituted either by an ATPase domain (as happens in RevU) or no amino-terminal domain was identified. Three of these sequences clustered with RevU from Streptomyces and LuxR or LuxR related to Streptomyces iranensis, Streptosporangium roseum A. terricola, and Arthrobacter spp., supporting the involvement of these proteins in the LuxR family. A separate group (cluster II of Figure 4) was found in which the canonical DNA-binding HTH domain was substituted with an OmpR/PhoB-type DNA-binding domain together with the same signal transduction response regulator receiver domain present in most of the cluster I AirR sequences (Figures 3, 4). We designated the auto-inducer analog regulator (AiaR) to this group because they form a sister cluster to AirR sequences deep inside the actinobacteria LuxR protein group but do not pose the HTH domain that defines LuxR protein, in addition to the analogous function of both domains with those present in canonical LuxR. Because no Lux-I homologous proteins were found in Arthrobacter sp. UMCV2, all AirR constitute “solo” proteins inside the luxR family.
Arthrobacter sp. UMCV2’s compound N, N-dimethylhexadecylamine induces auto-inducer-related regulators
LuxR solos have been extensively studied in Gram-negative bacteria. Some LuxR solos bind AHL from other bacterial species, probably as a strategy to expand their regulatory network by utilizing the existing components of the resident quorum-sensing systems (Martínez et al., 2015; Mosquito et al., 2020). Also, other LuxR solos have evolved to respond to different molecules including signals from plants (Patel et al., 2013). Although the functions of these proteins are under-studied in Gram-positive bacteria, especially in actinobacteria other than Streptomyces, the phylum has been phylogenetically explored by Lopes-Santos et al. (2012) and revised by Polkade et al. (2016). In A. aurescens and A. chlorophenolicus, multiple sequences of luxR homologs have been reported (16 and 17 sequences, respectively). However, no experimental work has been conducted (Lopes-Santos et al., 2012).
Because AirR proteins do not show the canonical LuxR autoinducer-binding domain, AHL is not expected to be the signal molecule for AirR proteins. However, a previous study revealed that DMHDA induced swarming motility in Arthrobacter sp. UMCV2 and simultaneously upregulates the expression of marker genes of the flagellar apparatus (Martínez-Cámara et al., 2020). Since swarming motility is frequently regulated by QS (Krishnan et al., 2012; Nickzad et al., 2015), in this study, swarming motility induction by DMHDA was reproduced and the expression of the airR genes was determined. It was found that 10 of the 16 luxR-related genes were statistically upregulated (Figure 6), but airR3 and airR5 (rich proteins clustered with RevU), and aiR1, airR2, and aiR7 were induced over 10-fold. Multiple inductions of airR genes suggest the existence of a hierarchical network of airR gene induction. In P. aeruginosa PAO1, four individual QS circuits have been described. Individual circuits are highly interconnected and involve auto-induction. The induction of an individual circuit by its auto-inductor results in the induction (or repression) of one or more of the other three (Papenfort and Bassler, 2016). Similarly, it is possible that DMHDA induction of an airR gene directly or indirectly upregulates other airR genes via unknown mechanisms. Therefore, additional studies are required to elucidate the function and regulation of airR genes.
In this study, the genomic analysis indicated that Arthrobacter sp. UMCV2 forms a separate branch within the genus Arthobacter, potentially constituting a new species. Genomic analysis also identified a group of 16 genes designed as airR, 10 of which were induced by Arthrobacter sp. UMCV2 compound DMHDA, which simultaneously induces QS behavior. To the best of our knowledge, this is the first report about the induction of the luxR gene family in the Arthrobacter genus by a bacterial self-compound.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, CP024915.1.
Author contributions
EV-C and GS: conceptualization. GM-H, EV-C, and JH-S: methodology. GS and GM-H: validation. MC-M, RM-C, and JH-S: formal analysis. MC-M, RM-C, and EV-C: investigation and writing – original draft preparation. EV-C: resources, supervision, project administration. EV-C, GS, and GM-H: writing – review and editing. All authors have read and agreed to the published.
