 Ru-teng Wei1†
Ru-teng Wei1† Ning Chen1†Yin-ting Ding1Lin Wang1Fei-fei Gao1Liang Zhang1Yi-hui Liu1Hua Li1,2,3*‡Hua Wang1,2,3*‡
Ning Chen1†Yin-ting Ding1Lin Wang1Fei-fei Gao1Liang Zhang1Yi-hui Liu1Hua Li1,2,3*‡Hua Wang1,2,3*‡- 1College of Enology, Northwest A&F University, Xianyang, China
- 2Shaanxi Engineering Research Center for Viti-Viniculture, Xianyang, China
- 3China Wine Industry Technology Institute, Zhongguancun Innovation Center, Yinchuan, China
Grapevine-related microorganisms affect the health and yield of grapes, the metabolic pathways of the fermentation process, and the regional characteristics of wine. However, the diversity of epidermal microorganisms during the development of berries under the ecological viticulture model has not been described in detail. In this study, high-throughput amplicon sequencing technology was used to perform ITS and 16S sequencing of Cabernet Sauvignon epidermal microbes at different developmental stages in the Wuhai region to investigate the succession of epidermal microbes and their response to developmental stages and vineyard weather. The results showed that the diversity of fungi and bacteria decreased during development. Epidermal microorganisms recruited members according to their developmental stages, but retained the core taxa, such as the fungi genera Alternaria, Jattaea, and Jattaea and the bacteria genera Brevundimonas, Sphingomonas, Acinetobacter, and Pseudomonas. In addition, the microbial diversity was associated with specific meteorological parameters, implying that there was a connection between the environmental conditions of the vineyard and the microbial distribution pattern such as the fungus genus Filobasidium was positively correlated with relative humidity and negatively correlated with average high temperature, average low temperature, and average ground temperature; the bacterium genus Lactobacillus was positively correlated with sunlight time, and negatively correlated with relative humidity. In conclusion, this study can help vineyard managers understand the microbial consortia associated with particular diseases, and also the dynamics of infection processes in order to take preventive actions, especially at the most critical moments.
Introduction
Wine is unique because it is essentially a natural product, which is a result of ordered and complex biochemical transformations including the ripening of grape and the metabolism of microbes from fermentation to aging (Liu et al., 2017; Carpena et al., 2020; Echave et al., 2021). In winemaking, “microbial terroir” refers to the contribution of microorganisms in a region to the characteristics of wine, and is a process that starts in the vineyards and then develops along the different stages of fermentation (Bokulich et al., 2014; Comitini et al., 2017; Wei et al., 2022a). Thus, the microbiological aspects of wine production are influenced by the vineyard ecosystem and not simply by the winery and fermentative processes (Grangeteau et al., 2017; Liu et al., 2019).
Grapevine hosts a variety of microorganisms (fungi, yeast, and bacteria) on and inside organs and their surrounding soil (Wei et al., 2022b). Among these, inhabitants are both harmful and beneficial microbes that are involved in crucial functions such as plant nutrition and plant resistance to biotic and abiotic stresses, hence in plant growth promotion, fruit yield, disease resistance, and survival (Barata et al., 2012; Wei et al., 2022d). Most studies have focused on a series of plant pathogenic fungi that affect grapes, including Erysiphe necator, Botrytis cinerea, and Plamospara viticola – the causative agents of grapevine powdery mildew, gray rot, and downy mildew, respectively. In addition, grapes may bear saprophytic molds such as Aspergillus spp., Cladosporium spp., and Penicillium spp., which are directly responsible for several grape rots and are indirectly involved in food spoilage because they produce mycotoxins (Martins et al., 2014). These fungi are unable to grow in wines, and their effect on wine quality is due to grape damage (Barata et al., 2012). In addition, some of these microorganisms are even considered as natural biocontrol agents due to their ability to protect the plant against phytopathogens and enhance the natural plant defenses (Pinto et al., 2014). Trichoderma is one of the most studied and applied fungal biocontrol agents. The benefits of these microorganisms to the plant include suppression of pathogens, growth promotion, enhanced nutrient availability, and induction of resistance. The biological activity is related to the variety of metabolites that they produce. These metabolites have been found to directly inhibit the pathogens, increase the disease resistance, and enhance the plant growth (Pascale et al., 2017).
In recent studies, a large number of literature have reported microbial diversity associated with vineyards. In addition, high-throughput sequencing technologies enable the detection and quantification of microorganisms present in vineyard soil and grapes, as well as its transformation later in winery (Bokulich et al., 2016; Morgan et al., 2017; Liang et al., 2019). High-throughput analysis of the grapevine phyllosphere, flowers and grape berry surfaces, demonstrated that the bacterial communities were predominated by Proteobacteria followed by Firmicutes, Actinobacteria, Acidobacteria, and Bacteroidetes (Perazzolli et al., 2014; Pinto et al., 2014, 2015; Portillo et al., 2016). The relative abundances of the groups vary depending on the plant tissue or organ. Dominant taxa include members of the genera Pseudomonas, Sphingomonas, Frigoribacterium, Curtobacterium, Bacillus, Enterobacter, Acinetobacter, Erwinia, Citrobacter, Pantoea, and Methylobacterium (Bokulich et al., 2014; Perazzolli et al., 2014; Pinto et al., 2015; Zarraonaindia et al., 2015; Portillo et al., 2016). The fungal diversity is very similar at the phylum level, mainly composed of Ascomycetes and Basidiomycetes. Other phyla such as Zygomycota and Chytridiomycota are only present in low abundance. Frequently encountered genera of filamentous fungi include Aspergillus, Alternaria, Penicillium, Cladosporium, Lewia, Davidiella, Erysiphe, and Botrytis and the yeast-like fungus, Aureobasidium pullulans, while the yeast genera include Hanseniaspora, Issatchenkia, Pichia, Candida, Rhodotorula, Lachancea, Metschnikowia, Cryptococcus, Filobasidiella, Sporobolomyces, and Torulaspora (Bokulich et al., 2014; Pinto et al., 2014; Wang et al., 2015; Filippis et al., 2017; Liu and Howell, 2021). The grape berry surface is, nevertheless, a natural habitat of microorganisms. Diversity and stability of the grape epidermal microorganisms are strongly associated with numerous factors, such as vineyard geography (altitude, latitude, and longitude; Gao et al., 2019), climatic conditions (rain, temperature, humidity, and maturity period; Liu and Howell, 2021; Wei et al., 2022c), grape variety, and viticultural practice (herbicides, fertilizers, pesticides, and fungicides used; Pinto et al., 2014).
The new ecological approach to viticulture with emphasis on ecologically sound grape production views grapevines as part of a complex agroecosystem where many organisms co-exist and interact (Likar et al., 2017). In particular, this approach recognizes the importance of interactions between the microbial communities and the plants, as these influence the growth, physiology, and yield of the grapevines (Compant et al., 2019). Although the new ecological approach to viticulture recognizes the importance of grapevine interactions with microbial communities, there remains less knowledge available on diversity and dynamics of grape microbial communities under ecological viticulture models. Therefore, we collected Cabernet Sauvignon berries at different developmental stages from ecological vineyards located in the Wuhai region, characterized the fungal and bacterial diversity of grape epidermis by amplicon sequencing, and further investigated the succession of epidermal microorganisms and the co-occurrence model of meteorological parameters and epidermal microorganisms.
Materials and Methods
Location Description and Sampling
In this study, from June to October in 2020, berry samples were collected from the Sunshine Tianyu International Winery Cabernet Sauvignon vineyard located in the Wuhai region (Inner Mongolia, China; Supplementary Figure 1). The vineyard adopts an ecological viticulture management model. No chemical fertilizers or insecticides are applied for pest or disease control during the grape growing season, and weeds are controlled via manually weeding every month. The cultivation frame adopts Modified VSP (All the vines were trained to a slope trunk with a vertical shoot positioning trellis system), which is convenient for the main vines to be buried in the soil in winter (Supplementary Figure 1). The sunshine hours (h), precipitation (mm), average low temperature (°C), average high temperature (°C), average ground temperature (°C), and relative humidity (%) were measured weekly. Meteorological data originated from the vineyard’s meteorological equipment.
To evaluate the changes in the microbial community during grape development, berry samples from the following five stages were collected aseptically: fruit setting (A), early veraison (B), end veraison (C), mid maturity (D), and harvest (E), corresponding to stages 31, 35, 36, 37, and 38 in the improved E-L system. In the Cabernet Sauvignon vineyard, a relatively flat plot with an area of approximately 1.5 ha and a regular topography was selected for sampling. A total of 3 biological replicates were sampled at each developmental berry stage, and each replicate was collected from 5 sample points fixed in the sampling plot that could cover the area of the sampling plot. Considering the heterogeneity of the tested grapes, the berries were collected from the upper, central, and lower part of the cluster at each sample point, both from the sun-exposed and shaded side. A total of 15 healthy grape berry samples were collected. All berry samples were immediately stored in sterile bags, transported to the laboratory on dry ice, and stored at −80°C before molecular analysis.
