 Irene Sánchez-Andrea1*†
Irene Sánchez-Andrea1*† Charlotte M. van der Graaf1†
Charlotte M. van der Graaf1† Bastian Hornung2
Bastian Hornung2 Nicole J. Bale3
Nicole J. Bale3 Monika Jarzembowska1
Monika Jarzembowska1 Diana Z. Sousa1
Diana Z. Sousa1 W. Irene C. Rijpstra3
W. Irene C. Rijpstra3 Jaap S. Sinninghe Damsté3,4
Jaap S. Sinninghe Damsté3,4 Alfons J. M. Stams1,5
Alfons J. M. Stams1,5- 1Laboratory of Microbiology, Wageningen University & Research, Wageningen, Netherlands
- 2Laboratory of Systems and Synthetic Biology, Wageningen University & Research, Wageningen, Netherlands
- 3Department of Marine Microbiology and Biogeochemistry, NIOZ Royal Netherlands Institute for Sea Research, Den Burg, Netherlands
- 4Department of Earth Sciences, Faculty of Geosciences, Utrecht University, Utrecht, Netherlands
- 5Centre of Biological Engineering, University of Minho, Braga, Portugal
In acid drainage environments, biosulfidogenesis by sulfate-reducing bacteria (SRB) attenuates the extreme conditions by enabling the precipitation of metals as their sulfides, and the neutralization of acidity through proton consumption. So far, only a handful of moderately acidophilic SRB species have been described, most of which are merely acidotolerant. Here, a novel species within a novel genus of moderately acidophilic SRB is described, Acididesulfobacillus acetoxydans gen. nov. sp. nov. strain INE, able to grow at pH 3.8. Bioreactor studies with strain INE at optimum (5.0) and low (3.9) pH for growth showed that strain INE alkalinized its environment, and that this was more pronounced at lower pH. These studies also showed the capacity of strain INE to completely oxidize organic acids to CO2, which is uncommon among acidophilic SRB. Since organic acids are mainly in their protonated form at low pH, which increases their toxicity, their complete oxidation may be an acid stress resistance mechanism. Comparative proteogenomic and membrane lipid analysis further indicated that the presence of saturated ether-bound lipids in the membrane, and their relative increase at lower pH, was a protection mechanism against acid stress. Interestingly, other canonical acid stress resistance mechanisms, such as a Donnan potential and increased active charge transport, did not appear to be active.
Introduction
Acid rock and acid mine drainage (ARD and AMD) environments are among the most extreme habitats on Earth due to their high acidity and high metal concentrations. Acid drainage is generated by the oxidative dissolution of metal sulfides to sulfuric acid and dissolved metals upon exposure to water and oxidants such as oxygen (O2) or ferric iron (Fe3+; Sand et al., 2001; Johnson and Hallberg, 2005). This can be triggered naturally (ARD) or by mining operations (AMD; Lottermoser, 2010). The Tinto River, which flows through the massive sulfide deposits of the Iberian Pyrite Belt (IPB; Huelva, Southwestern Spain), is one of the best-known examples of ARD. Although mining activity reportedly occurred in the IPB already >5,000 years ago, the acid drainage does not exclusively originate on the surface by mining activities but also in the subsurface, as a result of chemolithotrophic microbial activity (Amils et al., 2011).
Microorganisms thriving in acid drainage environments are polyextremophiles, as they need to withstand high acidity (the mean pH of the water column of the Tinto River is 2.3), extremely high metal concentrations (e.g., Fe 2.26 g/L, Cu 0.11 g/L, and Zn 0.24 g/L; López-Archilla et al., 2001) and high salinity due to high sulfate concentrations. Although much emphasis has been placed on the extreme acidophiles involved in the iron cycle and the oxidative part of the sulfur cycle, studies of anoxic Tinto River sediments showed the importance of more moderately acidophilic microorganisms, most notably acidophilic sulfate-reducing bacteria (SRB; Sánchez-Andrea et al., 2011, 2012b). SRB have been shown to have an attenuating effect on the extreme characteristics of the Tinto River through the formation of sulfide (biosulfidogenesis), enabling the precipitation of metals as metal sulfides, and the consumption of protons required for sulfate reduction at low pH, resulting in alkalinization (Sánchez-Andrea et al., 2012a). In specific layers of Tinto River sediments, a higher pH (up to 6.2), a lower redox potential, and lower sulfate and metal concentrations were accompanied by a higher abundance of SRB from the family Peptococcaceae (Sánchez-Andrea et al., 2012a), representing up to 40% of the total cell counts in the sediment layers. Geomicrobiological studies of abandoned open pit mines filled with AMD revealed that, similar to observations in AMD sediments, biosulfidogenesis by SRB also had an attenuating effect on acidity and metal concentrations in the water column (Wendt-Potthoff et al., 2012; Santofimia et al., 2013; Falagán et al., 2014; Sánchez-España et al., 2020; van der Graaf et al., 2020).
The in-depth study of the physiology and dominant stress resistance mechanisms of acidophilic SRB requires pure culture isolates. So far, only a few moderately acidophilic or acidotolerant SRB species have been described, Thermodesulfobium narugense (pH 4.0–6.5; Mori et al., 2003) and T. acidiphilum (pH 3.7–6.5; Frolov et al., 2017), both from the genus Thermodesulfobium within the family Thermodesulfobiaceae; Desulfothermobacter acidiphilus (pH 2.9–6.5; Frolov et al., 2018) from the family Thermoanaerobacteraceae; and Desulfosporosinus acididurans (pH 3.8-7.0; Sánchez-Andrea et al., 2015), D. acidiphilus (pH 3.6–5.5; Alazard et al., 2010), and D. metallidurans (pH 4.0–7.0; Panova et al., 2021) from the genus Desulfosporosinus in the family Peptococcaceae of the Clostridia class in the Firmicutes phylum. Here, we report the characterization of a novel moderately acidophilic SRB from a novel genus, Acididesulfobacillus acetoxydans strain INE, enriched from Tinto River sediments (Sánchez-Andrea et al., 2013). We use comparative proteomics and cell membrane lipid analysis performed on cells grown at optimum (5.0) and low (3.9) pH of growth to determine the main strategies enabling A. acetoxydans to grow at acidic pH.
Materials and Methods
Microorganisms
Strain INE was isolated from an enrichment culture containing acidic sediments from JL dam (37.691207 N, 6.560587 W) in the Tinto River basin (southwestern Spain) by incubation with 5 mM of lactate and 10 mM sulfate at pH 4.5 (Sánchez-Andrea et al., 2013). Detailed physicochemical information on the site has been published previously (Sánchez-Andrea et al., 2012a). For comparison purposes, D. orientis DSM 765T, Desulfitobacterium dehalogenans DSM 9161T and D. acidiphilus DSM 22704T were obtained from the DSMZ (Braunschweig, Germany). Desulfosporosinus acididurans DSM 27692T was taken from the laboratory collection.
Phenotypic Characterization: Morphology and Physiology
Cell morphology, motility and spore formation were examined by phase contrast microscopy using a Leica DM2000 microscope (Leica, Hesse, Germany). Cell size was determined by SEM microscopy. To this end, samples were fixed by immersion in glutaraldehyde (2.5%) for 2 h, and subsequently washed twice in sodium cacodylate buffer (0.2 M, pH 7.1). Samples were dehydrated in a graded series (10%, 30%, 50%, 70%, 90%, and 100%) of ethanol/water mixtures, leaving them 20 min in each mixture. After dehydration, samples were critical point dried and mounted on stubs. After gold shadowing, samples were examined in a Phillips XL30 EDAX DX4i SEM (Philips, Netherlands). The lengths and widths of several cells were measured, and mean dimensions recorded. Gram staining was performed according to standard procedures (Doetsch, 1981), and Gram-structure was checked by mixing cells with a drop of 3% (w/v) solution of KOH.
Gram-structure was further checked by TEM microscopy. For this, cells were pelleted, incubated in fixative solution (2.5% glutaraldehyde, 2% paraformaldehyde in a 0.1 M phosphate citrate buffer pH 5.0) for 1 h at room temperature, washed twice in 0.1 M phosphate citrate buffer (wash buffer), resuspended in 100 ml 2% gelatin in 0.1 M phosphate citrate buffer and incubated at 4°C until gelatin solidified. The gelatin pellet was incubated for 15 min at room temperature in fixative solution, cut into small pieces (maximum 0.5 cm3), and incubated for 30 min at room temperature. Gelatin sections were washed six times with wash buffer, stained and fixed in a 1% osmium tetroxide solution for 1 h at room temperature. The samples were then washed three times in distilled water, dehydrated in successive ethanol incubations (30%, 50%, 70%, 80%, 90%, and 96%), with a final dehydration step in 100% ethanol. Samples were embedded in Spurr epoxy resin as described previously (Spurr, 1969), cut into ultrathin sections with a Leica EM UC7 ultramicrotome (Leica, Wetzlar, Hesse, Germany), and poststained with uranyl-acetate and lead citrate. Samples were imaged using a JEOL JEM-1400 series 120 kV TEM (JEOL, Tokyo, Japan).
Catalase activity was determined by reaction with 15% (w/v) solution of H2O2. An oxidase test was performed with a filter impregnated in 1% (w/v) solution of tetramethyl-p-phenylenediamine in dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO). Indole and urease formation as well as gelatin and esculin hydrolysis were determined with API® 20A (bioMérieux, France) according to manufacturer’s instructions. Analysis of respiratory quinones of biomass grown on glycerol was carried out by the DSMZ Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany; Tindall, 1990a,b).
Growth experiments were performed in triplicate, using 120 ml-serum bottles as described elsewhere (Sánchez-Andrea et al., 2013). Growth conditions were pH 5.0 and Ta = 30°C, unless indicated otherwise. Growth was monitored by measuring optical density at 600 nm (OD600) with a spectrophotometer (U-1500 Hitachi, Tokyo, Japan). Soluble substrates and intermediates (sugars and volatile fatty acids) were measured using a thermo electron spectrasystem high performance liquid chromatography (HPLC) equipped with an Agilent Metacarb 67H column. Gaseous compounds (H2) were analyzed using a Shimadzu GC-2014 Gas Chromatograph equipped with a Mol sieve 13X column. Sulfide was measured photometrically with the methylene blue method (Cline, 1969). Different electron donors and acceptors were tested at final concentrations of 5 or 10 mM, respectively, except sulfite, which was tested at 5 mM. Electron donors and acceptors were added to the media from sterile 1 M stock solutions. When electron donors were tested, sulfate (10 mM) was used as acceptor and when acceptors were tested, glycerol (5 mM) was used as donor. Growth was also studied at different incubation temperatures (from 10°C to 45°C), pH (from 3.0 to 7.5), salinity (up to 0.5 M NaCl), oxygen concentrations, and with different reducing agents: L-cysteine, sodium sulfide, titanium citrate, and FeCl2 (2 mM). Sulfate reduction rates were determined during the exponential phase of the cultures grown between pH 4 and 7 at 30°C.
Bioreactor Cultivation
Bioreactor cultivation was performed in duplicate in two pH-controlled batch reactors at pH 3.9 and pH 5.0. Basal medium was supplemented with 0.1 g yeast extract L−1, 5 mM glycerol, 10 mM sulfate, and 2 mM L-cysteine as reducing agent. Glass reactors (Applikon, Netherlands) of 5 L volume capacity filled with 4 L of medium were operated with an ADI 1010 Bio-controller and the ADI 1025 Bio-console (Applikon). Autoclavation of the whole system enabled reactor operation under sterile conditions. Stirring speed was 50 rpm, temperature was maintained at 30°C, and pH was maintained at pH 5.0 or pH 3.9 by addition of 0.5 M HCl. The growth rates at pH 3.9 and pH 5.0 were calculated from semi-logarithmic plots of changes in OD600 values against time at 30°C. Values were obtained by implementing the modified Gompertz model (Zwietering et al., 1990). Biomass for lipid and proteome analysis was obtained from the reactors at 80% of maximum OD600.
Membrane Lipid Analysis
For comparison of the cellular fatty acid composition all strains were grown at their optimum pH in the medium described elsewhere (Sánchez-Andrea et al., 2013). Bicarbonate buffer was added to the medium for the cultivation of neutrophilic strains. For A. acetoxydans strain INE, biomass grown at both pH 3.9 and 5.0 was analyzed. Fatty acid analyses were carried out by acid hydrolysis of total cell material with 5% HCl in methanol by refluxing for 3 h, and analysis by gas chromatography, gas chromatography–mass spectrometry, following procedures described previously (Sinninghe Damsté et al., 2011), and were performed in duplicate. Intact polar lipids (IPLs) were extracted from freeze-dried biomass using a modified Bligh–Dyer procedure and analyzed by ultra-high-pressure liquid chromatography–high resolution mass spectrometry (UHPLC-HRMS) under conditions described previously (Bale et al., 2019).
Phylogenetic Analysis
Cloning of the 16S rRNA gene was performed to determine the phylogenetic affiliation of strain INE. Total genomic DNA was extracted using the FastDNA® SPIN Kit for Soil and the FastPrep® Instrument (MP Biomedicals, Santa Ana, CA). The 16S rRNA genes were amplified with the primers set 27F-1492R (Ta = 57°C) for Bacteria and cloned in Escherichia coli DH5α competent cells by using the pGEM-T vector (Promega, Madison, WI). Sequences were assembled with the DNABaser software 3.5.3 and prior to phylogenetic analysis, vector sequences flanking the 16S rRNA gene inserts were identified using the VecScreen tool (NCBI) and removed.1 Clone sequences were checked for chimeras (Wright et al., 2012), aligned with SINA (v1.2.11; Pruesse et al., 2012), and added to a database of over 230,000 homologous prokaryotic 16S rRNA gene primary structures by using the merging tool of the ARB program package (Ludwig et al., 2004). Sequences were then manually corrected with the alignment tool of the same software and added by parsimony to the tree generated in the Living Tree Project (LTP; Yarza et al., 2008). Phylogenetic reconstruction was performed using the three algorithms as implemented in the ARB package. The maximum-likelihood method was used for the generation of the consensus tree and bootstrap analysis performance. The bar indicates 10% estimated sequence divergence. The 16S rRNA gene sequence is available in the EMBL database under accession number LN551924.