Funding
We gratefully acknowledge the Valencia-Macías Foundation (México, Grant 8.1) and the Coordinación de la Investigación Científica-UMSNH (México, Grant 2.22) for financial support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.1040932/full#supplementary-material
Footnotes
References
1
Aframian N. Eldar A. (2020). A bacterial tower of babel: Quorum-sensing signaling diversity and its evolution.Annu. Rev. Microbiol.74587–606. 10.1146/annurev-micro-012220-063740
2
Alquéres S. Meneses C. Rouws L. Rothballer M. Baldani I. Schmid M. et al (2013). The bacterial superoxide dismutase and glutathione reductase are crucial for endophytic colonization of rice roots by Gluconacetobacter diazotrophicus PAL5.Mol. Plant Microbe Interact.26937–945. 10.1094/MPMI-12-12-0286-R
3
Aziz R. K. Bartels D. Best A. A. DeJongh M. Disz T. Edwards R. A. et al (2008). The rast server: Rapid annotations using subsystems technology.BMC Genom.9:75. 10.1186/1471-2164-9-75
4
Bez C. Covaceuzach S. Bertani I. Choudhary K. S. Venturi V. (2021). LuxR solos from environmental fluorescent pseudomonads.mSphere6e1322–e1320. 10.1128/mSphere.01322-20
5
Blum M. Chang H. Y. Chuguransky S. Grego T. Kandasaamy S. Mitchell A. et al (2021). The interPro protein families and domains database: 20 years on.Nucleic Acids Res.49D344–D354. 10.1093/nar/gkaa977
6
Brachmann A. O. Brameyer S. Kresovic D. Hitkova I. Kopp Y. Manske C. et al (2013). Pyrones as bacterial signaling molecules.Nat. Chem. Biol.9573–578. 10.1038/nchembio.1295
7
Brameyer S. Kresovic D. Bode H. B. Heermann R. (2014). LuxR solos in Photorhabdus species.Front. Cell. Infect. Microbiol.4:166. 10.3389/fcimb.2014.00166
8
Brameyer S. Kresovic D. Bode H. B. Heermann R. (2015). Dialkylresorcinols as bacterial signaling molecules.Proc. Natl. Acad. Sci. U. S. A.112572–577. 10.1073/pnas.1417685112
9
Bramhachari P. V. Yugandhar N. M. Prathyusha A. M. V. N. Mohana Sheela G. Naravula J. Venkateswarlu N. (2018). “Quorum sensing regulated swarming motility and migratory behavior in Bacteria,” in Implication of Quorum Sensing System in Biofilm Formation and Virulence, ed.PallavalV. B. (Singapore: Springer), 49–66. 10.1007/978-981-13-2429-1_5
10
Brettin T. Davis J. J. Disz T. Edwards R. A. Gerdes S. Olsen G. J. et al (2015). RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes.Sci. Rep.5:8365. 10.1038/srep08365
11
Case R. J. Labbate M. Kjelleberg S. (2008). AHL-driven quorum-sensing circuits: Their frequency and function among the Proteobacteria.ISME J.2345–349. 10.1038/ismej.2008.13
12
Castulo-Rubio D. Y. Alejandre-Ramírez N. A. Orozco-Mosqueda M. C. Santoyo G. Macías-Rodríguez L. I Valencia-Cantero E. (2015). Volatile organic compounds produced by the rhizobacterium Arthrobacter agilis UMCV2 modulate Sorghum bicolor (strategy II plant) morphogenesis and SbFRO1 transcription in vitro.J. Plant Growth Regul.34611–623. 10.1007/s00344-015-9495-8.20
13
Choi S. Greenberg E. P. (1991). The C-terminal region of the Vibrio fischeri LuxR protein contains an inducer-independent lux gene activating domain.Proc. Natl. Acad. Sci. U. S. A.8811115–11119. 10.1073/pnas.88.24.11115
14