DNA Extraction and Sequencing
The sample was thawed at 28°C for 30 min. An amount of 10 g of the sample was aseptically transferred to a polypropylene test tube containing 100 ml of washing solution (0.1 M potassium phosphate buffer, pH 7.0), and ultrasonic treatment was performed in an ultrasonic cleaning tank at a frequency of 40 kHz for 10 min. The sediment filtered was used to extract the microbial genome DNA. The process was performed using the methods described by Gao et al. (2019). Genomic DNA was submitted to Gene Denovo company (Guangzhou, China) for amplification. The primers ITS3_KYO2F (GATGAAGAACGYAGYRAA) and ITS4-2409R (TCCTCCGCTTATTGATATGC) were used to amplify the fungal ITS2 region, and the primers 341F (CCTACGGGNGGCWGCAG) and 806R (GGACTACHVGGGTATCTAAT) were used to amplify the V3–V4 region of the bacterial 16S rDNA gene. The PCR process was performed under the following conditions: 94°C for 5 min, 94°C for 30 s, 55°C for 30 s, and 28 cycles of 72°C for 1 min, and the final extension was performed at 72°C for 10 min (Zhang et al., 2017). The amplicon was recovered from 2% agarose gel and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) according to the manufacturer’s instructions and quantified using an ABI StepOnePlus Real-Time PCR system (Life Technologies, Foster City, United States; Zhou et al., 2019). The purified amplicons were subjected to paired-end sequencing (HiSeq 2500, PE250) on the Illumina platform according to standard operations.
We used FASTP (version 0.18.0) and FLSAH (version 1.2.11) to perform quality filtering and sequence merging on the raw fastq files of 15 grape samples to obtain Tags. Then, according to the QIIME (version 1.9.1) Tags Quality Control process, low-quality tags were filtered to obtain high-quality Clean Tags. Based on the reference database (Quast et al., 2013), the UCHIME Algorithm was used to detect and delete the chimeras of tags to obtain effective Clean Tags (Edgar et al., 2011). Finally, Clean Tags were assigned to the same OTUs (Operational Taxonomic Units) according to the similarity of ≥97% by using UPARSE (version 9.2.64; Edgar, 2013). The tag sequence with the highest abundance was selected as the representative sequence of each OTU. For each representative sequence, the classification information was annotated using the SIlVA database based on the mothur algorithm (Quast et al., 2013). The sequence data are available in NCBI under BioProject number PRJNA766154.
Data Analysis
According to the alpha index, one-way analysis of variance (ANOVA) and the Duncan test (p < 0.05) were used to analyze whether the sample diversity at different stages contained statistically significant differences. The statistical analyses were performed using the SPSS 25.0 (IBM, United States) software. Principal component analysis (PCoA) was used to evaluate the distribution pattern of grape epidermis microorganisms in different stages based on beta-diversity calculated by the Bray-Curtis distance with the “labdsv” package. The relatively abundant species (average relative abundance > 0.01%) were selected for network analysis, and those statistically significant and robust (Spearman correlation coefficient, r ≥ 0.6; p < 0.05) correlations were considered effective co-occurrence by Spearman correlation coefficient and were visualized by Cytoscape. Linear discriminant analysis effect size (LEfSe) was used to investigate the significant taxonomic differences of fungi and bacteria at different developmental stages. The OTU table was filtered to contain only the OTU with the relative abundance greater than 0.1% to reduce the LEfSe complexity. The factorial Kruskal–Wallis sum-rank test was used to identify taxa with significant differential abundances between classes, followed by the logarithmic LDA score (threshold = 4.0) to estimate the effect size of each discriminative feature. The correlation between the environmental characteristics and microbial community composition was examined by canonical correspondence analysis (CCA). The co-occurrence/interaction patterns between grape epidermis microorganisms and weather conditions during the growing season were explored using network analysis with Cytoscape. Correlation matrices were calculated with all possible pair-wise Spearman’s rank correlations between selected taxa and weather indexes. Correlations with a Spearman correlation coefficient r ≥ 0.5 and p < 0.05 were considered statistically robust. After the basic analysis, the visualization diagram was finally drawn using the package in RStudio (version 2.15.3).
The core microbiome was determined by abundance-occupancy distribution, including highly abundant and ubiquitous taxa. For the core microbiome of grape epidermis, the core OTU was the top 10% abundance of all samples which accounts for 50% of all samples or 100% at any developmental stage (average relative abundance ≥ 0.01%). Dynamics and successions of the core microbiome were illustrated by alluvial diagrams using the “ggalluvial” package in “ggplot2”.
Results
Evaluation of the Diversity and Richness of Microbial Populations in Grape Epidermis
The deep sequencing of microbial communities was originated from a total of 1,286,519 sequences, of which 1,283,205 sequences passed the Quality Control filters, which represented 99.7% of the obtained sequences (Table 1). For fungal microorganisms, a total of 648,234 sequences were obtained, while for bacterial microorganisms, a total of 634,971 sequences were obtained (Table 1). The number of reads per sample ranged from 122,648 to 132,602 sequences. All the high-quality sequence reads were grouped with 97% homology, and generated a total of 1,113 OTUs for fungi, and 2,175 for bacteria. On average, fungi and bacterial microorganisms generated 223 ± 17 and 435 ± 12 OTUs, respectively. The diversity of fungal and bacterial communities was compared between samples by rarefaction curve analysis (Figure 1). The rarefaction curve tended to be flat, indicating that deep sequencing provided good overall OTU coverage. For each sample, the expected richness (Chao1 index) has been determined. In the current analysis, we have predicted a total richness ranging from 223 ± 17 (fungal community) to 435 ± 12 (bacterial community). By comparing the obtained number of OTUs with its predicted Chao1, we were able to determine the coverage of our experiments. The richness estimators indicated that 80.6 ± 3.2 and 62.9 ± 1.5% of the fungal and bacterial community diversity were uncovered, respectively (Table 1). Therefore, we realized that despite revealing the complex and rich microbial structure, there still exists a hidden biodiversity in grapes that cannot be exposed.
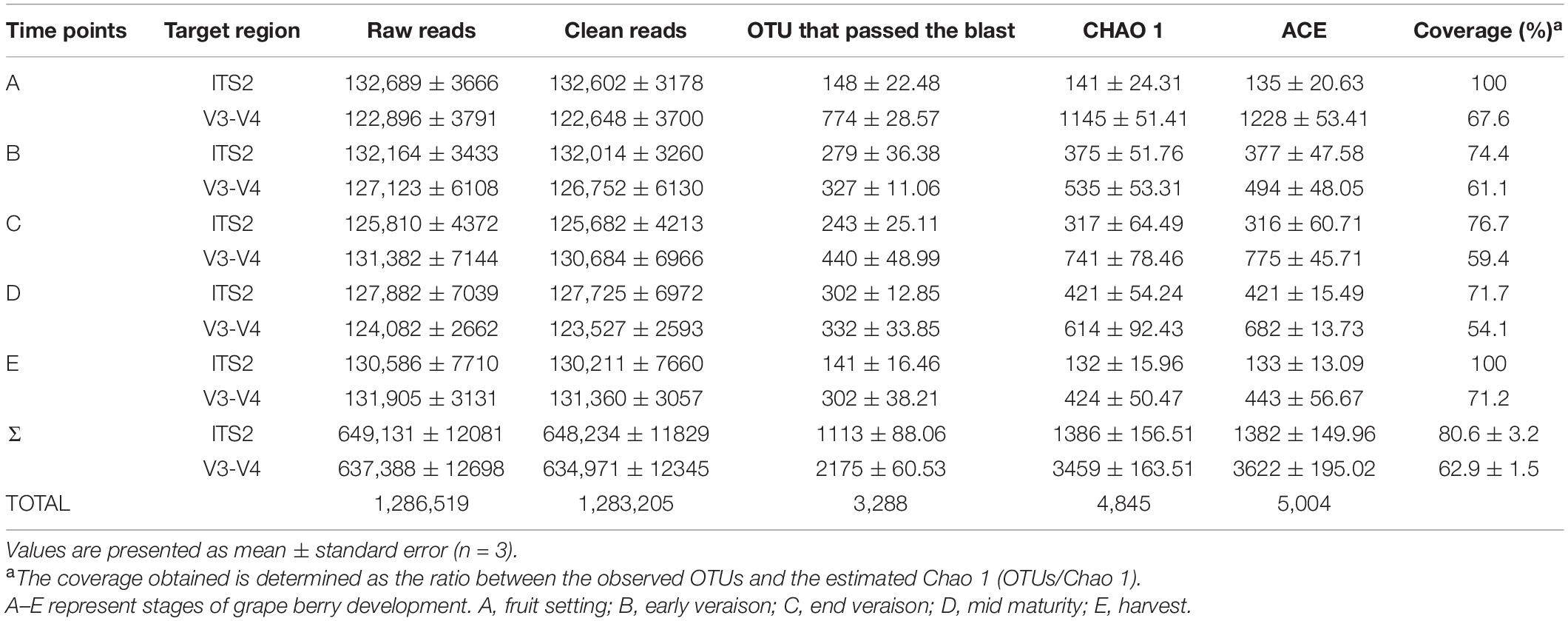
Table 1. Total sequences obtained for fungi and bacterial communities for all samples.