Genomic Analysis and Annotation Workflow
Genomic DNA was extracted with the MasterPure™ Gram Positive DNA Purification Kit (Epicentre, Madison, Wisconsin). Pacific Biosciences sequencing was performed on a PacBio RSII at GATC Biotech (Konstanz, Germany), and Illumina sequencing on a MiSeq sequencer (250 bp paired-end with 500 bp insert). The genome was assembled with the smrt analysis pipeline version 2.3.0 and the protocol HGAP version 3. After assembly, the taxonomy of all contigs was determined with megablast (Altschul et al., 1990) and a custom version of the LCA algorithm (Huson et al., 2007). In detail, a blast search was performed against various database of the NCBI (NCBI NT database, the draft bacteria genome database, the human genome, the protozoa database; NCBI Resource Coordinators, 2013), the Hungate 1,000 database (Seshadri et al., 2018) and the human microbiome database (Human Microbiome Project, 2012), using an e value of 0.0001. A custom script with the implementation of the LCA algorithm was used (Huson et al., 2007), with the exception that only the hits exceeding a bitscore of 50 and not deviating >10% in length from the longest hit were used to determine the taxonomy. After the taxonomy was obtained, all contigs not belonging to the family Peptococcaceae were discarded as contamination. A correction of the PacBio assembly was performed with Pilon 1.19 (Walker et al., 2014), and the Illumina MiSeq data, which was mapped to the assembly with bowtie2 (Langmead and Salzberg, 2012). The Illumina MiSeq data were additionally used as verification that the discarded contigs of the PacBio assembly were contaminating contigs. Average amino acid identity (AAI) was calculated with compareM v0.1.1.2
The annotation of the genome was performed with an early version of SAPP (Koehorst et al., 2018). Gene calling was performed with prodigal 2.6.3 (Hyatt et al., 2010). RNA genes were predicted with RNAmmer 1.2 (Lagesen et al., 2007), Aragorn 1.2.36 (Laslett and Canback, 2004), and the CRISPR Recognition tool version 1.2 (Bland et al., 2007). Protein coding genes were further annotated with InterproScan 5.19–58.0, PRIAM version March 2015 (Claudel-Renard et al., 2003), EnzDP version 1.0 (Nguyen et al., 2015), tmHMM version 2.0c (Krogh et al., 2001), and SignalP 4.1 (Petersen et al., 2011). Additional EC numbers were derived via Gene Ontology terms (Ashburner et al., 2000) predicted by InterproScan. Furthermore, blastp searches against the COG database (Galperin et al., 2015) and the Swissprot database (Release 06 Jul 2016; UniProt, 2014) were performed. The annotations were partially manually curated. To avoid repetition, locus ID is displayed without the locus tag “DEACI_.”
Proteomics
Duplicate samples at each pH value of the biomass grown in reactors (pH 3.5 A + B and pH 5.0 A + B) were taken at 80% of maximum OD600, centrifuged, washed, and transferred to 2-ml low-binding microcentrifuge tubes (Eppendorf, Netherlands) prior to protein extraction. Around 0.1 g of wet weight pellet was resuspended in 500 ml of 4% sodium dodecyl sulfate in 100 mM Tris-HCl pH 7.6. Cells were lysed by sonication with a 3-mm tip using a Branson sonifier in six pulses of 30 s. Around 40 μg of protein was separated by gel electrophoresis (SDS-PAGE) and cut into five slices per sample to fractionate proteins. The gel slices were subjected to in-gel tryptic digestion and peptides were loaded on STAGE-tips (STop And Go Elution, C18-reversed phase SPE) for desalting and concentration.
The resulting purified and concentrated peptide mixtures were analyzed by nanoflow C18 reversed phase HPLC coupled with a maXis 4G ETD ultra-high resolution Qq-TOF mass spectrometer (Bruker Daltonics) with collision-induced dissociation (CID) as fragmentation technique. Chromatographic separation was achieved via a linear gradient of 7–32% acetonitrile in 90 min using 0.1% formic acid as ion pair reagent. For data analysis, a custom Acididesulfobacillus database with added sequences for typical contaminant proteins (e.g., skin and hair proteins) was used. For database searches, validation, and relative quantification the MaxQuant software package was used (version 1.5.0.0).3 A false discovery rate of 1% was tolerated at both peptide and protein level.
In total, 1,258 Acididesulfobacillus proteins were identified. These proteins were quantified in both replicate samples of at least one of the conditions (pH 3.9 or 5.0). Known contaminants like human keratins were excluded from these lists. Please note that no correction for multiple testing has been applied due to the number of replicates (n = 2) for each condition.
Data Accessibility
All data has been uploaded to the European Nucleotide Archive under bioproject number PRJEB7665 composed by PacBio raw data (ERR3835955), Illumina MiSeq raw data (ERR665276), and genome assembly (ERZ1286184). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (Deutsch et al., 2020) via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD022508.
Results and Discussion
Characterization of Acididesulfobacillus acetoxydans gen. nov. sp. nov. strain INE
Morphology and Physiology
Acididesulfobacillus acetoxydans gen. nov. sp. nov. strain INE was isolated previously from enrichment cultures inoculated with Tinto River sediments, supplemented with lactate (5 mM) and sulfate (10 mM) at pH 4.5 (Sánchez-Andrea et al., 2013). Strain INE grew between pH 3.8 and 6.5, with a maximum specific growth rate at pH 5.0 (0.051 h−1). The temperature range of growth was between 25°C and 42°C, with an optimum at 30°C. Growth occurred in the presence of up to 15.3 g L−1 of NaCl. Cells of strain INE were single, straight rods, 4–7 μm long and ca. 0.6 μm wide (Figure 1A), with motility associated with observed lateral flagella (Figure 1B). Although strain INE stains Gram-negative, the double membrane characteristic of Gram-negative bacteria is absent (Figure 1C). Its Gram-positive cell wall structure was further supported by the negative KOH test and its susceptibility to vancomycin, as well as the membrane structure observed by TEM (Figure 1C).
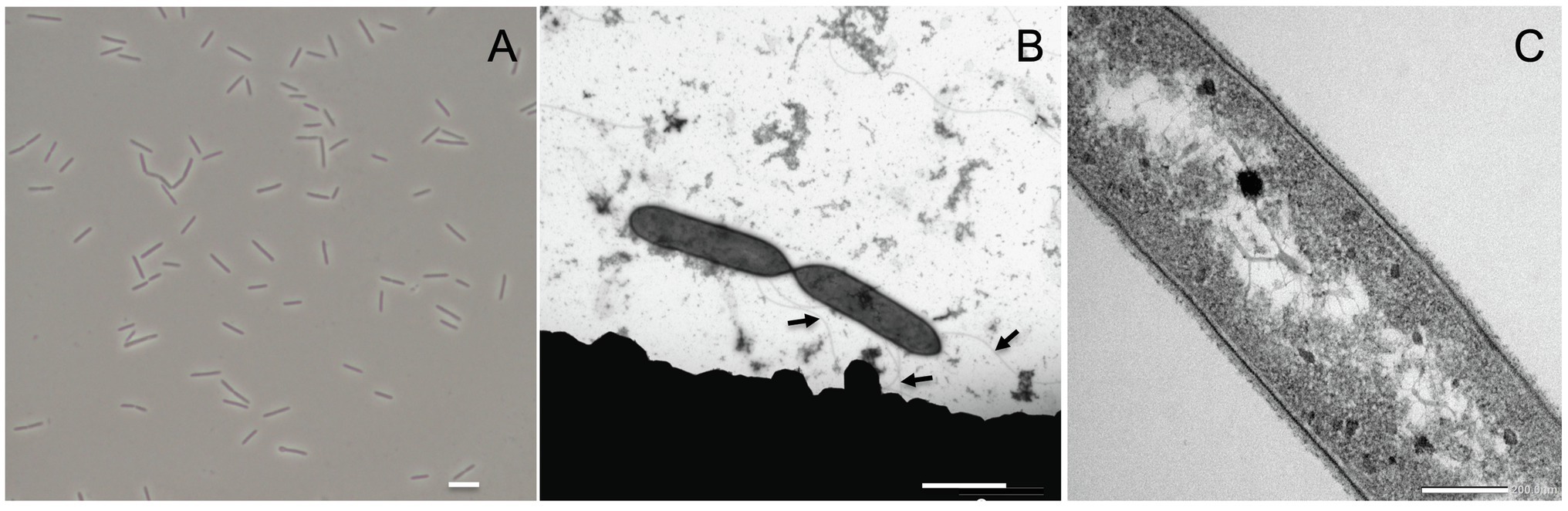
Figure 1. Acididesulfobacillus acetoxydans gen. nov. sp. nov. strain INE microscopy images: (A) phase contrast microscopy and (B,C) transmission electron microscopy. Scale bars represent 5 mm, 2 mm, and 200 nm, respectively. Arrows in (B) indicate flagella.
Strain INE is a strict anaerobe as it grew only in the presence of L-cysteine, ferrous iron, titanium citrate, or sulfide as reducing agents. With sulfate as electron acceptor, strain INE utilized a wide range of compounds as electron donors (Table 1): hydrogen, complex substrates such as yeast extract, carbohydrates such as xylose, glucose, fructose, maltose, sucrose, and raffinose, a variety of C1–C4 organic acids (formate, acetate, glycolate, pyruvate, lactate, malate, fumarate, butyrate, and succinate), the amino acid L-cysteine, and alcohols, such methanol, ethanol, glycerol, and 1- and 2-propanol. Strain INE was capable of the complete oxidation of substrates to CO2. No growth was observed with propionate, citrate, or benzoate as electron donor. With glycerol as electron donor, strain INE used a range of compounds as electron acceptor in addition to sulfate including elemental sulfur, thiosulfate, arsenate, selenate, nitrate, and DMSO. It could not reduce ferric iron as Fe(III)NTA, sulfite, dithionate, polysulfide, perchlorate, perchloroethylene, fumarate, humic acids, chromate, molybdate, manganese, or AQDS. Strain INE was also capable of disproportionation of elemental sulfur and thiosulfate, and of fermentation of cysteine, glucose, pyruvate, and yeast extract. The only detected menaquinone of strain INE was MK-7 (100%), as also detected for the closely related Desulfosporosinus spp., except for Ds. lacus which contained up to 36% MK-5 (Ramamoorthy et al., 2006).

Table 1. Main characteristics differentiating strain INE from its closest phylogenetic relatives.
Phylogenetic and Genomic Analysis
Genome assembly indicated the presence of one chromosome with a size of 4.52 Mbp, with a GC content of 53.65%, and 4,104 predicted protein coding sequences, of which 3,443 contained at least one protein domain, and 639 were identified as hypothetical proteins. In total 53 tRNAs and 3 full rRNA operons were identified. The phylogenetic position of the complete 16S rRNA gene sequence of strain INE (LN551924) showed that the strain is related to the Desulfosporosinus and Desulfitobacterium genera (Figure 2): the 16S rRNA gene sequence identity with its closest relatives, Ds. meridiei DSM 13257T, Ds. auripigmenti DSM 13351T and D. metallireducens DSM 15288T was 93.8%, 93.5%, and 93.5%, respectively. This is below the minimum similarity threshold of 94.5% for genus-delineation (Yarza et al., 2014), indicating that strain INE belongs to a new genus. This is further supported by the average amino acid identity (AAI) derived from the genome comparison of strain INE with Desulfosporosinus and Desulfitobacterium spp. (Supplementary Table S1). The highest similarity was observed with Peptococcaceae bacterium CEB3 (89.3%; Ňancucheo and Johnson, 2012), followed by Ds. acidiphilus DSM 22704 (64.4%). Based on the observed AAI values of more than 96% in most strains within one species and 62–64% within the same genus, respectively (Konstantinidis and Tiedje, 2005), AAI values also suggest that A. acetoxydans strain INE forms a new genus. Although the AAI with Ds. acidiphilus is close to the threshold of 64%, the 16S rRNA gene similarity is 92.7%, supporting the separation in a new genus. The 16S rRNA sequence identity further indicates that the novel genus Acididesulfobacillus encompasses the isolates CL4 and CEB3 (Jameson et al., 2010; Ňancucheo and Johnson, 2012). Although strain CL4 was formerly proposed as “Desulfobacillus acidavidus” (Jameson et al., 2010), it was never formally described, and current nomenclature regulations prevented us from using the proposed name. We therefore hereby propose the alternative name Acididesulfobacillus.