Chun J. Oren A. Ventosa A. Christensen H. Arahal D. R. da Costa M. S. et al (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes.Int. J. Syst. Evol. Microbiol.68461–466. 10.1099/ijsem.0.002516
15
Coutinho B. G. Mevers E. Schaefer A. L. Pelletier D. A. Harwood C. S. Clardy J. et al (2018). A plant-responsive bacterial-signaling system senses an ethanolamine derivative.Proc. Natl. Acad. Sci. U. S. A.1159785–9790. 10.1073/pnas.1809611115
16
De Schrijver A. De Mot R. (1999). A subfamily of MalT-related ATP-dependent regulators in the LuxR family.Microbiology1451287–1288. 10.1099/13500872-145-6-1287
17
Dufour D. Lévesque C. M. (2013). Bacterial behaviors associated with the quorum-sensing peptide pheromone (‘alarmone’) in streptococci.Future Microbiol.8593–605. 10.2217/fmb.13.23
18
Enkataraman A. Rosenbaum M. A. Werner J. J. Winans S. C. Angenent L. T. (2014). Metabolite transfer with the fermentation product 2, 3-butanediol enhances virulence by Pseudomonas aeruginosa.ISME J.81210–1220. 10.1038/ismej.2013.232
19
Farris J. S. (1972). Estimating phylogenetic trees from distance matrices.Am. Nat.106645–667. 10.1086/282802
20
Felsenstein J. (1985). Confidence limits on phylogenies: An approach using the bootstrap.Evolution39783–791. 10.1111/j.1558-5646.1985.tb00420.x
21
Ferluga S. Venturi V. (2009). OryR is a LuxR-family protein involved in interkingdom signaling between pathogenic Xanthomonas oryzae pv. oryzae and rice.J. Bacteriol.191890–897. 10.1128/JB.01507-08
22
Flegler A. Runzheimer K. Kombeitz V. Mänz A. T. von Heilborn D. H. Etzbach L. et al (2020). Arthrobacter bussei sp. nov., a pink-coloured organism isolated from cheese made of cow’s milk.Int. J. Syst. Evol. Microbiol.703027–3036. 10.1099/ijsem.0.004125
23
Fuqua C. (2006). The QscR quorum-sensing regulon of Pseudomonas aeruginosa: An orphan claims its identity.J. Bacteriol.1883169–3171. 10.1128/JB.188.9.3169-3171.2006
24
Fuqua C. Parsek M. R. Greenberg E. P. (2001). Regulation of gene expresión by cell-to-cell communication:acyl-homoserine lactone quorum sensing.Annu. Rev. Genet.35439–468. 10.1146/annurev.genet.35.102401.090913
25
Fuqua C. Winans S. C. Greenberg E. P. (1996). Census and consensus in bacterial ecosystems: The LuxR-LuxI family of quorum-sensing transcriptional regulators.Annu. Rev. Microbiol.50727–751. 10.1146/annurev.micro.50.1.727
26
Fuqua W. C. Winans S. C. Greenberg E. P. (1994). Quorum sensing in bacteria: The LuxR-LuxI family of cell density-responsive transcriptional regulators.J. Bacteriol.176269–275. 10.1128/jb.176.2.269-275.1994
27
Galet J. Deveau A. Hôtel L. Frey-Klett P. Leblond P. Aigle B. (2015). Pseudomonas ?uorescens pirates both ferrioxamine and ferricoelichelin siderophores from Streptomyces ambofaciens.Appl. Environ. Microbiol.813132–3141. 10.1128/AEM.03520-14
28
González J. F. Venturi V. (2013). A novel widespread interkingdom signaling circuit.Trends Plant Sci.18167–174. 10.1016/j.tplants.2012.09.007
29
González J. F. Myers M. P. Venturi V. (2013). The inter-kingdom solo OryR regulator of Xanthomonas oryzae is important for motility.Mol. Plant Pathol.14211–221. 10.1111/j.1364-3703.2012.00843.x
30