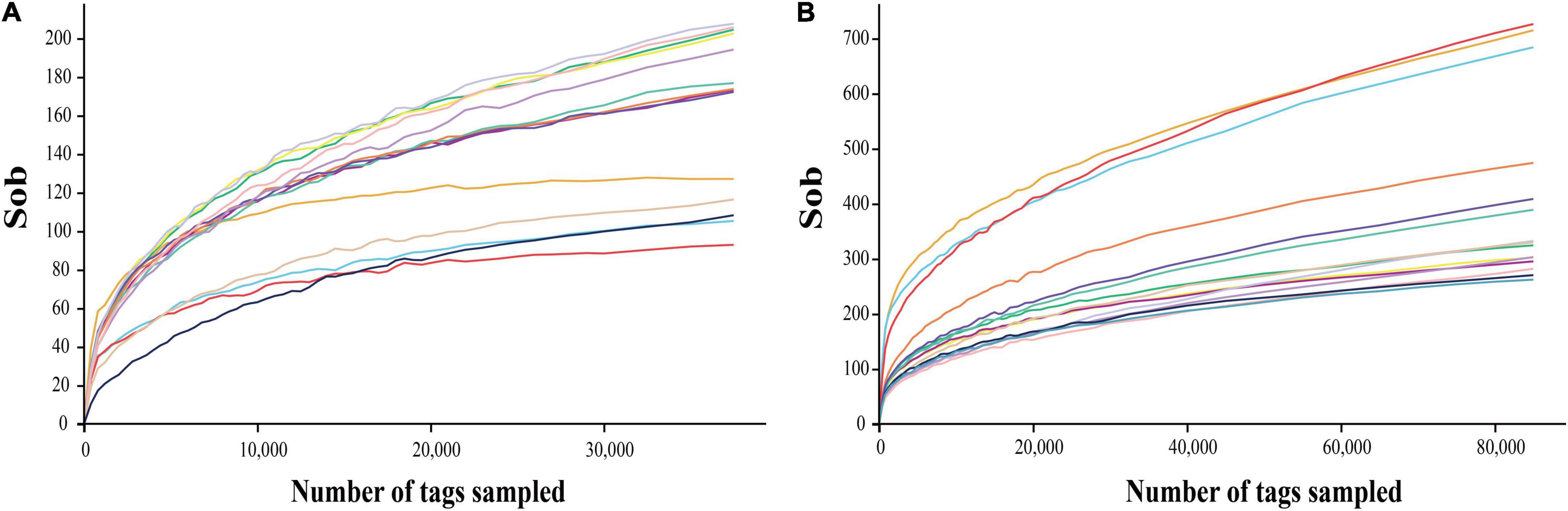
Figure 1. Rarefaction curves of fungi (A) and bacteria (B) in each sample with 97% similarity.
Diversity Changes During Grape Development
Grape microbial community compositions evolved over time. At the phylum level for fungal, most significant was the massive colonization of the grapes by Ascomycota, and it existed in high abundance at each stage, especially the veraison (B stage: 99.19%, C stage: 99.26%); the second highest abundance was shown by Basidiomycota, especially at the harvest (31.48%), but the veraison was the lowest (Figure 2A). At the genus level, the dominant taxa Alternaria was found to be massively enriched in the grape epidermis, which increased as the development stage proceeded (Figure 2C). At the phylum level for bacterial, most significant was the massive colonization of the grapes by Proteobacteria, and it existed in high abundance at each stage; the second highest abundance was shown by Firmicutes, mainly enriched at the fruit setting (30.89%; Figure 2B). At the genus level, the dominant taxa Brevundimonas was found to be massive enriched in the grape epidermis, which increased as the development stage proceeded, but decreased at the harvest (Figure 2D).
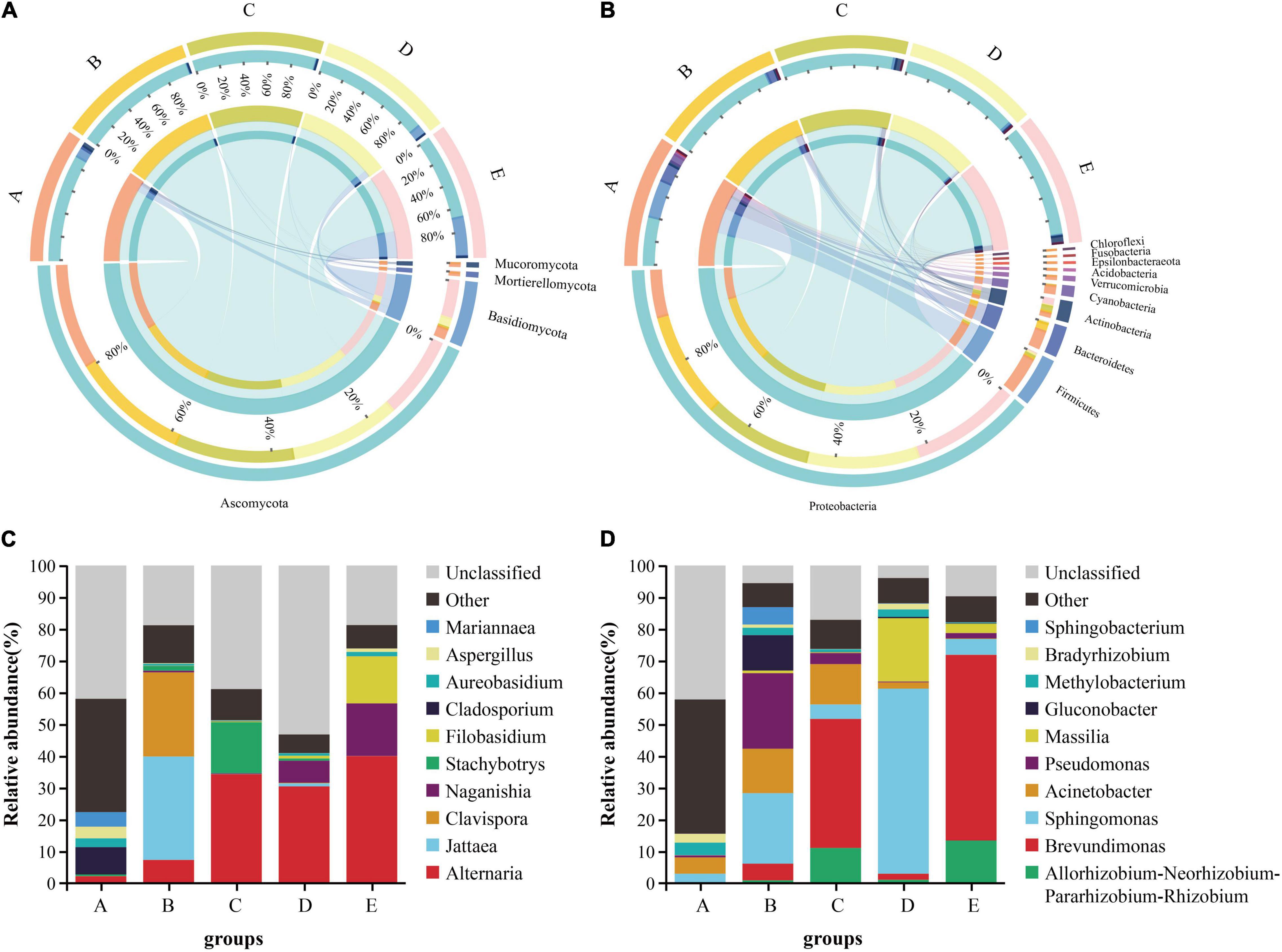
Figure 2. The differences in the microbial communities of Cabernet Sauvignon grapes at different growth stages. The difference between fungi (A) and bacteria (B) communities at the phylum level (relative abundance > 0.01%). The relative abundance of fungi (C) and bacteria (D) communities at the genus level (Shown top10). A, fruit setting; B, early veraison; C, end veraison; D, mid maturity; E, harvest.
Microbial diversity and similarity of grape epidermis changed with the growth stage. According to the measurement of the Shannon index, the diversity of fungal and bacterial communities in this study tended to decline over time. The Shannon index integrates the richness and evenness of the community. The fungal community declined significantly from the A stage to the B stage, and then the decline was not significant (Figure 3A). The bacterial community also decreased significantly from the A stage to the B stage, and did not change significantly during the veraison process. When it came to maturity, it first declined and then increased (Figure 3A). Interestingly, the diversity of fungal and bacterial communities changed significantly at the B stage.