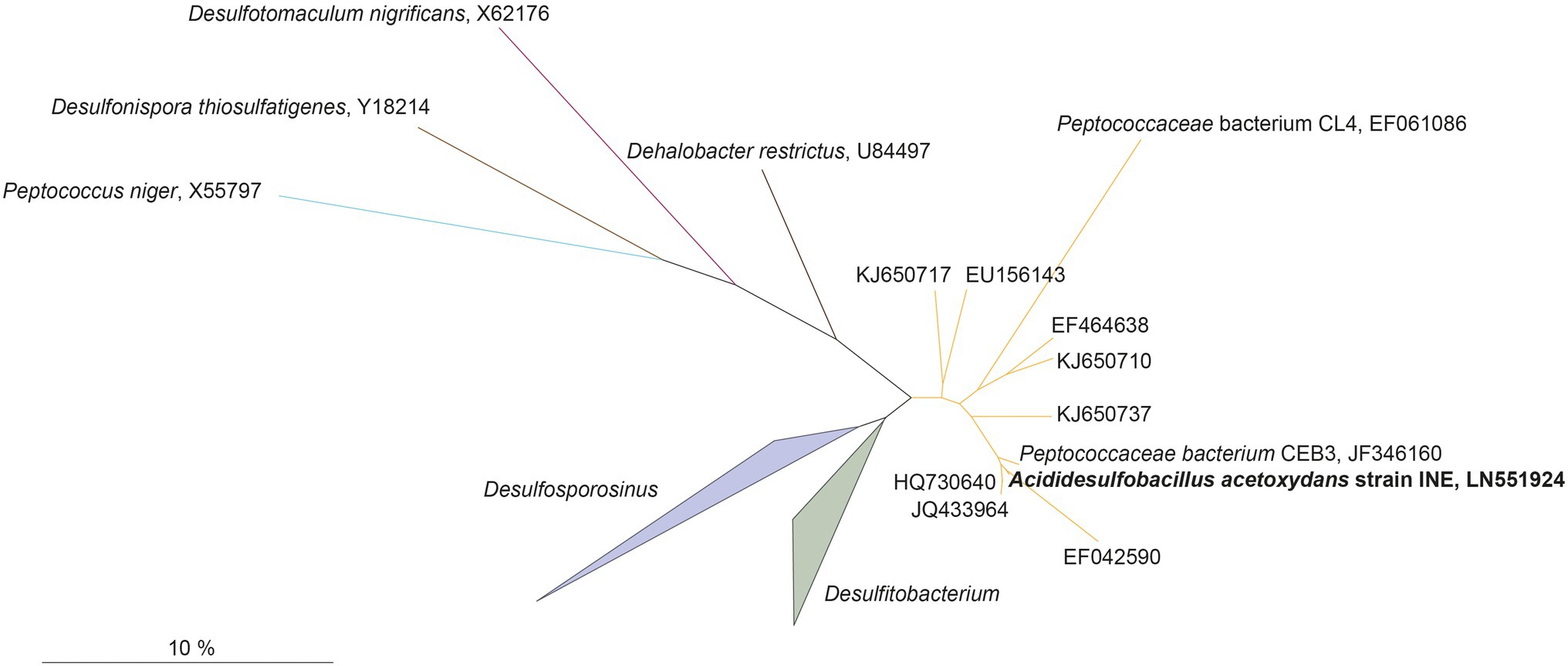
Figure 2. Maximum-likelihood tree showing phylogenetic affiliation of 16S rRNA gene sequences of Acididesulfobacillus acetoxydans (in bold) with related environmental sequences found in acidic environments, and closely related species in the Peptococcaceae family of the Firmicutes phylum. The accession numbers represent sequences retrieved from different acid drainage environments. KJ650710, KJ650737, or KJ650717: sulfidic mine tailings dumps in Botswana; EF042590: an abandoned copper mine in Odiel (Huelva, Spain); HQ730640, JQ433964: Tinto River sediments; EU156143: a thermal spring in Yellowstone National Park (WY, United States); EF464638: acidic enrichment cultures. JF346160 and EF061086: Peptococcaceae bacteria CEB3 and CL4. Scalebar indicates 10% sequence difference.
The Desulfosporosinus and Desulfitobacterium genera and the novel genus Acididesulfobacillus form a separate cluster from the other recognized genera and species of the family Peptococcaceae, order Clostridiales of the phylum Firmicutes. Members of the Desulfosporosinus and Desulfitobacterium genera are rod-shaped, Gram-positive, free-living, strictly anaerobic, and spore-forming bacteria. They display a diversity of metabolic traits relevant to bioremediation of polluted sites, e.g., reductive dehalogenation and reduction of metals such as arsenic or ferric iron, sulfur compounds, or nitrate (Spring and Rosenzweig, 2006; Alazard et al., 2010; Pester et al., 2012; Petzsch et al., 2015; Sánchez-Andrea et al., 2015; Mardanov et al., 2016). Members of Desulfosporosinus are known sulfate reducers in AMD environments (Nevin et al., 2003; Petrie et al., 2003; Suzuki et al., 2004; Kimura et al., 2006; Rowe et al., 2007; Cardenas et al., 2008; Lee et al., 2009; Madden et al., 2009; Senko et al., 2009; Ňancucheo and Johnson, 2014), and other low pH ecosystems such as peatlands (Hausmann et al., 2016, 2019). Three moderately acidophilic or acidotolerant Desulfosporosinus species were isolated so far: Ds. acidiphilus (pH 3.6–5.5, optimum pH 5.2; Alazard et al., 2010), Ds. acididurans (pH 3.8–7.0, optimum 5.5; Sánchez-Andrea et al., 2015), and Ds. metallidurans (pH 4.0–7.0, optimum 5.5; Panova et al., 2021).
All 16S rRNA gene amplicon sequences found in the public databases (SILVA and NCBI) that cluster together with A. acetoxydans strain INE were identified in AMD or ARD environments (Figure 2): in sulfidic mine tailings dumps in Botswana (pH 3.2–3.5; Korehi et al., 2014), in a stream of an abandoned copper mine in Odiel (Huelva, Spain; pH 2.5–2.75; Rowe et al., 2007), in Tinto River sediments (pH 5; Sánchez-Andrea et al., 2011) and in a thermal spring in Yellowstone National Park (pH 5.75–6.91; Hall et al., 2008). In addition, they were detected in enrichment cultures inoculated with samples from acid mine tailings (Winch et al., 2009). The two isolates included in the cluster, Peptococcaceae bacteria CL4 and CEB3 (Jameson et al., 2010; Ňancucheo and Johnson, 2012), were both obtained from reactors treating acid mine waters at a pH of about 2.5. The genome sequence from CEB3 is publicly available, but the two isolates were not characterized further. Interestingly, sequences of CEB3, which according to 16S rRNA gene sequence similarity corresponds to the same species as strain INE, became dominant in a reactor inoculated with an acidophilic consortium when the operating pH of the reactor was lowered, showing their predominance at more acidic conditions over other acidophilic SRB such as Ds. acididurans (Ňancucheo and Johnson, 2012). Its inability to grow at pH 7, combined with the lower pH optimum of strain INE (5.0), compared to Ds. acididurans (5.5) and Ds. acidiphilus (5.2; Table 1), this indicates that the SRB in this novel cluster are more acidophilic than the SRB described so far.
Chemotaxonomy
The lipid composition, including fatty acids (FAs), of strain INE grown at optimal pH (5.0) was compared with the type strains of the phylogenetically closest genera, Ds. orientis (Adams and Postgate, 1959; Stackebrandt et al., 1997) and D. dehalogenans (Utkin et al., 1994; Table 1). As these type strains are neutrophilic, lipid profiles were also compared with two acidophilic/acidotolerant strains from the Desulfosporosinus genus: Ds. acididurans (Sánchez-Andrea et al., 2015) and Ds. acidiphilus (Alazard et al., 2010).
The lipid profile of strain INE was dominated by the iso-C15:0 FA (representing almost 60%; Table 2), followed by iso-C15:0 dimethylacetal (DMA) as the second most abundant lipid. DMAs are formed upon acid hydrolysis from ether lipids containing an alk-1-enyl ether substituent at the sn-1 carbon of glycerol, so called plasmalogens (Jackson et al., 2021). Interestingly, the iso-C15:0 FA was also abundant in the acidophilic/acidotolerant strains Ds. acididurans (25.8%) and Ds. acidiphilus (16.6%), but absent in both the neutrophilic strains, Ds. orientis and D. halogenans. Iso-C17:0 and 10-methyl n-C17:0 FA and corresponding DMAs were also present in strain INE. Small amounts (i.e., 5% of total lipids) of 1-alkyl glycerol ethers with iso-C15 and 10-methyl n-C17 groups were also detected in strain INE, but saturated glycerol monoethers were absent in any of the other strains. The two most abundant FAs in Ds. orientis were n-C16:0 FA (35.7%) and the n-C18:1 DMA (16.4%). Both lipids are absent in strain INE and the latter is missing in all three acidophilic strains. Similarly, the abundant FAs n-C16:0 (23.6%), n-C16:1 (15.9%), and n-C14:0 (15.8%) in D. dehalogenans, the (neutrophilic) type strain of the second closest phylogenetic genus Desulfitobacterium, also showed a negligible presence in strain INE. The saturated branched-chain acid iso-C15:0 and the 1-alkyl glycerol ether lipids may therefore be characteristic for acidophilic SRB.
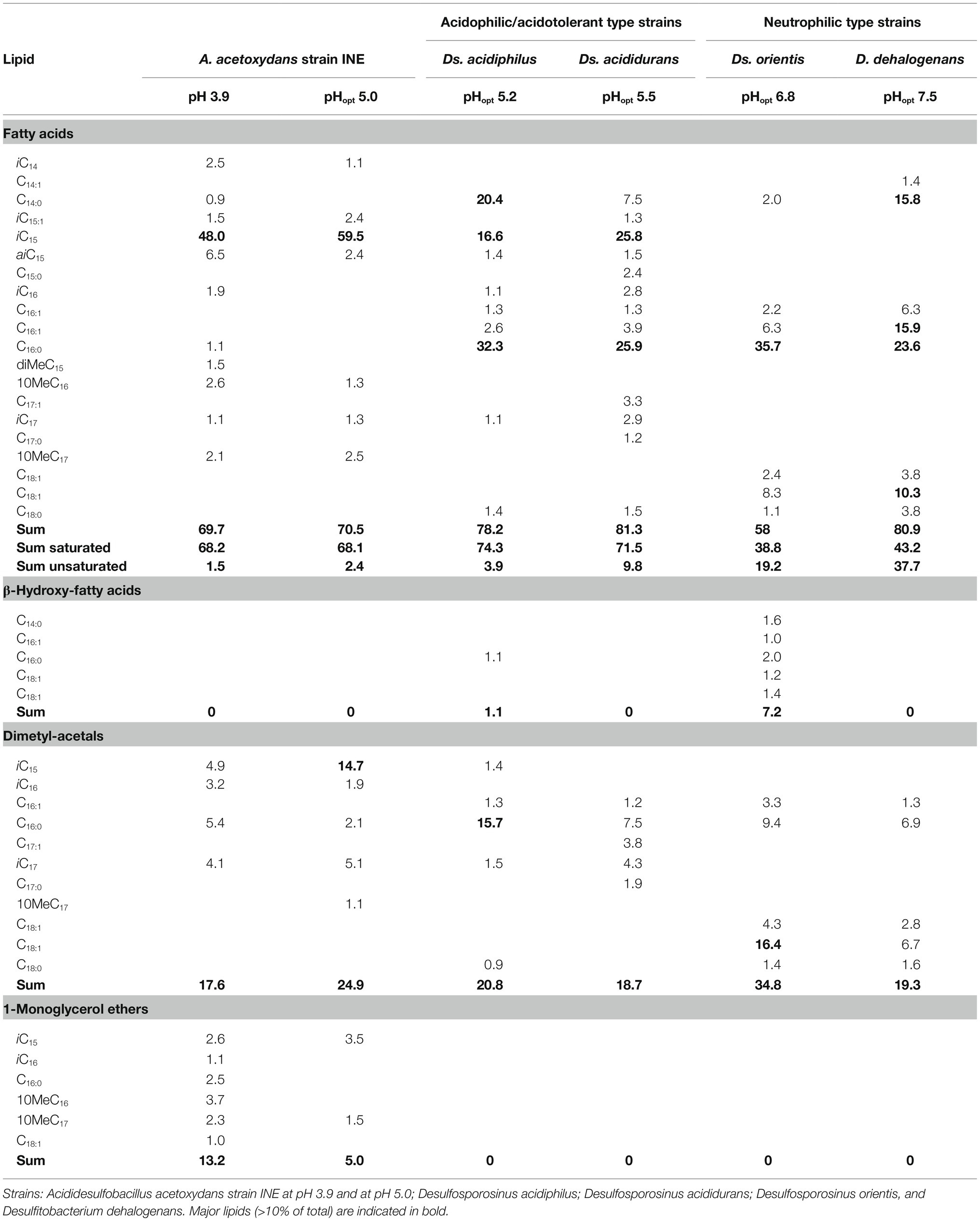
Table 2. Relative abundance (% of total) of fatty acids and ether lipids of Acididesulfobacillus acetoxydans strain INE and its phylogenetically closest relatives grown on glycerol and sulfate.
Analysis of the IPL of strain INE showed a high diversity of polar head groups (Supplementary Table S2), including sulfoquinovosyls (SQs), phosphoglycerols (PGs), phosphoethanolamines (PE), and diphosphatidylglycerols (DPG; commonly known as cardiolipins). Additionally, a series of glycolipids with hexosamine moieties were present. The dominant group was tentatively assigned a structure with a hexose group bound to the glycerol backbone and a hexosamine bound to this. The assignment was made based on their MS2 fragmentation and their accurate masses (Supplementary Figure S1; Supplementary Table S2). The second group also contained a hexosamine head group but the complete structure could not be elucidated (Supplementary Figure S1; Supplementary Table S2). Amino sugars are not routinely reported as components of polar headgroups in bacterial lipids but have been reported in a range of Archaea (Koga et al., 1993, 1998; Oger and Cario, 2013; Meador et al., 2014). Lipid-A, found in the outer-membrane of most Gram-negative bacteria, consists of two glucosamine moieties. However, the mass spectra of the hexosamine components detected in this study did not agree with published spectra of lipid A (Larrouy-Maumus et al., 2016; Crittenden et al., 2017). The IPL-bound core lipids (Supplementary Table S2) were in agreement with the hydrolysis-derived core lipid analysis (Table 2) and included diacylglycerols (DAGs) and mixed acyl/ether glycerols (AEGs; including plasmalogen lipids).