Hawver L. A. Jung S. A. Ng W. L. (2016). Specificity and complexity in bacterial quorum-sensing systems.FEMS Microbiol. Rev.40738–752. 10.1093/femsre/fuw014
31
Hernández-Soberano C. Ruiz-Herrera L. F. Valencia-Cantero E. (2020). Endophytic bacteria Arthrobacter agilis UMCV2 and Bacillus methylotrophicus M4-96 stimulate achene germination, in vitro growth, and greenhouse yield of strawberry (Fragaria × ananassa).Sci. Hortic.261:109005. 10.1016/j.scienta.2019.109005
32
Jones D. T. Taylor W. R. Thornton J. M. (1992). The rapid generation of mutation data matrices from protein sequences.Bioinformatics8275–282. 10.1093/bioinformatics/8
33
Kan J. Fang R. Jia Y. (2017). Interkingdom signaling in plant-microbe interactions.Sci. China Life Sci.60785–796. 10.1007/s11427-017-9092-3
34
Kim K. S. Lee S. Ryu C. (2013). Interespecific bacterial sensing through airbone signal modulates locomotion and drung resistance.Nat. Commun.4:1809. 10.1038/ncomms2789
35
Kim Y. C. Miller C. D. Anderson A. J. (2000). Superoxide dismutase activity in Pseudomonas putida affect utilization of sugars and growth on root surfaces.Appl. Environ. Microbiol.661460–1467. 10.1128/AEM.66.4.1460-1467.2000
36
Koster W. (1991). Iron (lll) hydroxamate transport across the cytoplasmic membrane of Escherichia coli.Biol. Met.423–32. 10.1007/BF01135553
37
Krishnan T. Yin W. F. Chan K. G. (2012). Inhibition of quorum sensing-controlled virulence factor production in Pseudomonas aeruginosa PAO1 by ayurveda spice clove (Syzygium Aromati cum) bud extract.Sensors124016–4030. 10.3390/s120404016
38
Kumar S. Stecher G. Li M. Knyaz C. Tamura K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms.Mol. Biol. Evol.351547–1549. 10.1093/molbev/msy096
39
Lee J. Y. Hyun D. W. Kim P. S. Kim H. S. Shin N. R. Yun J. H. et al (2016). Arthrobacter echini sp. nov., isolated from the gut of a purple sea urchin, Heliocidaris crassispina.Int. J. Syst. Evol. Microbiol.661887–1893. 10.1099/ijsem.0.000965
40
Lefort V. Desper R. Gascuel O. (2015). FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program.Mol. Biol. Evol.322798–2800. 10.1093/molbev/msv150
41
Lin P. Yan Z. F. Li C. T. (2020). Arthrobacter sedimenti sp. nov., isolated from river sediment in Yuantouzhu park, China.Arch. Microbiol.2022551–2556. 10.1007/s00203-020-01968-y
42
Liu Q. Xin Y. H. Chen X. L. Liu H. C. Zhou Y. G. Chen W. X. (2018). Arthrobacter ruber sp. nov., isolated from glacier ice.Int. J. Syst. Evol. Microbiol.681616–1621. 10.1099/ijsem.0.002719
43
Livak K. J. Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method.Methods25402–408. 10.1006/meth.2001.1262
44
Lopes-Santos C. Correia-Neves M. Moradas-Ferreira P. Mendes M. V. (2012). A walk into the LuxR regulators of actinobacteria: Phylogenomic distribution and functional diversity. PLoS One7:e46758. 10.1371/journal.pone.004675
45
Lyon G. J. Novick R. P. (2004). Peptide signaling in Staphylococcus aureus and other Gram-positive bacteria.Peptides251389–1403. 10.1016/j.peptides.2003.11.026
46
Mansurova M. Ebert B. E. Blank L. M. Ibañez A. J. (2018). A breath of information: The volatilome.Curr. Genet.64959–964. 10.1007/s00294-017-0800-x
47