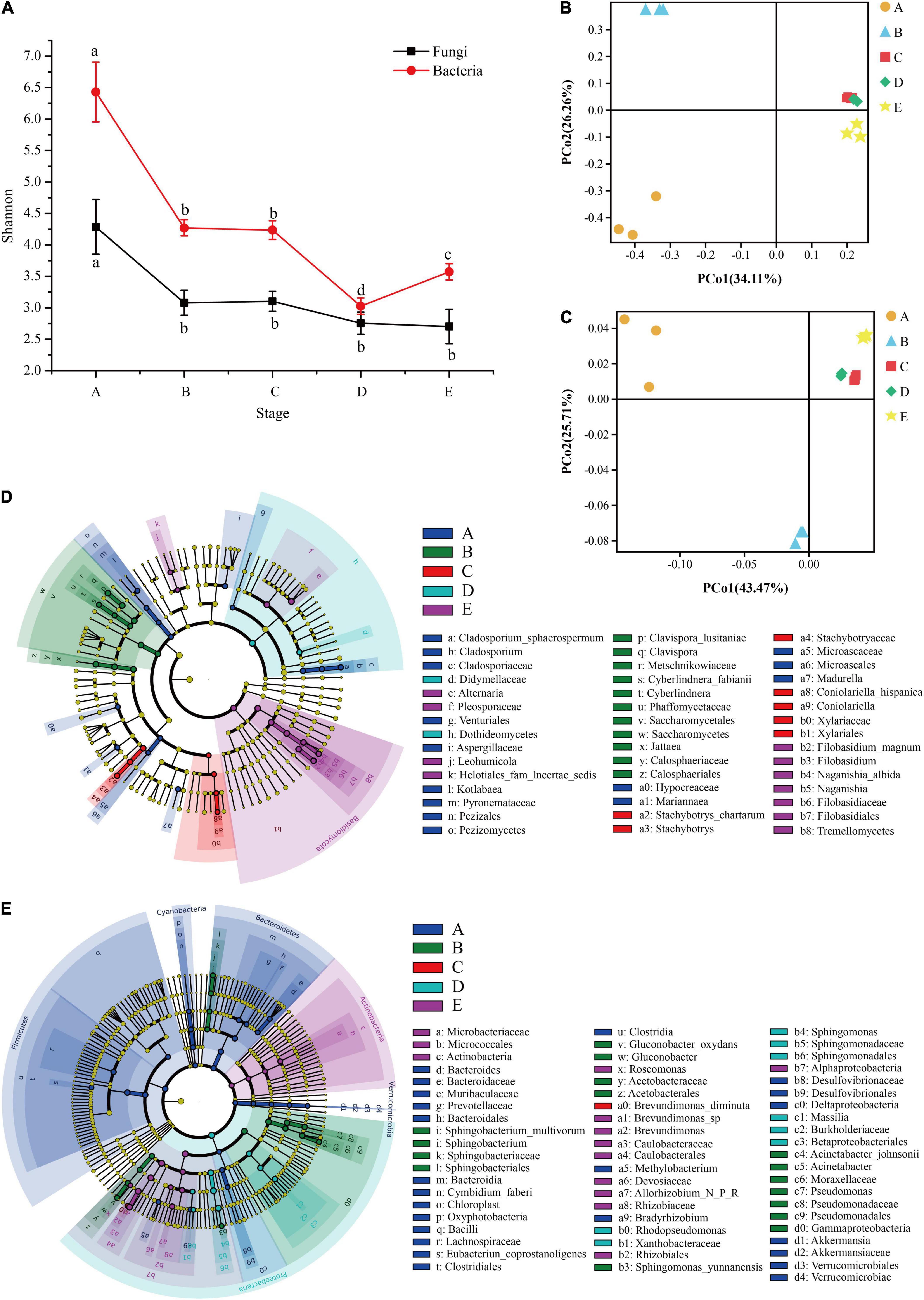
Figure 3. The microbial diversity of the epidermis changes during grape development. The line graph (A) shows the changing trend of fungal and bacterial α-diversity (Shannon index) during grape growth. PCoA plots based on the fungal Bray-Curtis distance (B) and the bacterial weighted UniFrac distance (C) of samples at different stages. LEfSe analysis revealed fungal (D) and bacterial (E) taxa with significant differences at different stages. A, fruit setting; B, early veraison; C, end veraison; D, mid maturity; E, harvest.
In addition, it was found that there were significant differences in the composition of fungal and bacterial communities over time (Supplementary Table 1, β-diversity, PERMANOVA; Bray-Curtis, R2 = 0.7036, and P = 0.001; Bacterial weighted UniFrac, R2 = 0.8588, and P = 0.001). PCoA showed that the samples at different stages were clearly separated, and the first two principal axes (PC) of the fungal and bacterial communities explained 60.37 and 69.18% of the total variation, respectively (Figures 3B,C). LEfSe analysis further confirmed that the pattern was related to the significant association between the species taxa and sample (Kruskal–Wallis rank-test, α < 0.05). LEfSe showed that there were 46 fungi taxa with significant differences. Among them, there were 14 taxa for the A stage (e.g., Cladosporium, Aspergillaceae, and Microascales), 11 for the B stage (e.g., Clavispora, Saccharomycetes, and Calosphaeriales), 7 for the C stage (e.g., Stachybotrys, Coniolariella, and Xylariaceae), 2 for the D stage (e.g., Didymellaceae and Dothideomycetes), and 12 for the E stage (e.g., Alternaria, Filobasidium, and Filobasidiaceae; Figure 3D). For bacteria, LEfSe showed that 67 taxa had significant differences. Among them, there were 27 taxa for the A stage (e.g., Bacteroides, Bacilli, and Lachnospiraceae), 16 for the B stage (e.g., Sphingobacterium, Pseudomonas, and Gammaproteobacteria), 1 for the C stage (e.g., Brevundimonas diminuta), 9 for the D stage (e.g., Rhodopseudomonas, Sphingomonas, and Massilia), and 14 for the E stage (e.g., Microbacteriaceae, Actinobacteria, and Brevundimonas; Figure 3E). These results indicated that there were significant differences in grape epidermis microbes at different developmental stages.
Dynamics of Core Microbiome During Grape Development
Here, we prioritized the core microbiome to further investigate the fungal and bacterial community succession of grape epidermis. At the genus level, the most important taxa were first identified according to the abundance-occupancy distribution, as the core microorganisms of the grape epidermis. The relative abundance of the fungal core taxa reached 58.58 ± 2.39%, including some filamentous fungi (Aspergillus, Rhizopus, Alternaria, and Penicillium) and yeasts (Clavispora, Candida, and Cyberlindnera; Supplementary Table 2). The relative abundance of the bacterial core taxa reached 71.73 ± 3.08%, including the common lactic acid bacteria (Leuconostoc and Lactobacillus) and spoilage bacteria (Gluconobacter) in wine (Supplementary Table 2).
Tracing the core taxa revealed significant temporal dynamics and succession associated with berry development. Overall, veraison appeared to be a key stage where the core taxa were different from other stages. For example, the fungal core taxa, Clavispora and Jattaea only existed with higher abundance at the B stage, while Alternaria, Filobasidium, and Naganishia gradually accumulated abundance from the C stage (Figure 4A). It was noteworthy that a few fungal core taxa only appeared at specific developmental stages. Cyberlindnera and Comoclathris could be detected from the B stage. Rosellinia, Jattaea, and Coniolariella were detected only in the middle stage of berry development (Supplementary Table 2). For bacterial core taxa, the dominant genus Brevundimonas began to accumulate with higher abundance from the B stage. Acinetobacter existed with high abundance before the E stage, especially veraison (Figure 4B). Similarly, a few core taxa appeared only at specific development stages. Azorhizobium could only be detected from the B stage. Caedibacter only existed with low abundance in the middle stage of berry development (Supplementary Table 2).
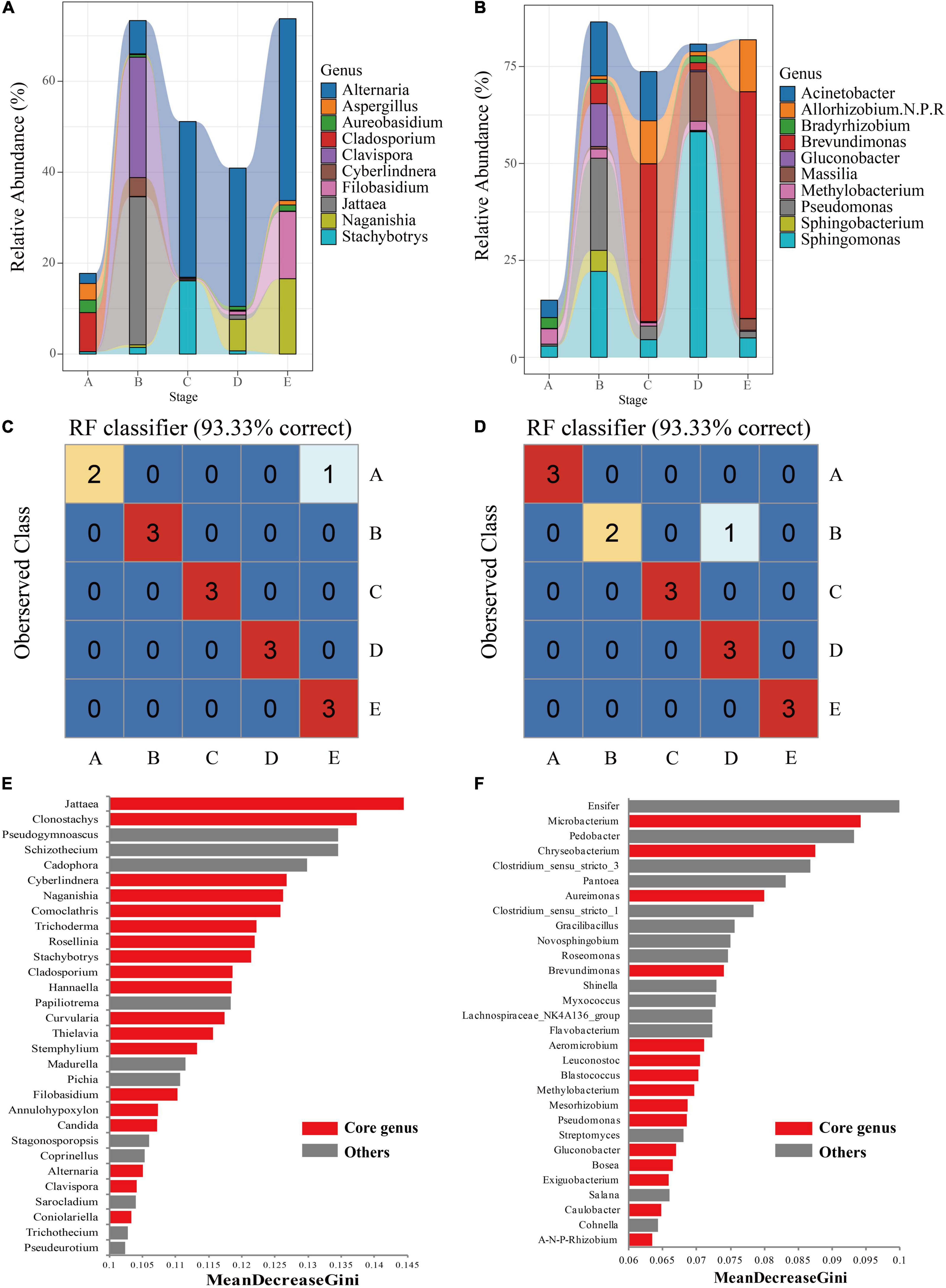
Figure 4. The core microbiome of grape epidermis at different developmental stages. The relative abundance of the core taxa of fungi (A) and bacteria (B) changed with development (relative abundance top 10 shown). The results of the Random Forest model showed that the microbiota of fungi (C) and bacteria (D) could distinguish the developmental stages of grapes. Important taxa of fungi (E) and bacteria (F) are shown based on the MeanDecreaseGini value. A, fruit setting; B, early veraison; C, end veraison; D, mid maturity; E, harvest.