Dominant Acid Stress Resistance Mechanisms in Acididesulfobacillus acetoxydans
Proton Consumption and Complete Organic Acid Oxidation at Optimum and Minimum pH
The pH dependence of growth of A. acetoxydans strain INET (pH 3.8–6.5 with pH optimum at 5.0) indicated that it is moderately acidophilic rather than acid-tolerant, since it is unable to grow at pH 7.0. As aforementioned, members of Acididesulfobacillus spp. were found to be more tolerant to low pH than other acidophilic/acidotolerant SRB such as Desulfosporosinus spp. The capacity for complete oxidation of organic acids to CO2 is not common in other acidophilic/acidotolerant SRB described so far (Alazard et al., 2010; Sánchez-Andrea et al., 2013, 2015) and was only recently reported to promote limited growth in Ds. metallidurans (Panova et al., 2021). Because of the increasing toxicity of organic acids at pH values below their pKa, lowering their concentration by complete oxidation likely constituted an important detoxification mechanism for A. acetoxydans strain INE. In order to elucidate the main strategies conferring increased robustness at low pH, we investigated this in more detail by monitoring its metabolism through quantifying proton consumption rates, and glycerol, sulfate, and acetic acid concentrations at optimum (5.0) and low (3.9) pH in pH-controlled batch bioreactors fed with glycerol.
By monitoring the acid addition required for pH control, the proton consumption rates could be calculated. A higher net consumption of protons was observed at pH 3.9 than at 5.0 (Table 3): 128 mM H+ g−1 biomass (dry weight) vs. to 58 mM H+ g−1 biomass (dry weight), respectively. Doubling times at pH 3.9 and 5.0 were 17.6 and 13.8 h, respectively. The maximum OD600 reached in the late exponential phase after complete oxidation of glycerol was 0.41 at pH 3.9 and 0.44 at pH 5.0, corresponding to a maximum biomass dry weight of 102 and 115 mg L−1, respectively (Table 3). This difference likely indicated increased energetic costs for cell maintenance at pH values below the optimum. The increased maintenance energy requirements could have been partly compensated for by the slightly larger theoretical Gibbs free energy change (ΔrGʹ) for sulfate reduction with glycerol at pH 3.9 (−1186.7 kJ/mol) compared to pH 5.0 (−1142.8 kJ/mol), as calculated with Equilibrator (Flamholz et al., 2012; Table 4).

Table 3. Summary parameters of bioreactor cultivation of Acididesulfobacillus acetoxydans gen. nov. sp. nov. strain INE at pH 3.9 and pH 5.0 when grown on 5 mM glycerol and 10 mM sulfate.
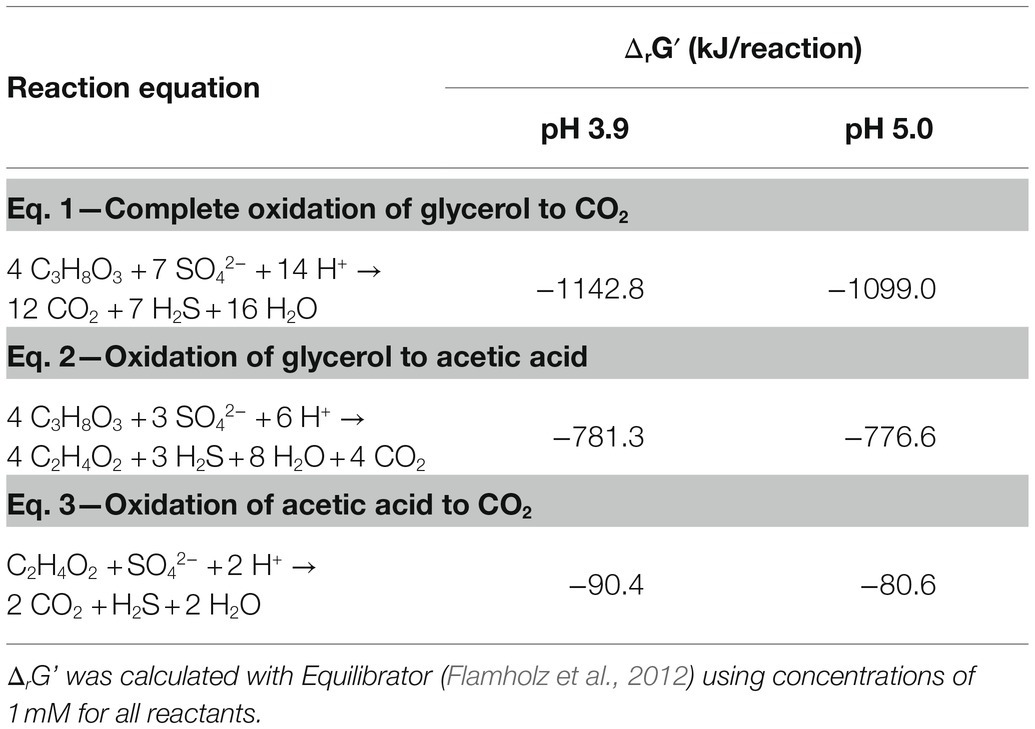
Table 4. Gibbs free energy of complete and incomplete oxidation of glycerol to CO2 and acetic acid, respectively.
Consumption of glycerol and sulfate was accompanied by an initial increase of total acetate concentrations to 2.4 mM at pH 3.9 and 2 mM at pH 5.0, followed by its complete removal (Figure 3). Total acetic acid concentrations started to decrease before all glycerol was consumed, indicating a co-consumption phase. Biomass concentrations remained approximately constant once glycerol was completely consumed, while total acetic acid concentrations kept decreasing, indicating that under these conditions, energy derived from acetate degradation was invested in cell maintenance rather than in net growth. This was also reflected in the continued addition of acid (HCl) required to maintain constant pH after the maximum optical density (OD600) was reached. Overall, approximately 30% of the carbon added as glycerol was not coupled to dissimilatory sulfate reduction but used for anabolism.
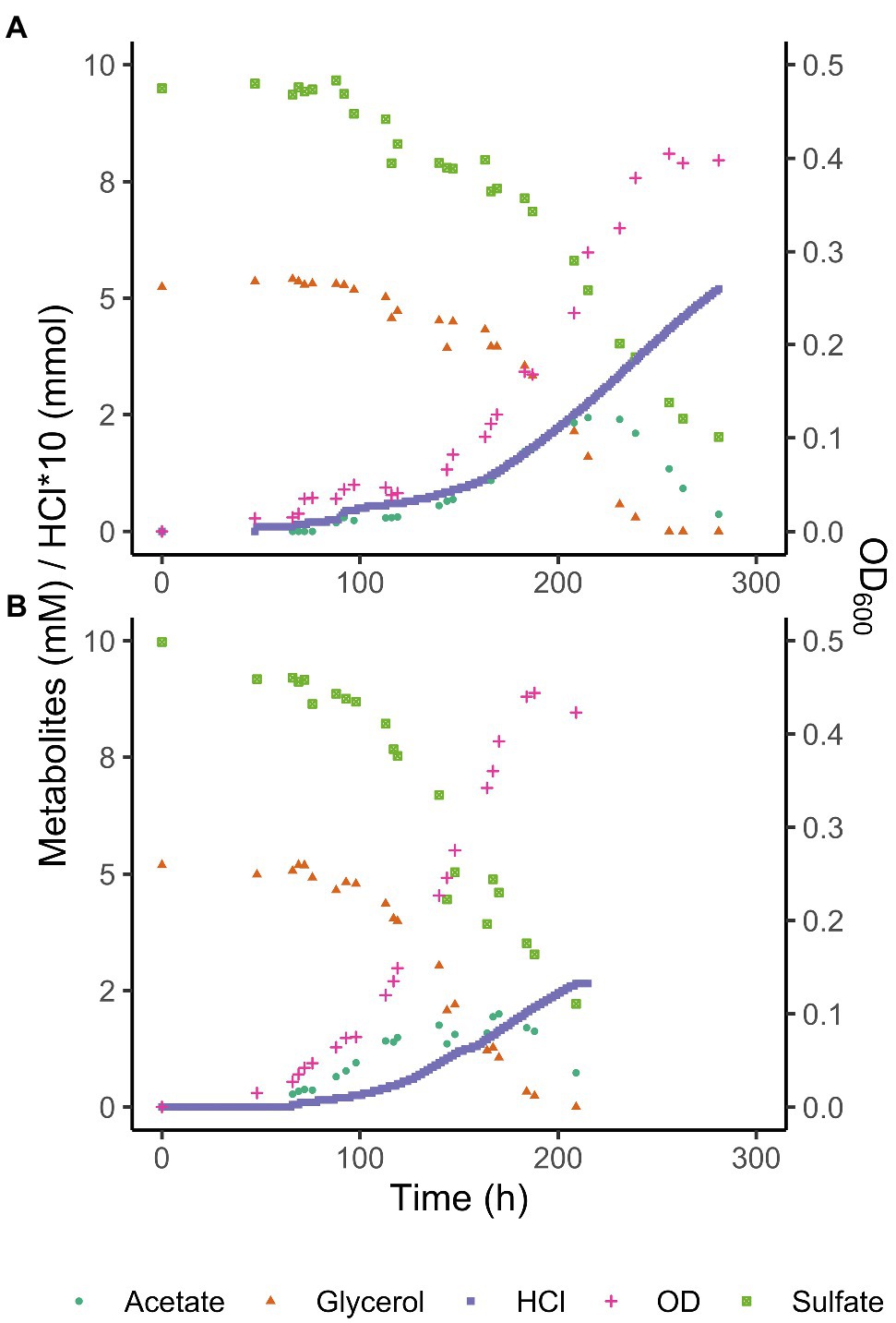
Figure 3. Glycerol, total acetate (undissociated + dissociated) and sulfate concentrations, and OD600 of Acididesulfobacillus acetoxydans strain INE grown in bioreactors at pH 3.9 (A) and pH 5 (B). Concentration profiles of glycerol, sulfate, acetic acid, as well as OD600 and HCl addition are shown for one reactor at each pH value for clarity.
The ability to use acetic acid as electron donor is advantageous in extreme environments such as AMD for several reasons. Firstly, acetic acid is a typical end product of other microorganisms such as fermenters present in AMD sediments (Sánchez-Andrea et al., 2018). The ability to metabolize a variety of organic acids, but especially a metabolic end product such as acetic acid (Table 1) increases the carbon and energy available to strain INE in nutrient-limited AMD/ARD environments. Furthermore, at pH values below their pKa, organic acids occur in their undissociated (protonated) form and can freely diffuse across the cell membrane, resulting in the acidification of the cytoplasm. Removal of intracellular protons is energetically costly and can be detrimental for growth. Complete oxidation of organic acids to CO2 prevents their accumulation in the environment, thereby lowering organic acid toxicity. For example, at the pH conditions compared in this study, maximum total extracellular acetic acid (dissociated and undissociated) concentrations reached 2.4 mM at pH 3.9 and 2.0 mM at pH 5.0 removal (Figure 3). Considering the pKa of acetic acid (4.8), its extracellular concentration (undissociated) at pH 3.9 is 2.1 mM, vs. 0.7 mM at pH 5.0. The higher extracellular acetic acid concentration measured at pH 3.9 could correspond to a higher consumption of acetic acid at the optimum pH 5.
Metabolic Pathways for Complete Acetic Acid Oxidation Coupled to Sulfate Reduction
To enable the more informed prediction of acetotrophic potential in acidophilic SRB from (meta)genomic data, the metabolic pathway for complete oxidation of acetic acid by A. acetoxydans was determined through proteome analysis. The Acetyl-CoA pathway seemed to be responsible for the complete oxidation of acetic acid to CO2, since all of its enzymes were present in the proteome (Figure 4). Additionally, the absence of citrate synthase indicated that acetyl-CoA could not be degraded through the tricarboxylic acid (TCA) cycle, the other potential pathway for acetate oxidation. Two genes encoding bifurcating enzymes related to the reverse Acetyl-CoA pathway were detected, the NADH-dependent reduced ferredoxin:NADP+ oxidoreductase (nfnAB, 2795-96), and the cytoplasmic NADH-dependent formate dehydrogenase (fdh, 1374–1377). NfnAB catalyses the NAD+-dependent reduction of ferredoxin by NADPH, acting possibly on the NADP+ generated in the conversion of methylene-THF to methenyl-THF in the Acetyl-CoA pathway. The nfnAB genes, both encoding iron–sulfur flavoproteins, are present in a diversity of microorganisms (Wang et al., 2010), but often incorrectly annotated as sulfide dehydrogenase. The nfnAB genes were detected in the proteome of strain INE and were found in the same operon as two genes encoding methylene-H4F-DH/methenyl-H4F cyclohydrolase and the methenyl-H4F cyclohydrolase (2793-94), both part of the Acetyl-CoA pathway. The second bifurcating enzyme detected in the proteome of strain INE, the cytoplasmic NADH-dependent formate dehydrogenase (Sebban et al., 1995; Costa et al., 1997), is a selenocysteine-containing Fdh that oxidizes formate while reducing ferredoxin and NAD+. The Fdh detected in the genome of strain INE resembled the Fdh protein encoded in the genome of Desulfotomaculum kuznetsovii (Deasku_2987-2991), and consisted of a catalytic subunit (fdhA, 1377), two subunits for oxidoreduction of NADH/NAD+ (fdhGH, 1374), the proton-translocating quinone (fdhE, 1375), and the 5-formyltetrahydrofolate cyclo-ligase (fdhH, 1373). This Fdh is predicted to act in the Acetyl-CoA pathway (Ragsdale and Pierce, 2008), and it was found next to a gene encoding a histidine kinase (1378), suggesting that it has a regulatory function (Pereira et al., 2011). Although the complete Acetyl-CoA pathway is also present in the genome of sulfate reducers from the genus Desulfosporosinus (Alazard et al., 2010; Pester et al., 2012; Sánchez-Andrea et al., 2015), complete oxidation of acetic acid in moderately acidophilic SRB was so far only observed in the recently described Ds. metallidurans, where weak growth on acetic acid was reported. However, acetate oxidation was proposed to occur through the TCA cycle instead of the Acetyl-CoA pathway based on genome analysis (Panova et al., 2021).