Martínez P. Huedo P. Martinez-Servat S. Planell R. Ferrer-Navarro M. Daura X. et al (2015). Stenotrophomonas maltophilia responds to exogenous AHL signals through the LuxR solo SmoR (Smlt1839).Front. Cell. Infect. Microbiol.5:41. 10.3389/fcimb.2015.00041
48
Martínez-Cámara R. Montejano-Ramírez V. Moreno-Hagelsieb G. Santoyo G. Valencia-Cantero E. (2020). The volatile organic compound dimethylhexadecylamine affects bacterial growth and swarming motility of bacteria.Folia Microbiol.65523–532. 10.1007/s12223-019-00756-6
49
Meier-Kolthoff J. P. Göker M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy.Nat. Commun.10:2182. 10.1038/s41467-019-10210-3
50
Meier-Kolthoff J. P. Auch A. F. Klenk H.-P. Göker M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions.BMC Bioinform.14:60. 10.1186/1471-2105-14-60
51
Meier-Kolthoff J. P. Sardà Carbasse J. Peinado-Olarte R. L. Göker M. (2022). TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes.Nucleic Acid Res.50D801–D807. 10.1093/nar/gkab902
52
Miller M. B. Bassler B. L. (2001). Quorum sensing in bacteria.Annu. Rev. Microbiol.55165–199. 10.1146/annurev.micro.55.1.165
53
Monnet V. Gardan R. (2015). Quorum-sensing regulators in gram-positive bacteria ‘cherchez le peptide’.Mol. Microbiol.97181–184. 10.1111/mmi.13060
54
Monnet V. Juillard V. Gardan R. (2014). Peptide conversations in gram-positive bacteria.Crit. Rev. Microbiol.42339–351. 10.3109/1040841X.2014.948804
55
Montejano-Ramírez V. García-Pineda E. Valencia-Cantero E. (2020). Bacterial compound N, N-dimethylhexadecylamine modulates expression of iron deficiency and defense response genes in Medicago truncatula independently of the jasmonic acid pathway.Plants9:624. 10.3390/plants9050624
56
Montejano-Ramírez V. Mártínez-Cámara R. García-Pineda E. Valencia-Cantero E. (2018). Rhizobacterium Arthrobacter agilis UMCV2 increases organ-specific expression of FRO genes in conjunction with genes associated with the systemic resistance pathways of Medicago truncatula.Acta Physiol. Plant.40:138. 10.1007/s11738-018-2712-x
57
Mosquito S. Meng X. Devescovi G. Bertani I. Geller A. M. Levy A. et al (2020). LuxR solos in the plant endophyte Kosakonia sp. strain KO348.Appl. Environ. Microbiol.86e622–e620. 10.1128/AEM.00622-20
58
Nickzad A. Lépine F. Déziel E. (2015). Quorum sensing controls swarming motility of Burkholderia glumae through regulation of rhamnolipids.PLoS One10:e0128509. 10.1371/journal.pone.0128509
59
Orozco-Mosqueda M. C. Velázquez-Becerra C. Macías-Rodríguez L. I. Santoyo G. Flores-Cortez I. Alfaro-Cuevas A. et al (2013). Arthrobacter agilis UMCV2 induces iron acquisition in Medicago truncatula (Strategy I Plant) in vitro via dimethylhexadecylamine Emission.Plant Soil36251–66. 10.1007/s11104-012-1263-y
60
Panthee S. Kito N. Hayashi T. Shimizu T. Ishikawa J. Hamamoto H. et al (2020). β-carboline chemical signals induce reveromycin production through a LuxR family regulator in Streptomyces sp. SN-593.Sci. Rep.10:10230. 10.1038/s41598-020-66974-y
61
Papenfort K. Bassler B. L. (2016). Quorum sensing signal-response systems in gram-negative bacteria.Nat. Rev. Microbiol.14576–588. 10.1038/nrmicro.2016.89
62