To verify the robustness of these observations in the berry growth cycle, random forest supervised learning models were used to classify the samples and determine which taxa explain the strongest variations in the grape development stage. Fungi and bacterial communities had a high discriminative power, with a resolution of 93.33% for different stages of the samples (Figures 4B,C). Among them, fungi and bacteria made misjudgments (class error = 33.33%) on the samples of the A stage and the B stage, respectively. The important features identified at different development stages of berries included not only core taxa but also non-core taxa. For fungal communities, filamentous fungi (e.g., Jattaea, Clonostachys, and Trichoderma) and fermenting yeasts (e.g., Cyberlindnera) within the core were top features in the classification models (Figure 4E). Some non-core taxa were also important for the models, such as Pseudogymnoascus and Pichia, which had relatively high abundance at the B stage compared with other stages, which was the key taxa distinguishing the B stage (Supplementary Table 3). Madurella could only be detected at the early stages of berry development, which was also an important feature of the classification model (Supplementary Table 3). For bacterial communities, Microbacterium, Chryseobacterium, and Aureimonas of the core taxa were top features (Figure 4F). Compared with other stages, non-core taxa Ensifer had the highest abundance at the C stage, which was the key taxa distinguishing the C stage; Pedobacter also had the highest abundance at the E stage (Supplementary Table 3). When the core taxa of fungi and bacteria were used to build a random forest classification model, all samples from different stages could be correctly identified (Supplementary Figure 2), indicating that the developmental stages of grapes could be distinguished based on the core taxa.
Co-occurrence Analysis of the Relationship Between Microorganisms During Grape Development
Microbial co-occurrence network analysis is a common tool to study the microbial community structure and the inside interactions. Figure 5 shows different topological structures of fungi and bacteria. The network of bacteria (network density = 0.253) was denser than that of fungi (network density = 0.202). For fungi taxa, Mariannaea had the highest connectivity (degree = 13). For example, Mariannaea was positively correlated with Malassezia, Madurella, Kotlabaea, Aspergillus, Cladosporium, Penicillium, Myceliophthora, and Infundichalara, and negatively correlated with Alternaria, Comoclathris, Curvularia, Naganishia, and Filobasidium (Figure 5). Trichoderma and Candida had the lowest connectivity (degree = 1). For example, Trichoderma was only negatively correlated with Alternaria, and Candida was only positively correlated with Cyberlindnera (Figure 5). For bacterial taxa, Bilophila and Negativibacillus had the highest connectivity (degree = 15), and there was a positive correlation between them. For example, Bilophila was positively correlated with Ruminiclostridium, Lactobacillus, Eubacterium, Bacteroides, Alloprevotella, and Akkermansia, and negatively correlated with Sphingobacterium, Roseomonas, Microbacterium, Mesorhizobium, Massilia, Gluconobacter, Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium, and Brevundimonas; Negativibacillus was positively correlated with Ruminiclostridium, Eubacterium, Bacteroides, Alloprevotella, Akkermansia, and Lactobacillus, and negatively correlated with Sphingobacterium, Microbacterium, Mesorhizobium, Massilia, Gluconobacter, Brevundimonas, Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium, and Roseomonas (Figure 5). Pseudomonas, Acinetobacter, Myxococcus, and Cupriavidus had the lowest connectivity (degree = 2). For example, Pseudomonas was positively correlated with Sphingobacterium and Sphingobacterium; Acinetobacter was positively correlated with Pseudomonas and Chryseobacterium; Myxococcus was positively correlated with Brevundimonas and Allorhizobium-Neorhizobium-Pararhizobium-Rhizobium; Cupriavidus was positively correlated with Brevundimonas and negatively correlated with Akkermansia.
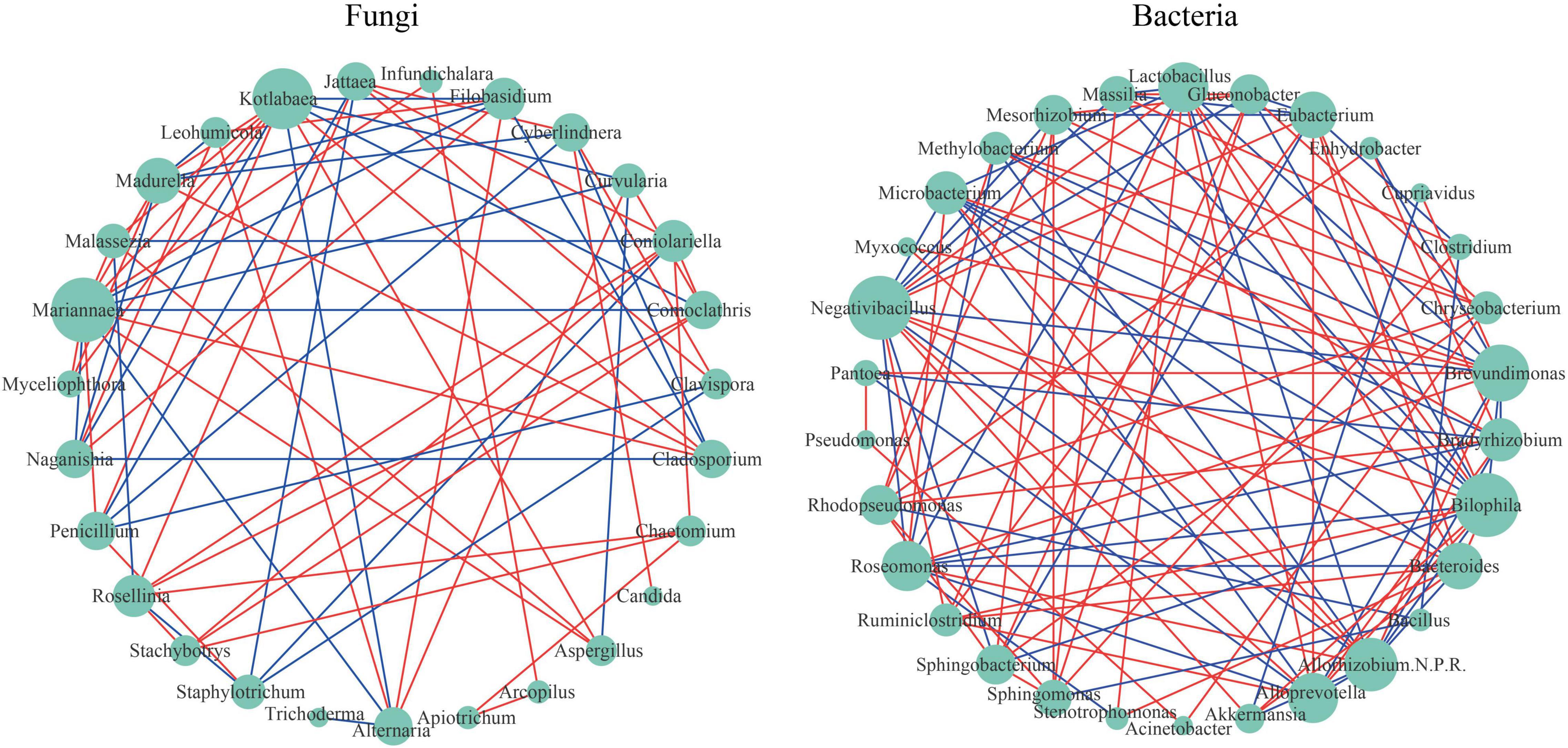
Figure 5. Network visualization shows co-occurrence and exclusion correlations among fungi and among bacteria at the genus level. The green circular nodes represent microbial taxa. Direct connections between nodes indicated strong correlations (Spearman correlation coefficient, r ≥ 0.6; p < 0.05). The color of the edges describes the positive correlation (red) or the negative correlation (blue). The size of the node is directly proportional to the degree of interconnection.
Effect of Vineyard Weather on Microbial Community of Grape Epidermis
The microbial community of the grape epidermis was closely associated with vineyard weather. During the grape growth cycle, all weather parameters displayed significant differences between the phenological stages (Supplementary Table 4). The phase-dependent pattern of the grape epidermal microbial community also indicated that longitudinal environmental conditions were likely responsible for structuring the microbial communities. CCA can be used to explore the influence of vineyard weather on the variation of grape epidermal microbial communities. Figures 6A,B show that vineyard weather had less effect on fungal and bacterial diversity of the A stage. The fungal diversity of the B stage was mainly affected by average high temperature and average low temperature; while the maturation process (C–E stage) was mainly affected by relative humidity and precipitation (Figure 6A). The bacterial diversity of the B and D stages was mainly affected by precipitation; the C and E stages were mainly affected by relative humidity (Figure 6B).