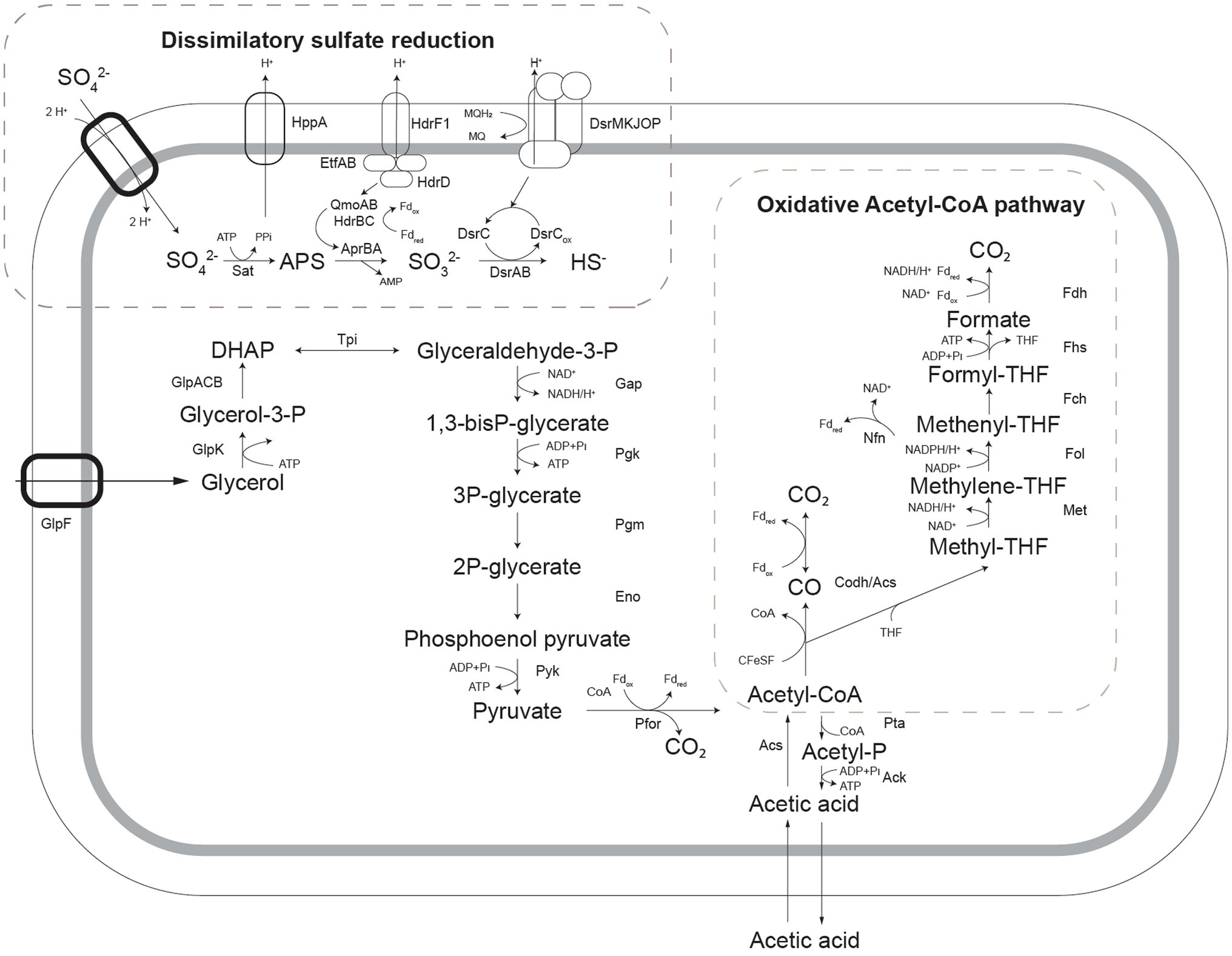
Figure 4. Schematic representation of pathways for glycerol oxidation and sulfate reduction as detected in the genome of Acididesulfobacillus acetoxydans. Sat: ATP sulfurylase, Apr: APS reductase, Dsr: dissimilatory sulfite reductase, Qmo: quinone-interacting membrane oxidoreductase complex, Hdr: heterodisulfide reductase, GlpF: glycerol transporter, GlpK: glycerol kinase, Tpi: triose phosphate isomerase, Gap: glyceraldehyde-3-phosphate dehydrogenase, Pgk: phosphoglycerate kinase, Pgm: phosphoglycerate mutase, Eno: phosphoenolpyruvate hydratase, Pyk: pyruvate kinase, Pfor: pyruvate:ferredoxin oxidoreductase, Pta: Phosphate acetyltransferase, Ack: acetate kinase, Acs: acetyl-coA synthase, Codh: carbon monoxide dehydrogenase, Met: methylene-tetrahydrofolate reductase, Fol: methylene-tetrahydrofolate dehydrogenase/methenyl-tetrahydrofolate cyclohydrolase, Fch: 5-formyltetrahydrofolate_cyclo-ligase, Fhs: Formate-tetrahydrofolate ligase/ formyl-H4F synthethase, Fdh: formate dehydrogenase, Nfn: NADH-dependent reduced ferredoxin:NADP+ oxidoreductase.
Enzymes of the metabolic pathways used for oxidation of glycerol coupled to sulfate reduction were also identified in the proteome (Figure 4). Glycerol is taken up by a glycerol transporter (DEACI_1309-DEACI_1310, hereafter “DEACI_” will be omitted for simplicity) and oxidized to dihydroxyacetone-P (DHAP) by glycerol kinase (0310) and glycerol-3-phosphate dehydrogenase (1301-1303). DHAP is converted to pyruvate involving enzymes of the Embden-Meyerhoff pathway (0305-0308, 2591-2592, 3679) and subsequently to acetyl-coA by pyruvate:ferredoxin oxidoreductase (0330, 0621, 1130, 1144, 2105, 2107, 2770). Acetyl-CoA is converted to acetate through Pi acetyltransferase (3443) and acetate kinase (3414), or completely oxidized to CO2 through the reverse Acetyl-CoA pathway, discussed above. Reducing equivalents are transferred to NAD+ and oxidized ferredoxin. Electrons flow to sulfate through the respiratory chain, for which the key enzymes were identified. Sulfate is activated with ATP to adenosine-phosphosulfate (APS) and pyrophosphate by ATP sulfurylase (Sat, 0463). Heterodisulfide reductase (HdrF1, 0461) is predicted to receive electrons from the quinone pool and direct them to the cytoplasmic QmoAB/HdrBC complex (0456, 0457, 0470, 0471; Rabus et al., 2015), reducing APS to sulfite through the AprAB (0464, 0465) complex, as described for other SRB (Ramos et al., 2012). The sulfur in sulfite is then reduced and transferred to DsrC (0677) forming DsrC trisulfide by DsrAB (0668, 0669; Santos et al., 2015), which is finally reduced to sulfide by electrons received from the DsrMKJOP complex (0672-0676; Pires et al., 2006).
The increased specific proton consumption rate at pH 3.9 compared to pH 5.0, and the ability to oxidize organic acids to CO2 has important effects on the biogeochemistry of ARD/AMD environments, as it contributes to the natural attenuation of the extreme conditions. In the Tinto River, for example, the pH values can be as low as 2 in the water column (Sánchez-Andrea et al., 2012; Amils et al., 2014). Consequently, the microbial diversity in the water column of the Tinto River is generally low, and dominated by only a few species belonging to the Acidithiobacillus, Leptospirillum, and Acidiphilium genera (López-Archilla et al., 2001). Microbial diversity is higher in the sediments (Sánchez-Andrea et al., 2011), partly due to the wider range of physicochemical characteristics found in the different layers. As strain INE was isolated from Tinto River sediments, its detoxifying characteristics indicate that it could assist in the creation of milder conditions, where less acidophilic microorganisms can thrive. This could facilitate the higher microbial diversity found in the sediments compared to the water column, where the environment is more dynamic due to the water flow.
Additional Stress Resistance Mechanisms at Low pH
Other possible pH homeostasis mechanisms enabling A. acetoxydans to grow at low pH besides acetate oxidation were also investigated. Protein abundance at optimum and minimum pH were compared to detect increased translation of proteins involved in acid stress response mechanisms at minimum pH. Protein abundances in cultures grown at optimum (5.0) and low (3.9) pH showed 1258 proteins present in at least one of the growth conditions, of which 949 were present in both. Using a value of p cut-off of 0.05 (student t-test), 30 proteins were present in at least two-fold higher abundance at pH 3.9 compared to pH 5.0, with 15 proteins unique for pH 3.9. At pH 5.0, 56 proteins were present in at least 2-fold higher abundance, of which 32 were uniquely present at pH 5.0 (Supplementary Table S3).
Donnan Potential and Transporters
A Donnan potential serves as a chemiosmotic barrier to the influx of protons at low extracellular pH as described for extreme acidophiles (Baker-Austin and Dopson, 2007). A key element in creating a positive membrane potential is the K+-transporting ATPase, encoded by the kdpABC operon, which was indeed detected in the genome of A. acetoxydans (3037-39). However, this K+ ATPase was not detected in the proteome of cells grown at either pH 3.9 or 5.0. Therefore, despite its apparent importance in other acidophiles (Baker-Austin and Dopson, 2007), it did not appear to play an important role in acid stress resistance of A. acetoxydans. Although the detection of membrane proteins such as ATPases with standard proteomics methods is difficult, and their presence cannot be completely dismissed based on these results alone, comparative transcriptomics analysis also verified the absence of the K+-transporting ATPase from the transcriptome (Sánchez-Andrea, unpublished data).
In addition to K+-ATPase, genes encoding secondary cation transporters such as Na+/Pi co-transporters (3752, 0312, 1305) and solute symporters (1779, 2169, 2179, 2830, 3466, 3909) were identified in the genome, and the corresponding proteins (1305, 2179, 3466, and 2830) were present in the proteome. Interestingly, a bile acid:Na+ symporter (2179) and a transporter from the major facilitator superfamily (0284) were only present at pH 3.9, indicating a potentially important role in ion transport at low pH. Build-up of excess protons in the cytoplasm can be further countered by active H+ export by ATPase. The proton ATPase found in strain INE (0352-0361) was present in the proteome at both pH values, but not in significantly different abundance at lower pH, suggesting that this system is not upregulated in response to increased acid stress. Antiporters such as the Na+/H+ antiporter (2829) or the H+/Cl− exchange transporter (clcA, 0494) also contribute to H+ removal. ClcA was detected in the proteome, albeit not in significantly different abundance at lower pH.
Cytoplasmic Buffering Capacity
The modification of the cytoplasmic buffering capacity through a change in the metabolism of certain amino acids could mitigate acid stress (Baker-Austin and Dopson, 2007; Slonczewski et al., 2009). The decarboxylation of arginine, glutamate and aspartate, for example, releases CO2 and amines, and consumes protons (Slonczewski et al., 2009). This would be net H+-consuming when the amino acids can be taken up from the environment, and it could be speculated that the yeast extract present in the medium served as an external source of amino acids. L-Aspartate decarboxylase (2148) was found in the proteome at pH 5.0, but not at pH 3.9, indicating that this reaction does not contribute to pH homeostasis at increased proton stress. Glutamate decarboxylase (2571), catalyzing the H+ consuming conversion of glutamate to g-aminobutyrate (GABA; Castanie-Cornet et al., 1999), and L-arginine decarboxylase (0577), catalyzing the H+-consuming conversion of L-arginine to agmatine, were detected in the proteome at both conditions, albeit not at a significantly different abundance. Furthermore, glutamate decarboxylase forms the GABA shunt together with GABA aminotransferase and succinate semialdehyde dehydrogenase, which is also proposed to be involved in acid stress resistance in bacteria (Feehily and Karatzas, 2013). Through the GABA shunt, glutamate is converted to GABA by glutamate decarboxylase (2571), consuming a proton, and GABA is converted to succinate semialdehyde (SSA) by GABA aminotransferase (4212, 3258), regenerating the glutamate. Succinate semialdehyde dehydrogenase then converts SSA to succinate (3872, 3892). Glutamate decarboxylase (2571) and GABA aminotransferase (3258, 4212) were detected in the proteome of strain INE, albeit not at a significantly different abundance at pH 5.0 or 3.9. Succinate semialdehyde dehydrogenase was not detected in the proteome; however, and the role of the GABA shunt in resistance to acid stress in strain INE could, therefore, not be clearly established.