Patankar A. V. González J. E. (2009). Orphan LuxR regulators of quorum sensing.FEMS Microbiol. Rev.33739–756. 10.1111/j.1574-6976.2009.00163.x
63
Patel H. K. Suárez-Moreno Z. R. Degrassi G. Subramoni S. González J. F. Venturi V. (2013). Bacterial LuxR solos have evolved to respond to different molecules including signals from plants.Front. Plant Sci.4:447. 10.3389/fpls.2013.00447
64
Polkade A. V. Mantri S. S. Patwekar U. J. Jangid K. (2016). Quorum Sensing: An under-explored phenomenon in the Phylum Actinobacteria.Front. Microbiol.7:131. 10.3389/fmicb.2016.00131
65
Rajput A. Kumar M. (2017). In silico analyses of conservational, functional and phylogenetic distribution of the LuxI and LuxR homologs in gram-positive bacteria.Sci. Rep.7:6969. 10.1038/s41598-017-07241-5
66
Schulz-Bohm K. Martin-Sanchez L. Garbeva P. (2017). Microbial volatiles: Small molecules with an important role in intra- and interkindom interactions.Front. Microbiol.8:2484. 10.3389/fmicb.2017.02484
67
Shadel G. S. Young R. Baldwin T. O. (1990). Use of regulated cell lysis in a lethal genetic selection in Escherichia coli: Identification of the autoinducer-binding region of the LuxR protein from Vibrio fischeri ATCC7744.J. Bacteriol.1723980–3987. 10.1128/jb.172.7.3980-3987.1990
68
Slock J. VanRiet D. Kolibachuk D. Greenberg E. P. (1990). Critical regions of the Vibrio fischeri LuxR protein defined by mutational analysis.J. Bacteriol.1723974–3979. 10.1128/jb.172.7.3974-3979.1990
69
Swift S. Bainton N. J. Winson M. K. (1994). Gram-negative bacterial communication by N-acyl homoserine lactones: A universal language?Trends Microbiol.2193–198. 10.1016/0966-842x(94)90110-q
70
Valencia-Cantero E. Hernández-Calderón E. Velázquez-Becerra C. López-Meza J. E. Alfaro-Cuevas R. López-Bucio J. (2007). Role of dissimilatory fermentative iron-reducing bacteria in Fe uptake by common bean (Phaseolus vulgaris L.) plants grown in alkaline soil.Plant Soil291263–273. 10.1007/s11104-007-9191-y
71
Vázquez-Chimalhua E. Barrera-Ortiz S. Valencia-Cantero E. López-Bucio J. Ruiz-Herrera L. F. (2021a). The bacterial volatile N,N-dimethyl-hexadecylamine promotes Arabidopsis primary root elongation through cytokinin signaling and the AHK2 receptor.Plant Signal. Behav.16:1879542. 10.1080/15592324.2021.1879542
72
Vázquez-Chimalhua E. Ruíz-Herrera L. F. Barrera-Ortiz S. Valencia-Cantero E. López-Bucio J. (2019). The bacterial volatile dimethyl-hexa-decilamine reveals an antagonistic interaction between jasmonic acid and cytokinin in controlling primary root growth of Arabidopsis seedlings.Protoplasma256643–654. 10.1007/s00709-018-1327-9
73
Vázquez-Chimalhua E. Valencia-Cantero E. López-Bucio J. Ruiz-Herrera L. F. (2021b). N, N, dimethyl-hexadecylamine modulates Arabidopsis root growth through modifying the balance between stem cell niche and jasmonic acid-dependent gene expression.Gene Expr. Patterns41:119201. 10.1016/j.gep.2021.119201
74
Velázquez-Becerra C. Macias-Rodriguez L. I. Lopez-Bucio J. Altamirano-Hernandez J. Flores-Cortez I. Valencia-Cantero E. (2011). A volatile organic compound analysis from Arthrobacter agilis identifies dimethylhexadecylamine, an amino-containing lipid modulating bacterial growth and Medicago sativa morphogenesis in vitro.Plant Soil339329–340. 10.1007/s11104-010-0583-z