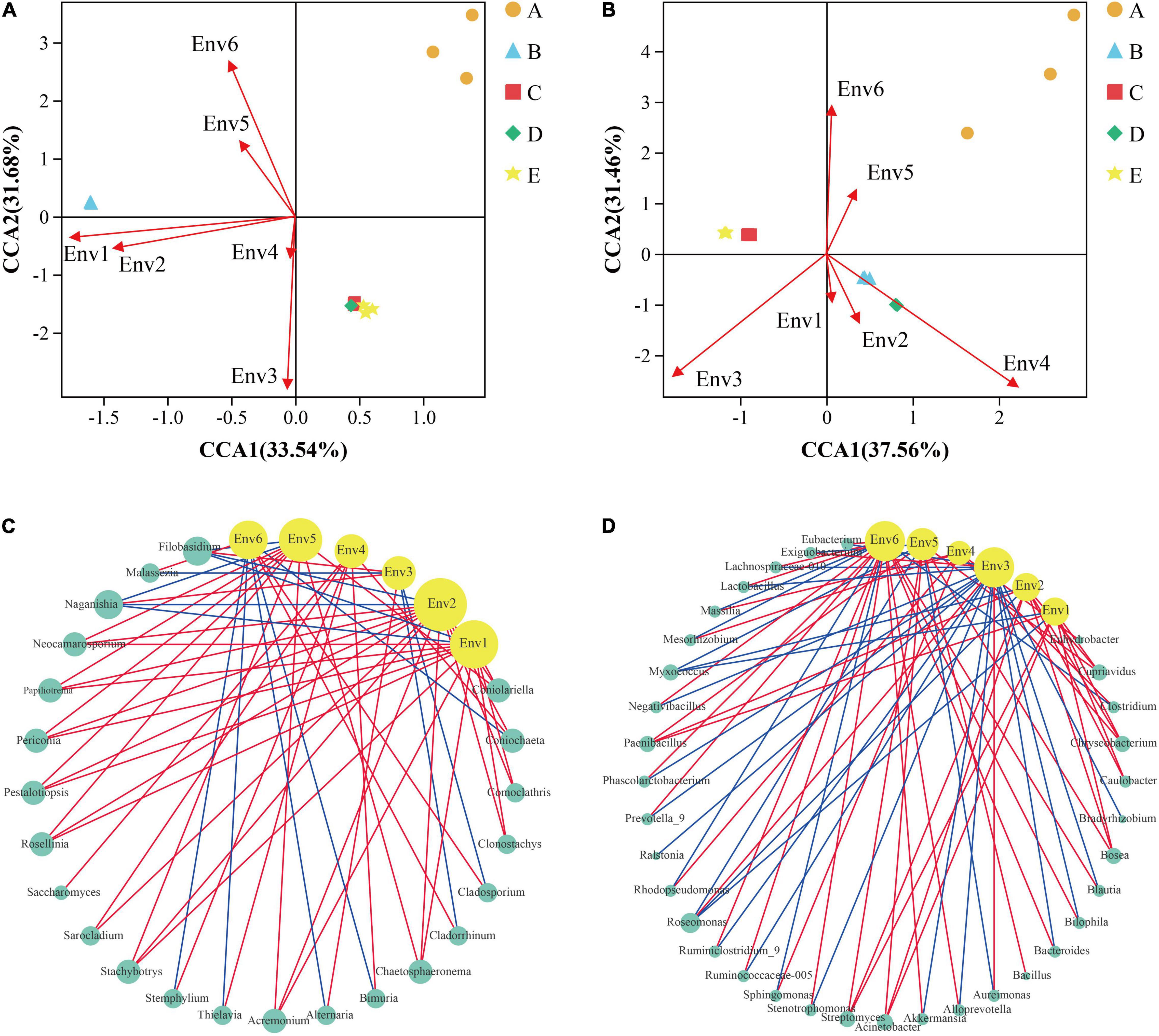
Figure 6. Statistically correlation among grape microorganisms and vineyard weather was observed during grape development. CCA reflects the relationship between meteorological parameters and the diversity of fungi (A) and bacteria (B) during grape development. Network analysis reflected the co-occurrence relationship between the fungi (C) and bacteria (D) taxa and meteorological parameters. The yellow circular nodes represent meteorological parameters. The green circular nodes represent microbial taxa. Direct connections between nodes indicated strong correlations (Spearman correlation coefficient, r ≥ 0.5; p < 0.05). The color of the edges represents positive correlation (red) or negative correlation (blue). The size of the node is directly proportional to the degree of interconnection. Env1, average high temperature; Env2, average low temperature; Env3, relative humidity; Env4, precipitation; Env5, average ground temperature; Env6, sunshine time. A, fruit setting; B, early veraison; C, end veraison; D, mid maturity; E, harvest.
The microbial community of grape epidermis was closely associated with each meteorological parameter. Network analysis was conducted to explore the co-occurrence patterns between grape epidermis microorganisms and meteorological parameters, based on the strong correlation coefficients (Spearman correlation coefficient, r ≥ 0.5; p < 0.05). The connected module between bacteria (average degree = 12.2, Figure 6D) and meteorological parameters was observed more closely than fungi (average degree = 9.8, Figure 6C). For the fungal taxa, the average high temperature, the average low temperature, and the average ground temperature had a higher connection density with the fungi taxa (degree > 10). For example, the average high temperature was positively correlated with Stachybotrys and negatively correlated with Naganishia; the average low temperature was positively correlated with Coniolariella and negatively correlated with Flobasidium; the average ground temperature was positively correlated with Rosellinia and negatively correlated with Naganishia (Figure 6C). For bacterial taxa, the relative humidity, the average ground temperature, and sunshine hours had a higher connection density with the bacterial taxa (degree > 10). For example, relative humidity was positively correlated with Aureimonas and negatively correlated with Lactobacillus; average ground temperature was positively correlated with Acinetobacter and negatively correlated with Roseomonas; sunshine hours were positively correlated with Stenotrophomonas and negatively correlated with Sphingomonas (Figure 6D).
Discussion
Microbial Diversity in Grape Epidermis
The grape surface is conducive to the growth of different types of microorganisms, so it is very important to understand the diversity and community functions of grape microflora. In this study, a total of 5 fungal phyla were detected, of which Ascomycota (88.89%) and Basidiomycota (9.71%) were the main ones, followed by Mortierellomycota (0.52%), Mucoromycota (0.47%), and Rozellomycota (<0.01%). It has been reported in the literature that Ascomycota and Basidiomycota were the dominant groups of grape epidermal microorganisms. Gao et al. (2019) studied the microbial diversity of wine grape epidermis in different regions of Xinjiang, and detected 3 fungal phyla, mainly Ascomycota and Basidiomycota, followed by Zygomycota. Compared to those at the phylum level, our study detected more fungal groups. Fungi were the main causes of common grape diseases, such as Phylloxera (Daktulosphaera vitifoliae), Down mildews (Plasmopara viticola), Powder mildews (Erysiphe Necator), Grey mold (Botryotinia fuckelina), and Anthracnose (Colletotrichum gloeosporides). In this study, Colletotrichum gloeosporoides and Erysiphe Necator (relative abundance < 0.01%) were detected at the B and E stages, respectively, but no disease was observed in vineyards. In addition, many types of yeasts were detected, such as Hanseniapora, Candida, Pichia, Saccharomyces, Rhodotorula, and Wickerhamomyces. For bacteria, a total of 27 phyla were detected, mainly Proteobacteria (82.31%), Firmicutes (7.38%), Bacteroidetes (4.56%), Actinobacteria (2.99%), and Cyanobacteria (1.40%). Portillo et al. (2016) studied the bacterial diversity of Grenache and Carignan epidermal bacteria in Priorat area; only 14 phyla were detected, but the main bacteria were Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. Bacteria are also the main cause of common grape diseases, such as Pierce’s Disease (Xylella fastidiosa). The bacterial pathogens in grapes were not detected in this study. In addition, many types of lactic acid bacteria, such as Lactobacillus, Leuconostoc, Pediococcus, and Weissella, were detected. Although our data revealed the complex and abundant grape microbial structure, there are still hidden microbial species that cannot be exposed, as confirmed by the rarefaction curve (Figure 1).
Biological control is becoming the main method of current plant disease management. In this study, the most potential fungal antagonist Trichoderma was detected, and Pichia, Aspergillus, and Penicillium are being studied. Alternaria, as the dominant genus in this study, was also found among the dominant members of the grapevine community in different regions and varieties (Gavin et al., 2017; Knapp et al., 2021). Many of its species are considered to be cosmopolitan saprobes, endophytes, pathogens, or the causal agents of postharvest rots of numerous agronomic plants (Armitage et al., 2015). In the analysis of the interaction between microorganisms, it was found that Trichoderma and Alternaria were negatively correlated during grape development (Figure 5). This phenomenon was supported by the test results of Bhattacharya and Chakraborty (2020). In addition, bacteria also have a biological control effect. According to previous studies, the most well-known and reported bacterial antagonists are species of Pseudomonas, Burkholderia, Bacillus, Serratia, and Pantoea (Trotel-Aziz et al., 2008; Bulgari et al., 2011). For example, Sawant et al. (2016) isolated multiple Bacillus species from vineyards that effectively inhibited the enrichment of Colletortrichum gloeosporiodes, Erysiphe necator, and Plasmopara viticola. This also explained why anthracnose and powdery mildew disease phenomena were not observed in this study. Our data contribute to the characterization of the grape biodiversity and to the analysis of biomarkers with the potential to unveil the vine health status. This will have a significant influence on the vine performance and also on the wine quality.