Interestingly, agmatinase (0579), converting agmatine to urea and putrescine, was detected in the proteome at both pH values. Urea may play a role in pH homeostasis through its degradation into two molecules of ammonium and one CO2, catalyzed by urease. This requires incorporation of 2 H+ and is known to play a role in acid stress resistance (Young et al., 1996). However, the gene encoding urease was not detected in the genome of strain INE. An alternative route for urea degradation could be through urea amidolyase; genes (0170, 0171) were detected in the genome, but the corresponding proteins were absent in the proteome. Urea does seem to play a role in pH homeostasis of strain INE at low pH—four out of the five enzymes of the urea cycle are present in increased abundance at pH 3.9. A carbamoyl phosphate synthase (0932) was found only at pH 3.9, and ornithine carbamoyltransferase (0934), and argininosuccinate synthase (0935) were present in twofold higher abundance. Argininosuccinate lyase (0936) was present in the proteome at both conditions in similar abundance. Arginase (0853), converting arginine to urea and ornithine and completing the urea cycle, was, however, not detected in the proteome, and the role of the urea cycle in acid stress resistance of strain INE, therefore, remains enigmatic.
DNA, Protein, and Lipid Repair Mechanisms
The extreme acidity and high metal concentrations in acid drainage environments increase the risk of damage to DNA, RNA, and proteins. A number of DNA, protein, and lipid repair systems were present in the proteome of strain INE. Ferroxidase (0081), linked to protection of DNA upon exposure to acid stress in Helicobacter pylori (Huang et al., 2010), was present in the proteome at both conditions. Several chaperones involved in protein refolding (0004, 2293, 3249, 0692, 2491, 2469, 1389, 1390) were present in the proteome, and a ferredoxin signature (3253) upstream of a chaperone (3249) was ca. 6-fold more abundant at pH 3.9 than at pH 5.0. An upregulation of chaperones was found to accompany growth at lower pH in many acidophiles (Jerez et al., 1988). Furthermore, six peroxiredoxins, potentially involved in lipid repair (Cárdenas et al., 2012), were detected in the proteomes at both conditions (2671, 0858, 0888, 0966, 1064, 2671, 3052). One of these (2671) was ca. twofold more abundant at low pH.
Altered Membrane Permeability
Comparison of the lipid composition in A. acetoxydans strain INE at pH 5.0 and at pH 3.9 showed a transition, with decreasing pH, from acyl/ether glycerol (AEG) lipids with an unsaturated ether moiety (plasmalogens) to AEG lipids with a saturated ether moiety (Table 2). This was evident in the core lipids through an increase in saturated monoalkyl glycerol ether lipids from 5.0% at pH 5.0 to 13.0% at pH 3.9 and a concomitant decrease in DMAs (from 24.9% to 17.6%). The increase in saturated ether moieties may represent an adaptation to maintain membrane homeostasis at lower pH. Increasing saturation of FAs in response to lower pH has been described some in bacterial species, but the opposite effect has been observed in others (Siliakus et al., 2017; Sohlenkamp, 2017). It should be noted that there was no change in the saturation of the diacyl glycerols (DAGs), as seen from the FAs (Table 2). With decreasing growth pH there was also a clear diversification in acyl and ether moieties (Table 2). Bacterial glycerol ether lipids likely play a role in cell resistance or adaptation to adverse environmental conditions (Grossi et al., 2015) since ether linkages are less sensitive to acid hydrolysis than ester bonds. Indeed, AEG lipids are thought to improve cell resistance to extreme external conditions, relative to DAGs (Grossi et al., 2015).
At both pH values, the A. acetoxydans fatty acids were dominated by branched-chain FAs (97% of FAs at pH 3.9, 100% at pH 5.0), including iso, anti-iso and mid-chain methylated FAs. Similarly, the percentage of FAs that were branch-chained was moderate to high in the other acidophilic strains, Ds. acididurans (26%) and Ds. acidiphilus (42%), while they were absent in both neutrophilic strains Ds. orientis and Ds. halogenans. In particular, the branched-chain acid iso-C15:0 was abundant in strain INE (48.1% at pH 3.9, 60.7% at pH 5.0) and in the acidophilic strains Ds. acididurans (25.8%) and Ds. acidiphilus (16.6). An increasing proportion of branched-chain FAs is a known adaption to maintain membrane homeostasis under external stress, and a number of extreme acidophilic bacteria have been reported to have high levels of branched-chain fatty acids (Siliakus et al., 2017).
In addition to an altered membrane FAs composition, a putative poly-gamma-glutamate (PGA) bacterial capsule synthesis protein CapA (2151) was found in 2-fold higher abundance at low pH in the proteome, providing another means to create physical separation between the cell and the acidic environment as a defense mechanism. PGA is a natural polymer that may be involved in avoiding dehydration in high salt concentrations and neutralizing pH in alkaliphiles (Ashiuchi and Misono, 2002) by forming a protective PGA capsule. Furthermore, spermidine is hypothesized to play a role in the protection against acid stress through lowering the permeability of the membrane to protons (Christel et al., 2017). Spermidine synthase (0578) was detected in both proteome sets at similar abundances, indicating spermidine could play a role in acid stress resistance of A. acetoxydans at both pH values tested.
Conclusion
Acididesulfobacillus acetoxydans strain INE, isolated from Tinto River sediments, represents a novel species of a novel genus of moderately acidophilic SRB. The increased specific proton consumption rates at low pH (3.9) compared to optimum pH (5.0) when grown with glycerol and sulfate demonstrated the alkalinizing effect of its metabolism in acidic environments. The capacity for complete oxidation of organic acids to CO2 can reduce organic acid toxicity and provide increased access to organic carbon compounds in nutrient-limited acid drainage environments. Interestingly, canonical acid stress resistance mechanisms such as an inverted membrane (Donnan) potential, increased abundance of charge transporters, and cytoplasmic buffering capacity were not detected in the proteome during growth at low pH. Instead, an increased fraction of ether-bound fatty acids in the overall membrane lipid composition appeared to be an important acid stress resistance mechanism for A. acetoxydans during growth at pH below optimum. In addition, the increased abundance of CapA could suggest that the response to proton stress in A. acetoxydans resembles the response to high salinity.
Description of Acididesulfobacillus gen. nov.
A.ci.di.de.sul.fo.ba.cil’lus. L. masc. adj. acidus, sour; N.L. masc. n. ‘Desulfobacillus’, a (not validly published) bacterial genus name; N.L. masc. n. Acididesulfobacillus, an acid-loving relative of the genus ‘Desulfobacillus’.
Cells present sub-terminal spores and are rod-shaped, strictly anaerobic, motile, and stain Gram-negative, but lack the characteristic double membrane. The genus is closely related to Desulfosporosinus and Desulfitobacterium. The type species is A. acetoxydans.
Description of Acididesulfobacillus acetoxydans sp. nov.
Acididesulfobacillus acetoxydans: a.cet.o’xy.dans. L. neut. n. acetum, vinegar; N.L. v. oxydo (from Gr. masc. adj. oxys, acid, sour), to oxidize; N.L. part. adj. acetoxydans, oxidizing acetate
Straight single rods are 4–7 μm long and about 0.6 μm wide. Motile in the exponential phase by lateral flagellation. Subterminal, oval endospores are formed in the stationary growth phase. Cells are Gram-positive that stain Gram-negative. The pH range for growth is pH 3.8–6.5, with an optimum at pH 5.0. The temperature range for growth is 25–42°C, with an optimum at 30°C. The upper limit for salt tolerance is 15.3 g L−1 NaCl. Strain INE uses a wide range of electron donors in the presence of sulfate such as H2; organic acids (formate, acetate, pyruvate, lactate, malate, fumarate, butyrate, and succinate); alcohols (methanol, ethanol, propanol, and glycerol); the amino acid L-cysteine; sugars (xylose, fructose, glucose, maltose, sucrose, and raffinose) and yeast extract or starch; but not propionate, citrate, or benzoate. Organic substrates are oxidized completely to CO2. Strain INE uses selenate, arsenate, nitrate, sulfate, elemental sulfur, thiosulfate, and DMSO as electron acceptors; but not iron, fumarate, PCE, perchlorate, sulfite, or dithionate. It also ferments some substrates such as cysteine, pyruvate, glucose, and yeast extract. It couples the disproportionation of sulfur and thiosulfate to growth, but not sulfite. Strain INE is susceptible to vancomycin. The predominant whole cell membrane lipids of the type strain INE at pH 5 are iso-C15:0 fatty acid (60.7%), and iso-C15:0 DMA (14.3%). Its genomic G + C content is 53.65 mol %. Phylogenetically, it is a member of the family Peptococcaceae, order Clostridiales within the Firmicutes phylum. The type strain A. acetoxydans, strain INE (=DSM 29876T = JCM 30553T), was isolated from the Tinto River, Spain.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/ena, PRJEB7665; https://www.ebi.ac.uk/pride/archive/, PXD022508.
Author Contributions
IS-A and AS initiated the study and designed the experiments. IS-A and MJ performed experiments. IS-A and CG processed and analyzed the reactor, genome, and proteome data and wrote the draft manuscript. BH assembled and compared genome data. NB, JS, and WR performed membrane lipid analysis. All authors contributed to the article and approved the submitted version.
Funding
This work was financed by ERC grants to AS (project 323009) and JS (project 694569), the research program TTW under project number 14797, which is financed by the Dutch Research Council (NWO) to IS-A, and a Gravitation grant (SIAM 024.002.002) of the Netherlands Ministry of Education, Culture and Science to AS and JS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Hans Wessels from Radboud University Nijmegen for the proteomics services, Marcel Giesbers and Jelmer Vroom from the Wageningen Electron Microscopy Center for TEM imaging, and Ellen Hopmans for advice on lipid identification. We thank Aharon Oren for fruitful discussion over nomenclature regulations and Barrie Johnson for his pioneering work on acidophilic SRB.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb. 2022.816605/full#supplementary-material
Supplementary Table S1 | AAI matrix.
Supplementary Table S2 | The intact polar lipid (IPL) composition of Acididesulfobacillus acetoxydans gen. nov. sp. nov. grown at pH 5.0 (growth optimum) as determined by UHPLC-HRMS analysis.
Supplementary Table S3 | Proteins detected at 2-fold or higher increased abundance at pH 3.9 or pH 5.0 (p-value < 0.05, student t-test). Locus tag DEACI_**** is followed by the annotation. U: unique at this condition.
Supplementary Table S4 | (A) Example MS2 spectra of a hexosamine-hexose lipid. [M+H]+ of parent ion in MS1 = 864.604. Placement of amine group in structure is tentative. Table insert of ion assignments, mmu = milli mass unit, Δ mmu = (measured mass – calculated mass) x 1000. (B) Example MS2 spectra of one of the unknown hexosamine-containing lipids. [M+H]+ of parent ion in MS1 = 880.599.
Footnotes
References
Adams, M. E., and Postgate, J. R. (1959). A new sulphate-reducing vibrio. J. Gen. Microbiol. 20, 252–257. doi: 10.1099/00221287-20-2-252
Alazard, D., Joseph, M., Battaglia-Brunet, F., Cayol, J. L., and Ollivier, B. (2010). Desulfosporosinus acidiphilus sp. nov.: a moderately acidophilic sulfate-reducing bacterium isolated from acid mining drainage sediments. Extremophiles 14, 305–312. doi: 10.1007/s00792-010-0309-4
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Amils, R., Fernández-Remolar, D., and IPBSL, Team (2014). Río Tinto: a geochemical and mineralogical terrestrial analogue of Mars. Life. 4, 511–534. doi: 10.3390/life4030511
Amils, R., González-Toril, E., Aguilera, A., Rodríguez, N., Fernández-Remolar, D., Gómez, F., et al. (2011). From Río Tinto to Mars: the terrestrial and extraterrestrial ecology of acidophiles. Adv. Appl. Microbiol. 77, 41–70. doi: 10.1016/B978-0-12-387044-5.00002-9
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Ashiuchi, M., and Misono, H. (2002). Biochemistry and molecular genetics of poly-γ-glutamate synthesis. Appl. Microbiol. Biotechnol. 59, 9–14. doi: 10.1007/s00253-002-0984-x
Baker-Austin, C., and Dopson, M. (2007). Life in acid: pH homeostasis in acidophiles. Trends Microbiol. 15, 165–171. doi: 10.1016/j.tim.2007.02.005
Bale, N. J., Sorokin, D. Y., Hopmans, E. C., Koenen, M., Irene Rijpstra, W. C., Villanueva, L., et al. (2019). New insights into the polar lipid composition of extremely halo(alkali)philic euryarchaea from hypersaline lakes. Front. Microbiol. 10:377. doi: 10.3389/fmicb.2019.00377
Bland, C., Ramsey, T. L., Sabree, F., Lowe, M., Brown, K., Kyrpides, N. C., et al. (2007). CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8:209. doi: 10.1186/1471-2105-8-209
Campbell, L. L., and Postgate, J. R. (1965). Classification of the spore-forming sulfate-reducing bacteria. Microbiol. Mol. Biol. Rev. 29, 359–363. doi: 10.1128/br.29.3.359-363.1965
Cardenas, E., Wu, W. M., Leigh, M. B., Carley, J., Carroll, S., Gentry, T., et al. (2008). Microbial communities in contaminated sediments, associated with bioremediation of uranium to submicromolar levels. Appl. Environ. Microbiol. 74, 3718–3729. doi: 10.1128/AEM.02308-07
Cárdenas, J. P., Moya, F., Covarrubias, P., Shmaryahu, A., Levicán, G., Holmes, D. S., et al. (2012). Comparative genomics of the oxidative stress response in bioleaching microorganisms. Hydrometallurgy 127-128, 162–167. doi: 10.1016/j.hydromet.2012.07.014
Castanie-Cornet, M.-P., Penfound, T. A., Smith, D., Elliott, J. F., and Foster, J. W. (1999). Control of acid resistance in Escherichia coli. J. Bacteriol. 181, 3525–3535. doi: 10.1128/JB.181.11.3525-3535.1999
Christel, S., Herold, M., Bellenberg, S., El Hajjami, M., Buetti-Dinh, A., Pivkin, I. V., et al. (2017). Multi-omics reveals the lifestyle of the acidophilic, mineral-oxidizing model species Leptospirillum ferriphilum. Appl. Environ. Microbiol. 84, e02091–e02017. doi: 10.1128/AEM.02091-17
Claudel-Renard, C., Chevalet, C., Faraut, T., and Khan, D. (2003). Enzyme-specific profiles for genome annotation – PRIAM. Nucleic Acids Res. 31, 6633–6639. doi: 10.1093/nar/gkg847
Cline, J. (1969). Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14, 454–458. doi: 10.4319/lo.1969.14.3.0454
Costa, C., Teixeira, M., LeGall, J., Moura, J. J. G., and Moura, I. (1997). Formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774: isolation and spectroscopic characterization of the active sites (heme, iron- sulfur centers and molybdenum). J. Biol. Inorg. Chem. 2, 198–208. doi: 10.1007/s007750050125
Crittenden, C. M., Akin, L. D., Morrison, L. J., Trent, M. S., and Brodbelt, J. S. (2017). Characterization of lipid A variants by energy-resolved mass spectrometry: impact of acyl chains. J. Am. Soc. Mass Spectrom. 28, 1118–1126. doi: 10.1007/s13361-016-1542-6
Deutsch, E. W., Bandeira, N., Sharma, V., Perez-Riverol, Y., Carver, J. J., Kundu, D. J., et al. (2020). The ProteomeXchange consortium in 2020: enabling “big data” approaches in proteomics. Nucleic Acids Res. 48, D1145–D1152. doi: 10.1093/nar/gkz984
Doetsch, R. N. (1981). “Determinative methods of light microscopy,” in Manual of Methods for General Bacteriology (Washington, D.C.: American Society for Microbiology), 21–33.