75
Velázquez-Becerra C. Macías-Rodríguez L. I. Lopez-Bucio J. Flores-Cortez I. Santoyo G. Hernandez-Soberano C. et al (2013). The rhizobacterium Arthrobacter agilis produces dimethylhexadecylamine, a compound that inhibits growth of phytopathogenic fungi in vitro.Protoplasma2501251–1262. 10.1007/s00709-013-0506-y
76
Venturi V. Fuqua C. (2013). Chemical signaling between plants and plant pathogenic bacteria.Annu. Rev. Phytopathol.5117–37. 10.1146/annurev-phyto-082712-102239
77
Wang L. Jeon B. Sahin O. Zhang Q. (2009). Identification of an arsenic resistance and arsenic-sensing system in Campylobacter jejuni.Appl. Environ. Microbiol.755064–5073. 10.1128/AEM.00149-09
78
Waters C. M. Bassler B. L. (2005). Quorum sensing: Cell-to-cell communication in bacteria.Annu. Rev. Cell Dev. Biol.21319–346. 10.1146/annurev.cellbio.21.012704.131001
79
Whiteley M. Diggle S. P. Greenberg E. P. (2017). Progress in and promise of bacterial quorum sensing research.Nature551313–320. 10.1038/nature24624
80
Xie S. Zang M. Wu H. Rajer F. U. Gao X. (2018). Antibacterial effects of volatiles produced by Bacillus strain D13 against Xanthomonas oryzae pv oryzae.Mol. Plant Pathol.1949–58. 10.1111/mpp.12494
81
Xu G. (2020). Evolution of LuxR solos in bacterial communication: Receptors and signals.Biotechnol. Lett.42181–186. 10.1007/s10529-019-02763-6
82
Yang L. L. Liu H. C. Liu Q. Xin Y. H. (2021). Arthrobacter cheniae and Arthrobacter frigidicola sp. nov., isolated from a glacier.Int. J. Syst. Evol. Microbiol.71:5177. 10.1099/ijsem.0.005177
83
Zavilgelsky G. B. Manukhov I. V. (2001). Quorum sensing, or how bacteria “talk” to each other.Mol. Biol.35224–232. 10.1023/A:1010439501530
84
Zhang L. Jia Y. Wang L. Fang R. (2007). A proline iminopeptidase gene upregulated in planta by a LuxR homologue is essential for pathogenicity of Xanthomonas campestris pv. campestris.Mol. Microbiol.65121–136. 10.1111/j.1365-2958.2007.05775.x
Summary
Keywords
actinobacteria, LuxR solos, domain approach, airR genes, aiaR genes, swarming motility
Citation
Chávez-Moctezuma MP, Martínez-Cámara R, Hernández-Salmerón J, Moreno-Hagelsieb G, Santoyo G and Valencia-Cantero E (2022) Comparative genomic and functional analysis of Arthrobacter sp. UMCV2 reveals the presence of luxR-related genes inducible by the biocompound N, N-dimethylhexadecilamine. Front. Microbiol. 13:1040932. doi: 10.3389/fmicb.2022.1040932
Received
09 September 2022
Accepted
10 October 2022
Published
31 October 2022
Volume
13 - 2022
Edited by
Manoj Kumar Solanki, University of Silesia in Katowice, Poland
Reviewed by
Bipransh Kumar Tiwary, North Bengal St. Xavier’s College, India; Anton Hartmann, Ludwig Maximilian University of Munich, Germany
Updates
Copyright
© 2022 Chávez-Moctezuma, Martínez-Cámara, Hernández-Salmerón, Moreno-Hagelsieb, Santoyo and Valencia-Cantero.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduardo Valencia-Cantero, eduardo.cantero@umich.mx
This article was submitted to Microbe and Virus Interactions with Plants, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.