Grape Epidermal Microbes in Response to the Development Stages of Berries
Microbiome succession is associated with plant development. Studies have shown that phyllosphere and rhizospheric fungal and bacterial communities of a wide range of plants, such as lettuce, alfalfa, switchgrass, and miscanthus, change according to plant development stages (Mougel et al., 2006; Rastogi et al., 2012; Grady et al., 2019). The nutrient composition of grapes is one of the important factors determining microorganisms on the grape surface (Ranade et al., 2021). In the early stages of development, the metabolism of berries is active, the acid content increases rapidly, and a small amount of sugar (not more than 10–20 g/L) appears. The acidic conditions in the early stage of grape growth are favorable for the colonization of most bacteria and a few fungi (Setati et al., 2012). In this study, the bacterial diversity of the A stage was significantly higher than that of fungi (Figure 3A). Veraison marks the beginning of grape ripening, which is a critical time point for grape metabolism and growth. Besides color changes caused by the production and accumulation of anthocyanins, the berry softens with the degradation of pectin and cellulose, the decrease of acidity, and the accumulation of sugar (Fasoli et al., 2016). All these factors create a more favorable environment for the colonization of grape epidermal microorganisms. In this study, veraison was the key stage of the change of epidermal microbial diversity. Compared with the A stage, the total relative abundance of fungal and bacterial core communities at the B stage was significantly higher. For example, the total relative abundance of the fungal core community was 30.76 ± 5.54 and 79.74 ± 2.63% at the A and B stages, respectively, and the bacterial core community was 19.11 ± 3.93 and 91.50 ± 1.08%, respectively (Supplementary Table 2). Similarly, the core members had undergone significant changes in Veraison. For example, the relative abundance of Alternaria at the A and C stages was 2.21 and 34.24%, and the relative abundance of Brevundimonas at the A and C stages was 0.05 and 40.64% (Supplementary Table 2). In addition, some taxa existed only at specific stages, such as Mariannaea and Negativibacillus only at the A stage. Studies have shown that plants can recruit microbes to participate in the key physiological processes and drive microbial assemblages to respond to biotic or abiotic stresses and improve environmental fitness (Hu et al., 2018). Our data may indicate how plant-driven microbes respond to environmental changes.
Grape Epidermal Microbes in Response to Vineyard Weather
Alongside the influences of changing metabolism and physiology during plant development, the grape epidermal microbiome is also subject to dynamic ecological conditions (Liu et al., 2019; Ranade et al., 2021). The microbial biogeography of grape berries is affected by climate, and populations are associated with specific climatic conditions, indicating a link between vineyard environmental conditions and microbial communities (Bokulich et al., 2014). In this study, water status (relative humidity and precipitation) had the greatest impact on fungal and bacterial diversity throughout the berry phenology period (Figure 6). For example, higher solar radiation and temperature, decreased precipitation, increased evaporation and transpiration, and lower relative soil moisture were observed from the A stage to the B stage in the vineyard, and this water stress event coincided with dramatic changes in the microbiome during the period discussed above (Figure 3A). Water status in the vineyard can condition the microbial populations of grape and must be at regional scales. Studies have shown that precipitation and humidity correlate with the presence of filamentous fungi (e.g., Penicillium) and yeasts (e.g., Saccharomyces, Hanseniaspora, and Metschnikowia; Bokulich et al., 2014; Jara et al., 2016). In this study, relative humidity was positively correlated with Alternaria and Aureimonas, and negatively correlated with Malassezia and Lactobacillus; precipitation was positively correlated with Saccharomyces and Sphingomonas (Figures 6C,D). In addition, water stress can trigger a large number of physiological reactions in grapevines, and disturb the water supply relationship of grapevines, the accumulation of grape chemical components, and the synthesis of secondary metabolites (Anjum et al., 2011; Hochberg et al., 2013), which may also affect the colonization of grape epidermis-related species, but further research is needed.
In summary, the microbial diversity of the Cabernet Sauvignon epidermis in the Wuhai region of China showed a downward trend during the development of berries. This diversity was mainly associated with berry development and the weather in the vineyard. Grapes recruited microbes according to their developmental stages, but retained the core species. In conclusion, this study provides in-depth information regarding the differences in the microbial communities on the surface of grapes during the developmental stages. Future research should further investigate the changes and functions of microbial communities in other habitats of grapevines (root zone soil, root system, leaves, and flowers) in order to increase our understanding of vineyard microbial ecology. This study will help improve vineyard management techniques, such as the use of biological control strategies to reduce pesticide costs, in order to maintain and produce healthy, high-quality grapes, allowing the expression of the regional character of the wine.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors. The data presented in the study are deposited in the NCBI repository, accession number: PRJNA766154.
Author Contributions
RW and NC collected wine grape samples, compiled the figures and table, and wrote the manuscript. YD and LW conceived the framework of the manuscript. YL, FG, and LZ conducted the bioinformatic analysis of data and provided advice and constructive critiques. HL and HW supervised the research activities. All authors reviewed the manuscript.
Funding
This study was supported by the National Key Research and Development Project (2019YFD1002500), the Ningxia Hui Autonomous Region Key Research and Development Project (2020BCF01003), and the Key Research and Development Project of Shaanxi Province (2020ZDLNY07-08).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Jianqing Huang for giving us access to the Sunshine Tianyu International Winery vineyards in this study and assisting with the sampling.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.935647/full#supplementary-material
References
Anjum, S. A., Xie, X. -Y., Wang, L. -C., Saleem, M. F., Man, C., and Lei, W. (2011). Morphological, physiological and biochemical responses of plants to drought stress. African J. Agric. Res. 6, 2026–2032. doi: 10.5897/AJAR10.027
Armitage, A. D., Barbara, D. J., Harrison, R. J., Lane, C. R., Sreenivasaprasad, S., Woodhall, J. W., et al. (2015). Discrete lineages within Alternaria alternata species group: identification using new highly variable loci and support from morphological characters. Fungal Biol. 119, 994–1006. doi: 10.1016/j.funbio.2015.06.012
Barata, A., Malfeito-Ferreira, M., and Loureiro, V. (2012). The microbial ecology of wine grape berries. Int. J. Food Microbiol. 153, 243–259. doi: 10.1016/j.ijfoodmicro.2011.11.025
Bhattacharya, D., and Chakraborty, M. (2020). Evaluation of Trichoderma species against Alternaria for management of blight disease of vigna unguiculata. Plant Arch. 20, 2018–2022.
Bokulich, N. A., Collins, T., Masarweh, C., Allen, G., Heymann, H., Ebeler, S. E., et al. (2016). Associations among wine grape microbiome, metabolome, and fermentation behavior suggest microbial contribution to regional wine characteristics. mBio 7:e00631-16. doi: 10.1128/mBio.00631-616
Bokulich, N. A., Thorngate, J. H., Richardson, P. M., and Mills, D. A. (2014). Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. U S A. 111, E139–E148. doi: 10.1073/pnas.1317377110
Bulgari, D., Casati, P., Crepaldi, P., Daffonchio, D., Quaglino, F., Brusetti, L., et al. (2011). Restructuring of endophytic bacterial communities in grapevine yellows-diseased and recovered Vitis vinifera L. plants. Appl. Environ. Microbiol. 77, 5018–5022. doi: 10.1128/AEM.00051-11
Carpena, M., Pereira, A. G., Prieto, M. A., and Simal-Gandara, J. (2020). Wine aging technology: fundamental role of wood barrels. Foods 9:1160. doi: 10.3390/foods9091160
Comitini, F., Capece, A., Ciani, M., and Romano, P. (2017). New insights on the use of wine yeasts. Curr. Opin. Food Sci. 13, 44–49. doi: 10.1016/j.cofs.2017.02.005
Compant, S., Samad, A., Faist, H., and Sessitsch, A. (2019). A review on the plant microbiome: ecology, functions, and emerging trends in microbial application. J. Adv. Res. 19, 29–37. doi: 10.1016/j.jare.2019.03.004
Echave, J., Barral, M., Fraga-Corral, M., Prieto, M. A., and Simal-Gandara, J. (2021). Bottle aging and storage of wines: a review. Molecules 26:713.