Falagán, C., Sánchez-España, J., and Johnson, D. B. (2014). New insights into the biogeochemistry of extremely acidic environments revealed by a combined cultivation-based and culture-independent study of two stratified pit lakes. FEMS Microbiol. Ecol. 87, 231–243. doi: 10.1111/1574-6941.12218
Feehily, C., and Karatzas, K. A. G. (2013). Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 114, 11–24. doi: 10.1111/j.1365-2672.2012.05434.x
Finneran, K. T., Forbush, H. M., VanPraagh, C. V., and Lovley, D. R. (2002). Desulfitobacterium metallireducens sp. nov., an anaerobic bacterium that couples growth to the reduction of metals and humic acids as well as chlorinated compounds. Int. J. Syst. Evol. Microbiol. 52, 1929–1935. doi: 10.1099/ijs.0.02121-0
Flamholz, A., Noor, E., Bar-Even, A., and Milo, R. (2012). eQuilibrator—The biochemical thermodynamics calculator. Nucleic Acids Res. 40, D770–D775. doi: 10.1093/nar/gkr874
Frolov, E. N., Kublanov, I. V., Toshchakov, S. V., Samarov, N. I., Novikov, A. A., Lebedinsky, A. V., et al. (2017). Thermodesulfobium acidiphilum sp. nov., a thermoacidophilic, sulfate-reducing, chemoautotrophic bacterium from a thermal site. Int. J. Syst. Evol. Microbiol. 67, 1482–1485. doi: 10.1099/ijsem.0.001745
Frolov, E. N., Zayulina, K. S., Kopitsyn, D. S., Kublanov, I. V., Bonch-Osmolovskaya, E. A., and Chernyh, N. A. (2018). Desulfothermobacter acidiphilus gen. Nov., sp. nov., a thermoacidophilic sulfate-reducing bacterium isolated from a terrestrial hot spring. Int. J. Syst. Evol. Microbiol. 68, 871–875. doi: 10.1099/ijsem.0.002599
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Grossi, V., Mollex, D., Vinçon-Laugier, A., Hakil, F., Pacton, M., and Cravo-Laureau, C. (2015). Mono- and dialkyl glycerol ether lipids in anaerobic bacteria: biosynthetic insights from the mesophilic sulfate reducer Desulfatibacillum alkenivorans PF2803 T. Appl. Environ. Microbiol. 81, 3157–3168. doi: 10.1128/AEM.03794-14
Hall, J. R., Mitchell, K. R., Jackson-Weaver, O., Kooser, A. S., Cron, B. R., Crossey, L. J., et al. (2008). Molecular characterization of the diversity and distribution of a thermal spring microbial community by using rRNA and metabolic genes. Appl. Environ. Microbiol. 74, 4910–4922. doi: 10.1128/AEM.00233-08 [doi]
Hausmann, B., Knorr, K. H., Schreck, K., Tringe, S. G., Glavina Del Rio, T., Loy, A., et al. (2016). Consortia of low-abundance bacteria drive sulfate reduction-dependent degradation of fermentation products in peat soil microcosms. ISME J. 10, 2365–2375. doi: 10.1038/ismej.2016.42
Hausmann, B., Pelikan, C., Rattei, T., Loy, A., and Pester, M. (2019). Long-term transcriptional activity at zero growth of a cosmopolitan rare biosphere member. MBio 10, 1–16. doi: 10.1128/mBio.02189-18
Huang, C. H., Lee, I. L., Yeh, I. J., Liao, J. H., Ni, C. L., Wu, S. H., et al. (2010). Upregulation of a non-heme iron-containing ferritin with dual ferroxidase and DNA-binding activities in helicobacter pylori under acid stress. J. Biochem. 147, 535–543. doi: 10.1093/jb/mvp200
Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. doi: 10.1186/1471-2105-11-119
Jackson, D. R., Cassilly, C. D., Plichta, D. R., Vlamakis, H., Liu, H., Melville, S. B., et al. (2021). Plasmalogen biosynthesis by anaerobic bacteria: identification of a two-gene operon responsible for Plasmalogen production in Clostridium perfringens. ACS Chem. Biol. 16, 6–13. doi: 10.1021/acschembio.0c00673
Jameson, E., Rowe, O. F., Hallberg, K. B., and Johnson, D. B. (2010). Sulfidogenesis and selective precipitation of metals at low pH mediated by Acidithiobacillus spp. and acidophilic sulfate-reducing bacteria. Hydrometallurgy 104, 488–493. doi: 10.1016/j.hydromet.2010.03.029
Jerez, C., Chamorro, D., Peirano, I., and Toledo, H. (1988). Studies of the stress response in chemolithotrophic acidophilic bacteria. Biochem. Int. 17, 989–999.
Johnson, D. B., and Hallberg, K. B. (2005). Acid mine drainage remediation options: A review. Sci. Total Environ. 338, 3–14. doi: 10.1016/j.scitotenv.2004.09.002
Kimura, S., Hallberg, K. B., and Johnson, D. B. (2006). Sulfidogenesis in low pH (3.8-4.2) media by a mixed population of acidophilic bacteria. Biodegradation 17, 159–167. doi: 10.1007/s10532-005-3050-4
Klemps, R., Cypionka, H., Widdel, F., and Pfennig, N. (1985). Growth with hydrogen, and further physiological characteristics of Desulfotomaculum species. Arch. Microbiol. 143, 203–208. doi: 10.1007/BF00411048
Koehorst, J. J., van Dam, J. C. J., Saccenti, E., Martins Dos Santos, V. A. P., Suarez-Diez, M., and Schaap, P. J. (2018). SAPP: functional genome annotation and analysis through a semantic framework using FAIR principles. Bioinformatics 34, 1401–1403. doi: 10.1093/bioinformatics/btx767
Koga, Y., Morn, H., Akagawa-Matsushita, M., and Ohga, M. (1998). Correlation of polar lipid composition with 16S rRNA phylogeny in methanogens. Further analysis of lipid component parts. Biosci. Biotechnol. Biochem. 62, 230–236. doi: 10.1271/bbb.62.230
Koga, Y., Nishihara, M., Morii, H., and Akagawa-Matsushita, M. (1993). Ether polar lipids of methanogenic bacteria: structures, comparative aspects, and biosyntheses. Microbiol. Rev. 57, 164–182. doi: 10.1128/mmbr.57.1.164-182.1993
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Korehi, H., Blöthe, M., and Schippers, A. (2014). Microbial diversity at the moderate acidic stage in three different sulfidic mine tailings dumps generating acid mine drainage. Res. Microbiol. 165, 713–718. doi: 10.1016/j.resmic.2014.08.007
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Larrouy-Maumus, G., Clements, A., Filloux, A., McCarthy, R. R., and Mostowy, S. (2016). Direct detection of lipid A on intact gram-negative bacteria by MALDI-TOF mass spectrometry. J. Microbiol. Methods 120, 68–71. doi: 10.1016/j.mimet.2015.12.004
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Lee, Y. J., Romanek, C. S., and Wiegel, J. (2009). Desulfosporosinus youngiae sp. nov., a spore-forming, sulfate-reducing bacterium isolated from a constructed wetland treating acid mine drainage. Int. J. Syst. Evol. Microbiol. 59, 2743–2746. doi: 10.1099/ijs.0.007336-0
López-Archilla, A. I., Marin, I., and Amils, R. (2001). Microbial community composition and ecology of an acidic aquatic environment: the Tinto River, Spain. Microb. Ecol. 41, 20–35. doi: 10.1007/s002480000044
Lottermoser, B. G. (2010). “Tailings” in Mine Wastes. (Verlag Berlin Heidelberg: Springer), 205–241.
Ludwig, W., Strunk, O., Westram, R., Richter, L., and Meier, H. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Madden, A. S., Palumbo, A. V., Ravel, B., Vishnivetskaya, T. A., Phelps, T. J., Schadt, C. W., et al. (2009). Donor-dependent extent of uranium reduction for bioremediation of contaminated sediment microcosms. J. Environ. Qual. 38, 53–60. doi: 10.2134/jeq2008.0071
Mardanov, A. V., Panova, I. A., Beletsky, A. V., Avakyan, M. R., Kadnikov, V. V., Antsiferov, D. V., et al. (2016). Genomic insights into a new acidophilic, copper-resistant Desulfosporosinus isolate from the oxidized tailings area of an abandoned gold mine. FEMS Microbiol. Ecol. 92, 1–14. doi: 10.1093/femsec/fiw111
Meador, T. B., Gagen, E. J., Loscar, M. E., Goldhammer, T., Yoshinaga, M. Y., Wendt, J., et al. (2014). Thermococcus kodakarensis modulates its polar membrane lipids and elemental composition according to growth stage and phosphate availability. Front. Microbiol. 5:10. doi: 10.3389/fmicb.2014.00010
Mori, K., Kim, H., Kakegawa, T., and Hanada, S. (2003). A novel lineage of sulfate-reducing microorganisms: Thermodesulfobiaceae fam. Nov., Thermodesulfobium narugense, gen. nov., sp. nov., a new thermophilic isolate from a hot spring. Extremophiles 7, 283–290. doi: 10.1007/s00792-003-0320-0
Ňancucheo, I., and Johnson, D. B. (2012). Selective removal of transition metals from acidic mine waters by novel consortia of acidophilic sulfidogenic bacteria. Microb. Biotechnol. 5, 34–44. doi: 10.1111/j.1751-7915.2011.00285.x
Ňancucheo, I., and Johnson, D. B. (2014). Removal of sulfate from extremely acidic mine waters using low pH sulfidogenic bioreactors. Hydrometallurgy 150, 222–226. doi: 10.1016/j.hydromet.2014.04.025
NCBI Resource Coordinators (2013). Database resources of the National Center for biotechnology information. Nucleic Acids Res. 41, D8–D20. doi: 10.1093/nar/gks1189
Nevin, K. P., Finneran, K. T., and Lovley, D. R. (2003). Microorganisms associated with uranium bioremediation in a high-salinity subsurface sediment. Appl. Environ. Microbiol. 69, 3672–3675. doi: 10.1128/AEM.69.6.3672-3675.2003
Nguyen, N.-N., Srihari, S., Leong, H. W., and Chong, K.-F. (2015). EnzDP: improved enzyme annotation for metabolic network reconstruction based on domain composition profiles. J. Bioinforma. Comput. Biol. 13:1543003. doi: 10.1142/S0219720015430039
Oger, P. M., and Cario, A. (2013). Adaptation of the membrane in Archaea. Biophys. Chem. 183, 42–56. doi: 10.1016/j.bpc.2013.06.020
Panova, I. A., Ikkert, O., Avakyan, M. R., Kopitsyn, D. S., Mardanov, A. V., Pimenov, N. V., et al. (2021). Desulfosporosinus metallidurans sp. Nov., an acidophilic, metal-resistant sulfate-reducing bacterium from acid mine drainage. Int. J. Syst. Evol. Microbiol. 71. doi: 10.1099/ijsem.0.004876
Pereira, I. A. C., Ramos, A. R., Grein, F., Marques, M. C., da Silva, S. M., Venceslau, S. S., et al. (2011). A Comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front. Microbiol. 2:69 doi: 10.3389/fmicb.2011.00069
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Pester, M., Brambilla, E., Alazard, D., Rattei, T., Weinmaier, T., Han, J., et al. (2012). Complete genome sequences of Desulfosporosinus orientis DSM765T, Desulfosporosinus youngiae DSM17734T, Desulfosporosinus meridiei DSM13257T, and Desulfosporosinus acidiphilus DSM22704T. J. Bacteriol. 194, 6300–6301. doi: 10.1128/JB.01392-12
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Petrie, L., North, N. N., Dollhopf, S. L., Balkwill, D. L., and Kostka, J. E. (2003). Enumeration and characterization of iron(III)-reducing microbial communities from acidic subsurface sediments contaminated with uranium(VI). Appl. Environ. Microbiol. 69, 7467–7479. doi: 10.1128/AEM.69.12.7467-7479.2003
Petzsch, P., Johnson, D. B., Daniel, R., and Schlömann, M. (2015). Genome sequence of the moderately acidophilic sulfate-reducing firmicute Desulfosporosinus acididurans (strain M1T). Genome Announc. 3, 5–6. doi: 10.1128/genomeA.00881-15.Copyright
Pires, R. H., Venceslau, S. S., Morais, F., Teixeira, M., Xavier, A. V., and Pereira, I. A. C. (2006). Characterization of the Desulfovibrio desulfuricans ATCC 27774 DsrMKJOP complex - A membrane-bound redox complex involved in the sulfate respiratory pathway. Biochemistry 45, 249–262. doi: 10.1021/bi0515265
Pruesse, E., Peplies, J., and Glöckner, F. O. (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. doi: 10.1093/bioinformatics/bts252
Rabus, R., Venceslau, S. S., Wöhlbrand, L., Voordouw, G., Wall, J. D., and Pereira, I. A. C. (2015). A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv. Microb. Physiol. 66, 55–321. doi: 10.1016/bs.ampbs.2015.05.002
Ragsdale, S. W., and Pierce, E. (2008). Acetogenesis and the wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 1784, 1873–1898. doi: 10.1016/j.bbapap.2008.08.012
Ramamoorthy, S., Sass, H., Langner, H., Schumann, P., Kroppenstedt, R. M., Spring, S., et al. (2006). Desulfosporosinus lacus sp. nov., a sulfate-reducing bacterium isolated from pristine freshwater lake sediments. Int. J. Syst. Evol. Microbiol. 56, 2729–2736. doi: 10.1099/ijs.0.63610-0
Ramos, A. R., Keller, K. L., Wall, J. D., and Pereira, I. A. (2012). The membrane QmoABC complex interacts directly with the dissimilatory adenosine 5′-phosphosulfate reductase in sulfate reducing bacteria. Front. Microbiol. 3:137. doi: 10.3389/fmicb.2012.00137
Robertson, W. J., Bowman, J. P., Franzmann, P. D., and Mee, B. J. (2001). Desulfosporosinus meridiei sp. nov., a spore-forming sulfate-reducing bacterium isolated from gasolene-contaminated groundwater. Int. J. Syst. Evol. Microbiol. 51, 133–140. doi: 10.1099/00207713-51-1-133
Rowe, O. F., Sánchez-España, J., Hallberg, K. B., and Johnson, D. B. (2007). Microbial communities and geochemical dynamics in an extremely acidic, metal-rich stream at an abandoned sulfide mine (Huelva, Spain) underpinned by two functional primary production systems. Environ. Microbiol. 9, 1761–1771. doi: 10.1111/j.1462-2920.2007.01294.x
Sánchez-Andrea, I., Florentino, A. P., Semerel, J., Strepis, N., Sousa, D. Z., and Stams, A. J. M. (2018). Co-culture of a novel fermentative bacterium, Lucifera butyrica gen. Nov. sp. nov., with the sulfur reducer Desulfurella amilsii for enhanced sulfidogenesis. Front. Microbiol. 9:3108. doi: 10.3389/fmicb.2018.03108
Sánchez-Andrea, I., Knittel, K., Amann, R., Amils, R., and Sanz, J. L. (2012a). Quantification of Tinto River sediment microbial communities: importance of sulfate-reducing bacteria and their role in attenuating acid mine drainage. Appl. Environ. Microbiol. 78, 4638–4645. doi: 10.1128/AEM.00848-12
Sánchez-Andrea, I., Rodriguez, N., Amils, R., and Sanz, J. L. (2011). Microbial diversity in anaerobic sediments at Rio Tinto, a naturally acidic environment with a high heavy metal content. Appl. Environ. Microbiol. 77, 6085–6093. doi: 10.1128/AEM.00654-11
Sánchez-Andrea, I., Rojas-Ojeda, P., Amils, R., and Sanz, J. L. (2012b). Screening of anaerobic activities in sediments of an acidic environment: Tinto River. Extremophiles 16, 829–839. doi: 10.1007/s00792-012-0478-4
Sánchez-Andrea, I., Stams, A. J. M., Amils, R., and Sanz, J. L. (2013). Enrichment and isolation of acidophilic sulfate-reducing bacteria from Tinto River sediments. Environ. Microbiol. Rep. 5, 672–678. doi: 10.1111/1758-2229.12066
Sánchez-Andrea, I., Stams, A. J. M., Hedrich, S., Ňancucheo, I., and Johnson, D. B. (2015). Desulfosporosinus acididurans sp. nov.: an acidophilic sulfate-reducing bacterium isolated from acidic sediments. Extremophiles 19, 39–47. doi: 10.1007/s00792-014-0701-6
Sánchez-España, J., Yusta, I., Ilin, A., van der Graaf, C. M., and Sánchez-Andrea, I. (2020). Microbial geochemistry of the acidic saline pit lake of Brunita mine (La Unión, SE Spain). Mine Water Environ. 39, 535–555. doi: 10.1007/s10230-020-00655-0
Sand, W., Gehrke, T., Jozsa, P.-G., and Schippers, A. (2001). (bio)chemistry of bacterial leaching—direct vs. indirect bioleaching. Hydrometallurgy 59, 159–175. doi: 10.1016/S0304-386X(00)00180-8
Santofimia, E., González-Toril, E., López-Pamo, E., Gomariz, M., Amils, R., and Aguilera, Á. (2013). Microbial diversity and its relationship to physicochemical characteristics of the water in two extreme acidic pit lakes from the Iberian Pyrite Belt (SW Spain). PLoS One 8, 1–12. doi: 10.1371/journal.pone.0066746
Santos, A. A., Venceslau, S. S., Grein, F., Leavitt, W. D., Dahl, C., Johnston, D. T., et al. (2015). A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science 350, 1541–1545. doi: 10.1126/science.aad3558
Sebban, C., Blanchard, L., Bruschi, M., and Guerlesquin, F. (1995). Purification and characterization of the formate dehydrogenase from Desulfovibrio vulgaris Hildenborough. FEMS Microbiol. Lett. 133, 143–149. doi: 10.1016/0378-1097(95)00339-7
Senko, J. M., Zhang, G., McDonough, J. T., Bruns, M. A., and Burgos, W. D. (2009). Metal reduction at low pH by a Desulfosporosinus species: implications for the biological treatment of acidic mine drainage. Geomicrobiol J. 26, 71–82. doi: 10.1080/01490450802660193
Seshadri, R., Leahy, S. C., Attwood, G. T., Teh, K. H., Lambie, S. C., Cookson, A. L., et al. (2018). Cultivation and sequencing of rumen microbiome members from the Hungate1000 collection. Nat. Biotechnol. 36, 359–367. doi: 10.1038/nbt.4110
Siliakus, M. F., van der Oost, J., and Kengen, S. W. M. (2017). Adaptations of archaeal and bacterial membranes to variations in temperature, pH and pressure. Extremophiles 21, 651–670. doi: 10.1007/s00792-017-0939-x
Sinninghe Damsté, J., Rijpstra, W. I., Hopmans, E. C., Weijers, J. W., Foesel, B. U., Overmann, J., et al. (2011). 13,16-dimethyl octacosanedioic acid (iso-diabolic acid), a common membrane-spanning lipid of Acidobacteria subdivisions 1 and 3. Appl. Environ. Microbiol. 77, 4147–4154. doi: 10.1128/AEM.00466-11
Slonczewski, J. L., Fujisawa, M., Dopson, M., and Krulwich, T. A. (2009). “Cytoplasmic pH measurement and homeostasis in bacteria and Archaea,” in Advances in Microbial Physiology (Elsevier), 1–317.
Sohlenkamp, C. (2017). Biogenesis of fatty acids, lipids and membranes. Biog. Fat. Acids, Lipids Membr. 1–13. doi: 10.1007/978-3-319-43676-0
Spring, S., and Rosenzweig, F. (2006). “The genera Desulfitobacterium and Desulfosporosinus: taxonomy,” in The Prokaryotes. 3rd Edn. eds. M. Dworkin (Editor-in-Chief), S. Falkow, E. Rosenberg, K.-H. Schleifer and E. Stackebrandt (NY: Springer).
Spurr, A. R. (1969). A low-viscosity epoxy resin embedding medium for electron microscopy. J. Ultrastruct. Res. 26, 31–43. doi: 10.1016/S0022-5320(69)90033-1
Stackebrandt, E., Sproer, C., Rainey, F. A., Burghardt, J., Pauker, O., and Hippe, H. (1997). Phylogenetic analysis of the genus Desulfotomaculum: evidence for the misclassification of Desulfotomaculum guttoideum and description of Desulfotomaculum orientis as Desulfosporosinus orientis gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 47, 1134–1139. doi: 10.1099/00207713-47-4-1134
Suzuki, Y., Kelly, S. D., Kemner, K. M., and Banfield, J. F. (2004). Enzymatic U(VI) reduction by Desulfosporosinus species. Radiochim. Acta 92, 11–16. doi: 10.1524/ract.92.1.11.25404
Tindall, B. J. (1990a). A comparative study of the lipid composition of Halobacterium saccharovorum from various sources. Syst. Appl. Microbiol. 13, 128–130. doi: 10.1016/S0723-2020(11)80158-X
Tindall, B. J. (1990b). Lipid composition of Halobacterium lacusprofundi. FEMS Microbiol. Lett. 66, 199–202. doi: 10.1111/j.1574-6968.1990.tb03996.x
UniProt, C. (2014). Activities at the universal protein resource (UniProt). Nucleic Acids Res. 42, D191–D198. doi: 10.1093/nar/gkt1140
Utkin, I., Woese, C. R., and Wiegel, J. (1994). Isolation and characterization of Desulfitobacterium dehalogenans gen. Nov., sp. nov., an anaerobic bacterium which reductively dechlorinates chlorophenolic compounds. Int. J. Syst. Bacteriol. 44, 612–619. doi: 10.1099/00207713-44-4-612
van der Graaf, C. M., Sánchez-España, J., Yusta, I., Ilin, A., Shetty, S. A., Bale, N. J., et al. (2020). Biosulfidogenesis mediates natural attenuation in acidic mine pit lakes. Microorganisms 8, 1–26. doi: 10.3390/microorganisms8091275
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, S., Huang, H., Moll, J., and Thauer, R. K. (2010). NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in clostridium kluyveri. J. Bacteriol. 192, 5115–5123. doi: 10.1128/JB.00612-10
Wendt-Potthoff, K., Koschorreck, M., Diez Ercilla, M., and Sánchez España, J. (2012). Microbial activity and biogeochemical cycling in a nutrient-rich meromictic acid pit lake. Limnologica 42, 175–188. doi: 10.1016/j.limno.2011.10.004
Winch, S., Mills, H. J., Kostka, J. E., Fortin, D., and Lean, D. R. S. (2009). Identification of sulfate-reducing bacteria in methylmercury-contaminated mine tailings by analysis of SSU rRNA genes. FEMS Microbiol. Ecol. 68, 94–107. doi: 10.1111/j.1574-6941.2009.00658.x
Wright, E. S., Yilmaz, L. S., and Noguera, D. R. (2012). DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl. Environ. Microbiol. 78, 717–725. doi: 10.1128/AEM.06516-11
Yarza, P., Richter, M., Peplies, J., Euzeby, J., Amann, R., Schleifer, K.-H., et al. (2008). The all-species living tree project: A 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst. Appl. Microbiol. 31, 241–250. doi: 10.1016/j.syapm.2008.07.001
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K. H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645. doi: 10.1038/nrmicro3330
Young, G. M., Amid, D., and Miller, V. L. (1996). A bifunctional urease enhances survival of pathogenic Yersinia enterocolitica and Morganella morganii at low pH. J. Bacteriol. 178, 6487–6495. doi: 10.1128/jb.178.22.6487-6495.1996
Keywords: acid rock/mine drainage, acidophiles, sulfate-reducing bacteria, Acididesulfobacillus, Desulfosporosinus, Desulfitobacterium, acetate oxidation, bioremediation
Citation: Sánchez-Andrea I, van der Graaf CM, Hornung B, Bale NJ, Jarzembowska M, Sousa DZ, Rijpstra WIC, Sinninghe Damsté JS and Stams AJM (2022) Acetate Degradation at Low pH by the Moderately Acidophilic Sulfate Reducer Acididesulfobacillus acetoxydans gen. nov. sp. nov. Front. Microbiol. 13:816605. doi: 10.3389/fmicb.2022.816605
Edited by:
Claudia P. Saavedra, Andres Bello University, ChileReviewed by:
Axel Schippers, Federal Institute For Geosciences and Natural Resources, GermanyShawn E. McGlynn, Tokyo Institute of Technology, Japan
Copyright © 2022 Sánchez-Andrea, van der Graaf, Hornung, Bale, Jarzembowska, Sousa, Rijpstra, Sinninghe Damsté and Stams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irene Sánchez-Andrea, aXJlbmUuc2FuY2hlemFuZHJlYUB3dXIubmw=
†These authors have contributed equally to this work