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fasoli, M., Dell’Anna, R., Dal Santo, S., Balestrini, R., Sanson, A., Pezzotti, M., et al. (2016). Pectins, hemicelluloses and celluloses show specific dynamics in the internal and external surfaces of grape berry skin during ripening. Plant Cell Physiol. 57, 1332–1349. doi: 10.1093/pcp/pcw080
Filippis, F. D., Storia, A. L., and Blaiotta, G. (2017). Monitoring the mycobiota during Greco di Tufo and Aglianico wine fermentation by 18S rRNA gene sequencing. Food Microbiol. 63, 117–122. doi: 10.1016/j.fm.2016.11.010
Gao, F., Chen, J., Xiao, J., Cheng, W., Zheng, X., Wang, B., et al. (2019). Microbial community composition on grape surface controlled by geographical factors of different wine regions in Xinjiang, China. Food Res. Int. 122, 348–360. doi: 10.1016/j.foodres.2019.04.029
Gavin, K., Mayerhofer, M., and Griffin, A. (2017). Fungal endophytes of wild and hybrid Vitis leaves and their potential for vineyard biocontrol. Can. J. Microbiol. 63, 583–595. doi: 10.1139/cjm-2016-0740
Grady, K. L., Sorensen, J. W., Stopnisek, N., Guittar, J., and Shade, A. (2019). Assembly and seasonality of core phyllosphere microbiota on perennial biofuel crops. Nat. Commun. 10:4135. doi: 10.1038/s41467-019-11974-11974
Grangeteau, C., Roullier-Gall, C., Rousseaux, S., Gougeon, R. D., Schmitt-Kopplin, P., Alexandre, H., et al. (2017). Wine microbiology is driven by vineyard and winery anthropogenic factors. Microb. Biotechnol. 10, 354–370. doi: 10.1111/1751-7915.12428
Hochberg, U., Degu, A., Toubiana, D., Gendler, T., Nikoloski, Z., Rachmilevitch, S., et al. (2013). Metabolite profiling and network analysis reveal coordinated changes in grapevine water stress response. BMC Plant Biol. 13:184. doi: 10.1186/1471-2229-13-184
Hu, L., Robert, C. A. M., Cadot, S., Zhang, X., Ye, M., Li, B., et al. (2018). Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 9:2738. doi: 10.1038/s41467-018-05122-5127
Jara, C., Laurie, V. F., Mas, A., and Romero, J. (2016). Microbial terroir in chilean valleys: diversity of non-conventional yeast. Front. Microbiol. 7:663. doi: 10.3389/fmicb.2016.00663
Knapp, D. G., Lazar, A., Molnar, A., Vajna, B., Karacsony, Z., Vaczy, K. Z., et al. (2021). Above-ground parts of white grapevine Vitis vinifera cv. Furmint share core members of the fungal microbiome. Environ. Microbiol. Rep. 13, 509–520. doi: 10.1111/1758-2229.12950
Liang, H., Wang, X., Yan, J., and Luo, L. (2019). Characterizing the intra-vineyard variation of soil bacterial and fungal communities. Front. Microbiol. 10:1239. doi: 10.3389/fmicb.2019.01239
Likar, M., Stres, B., Rusjan, D., Potisek, M., and Regvar, M. (2017). Ecological and conventional viticulture gives rise to distinct fungal and bacterial microbial communities in vineyard soils. Appl. Soil Ecol. 113, 86–95. doi: 10.1016/j.apsoil.2017.02.007
Liu, D., and Howell, K. (2021). Community succession of the grapevine fungal microbiome in the annual growth cycle. Environ. Microbiol. 23, 1842–1857. doi: 10.1111/1462-2920.15172
Liu, D., Zhang, P., Chen, D., and Howell, K. (2019). From the vineyard to the winery: how microbial ecology drives regional distinctiveness of wine. Front. Microbiol. 10:2679. doi: 10.3389/fmicb.2019.02679
Liu, Y., Rousseaux, S., Tourdot-Maréchal, R., Sadoudi, M., Gougeon, R., Schmitt-Kopplin, P., et al. (2017). Wine microbiome: a dynamic world of microbial interactions. Crit. Rev. Food Sci. Nutr. 57, 856–873.
Martins, G., Vallance, J., Mercier, A., Albertin, W., Stamatopoulos, P., Rey, P., et al. (2014). Influence of the farming system on the epiphytic yeasts and yeast-like fungi colonizing grape berries during the ripening process. Int. J. Food Microbiol. 177, 21–28. doi: 10.1016/j.ijfoodmicro.2014.02.002
Morgan, H. H., Toit, M. D., and Setati, M. E. (2017). The grapevine and wine microbiome: insights from high-throughput amplicon sequencing. Front. Microbiol. 8:820. doi: 10.3389/fmicb.2017.00820
Mougel, C., Offre, P., Ranjard, L., Corberand, T., Gamalero, E., Robin, C., et al. (2006). Dynamic of the genetic structure of bacterial and fungal communities at different developmental stages of Medicago truncatula Gaertn. cv. Jemalong line J5. New Phytol. 170, 165–175. doi: 10.1111/j.1469-8137.2006.01650.x
Pascale, A., Vinale, F., Manganiello, G., Nigro, M., Lanzuise, S., Ruocco, M., et al. (2017). Trichoderma and its secondary metabolites improve yield and quality of grapes. Crop Protection 92, 176–181. doi: 10.1016/j.cropro.2016.11.010
Perazzolli, M., Antonielli, L., Storari, M., Puopolo, G., Pancher, M., Giovannini, O., et al. (2014). Resilience of the natural phyllosphere microbiota of the grapevine to chemical and biological pesticides. Appl. Environ. Microbiol. 80, 3585–3596. doi: 10.1128/AEM.00415-14
Pinto, C., Pinho, D., Cardoso, R., Custódio, V., Fernandes, J., Sousa, S., et al. (2015). Wine fermentation microbiome: a landscape from different Portuguese wine appellations. Front. Microbiol. 6:905. doi: 10.3389/fmicb.2015.00905
Pinto, C., Pinho, D., Sousa, S., Pinheiro, M., Egas, C., and Gomes, A. C. (2014). Unravelling the diversity of grapevine microbiome. PLoS One 9:e85622. doi: 10.1371/journal.pone.0085622
Portillo, M. D. C., Franquès, J., Araque, I., Reguant, C., and Bordons, A. (2016). Bacterial diversity of Grenache and Carignan grape surface from different vineyards at Priorat wine region (Catalonia, Spain). Int. J. Food Microbiol. 219, 56–63. doi: 10.1016/j.ijfoodmicro.2015.12.002
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ranade, Y., Sawant, I., Saha, S., Chandrashekar, M., and Pathak, P. (2021). Epiphytic microbial diversity of vitis vinifera fructosphere: present status and potential applications. Curr. Microbiol. 78, 1086–1098. doi: 10.1007/s00284-021-02385-2380
Rastogi, G., Sbodio, A., Tech, J. J., Suslow, T. V., Coaker, G. L., and Leveau, J. H. (2012). Leaf microbiota in an agroecosystem: spatiotemporal variation in bacterial community composition on field-grown lettuce. ISME J. 6, 1812–1822. doi: 10.1038/ismej.2012.32
Sawant, I. S., Wadkar, P. N., Rajguru, Y. R., Mhaske, N. H., Salunkhe, V. P., Sawant, S. D., et al. (2016). Biocontrol potential of two novel grapevine associated Bacillus strains for management of anthracnose disease caused by Colletotrichum gloeosporioides. Biocontrol Sci. Technol. 26, 964–979. doi: 10.1080/09583157.2016.1174770
Setati, M. E., Jacobson, D., Andong, U. -C., and Bauer, F. (2012). The vineyard yeast microbiome, a mixed model microbial map. PLoS One 7:e52609. doi: 10.1371/journal.pone.0052609
Trotel-Aziz, P., Couderchet, M., Biagianti, S., and Aziz, A. (2008). Characterization of new bacterial biocontrol agents Acinetobacter, Bacillus, Pantoea and Pseudomonas spp. mediating grapevine resistance against Botrytis cinerea. Environ. Exp. Bot 64, 21–32. doi: 10.1016/j.envexpbot.2007.12.009
Wang, C., García-Fernández, D., Mas, A., and Esteve-Zarzoso, B. (2015). Fungal diversity in grape must and wine fermentation assessed by massive sequencing, quantitative PCR and DGGE. Front. Microbiol. 6:1156. doi: 10.3389/fmicb.2015.01156
Wei, R. T., Chen, N., Ding, Y. T., Wang, L., Liu, Y. H., Gao, F. F., et al. (2022a). Correlations between microbiota with physicochemical properties and volatile compounds during the spontaneous fermentation of Cabernet Sauvignon (Vitis vinifera L.) wine. LWT 163:113529. doi: 10.1016/j.lwt.2022.113529
Wei, R. T., Ding, Y. T., Chen, N., Wang, L., Gao, F. F., Zhang, L., et al. (2022b). Diversity and dynamics of microbial communities during spontaneous fermentation of Cabernet Sauvignon (Vitis vinifera L.) from different regions of China and their relationship with the volatile components in the wine. Food Res. Int. 156:111372. doi: 10.1016/j.foodres.2022.111372
Wei, R. T., Ding, Y. T., Gao, F. F., Zhang, L., Wang, L., Li, H., et al. (2022c). Community succession of the grape epidermis microbes of cabernet sauvignon (Vitis vinifera L.) from different regions in China during fruit development. Int. J. Food Microbiol. 362:109475. doi: 10.1016/j.ijfoodmicro.2021.109475
Wei, R. T., Wang, L., Ding, Y. T., Zhang, L., Gao, F. T., Chen, N., et al. (2022d). Natural and sustainable wine: a review. Crit. Rev. Food Sci. Nutr. Online ahead of print doi: 10.1080/10408398.2022.2055528
Zarraonaindia, I., Owens, S. M., Weisenhorn, P., West, K., Hampton-Marcell, J., Lax, S., et al. (2015). The soil microbiome influences grapevine-associated microbiota. mBio 6, 2527–2514. doi: 10.1128/mBio.02527-2514.
Zhang, S., Chen, X., Zhong, Q., Huang, Z., and Bai, Z. (2017). Relations among epiphytic microbial communities from soil, leaves and grapes of the grapevine. Front. Life Sci. 10, 73–83. doi: 10.1080/21553769.2017.1365776
Keywords: high throughput sequencing, ecological viticulture, grape, microbial diversity, developmental stage, vineyard weather
Citation: Wei R-t, Chen N, Ding Y-t, Wang L, Gao F-f, Zhang L, Liu Y-h, Li H and Wang H (2022) Diversity and Dynamics of Epidermal Microbes During Grape Development of Cabernet Sauvignon (Vitis vinifera L.) in the Ecological Viticulture Model in Wuhai, China. Front. Microbiol. 13:935647. doi: 10.3389/fmicb.2022.935647
Received: 04 May 2022; Accepted: 24 May 2022;
Published: 30 June 2022.
Edited by:
Yu Ding, Jinan University, ChinaReviewed by:
Tingting Liang, Guangdong Institute of Microbiology, Guangdong Academy of Science, ChinaMaria Dimopoulou, Agricultural University of Athens, Greece
Copyright © 2022 Wei, Chen, Ding, Wang, Gao, Zhang, Liu, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Li, bGlodWF3aW5lQG53YWZ1LmVkdS5jbg==; Hua Wang, d2FuZ2h1YUBud2FmdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship